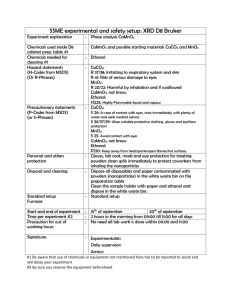
Gene Upstream Primer Downstream Primer Size (bp)
advertisement
")
Supplementary Materials and Methods Materials Antibodies: rabbit polyclonal anti ADH1 (sc-22750,Santa Cruz Biotechnology, California, USA); rabbit polyclonal phospho-Smad2 (Ser465/467) and phosphoSmad1/3 (9514, Cell Signaling Technology, Frankfurt, Germany); rabbit polyclonal Smad2 and Smad3 (51-1300 and 51-1500, Zymed Laboratory, Berlin, Germany); rabbit polyclonal Smad7 (24477, Abcam Ltd., Cambridge, UK); mouse monoclonal actin (A5441, Sigma-Aldrich Missouri, USA). siRNA for ADH1 was from Qiagen (Hilden, Germany). Chemicals: Human recombinant TGF-1, Peprotech (London, UK); β-Nicotinamide adenin dinucleotide hydrate from yeast, Oil red and 7-Methoxy4(trifluoromethyl)coumarin substrate (7-MFC), Williams medium E, SB431542, Insulin, Dexamethasone, Triton-X100 and acetylsalicylic acid, Sigma (Seelze, Germany ); Protease inhibitor cocktail, phosphatase inhibitor cocktail and rat tail collagen I, Roche (Mannheim, Germany); Penicillin/Streptomycin, L-glutamin, Cambrex (Taufkirchen, Germany); CellTrackerTM-Carboximethylfluorescein diacetate (CMFDA), Molecular Probes (Karlsruhe, Germany); Foetal Bovine Serum and Lipofectamin2000, Invitrogen (Karlsruhe, Germany); ethanol absolute, J.T. Baker (City, Holland) and 4-Methylpyrazol from Fluka, (Steinheim am Albuch, Germany). Cell preparation and treatment Mouse hepatocytes from male C57/BL-6 mice (100-150g) isolated by collagenase perfusion were plated on collagen coated 6-well-plates at a density of 5 x105 cells/well in Williams' medium E supplemented with 10 % FBS, 2 mmol L-glutamine, 1 % penicillin/streptomycin and 100 nmol dexamethasone. After 4 hrs of incubation in 5 % CO2 at 37 oC to facilitate attachment, medium was replaced with serum-free Williams' medium E supplemented with 1 % penicillin/streptomycin, 2 mmol Lglutamine and 100 nmol dexamethasone. The next day, medium was changed to serum-free Williams' medium E supplemented with 2 mMol L-glutamine and 1% penicillin/streptomycin [1]. Preparation of cell lysates and immunoblotting Cell lysates were prepared using RIPA buffer (1X Tris-buffered saline, 1 % Nonidet P-40, 0.5 % sodium deoxycholate, 0.1 % SDS). Protease inhibitor Mix and Phosphatase Inhibitor Cocktail II (Roche, Mannheim, Germany) were included before use. Protein concentration was determined with the DC Protein Assay (Biorad, München, Germany). Western blot analyses were performed as described.[5] Individual protein bands were quantified by densitometry using AIDA analysis software and normalized for -actin. All expression data were confirmed in three independent experiments. Antibodies are listed in Suppl.Table2. Isolation of Primary Human Hepatocytes. Human isolated primary hepatocyteswere obtained according to the institutional guidelines from liver resections of patients with primary or secondary liver tumors. Adenovirus infections and transient plasmid transfections Recombinant adenoviruses expressing constitutively active (ca) mutants for TGF-β type I receptors (Ad-caALK1 and Ad-caALK5), Smad7 (Ad-Smad7), or encoding ALK1 siRNA ) were prepared and used as previously described [5]. Briefly, mouse hepatocytes were infected (day 0) with 50-100 ifu/cell (infectious units ) adenovirus for 1 hr in William’s medium E containing 5 % FBS. Then, medium was changed to 2 serum free William’s medium E. After 36-48 hrs, target gene expression was investigated by Westernblot. Infectivity (ifu, infectious units) was determined using the Rapid Titre Kit from BD Bioscience and 50-100 ifu/cell were used. Generally, more than 90 % of hepatocytes were infected. Hepatocytes were transfected with 1µg ADH1 expression plasmids and empty vector pcDNA3.1 as a control (purchased from Qiagen). Briefly, cells were seeded at a density of 1x105/cm2 in 24 well plates. Expression plasmids were transfected with Lipofectamine™ 2000 (Cat. No. 11668-027 Invitrogen, Karlsruhe, Germany) according to the manufacturer’s instructions. 24 hrs after transfection, medium was changed and TGF- and or ethanol stimulations proceeded conform with the standard operating procedure. ADH1 protein overexpression was examined by Western blot analysis 48 hrs later. Ethanol metabolism Hepatocytes were transfected with ADH1 overexpression plasmids and control vectors, or ADH1 siRNAs and the corresponding scrambled siRNAs, followed by ethanol stimulation for 12 hrs. Residual ethanol in the medium was measured with an enzymatic method (Diasys, Germany), which photometrically determines NADH+ formation from NAD by alcohol dehydrogenase dependent oxidation at a wavelength of 340 nm. A standard curve was generated by immediate measurement of different ethanol concentrations in medium. Results are expressed as percentage from the initial ethanol concentration. Accumulation of lipids - Oil Red O staining Mouse hepatocytes were plated in 24- or 96-well-plates and stimulated for 3 days with 170 nM insulin in William’s Medium E (with 1% penicillin/streptomycin and 2 mM L-glutamine). Following stimulation, cells were fixed with 4% PFA and washed briefly with 60% isopropanol before staining for 10 min with 0.2% Oil Red O (Sigma-Aldrich Missouri, USA) in 60% isopropanol. Unbound Oil Red O was removed by washing with ddH2O. To quantify signals, bound Oil Red O was resolved in 100% isopropanol. After incubation for 10 min at RT, supernatant was transferred to a transparent 96well-plate and OD was measured at 500 nm with a RAINBOW Thermo ELISA reader (SLT Labinstruments, Achterwehr, Deutschland). Annexin-V staining Apoptosis in cultured mouse hepatocytes was assessed by Annexin-V staining as previously described [4]. Briefly, 6x104 hepatocytes per well were cultured on glass 8well-chamber slides (BD Biosciences, NJ, USA). After overnight serum starvation, cells were treated with 5 ng/ml TGF-, 100 mM ethanol or a combination of both for 48 hrs. Residual culture medium was washed off the cells with PBS (2 mM CaCl 2). Cells were stained for 5 min with 0.25 µg/ml Annexin-V-Cy3 (Abcam Ltd., Cambridge, UK) and 5 µg/ml Hoechst (Sigma) in PBS (2 mM CaCl2). Unbound stain was washed off the cells with PBS (2 mM CaCl2) and fluorescent signal is detected immediately. Lactate dehydrogenase assay The toxicity of TGF-β and ethanol was determined with a lactate dehydrogenase (LDH) leakage assay using a cytotoxicity detection kit (Roche, Mannheim, Germany). Cells (2×104/cm2) were seeded onto 12-well-plates and treated with 5 ng/ml TGF-β and 100 mM ethanol for 48 h. Cytotoxicity was expressed as percentage of LDH released into the culture medium in relation to total LDH. 3 Cytochrome P450 assay Fluorescence-based Cytochrome P450 assays were performed by incubation of intact cells (in 96-well plates) with selected substrates as previously reported [21]. Briefly, 100 µl reaction buffer (1 mM Na2HPO4, 137 mM NaCl, 5 mM KCl, 0.5 mM MgCl2, 2 mM CaCl2, 10 mM O-(+)-glucose, and 10 mM HEPES, pH 7.4) containing the appropriate amount of the fluorogenic substrate 7-Methoxy4(trifluoromethyl)coumarin (7-MFC) were added to each well. After 8 hrs iat 37°C, supernatants were transferred to white/black 96-well plates and cells were fixed for protein quantification by sulforhodamine B (SRB) staining. Potential metabolite conjugates formed were hydrolyzed by incubating supernatants with βglucuronidase/arylsulfatase (150 Fishman units/ml and 1,200 Roy units/ml for 2 h at 37.0°C. Samples were diluted (1:4) with the appropriate quenching solution. Formation of fluorescent metabolite was quantified with a Fluoroscan Ascent Fluorescence Microplate Reader. Results are given as picomoles of metabolite per minute, normalized to total protein content. Methanol-fixed cells were used for background subtraction. SRB Staining was performed as reported [21]. In brief, cells were covered with icecold fixation buffer (95% ethanol, 5% acetic acid) and kept at –20°C for 1 h. Fixed cells were stained with 0.4% SRB (w/v) in 1% acetic acid for 30 min. Unbound dye was removed by washing with 1% acetic acid. Bound SRB was resolved in 10 mM unbuffered Tris solution and optical densities were determined at 565 nm. From optical densities, total protein content was calculated with a standard curve obtained by plotting the optical density from SRB staining versus total protein contents measured with the DC Protein Assay Kit (Biorad, Muenchen, Germany). ROS production, glutathione depletion and lipid peroxidation Mouse hepatocytes were plated in 96-well-plates. After stimulation with 100 mM ethanol and/or 5 ng/ml TGF-, cells were washed 3 times with HBSS followed by incubation with 10 µM DCFH-DA (ROS measurement) or 10 µM monochlorobimane (MCB; GSH mesurement) in medium for 30 min at 37 °C. Thereafter, cells were washed three times with HBSS and lysed in 0.1 % Triton-X-100 in HBSS for 10 min at 37 °C. Cell lysates were transferred to a white 96-well plate (Nunc, Wiesbaden, Germany) and fluorescence was determined (ex/em 485/527; ROS or 355/460 nm; GSH) with a Fluoroskan Ascent fluorescence microplate reader (ThermoLabsystems, Engelsbach, Germany). For measuring lipid peroxidation, cells were treated in the same way for 48 hrs. Cell lysates were collected in ice-cold HBSS supplemented with 1/50 volume butylated alcohol (0.2 % 2,6-di-tert-butyl-4-methylphenol in ethanol) by sonication. 1 volume thiobarbituric acid (TBA) (5 mM in acetic acid) solution was added before boiling the reaction mixture for 60 minutes. Samples were chilled on ice and 0.5 volumes methanol were added. Samples were centrifuged (13,000 g, 5 min, RT) and supernatant was transferred to a white 96-well-plate to measure fluorescence (ex/em 530/590 nm). All in vitro data are presented as mean ± standard deviation of three measurements for each out of three separate experiments. In silico promoter analysis 1,500 bp upstream promoter region relative to the mouse Adh1 gene start codon were analyzed with Genomatix Software to identify putative binding elements (http://www.genomatix.de). 4 RNA based expression analyses Total RNA was purified from liver tissue after homogenization in TRIZOL reagent (GIBCO BRL, Eggenstein, Germany; 1 ml/50mg tissue) and from cultured hepatocytes using the High Pure RNA isolation kit (Roche, Mannheim, Germany) according to the manufacturer’s protocol. 1µg total RNA was transcribed to cDNA using Transcriptor First Strand cDNA synthesis kit (Roche). PCR amplifications were done for ADH1, Smad7 and rS6 (Suppl. Table 1). PCR products were obtained after 26 amplification cycles at annealing temperatures of 60 oC. Samples were run on 1.5 % agarose gels and visualized by ethidiumbromide staining (0.5 µg/ml) under UV light. The results were analysed by digital image analysis. Suppl. Table 1 Gene Up-stream Primer Down-stream Primer Product size (bp) rS6 5´-GTG CCT CGT CGG TTG GGA C-3´ 5´-GAC AGC CTA CGT CTC TTG GC-3´ 320 Adh1 5´-AGG AAG TTC TCC AGG AGA T-3´ 5´-TCT TAG CCA TGA AGT CAG CC-3´ 264 Microarray analysis Total RNA extraction and cDNA probe labelling was as described.[5] Hybridization to microarrays of type moe430_2 from Affymetrix (Santa Clara, CA, USA) and array scanning were performed according to manufacturers recommendations. Differential expression analysis was performed with ANOVA in BioConductor (http://www.bioconductor.org) using R V2.4.1. Raw fluorescence intensity values were normalized applying quantile normalization, and gene expression values were converted to a log2 scale. Significant differences in gene expression were analysed with limma package for R Multitest. Genes with mean expression changes greater than 1.7 fold (log2 greater than 0.7655 and lower than -0.7655) and which were significant at a false-discovery rate of q < 0.05 were selected as regulated by TGF-. CustomCDF with Entrez based gene/transcript definitions R package was used for gene annotation. Gene Ontology analysis was done with DAVID (Database for Annotation, Visualization, and Interpreted Discovery; http://apps1.niaid.nih.gov/david/. GeneChips microarray study followed MIAME guidelines issued by the Microarray Gene Expression Data group. We have deposited all files containing the raw data of the microarray analysis to ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) at the European Bioinformatics Institute (Hinxton). The data are available under accession number E-MEXP-1176. Suppl. Table 2 Antibodies and reagents used in this study: Antibodies Cat. No Clonality ab5825 RabbitPoly #3101 RabbitPoly #9514 RabbitPoly A-5441 Mouse- Supplied by Abcam Antibody, epitope, location MADH7 Species* h, m, r Dilution 1:500 (WB) Cell Signaling Cell Signaling P-Smad2, synthetic phosphopeptide, serine465/467, hSmad2 P-Smad1/3, synthetic phosphopeptide, serine 423/425, hSmad3 β-actin, N-terminal peptide h, m, r h, m, r 1:1,000 (WB) 1:100 (IHC) 1:1,000 (WB) h, m, r 1:10000 (WB) Sigma 5 51-1300 sc-22750 Mono RabbitPoly RabbitPoly Zymed Smad2 h, m, r 1:1,000 (WB) Santa Cruz ADH1 h, m, r 1:1,000 (WB) References 1 Klingmuller U, Bauer A, Bohl S, et al. Primary mouse hepatocytes for systems biology approaches: a standardized in vitro system for modelling of signal transduction pathways. Syst Biol (Stevenage) 2006;153:433-47. 2 Goumans MJ, Valdimarsdottir G, Itoh S, et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell 2003;12:817-28. 3 Dooley S, Delvoux B, Streckert M, et al. Transforming growth factor beta signal transduction in hepatic stellate cells via Smad2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. TGFbeta signal transduction during transdifferentiation of hepatic stellate cells. FEBS Lett 2001;502:4-10. 4 Dooley S, Hamzavi J, Ciuclan L, et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology 2008;135:642-59. 5 Ehnert S, Nussler AK, Lehmann A, Dooley S: Blood monocyte-derived neohepatocytes as in vitro test system for drug metabolism. Drug Metab Dispos 2008, 36(9):1922-1929. 6 Glynn Dennis Jr, Brad T Sherman, Douglas A Hosack, Jun Yang, Wei Gao, H Clifford Lane and Richard A Lempicki. DAVID: Database for Annotation, Visualization, and Integrated Discovery Genome Biology 2003, 4:R60 6 Supplementary Results TGF-β enhances ethanol induced toxicity and oxidative stress in mouse hepatocytes To determine appropriate concentrations for TGF-β and ethanol for measurable effects in hepatocytes, we established dose response curves for cellular damage as determined by the percentage of LDH release into the culture medium during 48 hrs treatment. The results indicate 1- 5ng/ml TGF-β1 and 100mM ethanol as lowest doses inducing a significant response (Suppl.Fig.2A,B). After the addition of ethanol, plates were sealed with parafilm to prevent evaporation. With this setting, time dependent variations in the ethanol concentration were due to its metabolism and not to different evaporation rates. For example, the concentration of ethanol in the culture medium 12 hours following ethanol addition was reproducibly reduced to ~ 50% in case of parafilm sealed plates, compared to ~ 33% for unsealed plates, when related to the initial ethanol concentration (Suppl.Fig.5A). Ethanol was added in fresh medium every 24 hours, to study long term effects. Control cultures were treated similarly. TGF-β decreases ADH1 expression in hepatocytes Affymetrix oligonucleotide microarray analysis covering the whole mouse genome was performed to obtain a comprehensive view on TGF-β effects in hepatocytes. We identified functional groups associated with signal transduction, fibrogenesis and cell death [5], as well as with transport, metabolism and oxidative stress (Suppl.Fig.3). In the metabolism related group, 57 genes belonging to lipid- and 11 to glucosemetabolism. Among them such involved in triacylglycerol metabolism, among others, the triacylglycerol degradation pathway, lipoprotein lipase (Lpl), hepatic lipase (Lipc), monoglyceride lipase (Mgll) and liver specific fatty acid binding protein 1 (Fabp1) were down-regulated. In contrast to this, one of the enzymes synthesizing acyl CoA, acyl-CoA synthetase bubblegum family member 1 (Acsbg1), and one lipoprotein transporter, low density lipoprotein receptor-related protein 11 (Lrp11) were upregulated. Several detoxifying and electron transporting cytochromes were downregulated (e.g. CYP27A1, CYP2B10, CYP2C70, CYP2D9, CYP39A1, CYP3A13, CYP4F14 and CYP7B1), whereas the expression of CYP21A1, an enzyme involved in glucocorticoid production and fat accumulation, was increased. Moreover, mRNA abundance of several genes involved in the anti-oxidant defense mechanism was markedly decreased, for example glutathione S-transferases (Gsta2, Gstt1, Gsta4, Gstt2, Gstm4, Gstm7 and Mgst3), glutathione synthetase (Gss), whereas glutathione peroxidase 3 (Gpx3) was up-egulated. Consistently with this, expression of monoamine oxidase A (Maoa), a repressor of the ROS producing system was down-regulated. Furthermore, some genes involved in glucose metabolism were changed by TGF- treatment. These include up-regulated hexokinase 2 (Hk2), insulin-like growth factor 1 (Igf1) and phosphoglucomutase 1 (Pgm1). Other enzymes involved in glycolysis, gluconeogenesis and their regulation were down-regulated (Aldob, Onecut1, Pklr, Gpd1, Pdk1, Pdk2, Pdk4, Tpi1 and Atf3) (Fig.3A and Suppl.Fig.3). In culture, mouse and human hepatocytes share similar phenotypes as seen by light microscopy photographs (Suppl.Fig.4B). TGF-β down-regulates ADH1 expression in hepatocytes via activation of the ALK5 pathway 7 As Smad independent TGF-β signaling occurs in hepatocytes, but inhibition of MAPK pathway, showed no significant effect on Adh1 expression (data not shown), we assume that TGF- dependent ADH1 expression reduction in hepatocytes solely relies on activation of the ALK5/Smad2/3 signaling pathway. The effect of ADH1 deficiency/ADH1 overexpression on ethanol metabolism To determine the effect of ADH1 deficiency/ADH1 overexpression on ethanol metabolism, the residual ethanol concentration was determined in the culture medium of alcohol treated mouse hepatocytes after 12 hrs of incubation. Hepatocytes were transfected with ADH1 overexpression plasmids and control vectors, or ADH1 siRNAs and the corresponding scrambled siRNAs, followed by ethanol stimulation for 12 hrs. In this setting, the medium ethanol concentration is reproducibly decreased to ~ 20% in ADH1 overexpressing hepatocytes as compared to ~ 50% in controls. On the other hand, reducing ADH1 expression with RNAi, increased ethanol levels to ~ 85% (Suppl.Fig.5B), suggesting significantly diminished oxidation of ethanol to acetaldehyde. Furthermore, these data confirm that the measured changes in ethanol levels are due to metabolic effects and not to evaporation. We also investigated the combined effect of ethanol and TGF-β. Challenge with 5ng/ml TGF-β for 24 hrs reduced Adh1 mRNA expression with and without ethanol stimulation (48 hrs, 100mM), although with ethanol the effect was weakened (Suppl.Fig.5C), which might be due to a stimulatory impact of ethanol for Adh1 [12]. Supplementary figure legends Suppl.Fig.1. Quantitative analysis of Annexin V staining before and 48 hrs after treatment with 5 ng/ml TGF-β and/or 100 mM ethanol . Results are presented as arbitrary units of the measured fluorescence. Suppl.Fig.2. Cell induced by dose response curves of TGF-β and alcohol dependent toxicity in hepatocytes. (A,B) Hepatocytes were stimulated or not with 1, 5, 10 ng/ml TGF- and 50, 100, 200mM ethanol for 48 hrs. Cellular damage was determined by measuring the percentage of LDH released into the culture medium in relation to total LDH. (three different mouse hepatocyte preparations were investigated in triplicate measurements; **p<0.01, *p<0.05 vs control). Suppl.Fig.3. TGF-β regulates expression of genes involved in its signal transduction, fibrogenesis, apoptosis, intracellular transport and metabolic processes in hepatocytes. (A) Mouse hepatocytes were stimulated or not with 5 ng/ml TGF- for 24 hrs and total RNA was analyzed using Affymetrix GeneChips. Genes whose expression levels were significant at a false-discovery rate of q < 0.05 and whose fold change relative to untreated controls was higher than 1.7 are shown. The DAVID tool was used to elucidate biological functions that are enriched. The lengths of the bars represents the number of genes in each modul. Detailed descriptions of the terms can be found at the homepage of the Gene Ontology project (http://www.geneontology.org/index.shtml). 8 Suppl.Fig.4. Adh1 mRNA expression remains unchanged during culture of mouse hepatocytes and TGF-β down-regulates ADH1 expression in the same manner in human hepatocytes. (A,B) Mouse and human hepatocytes were cultured in serum-free medium on collagen-coated plates. Human liver cells were stimulated or not with 5 ng/ml TGF- for 24 hrs. Adh1 mRNA expression was examined by PCR and rS6 was used as a loading control. Primers are listed in Suppl.Table 1. (B) Light microscopy photographs of mouse and human hepatocytes cultured in serum-free medium on collagen-coated plates for two and five days, respectively. (magnification: 200X). Suppl.Fig.5. Adh1 dependent ethanol metabolism in cultured hepatocyes (A) Hepatocytes were stimulated with 100 mM ethanol for 6-24 hrs as indicated. Alcohol treated plates were sealed or not with parafilm, concentration was measured in medium at 340 nm and related to a standard curve generated from known ethanol concentrationsmixed with medium. (B) Hepatocytes were transfected with control vectors and the corresponding ADH1 siRNAs or ADH1 overexpression plasmids, followed by ethanol (100 mM) stimulation for 12 hrs (as presented in Supplementary Material and methods). (C) Real-time RTPCR of Adh1 mRNA expression in cultured mouse hepatocytes treated with ethanol (48hrs) and/or TGF-β (24 hrs) as indicated in supplementary results. Suppl.Table 3 Serum TGF- concentration in control and TGF- transgenic mice that were used to analyse Adh1 mRNA expression. Number 1 2 3 4 5 6 7 8 9 10 wt wt wt wt wt TGF- tg TGF- tg TGF- tg TGF-tg TGF- tg Serum TGF- concentration (ng/ml) 50 51 30 40 57 466 240 1017 223 250