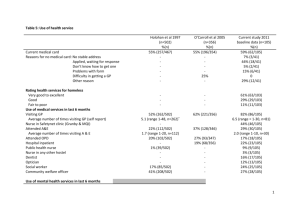
G 5i1.2 March 2014
advertisement

UK Standards for Microbiology Investigations Investigation of Hepatitis Issued by the Standards Unit, Microbiology Services, PHE Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 1 of 44 © Crown copyright 2014 Investigation of Hepatitis Acknowledgments UK Standards for Microbiology Investigations (SMIs) are developed under the auspices of Public Health England (PHE) working in partnership with the National Health Service (NHS), Public Health Wales and with the professional organisations whose logos are displayed below and listed on the website http://www.hpa.org.uk/SMI/Partnerships. SMIs are developed, reviewed and revised by various working groups which are overseen by a steering committee (see http://www.hpa.org.uk/SMI/WorkingGroups). The contributions of many individuals in clinical, specialist and reference laboratories who have provided information and comments during the development of this document are acknowledged. We are grateful to the Medical Editors for editing the medical content. For further information please contact us at: Standards Unit Microbiology Services Public Health England 61 Colindale Avenue London NW9 5EQ E-mail: standards@phe.gov.uk Website: http://www.hpa.org.uk/SMI UK Standards for Microbiology Investigations are produced in association with: Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 2 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis Contents ACKNOWLEDGMENTS .......................................................................................................... 2 CONTENTS ............................................................................................................................. 3 AMENDMENT TABLE ............................................................................................................. 4 UK STANDARDS FOR MICROBIOLOGY INVESTIGATIONS: SCOPE AND PURPOSE ....... 5 SCOPE OF DOCUMENT ......................................................................................................... 8 INTRODUCTION ..................................................................................................................... 8 1 HEPATITIS A ............................................................................................................. 12 2 HEPATITIS B ............................................................................................................. 14 3 HEPATITIS C ............................................................................................................. 20 4 HEPATITIS D ............................................................................................................. 23 5 HEPATITIS E ............................................................................................................. 25 6 HEPATITIS G ............................................................................................................. 29 7 CYTOMEGALOVIRUS (CMV) .................................................................................... 29 8 EPSTEIN BARR VIRUS (EBV) .................................................................................. 30 9 YELLOW FEVER ....................................................................................................... 31 10 Q FEVER.................................................................................................................... 33 REFERENCES ...................................................................................................................... 35 Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 3 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis Amendment Table Each SMI method has an individual record of amendments. The current amendments are listed on this page. The amendment history is available from standards@phe.gov.uk. New or revised documents should be controlled within the laboratory in accordance with the local quality management system. Amendment No/Date. 2/03.03.14 Issue no. discarded. 1.1 Insert Issue no. 1.2 Section(s) involved Amendment Document has been transferred to a new template to reflect the Health Protection Agency’s transition to Public Health England. Front page has been redesigned. Whole document. Status page has been renamed as Scope and Purpose and updated as appropriate. Professional body logos have been reviewed and updated. Scientific content remains unchanged. Amendment No/Date. 1/11.11.11 Issue no. discarded. 1 Insert Issue no. 1.1 Section(s) involved Amendment Whole document. Formerly QSOP 54 now G 5. Document presented in a new format. Relevant NSM and Glossary of terms sections. Removed. References. Some references updated. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 4 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis UK Standards for Microbiology Investigations: Scope and Purpose Users of SMIs SMIs are primarily intended as a general resource for practising professionals operating in the field of laboratory medicine and infection specialties in the UK. SMIs provide clinicians with information about the available test repertoire and the standard of laboratory services they should expect for the investigation of infection in their patients, as well as providing information that aids the electronic ordering of appropriate tests. SMIs provide commissioners of healthcare services with the appropriateness and standard of microbiology investigations they should be seeking as part of the clinical and public health care package for their population. Background to SMIs SMIs comprise a collection of recommended algorithms and procedures covering all stages of the investigative process in microbiology from the pre-analytical (clinical syndrome) stage to the analytical (laboratory testing) and post analytical (result interpretation and reporting) stages. Syndromic algorithms are supported by more detailed documents containing advice on the investigation of specific diseases and infections. Guidance notes cover the clinical background, differential diagnosis, and appropriate investigation of particular clinical conditions. Quality guidance notes describe laboratory processes which underpin quality, for example assay validation. Standardisation of the diagnostic process through the application of SMIs helps to assure the equivalence of investigation strategies in different laboratories across the UK and is essential for public health surveillance, research and development activities. Equal Partnership Working SMIs are developed in equal partnership with PHE, NHS, Royal College of Pathologists and professional societies. The list of participating societies may be found at http://www.hpa.org.uk/SMI/Partnerships. Inclusion of a logo in an SMI indicates participation of the society in equal partnership and support for the objectives and process of preparing SMIs. Nominees of professional societies are members of the Steering Committee and Working Groups which develop SMIs. The views of nominees cannot be rigorously representative of the members of their nominating organisations nor the corporate views of their organisations. Nominees act as a conduit for two way reporting and dialogue. Representative views are sought through the consultation process. Microbiology is used as a generic term to include the two GMC-recognised specialties of Medical Microbiology (which includes Bacteriology, Mycology and Parasitology) and Medical Virology. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 5 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis SMIs are developed, reviewed and updated through a wide consultation process. Quality Assurance NICE has accredited the process used by the SMI Working Groups to produce SMIs. The accreditation is applicable to all guidance produced since October 2009. The process for the development of SMIs is certified to ISO 9001:2008. SMIs represent a good standard of practice to which all clinical and public health microbiology laboratories in the UK are expected to work. SMIs are NICE accredited and represent neither minimum standards of practice nor the highest level of complex laboratory investigation possible. In using SMIs, laboratories should take account of local requirements and undertake additional investigations where appropriate. SMIs help laboratories to meet accreditation requirements by promoting high quality practices which are auditable. SMIs also provide a reference point for method development. The performance of SMIs depends on competent staff and appropriate quality reagents and equipment. Laboratories should ensure that all commercial and inhouse tests have been validated and shown to be fit for purpose. Laboratories should participate in external quality assessment schemes and undertake relevant internal quality control procedures. Patient and Public Involvement The SMI Working Groups are committed to patient and public involvement in the development of SMIs. By involving the public, health professionals, scientists and voluntary organisations the resulting SMI will be robust and meet the needs of the user. An opportunity is given to members of the public to contribute to consultations through our open access website. Information Governance and Equality PHE is a Caldicott compliant organisation. It seeks to take every possible precaution to prevent unauthorised disclosure of patient details and to ensure that patientrelated records are kept under secure conditions. The development of SMIs are subject to PHE Equality objectives http://www.hpa.org.uk/webc/HPAwebFile/HPAweb_C/1317133470313. The SMI Working Groups are committed to achieving the equality objectives by effective consultation with members of the public, partners, stakeholders and specialist interest groups. Legal Statement Whilst every care has been taken in the preparation of SMIs, PHE and any supporting organisation, shall, to the greatest extent possible under any applicable law, exclude liability for all losses, costs, claims, damages or expenses arising out of or connected with the use of an SMI or any information contained therein. If alterations are made to an SMI, it must be made clear where and by whom such changes have been made. The evidence base and microbial taxonomy for the SMI is as complete as possible at the time of issue. Any omissions and new material will be considered at the next Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 6 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis review. These standards can only be superseded by revisions of the standard, legislative action, or by NICE accredited guidance. SMIs are Crown copyright which should be acknowledged where appropriate. Suggested Citation for this Document Public Health England. (2014). Investigation of Hepatitis. UK Standards for Microbiology Investigations. G 5 Issue 1.2. http://www.hpa.org.uk/SMI/pdf. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 7 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis Scope of Document Type of Specimen N/A Scope This SMI describes the examination of samples for Hepatitis. This SMI should be used in conjunction with other SMIs. Introduction Background The term "hepatitis" means inflammation of the liver. Hepatitis may be caused by viruses, bacteria, drugs, toxins, or excess alcohol intake. Viral hepatitis was first recognized as a distinct clinical entity during the late 18th and early 19th centuries. At that time, reports of scattered outbreaks of this syndrome, referred to as infectious hepatitis, epidemic hepatitis, or catarrhal jaundice, began to appear in the medical literature. The viruses associated with the main clinical feature of hepatitis or jaundice are known as the hepatitis viruses, of which there are currently five described - hepatitis A, B, C, E and D viruses, although other viruses can cause hepatitis as part of a wider illness, for example, CMV and EBV. There are likely to be others, as yet uncharacterised, hepatitis viruses. Viral hepatitis is the most common of the serious contagious diseases, with significant morbidity and mortality resulting from acute illness (hepatitis A, B, C, D, E) and cirrhosis and liver cancer associated with chronic infection (hepatitis B, C, D) in addition to adverse economic effects. Viral hepatitis can be transmitted enterically and parenterally. Enterically Transmitted Hepatitis This occurs when a sufficient amount of the virus enters the mouth to cause infection. This can occur through direct contact with an infected person’s faeces or indirect faecal contamination of food, water supply, shellfish or utensils. This is the main route of transmission for hepatitis A and E. Parenterally Transmitted Hepatitis This is when the disease is transmitted from one person to another by infectious blood and body fluids. The people most likely to become infected include: The family of infected persons (including newborn infants) Injecting drug users (current or previous) Recipients of transfusions, blood or blood products, or transplanted organs prior to 1990 when blood banks began testing for hepatitis C Persons with needle-stick injury Institutionalised persons Persons with multiple sex partners Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 8 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis Persons with tattoos Healthcare professionals – rare in the UK Hepatitis B, C and D are transmitted parenterally, hence including them in the bloodborne viruses. The Liver The liver weighs approximately 1.5 kilograms in the normal adult and it is located in the right side of the abdomen, immediately under the diaphragm. Liver cells perform many diverse chemical activities including: Production of bile, which aids digestion through emulsification of lipids Storage of vital substances such as glycogen, iron, copper, and certain vitamins especially vitamins A, B12 and D Disposal of amino acid wastes, producing urea from ammonia Production of energy through production of glucose from amino acids and breakdown of glycogen, and breakdown of fats Production of protein substances including coagulation factors that regulate blood clotting and plasma proteins notably albumin Filtration of toxic substances that could damage the body if allowed to accumulate All the above aspects of normal liver cell function may be affected by hepatitis, resulting in general systemic symptoms as well as more specific infection and hepatic damage related symptoms and signs (see below). However, the liver is one of the most remarkable and versatile organs in the human body. In fact, it is the only organ able to regenerate itself – up to 80% to 85% of the liver can be destroyed and the remaining tissue may recover. Symptoms of Hepatitis Early symptoms of infective hepatitis are similar to common flu symptoms—general fatigue, joint and muscle pain, and loss of appetite. Nausea, vomiting, and diarrhoea or constipation may follow with a low-grade fever (up to 39°C). As the disease progresses, the liver may enlarge and become tender. Chills, weight loss, and distaste for food and, curiously, cigarettes may occur. Occasionally, the urine and faeces will change colour. There is a spectrum of clinical features found in all types of viral hepatitis, ranging from asymptomatic or subclinical to classical jaundice, and through to acute liver failure and death. Any combination of fatigue, fever, loss of appetite, nausea, vomiting, diarrhoea, or abdominal discomfort may occur. A proportion of patients notice dark urine and light-coloured stools, followed by jaundice, in which the skin and whites of the eyes appear yellow. Itching of the skin may be present. With the onset of jaundice, other symptoms tend to subside. Jaundice is due to accumulation of bilirubin in the blood. It is formed primarily from the breakdown of “haem” in red blood cells and is a waste product. In a healthy individual, there is only a small amount of bilirubin circulating in the blood. However, increased levels of serum bilirubin are found in patients that are suffering from conditions that increase the destruction of red blood cells, and conditions such as Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 9 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis liver disease, where there is a decreased removal of the substance from the blood stream. Bilirubin is a yellow pigment and when accumulated in the blood in abnormally high levels, will make the skin and the whites of the eyes appear yellow. Jaundice (or icterus) is the clinical term for this condition. In some cases the cause of acute hepatitis can be suggested by clinical features and the patient’s history. However, specific laboratory tests must be used to establish a diagnosis. Biochemical Tests “Liver function tests” (LFTs) is a commonly used term to describe a group of blood tests that assess the general state of the liver and biliary system. Routine blood tests can be divided into those tests that are true LFTs, such as for serum albumin, prothrombin time, or serum bilirubin, and those tests that are simply markers of liver or biliary tract disease, such as for the various liver enzymes. Serum bilirubin is generally considered a true test of liver function, since it reflects the liver’s ability to take up, process, and secrete bilirubin into the bile. It is relatively non-specific, however, because many forms of liver or biliary tract disease can result in elevated levels of serum bilirubin. In addition to the usual liver tests obtained on routine automated chemistry panels, physicians may order more specific tests such as viral serologic tests, or tests for autoimmunity, to determine the specific cause of a liver disease. Liver Enzymes There are two general categories of “liver enzymes”. The first group includes alanine aminotransferase (ALT) and aspartate aminotransferase (AST), formerly referred to as SGPT and SGOT. Presence of high levels of these enzymes is an indicator of liver cell damage. The other frequently used liver enzyme tests are for alkaline phosphatase and gamma-glutamyltranspeptidase (GGT). High levels of the latter enzymes indicate obstruction to the biliary system, either within the liver or in the large bile channels outside the liver. ALT and AST enzymes are found in liver cells (hepatocytes). When liver cells are injured, these enzymes are released into the bloodstream. In general, the level of ALT is thought to be a more specific indicator of liver inflammation, since AST may be elevated in diseases of other organs such as the heart. In acute liver injury, such as acute viral hepatitis, ALT and AST are often dramatically elevated; in chronic hepatitis or cirrhosis, the elevation of these enzymes may be minimal, ie 2-3 times normal, or moderate. Mild or moderate elevations of ALT may be caused by a wide range of liver diseases. Determination of ALT and AST level is often used to monitor the course of chronic hepatitis and the response to treatments such as interferon and other antivirals. Alkaline phosphatase and GGT are elevated in a large number of disorders that affect the drainage of bile. Because alkaline phosphatase is also found in other areas, such as bone, placenta, and intestine, an elevated level is not specific for liver disease; measurement of GGT is utilized as a supplementary test to be sure that the elevation of alkaline phosphatase is indeed coming from the liver or the biliary tract, since GGT is not elevated in diseases of bone, placenta, or intestine. Mild or moderate elevation of GGT in the presence of a normal level of alkaline phosphatase Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 10 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis is difficult to interpret and is often caused by changes in the liver cell enzymes induced by alcohol or medications but not associated with injury to the liver. Albumin and Prothrombin Time Two other commonly used indicators of liver function are the serum albumin level and prothrombin time. Albumin is a major protein formed by the liver, and chronic liver disease causes a decrease in the amount of albumin produced. Therefore, in more advanced liver disease, the level of the serum albumin is reduced. The prothrombin time or PT is a test that is used to assess blood clotting. Blood clotting factors are proteins made by the liver; when the liver is significantly injured, these proteins are not normally produced. The prothrombin time is a useful test of liver function, since there is good correlation between abnormalities in coagulation measured by the prothrombin time and the degree of liver dysfunction. Prothrombin time is usually expressed in seconds and is compared to a normal control patient’s PT. Other Biochemical Tests There are many other specialised biochemical tests that may be used to diagnose the cause of liver disease. In addition, there are also specific blood tests used to diagnose the cause of viral hepatitis. Histological Assessment Liver biopsy to remove samples of liver cells (hepatocytes) is generally used to assess liver cell damage. Assessment of these biopsies is highly subjective and qualitative. In an attempt to qualify comparisons of liver histology, scoring systems to quantify all aspects of the necroinflammatory-fibrotic process of chronic hepatitis are used such as the Ishak, Knodell and Metavir scores. Serologic and molecular testing has made liver biopsy less valuable as a diagnostic tool, but it remains important to exclude other liver diseases and to assess the extent of injury. Other, non-invasive, methods of assessing liver fibrosis have recently been introduced, such as Fibroscan (a measure of liver elasticity) and Hepascore based on blood parameters. Notification of Hepatitis Suspected acute infectious hepatitis must be notified by a registered medical practitioner to the proper officer of the local authority in which the patient was seen, as stated in the Department of Health. Health Protection Legislation (England) Guidance 2010; this notification does not need to await laboratory confirmation1. Laboratories are to notify the PHE of cases of hepatitis A, and both acute and chronic cases of hepatitis B and C. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 11 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 1 Hepatitis A 1.1 Basic Virology and Taxonomy The enterically transmitted disease known as ‘infectious hepatitis’ was known for many years before its causal agent was identified in 1973 by demonstration of a 27nm icosahedral virus in patient faeces using immune electron microscopy2. Biophysical and biochemical studies later established Hepatitis A (HAV) as a member of the family Picornaviridae. In 1992 HAV was assigned to a separate genus Hepatovirus, on the basis of a number of differences from the enteroviruses, including stability of the virion at 60°C, lack of reactivity with an enterovirus groupspecific monoclonal antibody, low percentage nucleotide homology with the genome of the enteroviruses and certain differences in the replication cycle. The genome is single stranded positive sense RNA in an unenveloped icosahedral particle3. The capsid comprises 60 copies of each of the four structural proteins VP1-4. A single copy of the genome-linked protein, VPg, is attached covalently to the 5’ end of the RNA. All isolates of HAV are of a single serotype with seven genotypes recognised on the basis of less than 85% nucleotide identity in selected regions of the genome. 1.2 Laboratory Diagnosis HAV is present in stools before the onset of clinical symptoms and can be demonstrated by electron microscopy. Isolation in cell cultures is difficult but when necessary for research purposes virus can be recovered from faeces in primary or continuous lines of primate cells, derived from monkey kidney or from human fibroblasts or hepatoma. Detection of nucleic acid with PCR can be useful as an epidemiological tool and in environmental studies. Antibodies can be demonstrated from the onset of the clinical symptoms. Diagnosis of acute infection requires demonstration of anti-HAV IgM antibodies or seroconversion. IgM antibodies disappear about 3-6 months after the onset of disease. IgG antibodies persist for life and indicate immunity against reinfection. Antibodies are demonstrated by means of EIA. Hepatitis A IgM is also seen after vaccination, and false positive results are not uncommon, especially in older age groups. 1.3 Clinical Symptoms The incubation period is usually 4 weeks (2-6 weeks). The site of primary infection is in the alimentary tract, although the sequence of events that eventually results in hepatitis is not well understood. The short prodromal phase, varying from 2 to 7 days, usually precedes the onset of jaundice. The most prominent symptoms of this phase are fever, headache, muscular and abdominal pain, anorexia, nausea, vomiting and sometimes arthralgia. Hepatomegaly and leukopenia are often present during this period followed by bilirubinuria, then pale faeces and jaundice about 10 days later. The liver is usually enlarged and liver function tests are abnormal with elevated levels of serum alanine aminotransaminase (ALT) and serum aspartate amino transaminase (AST). Jaundice is sometimes accompanied by itching and by urticarial or papular rashes4. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 12 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis Worldwide, most infections occur in young children and are subclinical. The disease is usually mild in older children and young adults with severity increasing with age 5. Symptoms are likely to disappear after 10-14 days except in adults where they may last up to 4-5 weeks. Liver function tests rapidly return to normal when clinical symptoms disappear, but in some cases may persist for several months. Prolonged excretion of virus in faeces has been observed in neonates nosocomially infected in an intensive care unit6. There are no long term sequelae and no chronic carrier state. The infection may produce a relapsing course, and less often an illness with cholestasis as the principal feature4. Acute hepatitis A is a major cause of fulminant hepatitis and has been reported to account for 10% of liver transplants in children4,7. 1.4 Transmission Transmission can be via various routes: The most important means of transmission is via the faecal-oral route from person to person. Transmission is generally limited to close contact especially within families In many countries an important mechanism of transmission is consumption of raw or partially cooked shell fish, for example oysters harvested from waters that have been contaminated with human sewage8. Other foods have been implicated in outbreaks of HAV including milk, strawberries or salad 9. Various waterborne outbreaks have also been noted Several large outbreaks have occurred in injecting drug users (IDUs) and among men who have sex with men10,11 1.5 Epidemiology Hepatitis A is present in a worldwide distribution3. Epidemiological patterns of hepatitis A infection vary in different parts of the world although the differences are linked more to socio-economic conditions than to geographic regions. In developing countries where sanitation and hygiene are poor, subclinical childhood infection is common and 90% of the adult population is immune. In industrialised countries, most people are susceptible to infection and unimmunised travellers to developing countries thus risk contracting the infection. 1.6 Prevention and Control As hepatitis A is transmitted via the faecal-oral route, high standards of public and personal hygiene are still the cornerstone of control. Clean water supplies and efficient modern methods of collection, treatment and disposal of sewage are important. Those employed in the food industry should be required to observe high standards of hygiene. Passive immunisation against hepatitis A is a well established practice. Administration of 0.02-0.06mL/kg normal immunoglobulin before exposure gives 8090% protection for a period of 4-6 months and has been used for travellers to endemic areas. Currently, four inactivated vaccines against HAV are internationally available. All four vaccines are safe and effective, with long-lasting protection. All four vaccines are Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 13 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis similar in terms of efficacy and side-effect profile. The vaccines are given parenterally, as a dose plus booster series, 6-18 months apart. The dose of vaccine, vaccination schedule, ages for which the vaccine is licensed, and whether there are paediatric and adult formulations varies from manufacturer to manufacturer. No vaccine is licensed for children younger than one year of age. Hepatitis A vaccines are all highly immunogenic. Nearly 100% of adults develop protective levels of antibody within one month after a single dose of vaccine. Although one dose of vaccine provides at least short-term protection, the manufacturers currently recommend two doses to ensure long-term protection. Contraindications to hepatitis A vaccination include a known allergy to any of the vaccine components. Combined hepatitis A/hepatitis B vaccines are also available. Recommendations for hepatitis A vaccination in outbreak situations depend on the epidemiology of hepatitis A in the community, and the feasibility of rapidly implementing a widespread vaccination programme. The use of hepatitis A vaccine to control community-wide outbreaks has been most successful in small, selfcontained communities, when vaccination is started early in the course of the outbreak, and when high coverage of multiple-age cohorts is achieved. Vaccination efforts should be supplemented by health education and improved sanitation12,13. Human normal immunoglobulin should be given as well as vaccine to those over 50 years of age and to individuals with chronic liver disease14. At present no specific antiviral therapy is available for hepatitis A. 2 Hepatitis B 2.1 Basic Virology and Taxonomy The hepatitis B virus (HBV) is the human representative of the Hepadnaviridae family, a group of small DNA viruses which infect a range of animal species. In many of these animals eg the Eastern woodchuck, the Beechey ground squirrel, and the Pekin duck, the viruses are hepatotropic15. The hepatitis B virus is an enveloped virus with a partially double-stranded circular DNA genome of 3.2 kb; the plus strand is incomplete prior to replication. Eight genotypes of HBV, designated A-H, are described by sequence analysis, based on nucleotide variation of over 8% across the genome16,17. The genome of HBV contains four overlapping open reading frames: S, C, P, X. The S ORF codes for surface (envelope) proteins, which can be translated from three initiation points to give three proteins of varying size – L (large) includes all three regions of the S ORF ie pre-S1, pre-S2, S, M (major) includes pre-S2 and S, and S (small) includes the product of the S region of the S ORF. The surface proteins together are designated hepatitis B surface antigen HBsAg. Four principal subtypes of virus can be recognised by HBsAg heterogeneity in response to anti-HBs binding, subtypes adw, ayw, adr, ayr15,18. The C gene similarly has two initiation points, the longer comprising the products of pre-C and C (this long translational product is cleaved to produce the soluble core component HBeAg) and the shorter the nucleocapsid C (core) protein HBcAg. The P ORF codes for viral polymerase which includes a reverse transcriptase function. The X gene has multiple functions but its role is not well characterized; it may have a role in oncogenicity of HBV. The replication cycle begins with closure of the gap in the + DNA strand and the DNA moves to the cell nucleus in a covalently closed circular form (ccc DNA). The Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 14 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis negative strand of ccc DNA is transcribed by cellular RNA polymerase II to pregenome RNA (longer than genome length) and shorter subgenomic transcripts, which serve as mRNA producing P and C proteins and the pre-S and S envelope proteins respectively. The polymerase P has reverse transcriptase activity and transcribes DNA from the RNA template in the new virions. In the nucleus ccc DNA copies accumulate and remain19,20. 2.2 Laboratory Diagnosis Testing for hepatitis B infection is based on detection of HBsAg in the first instance. HBsAg will be detectable in the blood if there is current hepatitis B infection. For many years very sensitive immunoassays have been available for this purpose including radioimmunoassay (RIA) and a range of enzyme immunoassays (EIA). Many tests now are done using EIA tests on automated analysers, for example where HBsAg bound by anti-HBs antibody on microparticles is detected by a chemiluminescent reaction21. Mutations in the ‘S’ gene can lead to reduced sensitivity or failure to detect HBsAg, especially if monoclonal anti-HBs is used for both capture and probe in the immunoassay22. Although the published specification for the minimum level of sensitivity for HBsAg detection in the UK National Blood Service is at present 0.2 IU/mL there is general acceptance that assays should detect 0.05 IU/mL HBsAg or less23. Assays should be CE marked. Because of the important implications of a positive finding the initial HBsAg reactivity should be confirmed by a neutralisation test or by repeat HBsAg reactivity in a second assay, and a second sample tested as well. HBsAg appears about 2-4 weeks before the ALT rises. However, HBV DNA can be detected about 3-4 weeks earlier24. Detection of HBsAg by immunoassay at a single time point does not give information on the duration of infection. Its presence in samples 6 months or more apart defines ‘chronic’ hepatitis B infection. Presence of detectable IgM antibody to hepatitis B core antigen (anti-HBc IgM) is used to help determine whether the HBsAg is associated with an acute or a chronic infection. In acute hepatitis B anti-HBc IgM appears in high levels (>50 Paul Ehrlich units/mL) in both symptomatic and asymptomatic individuals and is commonly said to be detectable for about 3-6 months. However, this antibody however is a marker of HBV activity and its appearance and disappearance are quite variable. In a study using RIA anti-HBc IgM appeared in most within a week of symptoms, but was delayed to 2 weeks in about 8%25. Median duration of anti-HBc IgM was 32 weeks, with a range from two weeks to over two years; 14% had detectable IgM for over one year25. Thus anti-HBc IgM results need to be interpreted with caution and with the clinical and biochemical information. Because it can be found in chronic hepatitis B with active viral replication, quantitation of anti-HBc IgM is also important in differentiating acute and chronic hepatitis B with high levels of anti-HBc IgM correlating with acute infection rather than chronic infection with exacerbation of symptoms26,27. HBsAg levels tend to be higher in acute infection, and in early infection determination of anti-HBc avidity might also be a useful test27. Antibody to hepatitis B core antigen (anti-HBc) was recognised as persisting after hepatitis B infection by counterimmunoelectrophoresisand, as it persists generally for lifeis used as a marker of infection with HBV at some time28,29. Most commercial tests are total antibody tests detecting both IgG and IgM, most are competitive assays. It is a useful test for validating a positive HBsAg result, and is found together with anti-HBs antibody in past resolved infections. Isolated anti-HBc found in the Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 15 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis absence of HBsAg or any other serological markers of hepatitis B infection is difficult to interpret. Detection of circulating HBeAg, a soluble derivative of the core ORF product, occurs when virus is actively replicating in the liver, so it is associated with high levels of HBV DNA in the blood and high potential infectivity. Its association with progressive liver disease varies with the stage in the natural history, so both HBeAg and antiHBe antibody are monitored to follow the stage of infection and response to treatment. Both HBeAg and anti-HBe antibody are detected using enzyme immunoasays, with anti-HBe tests usually in a competitive format24. Both HBeAg and anti-HBe may coexist30. Anti-HBs assays use HBsAg bound to solid phase to capture the antibody. Automated assays usually use recombinant antigen as capture antigen and for the labelled probe24. Anti-HBs is used to monitor post-vaccination immunity, where an initial level of 10 mIU/mL is recognised as conferring protection against HBV31. It is also used as a marker of resolution of infection (absent HBsAg, positive anti-HBc) but HBsAg and anti-HBs can coexist, particularly where the subtype specificities are different, so the presence of anti-HBs cannot exclude chronic hepatitis B infection30,32. Hepatitis B DNA is measured by PCR (both in house and commercial assays are available) or by other amplification assays such as branched chain DNA. Assays may not be directly comparable and vary in performance and dynamic range. Detection of HBV DNA is useful in early diagnosis in at risk individuals before HBsAg appears, and for monitoring viral load during therapy; the best endpoint for management is to reach a level of HBV DNA which is undetectable by current methods with a sensitivity of 10-15 IU/mL33,34. HBV DNA is also a significant prognostic marker for cirrhosis. In the UK, health care workers (HCW) who are HBsAg positive and HBeAg negative must be tested for HBV DNA level before doing blood exposure prone procedures. A level below 103 genome equivalents/mL is acceptable but must be rechecked yearly35. If viral load is between 103 to 105 geq/mL, the HCW may work while taking antiviral therapy provided the HBV DNA level is less than 103 geq/mL on treatment and HBV DNA monitoring is done at 3monthly intervals36. Assays for detecting ccc DNA in blood have been developed but as yet their clinical relevance is uncertain37. 2.3 Transmission HBV is transmitted by blood and blood products and by other secretions. Blood represents a particular risk, as there may be over 108 virions/mL, which can be directly visualised by electron microscopy38. In the past, blood transfusion was an important route of transmission, with 6% of multiply transfused individuals acquiring hepatitis B infection prior to the reduction in risk resulting from the introduction of screening for HBV in the early 1970s39. Screening for HBsAg alone does not entirely eliminate HBV transmission since there are occasional blood donations where the donor has early hepatitis B before appearance of HBsAg. In an experimental animal model, blood taken early in infection prior to appearance of anti-HBc is significantly more infectious than that taken later in the acute phase 40. Infectivity persists throughout the course of active infection. Transmission from a surgeon with chronic Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 16 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis HBV to a patient in the UK has been associated with levels of circulating HBV DNA as low as 4 x 104 copies/mL40. Those with chronic hepatitis B with undetectable HBsAg, so called ‘occult’ hepatitis B, are generally positive for anti-HBc antibody and sometimes anti-HBs, but with low levels of HBV DNA consistent with ongoing viral replication40. Measures to exclude such individuals from the blood donor pool require use of anti-HBc testing and HBV DNA testing39. Vertical transmission from mother to baby of HBV is one of the most important routes of HBV transmission worldwide. Most transmission occurs in the intrapartum period, with exposure of the baby to maternal blood and genital secretions. Less than 3% of cases of neonatal infection arose from transmission in utero in a Taiwanese study39. Although possible micro-leakage of blood across the placenta in threatened abortion or premature labour might be a factorthis was not noted in other studies 41,42. The principal risk of vertical transmission is from HBeAg positive mothers from whom transmission to the neonate occurs in about 85% of cases, compared to 31% from HBeAg negative women43. There is an increased risk of transmission to the neonate in acute hepatitis B during pregnancy, particularly with hepatitis B acquired later in pregnancy44. Breast milk contains HBV in some 70% of mothers with circulating HBsAgand can presumably transmit infection to the neonate45. In general the risk from this route is relatively unimportant as the risk to the baby during the birth process is greater, and prophylaxis is given to the at risk baby. In the developed countries, hepatitis B is principally associated with sexual intercourse, both heterosexual and between men, and injecting drug use (IDU). Incidence of acute hepatitis B in IDU may have fallen in parts of the UK where active vaccination programmes have been pursued but overall in the UK this is not the case, as vaccination uptake among IDU varies widely46. In 2003, drug use was the commonest risk factor and an increase in seroprevalence to anti-HBc from 3.4% in 1997 to 10% in 2006 suggests that this is a continuing risk47. In the North West of England, for example, the most likely transmission route was not known in two thirds of cases; of clear risk factors homosexual sex was the most common (17%), followed by heterosexual exposure (9%)46. The majority of cases are in males, predominantly in the 25 to 45 year age group46,48. 2.4 Clinical Features The incubation period for hepatitis B infection is between 2 and 6 months, with higher viral inocula associated with shorter incubation periods. Symptoms of infection vary with the age at which the individual is infected. Acute infection is usually asymptomatic in the infected neonate and young child, but even in infections acquired in adult life, asymptomatic infection is common (around 50%). If symptoms occur, the patient may have jaundice, feel nauseated and lethargic, and may have right upper quadrant discomfort or pain. Jaundice is seldom marked but there may be lightening of faeces colour and darkening of urine, and sometimes pruritus towards the end of the symptomatic illness. Acute hepatitis B is preceded by a serum sickness-like prodrome with fever, arthralgia or arthritis, and rash, in up to 10% of cases. These symptoms occur about 2 weeks prior to the onset of jaundice, and usually subside soon after jaundice Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 17 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis appears. Another characteristic rash, papular acrodermatitis (Gianotti-Crosti syndrome), is seen in childhood in association with hepatitis B; maculopapular erythematous nonpruritic lesions occur on the face and extremities, and persist for 15-20 days19. Also included among the extrahepatic manifestations of hepatitis B is membranous glomerulonephritis49. Although more common in children it is more likely (30-60%) to spontaneously resolve in childhood, whereas one third of cases in adults progress to renal failure. Resolution is generally associated with HBeAg clearance 49. Fulminant hepatitis occurs in less than 0.5% of acute cases but is a severe lifethreatening condition. The proportion of cases of acute liver failure due to hepatitis B has been declining in recent years and it now causes less than 10% of cases50. It is important to note that HBsAg may be weak or undetectable in some cases of fulminant infection, but that anti-HBc IgM and HBeAg are generally found51. This is usually due to an enhanced immune response, but a proportion may be due to failure of HBsAg assays to detect ‘S’ mutant virus52. Chronic hepatitis B is defined (arbitrarily) as persistence of detectable HBsAg for over 6 months. This generally correlates with the persistence of raised liver transaminases for over 6 months, consistent with ongoing viral replication and liver damage. The course of chronic infection can be divided into several phases, reflecting the dynamics of virus replication and host immune response. These are not necessarily sequential. The phases are: 1. immune tolerance; 2. immune elimination (immune reactive or immune clearance); 3. low replication (inactive); 4. HBeAg-negative chronic hepatitis B (reactivation)33,53. In the phase of immune tolerance, high levels of HBV DNA and HBeAg are seen in the blood but there is little or no liver damage. The ALT remains normal and there is little or no inflammation seen on liver biopsy. This phase is characteristic of hepatitis B acquired in the perinatal period, generally by vertical transmission, and it may last 10-30 years54. After some time immune tolerance is lost and an immune response occurs against hepatitis B infected hepatocytes. This immune elimination (immune clearance or immune reactive) phase, which can last weeks or years, is characterised by falling HBV DNA levels, raised or fluctuating ALT, and histological evidence of necroinflammation with progression of fibrosis54. There is increased loss of HBeAg with seroconversion to anti-HBe antibody positivity; such seroconversion is often accompanied by a symptomatic exacerbation of hepatitis. The low replication inactive carrier state may follow immune clearance and seroconversion to anti-HBe positivity. Liver enzymes – ALT and AST – are normal and HBV DNA level is low or undetectable. Prognosis is favourable with low rate of progression to cirrhosis and HCC. Loss of HBsAg occurs in about 1-3% of chronic carriers per year. HBeAg negative chronic hepatitis B occurs in patients in whom mutations appear in the pre-C or basal core promoter regions giving rise to virus that cannot express HBeAg. The most common mutation is the A1896G pre-C mutation55. This cannot occur in genotype A viruses, although basal core promoter mutations (notably the double mutation A1762T plus G1764A) are seen as in other genotypes 56. As these mutations are selected by anti-HBe antibody, the typical serological pattern is HBsAg positive, anti-HBc positive, HBeAg negative, anti-HBe positive. During this phase reactivation can occur, giving fluctuating liver enzyme levels and HBV DNA levels, so Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 18 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis monitoring regularly throughout this period is essential in differentiating individuals with active disease with risk of progression to cirrhosis and those with inactive antiHBe positive disease. In what is termed by some a fifth phase, the state after loss of HBsAg, prognosis is generally good. However, low level HBV replication can persist in the liver despite undetectable HBV DNA in the blood. These individuals are HBsAg negative, are positive for anti-HBc and may or may not have anti-HBs antibody. Reactivation of infection may occur if the patient is immunosuppressed, particularly if monoclonal antibodies targeting the immune system eg rituximab are given57,58. Cirrhosis is the result of progressive liver damage, a liver with abnormal architecture and loss of function, characterised by proliferation of fibrous tissue and scarring, with nodular regeneration of hepatocytes. Principal causes include hepatitis B, hepatitis C, and alcohol. Some 15-40% of patients with chronic hepatitis B develop liver damage16. A sustained high HBV DNA viral load seems to be a major predictor of progression to cirrhosis and hepatocellular carcinoma59-61. Hepatocellular carcinoma (HCC) may arise after many years of chronic infection with hepatitis B. In a large Taiwanese study HCC developed at a rate of 1169 per 100000 person years for those who were HBeAg positive and at 324 cases for those with HBeAg negative HBV. The pathogenesis of HCC is not entirely clear, but the long period required for its development suggests that several low probability events must occur. Continued proliferation of hepatocytes in response to loss of infected cells may be one factor, and it is likely that the X protein of HBV has oncogenic properties19,62. Risk of HCC increases with higher HBV viral load, especially for viral load above 20000 IU/mL60. Risk may vary among hepatitis B genotypes. Cofactors are also important in development of HCC in chronic hepatitis B. These include aflatoxins, alcohol and coinfection with hepatitis C63. 2.5 Epidemiology The prevalence of hepatitis B varies markedly around the world, reflecting to a large extent differing modes of transmission. Rates of seroprevalence for anti-HBc antibody, a marker of infection at any time, vary for instance from 1-2% in the UK, 0.5-1% in the Netherlands, to 96% in China and South Korea, while Ag positive rates can reach over 5-10% in some regions, particularly in China and South East Asia, Africa and Oceania64. In the Far East, transmission occurs in the neonatal or perinatal period, whilst in African there is a more complex pattern involving perinatal transmission, and horizontal transmission in early childhood through close contact with abraded skin and during tribal rituals65. Infection around the time of birth is highly efficient (around 90% transmission rate from an HBeAg positive mother to the infant and is likely to result in chronic infection in the child66. Transmission in utero is relatively uncommon (less than 5% of mother/infant transmissions)66,67. Genotype distribution varies geographically; genotype A predominates in Northern Europe and North America, B in the Far East, C in West and Central Africa, D especially in North Africa, the Mediterranean littoral, the Middle East, and Asia, F in indigenous peoples of North and South America, G in France and the USA (rarely found and then usually in association with genotype A virus), H in Central America17. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 19 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 2.6 Prevention and Control Hepatitis B is notifiable in the UK; both acute cases and chronic cases should be notified, so that risk factors can be determined and screening and prophylaxis may be offered where appropriate14. 2.6.1 Vaccination Spread of hepatitis B infection can be controlled by vaccination. Given the global importance of hepatitis B infection, most countries have opted for universal vaccination against this virus68,69. Countries with very low overall prevalence such as the UK have preferred targeted vaccination of high risk groups. A number of commercial vaccine preparations are available, generally containing a recombinant yeast-derived HBs protein segment which includes the major immunodominant ‘a’ epitope. The failure of currently licensed vaccines to include pre-S may be one factor in suboptimal responses but other important factors include increasing age, injection into fat, smoking, alcohol use, renal impairment and HIV infection70-72. Hepatitis B Immunoglobulin (HBIG) provides passive immunity for post exposure prophylaxis for known high risk exposures. It is used as an adjunct to vaccination for example in non immune adults, or for individuals at risk who have failed to mount a satisfactory immune response after a vaccine course. HBIG should ideally be given intramuscularly within 48 hours of exposure but this can be extended to a week particularly after sexual exposure73. For babies born to mothers who are HBeAg positive, HBeAg negative and anti-HBe negative, or anti-HBe positive with HBV DNA >106 IU/mL HBIG is offered together with vaccine in the UK; in this situation HBIG should be given within 24 hours of delivery at a site different from that used for the first dose of vaccine73,74. HBIG is also used for prevention of recurrence of hepatitis B after liver transplantation for chronic hepatitis B infection, together with antiviral agents such as lamivudine or adefovir33. 2.6.2 Treatment Treatment is considered for severe acute hepatitis B and for chronic infection. A detailed description of treatment regimens and monitoring response is outside the remit of this document. There are a number of current published guidelines which should be consulted on this subject. They include recommendations on specific antivirals from the National Institute for Health and Clinical Excellence (NICE), and comprehensive guidelines on management from bodies including the European Association for the Study of the Liver, the Asian-Pacific Association for the Study of the Liver, and the American Association for the Study of Liver Diseases 33,65,75. 3 Hepatitis C 3.1 Basic Virology and Taxonomy Hepatitis C virus (HCV) is a blood-borne virus of the Flaviviridae family and the only member of the hepacivirus genus76,77. It is a single-stranded, positive sense enveloped RNA virus with a genome of approximately 9600 bases76. The virus was discovered in 1989 and subsequently the genome was patented by the Chiron Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 20 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis Corporation, the first virus to have ever been patented. Prior to this discovery, a class of blood-borne hepatitis transmissible by blood known to be negative for hepatitis A and B and known as non-A, non-B hepatitis (NANBH), was recognised to cause hepatitis after blood transfusion and use of blood products78. Only a small percentage of HCV early infections cause the patient to seek medical intervention79. It is estimated that in 50-85% of all cases the virus is not cleared, leading to chronic infection, even though there is evidence of seroconversion 78,79. In the case of HCV, the relationship between neutralising antibodies and the control of viraemia is more complex due to the enormous genetic variability of the virus (much greater than the variability seen with HIV), particularly in the E2 envelope glycoprotein region where many antibodies are targeted79. This variability leads to HCV being regarded as existing in an individual as a ‘quasispecies’, as essentially HCV is likened to a horde of closely-related variants circulating in the one infected host76. The window between detection of HCV RNA and antibodies to HCV can be months, with an average of 60 days80. There are six genotypes of HCV (1-6) with multiple subtypes, although a new genotype has been proposed as genotype 7 81,82. Transmission of hepatitis C virus is through contact with blood or blood derived products. Therefore, the main risk factors are contaminated blood supply, injecting drug use, unsafe therapeutic needle use, or medical procedures83. There is a clear divide between the modes of transmission depending on whether in the developed or developing world83. In the developed world, the primary mode of transmission is through injecting drug use, particularly since blood and blood derived products are so thoroughly screened for HCV. In developing countries, where supplies of sterile syringes are short or non-existent, unsafe therapeutic needle use is much higher than in the developed world, where a person may receive multiple injections adding a cumulative risk of acquiring HCV83. An example of this is seen in countries such as Egypt, where prevalence of HCV is extremely high in the older population, probably due to shared syringes during nationwide schistosomiasis treatment policies that date back to the 1920’s84. Blood transfusion related HCV incidence in Egypt has become almost non-existent since the introduction of all-volunteer donor systems, screening of blood using highly sensitive and specific assays for anti-HCV and HCV RNA, and detailed screening of donors for HIV risk factors83. In fact, the safety of donated blood in the developed world is at a level where methods to measure risk are not sensitive enough to be useful in the estimation of transfusion-related transmission of HCV85. Most countries in the developing world, however, do not screen blood donors for HCV83. Other sources are occupational, perinatal and sexual exposures, but these occur very infrequently. Prevalence of HCV infection is higher in persons having received blood transfusions, tissue/organ transplants or blood products such as haemophiliacs, prior to 1992 before sensitive screening tests were available86. 3.2 Laboratory Diagnosis Laboratory diagnosis is based upon the detection of antibodies for the virus using serological methods followed by the detection of HCV RNA using molecular methods to determine viraemia76,86. Antibodies to HCV are detected using EIA. Assays have progressed in development to second generation (core proteins and non-structural proteins 3 and 4) and third generation (inclusion of antigen non-structural protein 5)81. Assays are now available that will detect free HCV core antigen in serum/plasma and display similar sensitivity to some molecular methods 87. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 21 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis The presence of HCV in blood is a good marker of replicating virus80. Detection is possible 1-3 weeks after infection with the use of molecular methods to detect HCV RNA. Molecular methods can also be used to distinguish between the genotypes and subtypes, of particular importance as some genotypes are more prevalent than others in different global regions81. Knowledge of the genotype is crucial in the treatment selection process81. Real-time PCR provides an excellent quantitative range which allows determination of viral load up to 7-8 Log10 IU/mL and as low as 10-15 IU/mL to assess treatment efficacy80. Qualitative PCR is not often used even though sensitivity is often considered superior to quantitative real-time PCR; there may be a role for a qualitative assay in cases where extremely low levels of viraemia are suspected 86. The presence of RNA and antibodies to HCV do not give an indicator as to whether it is acute or chronic infection80. However, antibodies do take a long time to develop and are therefore only detectable in 50-70% of symptomatic acute infections88. 3.3 Clinical Features HCV is often asymptomatic (85-90% of cases) and therefore is rarely diagnosed during the acute phase of infection81,88. Symptoms may include jaundice, nausea and malaise; however, although fulminant hepatitis is extremely rare, it has been described during the first 8 weeks of infection81. A majority of acute cases go on to become chronic and, without treatment, the majority of chronic cases do not clear the virus81. HCV RNA detectable for longer than 6 months is considered chronic infection with sequelae including end-stage cirrhosis and hepatocellular carcinoma (HCC), although the length of time this may take can vary from less than 20 years to greater than 30 years81. Co-infection with HIV displays accelerated disease progression as HCV behaves as an opportunistic infection83. Co-infection with hepatitis B virus displays more severe liver disease progression than either single infection83. Increased alcohol intake also accelerates the progression of chronic HCV towards end-stage cirrhosis and HCC83. Transplantation is often necessary due to HCV infection related liver failure, leading to further complications with progression towards cirrhosis in over 25% of patients within 5 years of transplantation89. 3.4 Epidemiology HCV is a leading cause of chronic liver disease and a serious public health problem with high global prevalence estimates at around 2-3%, or around 120-180 million people90,91. Approximately 1.5 million people die of viral hepatitis related chronic liver disease80. This includes end-stage cirrhosis and HCC. In the UK, data shows that HCV remains common in the injecting drug use population, with 10% and 70% antibody positivity prevalence for those injecting for 2 years and 15 years respectively92. There is an estimated 10 million people worldwide believed to be co-infected with HIV, possibly linked to high-risk traumatic sexual practices and drug use in men who have sex with men (MSM)93. 3.5 Prevention and Control Knowledge of the genotype can be critical in the treatment schedule chosen, as well as the rate of success of treatment81. Treatment involves use of interferon usually in Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 22 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis combination of ribavirin88. Of the six major genotypes described genotypes 2 and 3 respond more favourably to treatment with interferon and ribavirin than genotype 1. The recommended length of treatment in the UK is 24 weeks for genotypes 2 and 3, and 48 weeks treatment for genotype 1 and remaining genotypes. Pegylated interferon is often used to increase the time it takes to clear the drug and therefore reduce the number of times interferon needs to be taken to once per week78. Ribavirin is required to be taken daily88. Infection is considered eradicated when there is no HCV RNA detected for more than 6 months after cessation of therapy (also known as a sustained virological response)86,91,94. All the major genotypes have multiple subtypes but there is little evidence of differences in response among these. Other antivirals specific for hepatitis C are being trialled. The protease inhibitors telaprevir and boceprevir (which inhibit the NS3/4A protease) appear to be very active. As resistance develops readily with monotherapy, telaprevir must be combined with ribavirin and interferon95. Vaccines that induce protection via the production of neutralising antibodies have two hurdles to overcome – heterogeneity (the various genotypes) and mutability (the rapid generation of antigenically diverse E2 envelope glycoproteins) of HCV79. There are a host of vaccine development initiatives that aim to prevent initial infection, viral persistence or clear the virus entirely. 4 Hepatitis D96,97 4.1 Basic Virology and Taxonomy HDV is closely associated with HBV because it is a defective virus that requires a helper function provided by HBV. It is unique among animal viruses and has similarities with both viroids and virusoids of plants in its structure and mode of replication. It is classified in the floating genus Deltavirus. The virus is an enveloped, spherical particle with an average diameter of 36 nm. The outer capsid is HBsAg, and the interior nucleocapsid is the genome complexed with hepatitis delta antigen (HDAg) which is the product of the sole structural gene. The HBsAg is responsible for attachment to the host cell and the HDAg for nuclear localisation. The genome is single stranded, circular negative-sense RNA of 1679 nucleotides. The genome has extensive secondary structure due to intramolecular base pairing. The HDV genome can function as a ribozyme, capable of self-cleavage. This is a necessary part of the complex rolling circle mechanism of replication. The virus exists as various genotypes, originally three major genotypes and recently seven genotypes are described. Within an infected patient the virus is a mixture of different related species known as quasispecies. 4.2 Laboratory Diagnosis The most sensitive method of laboratory confirmation is detection of HDV RNA by PCR using primers from the most conserved sequences. HDAg and HDV antibodies (IgG or IgM) in the serum can be detected by EIA. High titres of HDV IgG are found during chronic infection, and IgM may also persist; but EIA is less sensitive than PCR in the acute phase of infection. Quantitation of the viral load has been used in studies of antiviral therapy. Simultaneous infection with HBV and HDV can be differentiated from super-infection of a chronically HBV infected patient by the appearance of anti-HBV core IgM with anti-HDV IgM, or by the appearance of HDV RNA without detectable HDV antibody. Immunohistochemical staining for HDAg is Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 23 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis still used, and this is the technique that was responsible for the discovery of hepatitis D in the 1970s. 4.3 Clinical Symptoms HDV may be transmitted with HBV to a susceptible person, this is known as coinfection. Super-infection is infection of an individual already chronically infected with HBV. Co-infection may result in an acute HBV / HDV infection that is mild to severe. Most cases are clinically indistinguishable from acute B virus infection. However, acute HDV / HBV co-infections have a much greater risk of evolving into acute liver failure than acute HBV infection. After super-infection, chronic co-infection causes a rapidly progressive chronic liver disease in 90% of cases with spontaneous resolution being rare. Thus superinfection has a poorer prognosis than chronic HBV infection alone, with higher rates of cirrhosis, hepatocellular carcinoma and death. It may present as an exacerbation of an already diagnosed HBV disease or lead to the presentation and diagnosis of a previously asymptomatic and undetected HBV infection. The latter situation may be initially considered a case of acute hepatitis B until negative anti-HBc IgM test results and positive HDV test results indicate the true picture. Most patients with chronic HDV infection have antibodies to HBeAg and low levels of HBV DNA. Different from HDV negative chronic hepatitis B, there is no correlation between serum HBV DNA and ALT levels in patients with chronic hepatitis D, suggesting that the liver damage in these patients is mainly caused by HDV. High doses of interferon have been reported in a randomised clinical trial to improve survival, reduce fibrosis and reduce viral replication98. Nucleoside analogues given without interferon have so far been ineffective. HDV genotype I has been independently associated with a poorer prognosis99. 4.4 Epidemiology It is reported than 15 to 23 million of the estimated 460 million people chronically infected with HBV are also infected chronically with HDV, that is up to 5% 100. The prevalence of HDV varies widely geographically, and does not always mirror the prevalence of HBV. The literature suggests a declining trend at least in southern Europe. Increasing use of universal hepatitis B vaccination will be expected to reduce the prevalence of HDV also. HDV can be spread by blood contact in injecting drug users, though blood product screening for HBV has reduced the iatrogenic route. Molecular epidemiology has confirmed sexual transmission and an inapparent parenteral route is suggested to be important in families in endemic areas. Foci of infection are identified in southern Europe (including Italy and Albania), the Middle East, Saudi Arabia, northern India, Russia, West Africa, Japan, Taiwan, some South Pacific islands and South America. 4.5 Prevention and Control There is currently no vaccine against HDV itself, but vaccination against HBV also protects against HDV. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 24 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 5 Hepatitis E Hepatitis E infection is increasingly recognised in the UK, including a high proportion of cases without the traditional risk factor of travel abroad to a high incidence area. It is important to consider hepatitis E as a potential cause of viral hepatitis early on in the assessment of the patient. 5.1 Basic Virology and Taxonomy Following the development of sensitive and specific tests for acute hepatitis A and B infections, it became clear from clinical, epidemiological, and laboratory data that another enterically transmitted viral hepatitis agent existed101. This agent was descriptively called epidemic non-A non-B hepatitis virus. Characterisation of this new virus began in 1983 when Dr Balayan infected himself using stool associated with non-A non-B hepatitis from an outbreak in the former Soviet Union 102. He subsequently developed hepatitis and analysed his own samples to visualise the virus by immune electron microscopy, and to reproduce hepatitis in Macaca fasicularis. Cloning and sequencing of the virus was reported in 1990 and 1991respectively, and it was subsequently named hepatitis E virus103,104. Hepatitis E virus is classified as the sole member in the family Hepeviridae, genus Hepevirus105. A typical virion is non-enveloped, 32-34nm diameter containing a positive sense single stranded RNA genome of around 7.2Kb. The genome has three open reading frames encoding non-structural proteins (ORF1), the capsid (ORF2), and a small protein of unclear function (ORF3). There are four main genotypes (G1-4), G1 and 2 are likely to be exclusive human pathogens, whereas G3 and 4 are probably zoonotic with swine as the main host. An avian HEV is proposed as a fifth genotype106. G1-4 show geographical variation in prevalence. 5.2 Laboratory Diagnosis The clinical presentation of acute symptomatic hepatitis E is difficult to distinguish from that of any other viral hepatitis. Whilst epidemiological features may suggest HEV infection, laboratory tests should be performed to confirm any clinical diagnosis. Virus can be detected in stool by immune electron microscopy, and viral nucleic acid can be detected in stool and blood by RT-PCR. Some cell lines support HEV replication but virus isolation is not used as a diagnostic technique. Non-human primates are susceptible to HEV and have been used in research only; no animal model shows the spectrum of HEV infection seen in humans. A number of antibody detection formats have been developed but the commercially available systems are solid phase EIA (IgG and IgM). These assays are usually based upon antigens derived from G1 and 2 viruses and although there is probably a single HEV serotype for human infection, they may be suboptimal for G3 virus endemic in the UK. In common with many of the other viral hepatitides, the maximal liver injury appears to coincide with development of the immune response, leading to a typical picture of the relationship between the timings of virus detection, liver injury, and antibody detection. Available data is potentially complicated by variable assay performances, in general and via genotype specific differences. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 25 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis HEV is detectable in the stool from around 1 week before, and up to three weeks after, symptoms appear. There are reports of more prolonged faecal shedding of virus107,108. Viraemia detectable by RT-PCR probably mirrors the period of faecal shedding, but may persist for much longer107,108. IgM antibody becomes detectable just prior to the maximal liver injury, potentially coinciding with the onset of symptoms, and remains at detectable levels for several months. Some patients do not mount a detectable IgM response. There is a spectrum of IgM persistence but over 50% of patients will be negative six months after onset. IgG antibody appears shortly after IgM (when present), persisting for several years in the majority. Laboratory diagnostic criteria can be drawn up to account for the variability in natural immune responses and assay performance, particularly in the setting of low endemicity. An acute case (symptomatic presentation) is best defined by being HEV RNA positive or by showing IgG seroconversion. Other combinations of IgG and IgM results may be best interpreted according to antibody titre/ reactivity levels. IgG avidity may be useful in inconclusive cases109. The histopathological appearance of acute infection occurring in hyperendemic regions is of focal hepatic necrosis, ballooned hepatocytes and a lymphocytic inflammatory infiltrate110. A cholestatic picture may occur but it is uncommon. The histopathological appearance of acute infection acquired in the UK may be different. 5.3 Clinical Features There is a spectrum of outcomes after HEV infection, ranging from asymptomaticand also implied by seroprevalence data to acute liver failure111. Although it is not possible to predict the course of the infection using genotype data or host features, a striking feature of hepatitis E is the enhanced severity in late pregnancy. HEV infection in the immunocompetent is believed to be self-limiting, without chronic infection. Several reports have been published describing likely chronic hepatitis E in immunocompromised organ transplant recipients112-114. The extent and clinical relevance of this condition are yet to be fully defined. Data from volunteer studies, case reports, and outbreaks have defined the typical features of symptomatic HEV infection occurring in hyperendemic areas115,116. Although these features can be applied to hepatitis E indigenous to developed countries, there may not be complete concordance. Incubation period: 15 to 60 days The spectrum of illness is wide. Typically a self-limiting viral hepatitis occurs with recovery after 2-3 weeks. Asymptomatic and fulminant hepatitis are both described. Rarely there is a prolonged illness lasting a few months, or a cholestatic picture. A prodrome or pre-icteric phase may occur lasting 3-4 days on average (range 1-10) characterised by epigastric pain, nausea, malaise, mild pyrexia (<38°C). The icteric phase starts abruptly with jaundice, pale stools and dark urine, often accompanied by fever and arthralgia. The overall mortality rate for HEV is 1-3% but in pregnancy this increases to 5–25%. Pregnant women appear to be more frequently affected by hepatitis E during an outbreak, and the disease is often more severe, especially in late Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 26 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis pregnancy117,118. Vertical transmission may be relatively common and associated with poor fetal outcome117,119. The reasons for this excess mortality have not been clearly defined, complicated by the lack of an appropriate animal model. Co-infections with hepatitis A and superinfection on a background of chronic viral hepatitis (HBV and HCV) are likely to be common in areas of endemicity and they may not run a typical course or may lead to hepatic decompensation 120. However, it is unclear whether the severity is always increased. The clinical management of hepatitis E is based upon assessment of severity of liver damage and associated effects, influencing the degree of supportive measures needed. No specific antiviral therapy is available. Liver transplant is an option for acute liver failure. Acute hepatitis E is a notifiable disease14. 5.4 Epidemiology HEV causes epidemics and sporadic cases, and is responsible for the majority of acute clinically apparent cases of hepatitis in central and south-east Asia. Transmission is faecal-oral, often by contaminated water, and potentially via food. Epidemics associated with poor water quality have occurred in many areas of the world (Pakistan, India, South-East Asia, Nepal, Africa). The largest epidemic described occurred in China, with over 119,000 cases of jaundice121. No outbreaks of hepatitis E have been described in the USA, Canada, Europe, or developed areas of Asia. Sporadic cases of infection occur worldwide, often as the result of travel to an endemic area but increasingly recognised as indigenous. Some countries, such as Egypt appear to have a high number of sporadic cases yet have never reported an epidemic122. Estimates implicate HEV as the cause of around 2 million cases of hepatitis a year in India, where it is a common cause of jaundice123. Blood-borne transmission is described, reflecting the viraemic phase of the illness, however, this is thought to be an uncommon event124. Seroprevalence studies investigating the potential for bloodborne transmission involving injecting drug users, haemodialysis patients, and patients infected with HBV or HCV have been inconclusive. There is no convincing evidence for sexual transmission. In broad terms, genotype 1 is the predominant type found throughout Asia and North Africa. Genotype 2 is represented by the prototype strain from an outbreak in Mexico, with variants detected in parts of Africa. Genotype 3 has been recovered from humans in North and South America, Japan and many countries in Europe. Genotype 4 has been recovered from humans in China, Japan and Taiwan. The prevalent genotype impacts on the epidemiology (age at infection), influenced by the likely source of infection (human or zoonotic)125. Genotypes 3 and 4 have been recovered from swine throughout the world. Waterborne epidemics in developing countries due to genotypes 1 and 2 exhibit an attack rate of 1-15%, with a typical age related bias for clinically apparent hepatitis towards older children and young adults (15-40 years). Sporadic cases caused by genotypes 3 and 4 in developed countries affect an older age group on average. Given that the person to person transmission rate is low, rarely above 1-2%, it is likely that these G3 and G4 cases are acquired from an animal reservoir126. Indeed, there is increasing evidence for HEV as a zoonosis in developed parts of the world127. HEV is considered to be enzootic in swine, and anti-HEV can be found in many other species, including chickens, cats, dogs, goats, sheep, and rodents. Experimental transmission studies have shown G1 and G2 can infect some nonClinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 27 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis human primates but not other animals, whereas G3 viruses from humans or swine can infect non-human primates and other animals128,129. Some studies show a significantly higher anti-HEV prevalence amongst pig handlers than in age matched controls from the same areas130. Nucleotide sequencing of isolates from humans and swine from Taiwan, Japan and the USA show a high degree of similarity108,131,132. HEV appears to be endemic in other animals that may act as a reservoir (rats, cows, sheep, goats, monkeys, dogs), but there is as yet no clear evidence implicating them as a zoonotic source. Recent reports suggest food as a vector for transmission in some countries: shellfish, pig liver,venison, boar133-137. The reasons for these geographically linked differences are unclear, potentially reflecting variable viral virulence, host susceptibility to infection or propensity to develop disease. HEV IgG seroprevalence studies have shown the different clinico-epidemiological patterns mentioned previously, and suggest exposure to HEV may be more widespread than implied by clinical disease rates. There appears to be a background prevalence of seropositivity for HEV IgG worldwide, although the patterns in epidemic regions do not fit those expected for a waterborne agent, with rates in India of around 10% in children, reaching a maximum of around 40% in adulthood 138. The background rate of anti-HEV IgG in European blood donors is often reported to be between 1 and 9%, although there are reports of rates reaching around 16%139-141. In the USA, rates of between 2% and 26% have been found129. Interpretation of such seroprevalence data should be made taking into account variable assay performance, the use or not of confirmatory assays, the validity of low antibody indices for indicating true past infection, and the potential for pockets of high prevalence within countries. Data from the UK has been limited but does indicate a pattern of HEV disease consistent with that of the USA and most of Europe, with HEV endemic in the pig population. Reports of HEV hepatitis acquired in the UK have been available for many years142. The epidemiological profile of HEV acquired in England and Wales has been recently summarised143. This work showed that the typical patient was caucasian male and over 55 years of age, but a common source of infection could not be identified. Further work is required to define whether those patients presenting with a clinical illness subsequently identified as hepatitis E have been more at risk of infection or are simply more likely to be symptomatic. 5.5 Prevention and Control In regions where outbreaks occur, prevention strategies should focus upon improving the quality of the water supply and sewage disposal, identification of infectious sources, and vaccine development. In countries such as the UK where no common source of infection is known, improved case ascertainment and targeted vaccination may be more appropriate. Travellers to hyperendemic regions should be advised to take precautions against enteric infections, including HEV. Boiling drinking water is likely to inactivate HEV. Immune serum globulin has not been shown to protect against infection in endemic areas, possibly due to the relatively low titre of HEV IgG in such preparations. The results from trials of active immunisation are promising. A recombinant vaccine comprising a truncated capsid protein expressed in insect cells was evaluated by a Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 28 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis randomised controlled trial including 1794 subjects based in Nepal144. The vaccine was found to be safe and efficacious, with an intention to treat efficacy of 88.5%. Further studies are required to define the value of such a vaccine in alternative settings. 6 Hepatitis G Hepatitis G virus is a blood-borne human virus of the Flaviviridae family. The virus was first discovered in 1995, the same year GB virus C was discovered 145. Both GBV-C and hepatitis G virus were showed to be the same virus and the two have since been known as GBV-C. There is no supporting evidence that GBV-C causes chronic hepatitis possibly due to poor replication in hepatocytes (replication appears to occur in lymphocytes). There is, however, strong evidence to suggest that persistent GBV-C co-infection with HIV reduces disease progression of HIV disease thus reducing the hazard for mortality due to HIV infection145. 7 Cytomegalovirus (CMV) 7.1 Basic Virology and Taxonomy146 Cytomegalovirus (CMV) was first thought of solely as a human salivary gland virus until 1956, when it was isolated from the adenoids of children undergoing tonsillectomies and adenoidectomies. The virus itself belongs to the family Herpesviridae and is a β herpes virus. As with all herpes viruses, it has the ability to establish latency in the host after recovery. The CMV viral particle is about 102nm in diameter and has a lipid envelope that is studded with glycoproteins, surrounding a matrix of phosphoproteins, an icosahedral nucleocapsid and a DNA genome with a molecular mass of 155kDa146. 7.2 Laboratory Diagnosis The diagnosis of CMV hepatitis is made from a combination of excluding other viral causes of hepatitis, such as HBV, HAV and EBV and the demonstration of IgM antibody to CMV or sero-conversion for CMV. The presence of IgM however may be due not only to primary infection but also to reinfection or reactivation of latent virus, as well as the possibility of heterotypic responses and false IgM reactivity. A new CMV infection will give rise to a four fold increase in complement fixing antibodies or a single high titre; importantly in the three months after a primary infection the CMV IgG will have low avidity147. The role of a molecular assay is unclear at the present time for diagnosis, but the presence of CMV DNA in blood is consistent with an active CMV infection, which may need to be monitored especially in the immunocompromised. 7.3 Clinical Features Symptomatic primary CMV infection is uncommon; it can be associated with hepatitis in immunocompetent adults, usually as part of a heterophile antibody-negative mononucleosis syndrome. This is usually accompanied by hepatosplenomegaly, minor elevations of serum aminotransferases and aytypical lymphocytes. It may rarely be associated with a pure hepatitis. CMV hepatitis in this context is self limiting Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 29 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis and treatment is supportive. There is no role for antiviral agents. CMV hepatitis is a feature of symptomatic congenital infection. 7.4 Epidemiology Human CMV is endemic and infects humans of all ages, the peak of acquisition occurring in childhood. The sero-prevalence of CMV in young adult populations depends upon socio-economic status ranging from 40% to 80% (high to low). The virus is well adapted and in patients who are immunocompetent primary infection is often asymptomatic (90-95%). The virus causes more significant disease in an immunocompromised host, such as patients that have undergone transplant operations and those that are infected with HIV. It is also an important cause of congenital infection when it may be either symptomatic or asymptomatic. 7.5 Prevention and Control In patients that are immunocompromised, both primary CMV infection and reactivation, can give rise to CMV disease. As part of CMV disease, there may be liver involvement. There is a role for antiviral therapy in preventing and treating CMV in this context. Antiviral agents that can be used to treat CMV disease include ganciclovir, foscarnet and cidofovir. 8 Epstein Barr Virus (EBV) 8.1 Basic Microbiology and Taxonomy148 The Epstein-Barr virus was first discovered in 1964 in cells that had been cultured from tumour specimens from patients in Kampala, Uganda. EBV belongs to the Herpesviridae family and has morphologic features that are characteristic of this family. It has a hexagonal nucleocapsid that is surrounded by a complex envelope and is approximately 180 – 200nm in diameter. 8.2 Laboratory Diagnosis This section should be read in conjunction with V 26 - Epstein-Barr Virus Serology. Heterophile antibodies are used to diagnose infectious mononucleosis (IM) and specific EBV serology to diagnose EBV hepatitis and IM with negative heterophile antibody such as in children: 8.2.1 Primary EBV infection Antibodies to viral capsid antigen (VCA): IgG, IgM and IgA are all typically detectable in the serum of patients with EBV infection by the time of onset of symptoms IgA and IgM decline to undetectable levels during convalescence IgM detection by indirect immunofluorescence (IF) is a highly sensitive test for EBV infection Complexes of IgG and IgM class rheumatoid factor can cause false positive IgM results. EBV IgG can cause false negative IgM results by competing with EBV IgM. Serum samples should be pre-treated to remove IgG before the IF test. Non-specific anti-cell fluorescence may be seen but should be readily differentiated from specific Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 30 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis fluorescence by an experienced observer. ELISA tests for EBV IgM are less sensitive, but are more widely used. Weak cross-reactions may cause false positive EBV VCA IgM results, particularly during primary CMV. Antibodies to EBNA-1 IgG is not usually detectable until the convalescent period. Its detection excludes recent or current IM. Commercial EBNA-1 IgG ELISAs are available and are widely used. EBV DNA tests (quantitative PCR assays) are used in the monitoring of patients who are immunocompromised and at risk of EBV associated lymphoproliferative disease, where very high levels may be observed. Lower levels are seen in uncomplicated IM. A role for EBV DNA quantitation in the diagnosis and monitoring of EBV infection and its complications has been suggested. Levels are higher in IM than in healthy carriers and higher still in complicated IM. 8.3 Clinical Features IM, or glandular fever, is one of the commonest causes of prolonged illness in adolescents and young adults in developed countries. Signs of IM are typified by fever, pharyngitis, lymphadenopathy, splenomegaly, and hepatocellular dysfunction. Jaundice occurs in 5% of cases, although hepatocellular enzymes are raised in 80 to 90% of cases149. Most cases resolve spontaneously over a 2 to 3 week period. Malaise may occur for weeks or months in some patients. Rarely primary EBV in a person who is immunocompetent may be complicated by fulminant IM associated with haemophagocytosis caused by activation of the monophagocytic system in multiple organs150. Liver failure is the cause of death in about half of patients with fatal infectious mononucleosis, the remaining deaths resulting from opportunistic infections. Chronic active EBV (CAEBV) infection is a rare condition, distinct from chronic fatigue, that is typified by severe, chronic, or recurrent IM-like symptoms after a well documented primary EBV infection. There is a marked increase of EBV load in peripheral blood, often with infection of T and/or natural killer cells, and evidence of major organ involvement, eg hepatitis. CAEBV has a high morbidity and mortality from hepatic failure, lymphoma, sepsis, or haemophagocytic syndrome149. Some cases of autoimmuneor granulomatous hepatitishave also been associated with EBV infection151,152. Clinically significant EBV liver damage is mostly observed in the immunocompromised states, including HIV infection, post-transplant and X-linked lymphoproliferative disorders. 8.4 Epidemiology Transmission occurs predominantly sub-clinically during childhood, often by spread via salivary contact. In developed countries, primary infection often occurs during adolescence, presenting as infectious mononucleosis (IM). More than 90% of adults worldwide have been infected with EBV and carry the virus as a life-long persistent infection, with latent infection of B lymphocytes. 9 Yellow Fever 9.1 Basic Virology and Taxonomy Yellow fever virus, isolated in West Africa in 1927, is the prototype member of the family Flaviviridae, and is an enveloped virus with a single positive sense RNA Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 31 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis genome, containing approximately 11,000 nucleotides153. The family Flaviviridae is divided into the genera Flavivirus, Pestivirus and Hepacivirus, and the genus Flavivirus is further classified antigenically into 8 complexes (yellow fever has not, however, been assigned to an antigenic complex)154. Flavivirus particles are 40-50 nm in diameter and have a spherical nucleocapsid surrounded by a lipid bilayer envelope. Analysis of the yellow fever virus Pre-Membrane/Membrane and a portion of the E (envelope) gene sequence and 3’-non-coding region has distinguished at least 7 genotypes, with homology >90% across the genotypes. These include two South American genotypes, two West African genotypes (I and II), a single Central/South African genotype (Angola), and two East African genotypes (East and East/Central)155,156. 9.2 Laboratory Diagnosis In the UK, testing is performed at the Special Pathogens Reference Unit (SPRU) at CEPR, Porton Down. The information they require in all cases is the onset date of illness, the countries and dates of visit, the date of return to the UK, and whether yellow fever vaccination has been given. In the first 7-10 days post onset of symptoms, an EDTA sample can be tested for yellow fever RNA detection. Serologic testing on serum is also performed to determine virus-specific IgM and IgG antibodies (ELISA, Immunofluorescence). The IgM response is very specific but may be short-lived. The IgG response can show cross-reactivity between the flavivirus complexes/strains (JE, WN, Dengue, TBE, YF), thus other serologic assays (eg neutralisation tests) need then be utilised to determine the strain involved. There is also the facility to culture the virus from tissue samples or to perform PCR on tissue samples. 9.3 Clinical Features and Transmission Yellow fever is spread by the bite of an infected mosquito. Humans and monkeys are the main virus reservoir, with the principle mosquito vector either Aedes Aegypti or Haemagogus species. There are 3 main transmission cycles, recently reviewed157: 1. The sylvatic (jungle or forest) transmission cycle causes sporadic cases in South America and Africa. The cycle occurs between monkeys and mosquitoes, with humans infected when they live or work in the cycle areas 2. An intermediate sylvatic cycle occurs in the moist African savannah where semi-domestic mosquitoes infect animals and humans to cause small epidemics in rural villages 3. Urban transmission can occur between humans via domestic (ie breed around human habitation) Aedes aegypti and can lead to large epidemics if the population is unvaccinated The Aedes mosquito is active during the day and once infected with virus remains infectious for life (2-3 months). The virus can survive over the dry season in mosquito eggs. The incubation period is 3-6 days and there can be three distinct phases to the illness. Initial symptoms are fever, prostration, extremity pain (including knee joint), headache, photophobia, lumbosacral pain, anorexia, epigastic pain and vomiting. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 32 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis Laboratory abnormalities include leukopenia from the onset and elevation of serum transaminases on days 2-3. In many patients gradual recovery occurs after 3-4 days. However, in approximately 15-25% patients, the second phase is an abatement of fever for 2-24 hrs, followed by a more serious illness in the third phase. Hepatic coagulopathy produces haemorrhagic symptoms, including haematemesis, epistaxis, bleeding, petechial and purpuric haemorrhage. There is a marked thrombocytopenia, a deepening jaundice and proteinuria. Patients may develop shock, metabolic acidosis, acute tubular necrosis, myocardial dysfunction, cardiac arrhythmias, confusion, seizures and coma. Death rates of 20-50% occur and there is often secondary bacterial infection and renal failure158. Survival gives lifelong immunity. 9.4 Epidemiology The disease is endemic in 10 South American countries and over 30 countries in sub-Saharan Africa. WHO estimates approximately 200,000 cases of yellow fever annually with 30,000 deaths (although there is under-reporting of disease). Yellow fever can reappear with outbreaks after long periods of apparent quiescence 157. 9.5 Prevention and Control There is no specific antiviral available, thus good supportive management is essential. Prevention entails avoidance of mosquito bites, control of mosquito populations and immunisation. A live attenuated vaccine has been available for more than 50 years and is recommended for laboratory workers handling infective material and for everyone visiting countries with a risk of yellow fever transmission. With the recent recognition of rare severe adverse events related to yellow fever vaccine, it is essential to make a careful risk assessment prior to administering vaccine159. Currently yellow fever is the only disease for which an ICVP (international certificate of vaccine or prophylaxis) may be required for entry. There is only one licensed yellow fever vaccine in the UK which contains an attenuated strain of the 17D strain of yellow fever virus that protects against all yellow fever genotypes (Stamaril, manufactured by Sanofi Pasteur MSD). A single dose of vaccine, correctly administered, confers immunity in 95-100% of recipients and this immunity persists for at least 10 years160162. Vaccination should be performed at least ten days prior to travel to an endemic area to allow protective immunity to develop and for the ICVP to become valid. International Heath Regulations (2005) require re-vaccination at 10 year intervals to retain a valid ICVP. 10 Q Fever 10.1 Basic Microbiology and Taxonomy Coxiella burnetii, the causative agent of Q fever, a zoonotic infection, is a Gram negative, pleomorphic coccobacillus. It is an obligate intracellular gamma proteobacterial organism. 10.2 Laboratory Diagnosis Q fever diagnosis is commonly based upon serological methods. Both the complement fixation test (CFT) and the indirect immunofluorescence assay (IFA) may be utilized. In acute illness, antibodies to phase II antigen are higher than those Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 33 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis of phase I, whereas, in chronic Q fever, antibodies to phase I predominate163. C. burnetii PCR offers a sensitive direct detection approach to the diagnosis of acute Q fever164. 10.3 Clinical Features Acute Q fever is mostly a self-limiting febrile illness and may present with a wide variety of symptoms. The fever normally lasts for 1 to 2 weeks and many patients will develop pneumonia. In addition a majority of patients will have abnormal liver function tests and some will develop hepatitis163. Q fever hepatitis usually manifests with a moderate rise in liver enzyme levels rising to around two to three times the upper limit of normal163. The hepatitis may occur in the absence of pulmonary involvement165. 10.4 Transmission Transmission of Q fever is primarily via the aerosol route by inhalation of contaminated material. The most infectious source is parturient fluids from animals, usually infected sheep, cattle or goats, but other means of infection include animal blood, milk, urine and faeces. The organism is very robust and may survive in the environment for many weeks163. 10.5 Epidemiology Q fever is found worldwide, but is relatively rare in the UK3. The infection is associated with rural living and is most common in those regions associated with agriculture. Q fever is capable of causing outbreaks and should be considered in certain circumstances166. 10.6 Prevention and Control Prevention and control measures are directed at those handling possibly infected animals with emphasis on appropriate disposal of animal birth products. Acute Q fever usually resolves without treatment in about two weeks, although treatment of Q fever with tetracycline or doxycycline or a fluoroquinolone is normally effective in reducing the duration of illness167,168. At present there is no vaccine against Q fever available in the UK. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 34 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis References 1. Department of Health. Health Protection Legislation (England) Guidance. 2010. p. 1-112. 2. Koff RS. Feinstone SM,Kapikian AZ,Purcell RH. Hepatitis A: detection by immune electron microscopy of a virus like antigen associated with acute illness [Science 1973;182:1026-1028]. J Hepatol 2002;37:2-6. 3. Lemon SM. Type A viral hepatitis: epidemiology, diagnosis, and prevention. Clin Chem 1997;43:1494-9. 4. Schiff ER. Atypical clinical manifestations of hepatitis A. Vaccine 1992;10 Suppl 1:S18-S20. 5. Lednar WM, Lemon SM, Kirkpatrick JW, Redfield RR, Fields ML, Kelley PW. Frequency of illness associated with epidemic hepatitis A virus infections in adults. Am J Epidemiol 1985;122:226-33. 6. Rosenblum LS, Villarino ME, Nainan OV, Melish ME, Hadler SC, Pinsky PP, et al. Hepatitis A outbreak in a neonatal intensive care unit: risk factors for transmission and evidence of prolonged viral excretion among preterm infants. J Infect Dis 1991;164:476-82. 7. Ciocca M. Clinical course and consequences of hepatitis A infection. Vaccine 2000;18 Suppl 1:S71-S74. 8. Conaty S, Bird P, Bell G, Kraa E, Grohmann G, McAnulty JM. Hepatitis A in New South Wales, Australia from consumption of oysters: the first reported outbreak. Epidemiol Infect 2000;124:12130. 9. From the Centers for Disease Control and Prevention. Hepatitis A associated with consumption of frozen strawberries--Michigan, March 1997. JAMA 1997;277:1271. 10. Harkness J, Gidon B, Istre GR. Outbreaks of Hepatitis A amongst illicit drug abusers, Oklahoma 1984 - 87. Am J Public H 1989;79:463-6. 11. Corey L, Holmes KK. Sexual transmission of hepatitis A in homosexual men: incidence and mechanism. N Engl J Med 1980;302:435-8. 12. Crowcroft NS, Walsh B, Davison KL, Gungabissoon U, on behalf of PHLS Advisory Committee on Vaccination and Immunisation. Guidelines for the control of hepatitis A virus. Infect Commun Dis Public Health 2001;213-27. 13. Feinstone SM, Gust ID. Hepatitis A Vaccine. In: Plotkin SA, Orenstein WA, editors. Vaccines. 3rd ed. Philadelphia: WB Saunders Company; 1999. p. 650-71. 14. Thomas L and the Hepatitis A Guidelines Group. Thomas L and the Hepatitis A Guidelines Group. Guidance for the prevention and control of hepatitis A infection Health Protection Agency. 2009. 15. Kidd-Ljunggren K, Miyakawa Y, Kidd AH. Genetic variability in hepatitis B viruses. J Gen Virol 2002;83:1267-80. 16. Niesters HG, Pas S, de Man RA. Detection of hepatitis B virus genotypes and mutants: current status. J Clin Virol 2005;34 Suppl 1:S4-S8. 17. McMahon BJ. The influence of hepatitis B virus genotype and subgenotype on the natural history of chronic hepatitis B. Hepatol Int 2009;3:334-42. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 35 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 18. Robinson W, Marion P, Feitelson M, et al. The hepadnavirus group:Hepatitis B and related viruses. In: Szmuness W, Alter H, Maynard JE, editors. Viral Hepatitis: 1981 International symposium. Philadelphia: Franklin Institue Press; 1982. 19. Liang TJ. Hepatitis B: the virus and disease. Hepatology 2009;49:S13-S21. 20. Flint SJ, Enquist LW, Racanielo VR, Skalka AM. Family Hepadnaviridae. Principles of Virology. 3rd ed. Washington DC: Molecular Biology ASM Press; 2009. p. 505-7. 21. Burgess,C, Perry,K, Newham,J, Kitchen,A. Evaluation of Abbott Architect HBsAg assay Product Code 6C36 HPA-MiDAS and NBS-NTMRL. 2008. 22. Weber B. Genetic variability of the S gene of hepatitis B virus: clinical and diagnostic impact. J Clin Virol 2005;32:102-12. 23. UK Blood Transfusion and Tissue Transplantation Service. Guidelines for the Blood Transfusion Services in the United Kingdom. 2005. 24. Horvat RT, Tegtmeier GE. Hepatitis B and D viruses. In: Murray PR, Baron EJ, Jorgensen JH, Landry ML, Pfaller MA, editors. Manual of Clinical Microbiology. 9th ed. Washington DC: ASM Press; 2007. p. 1641-59. 25. Lindsay KL, Nizze JA, Koretz R, Gitnick G. Diagnostic usefulness of testing for anti-HBc IgM in acute hepatitis B. Hepatology 1986;6:1325-8. 26. Kumar M, Jain S, Sharma BC, Sarin SK. Differentiating acute hepatitis B from the first episode of symptomatic exacerbation of chronic hepatitis B. Dig Dis Sci 2006;51:594-9. 27. Rodella A, Galli C, Terlenghi L, Perandin F, Bonfanti C, Manca N. Quantitative analysis of HBsAg, IgM anti-HBc and anti-HBc avidity in acute and chronic hepatitis B. J Clin Virol 2006;37:206-12. 28. Hansson BG. Persistence of serum antibody to hepatitis B core antigen. J Clin Microbiol 1977;6:209-11. 29. Bowden S. Serological and molecular diagnosis. Semin Liver Dis 2006;26:97-103. 30. Thompson A, Bell SJ, Locarnini S. Hepatitis B virus. In: Richman DD, Whitley RJ, Hayden FG, editors. Clinical Virology. 3rd ed. Washington DC: ASM Press; 2009. 31. World Health Organization. Hepatitis B. 2009. 32. Zhang JM, Xu Y, Wang XY, Yin YK, Wu XH, Weng XH, et al. Coexistence of hepatitis B surface antigen (HBsAg) and heterologous subtype-specific antibodies to HBsAg among patients with chronic hepatitis B virus infection. Clin Infect Dis 2007;44:1161-9. 33. European association for the Study of the Liver. EASL Clinical Practice Guidelines: management of chronic hepatitis B. J Hepatol 2009;227-42. 34. Andersson KL, Chung RT. Monitoring during and after antiviral therapy for hepatitis B. Hepatology 2009;49:S166-S173. 35. DH 2000 Health Service Circular 2000/020. Hepatitis B infected health care workers. 2009. 36. Department of Health. Hepatitis B infected healthcare workers antiviral therapy. 2007. 37. Takkenberg RB, Zaaijer HL, Molenkamp R, Menting S, Terpstra V, Weegink CJ, et al. Validation of a sensitive and specific real-time PCR for detection and quantitation of hepatitis B virus Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 36 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis covalently closed circular DNA in plasma of chronic hepatitis B patients. J Med Virol 2009;81:98895. 38. Dane DS, Cameron CH, Briggs M. Virus-like particles in serum of patients with Australia-antigenassociated hepatitis. Lancet 1970;1:695-8. 39. Candotti D, Allain JP. Transfusion-transmitted hepatitis B virus infection. J Hepatol 2009;51:798809. 40. Tabuchi A, Tanaka J, Katayama K, Mizui M, Matsukura H, Yugi H, et al. Titration of hepatitis B virus infectivity in the sera of pre-acute and late acute phases of HBV infection: transmission experiments to chimeric mice with human liver repopulated hepatocytes. J Med Virol 2008;80:2064-8. 41. Lin HH, Lee TY, Chen DS, Sung JL, Ohto H, Etoh T, et al. Transplacental leakage of HBeAgpositive maternal blood as the most likely route in causing intrauterine infection with hepatitis B virus. J Pediatr 1987;111:877-81. 42. Tang JR, Hsu HY, Lin HH, Ni YH, Chang MH. Hepatitis B surface antigenemia at birth: a longterm follow-up study. J Pediatr 1998;133:374-7. 43. Beasley RP, Trepo C, Stevens CE, Szmuness W. The e antigen and vertical transmission of hepatitis B surface antigen. Am J Epidemiol 1977;105:94-8. 44. Jonas MM. Hepatitis B and pregnancy: an underestimated issue. Liver Int 2009;29 Suppl 1:133-9. 45. Chaudhary RK. Perinatal transmission of hepatitis B virus. Can Med Assoc J 1983;128:664-6. 46. HPA North West Region. Acute Hepatitis B in the North West:2007 Liverpool. 2008. 47. Health Protection Agency. Infections among injecting drug users in the United Kingdom. London 2008. 48. Health Protection Agency. Acute hepatitis B infection laboratory reports: England and Wales, by Age Group, 1990-2003. 2004. 49. Naicker S, Fabian J, Naidoo S, Wadee S, Paget G, Goetsch S. Infection and glomerulonephritis. Semin Immunopathol 2007;29:397-414. 50. Ichai P, Samuel D. Etiology and prognosis of fulminant hepatitis in adults. Liver Transpl 2008;14 Suppl 2:S67-S79. 51. Gimson AE, Tedder RS, White YS, Eddleston AL, Williams R. Serological markers in fulminant hepatitis B. GUT 1983;24:615-7. 52. Carman WF, Korula J, Wallace L, MacPhee R, Mimms L, Decker R. Fulminant reactivation of hepatitis B due to envelope protein mutant that escaped detection by monoclonal HBsAg ELISA. Lancet 1995;345:1406-7. 53. Shi YH, Shi CH. Molecular characteristics and stages of chronic hepatitis B virus infection. World J Gastroenterol 2009;15:3099-105. 54. Fattovich G, Bortolotti F, Donato F. Natural history of chronic hepatitis B: special emphasis on disease progression and prognostic factors. J Hepatol 2008;48:335-52. 55. Carman WF, Jacyna MR, Hadziyannis S, Karayiannis P, McGarvey MJ, Makris A, et al. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet 1989;2:588-91. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 37 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 56. Hunt CM, McGill JM, Allen MI, Condreay LD. Clinical relevance of hepatitis B viral mutations. Hepatology 2000;31:1037-44. 57. Yeo W, Johnson PJ. Diagnosis, prevention and management of hepatitis B virus reactivation during anticancer therapy. Hepatology 2006;43:209-20. 58. Hoofnagle JH. Reactivation of hepatitis B. Hepatology 2009;49:S156-S165. 59. Iloeje UH, Yang HI, Su J, Jen CL, You SL, Chen CJ. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006;130:678-86. 60. Chen CJ, Yang HI, Su J, Jen CL, You SL, Lu SN, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006;295:65-73. 61. Chen CJ, Yang HI, Iloeje UH. Hepatitis B virus DNA levels and outcomes in chronic hepatitis B. Hepatology 2009;49:S72-S84. 62. Flint SJ, Enquist LW, Racanielo VR, Skalka AM. Transformation and Oncogenesis. Principles of Virology. 3rd ed. Washington DC: Pathogenesis and Control ASM Press; 2009. p. 200-48. 63. Yu M.C., Yuan J.M, Lu SC. Alcohol, cofactors and genetics of hepatocellular carcinoma. Journal of Gasteroenterology and Hepatology 2008;23:S92-S97. 64. Maynard JE. Hepatitis B: global importance and need for control. Vaccine 1990;8 Suppl:S18-S20. 65. Kiire CF. The epidemiology and prophylaxis of hepatitis B in sub-Saharan Africa: a view from tropical and subtropical Africa. GUT 1996;38 Suppl 2:S5-12. 66. Stevens CE, Toy PT, Tong MJ, Taylor PE, Vyas GN, Nair PV, et al. Perinatal hepatitis B virus transmission in the United States. Prevention by passive-active immunization. JAMA 1985;253:1740-5. 67. Chang MH. Hepatitis B virus infection. Semin Fetal Neonatal Med 2007;12:160-7. 68. Kao JH, Chen DS. Global control of hepatitis B virus infection. Lancet Infect Dis 2002;2:395-403. 69. Zanetti AR, Van Damme P, Shouval D. The global impact of vaccination against hepatitis B: a historical overview. Vaccine 2008;26:6266-73. 70. Young MD, Gooch WM, III, Zuckerman A.J., Du W, Dickson B, Maddrey WC. Comparison of a triple antigen and a single antigen recombinant vaccine for adult hepatitis B vaccination. J Med Virol 2001;64:290-8. 71. Kim HN, Harrington RD, Van Rompaey SE, Kitahata MM. Independent clinical predictors of impaired response to hepatitis B vaccination in HIV-infected persons. Int J STD AIDS 2008;19:600-4. 72. Fisman DN, Agrawal D, Leder K. The effect of age on immunologic response to recombinant hepatitis B vaccine: a meta-analysis. Clin Infect Dis 2002;35:1368-75. 73. Department of Health. Immunisation against infectious disease Hepatitis B. 2006. 74. Department of Health. Immunisation against infectious disease Update to Chapter 18 Hepatitis B. 2006. 75. Lok ASF MB. AASLD Practice Guidelines Chronic hepatitis B. Hepatology 2007;507-39. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 38 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 76. Jardim AC, Yamasaki LH, de Queiroz AT, Bittar C, Pinho JR, Carareto CM, et al. Quasispecies of hepatitis C virus genotype 1 and treatment outcome with Peginterferon and Ribavirin. Infect Genet Evol 2009;9:689-98. 77. World Health Organization. Hepatitis C. 2009. 78. Davis GL, Lindsay KL. Treatment of chronic hepatitis C infection: one step at a time. Lancet Infect Dis 2005;5:524-6. 79. Strickland GT, El Kamary SS, Klenerman P, Nicosia A. Hepatitis C vaccine: supply and demand. Lancet Infect Dis 2008;8:379-86. 80. Chevaliez S, Pawlotsky JM. Diagnosis and management of chronic viral hepatitis: antigens, antibodies and viral genomes. Best Pract Res Clin Gastroenterol 2008;22:1031-48. 81. Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med 2001;345:41-52. 82. Murphy D, Chamberland J, Dandavino R, et al. A new genotype of Hepatitis C Virus originating from central Africa. Hepatology 2007;46:623A. 83. Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 2005;5:558-67. 84. Frank C, Mohamed MK, Strickland GT, Lavanchy D, Arthur RR, Magder LS, et al. The role of parenteral antischistosomal therapy in the spread of hepatitis C virus in Egypt. Lancet 2000;355:887-91. 85. Busch MP, Kleinman SH, Nemo GJ. Current and emerging infectious risks of blood transfusions. JAMA 2003;289:959-62. 86. Strader DB, Wright T, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C. Hepatology 2004;39:1147-71. 87. Veillon P, Payan C, Picchio G, Maniez-Montreuil M, Guntz P, Lunel F. Comparative evaluation of the total hepatitis C virus core antigen, branched-DNA, and amplicor monitor assays in determining viremia for patients with chronic hepatitis C during interferon plus ribavirin combination therapy. J Clin Microbiol 2003;41:3212-20. 88. Pawlotsky JM. Molecular diagnosis of viral hepatitis. Gastroenterology 2002;122:1554-68. 89. Mukherjee S, Sorrell MF. Controversies in liver transplantation for hepatitis C. Gastroenterology 2008;134:1777-88. 90. Perz JF, Farringtom L.A., Pecoraro C, Hutin Y.J.F, Armstrong G.L. Estimated global prevalence of Hepatitis C virus infection. 42nd Annual Meeting of the infectious Diseases Society of America 2004. 91. Craxi A, Laffi G, Zignego AL. Hepatitis C virus (HCV) infection: a systemic disease. Mol Aspects Med 2008;29:85-95. 92. Craine N., Walker M., Carnworth T., Klee H. Hepatitis C testing injecting risk behaviour: the results of a uk based pilot study. International Journal of Drug Policy 2004;15:115-22. 93. Danta M, Brown D, Bhagani S, Pybus OG, Sabin CA, Nelson M, et al. Recent epidemic of acute hepatitis C virus in HIV-positive men who have sex with men linked to high-risk sexual behaviours. AIDS 2007;21:983-91. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 39 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 94. Gordon CE, Uhlig K, Lau J, Schmid CH, Levey AS, Wong JB. Interferon treatment in hemodialysis patients with chronic hepatitis C virus infection: a systematic review of the literature and meta-analysis of treatment efficacy and harms. Am J Kidney Dis 2008;51:263-77. 95. Kronenberger B, Zeuzem S. Current and future treatment options for HCV. Ann Hepatol 2009;8:103-12. 96. Farci P. Delta hepatitis: an update. J Hepatol 2003;39 Suppl 1:S212-S219. 97. Harrison T.J., Dushieko G.M., Zuckerman A.J. Hepatitis Viruses. In: Zuckerman AJ, Banatvala JE, Pattison JR, Griffiths PD, Schoub BD, editors. Principles and Practice of Clinical Virology. 6th ed. Chichester: Wiley; 2009. p. 273-320. 98. Farci P, Roskams T, Chessa L, Peddis G, Mazzoleni AP, Scioscia R, et al. Long-term benefit of interferon alpha therapy of chronic hepatitis D: regression of advanced hepatic fibrosis. Gastroenterology 2004;126:1740-9. 99. Su CW, Huang YH, Huo TI, Shih HH, Sheen IJ, Chen SW, et al. Genotypes and viremia of hepatitis B and D viruses are associated with outcomes of chronic hepatitis D patients. Gastroenterology 2006;130:1625-35. 100. Taylor JM. Hepatitis delta virus. Virology 2006;344:71-6. 101. Wong DC, Purcell RH, Sreenivasan MA, Prasad SR, Pavri KM. Epidemic and endemic hepatitis in India: evidence for a non-A, non-B hepatitis virus aetiology. Lancet 1980;2:876-9. 102. Balayan MS, Andjaparidze AG, Savinskaya SS, Ketiladze ES, Braginsky DM, Savinov AP, et al. Evidence for a virus in non-A, non-B hepatitis transmitted via the fecal-oral route. Intervirology 1983;20:23-31. 103. Reyes GR, Purdy MA, Kim JP, Luk KC, Young LM, Fry KE, et al. Isolation of a cDNA from the virus responsible for enterically transmitted non-A, non-B hepatitis. Science 1990;247:1335-9. 104. Tam AW, Smith MM, Guerra ME, Huang CC, Bradley DW, Fry KE, et al. Hepatitis E virus (HEV): molecular cloning and sequencing of the full-length viral genome. Virology 1991;185:120-31. 105. Emerson SU, Anderson D, Arankalle A, et al. Hepevirus. In: Fauquet CM, editor. Elsevier/ Academic Press; 2005. p. 853-7. 106. Huang FF, Sun ZF, Emerson SU, Purcell RH, Shivaprasad HL, Pierson FW, et al. Determination and analysis of the complete genomic sequence of avian hepatitis E virus (avian HEV) and attempts to infect rhesus monkeys with avian HEV. J Gen Virol 2004;85:1609-18. 107. Nanda SK, Ansari IH, Acharya SK, Jameel S, Panda SK. Protracted viremia during acute sporadic hepatitis E virus infection. Gastroenterology 1995;108:225-30. 108. Takahashi M, Tanaka T, Azuma M, Kusano E, Aikawa T, Shibayama T, et al. Prolonged fecal shedding of hepatitis E virus (HEV) during sporadic acute hepatitis E: evaluation of infectivity of HEV in fecal specimens in a cell culture system. J Clin Microbiol 2007;45:3671-9. 109. Bendall R, Ellis V, Ijaz S, Thurairajah P, Dalton HR. Serological response to hepatitis E virus genotype 3 infection: IgG quantitation, avidity, and IgM response. J Med Virol 2008;80:95-101. 110. Malcolm P, Dalton H, Hussaini HS, Mathew J. The histology of acute autochthonous hepatitis E virus infection. Histopathology 2007;51:190-4. 111. Stoszek SK, Engle RE, Abdel-Hamid M, Mikhail N, Abdel-Aziz F, Medhat A, et al. Hepatitis E antibody seroconversion without disease in highly endemic rural Egyptian communities. Trans R Soc Trop Med Hyg 2006;100:89-94. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 40 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 112. Gerolami R, Moal V, Colson P. Chronic hepatitis E with cirrhosis in a kidney-transplant recipient. N Engl J Med 2008;358:859-60. 113. Haagsma EB, van den Berg AP, Porte RJ, Benne CA, Vennema H, Reimerink JH, et al. Chronic hepatitis E virus infection in liver transplant recipients. Liver Transpl 2008;14:547-53. 114. Kamar N, Selves J, Mansuy JM, Ouezzani L, Peron JM, Guitard J, et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N Engl J Med 2008;358:811-7. 115. Chauhan A, Jameel S, Dilawari JB, Chawla YK, Kaur U, Ganguly NK. Hepatitis E virus transmission to a volunteer. Lancet 1993;341:149-50. 116. Tsega E, Krawczynski K, Hansson BG, Nordenfelt E, Negusse Y, Alemu W, et al. Outbreak of acute hepatitis E virus infection among military personnel in northern Ethiopia. J Med Virol 1991;34:232-6. 117. Khuroo MS, Kamili S, Jameel S. Vertical transmission of hepatitis E virus. Lancet 1995;345:10256. 118. Tsega E, Hansson BG, Krawczynski K, Nordenfelt E. Acute sporadic viral hepatitis in Ethiopia: causes, risk factors, and effects on pregnancy. Clin Infect Dis 1992;14:961-5. 119. Kumar RM, Uduman S, Rana S, Kochiyil JK, Usmani A, Thomas L. Sero-prevalence and motherto-infant transmission of hepatitis E virus among pregnant women in the United Arab Emirates. Eur J Obstet Gynecol Reprod Biol 2001;100:9-15. 120. Kumar A, Agarwal R, Naik SR, Saraswat V, Ghoshal UC, Naik S. Hepatitis E virus is responsible for hepatic decompensation of chronic liver disease in an endemic region. Indian J Gastro Enterol 2004;23:59-62. 121. Zhuang H, Cao XY, Liu CB, Wang GM. Enterically transmitted non-a, non-B hepatitis in China. In: Shikata Tea, editor. Viral Hepatitis C, D, E. New York: Elsevier; 2008. p. 277-85. 122. el Zimaity DM, Hyams KC, Imam IZ, Watts DM, Bassily S, Naffea EK, et al. Acute sporadic hepatitis E in an Egyptian pediatric population. Am J Trop Med Hyg 1993;48:372-6. 123. Datta R, Panda SK, Tandon BN, Madnagopalan N, Bose SLea. Acute sporadic non-A, non-B viral hepatitis of adults in India, epidemiological and immunological studies. J Gastroenterol 1987;33345. 124. Boxall E, Herborn A, Kochethu G, Pratt G, Adams D, Ijaz S, et al. Transfusion-transmitted hepatitis E in a 'nonhyperendemic' country. Transfus Med 2006;16:79-83. 125. Purcell RH, Emerson SU. Hepatitis E: an emerging awareness of an old disease. J Hepatol 2008;48:494-503. 126. Singh V, Singh V, Raje M, Nain CK, Singh K. Routes of transmission in the hepatitis E epidemic of Saharanpur. Trop Gastroenterol 1998;19:107-9. 127. Teo CG. Hepatitis E indigenous to economically developed countries: to what extent a zoonosis? Curr Opin Infect Dis 2006;19:460-6. 128. Meng XJ, Halbur PG, Shapiro MS, Govindarajan S, Bruna JD, Mushahwar IK, et al. Genetic and experimental evidence for cross-species infection by swine hepatitis E virus. J Virol 1998;72:9714-21. 129. Meng XJ, Wiseman B, Elvinger F, Guenette DK, Toth TE, Engle RE, et al. Prevalence of antibodies to hepatitis E virus in veterinarians working with swine and in normal blood donors in the United States and other countries. J Clin Microbiol 2002;40:117-22. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 41 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 130. Drobeniuc J, Favorov MO, Shapiro CN, Bell BP, Mast EE, Dau Eea. Hepatitis E virus antibody prevalence among persons who work with swine. J Clin Microbiol 2001;184:1594-7. 131. Hsieh SY, Meng XJ, Wu YHea. Identity of a novel swine hepatitis E virus in Taiwan forming a monophylogenetic group with Taiwanese isolates of human hepatitis E. J Clin Microbiol 1999;37:3828-34. 132. Schlauder GG, Dawson GJ, Erker JC, Kwo PY, Knigge MF, Smalley DL, et al. The sequence and phylogenetic analysis of a novel hepatitis E virus isolated from a patient with acute hepatitis reported in the United States. J Gen Virol 1998;79 ( Pt 3):447-56. 133. Ijaz S, Arnold E, Banks M, Bendall RP, Cramp ME, Cunningham R, et al. Non-travel-associated hepatitis E in England and Wales: demographic, clinical, and molecular epidemiological characteristics. J Infect Dis 2005;192:1166-72. 134. Koizumi Y, Isoda N, Sato Y, Iwaki T, Ono K, Ido K, et al. Infection of a Japanese patient by genotype 4 hepatitis e virus while traveling in Vietnam. J Clin Microbiol 2004;42:3883-5. 135. Feagins AR, Opriessnig T, Guenette DK, Halbur PG, Meng XJ. Detection and characterisation of infectious hepatitis E virus from commerical pig livers sold in local grocery stores in the USA. J Gen Virol 2007;88:912-7. 136. Takahashi K, Kitajima N, Abe N, Mishiro S. Complete or near-complete nucleotide sequences of hepatitis E virus genome recovered from a wild boar, a deer, and four patients who ate the deer. Virology 2004;330:501-5. 137. Li TC, Chijiwa K, Sera N, Ishibashi T, Etoh Y, Shinohara Y, et al. Hepatitis E virus transmission from wild boar meat. Emerg Infect Dis 2005;11:1958-60. 138. Arankalle VA, Tsarev SA, Chadha MS, Alling DW, Emerson SU, Banerjee Kea. Age specific prevalence of antibodies to hepatitis Aand E in Pune, India, 1982 and 1992. J Infect Dis 1995;171:447-50. 139. Boutrouille A, Bakkali-Kassimi L, Crucière C, Pavio NJ. Prevalence of anti-hepatitis E virus antibodies in French blood donors. Clin Microbiol 2007;45:2009-10. 140. Mansuy JM, Legrand-Abravanel F, Calot JP, Peron JM, Alric L, Agudo S, et al. High prevalence of anti-hepatitis E virus antibodies in blood donors from South West France. J Med Virol 2008;80:289-93. 141. Dalton HR, Stableforth W, Hazeldine S, Thurairajah P, Ramnarace R, Warshow U, et al. Autochthonous hepatitis E in Southwest England: a comparison with hepatitis A. Eur J Clin Microbiol Infect Dis 2008;27:579-85. 142. McCrudden R, O'Connell S, Farrant T, Beaton S, Iredale JP, Fine D. Sporadic acute hepatitis E in the united kingdom: an underdiagnosed phenomenon? GUT 2000;46:732-3. 143. Lewis HC, Boisson S, Ijaz S, Hewitt K, Ngui SL, Boxall E, et al. Hepatitis E in England and Wales. Emerg Infect Dis 2008;14:165-7. 144. Shrestha MP, Scott RM, Joshi DM, Mammen MP, Jr., Thapa GB, Thapa N, et al. Safety and efficacy of a recombinant hepatitis E vaccine. N Engl J Med 2007;356:895-903. 145. Zhang W, Chaloner K, Tillmann HL, Williams CF, Stapleton JT. Effect of early and late GB virus C viraemia on survival of HIV-infected individuals: a meta-analysis. HIV Med 2006;7:173-80. 146. Cheeseman SH. Cytomegalovirus. In: Gorback SL, Bartlett JG, Blacklow NR, editors. Infectious Diseases. 2nd ed. W.B. Saunders company; 2009. p. 2084-91. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 42 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 147. Bodeus M, Feyder S, Goubau P. Avidity of IgG antibodies distinguishes primary from non-primary cytomegalovirus infection in pregnant women. Clin Diagn Virol 1998;9:9-16. 148. Schooley RT. Epstein Barr Virus (Infectious Mononucleosis). In: Mandell GL, Bennett JE, Dolin R, editors. Douglas and Bennett's Principles and Practice of Infectious Diseases. 5th ed. Vol 2. Edinburgh: Churchill Livingstone; 2000. p. 1599-613. 149. Macsween KF, Crawford DH. Epstein-Barr virus-recent advances. Lancet Infect Dis 2003;3:13140. 150. Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis 2007;7:814-22. 151. Chiba T, Goto S, Yokosuka O, Imazeki F, Tanaka M, Fukai K, et al. Fatal chronic active EpsteinBarr virus infection mimicking autoimmune hepatitis. Eur J Gastroenterol Hepatol 2004;16:225-8. 152. Biest S, Schubert TT. Chronic Epstein-Barr virus infection: a cause of granulomatous hepatitis? J Clin Gastroenterol 1989;11:343-6. 153. Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 1990;44:649-88. 154. Calisher CH, Karabatsos N, Dalrymple JM, Shope RE, Porterfield JS, Westaway EG, et al. Antigenic relationships between flaviviruses as determined by cross-neutralization tests with polyclonal antisera. J Gen Virol 1989;70 ( Pt 1):37-43. 155. Mutebi JP, Wang H, Li L, Bryant JE, Barrett AD. Phylogenetic and evolutionary relationships among yellow fever virus isolates in Africa. J Virol 2001;75:6999-7008. 156. Mutebi JP, Rijnbrand RC, Wang H, Ryman KD, Wang E, Fulop LD, et al. Genetic relationships and evolution of genotypes of yellow fever virus and other members of the yellow fever virus group within the Flavivirus genus based on the 3' noncoding region. J Virol 2004;78:9652-65. 157. Ellis BR, Barrett AD. The enigma of yellow fever in East Africa. Rev Med Virol 2008;18:331-46. 158. Monath TP, Cetron MS. Prevention of yellow fever in persons traveling to the tropics. Clin Infect Dis 2002;34:1369-78. 159. Centers for Disease Control and Prevention. Adverse events associated with 17D-derived yellow fever vaccination--United States, 2001-2002. MMWR Morb Mortal Wkly Rep 2002;51:989-93. 160. Groot H, RIBERIRO RB. Neutralizing and haemagglutination-inhibiting antibodies to yellow fever 17 years after vaccination with 17D vaccine. Bull World Health Organ 1962;27:699-707. 161. Rosenzweig EC, Babione RW, Wisseman CL, Jr. Immunological studies with group B arthropodborne viruses. IV. Persistence of yellow fever antibodies following vaccination with 17D strain yellow fever vaccine. Am J Trop Med Hyg 1963;12:230-5. 162. Poland JD, Calisher CH, Monath TP, Downs WG, Murphy K. Persistence of neutralizing antibody 30-35 years after immunization with 17D yellow fever vaccine. Bull World Health Organ 1981;59:895-900. 163. Maurin M., Raoult D. Q fever. Clinical Microbiology Reviews 1999;12:518-53. 164. Schneeberger PM, Hermans MH, van Hannen EJ, Schellekens JJ, Leenders AC, Wever PC. Real-time PCR with serum samples is indispensable for early diagnosis of acute Q fever. Clin Vaccine Immunol 2010;17:286-90. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 43 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Investigation of Hepatitis 165. Yale SH, de Groen PC, Tooson J.D., Kurtin P.J. Unusual aspects of acute Q fever associated hepatitis. Mayo Clinical protocols 1994;69:769-73. 166. Wilson LE, Couper S, Prempeh H, Young D, Pollock KG, Stewart WC, et al. Investigation of a Q Fever Outbreak in a Scottish Co-Located Slaughterhouse and Cutting Plant. Zoonoses Public Health 2009. 167. Raoult D. Treatment of Q fever. Antimicrob Agents Chemother 1993;37:1733-6. 168. Million M, Thuny F, Richet H, Raoult D. Long-term outcome of Q fever endocarditis: a 26-year personal survey. Lancet Infect Dis 2010;10:527-35. Clinical Guidance | G 5 | Issue no: 1.2 | Issue date: 03.03.14 | Page: 44 of 44 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England