The following text details the analysis we performed to
advertisement

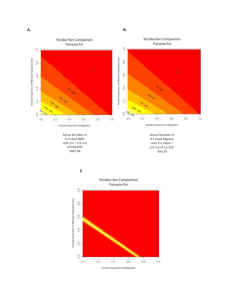
The following text details the analysis we performed to explore whether the overlap of 40,856 bp (including only one exon, of 433 bp) at the 3' end of the ~1-Mb region we have analyzed of the LARGE gene, which has been suggested to be under positive selection in Subsaharan Africans1 could have distorted the patterns of interpopulation differentiation in the Iberian and N. African samples we have genotyped. Linkage disequilibrium We plotted LD in the region analyzed using the default color scheme in Haploview 4.02. As can be seen in Figs. 1 and 2, in all populations the longest LD blocks lie at the 5' end of the region, that is, away from LARGE. LD with LARGE does not seem to extend far into the region. It is not apparent from these LD patterns that a selective sweep caused by variation at LARGE has affected the region we have genotyped in these populations. Additionaly, LD in Subsaharan Africans in this region is smaller than elsewhere3. Variation at LARGE is the most obvious candidate for natural selection in this region; the observed LD patterns do not rule out, though, that selection has occurred at the 5' end of the region, where functional variation is either absent or poorly characterized. Expected FST values An island model4 can be used to model expected, neutral FST values according to heterozygosity. We used fdist24 to generate neutral FST distributions with the same mean as that observed in our data. We plotted the simulated neutral FST median and the empirical 99% confidence interval, as well as the FST values for each of the 123 SNPs we genotyped (Fig. 3). The number of islands simulated did not change noticeably the results; here we present the outcome of 1,000,000 simulations for 20 islands. Two SNPs, rs2032461 and rs2899222, have FST values that exceed the 99.5% percentile of the simulated distribution; they are indicated as orange arrows in Fig. 4. rs2032461 shows a higher frequency of its major allele in Andalusians; rs2899222 has an irregular pattern, with major allele frequencies 0.6-0.7 in Andalusians, Catalans, and N. Moroccans, and 0.4-0.5 in French and Spanish Basques, and Extremadurans. Thus, the relatively high differentiation of these two SNPs does not seem to be caused by a single selection event, and is more likely to reflect random variation5. Six SNPs showed FST values below the simulated 0.5% percentile; they are indicated as purple arrows in Fig. 4. They seem to be scattered throughout the region studied, and, in particular, they do not cluster close to the overlap with the LARGE gene. In summary, the expected FST distribution according to an island model did not reveal SNPs in the region with extreme FST values that could be attributed to natural selection. Empirical FST distributions. Finally, we compared FST in the region studied with that across the genome. Li et al.6 genotyped the Illumina HumanHap650K Beadchips in the CEPH HGDP samples. We compared FST among Europeans in our region compared to the rest of the genome with their data. The region in chr. 22 we analyzed was covered with 284 SNPs in ref. 6, of which 251 presented less than 5% missing genotypes. The average FST among Europeans in those was 0.0121, and for 587,119 SNPs in the rest of the autosomal genome it was 0.0129. A permutation test with 10,000 iterations showed that both averages were not significantly different with P=0.41. A dataset with a SNP density greater than the one we used (and, thus, presumably statistically more powerful) showed no difference in FST patterns between the region in chr. 22 and the rest of the genome among Europeans. Obviously, this does not rule out that natural selection could have still distorted FST among Spanish populations, but, in that case, this putative adaptation should be restricted to Iberia and would have spared other Mediterranean populations represented in the CEPH HGDP panel such as Sardinia and Peninsular Italy. Alternatively, one can assume that natural selection, if present in this genomic region, has not biased interpopulation differentiation to the point of obscuring the imprint left by demographic events. Figure 1: Linkage disequilibrium in the region analyzed. Under the physical scale and in blue, the LARGE gene (line, intron; box, exon) Figure 2: Linkage disequilibrium in the region analyzed. Under the physical scale and in blue, the LARGE gene (line, intron; box, exon) 0.08 0.07 0.06 0.05 0.04 0.03 0.02 0.01 0 0 0.1 0.2 0.3 0.4 0.5 -0.01 -0.02 Figure 3. FST against heterozygosity. Lines are the 99.5%, median, and 0.5% percentiles obtained with an island-model simulation, for each heterozygosity class (0-0.01, 0.01-0.02, ..., 0.49-0.50). Dots are the actual values observed in 123 SNPs. 0.2 0.1 0 32.6 32.7 32.8 32.9 33.0 33.1 33.2 33.3 33.4 33.5 33.6 Position in Mb Figure 4. Position of SNPs with outlier FST values according to the island-model simulation. Dots: all SNPs; purple arrows: SNPs with lower FST than expected; orange arrows: SNPs with higher FST tahn expected. Blue bar: position of the LARGE gene. References 1 2 3 4 5 6 Sabeti PC, Varilly P, Fry B et al: Genome-wide detection and characterization of positive selection in human populations. Nature 2007; 449: 913-918. Barrett JC, Fry B, Maller J, Daly MJ: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263-265. González-Neira A, Ke XY, Lao O et al: The portability of tagSNPs across populations: A worldwide survey. Genome Research 2006; 16: 323-330. Beaumont MA, Nicholson J: Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society of London. Series B: Biological Sciences 1996; 263: 1619-1626. Gardner M, Williamson S, Casals F et al: Extreme individual marker F(ST )values do not imply population-specific selection in humans: the NRG1 example. Hum Genet 2007; 121: 759-762. Li JZ, Absher DM, Tang H et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100-1104.