Supplementary methods - Word file (26 KB )
advertisement
")
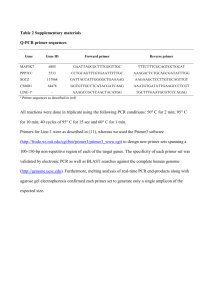
Supplementary Methods Extraction of GTs from Rose flowers Flowers from rose (Rosa hybrida) cultivars of the Hybrid Tea group ‘Crimson Glory’, ‘Carl Red’, ‘Rote Rose’ and ‘Sterling Silver’, which bear cyanic flowers, were powdered in liquid nitrogen. The powder was homogenized gently in 0.15M potassium phosphate buffer (pH 7.0) containing 14mM 2-mercaptoethanol (2-ME), 100mM NaCl, 5mM EDTA, 5mM ascorbate, 1% polyvinylpyrrolidone K-30, 0.1% Triton X-100 and 10% glycerol. The homogenate was filtered through four layers of gauze, and the filtrate was centrifuged. Solid (NH4)2SO4, was added to the supernatant, bringing it to 10% saturation, stirred, and centrifuged. The supernatant fraction was subsequently brought to 70% saturation of solid (NH4)2SO4 and re-centrifuged. The pellet was dissolved in a minimum volume of Tris-HCl buffer (pH 8.0) containing 7mM 2-ME and desalted through a Sephadex G-25 (Amersham Bioscience). To remove phenolic compounds, desalted solutions were applied to a DEAE-cellulose (Whatman) column and eluted with DEAE elution buffer (Tris-HCl buffer (pH 8.0) containing 7mM 2-ME and 0.5M NaCl). The eluant fraction was desalted on a PD-10 column (Amersham Bioscience) and re-eluted with Tris-HCl buffer (pH 8.0) containing 7mM 2-ME. Glucosyltransferase assay To assay GT enzyme activity, 50μl reaction mixture consisted of 30μl crude enzyme preparation (100-150μg protein in 50mM Tris-HCl buffer (pH 8.0) containing 7mM 2-ME) as described above, 5nmol of the glucosyl acceptor (anthocyanidins or anthocyanins) and 10nmol UDP-glucose. The mixture was incubated for 10 min at 30°C and stopped by the addition of 50 μl CH3Cl:CH3OH, 2:1(v/v; plus 10% HCOOH). Identification of reaction products was carried out by HPLC analysis equipped with a reverse-phase column (Wakosil-II 5C18 AR, Wako), was used a linear elution gradient (1ml/min) from 5 to 30% solvent B (0.5% TFA in CH3CN) in solvent A (0.5% TFA, 5% CH3CN in H2O) over 20min with monitoring of absorbance at 520nm as a standard method. Products were identified by co-chromatography with authentic anthocyanins. Isolation of RhGT and expression of recombinant protein in E. coli. Total RNA was extracted from petals using a modified CTAB method1. cDNA was synthesized with a First Strand cDNA Synthesis kit (Amersham). A degenerate primer, GTSPFd (WCICAYTGYGGITGGAAYTC) was designed on the basis of the conserved PSPG-box. The GT gene was amplified by PCR using the GTSPFd and NotI R1 primer (AACTGGAAGAATTCGCGGC). A second PCR amplification was carried out using the GTSPFd and NotI R2 primer (GAACGCGGCCGCAGGAAT). NotI R1 and R2 primer sequences included in the NotId(T)18 primer (Amersham) were used for cDNA synthesis. PCR were performed using the Expand High Fidelity PCR system (Boehringer Mannheim). Second amplification PCR products were cloned in pGEM Easy T-vector (Promega) and sequenced, and a GT-homologous clone was identified using BLAST X algorithm software. Full length cDNA from petal mRNA was synthesized using a GeneRacer kit (Invitrogen) and the 5’ terminal region of the GT-like gene was amplified using primer R1 (TTATGCGGGCCCAACATGCC) for a first PCR amplification, primer R2 (CCACAGCTGAGCCAACTTGG) for a second amplification. Amplified products were cloned in pGEM Easy T-vector (Promega) and sequenced. The coding region of the GT-like gene was amplified by RT-PCR using GT-like specific primers RhGTFd (GACGACGACAAGATGGGTGGTGATGCTATAGTTTG) and RhGTRv (GAGGAGAGCCCGGTTCATTTTTGCTTCCACAGCTGAGCC), which contain start and stop codons respectively, and a sequence for cloning in the pET vector (Novagen). RT-PCR products were cloned in the pET vector (Promega) and sequenced. Recombinant proteins were expressed in E. coli BL21(DE3)(Novagen). Transformed E. coli expressing the recombinant GT protein was cultured overnight in 3ml of liquid 2YT medium containing kanamycin (50μg/ml) at 37℃, and then for 6h at 30℃ with 400μM IPTG. Cells were harvested by centrifugation, re-suspended in 300μl of 50mM Tris-HCl buffer (pH8.0) containing 7mM 2-ME, and the bacteria were lysed with an ultrasonic disruptor. Cell debris was removed by centrifugation. Enzyme assays were carried out as for crude enzyme extracts from rose flowers. Northern blot analysis of RhGT1 RNA extracted from sepals, stems, young leaves and mature leaves and five rose developmental stages were used for northern blot analysis. The coding region of RhGT1 from ‘Crimson Glory’ was labelled with 32P using RediPrimeII DNA labelling kit (Amersham) and used as a probe. GenBank accession numbers of the GTs; Rauvolfia serpentiana Hydroquinone GT (AJ310148); Dorotheanthus bellidiformis Betanidin 6-GT (AF374004); Medicago truncatula Triterpene GT (AY747627); Scutellaria baicalensis Baicalein 7-GluT (AB042277); Phaseolus lunatus Zeatin GT (AF101972); Zea mays Zeatin GT (AF318075); Vigna angularis ABA GT (AB065190); Scutellaria baicalensis 7-GT (AB031274); Dorotheanthus bellidiformis Betanidin 5-GT (Y18871); Gentiana triflora 3’-GT (AB076697); Hordeum vulgare 3-GT (X15694); Zea mays 3-GT (X13501); Dianthus caryophyllus 3-GT (AB191245); Vitis vinifera 3-GT (AF000371); Gentiana triflora 3-GT (D85186); Perilla flutescens 3-GT (AB002818); Petunia hybrida 3-GT (AB027454); Solanum melongena 3-GT (X77369); Zea mays IAA GT (L34847); Citrus unshiu Limonoid GT (AB033758); Eucalyptus perriniana Monoterpene GT (AB166766); Petunia hybrida 5-GT (AB027455); Varbena hybrida 5-GT (AB013598); Perilla flutescens 5-GT (AB013596); Torenia hybrida 5-GT (AB076698); Citrus maxima RhaT (AY048882); Petunia hybrida RhaT (X71059). Reference 1. Chang, S., Pryear, J. & Cairney, J. Plant Mol. Biol. Rep. 11, 113-117 (1993)