PrakashProp - Shiu Lab - Michigan State University
advertisement

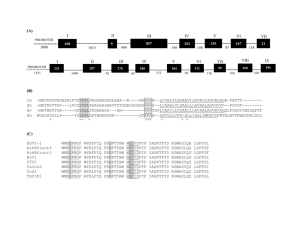
TITLE: Survey of Misannotations and Pseudogenes in the Rice Genome Author: Tanmay Prakash, Novi High School Mentor: Kousuke Hanada and Shin-Han Shiu, Department of Plant Biology, Michigan State University Abstract There are occasions where there are misannotations that sometimes are due to the existence of pseudogenes. This makes it difficult to conduct accurate research with this data. In the preliminary research, misannotations in the introns of Arabidopsis thaliana (Arabidopsis Genome Initiative, 2000) have been assessed using the protein kinase domains. The protein kinase family was chosen for pilot study because of its large size with more than 1000 genes in Arabidopsis thaliana and thus large potential for finding misannotations. No misannotations or pseudogenes were found. However, sequence with significant similarities (BLAST E value < 1e-5) were found in the introns of 5 Arabidopsis genes. This is most likely due to the extensive analysis done on the Arabidopsis Genome. I plan to identify the misannotations and those that are pseudogenes in the rice introns. Doing this will improve the quality of the rice genome annotations and thus the research done that utilizes the rice genome. It also provides more pseudogenes to study such things as neutral selection. Introduction Pseudogenes are DNA sequences that no longer function but resemble the functional genes they once were (Torrents et al., 2003). There are two types of pseudogenes, processed and non-processed. Processed pseudogenes are formed by retrotransposition and comprise most of the pseudogenes in mammals. Non-processed pseudogenes are products of duplication of the entirety of portion of a segment of genes followed by mutations. Because polyploidiszation (the process of having more one sets of chromosomes) is common in plants, the majority of pseudogenes in plants are nonprocessed (Blanc and Wolfe, 2004). Pseudogenes are mainly identified by the existence of premature stop codons or frameshift (Zhang et al., 2004). They can also be identified by their lack of selective pressure. In functional genes selective pressure results in mostly synonymous substitutions (mutation in a codon that produces the same amino acid) (Torrents et al., 2003). In pseudogenes, however, there is no selective pressure so substitutions can be synonymous or nonsynonymous (mutations result in different amino acids). Based on these properties, the rates of nonsynonymous (KA) and synonymous (KS) substitutions can be used as a measure of selection pressure. Functional genes normally have KA/KS values that are significantly less than one, a property that can be used to distinguish functional and pseudogenes. The annotation of a gene is the process of assigning its introns, exons, and untranslated regions. One common type of misannotation is labeling a pseudogene as functional gene due to mis-assignment of introns, which is problematic because the misannotated genes produce erroneous results if used for research. The proposed studies focus on the pseudogenes that are misannotated introns (Figure 1, next page). If part of a protein domain (folds of a protein that play a certain role and can appear in many Figure 1 Possible Sequence Similarities exon intron exon exon intron domain domain Case 1 Case 2 exon intron exon exon domain Case 3 exon intron exon exon intron domain domain Case 4 Case 5 exon different proteins) is found in the exons but not in the intron between the exons then for the intron is likely correctly annotated (Figure1, Case 1). However, if an intron does contains part of a protein domain and the flanking exons contain parts of the same domain (Figure 1, Case 2), this intron is likely misannotated. If any stop codons are found in the introns, the misannotation is a potential pseudogene. These criteria together with an examination of signature of purifying selection will be used to accomplish the objectives of my research. Objectives 1. Identify any misannotated regions in rice introns. We plan to do identify the misannotations in the rice introns by checking for sequence similarity to any domains in the introns and then in the genes exons. If the sequence similarity to a domain is found in the intron, then this region is a possible misannotation and could be a pseudogene. By accomplishing this, we will have a more accurate understanding of the genes analyzed. Also, working with misannotated sequences could be disastrous for other research. 2. Check if the misannotated regions represent pseudogenes. We do this because pseudogenes can hold a wealth of information, such as how neutral selection works. In addition, locating pseudogenes can make future annotation easier and present annotation more accurate. The pseudogenes will be found using the two methods (a) premature stop codons, or frameshift mutations in the misannotated genes and (b) signature of negative selection (KA/KS). By using two independent methods to identify pseudogenes, a greater level of accuracy can be achieved. I have carried out a pilot study on the protein kinase domain to determine the feasibility of my planned approach with the exception of (2b). The results are presented in the next section. Preliminary Results There are more than 8296 protein domains (Robert D. et al., 2006). The first domain sequence checked was that of protein kinase. This is a family of over 1000 genes in Arabidopsis (Arabidopsis Genome Initiative, 2000), which made it more likely to be misannotated and a good test of the process that is to be used. The procedures followed for the preliminary research are outlined in Figure 2 Figure 2 Automated Pipeline Query Rice Protein Domain Blastall BLAST search Matching genes Subject Database of Rice Genome Introns Exons from Matching Genes Formatdb Exons formatted into a database Query Rice Protein Domain Subject Blastall BLAST search Check KA/KS value Possible Pseudogenes Check for Stop Codons and Frameshift Mutations Possibly Misannotated Genes To find the misannotated introns containing kinase domain sequences, I first conducted searches to see if there was any sequence similarity to the protein kinase domain in Arabidopsis thaliana introns with BLAST (Basic Local Alignment Search Tool, Expected value < 1e-5, Altschul et al). For genes with intron matching kinase domains, the exon flanking introns were then checked for sequence similarity to the protein kinase domain with BLAST (E value < 1e-5). The results of the BLAST search with the protein kinase domain and exons were also evaluated with a HMMER, a program that searches for protein homology in amino acid sequences utilizing Hidden Markov models, (Wistrand, Sonnhammer, 2005) search to get a better assessment on whether there truly is a sequence similarity. There are five expected outcomes as shown in Figure 1. Among about 25,000 Arabidopsis genes, almost all belong to Case 1 with correct annotations (Arabidopsis Genome Initiative, 2000). The AT3G45390.1 and AT4G25390.2 89-348 (39 sequence similarities in this region) KPRO_MAIZE/534-812 141-461 KPRO_MAIZE/534-812 105-249 (one of the 39 similarities) AT3G01830.2 KPRO_MAIZE/534-812 CDC15_YEAST/25-272 CDC5_YEAST/82-337 MOS_CERAE/60-338 MIL_AVIMH/82-339 M3K9_HUMAN/144-403 MKK1_YEAST/221-488 PHKG2_RAT/24-291 KPK2_PLAFK/111-364 STE7_YEAST/191-466 KIN1_SCHPO/125-395 MK04_HUMAN/20-312 KGP1_DROME/457-717 1040-276 1040-498 920-498 1040-498 758-498 1040-498 1040-498 1013-498 869-498 926-498 869-498 722-345 869-498 AT3G45390.1 KPRO_MAIZE/534-812 357-536 KPRO_MAIZE/534-812 2-229 BUR1_YEAST/60-366 351-411 AT1G24040.2 KPRO_MAIZE/534-812 1421-1221 AT1G24040.1 KPRO_MAIZE/534-812 1421-1221 Region of Sequence Similarity Figure 3 Gene with sequence similarity to the protein kinase domain in the introns 5’UTR 3’UTR Coding Region Intron AT4G25390.2 genes resembled Case 4 and Case 5 of Figure 1 respectively with respect to the protein kinase domain. The AT3G01830.2, AT1G24040.2, and AT1G24040.1 genes all resembled Case 3 of Figure 1 with respect to the protein kinase domain. None of the five genes whose introns had sequence similarities to the protein kinase domains had sequence similarities located as in Case 2 of Figure 1, so I didn’t find any misannotations or pseudogenes. There are a substantial number of full-length cDNA and ESTs for Arabidopsis thaliana and these sequences have been used to improve its annotation significantly (Yamada et al., 2003 Science). Therefore, this is likely the reason why I did not find any mis-annotated kinases. Research Plan The objectives of this project are to (1) find any misannotated regions in the introns of the rice genome and (2) to check if the misannotated regions represent pseudogenes. Finding any misannotated regions provides more accurate data for use by other researchers and provides candidates for pseudogenes. Finding pseudogenes can help with future annotations and can be used to study things like neutral selection. These objectives will be completed by the following methods. 1. Find any misannotated regions in rice introns. This is done using an automated pipeline I am developing (Figure 2) in UNIX that first searches a domain against a database of what introns using BLAST (HMMER as well). The genes whose introns match the domain have their exons put into a new database. The domain is then searched against this database using HMMER. The queries and databases for each next step will be extracted from the previous search results using computer programs written in Perl using methods such as regular expression matching. The necessary programs shall be run on the Calculon computer system in the Plant Biology Department of Michigan State University. If an intron and its flanking exons have matches to the same domain, an argument for a misannotation can be made. This can also be analyzed for a possible pseudogene. If matches to the domain are found in introns and non-flanking exons, or if a match to the domain is found in the intron but to a different domain in the exons, the result will be further analyzed at a later time. This processes will be repeated for 8296 domains from the Pfam database (Robert D. et al., 2006). 2. Check if the misannotated regions represent pseudogenes A computer will check the genes for premature stop codons, frameshift mutations and signatures of negative selection using programs written in Perl in the introns that have sequence similarity to a protein domain. The computer will look for “taa”, “tag” or “tga” in a nucleotide sequence and an asterisk in an amino acid sequence to find the premature stop codon in the introns that have sequence similarity to a protein domain. The frameshifts will be detected by searching for insertions or deletions the introns that have sequence similarity to a protein domain using alignment methods. The signatures of negative selection will be calculated by aligning the genes and then counting how many synonymous and nonsynonymous substitutions there are and then using those numbers in the formula to calculate the KA/KS value. If the KA/KS value is significantly less than 1, the gene is most likely functional. If the KA/KS value is closer to 1, the gene may be a pseudogene. Pseudogenes that are found will be posted on the homepage of Shiu Lab, http://shiulab.plantbiology.msu.edu/wiki/index.php/Main_Page Reference Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. (1990) "Basic local alignment search tool." J. Mol. Biol. 215:403-410 Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. David Torrents, Mikita Suyama, Evgeny Zdobnov and Peer Bork. A Genome-Wide Survey of Human Pseudogenes. Genome Research 13:2559-2567, 2003 Guillaume Blanc and Kenneth H. Wolfe. Functional Divergence of Duplicated Genes Formed by Polyploidy during Arabidopsis Evolution. Plant Cell. 2004 July; 16(7): 1679–1691. Robert D. Finn, Jaina Mistry, Benjamin Schuster-Böckler, Sam Griffiths-Jones, Volker Hollich, Timo Lassmann, Simon Moxon, Mhairi Marshall, Ajay Khanna, Richard Durbin, Sean R. Eddy, Erik L. L. Sonnhammer and Alex Bateman Nucleic Acids Research (2006) Database Issue 34:D247-D251 Wistrand M, Sonnhammer EL. Improved profile HMM performance by assessment of critical algorithmic features in SAM and HMMER. BMC Bioinformatics. 2005 Apr 15;6-99. Yamada K, Lim J, Dale JM. Empirical analysis of transcriptional activity in the Arabidopsis genome. Science. 2003 Oct 31;302(5646):842-6. Zhaolei Zhang, Nick Carriero and Mark Gerstein. Comparative analysis of processed pseudogenes in the mouse and human genomes. Trends in Genetics 2004 Feb;62-67 Li WH, Gojobori T, Nei M. Pseudogenes as a paradigm of neutral evolution. Nature. 1981 Jul 16;292(5820):237-9