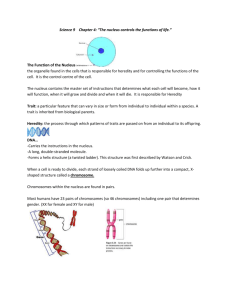
unit-2 genetics of prokaryotes and eukaryotic organelles and gene
advertisement

UNIT 4 CYTOGENETICS OF ANEUPLOIDS AND STRUCTURAL HETEROZYGOTES Structure 4.1. Introduction 4.2. Objective 4.3. Aneuploidy 4.4. Monosomics 4.4.1. Methods of production of monosomics 4.4.2. Description and Identification of monosomics 4.4.3. – 4.4.4. Meiotic behaviour of monosomics 4.4.5. Transmission of monosomics 4.5. Trisomics 4.5.1. Trisomics in diploids 4.5.2. Trisomics are classified into 3 categories 4.5.3. Origin and source of trisomics 4.5.4. Use of monosomics and trisomics in chromosomal mapping of diploid and polyploidy species 4.6. Euploidy 4.6.1. Haploidy 4.6.2. Polyploidy 4.6.2.1. Autopolyploidy 4.6.2.2. Allopolyploidy 4.1 INTRODUCTION The expression and inheritance of the sum total of characters of an organism is determined by the number and sequence of genes of its cells. Most of the plant and animal species are diploid and in their somatic tissue chromosomes are found in pairs. The number of chromosomes in a species normally remains constant through successive generations and their results into the constancy of characters. Gametes produced as a result of meiosis possess half as many chromosomes as the somatic cells and in them each chromosome is represented only once. Fusion of gametes brings together homologous chromosomes from the two parents and restores the diploid number. Alternation of haploid and diploid generations occurs in all sexual organisms. There is a great diversity in the number of chromosomes in different species. But this number is fixed in a particular species. Chromosome number is one of the characters that differentiate one species from another. In plants the somatic chromosome number ranges from four (two pairs) in Haplopappus to as many as 1260 in Ophioglossum. Constancy of the genetic material through successive generations is essential for maintaining the identity of a species. But variation is necessary if evolution has to occur. Sometimes such [1] variations involve whole chromosome or chromosome sets and can be seen cytologically. Variations that involve changes in number are of two types – Euploidy- variations that involve entire sets of chromosome and, Aneuploidy-variations that involve only single chromosome of a set. Numerical changes in chromosomes or variations in chromosome number means heteroploidy can be mainly of two types namely (i) Aneuploidy (ii) Euploidy Aneuploidy- means presence of chromosome number which is different than a multiple of basic chromosome number, or variants that involve one or only a few chromosomes but not the entire set. Euploidy- on the other hand, means presence of chromosome number which is similar multiple of basic chromosome number, or variation involve in entire sets of chromosomes. In above both alteration in chromosomes number, addition (hyperpoidy) or loss of chromosomes, entire set or individually (hypoploidy) present. Fig 4.1A diagrammatic representation of different types of numerical changes given below – Numerical alteration in chromosomes Aneuploidy Hypolpoidy Euploidy Hyperploidy Hyperploidy Haploidy 1x Nullisomy Monosomy 2n-(1x2) 2n-1 (pair) Trisomy 2n+1 Hypolpoidy Tetrasomy 2n+(1x2) (pair) Diploidy 2x Triploidy 3x Tetraploidy 4x * Different types of numerical changes in chromosomes. (x = basic chromosome number, 2n = somatic chromosome number, n = chromosome number of a gamete) [2] 4.2. OBJECTIVES This unit aims: To study the origin, breeding behavior and the phenotypic effects resulting from the numeric alterations in chromosomes. To study the role of numeric alterations of chromosome in agriculture and evolution. 4.3. ANEUPLOIDY Aneuploidy can be either due to loss of any one or more chromosomes to complete chromosome set (Hypoploidy). Hypoploidy is due to loss of single chromosome is known as monosomy, and the individual that carries such number is known as monosomics. The formula for monosomy is 2n-1. Another type of hypo aneuploidy is nullisomy – where the loss of a pair chromosome from diploid set. Both chromosome of a diploid set is absent. The general formula for nullisomy is 2n-(1x2) or 2n-2. Origin of aneuploidy (i) Monosomy – When monosomy is present in diploid organism it can not be tolerated because one complete chromosome is lacking. Numbers of genes present in this chromosome were affected. This unbalancing creates a problem, but when monosomy is present in polyploid organism these could be easily produced and tolerable because more than two chromosome of same combination is present. Meiosis in monosomics behaves like haploids. In monosomics one of the chromosomes will go to only one pole during division and other pole will be devoid of it. Other chromosomes will asset normally. In other words, monosomics behave like a haploid for one chromosome and as a diploid for others. (ii) Nullisomy – Another type of hypo-aneuploidy where a loss of pair of homologous chromosome is seen. When it found in diploid organism the organism doesn’t tolerate. The gametes produced by these individual are devoid of one chromosome – means they are aneuploids. The frequency of sterility is more in animals than in plants because the plants are able to tolerate loses of chromosomes much better than animals. 4.4 MONOSOMICS [3] Numbers of chromosomes in Nicotiana tabacum are 24, and all the 24 monosomics for that plant were reported by Clausenand Camerson (1944). Same work was done by Sears (1954) for hexaploid bread wheat. A set of monosomic for Gossypium hirsutum was reported by Endrizzi. 4.4.1 Methods of production of monosomics (i) From haploids – Sears (1939) got accidentally two haploids when he crossed two Geneva – (Secale cereale and Chinese springwheat). That haploid is used as female parent and crossed with normal hexaploid male. The seeds produced by this method showing monosomics. When these monosomics are selfed (inter crossing) nullisomic, trisomic and then later on tetrasomics were found by selfing. (ii) From backcrosses of inter specific hybrids – In N. tabacum monosomics have been produced by crossing the polyploidy species with one of its progenitors. The resultant F1 plant is backcrossed with polyploidy crop by which monosomics are found. Eg: In this eg. The haploid (n = 24) N. tabacum having 12 chromosomes of N. Sylvestris (n = 12) and 12 chromosomes of N. tomentosa (n = 12). This crop i.e. N. tabacum is crossed with N. sylvestris or (n = 24) develops F1 plant. Obtained F1 plants has 12 pairs chromosomes of N. sylvestris species and 12 single chromosomes of N. tomentosa. The gametes of this crop contain n = 12 to 24 as they have 21 chromosomes from N. sylvestris but the no. of single 12 chromosomes of N. tomentosa varies from gametes to gametes resulting in n = 12 to 24. These are then backcrossed with N. tabacum (n = 24) to develop monosomics for N. tomentosa. (a) N. tabacum X N. sylvestris (n = 24) (n = 12) F1 [12II (sylvestris) + 12I (tomentosa)] X N. tabacum (n = 24) (n = 12 to 24) (b) N. tabacum X N. tomentosa (n = 24) (n = 12) 23II + 1I (tomentosa) (among other constitutions) F1 [12II (tomentosa) + 12I (sylvestris)] X N. tabacum (n = 24) (n = 12 to 24) [4] 23II + 1I (sylvestris) (among other constitutions) Fig.4.2 Production of monosomics through interspecific hybridization in tobacco. (iii) From partially asynaptic plants – In this type during meiotic metaphase I, perfect bivalent formation is not observed, a variable number of univalents are observed. These univalents will be distributed to the two poles randomly during anaphase I. (iv) Irradiation treatment – In case of Gossypium hirsutum (cotton) and Avena sativa (Oats), irradiation of inflorescence to the production of gametes with n+1 or n-1. It is a type of artificial treatment. (v) Spontaneous production – Monosomics have been observed in wheat, tobacco, cotton by natural occasional non-disjunction of a bivalent during meiosis. 4.4.2 Description and Identification of monosomics Sometimes monosomics are morphologically different or sometimes it may not. It depends on genes present on that particular chromosome for which plant is monosomic. Monosomic condition is confirmed through chromosome count during mitotic metaphase and by the use of a univalent at meiosis. 4.4.4 Meiotic behaviour of monosomics Monosomics usually form bivalents in addition to a solitary univalent at end of prophase I and metaphase I. 4.4.5 Transmission of monosomics Breeding behaviour of monosomics has been studied by examining the progeny obtained on selfing them and on crossing them separately as female and male parents with the normal. On the basis of these studies, it is always possible to calculate the frequency of functional deficient (n = 2x-1 or 3x-1) gametes relative to normal (n = 2x or 3x) type of gametes. Although deficient gametes are produced at a frequency higher than 75%, but they function in the pollen at a very low frequency in wheat (4%), but at a variable frequency in oats (1%-74%). In tobacco, although deficient pollen may function at a low frequency, nullisomic zygotes, which normally do not survive. This situation leads to the production of nullisomics in the progenies of selfed monosomics, in variable frequency. [5] Eg, In case of wheat monosomics, (where normally n = 3x = 21), functional male gametes are of 2 types, i) n = 3x = 21, ii) n = 3x-1 = 20. The first category, male gametes are 96% while in second case only 4% gametes are produced. But in female gametes 25% gametes carrying n = 3x = 21 while remaining 75% having n = 3x-1 = 20. When these types of gametes crossed, percentage of nullisomics progeny is 3%, monosomics progeny is 73% and normal progeny is 24%. 4.5 TRISOMICS 4.5.1 Trisomics in Diploids The first aneuploid chromosome number was reported in Oenothera hybrids by Lutz (1909) and by Gates (1909) who recorded n = 15 while normal has n = 14. Trisomy – Trisomics is an example of hyperaneuploidy where an organism has an extra chromosome (2n+1). Since the extra chromosome may belong to anyone of the different chromosomes of a haploid complement. The number of possible trisomics in an organism will be equal to its haploid chromosome number. Eg, Barley the haploid chromosome no. is 7 (n = 7) consequently, 7 trisomics are possible. 4.5.2 Trisomics are classified into 3 categories i) ii) iii) Primary Trisomics – where an extra chromosome is identical to two homologous chromosomes (diploid) in their gene sequence. Secondary Trisomics – where the extra chromosome having same gene in multiple copies (i.e. isochromosome). The extra chromosome having duplicated and deficient gene. Tertiary Trisomics – The extra chromosome should be product of translocation i.e. this chromosome having genes of 2 or more other chromosomes. Trisomics were obtained for the first time in Datura stramonium by A. F. Blakeslee and his coworkers. The genomic number of this species is n = 12. Blakeslee and his coworkers found 12 primary trisomics, 24 secondary trisomics and large no. of possible tertiary trisomics. In Datura stramonium most of the trisomic were identified by the morphological features of fruit by shape and size. [6] Types of trisomy 2n + A 2n + B 2n + C 2n + D 2n + E 2n + F 2n + G 2n + H 2n + I 2n + J 2n + K 2n + L Name Rolled Glossy Buckling Elongate Echinus Cockleber Microcarpic Reduced Poinsettia Spinach Globe Ilex Figure 4.3 Different trisomics of Datura 4.5.3 Table-Different Origin and trisomics sourceofofDatura trisomics Trisomics may originate spontaneously due to production of (n+1) type of gametes; this gamete develops due to non-disjunction of a bivalent. Mostly trisomics are produced by artificial methods. Cytology of trisomics A trisomic has an extra chromosome, which is homologous to one of the chromosomes of the complement. During meiosis all of them synapse with each other and at metaphase I they arrange themselves from a peculiar shape of arrangement. (Fig 4.4) Somatic Chromosome Metaphase I configuration [7] Primary trisomics Secondary trisomics Tertiary trisomics Fig 4.4 Different types of trisomics and their meiotic configurations at metaphase I 4.5.4 Use of monosomics and trisomics in chromosomal mapping of diploid and polyploidy species The result of trisomic ratios can be utilized for locating genes on specific chromosome or for finding out distances of these genes from centromere, this technique is called chromosome mapping. If linkage groups are already established in an organism, trisomics can be effectively used for assigning these linkage groups to specific chromosomes. Since the segregation ratio for genes located on the chromosome involved in trisomic condition differs from that, genes located on other chromosomes, through these ratios one can found out on which chromosome a particular gene is located. This technique has been successfully applied in maize, tomato, barley, datura and Arabidopsis. [8] 4.6 EUPLOIDY It means presence of chromosome number which is similar multiple of basic chromosome number, or variation involve in entire sets of chromosomes. Each set of chromosomes is designated by the letter n and the numerical prefix refers to the number of sets or the level of ploidy. 4.6.1 Haploidy Haploid number of chromosomes is normally found in gametes of diploid (2n) organisms, but sometimes the whole organism may be haploid. Lower organism like bacteria and fungi are haploid for most of their life and become diploid or partially diploid for only a brief period. Monoploidy and haploidy A distinction should be made between monoploidy and haploidy. Monoploidy have a single basic set of chromosomes e.g. 2n = x = 7 in barley or 2n = x = 10 in corn. Haploids, on the other hand represent individuals having half the somatic chromosome number found in normal individual. Therefore, individuals having 2n = 3x = 21 in wheat would also be haploids. These latter kind of haploids obtained from polyploids are often called polyhaploids in order to distinguish them from monoploids. While reviewing the work on haploids in flowering plants in 1963, G. Kimber and R. Riley (now, John Inner Centre or JRC, Norwich, UK), gave a classification for haploids. They classified haploids in euhaploids and aneuhaploids which as the term indicate are derived from euploids and aneuploids respectively. A modified classification given by K. J. Kasha is presented. Euhaploids will include monohaploids as well as polyhaploids. Euhaploids Monohaploids Polyhaploids Allopolyhaploids Autopolyhaploids Aneuhaploids Disomic Haploids (n+1) Addition Haploids (n+1.etc) Nullisomic Haploids (n-1) Substitution Haploids (n-1+1) Misdivision Haploids Haploids [9] Origin, Occurence and Production of Haploids The haploids may originate at different points during the life cycle. Their spontaneous production may be facilitated by environmental factors and these may also be produced in the laboratory by artificial methods. Based on the point in the life cycle involved, haploid production systems can be broadly classified in three main categories namely: i) parthenogenesis and apogamy, ii) chromosome elimination and iii) culture methods 1. Production of haploids through parthenogenesis and apogamy The development of an embryo from the egg cell without the participation of a male gamete is parthenogenesis, and when the embryo develops from any other gametophytic cell other than egg cell without fertilization, it is apogamy. There are varieties of methods, which bring about parthenogenetic production of haploids like: a. Spontaneous- .It is largely due to parthenogenetic development of egg or any other cell of the embryo sac. b. Artificial treatments- The induction of haploids can be facilitated due to different treatments like irradiation treatment including X-rays and UV rays, hot and cold temperature shocks, wounds or injury, and chemical treatment c. Delayed pollination- A delay in pollination has also been found to induce parthenogenetic development of egg into a haploid embryo. d. Wide hybridization- In this method, when a suitable pollinator is used, pollen stimulates the development of embryo without fertilization. 2. Production of haploids through chromosome elimination Cases are known where either spontaneously or due to specific treatments, the chromosome number was reduced to half in the somatic tissues, a phenomenon described as somatic reduction or reductional mitosis. Swaminathan and Singh (1958) induced a haploid branch of watermelon by irradiation of the seed used. 3. Production of haploids through culture methods a. Through anther and pollen culture. It is one of the popular methods for production of haploids using artificial culture medium. This leads to the growth of microspores into sporophytes. [10] b. Through ovule culture. Haploids have also been successfully produced from cultured female gametophytes. Haploidy is common in plants but rare in animals except some diploid species of insects, rotifers, mites, etc., which produce haploid males parthenogenetically. In haploids each chromosome represented only once due to which there is no zygotene pairing and all the chromosomes appear as univalents on a metaphase plate at the time of meiosis. During anaphase each chromosome moves independently of the other and goes to either of the poles. According to the law of probability the chance that a particular chromosome will go to a particular pole is half and the chance that all the chromosomes of a haploid set will go to the same is ½ x ½ x ½…n times, where n = number of chromosomes in the haploid set. Therefore, the frequency of gametes with the haploid set or n number of chromosomes will be (½)n. This indicates that higher the number of chromosomes in a haploid set, lesser will be the frequency of all of them being included in the same gamete. Gametes with less than the haploid number of chromosomes are normally not viable. Haploid organism, therefore, are highly sterile. Male bees and such haploid animals are exceptions and in them there is no meiosis at the time of gamete formation. Cells arising as a result of mitosis get differentiated into sperms and are, therefore, haploid and viable. Haploid individuals usually develop abnormally in many diploid animals. Amphibian haploids have been induced by various means, but the embryos rarely reach the adult stage. In many plants, such as Nicotiana, Datura, Oiyza, Secale etc., fully viable haploids can be formed, but they turn out to be sterile because meiosis produces inviable gametes. In all organisms’ haploid and diploid generations alternate but in evolved organisms, like higher plants and animals, the diploid generation predominates and the haploid generation is transitory and short lived. As we go down the evolutionary scale, the balance shifts in favour of the haploid generation, so much so that in micro-organisms major part of the life span is haploid. During haploid state there is only one copy of each chromosome and only one allele of each gene. Consequently, each gene is expressed whether it is dominant or recessive. This facilitates genetic experiments and this is the reason why micro-organisms have been helpful in genetic studies. For the same reason scientists are trying to develop haploid strains of flowering plants. Success has been achieved in developing haploid strains of Nicotiana, Datura, Triticum, etc., but the problem of their poor growth and sterility has yet to be overcome. Haploid strains of crop are useful in another way. Present day diploid and polyploidy species are the result of evolution through ages and inconceivable [11] diversity and heterozygosity has been generated due to mutation and recombination. Heterozygosity is a hindrance in plant breeding because the recessive genes keep on appearing during successive generations. If the chromosome number of a haploid strain is duplicated, a diploid will arise which will be homozygous for all the characters and genes. Such a strain will be an ideal starting material for crop breeding. This is the reason why biologists are synthesizing haploid strains of crops like rice, wheat, etc. It was Maheshwari and Guha (1966) of Delhi University who, for the first time, were successful in obtaining haploid plants of tobacco by culturing anthers on suitable media. Since then this technique has been used for developing haploids in a number of plants. In the same department Sinha and Bhojwani have been able to induce haploidy in somatic cells of onion by growing the roots in a medium containing para-fluorophenylalanine. Chemical induction of haploidy in higher plants, if successful on a large scale, will be a very useful tool in the hands of plant breeders. 4.6.2 Polyploidy In polyploidy cells and organisms the number of chromosome sets exceeds that of the diploid. Depending on the number of chromosome sets, polyploidy can be classified as triploids, tetrapoloids, pentaploids, etc. diploid organisms possess polyploidy cells and tissue. Diploid human cells have 46 chromosomes each, but in cancer cells this number may reach 100 or more due to abnormal divisions. In the liver of mice about 40 percent of the cells are tetraploid and about five percent are octaploid. Polyploidy nuclei, cells and tissue are very common even in plants. Endosperm nuclei are generally triploid because of triple fusion. Sometimes they may be tetra-, penta-, hexa-, or polyploids of even higher order. Origin and Induction of Polyploidy Failure of normal mitotic divisions results into nuclei with increased sets of chromosomes. During mitosis in a diploid somatic cell, if the chromosomes duplicate and divide but cytokinesis fails to occur, a tetraploid cell arises. Normal mitotic division of this cell gives rise to a tetraploid tissue. Organs or branches that develop from such a tissue are tetraploid and flower borne on such branches produce diploid instead of haploid gametes. A doubling of the number of somatic chromosomes in a sex organ results into a doubling of the number of gametic chromosomes. A fusion of two diploid gametes produces a tetraploid individual. A fusion of a diploid and a haploid gamete results into a triploid individual. Even abnormal meiosis may result into a diploid gamete instead of a haploid in a diploid [12] organism. Polyploidy can also occur if an egg is fertilized by more than one male gamete. Polyploidy can be induced with the help of chemicals, by giving temperature shocks or by causing mechanical injuries to tissues. In tomato plants disorganized cell division occurs at cut ends of stem and a callus tissue develops. Most of the cells in such a tissue are tetraploid rather than diploid. About seven percent of the branches developed from such a tissue are polyploid. Similarly, octaploid branches can be developed on tetraploid tissues. In plants, like maize, polyploidy can be induced by temperature shocks which disorganize cytokinesis. Tissues developed from a polyploid cell are polyploid. In maize short pulses of high temperature are necessary for inducing polyploidy. In other organisms short pulses of low temperatures or alternating high and low temperatures are required for this purpose. Divided nuclei can be prevented from separating in Spirogyra by refrigeration or by treatment with ether or chloroform. If nuclei do not separate and cytokinesis occurs, the result is a cell with two nuclei and another cell without any nucleus. A cell with two nuclei divides to give rise to polyploidy cells. Chemicals, like indole acetic acid induce cell division in non-meristematic cells, thereby inducing polyploidy. If the rate of nuclear division is increased, polyploidy cells arise because cell wall synthesis, which is responsible for cytokinesis, is not able to cope with the increased division rate of the nucleus. When roots of leguminous plants are infected with nitrogen-fixing bacteria like Rhizobium, the level of indoleacetic acid increases in cortical cells and this causes polyploidy. It is a matter of dispute whether indoleacetic acid induces polyploidy or simply helps in division and multiplication of a polyploidy cell. Let us imagine that A, B1, B2, and C are four different haploid sets of chromosomes and that genomes B1 and B2 are related. (Fig 4.5) [13] AAAAB1B1B1B1 Autoallopolyploid 2n = 56 AB1B1B2 Hybrid 2n = 28 AAB1B1CC Allohexaploid 2n = 42 AAAA Autotetraploid 2n = 28 AAB1B1 Allotetraploid 2n = 28 B1B1B2B2 Segmental allotetraploid 2n = 28 B2B2B2B2 Autotetraploid 2n = 28 AB1C Allotriploid hybrid 2n = 21 AA Diploid 2n = 14 AB1 Hybrid 2n = 14 B1B1 Diploid 2n = 14 B1B2 Hybrid 2n = 14 B2B2 Diploid 2n = 14 CC Diploid 2n = 14 Figure 4.5 Different kinds of polyploids and their derivation from one or more basic diploid species [14] Chemicals like colchicines and acenapthene are believed to interfere with spindle formation and thus induce polyploidy. Colchicine is an alkaloid which is extracted from corms of Colchicum autumnale or Colchicum luteum of the family Liliaceae. The corm is used as a carminative, laxative, aphrodisiac and is useful in gout, rheumatism and disease of liver and spleen and also as an external application to lessen inflammation and pain. Eigsti (1937) noticed that if onion bulbs are allowed to root in a 0.05 percent aqueous solution of colchicines, some cells show polyploidy and if colchicines treatment is continued as many as 1000 chromosomes can be found in a cell. As colchgicine prevents the formation and organization of spindle fibre, the metaphase chromosomes of affected cells (C-metaphase or colchicines metaphase) do not move to a metaphase plate and remain scattered in the cytoplasm. Even cytokinesis is prevented and with duplications of chromosomes their number goes on increasing. Soon after Eigsti discovered the polyploidy inducing potentiality of colchicines, Blakeslee, Avery and Nebel (1937) used it for getting a polyploidy plant. Now-a-days colchicine is one of the most widely used chemicals for induction of polyploidy and for causing related effects. As colchicine interferes with spindle formation, its effects are limited to dividing or meristematic cells. Once a polyploidy cell is formed, its normal division gives rise to a polyploidy tissue, branch, flowers, fruits, seeds, etc., and a polyploidy variety is established. Polyploidy There are mainly three different kinds of polyploids, namely i) Autopolyploid, ii) Allopolyploids, and iii) Auto allopolyploids. [15] Species A 2n = AA Species B 2n = BB Species C 2n = CC F1 2n = AB Autotriploid 3n = AA Autotetraploid 4n = AAAA Triploid 3n = ABB Allotetraploid 4n = AABB Triploid 3n = ABC Autohexaploid 6n = AAAAAA Autoallohexaploid 6n = AABBBB Autopentaploid 5n = AAAAA Allohexaploid 6n = AABBCC Fig. 4.6. Interrelationships of auto- and allopolyploids. (After Stebbins, 1950). [16] 4.6.2.1 Autopolyploids Autopolyploids are those polyploids, which has the same basic set of chromosomes multiplied. For instance, if a diploid species has two similar sets of chromosomes or genomes (AA), an autotriploid will have three similar genomes (AAA) and an autotetraploid will have four such genomes (AAAA) as shown in figure. Origin and production of autopolyploids. The autopolyploids may occur in nature or may be artificially produced. When they are found in nature, their autopolyploid nature is inferred mainly by their meiotic behaviour. One of the very common examples of natural autopolyploidy relevant to northern India pertains to common ‘doob’ grass (Cynodon dactylon). In U.P. and Bihar, common ‘doob’ grass was found to be an autotriploid as inferred from its meiotic behaviour (Gupta and Srivastava, 1970). It is perhaps successful due to efficient vegetative reproduction, because, as will be seen, autopolyploids are normally triploids and set no seeds. Autotriploids are also known in watermelons, sugarbeet, tomato, grapes and banana; although in several of these cases the polyploids have been artificially produced. Similarly autopolyploids are known in rye (Secale cereale), corn (Zea mays), red clover (trifolium prantese), berseem (trifolium alexandrium), marigolds (Tagetes), snapdragons (Antirrhinum), Phlox, grapes, apples, etc. In Oenothera lamarckiana, an American plant, a giant mutant described by Hugo de Vries was later discovered to be an autotetraploid. Induced autopolyploids. In many cases listed above, autopolyploidy is artificially induced. Since polyploids are normally larger and more vigorous, their role in crop improvement has been realized and techniques developed for artificial induction of polyploidy. Polyploidy is mainly induced by treatment with aqueous solution of a drug called colchicine. This drug has the property of arresting and breaking the spindle so that a cell division without cell wall formation may be affected leading to doubling of chromosome number. The concentration of aqueous solution of colchicines may vary from 0.01% to 0.50% and the treatment may be given in one of the following manners. (I) Seed treatment may be mainly given by soaking seeds for different durations in aqueous solution of colchicines. (II) Injections of colchicines solution may also be given at seedling stage so as to inject solution into cortex tissue with the help of a hypodermic needle. (III) Axillary bud treatment is also effective. Since bud is meristematically active, placing cotton soaked in colchicine on the bud and continuous dropping of solution on cotton leads to induction of polyploidy in the branch arising from the treated bud. [17] (IV) Shoot apex treatment is brought about just like bud treatment and is fairly effective, but the shoot apex should come in direct contact of the solution. In order to facilitate this, young leaves covering the shoot apex may be removed. Colchicine is an alkaloid extracted from seed and corm of Colchicine autumnale. The action of colchicines and its use in inducing polyploidy was first studied in 1930’s. The successful doubling of chromosome number was described for the first time by Levan (1938) and by Eigsti (1938 - 40). It was surprising however, that colchicine does not affect colchicum, the plant from which it is extracted. This was attributed to the presence of an ‘anticolchicine’. The success of colchicine treatment depend on three prerequisites, namely (i) direct contact of growing tissue with solution, (ii) effective concentration of colchicine and (iii) The optimal stage of development. These three conditions are met with in the methods of treatment described above. Effects of chromosome doubling. One of the important effects of polyploidy is to produce ‘gigantism’. The autopolyploids may be normally larger in size. Sometimes plants may be smaller than diploids. But leaves, flowers, and the cells themselves may be bigger in size. Some of the important effects of polyploidy are as follows: With increase in cell size, water content increases leading to decrease in osmotic pressure. This results into loss of resistance against frost, etc. Growth rate decreases due to slower rate of cell division; this leads to a decrease in auxin supply and a decrease in respiration. In autopolyploids, time of blooming is delayed and prolonged due to slow growth rate. At higher ploidy level (autooctoploids or higher), the adverse effects are highly pronounced and lead to death of plants. Cytology of autopolyploid. In an autopolyploid, there will be more than two sets of homologous chromosomes. This leads to formation of multivalents instead of bivalent as found in diploids. An important difference exists even between autotriploids and autotetraploids, because while in the latter normal disjunction is possible giving rise to diploid gametes, in triploids it is not possible. In an autotriploid, there are three sets of homologous chromosomes. If these three sets are normally paired, trivalents can not disjoin normally and will either disjoin 2:1 chromosomes to two poles or will disjoin 1:1 leaving one chromosome as a laggard. The number of chromosomes in gametes of triploid organism, therefore, will vary from n to 2n. Most of these gametes are unbalanced leading to high degree of sterility. [18] In autotetraploids, since there are four sets of chromosomes, quadrivalents are formed, which disjoin in a normal 2:2 manner giving diploid gametes. Rarely however, a quadrivalent may disjoin 3:1 or may leave a chromosome as a laggard at anaphase I. Therefore autotetraploids also have a certain degree of sterility, although it will not be as high as on autotriploids. 4.6.2.2 Allopolyploids. Polyploidy may also result from doubling of chromosome number in a F1 hybrid which is derived from two distinctly different species. This will bring two different sets of chromosomes in F1 hybrid. The number of chromosomes in each of these two sets may differ. Let A represent a set of chromosomes (genome) in species X, and let B represent another genome in a species Y.The F1 will then have one A genome and another B genome. The doubling of chromosomes in this F1 hybrid (AB) will give rise to a tetraploid with two A and two B genomes. Such a polyploidy is called an allopolyploid or amphidiploid. Raphanobrassica is a classical example of allopolyploidy. In 1927, G. D. Karpechenko, a Russian geneticist, reported a cross between Raphanus sativus (2n = 18) and Brassica oleracea (2n = 8) to produce F1 hybrid which was completely sterile. This sterility was due to lack of chromosome pairing, since there is no homology between genomes from Raphanus sativus and Brassica oleracea. Among these sterile F1 hybrids, Karpechenko found certain fertile plants. On cytological examination these fertile plants were found to have 2n = 36 chromosomes, which showed normal pairing into 18 bivalents. Radish (9R + 9R) Gametes : F1 : Gametes : F1 : Cabbage (9C + 9C) 9R 9C (9R + 9C) Diploid hybrid (9R 9C) (9R 9C) (18R + 18C) Raphanobrassica [19] Fig 4.7 Example of an allopolyploidy. (a) Evolution of Wheat. Common cultivated wheat is another important example of allopolyploidy, although its allopolyploid nature has now been questioned. There are three different chromosomes numbers in the genus Triticum, namely 2n = 14, 2n = 28, 2n = 42. The common wheat is hexaploid with 2n = 42 and is derived from three diploid speices: (i) AA = Triticum aegilopoides (2n = 14), (ii) BB = Aegilops speltoides ? (2n = 14)(in the past evidence was made available, showing that Ae. Speltoides may not be the donor of E genome; it is also believed that the donor of E genome may never be discovered) and (iii) DD = Aegilops tauschii, earlier known as Ae.squarrosa (2n = 14). The hexaploid wheat, therefore, is designated as AABBDD, the tetraploid (2n = 28) as AABB and diploid (2n = 14) as AA. There is, however, evidence available now which suggests that A, B and D genomes from three diploid species mentioned above are not much different from one another, so that it is believed that the three diploid progenitors of common hexaploid wheat were derived from a common ancestor. For this reason, the common hexaploid wheat is now considered an autopolyploid rather than an allopolyploid. At best, it may be a segmental allopolyploid. (Fig.4.8) (b) Triticale (x Triticosecale), a new man made cereal. During the last decades, considerable emphasis has been laid on the possibility of utilizing a new man made cereal known as triticale, on a commercial scale. It is already grown in an estimated area of more than three million hectares and research at several places all over the world is in progress to improve this man made crop. Triticale is the first man made crop, in so far as it resulted as an artificial allopolyploid derived by crossing wheat (Triticum) and rye (Secale). Depending upon whether tetraploid (2n = 4x = 28) or hexaploid (2n = 6x = 42) wheat is utilized for the synthesis, one would get hexaploid triticale (2n = 6x = 42) or octoploid triticale (2n = 8x = 56) respectively. In each case, only diploid rye (2n = 2x = 14) was used. (a) Triticum durum X Secale cereale 2n = 28 2n = 14 F1 hybrid (sterile) 2n = 21 Chromosome doubling 2n = 42 Hexaploid triticale [20] (b) Triticum aestivum X Secale cereale 2n = 42 2n = 14 Hybrid 2n = 14 AB Chromosome doubling Aegilops speltoides X 2n = 14 Triticum dicoccoides BB 4n = 28 Wild AABB Triticum boeoticum 2n = 14 AA Wild Triticum monococcum 2n = 14 AA Cultivated Wild F1 hybrid (sterile) 2n = 28 Chromosome doubling 2n = 56 Octoploid triticale Fig.4.9 Artificial synthesis of a) Hexaploid triticale b) Octoploid triticale [21] Fig.4.8 Diagrammatic representation of the origin of tetraplooid and hexaploid cultivated wheat from their wild ancestors. Triticum durum Triticum dicoccum Aegilops sqarrosa X 4n = 28 4n = 28 2n = 14 AABB AABB DD Cultivated Cultivated Wild Check Your Progress Note: (a) Write your answer in the (b) Compare your answer with of the unit. Q.1 What is allopolyploidy?Describe the Raphanobrassica was produced. Hybrid 3n = 21 ABD space given below, key given at the end Chromosome doubling Triticum spelta, Triticum vulgare. Etc. 6n = 42 AABBDD Cultivated experiment by which ……………………………………………………………………………………… ……………………………………………………………………………………… ……………… Q.2 How many trisomics were found in Datura? [22] ……………………………………………………………………………………… ……… Q.3 What are secondary and tertiary trisomics? ……………………………………………………………………………………… ……… Q.4 How did triticum aestivum be produced artificially? ……………………………………………………………………………………… ……………………………………………………………………………………… ……………... Q.5 Differentiate between Aneuploid and euploid. ……………………………………………………………………………………… ……………………………………………………………………………………… ……………………………………………………………………………………… …………………….... Let’s Sum Up Change in the number and structure of chromosome may occur spontaneously or experimentally by the action of radiation or chemicals. Chromosomal changes in number are of two main kinds: in euploids, the set is kept balanced. In aneuploids there is a loss or gain of one or more chromosomes. In plants, polyploids (triploid, tetraploiid etc) are rather common. They originate by reduplication without cytokinesis. Polyploidy can be induced by colchicines, this substance is used in agriculture to improve certain plant species. Allopolyploidy consists of the formation of a hybrid with different sets of chromosomes. Among the aneuploid organisms there are the trisomic (i.e. with three similar chromosomes instead of normal pair) and the monosomic (missing one member of a pair of homologous chromosomes). Assignment – a) b) c) d) Role of aneuploidy and euploidy in agriculture. Meiotic behavior of autopolyploid and allopolyploids. Aneuploid abnormalities in Drosohila, Datura and Human. Behavior of metaphase configuration in different types of trisomics. [23] Check Your Progress: Key Q.1 A polyploidy containing two or more different genomes.By crossing radish and cabbage and in F1 hybrid the chromosome number is doubled to produce raphanobrassica. Q.2 12 primary trisomics, 24 secondary trisomics and a large number of tertiary trisomics. Q.3 Refer section 4.5.2 Q.4 Refer section 4.6.2.2 Q.5An aneuploid organism has a chromosome number that is not an exact multiple of the basic chromosome number while euploid has exact multiple of the basic number. References Cell and molecular biology-P.K.Gupta Cytogenetics, Plant Breeding and evolution-U Sinha and Sunita Sinha Cytogenetics- P.K.Gupta Cell Biology-P.S.Verma Chromosomes-M.S.Clark and W.J.Wall Cell and molecular biology-Ajay Paul Cell and molecular biology -De Robertis [24] UNIT-5 MOLECULAR CYTOGENETICS AND ALIEN GENE TRANSFER THROUGH CHROMOSME MANIPULATION Structure 5.0 Introduction 5.1 Objectives 5.2 Molecular Cytogenetics: 5.2.1 Nuclear DNA Content 5.2.2 C-value Paradox 5.2.3 Cot Curve and its Significance 5.2.4 Restriction Techniques 5.2.5 Multigene Families and their Evolution 5.2.6 In-Situ Hybridization – Concept and Techniques 5.2.7 Physical Mapping of Genes on Chromosomes 5.2.8 Computer Assisted Chromosome Analysis 5.2.9 Chromosome Microdissection and Microcloning 5.2.10 Flow Cytometry and Confocal Microscopy in Karyotype Analysis 5.3 Alien Gene Transfer Through Chromosome Manipulation: 5.3.1 Transfer of Whole Genome – Examples from Wheat, Archis & Brassica 5.3.2 Transfer of Individual Chromosomes and Chromosome Segments 5.3.3 Methods for Detecting Alien Chromatin 5.3.4 Production, Characterization and Utility of Alien Addition & Subtraction Lines RNA Splicing 5.3.5 Genetic Basis of Inbreeding and Heterosis 5.3.6 Exploitation of Hybrid Vigour 5.4 Let Us Sum Up 5.5 Check Your Progress 5.6 Check Your Progress: The Key 5.7 Assignment 5.8 References 5.0 INTRODUCTION Molecular cytogenetics is a branch of biology in which nucleic acid and other cytological molecules which being involve in genetic inheritance are studied. In eukaryotes nucleus consist DNA as genetic material which have different components like chromatin, nucleosomes, histone etc. Nucleolus also have important role. Various techniques are now available to measure the amount of genome, number and position of genes and many other such type of work for the research and applicability purposes. [25] Breeding programmes aiming at transferring desirable genes from one species to another through interspecific hybridization and backcrossing often produce monosomic and disomic addition as intermediate crossing products. Such aneuploids contain alien chromosomes added to the complements of the recipient parent and can be used for further introgression programmes, but lack of homoeologous recombination and inevitable segregation of the alien chromosome at meiosis make them often less ideal for producing stable introgression lines. Monosomic and disomic additions can have specific morphological characteristics, but more often do they need additional confirmation by molecular marker analyses and assessment by fluorescence in situ hybridization with genomic and chromosome specific DNA as probes. Their specific genetic and cytogenetic properties make them powerful tools for fundamental research elucidating regulation of homoeologous recombination, distribution of chromosome specific markers and repetitive DNA sequences, and regulation of heterologous gene expression. In above chapter it has been tried to present the major characteristics of such interspecific aneuploids highlighting their advantages and drawback for breeding and fundamental research. Therefore present chapter is being prepared to enlighten the role of nucleus in genetics along with techniques of genetic engineering which are also helpful to improve the quality of plants of agricultural, horticultural importance. 5.1 OBJECTIVE Purpose of this unit is to cover the following points to describe the utility of molecular cytogenetics: C-value genomic content in a haploid cell which is related to genome size and varies organism to organism, The Cot curve (an S-shaped curve) is obtained by plotting the fraction of singlestranded DNA remaining (C/C0) as a function of the logarithm of the product of the initial concentration and the elapsed time, Eukaryotic DNA consists of at least three types of sequences nonrepetitive (unique-sequence DNA), moderately repetitive DNA, highly repetitive DNA, Restriction techniques involve methods for locating specific DNA sequences, cutting DNA at precise locations, amplifying a particular DNA sequence, mutating and joining DNA fragments to produce desired sequences and procedures for transferring DNA sequences into recipient cells, A multigene family involves members of a family of related proteins encoded by a set of similar genes. Multigene families are believed to have arisen by duplication and variation of a single ancestral gene, In situ hybridization techniques locate specific nucleic acid sequences in cells or on chromosomes, The ultimate goal of gene mapping is to clone genes, especially disease genes. Once a gene is cloned, we can determine its DNA sequence and study its protein product, Computer programmes are very helpful to analyse the karyotype and chromosomal information, [26] Chromosome microdissection is a specialized way of isolating specific regions by removing the DNA from the band and making that DNA available for further study (microcloning), Flow cytometry is very fast and informative, quantitative and qualitative analysis method which objects include chromosomes and nuclei, Alien gene transfer is aiming to improve the crop quality by using the different properties of plant like heterosis, hybrid vigour etc. 5.2 MOLECULAR CYTOGENETICS 5.2.1 Nuclear DNA Content The nucleus mainly have chromosome and nucleoplasm. The molecule of DNA in a single human chromosome ranges in size from 50 x 106 nucleotide pairs in the smallest chromosome (stretched full-length this molecule would extend 1.7 cm) up to 250 x 106 nucleotide pairs in the largest (which would extend 8.5 cm). Stretched end-to-end, the DNA in a single human diploid cell would extend over 2 meters. In the intact chromosome, however, this molecule is packed into a much more compact structure. The packing reaches its extreme during mitosis when a typical chromosome is condensed into a structure about 5 µm long (a 10,000fold reduction in length). Chromatin - The nucleus contains Figure 5.1: Electron micrograph showing chromatin from the nucleus of a chicken red blood cell (birds, unlike most mammals, retain the nucleus in their mature red blood cells). The arrows point to the nucleosomes. You can see why the arrangement of nucleosomes has been likened to "beads on a string"(courtesy of David E. Olins and Ada L. Olins). the chromosomes of the cell. Each chromosome consists of a single molecule of DNA complex with an equal mass of proteins. Collectively, the DNA of the nucleus with its associated proteins is called chromatin (Figure 5.1). Most of the protein consists of multiple copies of 5 kinds of histones. These are basic proteins, bristling with positively charged arginine and lysine residues. (Both Arg and Lys have a free amino group on their R group, which attracts protons (H+) giving them a positive charge.) Just the choice of amino acids you would make to bind tightly to the negatively-charged phosphate groups of DNA. Chromatin also contains small amounts of a wide variety of nonhistone proteins. Most of these are transcription factors (e.g., the steroid receptors) and their association with the DNA is more transient. [27] Nucleosomes - Two copies of each of four kinds of histones H2A H2B H3 and H4 form a core of protein, the nucleosome core (Figure 5.2). Around this is wrapped about 147 base pairs of DNA. From 20–60 bp of DNA link Figure 5.2:Nuclosome, Histone and Linker DNA one nucleosome to the next. Each linker region is occupied by a single molecule of histone 1 (H1). The binding of histones to DNA does not depend on particular nucleotide sequences in the DNA but does depend critically on the amino acid sequence of the histone. Histones are some of the most conserved molecules during the course of evolution. Histone H4 in the calf differs from H4 in the pea plant at only 2 amino acids residues in the chain of 102. The formation of nucleosomes helps somewhat, but not nearly enough, to make the DNA sufficiently compact to fit in the nucleus. In order to fit 46 DNA molecules (in humans), totaling over 2 meters in length, into a nucleus that may be only 10 µm across requires more extensive folding and compaction. interactions between the exposed "tails" of the core histones cause nucleosomes to associate into a compact fiber 30 nm in diameter. these fibers are then folded into more complex structures whose precise configuration is uncertain and which probably changes with the level of activity of the genes in the region. Histone Modifications - Although their amino acid sequence (primary structure) is unvarying, individual histone molecules do vary in structure as a result of chemical modifications that occur later to individual amino acids. These include adding: acetyl groups (CH3CO-) to lysines phosphate groups to serines and threonines methyl groups to lysines and arginines Although 75–80% of the histone molecule is incorporated in the core, the remainder - at the N-terminal - dangles out from the core as a "tail" (not shown in the Figure 5.2). The chemical modifications occur on these tails, especially of H3 and H4. Most of theses changes are reversible. For example, acetyl groups are added by enzymes called histone acetyltransferases (HATs)(not to be confused with the "HAT" medium used to make monoclonal antibodies!) and removed by histone deacetylases (HDACs). More often than not, acetylation of histone tails occurs in regions of chromatin that become active in gene transcription. This makes a kind of intuitive sense as adding acetyl groups neutralizes the positive charges on Lys thus reducing the strength of the association between the highly-negative DNA and the highly-positive histones. But there is surely more to the story. [28] Acetylation of Lys-16 on H4 prevents the interaction of their "tails" needed to form the compact 30-nm structure of inactive chromatin. But this case involves interrupting protein-protein not protein-DNA interactions. Methylation, which also neutralizes the charge on lysines (and arginines), can either stimulate or inhibit gene transcription in that region. o methylation of lysine-4 in H3 is associated with active genes while o methylation of lysine-9 in H3 is associated with inactive genes. (These include those imprinted genes that have been permanently inactivated in somatic cells.) and Adding phosphates causes the chromosomes to become more - not less - compact as they get ready for mitosis and meiosis. In any case, it is now clear that histones are a dynamic component of chromatin and not simply inert DNA-packing material. Histone Variants - We have genes for 8 different varieties of histone 1(H1). Which variety is found at a particular linker depends on such factors as o the type of cell, o where it is in the cell cycle, and o its stage of differentiation. In some cases, at least, a particular variant of H1 associates with certain transcription factors to bind to the enhancer of specific genes turning off expression of those genes. Some other examples of histone variants: o H3 is replaced by CENP-A ("centromere protein A") at the nucleosomes near centromeres. Failure to substitute CENP-A for H3 in this regions blocks centromere structure and function. o H2A may be replaced by the variant H2A.Z at the boundaries between euchromatin and heterochromatin. o All the "standard" histones are replaced by variants as sperm develop. In general, the "standard" histones are incorporated into the nucleosomes as new DNA is synthesized during S phase of the cell cycle. Later, some are replaced by variant histones as conditions in the cell dictate. Euchromatin versus Heterochromatin - The density of the chromatin that makes up each chromosome (that is, how tightly it is packed) varies along the length of the chromosome. dense regions are called heterochromatin, less dense regions are called euchromatin (Figure 5.3). Figure 5.3: The diagram represents a hypothetical model of how euchromatin and heterochromatin may be organized during interphase in a vertebrate cell. Heterochromatin is found in parts of the chromosome where there are few or no genes, such as [29] o o centromeres and telomeres is densely-packed; is greatly enriched with transposons and other "junk" DNA; is replicated late in S phase of the cell cycle; has reduced crossing over in meiosis. Those genes present in heterochromatin are generally inactive; that is, not transcribed and show o increased methylation of the cytosines in CpG islands of the DNA; o decreased acetylation of histones and o increased methylation of lysine-9 in histone H3, which now provides a binding site for heterochromatin protein 1 (HP1), which blocks access by the transcription factors needed for gene transcription. Euchromatin is found in parts of the chromosome that contain many genes; is loosely-packed in loops of 30-nm fibers. These are separated from adjacent heterochromatin by insulators. In yeast, the loops are often found near the nuclear pore complexes. This would seem to make sense making it easier for the gene transcripts to get to the cytosol. However, in animal cells, gene transcription appears to be repressed near the inner surface of the nuclear envelope. The genes in euchromatin are active and thus show o decreased methylation of the cytosines in CpG islands of the DNA o increased acetylation of histones and o decreased methylation of lysine-9 in histone H3. Nucleosomes and Transcription - Transcription factors cannot bind to their promoter if the promoter is blocked by a nucleosome. One of the first functions of the assembling transcription factors is to either expel the nucleosome from the site where transcription begins or at least to slide the nucleosomes along the DNA molecule. Either action exposes the gene's promoter so that the transcription factors can then bind to it. The actual transcription of protein-coding genes is done by RNA polymerase II (RNAP II). In order for it to travel along the DNA to be transcribed, a complex of proteins removes the nucleosomes in front of it and then replaces them after RNAP II has transcribed that portion of DNA and moved on. The Nucleolus - During the period between cell divisions, when the chromosomes are in their extended state, 1 or more of them (10 in human cells) have loops extending into a spherical mass called the nucleolus. Here are synthesized three (of the four) kinds of RNA molecules (28S, 18S, and 5.8S) used in the assembly of the large and small subunits of ribosome. 28S, 18S, and 5.8S ribosomal RNA is transcribed (by RNA polymerase I) from hundreds to thousands of tandemly-arranged rDNA genes distributed (in humans) on 10 different chromosomes. The rDNA-containing regions of these 10 chromosomes cluster together in the nucleolus. (In yeast, the 5S rRNA molecules - as well as transfer RNA molecules are also synthesized (by RNA polymerase III) in the nucleolus. Once formed, rRNA [30] molecules associate with the dozens of different ribosomal proteins used in the assembly of the large and small subunits of the ribosome. But all proteins are synthesized in the cytosol - and all the ribosomes are needed in the cytosol to do their work - so there must be a mechanism for the transport of these large structures in and out of the nucleus. This is one of the functions of the nuclear pore complexes. Nucleoplasm - The term "nucleoplasm" is still used to describe the contents of the nucleus. However, the term disguises the structural complexity and order that seems to exist within the nucleus. For example, there is evidence that DNA replication and transcription occur at discrete sites within the nucleus. C-value is the mass of DNA (expressed, for example, in picograms per cell) in the haploid genome of a species. 5.2.2 C-value Paradox / Engima C-value - The term C-value refers to the amount of DNA contained within a haploid nucleus (e.g., in a gamete or one half the amount in a diploid somatic cell) of a eukaryotic organism. In some cases (notably among diploid organisms), the terms C-value and genome size are used interchangeably, however in polyploids the C-value may represent two genomes contained within the same nucleus. Greilhuber et al. (2005) have suggested some new layers of terminology and associated abbreviations to clarify this issue, but these somewhat complex additions have yet to be used by other authors. C-values are reported in picograms. Origin of the term - Many authors have incorrectly assumed that the "C" in "Cvalue" refers to "characteristic", "content", or "complement". Even among authors who have attempted to trace the origin of the term, there had been some confusion because Hewson Swift did not define it explicitly when he coined it in 1950. In his original paper, Swift appeared to use the designation "1C value", "2C value", etc., in reference to "classes" of DNA content (e.g., Gregory 2001, 2002); however, Swift explained in personal correspondence to Prof. Michael D. Bennett in 1975 that "I am afraid the letter C stood for nothing more glamorous than 'constant', i.e., the amount of DNA that was characteristic of a particular genotype" (quoted in Bennett and Leitch 2005). This is in reference to the report in 1948 by Vendrely and Vendrely of a "remarkable constancy in the nuclear DNA content of all the cells in all the individuals within a given animal species" (translated from the original French). Swift's study of this topic related specifically to variation (or lack thereof) among chromosome sets in different cell types within individuals, but his notation evolved into "C-value" in reference to the haploid DNA content of individual species and retains this usage today. C-value paradox history - In 1948, Roger and Colette Vendrely reported a "remarkable constancy in the nuclear DNA content of all the cells in all the individuals within a given animal species", which they took as evidence that DNA, rather than protein, was the substance of which genes are composed. The term C-value reflects this observed constancy. However, it was soon found that C-values (genome sizes) vary [31] enormously among species and that this bears no relationship to the presumed number of genes (as reflected by the complexity of the organism). For example, the cells of some salamanders may contain 40 times more DNA than those of humans. Given that C-values were assumed to be constant because DNA is the stuff of genes, and yet bore no relationship to presumed gene number, this was understandably considered paradoxical; the term C-value paradox was used to describe this situation by C.A. Thomas, Jr. in 1971. The discovery of non-coding DNA in the early 1970s resolved the C-value paradox. It is no longer a mystery why genome size does not reflect gene number in eukaryotes: most eukaryotic (but not prokaryotic) DNA is non-coding and therefore does not consist of genes, and as such total DNA content is not determined by gene number in eukaryotes. The human genome, for example, comprises only about 1.5% protein-coding genes, with the other 98.5% being various types of non-coding DNA (especially transposable elements) (International Human Genome Sequencing Consortium 2001). It is unclear why some species have a remarkably higher amount of non-coding sequences than others of the same level of complexity. Non-coding DNA may have many functions yet to be discovered. Though now it is known that only a fraction of the genome consists of genes, the paradox remains unsolved. The term "C-value enigma" represents an update of the more common but outdated term "C-value paradox" (Thomas 1971), being ultimately derived from the term "C-value" (Swift 1950) in reference to haploid nuclear DNA contents. The term was coined by Canadian biologist Dr. T. Ryan Gregory of the University of Guelph in 2000/2001. In general terms, the C-value enigma relates to the issue of variation in the amount of noncoding DNA found within the genomes of different eukaryotes. The C-value enigma, unlike the older C-value paradox, is explicitly defined as a series of independent but equally important component questions, including: a) What types of non-coding DNA are found in different eukaryotic genomes, and in what proportions? b) From where does this non-coding DNA come, and how is it spread and/or lost from genomes over time? c) What effects, or perhaps even functions, does this non-coding DNA have for chromosomes, nuclei, cells, and organisms? d) Why do some species exhibit remarkably streamlined chromosomes, while others possess massive amounts of non-coding DNA? Variation among species - C-values vary enormously among species. In animals they range more than 3,300-fold, and in land plants they differ by a factor of about 1,000 (Bennett and Leitch 2005; Gregory 2005). Protist genomes have been reported to vary more than 300,000-fold in size, but the high end of this range (Amoeba) has been called into question. Variation in C-values bears no relationship to the complexity of the organism or the number of genes contained in its genome, an observation that was deemed wholly counterintuitive before the discovery of non-coding DNA and which became known as the C-value paradox as a result. However, although there is no longer any paradoxical aspect to the discrepancy between C-value and gene number, this term remains in common usage. For reasons of conceptual clarification, the various puzzles that remain with regard to genome size variation instead have been suggested to more accurately comprise a complex but clearly defined puzzle known as [32] the C-value enigma. C-values correlate with a range of features at the cell and organism levels, including cell size, cell division rate, and, depending on the taxon, body size, metabolic rate, developmental rate, organ complexity, geographical distribution, and/or extinction risk. Calculating C-values - By using the data in Table 5.1, relative weights of nucleotide pairs can be Relative calculated as Nucleotide Chemical formula molecular follows: AT = weight 615.3830 and 2′-deoxyadenosine 5′-monophosphate C10H14N5O6P 331.2213 GC = 2′-deoxythymidine 5′-monophosphate C10H15N2O8P 322.2079 616.3711. 2′-deoxyguanosine 5′-monophosphate C10H14N5O7P 347.2207 Provided the 2′-deoxycytidine 5′-monophosphate C9H14N3O7P 307.1966 ratio of AT to *Source of table: Doležel et al., 2003* GC pairs is 1:1, the mean relative weight of one nucleotide pair is 615.8771 (±1%). The relative molecular weight may be converted to an absolute value by multiplying it by the atomic mass unit (1 u), which equals one-twelfth of a mass of 12C, i.e., 1.660539 × 10-27 kg. Consequently, the mean weight of one nucleotide pair would be 1.023 × 10-9 pg, and 1 pg of DNA would represent 0.978 × 109 base pairs. Table 5.1: Relative Molecular Weights of Nucleotides The formulas for converting the number of nucleotide pairs (or base pairs) to picograms of DNA and vice-versa are: Genome size (bp) = (0.978 x 109) x DNA content (pg) DNA content (pg) = genome size (bp) / (0.978 x 109) 1 pg = 978 Mb The current estimates for human female and male diploid genome sizes are 6.406 × 109 bp and 6.294 × 109 bp, respectively. By using the conversion formulas given above, diploid human female and male nuclei in G1 phase of the cell cycle should contain 6.550 and 6.436 pg of DNA, respectively. The phenomenon that, frequently, C values do not correlate with the evolutionary complexity of species; they are large in some small organisms. This is presumably due to the fact that sizeable portions of the DNA do not code for proteins and either have other regulatory functions or are functionless. 5.2.3 C0t Curve and its Significance C0t Curve - The curve obtained by plotting the data of a reassociation kinetics experiment. Since the reassociation of DNA is a bimolecular, second-order reaction, it follows that C/C0=17 (1 + A:C00 where ^_ is the second-order rate constant (L molds'"1), t is the time (s), C0 is the initial concentration of single-stranded DNA (moles of nucleotide per liter), and C is the concentration of single stranded DNA remaining in the reaction mixture at time / (moles of nucleotide per liter). The cot curve is obtained by plotting the fraction of single-stranded DNA remaining (C/C0) as a function of log (C0Oi that is, the logarithm of the product of the initial concentration and the elapsed time. The cot curve is an S-shaped curve. See also reassociation kinetics. [33] Variation in Eukaryotic DNA Sequences - Prokaryotic and eukaryotic cells differ dramatically in the amount of DNA per cell, a quantity termed an organism’s C value (Table 5.2). Each cell of a fruit fly, for example, contains 35 times the amount of DNA found in a cell of the bacterium E. Table 5.2: Genome sizes of various coli. In general, eukaryotic cells contain organisms more DNA than that of prokaryotes, but Approximate variability in the C values of different Organism Genome Size eukaryotes is huge. Human cells contain (bp) more than 10 times the amount of DNA λ -bacteriophage 50,000 found in Drosophila cells, whereas some E. coli (bacterium) 4,600,000 salamander cells contain 20 times as Saccharomyces cerevisiae 13,500,000 much DNA as that of human cells. (yeast) Clearly, these differences in C value Arabidopsis thaliana (plant) 100,000,000 cannot be explained simply by Drosophila melanogaster 140,000,000 (insect) differences in organismal complexity. Homo sapiens (human) 3,000,000,000 So what is all this extra DNA in Zea mays (corn) 4,500,000,000 eukaryotic cells doing? We do not yet Amphiuma (salamander) 765,000,000,000 have a complete answer to this question, but examination of DNA sequences has revealed that eukaryotic DNA has complexity that is absent from prokaryotic DNA. Denaturation and Renaturation of DNA - The first clue that the DNA of eukaryotes contains several types of sequences came from the results of studies in which double-stranded DNA was separated and then allowed to reassociate. When doublestranded DNA in solution is heated, the hydrogen bonds that Figure 5.4: The slow heating of DNA causes the two hold the two strands together are strands to separate (denature). weakened and, with enough heat, the two nucleotide strands separate completely, a process called denaturation or melting (Figure 5.4). DNA is typically denatured within a narrow temperature range. The midpoint of this range, the melting temperature (Tm), depends on the base sequence of a particular sample of DNA: G–C base pairs have three hydrogen bonds, whereas A–T base pairs only have two; so the separation of G–C pairs requires more energy than does the separation of A–T pairs. A DNA molecule with a higher percentage of G–C pairs will therefore have a higher Tm than that of DNA with more A–T pairs. The denaturation of DNA by heating is reversible; if single-stranded DNA is slowly cooled, single strands will collide and hydrogen bonds will again form between complementary base pairs, producing double-stranded DNA (Figure 5.4). This reaction, called renaturation or reannealing, takes place in two steps. First, single strands in solution collide randomly with their complementary strands. Second, hydrogen bonds [34] form between complementary bases. Two single-stranded molecules of DNA from different sources will anneal if they are complementary, a process termed hybridization. For hybridization to take place, the two strands do not have to be complementary at all their bases - just at enough bases to hold the two strands together. The extent of hybridization can be used to measure the similarity of nucleic acids from two different sources and is a common tool for assessing evolutionary relationships. The rate at which hybridization takes place also provides information about the sequence complexity of DNA. Renaturation Reactions and C0t Curves - In a typical renaturation reaction, DNA molecules are first sheared into fragments several hundred base pairs in length. Next, the fragments are heated to about 100° C, which causes the DNA to denature. The solution is then cooled slowly, and the amount of renaturation is measured by observing optical absorbance. Double-stranded DNA absorbs less UV light than does single-stranded DNA; so the amount of renaturation can be monitored by shining a UV light through the solution and measuring the amount of the light absorbed. The amount of renaturation depends on two critical factors: (1) initial concentration of singlestranded DNA (C0) and (2) amount of time allowed for renaturation (t). Other things being equal, there will be more renaturation at higher Figure 5.5: A C0t curve represents the fraction of DNA remaining single stranded in a renaturation concentrations of DNA, because reaction, plotted as a function of DNA concentration _ high concentrations increase the time (C0t). This graph is a typical C0t curve for a likelihood that the two prokaryotic organism. complementary strands will collide. There will also be more renaturation with increasing time, because there are more opportunities for two complementary sequences to collide. These two factors together form a parameter called Cot, which equals the initial concentration multiplied by the renaturation time (Co x t = Cot). A plot of the fraction of single-stranded DNA as a function of Cot during a renaturation reaction is called a C0t curve. A typical Cot curve for a prokaryotic organism is shown in figure 5.5. The upper left-hand side of the curve represents the start of the renaturation reaction, when all of the DNA is single stranded, and so the proportion of single-stranded DNA is 1. As the reaction proceeds, single stranded DNA pairs to form double-stranded DNA, represented by the decreasing fraction of single-stranded DNA. At the end of the reaction, the proportion of single stranded DNA is 0, because all of the DNA is now double stranded. The value at which half of the DNA is reannealed is called Cot ½. [35] The rate of renaturation also depends on the size and complexity of the DNA molecules used. Consider the following analogy. Suppose we distribute 100 cards equally among the students in a class. We ask each student to write his or her name on the cards, and we put all the cards in a hat. We then randomly draw two cards from the hat and see if the names on the two cards match. If they don’t match, we put them back in the hat; if they do match, we remove them, and we continue drawing until all the cards have been removed. If there are only four students in the class, each student will receive 25 cards. Because each student’s name is on 25 cards, the chance of drawing two cards that match is high, and we will quickly empty the hat. If we do the same exercise in another class with 50 students, again using 100 cards, each student’s name will appear on only two cards, and the chance of removing two cards with the same name is much lower. Thus, it will take longer to empty the hat. This exercise resembles what occurs in the renaturation reaction. If we start with the same total amount of DNA, but there are only a few different sequences in the DNA, a chance collision between two complementary fragments is more likely to occur than if there were many different sequences. Therefore DNA from organisms with larger genomes will have a larger C0t value. Thus far, we have considered renaturation reactions in which each DNA sequence is present only once in each molecule. If some sequences are present in multiple copies, these sequences will be more likely to collide with a complementary copy, and renaturation of these sequences Figure 5.6: A typical C0t curve for a eukaryotic will be rapid. Think about our organism contains several steps. The first step in the analogy of drawing names from a curve represents DNA renaturing at very low C 0t hat. Imagine that we have 50 values, because these sequences are present in many copies (highly repetitive). The second step represents students and 100 cards; each student DNA renaturing at intermediate C0t values; these gets two cards. This time, the sequences are present in an intermediate number of students write only their first names copies (moderately repetitive). The last step represents on the cards. Again, we place the DNA that renatures slowly; these sequences are cards in the hat and draw out two present singly or in few copies (unique). cards at random. If there are five students in the class named Scott, this name will appear on ten cards; so the chance of drawing out two cards at random bearing the name Scott is fairly high. On the other hand, if there is only one Susan in the class, this name will appear on only two cards, and the chance of drawing out two cards with the name Susan is low. The cards with Scott match up more quickly than the cards with Susan, because there are more copies with the name Scott. Similarly, in a renaturation reaction, if some sequences of DNA are present in multiple copies, they will renature more quickly. [36] Types of DNA Sequences in Eukaryotes - For most eukaryotic organisms, C0t curves similar to the one presented in figure 5.6 are produced and indicate that eukaryotic DNA consists of at least three types of sequences. Slowly renaturing DNA consists of sequences that are present only once or at most a few times, in the genome. This nonrepetitive, unique-sequence DNA includes sequences that code for proteins, as well as a great deal of DNA whose function is unknown. The more rapidly renaturing DNA represents two kinds of repetitive DNA - DNA sequences that exist in multiple copies. Although not identical, these copies are similar enough to reanneal. Moderately repetitive DNA typically consists of sequences from 150 to 300 bp in length (although they may be longer) that are repeated many thousands of times. Some of these sequences perform important functions for the cell; for example, the genes for ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs) make up a part of the moderately repetitive DNA. However, much of the moderately repetitive DNA has no known function in the cell. Moderately repetitive DNA itself is of two types of repeats. Tandem repeat sequences appear one after another and tend to be clustered at a few locations on the chromosomes. Interspersed repeat sequences are scattered throughout the genome. An example of an interspersed repeat is the Alu sequence, each of which consists of about 200 bp. The Alu sequence is present more than a million times in the human genome and makes up about 11% of each person’s DNA. Short repeats, such as the Alu sequences, are called SINEs (short interspersed elements). Longer interspersed repeats consisting of several thousand base pairs are called LINEs (long interspersed elements). Most interspersed repeats are transposable genetic elements, sequences that can multiply and move. The other major class of repetitive DNA is highly repetitive DNA. These short sequences, often less than 10 bp in length, are present in hundreds of thousands to millions of copies that are repeated in tandem and clustered in certain regions of the chromosome, especially at centromeres and telomeres. Highly repetitive DNA is sometimes called satellite DNA, because it has a different base composition from those of the other DNA sequences and separates as a satellite fraction when centrifuged at high speeds. Highly repetitive DNA is rarely transcribed into RNA. Although these sequences may contribute to centromere and telomere function, most highly repetitive DNA has no known function. 5.2.4 Restriction Techniques Restriction (Recombinant DNA) Techniques - In the sections that follow, we will examine some of the following techniques of recombinant DNA technology and see how they are used to create recombinant DNA molecules: (1.) Methods for locating specific DNA sequences, (2.) Techniques for cutting DNA at precise locations, (3.) Procedures for amplifying a particular DNA sequence billions of times, producing enough copies of a DNA sequence to carry out further manipulations, (4.) Methods for mutating and joining DNA fragments to produce desired sequences and (5.) Procedures for transferring DNA sequences into recipient cells [37] Cutting and Joining DNA Fragments - The key development that made recombinant DNA technology possible was the discovery in the late 1960s of restriction enzymes (also called restriction endonucleases) that recognizes and make doublestranded cuts in the sugar–phosphate backbone of DNA molecules at specific nucleotide sequences. These enzymes are produced naturally by bacteria, where they are used in defense against viruses. In bacteria, restriction enzymes recognize particular sequences in viral DNA and then cut it up. A bacterium protects its own DNA from a restriction enzyme by modifying the recognition sequence, usually by adding methyl groups to its DNA. Three types of restriction enzymes have been isolated from bacteria (Table 5.3). Type I restriction enzymes recognize specific sequences in the DNA but cut the DNA at random sites that may be some Table 5.3: Types of restriction enzymes distance (1000 bp or Activity of ATP Type Cleavage Site more) from the Enzyme Required I Cleavage and Yes Random sites distant recognition sequence. methylation from recognition site Type III restriction II Cleavage only No Within recognition site enzymes recognize III Cleavage and Yes Random sites near specific sequences and methylation recognition site cut the DNA at nearby sites, usually about 25 bp away. Type II restriction enzymes recognize specific sequences and cut the DNA within the recognition sequence. Virtually all work on recombinant DNA is done with type II restriction enzymes; discussions of restriction enzymes throughout this book, refers to type II enzymes. More than 800 different restriction enzymes that recognize and cut DNA at more than 100 different sequences have been isolated from bacteria. Many of these enzymes are commercially available; examples of some commonly used restriction enzymes are Figure 5.7: Action of given in Table 5.3. Each restriction enzyme is referred to by a HindIII producing sticky / short abbreviation that signifies its bacterial origin. The cohesive / staggered sequences recognized by restriction enzymes are usually from end DNA fragments. 4 to 8 bp long; most enzymes recognize a sequence of 4 or 6 bp. Most recognition sequences are palindromic - sequences that read the same forward and backward. Notice in Table 5.3 that the sequence on the bottom strand is the same as the sequence on the top strand, only reversed. All type II restriction enzymes recognize palindromic sequences. Some of the enzymes make staggered cuts in the DNA. For example, HindIII recognizes the sequence as given in figure 5.7, HindIII cuts the sugar-phosphate backbone of each strand at the point indicated by the arrow, generating fragments with short, singlestranded overhanging ends: Such ends are called cohesive ends Figure 5.8: Action of or sticky ends, because they are complementary to each other PvuII producing blunt end DNA and can spontaneously pair to connect the fragments. Thus DNA fragments. fragments can be “glued” together: any two fragments cleaved by the same enzyme will have complementary ends and will pair (Figure 5.8). When their [38] cohesive ends have paired, two DNA fragments can be joined together permanently by the enzyme DNA ligase, which seals nicks between the sugar–phosphate groups of the fragments. Not all restriction enzymes produce staggered cuts and sticky ends. PvuII cuts in the middle of its recognition site, producing blunt-ended fragments: Fragments with blunt ends must be joined together in other ways. Figure 5.8: Restriction enzymes make doublestranded cuts in the sugar–phosphate backbone of DNA, producing cohesive, or sticky, ends. The sequences recognized by a restriction enzyme occur randomly within genomic DNA. Consequently, there is a relation between the length of the recognition sequence and its frequency of occurrence: there are fewer long recognition sequences than short sequences because the probability of all the bases being in the required order is less. Restriction enzymes are the workhorses of recombinant DNA technology and are used whenever DNA fragments must be cut or joined. In a typical restriction reaction, a concentrated solution of purified DNA is placed in a small tube with a buffer solution and a small amount of restriction enzyme. The reaction mixture is then heated at the optimal temperature for the enzyme, usually 37o C. Within a few hours, the enzyme cuts all the restriction sites in the DNA, producing a set of DNA fragments (Figure 5.9). Restriction enzymes can cut linear as well as circular both type of DNA (Figure 5.10). DNA fragments obtain by the action of restriction enzyme can further use for gel electrophoresis and cloning purpose (Figure 5.11). DNA fragments can be inserted into the plasmid by 3 methods (a) restriction cloning (b) cloning by tailing and (c) cloning by using linkers, in each method restriction enzyme again play important role (Figure 5.11). Thus restriction enzymes are major components of restriction techniques. [39] Table 5.4: Characteristics of some common type II restriction enzymes used in recombinant DNA technology Note: The first three letters of the abbreviation for each restriction enzyme refer to the bacterial species from which the enzyme was isolated (e.g., Eco refers to E. coli). A fourth letter may refer to the strain of bacteria from which the enzyme was isolated (the “R” in EcoRI indicates that this enzyme was isolated from the RY13 strain of E. coli). Roman numerals that follow the letters allow different enzymes from the same species to be identified. For convenience, molecular geneticists have come up with idiosyncratic pronunciations of the names: EcoRI is pronounced “echo-R-one,” HindIII is “hin-D-three,” and HaeIII is “hay-three.” These common pronunciations obey no formal rules and simply have to be learned. [40] Figure 5.9: The number of restriction sites is related to the number of fragments produced when DNA is cut by a restriction enzyme. A B C D A Figure 5.10: Gel electrophoresis can be used to separate DNA molecules on the basis of their size and electrical charge. (Photo courtesy of Carol Eng.) [41] Figure 5.11 : A foreign DNA fragment can be inserted into a plasmid with the use of (a) restriction cloning, (b) tailing, or (c) linkers. [42] 5.2.5 Multigene Families and their Evolution The term “Multigene Families” is used to include groups of genes from the same organism that encode proteins with similar sequences either over their full lengths or limited to a specific domain. DNA duplications can generate gene pairs. If both copies are maintained in subsequent generations then a multigene family will exist. A multigene family is a member of a family of related proteins encoded by a set of similar genes. Multigene families are believed to have arisen by duplication and variation of a single ancestral gene. Examples of multigene families include those that encode the actins, hemoglobins, immunoglobulins, tubulins, interferons, histones etc. DNA duplications that involve one or more genes generate gene pairs. If both copies are maintained in subsequent generations then a multigene family will exist in the genome. Because most duplications occur adjacent to the original copy, a subsequent duplication encompassing both paralogs may generate a family of four chromosomal rearrangements disperse the multigene families throughout the genome. Dispersed members of the multigene family can still be recognized by sequence comparison. The significance of recognizing multigene families is that the members may have related functions. Genes that are identical or nearly identical in sequence and regulation can be considered to encode isoforms rather than members of a multigene family. In addition, genes that were derived from a common ancestral gene but have diverged extensively may not be recognized as related. The term “Super-Family” is used to describe a group of proteins with significant sequence similarity to each other but with clearly defined multigene families. The individual multigene families are likely to have distinct functions that select for shared sequences that vary from the global consensus sequence seen in the whole super-family. The term "clan" is used for related protein families that share some properties but display no clear phylogenetic relationship with each other. It covers cases of convergent evolution of proteins with similar functions but no convincing evidence of a common origin. Comparative genomics has increasingly shown that most eukaryotic genes are derived from genes that were present in one form or another in the eukaryotic ancestor. Subsequent gene loss or amplification led to quantitative and qualitative differences observed in distant phyla. (1) Origins and Evolution of the Formin Multigene Family that Is Involved in the Formation of Actin Filaments - In eukaryotes, the assembly and elongation of unbranched actin filaments is controlled by formins, which are long, multidomain proteins. These proteins are important for dynamic cellular processes such as determination of cell shape, cell division, and cellular interaction. Yet, no comprehensive study has been done about the origins and evolution of this gene family. We therefore performed extensive phylogenetic and motif analyses of the formin genes by examining 597 prokaryotic and 53 eukaryotic genomes. Additionally, we used three-dimensional protein structure data in an effort to uncover distantly related sequences. Our results suggest that the formin homology 2 (FH2) domain, which promotes the formation of actin filaments, is a eukaryotic innovation and apparently originated only once in eukaryotic evolution. Despite the high degree of FH2 domain [43] sequence divergence, the FH2 domains of most eukaryotic formins are predicted to assume the same fold and thus have similar functions. The formin genes have experienced multiple taxon-specific duplications and followed the birth-and-death model of evolution. Additionally, the formin genes experienced taxon-specific genomic rearrangements that led to the acquisition of unrelated protein domains. The evolutionary diversification of formin genes apparently increased the number of formin's interacting molecules and consequently contributed to the development of a complex and precise actin assembly mechanism. The diversity of formin types is probably related to the range of actin-based cellular processes that different cells or organisms require. Our results indicate the importance of gene duplication and domain acquisition in the evolution of the eukaryotic cell and offer insights into how a complex system, such as the cytoskeleton, evolved. (2) Molecular evolution of a multigene family in group A Streptococci - The emm genes are members of a gene family in group ‘A’ streptococci (GAS) that encode for antiphagocytic cell-surface proteins and/or immunoglobulin-binding proteins. Previously sequenced genes in this family have been named "emm," "fcrA," "enn," "arp," "protH," and "mrp"; herein they will be referred to as the "emm gene family." The genes in the emm family are located in a cluster occupying 36 kb between the genes mry and scpA on the chromosome of Streptococcus pyogenes. Most GAS strains contain one to three tandemly arranged copies of emm-family genes in the cluster, but the alleles within the cluster vary among different strains. Phylogenetic analysis of the conserved sequences at the 3' end of these genes differentiates all known members of this family into four evolutionarily distinct emm subfamilies. As a starting point to analyze how the different subfamilies are related evolutionarily, the structure of the emm chromosomal region was mapped in a number of diverse GAS strains by using subfamily-specific primers in the polymerase chain reaction. Nine distinct chromosomal patterns of the genes in the emm gene cluster were found. These nine chromosomal patterns support a model for the evolution of the emm gene family in which gene duplication followed by sequence divergence resulted in the generation of four major gene subfamilies in this locus. (3) Molecular Evolution of the Cecropin Multigene Family in Drosophila: Functional Genes vs. Pseudogenes - Multigene families are formed by genes originated by gene duplication that have retained a certain degree of similarity. The different members are often arranged in a compact cluster although they might be more or less dispersed in the genome, mostly due to chromosomal rearrangements subsequent to the gene duplications. Members of a family can be functional or nonfunctional (pseudogenes). Functional members can be very similar as the copies might have retained the same function and be redundant. However, one of the copies may have acquired a new function and suffered a certain degree of differentiation, which would be best explained by the action of Darwinian selection (Ohta 1994). On the other hand, pseudogenes can accumulate substitutions due to the lack of functional constraints. Concerted evolution of the different copies of a gene, which is facilitated by their compact clustering, can restrict the functional differentiation as well as the loss of function of the copies (Walsh 1987). Otherwise, members of a family where concerted [44] evolution is weak or absent have a higher probability to become pseudogenes (Walsh 1995). The cecropin multigene family of Drosophila melanogaster is a family with both functional genes and pseudogenes. The functional genes of this family code for cecropins, which are antibacterial peptides involved in the insect humoral immune response (Kylsten et al. 1990; Tryselius et al. 1992). In Drosophila this response is mediated by at least another eight different kinds of peptides: defensin, attacin, diptericin, drosocin, metnikowin, drosomycin, andropin, and lysozyme (Engstrom 1997; Hetru et al. 1997; Meister et al. 1997). The humoral response constitutes together with the cellular response the immune system in insects (Hultmark 1993). In D. melanogaster the Cecropin region was cloned and sequenced by Kylsten et al. 1990 and by Tryselius et al. 1992. In an ~7-kb region these authors detected four functional Cecropin genes (CecA1, CecA2, CecB and CecC) and two pseudogenes (Cec1 and Cec2). All functional genes are expressed upon bacterial infection, mainly in the fat body, although at different times during development: CecA1 and CecA2 are essentially expressed in larvae and adults while CecB and CecC are mainly expressed during the pupal stage (Hultmark 1993). 5.2.6 In-Situ Hybridization – Concept and Techniques In Situ Hybridization - In situ hybridization is another method for determining the chromosomal location of a particular gene. This method requires a DNA copy of the gene or its RNA product, which is used to make a molecule (called a probe) that is complementary to the gene of interest. The probe is made radioactive or is attached to a special molecule that fluoresces under ultraviolet (UV) light and is added to chromosomes from specially treated cells that have been spread on a microscope slide. The probe binds to the complementary DNA sequence of the gene on the chromosome. The presence of radioactivity or fluorescence from the bound probe reveals the location Figure 5.12 : In situ hybridization determine the chromosomal location of a gene. (a) FISH technique: in this case, the bound probe reveals sequences associated with the centromere. (b) SKY technique: 24 different probes, each specific for a different human chromosome and producing a different color, identify the different human chromosomes. (Courtesy of Dr. Hesed Padilla-Nash and Dr. Thomas Ried, NIH.) [45] of the gene on a particular chromosome (Figure 5.12a). The use of fluorescence in situ hybridization (FISH) has been widely used to identify the chromosomal location of human genes. In Spectral Karyotyping (SKY) (Figure 5.12b), a set of 24 FISH probes, each specific to a different human chromosome and attached to a molecule that fluoresces a different color, allows each chromosome in a karyotype to be identified. Figure 5.13 : In situ hybridization to locate specific genes on chromosomes. Here, six different DNA probes have been used to mark the location of their respective nucleotide sequences on human chromosome 5 at metaphase. The probes have been chemically labeled and detected with fluorescent antibodies. Both copies of chromosome 5 are shown, aligned side by side. Each probe produces two dots on each chromosome, since a metaphase chromosome has replicated its DNA and therefore contains two identical DNA helices. (Courtesy of David C. Ward.) Figure 5.14 : In situ hybridization for RNA localization in tissues. Autoradiograph of a section of a very young Drosophila embryo that has been subjected to in situ hybridization using a radioactive DNA probe complementary to a gene involved in segment development. The probe has hybridized to RNA in the embryo, and the pattern of autoradiographic silver grains reveals that the RNA made by the gene (called ftz) is localized in alternating stripes across the embryo that are three or four cells wide. At this stage of development (cellular blastoderm), the embryo contains about 6000 cells. (From E. Hafen, A. Kuriowa, and W.J. Gehring, Cell 37:833-841, 1984. © Cell Press.) [46] In Situ Hybridization Techniques Locate Specific Nucleic Acid Sequences in Cells or on Chromosomes - Nucleic acids, no less than other macromolecules, occupy precise positions in cells and tissues, and a great deal of potential information is lost when these molecules are extracted by homogenization. For this reason, techniques have been developed in which nucleic acid probes are used in much the same way as labeled antibodies to locate specific nucleic acid sequences in situ, a procedure called in situ hybridization. This can now be done both for DNA in chromosomes and for RNA in cells. Labeled nucleic acid probes can be hybridized to chromosomes that have been exposed briefly to a very high pH to disrupt their DNA base pairs. The chromosomal regions that bind the probe during the hybridization step are then visualized. Originally, this technique was developed using highly radioactive DNA probes, which were detected by autoradiography. The spatial resolution of the technique, however, can be greatly improved by labeling the DNA probes chemically instead of radioactively. For this purpose the probes are synthesized with special nucleotides that contain a modified side chain, and the hybridized probes are detected with an antibody (or other ligand) that specifically recognizes this side chain (Figure 5.13). In situ hybridization methods have also been developed that reveal the distribution of specific RNA molecules in cells in tissues. In this case the tissues are not exposed to a high pH, so the chromosomal DNA remains double-stranded and cannot bind the probe. Instead the tissue is gently fixed so that its RNA is retained in an exposed form that will hybridize when the tissue is incubated with a complementary DNA or RNA probe. In this way the patterns of differential gene expression can be observed in tissues. In the Drosophila embryo, for example, such patterns have provided new insights into the mechanisms that create distinctions between cells in different positions during development (Figure 5.14). 5.2.7 Physical Mapping of Genes on Chromosomes "Gene mapping" refers to the mapping of genes to specific locations on chromosomes. It is a critical step in the understanding of genetic diseases. There are two types of gene mapping: Genetic Mapping - using linkage analysis to determine the relative position between two genes on a chromosome. Physical Mapping - using all available techniques or information to determine the absolute position of a gene on a chromosome. The ultimate goal of gene mapping is to clone genes, especially disease genes. Once a gene is cloned, we can determine its DNA sequence and study its protein product. For example, cystic fibrosis (CF) is the most common lethal inherited disease in the United States. As many as 1 in 2500 Americans of Northern European descent carries a gene with CF. In 1985, the gene was mapped to chromosome 7q31-q32 by linkage analysis. Four years later, it was cloned by Francis Collins and his co-workers. We now know that the disease is caused by the defect of a chloride channel - the protein product of this disease gene. Physical mapping techniques which are most common are following two:- [47] 1) Somatic cell hybridization and 2) Fluorescent In situ Hybridization (FISH) The resolution of FISH is much higher than somatic cell hybridization. Mapping by DNA Sequencing - Another means of physically mapping genes is to determine the sequence of nucleotides in the DNA. With this technique, physical distances between genes are measured in numbers of base pairs. Continuous sequences can be determined for only relatively small fragments of DNA; so, after sequencing, some method is still required to map the individual fragments. This mapping is often done by using the traditional gene mapping that examines rates of crossing over between molecular markers located on the fragments. It can also be accomplished by generating a set of overlapping fragments, sequencing each fragment, and then aligning the fragments by using a computer program that identifies the overlap in the sequence of adjacent fragments. With these methods, complete physical maps of entire genomes have been produced. Physical Chromosome Mapping - Genetic maps reveal the relative positions of genes on a chromosome on the basis of frequencies of crossing over, but they do not provide information that can allow us to place groups of linked genes on particular chromosomes. Furthermore, the units of a genetic map do not always precisely correspond to physical distances on the chromosome, because a number of factors other than physical distances between genes (such as the type and sex of the organism) can influence rates of crossing over. Because of these limitations, physical-mapping methods that do not rely on rates of crossing over have been developed. Deletion Mapping - One method for determining the chromosomal location of a Figure 5.15 : Deletion mapping can be used to determine the chromosomal location of a gene. An individual homozygous for a recessive mutation in the gene of interest (aa) is crossed with an individual heterozygous for a deletion. [48] gene is deletion mapping. Special staining methods have been developed that make it possible to detect chromosome deletions, mutations in which a part of a chromosome is missing. Genes are assigned to regions of particular chromosomes by studying the association of a gene’s phenotype or product and particular chromosome deletions. In deletion mapping, an individual that is homozygous for a recessive mutation in the gene of interest is crossed with an individual that is heterozygous for a deletion (Figure 5.15). If the gene of interest is in the region of the chromosome represented by the deletion (the red part of chromosome in Figure 5.15), approximately half of the progeny will display the mutant phenotype (see Figure 5.15a). If the gene is not within the deleted region, all of the progeny will be wild type (see Figure 5.15b). Deletion mapping has been used to reveal the chromosomal locations of a number of human genes. For example, Duchenne Muscular Dystrophy is a disease that causes progressive weakening and degeneration of the muscles. From its X-linked pattern of inheritance, the mutated allele causing this disorder was known to be on the X chromosome, but its precise location was uncertain. Examination of a number of patients having Duchenne Muscular Dystrophy, who also possessed small deletions, allowed researchers to position the gene to a small segment of the short arm of the X chromosome. Somatic-Cell Hybridization - Another method used for positioning genes on chromosomes is somatic cell hybridization, which Figure 5.16 : Somatic-cell hybridization can be used to requires the fusion of different types determine which chromosome contains a gene of interest. of cells. Most mature somatic (non sex) cells can undergo only a limited number of divisions and therefore cannot be grown continuously. However, cells that [49] have been altered by viruses or derived from tumors that have lost the normal constraints on cell division will divide indefinitely; these types of cells can be cultured in the laboratory and are referred to as a cell line. Cells from two different cell lines can be fused by treating them with polyethylene glycol or other agents that alter their plasma membranes. After fusion, the cell possesses two nuclei and is called a heterokaryon. The two nuclei of a heterokaryon eventually also fuse, generating a hybrid cell that contains chromosomes from both cell lines. If human and mouse cells are mixed in the presence of polyethylene glycol, fusion results in human–mouse somatic-cell hybrids (Figure 5.16). The hybrid cells tend to lose chromosomes as they divide and, for reasons that are not understood, chromosomes from one of the species are lost preferentially. In human–mouse somatic-cell hybrids, the human chromosomes tend to be lost, whereas the mouse chromosomes are retained. Eventually, the chromosome number stabilizes when all but a few of the human chromosomes have been lost. Chromosome loss is random and differs among cell lines. The presence of these extra” human chromosomes in the mouse genome makes it possible to assign human genes to specific chromosomes. In the first step of this procedure, hybrid cells must be separated from original parental cells that have not undergone hybridization. This separation is accomplished by using a selection method that allows hybrid cells to grow while suppressing the growth of parental cells. The most Figure 5.17 : HAT medium can be commonly used method is called HAT used to separate human–mouse hybrid selection (Figure 5.17), which stands for cells from the original hybridized cells. hypoxanthine, aminopterin, and thymidine, three chemicals that are used to select for hybrid cells. In the presence of HAT medium, a cell must possess two enzymes to synthesize DNA: thymidine kinase (TK) and hypoxanthine-guanine phosphoribosyl transferase (HPRT). Cells that are tk - or hprt - cannot synthesize DNA and will not grow on HAT medium. The mouse cells used in the hybridization procedure are deficient in TK, but can produce HPRT (the cells are tk – hprt +); the human cells can produce TK but are deficient for HPRT (they are tk + hprt - ). On HAT medium, the mouse cells do not survive, because they are tk - ; the human cells do not survive, because they are hprt - . Hybrid cells, on the other hand, inherit the ability to make HPRT from the mouse cell and the ability to make [50] Figure 5.18 : Somatic-cell hybridization is used to assign a gene to a particular human chromosome. A panel of six cell lines, each line containing a different subset of human chromosomes, is examined for the presence of the gene product (such as an enzyme). A plus sign means that the gene product is present; a minus sign means that the gene product is missing. Four of the cell lines (A, B, D, and F) have the gene product, indicating that the gene is present on one of the chromosomes found in these cell lines. The only chromosome common to all four of these cell lines is chromosome 4, indicating that the gene is located on this chromosome. TK from the human cell; thus, they produce both enzymes (the cells are tk + hprt +) and will grow on HAT medium. To map genes using somatic-cell hybridization requires the use of a panel of different hybrid cell lines. The cell lines of the panel differ in the human chromosomes that they have retained. For example, one cell line might possess human chromosomes 2, 4, 7, and 8, whereas another might possess chromosomes 4, 19, and 20. Each cell line in the panel is examined for evidence of a particular human gene. The human gene can be detected either by looking for the protein that it produces or by looking for the gene itself with the use of molecular probes. Correlation of the presence of the gene with the presence of specific human chromosomes often allows the gene to be assigned to the correct chromosome. For example, if a gene was detected in both of the aforementioned cell lines, the gene must be on chromosome 4, because it is the only human chromosome common to both cell lines (Figure 5.18). Two genes determined to be on the same chromosome with the use of somatic-cell hybridization are said to be syntenic genes. This term is used Figure 5.19: 7.21 Genes can be localized to a specific part of a chromosome by using somatic-cell hybridization. Note: If the gene product is present in a cell line with an intact chromosome but missing from a line with a chromosome deletion, the gene for that product must be located in the deleted region. [51] because syntenic genes may or may not exhibit linkage in the traditional genetic senseremember that two genes can be located on the same chromosome but may be so far apart that they assort independently. Synthenic refers to genes that are physically linked, regardless of whether they exhibit genetic linkage. (Synteny is sometimes also used to refer to gene loci in different organisms located on a chromosome region of common evolutionary origin.) Sometimes somatic-cell hybridization can be used to position a gene on a specific part of a chromosome. Some hybrid cell lines carry a human chromosome with a chromosome mutation such as a deletion or a translocation. If the gene is present in a cell line with the intact chromosome but missing from a line with a chromosome deletion, the gene must be located in the deleted region (Figure 5.19) Similarly, if a gene is usually absent from a chromosome but consistently appears whenever a translocation (a piece of another chromosome that has broken off and attached itself to the chromosome in question) is present, it must be present on the translocated part of the chromosome. Fluorescent In situ Hybridization (FISH) - This technique has been described in section 5.2.6. 5.2.8 Computer Assisted Chromosome Analysis Chromosome Analysis - Comparing the DNA of different organisms can show how closely related they are. Since for each species the DNA information is organized in a characteristic number of chromosomes, the number of chromosomes is a reasonable indicator of the relatedness of two similar species. Sometimes the DNA information on a chromosome is reorganized. Chromosomes can sometimes fuse with each other or can exchange chromosome "arms". When this happens, DNA information is not always lost, but it can become mixed up. This sort of rearrangement may not cause problems for the individual who carries the change --- as long as all the DNA is still present. A mule is the product of two different species (a horse and a donkey) mating with each other. The fact that these two different types of animals can mate and produce viable offspring tells scientists that horses and donkeys are closely related. However, mules are always sterile. Why is this? Horses and donkeys have different chromosome numbers. The fact that horses and donkeys have different chromosome numbers tells scientists that these two are different species. Figure 5.20: One Utah species originally assigned to For the mule, having parents with Notholaena has 27 chromosomes. different chromosome numbers [52] isn't a problem. During mitotic cell division, each of the chromosomes copies itself and then distributes these two copies to the two daughter cells. In contrast, when the mule is producing sperm or egg cells during meiosis, each pair of chromosomes (one from Mom and one from Dad) need to pair up with each other. Since the mule doesn't have an even number of homologous pairs (his parents had different chromosome numbers), meiosis is disrupted and viable sperm and eggs are not formed. Using Chromosomes To Classify Plant Species - Variations in Figure 5.21: Species of Notholaena from chromosome number are even more common other states have multiples of 30 in the plant kingdom. In plants, chromosome chromosomes number is an important indicator for determining relationships between plant species. Scientists at the Utah Museum of Natural History recently used studies of chromosome number to show that a Utah fern was not the same species as a very similar fern found in other states. Studies of the Utah Jones Cloak Fern (originally thought to be a Notholaena species) showed that the cells of this plant have 27 chromosomes (Figure 5.20). Other species of Notholaena found in other states have 30 chromosomes (Figure 5.21). When combined with the results of other studies, the difference in chromosome number helped to prove that the Utah species actually belonged in the genus Argyrochosma, a very distant relative of Notholaena . This sort of information is important because it helps conservation biologists understand the distribution of each different species of plant. With this sort of information, scientists are better able to decide which plants are Figure 5.22 : A logical tree structure of the dynamic encoding rare and require protection by means process. (Note: When a branch terminates with "finish," a such as the endangered species list. correct decision can probably be made. When a branch terminates with a numbered "return," the data must be augmented, or modified if possible, then reconsidered starting at the corresponding auxiliary entry point of the decision tree.) [53] The purpose of the computer program is twofold: (i) to illuminate the basic mechanisms by which a human recognizes an object, such as a chromosome, and distinguishes it from other entities; and (ii) the employment of these mechanisms is an automatic and precise extraction of chromosome features. The computer program must locate a single entity- chromosome or nonchromosome-then identify as many of its characteristics as possible and employ them to make a decision. If the characteristics noted are sufficient to satisfy the observer that his decision will be correct, he will make a decision. Otherwise, he will investigate alternative ways of looking at the object. On a computer, this takes the form of a decision tree (Figure 5.22). The chromosome is defined and recognized by the presence or absence of "arms" and of their shape and size. Computer chromosome recognition has an extensive literature. For example : Ledley et al. describe syntax-directed methods using a special scanner for computer input of pictures. Hilditch and Rutovitz present a comprehensive discussion of shape-oriented chromosome recognition experiments using the same scanner. Rutovitz employs two of our concepts: "a series of increasingly complex alternatives" and "a polar coordinate representation of the boundary." Hilditch describes the separation of touching chromosomes, While Gallus and Neurath improve earlier programs by incorporating boundary analysis. Here is an example of a work which presents methods for data reduction and isolation of visual entities (chromosomes, blobs, touching and overlapping chromosomes) as well as boundary encoding via a scaled polar plot which enables analysis for overlap resolution. It has an experimental program which exemplifies feature encoding and decision making in hierarchical fashion. Few steps of this program are as follows which is explained here only by program generated pictures. (1) Data Reduction and Isolation of Separate Entities The first phase of the computer program modifies the data representing the picture so that it becomes as small a data base as possible. The scanned picture is input in the form of numbers representing gray levels, or Figure 5.23 : Points are assigned to an object as the boundary is traced. [54] relative darkness, corresponding to a particular small area of the original photograph. A picture of 1 000000 points is converted into a different form and reduced to some 10 000 pairs of coordinates (Figure 5.23). (2) Dynamic Encoding of Information for Feature Extraction - The actual encoding of information is the major task of the computer program. Two techniques are incorporated in the program. These are :(a) Integral projections or chord profiles (Figure 5.24) and (b) The production of a polar plot (Figure 5.25) of the suspected chromosome. These two methods are not parallel. The integral projection technique is used as a pretest to determine which branches of the tree should be followed. At a later time the polar plot is used for the more explicit decision making. Figure 5.24 detached part. : Chromosome with [55] (3) Analysis and Decision Making - Objects encountered by the decision making apparatus are being evaluated for possible membership in four categories: normal X chromosome, abnormal X chromosome, Y chromosome, and non-chromosome. (4) Classification - Figure 5.26 is an example of what is obviously a complete and probably normal chromosome, although it is not an excellent image. Figure 5.25 : Polar plot of chromosome with detached arm. Figure 5.26 : An example of a complete and probably normal chromosome. [56] (5) Overlap Resolution – Result could indicate an object of the type as shown in figure 5.27 and could be interpreted as two X chromosomes, one on top of the other. The polar plot does not make it immediately obvious that this is the case, but it does indicate that the possibility exists. Once the coordinates have been translated as shown in figure 5.28, the polar plot can be reexamined in a new light. It is assumed that the largest peaks in this profile are not part of the chromosome. The experimental pattern recognition program described here employs several feature extraction techniques to enable computer analysis of chromosome patterns. The program -performs data reduction to isolate objects from the scene in an efficient manner based on the line at a time input of scanned picture data from magnetic tape, and retaining the useful boundary information in this process. One test-a convexity indication eliminating blobs-is based on the easily computed integral projections/chord profiles used elsewhere in pattern recognition by Pavlidis and Ball. Boundary data is encoded and features obtained (peaks in a polar plot of the boundary indicating chromosome arms) as the program continues past this test through its decision tree. Features are summarized in a pattern vector which consists of the relative maxima and minima in the final polar plot of the chromosome, while the record of translations and deformation which led to this plot is Figure 5.27 : Two overlapping chromosomes. Figure 5.28 : Overlapping chromosomes reconsidered using a new origin. [57] retained. Methods for using the data to classify the chromosomes (normal X chromosome, abnormal X chromosome, Y chromosome, and nonchromosome) are described which could easily be incorporated in the existing program, to make it an operational program. Finally, a method for resolving cases of overlapping chromosomes by selecting subsets of the polar plot peaks was presented, as was a method for assigning a confidence level to each decision (Figure 5.29). The key concept presented here is a two-level logical structure of the pattern recognition program which allows it to select the procedures and criteria most likely to lead to a correct decision. In particular, the program uses a changing organization of the features of the object discovered as the decision-making task changes. The resulting pattern recognition concept we term dynamic encoding for feature extraction, while the overall task this program is intended to perform is that of flexible decision making needed for picture processing. 5.2.9 Chromosome Microdissection and Microcloning Chromosome microdissection is a technique that physically removes a large section of DNA from a complete chromosome (Figure 5.30). The smallest portion of DNA that can be isolated using this method comprises 10 million base pairs hundreds or thousands of individual genes. Scientists who study chromosomes are known as cytogeneticists. They are able to identify each chromosome based on its unique pattern of dark and light bands. Certain abnormalities, however, cause chromosomes to have unusual banding patterns. For example, one chromosome may have a piece of another chromosome inserted within it, creating extra bands. Or, a portion of a chromosome may be repeated over and over again, resulting in an unusually wide, dark band (known as a homogeneously staining region). Some chromosomal aberrations have been linked to cancer and inherited genetic disorders, and the chromosomes of many tumor cells exhibit irregular bands. To Figure 5.29 : Decomposition of the plot in order to distinguish two chromosomes. [58] understand more about what causes these conditions, scientists hope to determine which genes and DNA sequences are located near these unusual bands. Chromosome microdissection is a specialized way of isolating these regions by removing the DNA from the band and making that DNA available for further studies. To prepare cells for chromosome microdissection, a scientist first treats them with a chemical that forces them into metaphase: a phase of the cell's life-cycle where the chromosomes are tightly coiled and highly visible. Next, the cells are dropped onto a microscope slide so that the nucleus, which holds all of the genetic material together, breaks apart and releases the chromosomes onto the slide. Then, under a microscope, the scientist Figure 5.30 : Procedure of microdissection locates the specific band of interest, and, using a of a chromosome very fine needle, tears that band away from the rest of the chromosome. The researcher next produces multiple copies of the isolated DNA using a procedure called PCR (polymerase chain reaction). The scientist uses these copies to study the DNA from the unusual region of the chromosome in question. Microcloning is a process in which very small amount of chromosome/genome is cloned into a host. A little discussion about the microcloning process in different host is present here. Genome require isolation of large numbers of DNA probes from defined regions of the chromosomes. In addition, the effective use of genetic linkage analysis with polymorphic DNA probes from the human genome has localized more and more inherited diseases of unknown etiology, as well as specific forms of cancer, to refined regions of various chromosomes. Thus, the next major task is to isolate the genes underlying these diseases for better understanding, prevention, diagnosis, and treatment of the disease. A direct approach to this objective, as proposed by Edstrom and his colleagues, is to use the chromosome microdissection technique to remove physically the chromosomal region of interest and to clone the minute quantities of the dissected chromosomal DNA by a microcloning procedure. This approach has been successfully applied first with the Drosophila polytene chromosome and later with the mammalian metaphase chromosome (Figure 5.31). Recently, a modification of this method was described by Ludecke et al. in which significantly increased cloning efficiency was achieved by ligation of the dissected chromosomal DNA to a vector, followed by polymerase chain reaction (PCR) to amplify the dissected sequences. The PCR products were cloned into the plasmid vector pUC13. Using this method, these investigators obtained between 5000 and 20,000 clones from each of the dissected chromosome regions. The advantages of this method over the original microcloning procedure include (i) banded and stained chromosome preparations can be used, (ii) fewer dissected chromosome fragments are needed, and (iii) large numbers of microclones can be obtained. [59] Another method of PCR microcloning was described by Saunders et al. and by Johnson for a universal amplification of microdissected Drosophila polytene chromosome fragments. In this method, a polytene chromosome band was dissected, cleaved with either Sau3A or MboI, and ligated to a linker adaptor. The linker adaptor was constructed from oligonucleotides of 24-mer and 20-mer annealed to yield a 5' protruding Sau3A or MboI sequence. The 20-mer of the linker adaptor was used as a primer for PCR. The amplified products were digested again with Sau3A or Mbo I and cloned into the compatible BamHI site of a plasmid vector. Large numbers of clones were obtained by FIG. 5.31 : (a) Unstained normal human metaphase spread; the arrow indicates the location of a chromosome 2. (b) Same spread, showing the chromosome 2 after the removal of the distal half of the short arm with a fine glass needle. this method. More importantly, Saunders et al. also compared the PCR microcloning method with the conventional microcloning method and showed that the PCR method generated probes representing >90% of the dissected genomic region, whereas the probes from the conventional method covered only 3-4% of the same dissected region. Moreover, the PCR microcloning by the linker-adaptor method appears to offer additional useful features over the PCR microcloning method of Ludecke et al. : (i) several different linker sequences and restriction enzymes can be used, and (ii) a more efficient sticky-end ligation, instead of blunt-end ligation, is used. Several approaches have been used to obtain a number of clones specific to a subchromosomal region for use in mapping. The phenol-enhanced reassociation technique, as used for Duchenne muscular dystrophy, relies on the presence of a deletion which spans the region of interest. Hybrids containing only specific chromosomal fragments can be constructed by chromosome-mediated gene transfer, from which libraries can be prepared. However, this method depends on the natural presence or chance integration of a selectable marker. [60] An alternative and more generally applicable technique clones picogram amounts of DNA isolated by the direct microdissection of chromosomes. This method was first applied to DNA from individual bands of Drosophila melanogaster polytene chromosomes and was extended to mammalian chromosomes with the isolation of clones mapping to the mouse t complex and to a proximal region of the mouse X chromosome. In mouse genetics, the technique has fulfilled the promise of fine genetic and molecular mapping of individual chromosome regions and allows the isolation and characterization of previously unclonable genetic loci. With the use of Robertsonian translocations for chromosomal identification, every chromosomal region of mice is now available for dissection. Microdissection and microcloning of the euchromatin-heterochromatin transition region of the Drosophila melanogaster polytene X chromosome and part of the euchromatin of chromosome 4 reveals that they share certain features characteristic of (3heterochromatin, which is morphologically defined as the loosely textured material at the bases of some polytene chromosome arms. Both are mosaics of many different middlerepetitive DNA sequences interspersed with single-copy DNA sequences. Sixty percent of cloned inserts derived from division 20 and about 40 percent from subdivisions l9EF of the X chromosome harbor at least one repetitive DNA sequence in an average insert of 4.5 kilobases. No repeats have significant cross-hybridization to any of the eleven satellite DNAs, or to the clustered-scrambled sequences present in pDml. The repetitive elements are, in general, confined to the g3-heterochromatic regions of polytene chromosomes, but some are adjacent to nomadic elements. Chromosome 4, however, has some repeats spread throughout its entire euchromatin. These data have implications for the structure of transition zones between euchromatin and heterochromatin of mitotic chromosomes and also provide a molecular basis for reexamining some of the unusual classical properties of chromosome 4. 5.2.10 Flow Cytometry and Confocal Microscopy in Karyotype Analysis Flow Cytometry - Often the experimenter is not interested in keeping the cells but simply in analyzing the distribution of cell size and/or surface molecules on the cells in the suspension. The signals from the detectors can generate these sorts of data, a procedure known as flow cytometry. The data are analyzed by computer and can be plotted in several ways. The more elaborate flow cytometers can use two different lasers to detect as many as three different colors on the cells in the suspension. Here are some examples (all courtesy of Becton Dickinson Immunocytometry Systems). Counting T and B cells by flow cytometry - A sample of normal human blood was treated with a fluorescent monoclonal antibody specific for the T cell Figure 5.32: Flow Cytometry for counting T and B Cells (a) Different Peaks of T and B cell (b) Separation of T and B cells. [61] surface antigen CD3 and a monoclonal antibody conjugated to a different fluorescent dye and specific for the B cell surface antigen designated CD19. Fluorescence intensity (logarithmic) is plotted on the x axis; cell number on the y axis. Note that the number of B cells is substantially less than that of T cells. The right-hand panel shows another way of plotting the data. Counting T-cell subsets in normal human blood - Fluorescent monoclonal antibodies were directed against the CD4 molecule (left) and CD8 molecule (right). CD4+ T cells are responsible for several cell-mediated immune responses and giving help to B cells. CD8+ T cells are cytotoxic T lymphocytes (CTLs). Figure 5.33: Two subset of T cell : CD4 molecules (left) and CD8 molecules (right). The preponderance of CD4+ over CD8+ cells shown here is typical of healthy humans. In AIDS patients, this ratio becomes reversed and the CD4+ subset may eventually disappear. Flow Cytometry and Genome Analysis - Flow cytometry allows for fast and informative, quantitative and qualitative analysis of objects including chromosomes and nuclei, normally by measuring the fluorescence of molecules that are specifically bound to structures of interest (Figure 5.34; Melamed et al., 1990). The molecules measured are often fluorochrome-conjugated antibodies or fluorescent dyes binding specifically to DNA or proteins. Figure 5.34 : The optical and mechanical arrangement of a flow cytometer capable of measuring the fluorescence of stained particles, such as chromosomes of nuclei, in a fluid stream. After arrangement, the particles, by now contained in droplets, are sorted by charged plates depending on their fluorescence. Flow cytogenetics underpins much of the human genome project: the Department of Energy Human Genome Program reports that "among the resources most crucial to mapping progress are the libraries of clones representing each of the human chromosomes. This chromosome-specific clone library production from physically purified chromosomes depends on the unique chromosome-sorting facilities" at Los Alamos and Lawrence Livermore National Laboratories (Anon, 1993). Flow technology is also recognized as being critical to pig and bovine genome projects (Miller et al., 1992; Dixon et. al., 1992). [62] Joe Gray, from the University of California, San Francisco, and Scott Cram, Life Science Division Leader at Los Alamos National Laboratory, described some of the advantages of flow cytometry for plant molecular cytogenetics and genome analysis. "The analysis and sorting of plant chromosomes is of considerable economic interest. As is the case for mammalian chromosomes, flow karyotyping and chromosome sorting provide the opportunity for gene mapping and the construction of chromosome-specific libraries" (Gray and Cram, 1990). Since they wrote this, there have been successful applications of the methods in plants for genome size measurements (including the specific AT and GC base-pair content), cell cycle analysis, flow karyotyping (by measuring the DNA content of chromosomes), chromosome sorting and production of chromosome-enriched DNA libraries, although these analyses are not as yet extensively exploited. In this short review, I aim to highlight potential and recent applications of flow cytometry to plant genomes; the literature on chromosome analysis has been reviewed recently by a group of European collaborators from the Czech Republic, Italy and Germany (Dolezel et al. 1994), while many techniques for flow analysis of plants are discussed in the same paper and elsewhere (e.g., Heslop-Harrison and Schwarzacher, 1995). Genome size analysis - Changes between species, during differentiation and during the cell cycle - Analytical information about the physical size of plant genomes and their state of replication is easily obtainable from flow cytometry. Knowing the number of base pairs in a genome is valuable for studies of new species, and an extensive list based on flow cytometric estimates was published by Arumuganathan, now at the University of Lincoln, Nebraska, and Lisa Earle, from Cornell University, in 1991. Flow cytometry provides a fast and accurate way to look at changes in genome size during evolution and differentiation. Establishment of ploidy and aneuploidy changes during tissue culture, and examination of intra- and inter-specific variation of DNA content can all be important in plant hybridization, breeding, and genetic manipulation programs (Dolezel et al. 1994; Leitch et al. 1992). Perhaps, in contrast to animals, polyploidy often accompanies differentiation, and is an important part of plant development, with different cell types having characteristic ploidies (Galbraith et al. 1991; Bino et al. 1993). Such differentiation by polyploidization is important for understanding the regulation of gene expression in differentiated tissues, and for understanding the nature of tissues used for plant regeneration and transformation. Flow cytometry provides an accurate method for determining the proportions of cells in G1, S and G2/M stages of the cell cycle. These data can be used to calculate cell cycle times, which are needed in studies of the genetics and control of this process, and are useful for analysis of aspects of crop growth and development. Flow karyotyping - Flow karyotypes, giving the average sizes of chromosomes from mitotic cells, are quick, accurate and quantitative. To make a flow karyotype, a suspension of many thousands of chromosomes is made and stained with a fluorochrome which binds quantitatively to DNA. The fluorescence of chromosomes is measured as they pass individually through a cytometer (Figure 5.35), giving a histogram where each peak represents one or a group of chromosomes. [63] Flow methods enable differences as small as 1.5 to 4 Mb to be analyzed in humans, and both aneuploidy and many chromosome deletions can be detected easily. In plants, eight species have been flow karyotyped so far, including Haplopappus gracilis, a plant with only two pairs of chromosomes, four solanaceous species and Melandrium album (with sex chromosomes), wheat (Triticum aestivum), and field or broad bean (Vicia faba). Chromosome sorting for gene mapping and library construction - After identification of a Figure 5.35 : A "flow karyotype" of a wheat cell line. The continuous trace is a frequency distribution histogram showing the number of chromosomes with each particular fluorescence. The five sharp peaks correspond to single chromosomes or pairs of chromosomes, while the smoother curves are formed where many individual chromosome types are not resolved (right), or from broken chromosomes, nuclei, or organelles (left). This graph is derived from a two-wavelength analysis, shown as the diagonal line where density of dots is proportional to number of chromosomes detected of each fluorescence. Some peaks on the continuous line are now resolved as two peaks (arrow). The karyotype is derived from analysis of 50, 000 chromosomes, taking about 5 minutes. I thank Nigel Miller at the Babraham Institute, Cambridge, for assistance in operating the flow cylinder. polymerase chain reaction (PCR) can be mapped by using chromosome by its fluorescence, particular chromosomes can be sorted from others in a flow cytometer by either electrical (as in Figure 5.34) or mechanical deflection. Chromosome sorting has been achieved from important crop species including wheat, tomato (Lycopersicon esculentum), and field bean. Sorted chromosomes serve as a source of DNA for production of chromosome-specific and chromosome recombinant DNA libraries. A chromosome-specific recombinant DNA library from chromosome 4A showed 20-fold enrichment of clones as compared to a random genomic library (Wang et al. 1992). Partitioning the genome before cloning can reduce the effort required for screening a library. Dot blots, in which chromosomes are sorted onto membranes, have been used for gene mapping by probing with cDNA and other clones. Primers for the sorted chromosomes as the [64] DNA source (Macas et al. 1993), and subchromosomal localization is possible by probing sorted chromosomes from karyotypes with translocations. Sorted chromosomes also can be used for microinjection, bombardment, or electroporation into regenerable tissues to make, perhaps, chromosome-specific transfectants. Quantification - Flow cytometry can be used to measure small DNA molecules down to 20 kb, below the threshold of separation by normal electrophoresis. The ability to quantify accurately large (from 20 kb to chromosomal size) DNA molecules may be useful in analysis of large genomic features - the distribution of Notl or M1u1 islands, for example. Work at National Flow Cytometry Resource Center, Los Alamos, and elsewhere, is pushing the limits to base-pair resolution: DNA sequencing at rates of tens of base pairs per second may be feasible by Ba/31 exonuclease digestion of a single DNA molecular and cytometric identification of the bases as they are cut off. In situ hybridization methods in suspension may enable accurate quantification of numbers of copies of repetitive DNA sequences on different chromosomes in a species -- something often difficult to do by other methods. Conclusions - Applications of flow cytogenetics range from detection of aberrant cell cycles and changes in nuclear DNA amounts to sorting of chromosomes for gene mapping and library construction. The methods are used extensively in the human genome project, and are applicable to plants. The fast and quantitative results make the method an important analytical tool which has the potential to advance many aspects of plant genome analysis. CONFOCAL MICROSCOPY - Confocal microscopy is an optical imaging technique used to increase micrograph contrast and/or to reconstruct three-dimensional images by using a spatial pinhole to eliminate out-of-focus light or flare in specimens that are thicker than the focal plane. This technique has been gaining popularity in the scientific and industrial communities. Typical applications include life sciences and semiconductor Basic concept - The principle of confocal imaging was patented by Marvin Minsky in 1957. In a conventional (i.e., wide-field) fluorescence microscope, the entire specimen is flooded in light from a light source. Due to the conservation of light intensity transportation, all parts of the specimen throughout the optical path will be excited and the fluorescence detected by a photodetector or a camera. In contrast, a confocal microscope uses point illumination and a pinhole in an optically conjugate plane in front of the detector to eliminate out-of-focus information. Only the light within the focal plane can be detected, so the image quality is much better than that of wide-field images. As only one point is illuminated at a time in confocal microscopy, 2D or 3D imaging requires scanning over a regular raster (i.e. a rectangular pattern of parallel scanning lines) in the specimen. The thickness of the focal plane is defined mostly by the square of the numerical aperture of the objective lens, and also by the optical properties of the specimen and the ambient index of refraction. [65] Types - Three types of confocal microscopes are commercially available: Confocal laser scanning microscopes (Figure 5.36), spinning-disk (Nipkow disk) confocal microscopes and Programmable Array Microscopes (PAM). Confocal laser scanning microscopy yields better image quality than Nipkow and PAM, but the imaging frame rate was very slow (less than 3 frames/second) until recently; spinningdisk confocal microscopes can achieve video rate imaging - a desirable feature for dynamic observations such as live cell imaging. Confocal laser scanning microscopy has now been improved to provide better than video rate (60 frames/second) imaging by using MEMS based scanning mirrors. Biologists quickly realized that confocal microscopy offers one big advantage: the ability to perform Figure 5.36 : "Confocal Microscope" - Path for Light optical sectioning at the cellular level in a noninvasive way. It eliminates the need to embed material and cut serial sections. The confocal microscope offers biologists the chance to investigate the microscopic world in 3-dimensions. A second advantage is the ability to quantify the visual data that greatly enhanced the use of confocal microscopy. The scientific literature contains many confocal microscope studies ranging from the cellular to the macroscopic. Molecular biology in particular is exploiting the technology. It is being used to unravel the 3dimensional functioning of genes in nuclei, for example to show when they become active (Rowland and Nickless 1999). In fact, confocal microscopy is now applied worldwide not only in all avenues of the biological sciences but also the physical sciences The Confocal Microscope Facility at Massey University is a multi-user facility used by plant biologists, veterinary researchers, food technologists, dairy product researchers, microbiologists, cytogeneticists, molecular biologists and even technologists examining bitumen (asphalt like products). The potential of the machine is limited only by the type of questions one wishes to ask. Unquestionably, it has created a sense of inquiry where [66] features relating to growth and development, injury and disease, mode of infection of pathogens, chromosomal behavior, milk processing and product development all require some quantitative understanding of events that unfold. 5.3 ALIEN GENE TRANSFER THROUGH CHROMOSME MANIPULATION Genetic improvement of crops has traditionally been achieved through sexual hybridization between related species, which has resulted in numerous cultivars with high yields and superior agronomic performance. Conventional plant breeding, sometimes combined with classical cytogenetic techniques, continues to be the main method of cereal crop improvement. More recently, through the introduction of new tools of biotechnology, crossing barriers have been overcome, and genes from unrelated sources have become available to be introduced asexually into plants. Cereal crops were initially difficult to genetically engineer, mainly due to their recalcitrance to in vitro regeneration and their resistance to Agrobacterium infection. Systematic screening of cultivars and explant tissues for regeneration potential, development of various DNA delivery methods and optimization of gene expression cassettes have produced transformation protocols for the major cereals, although some elite cultivars still remain recalcitrant to transformation. Most of the transgenic cereals developed for commercial purpose exhibit herbicide and/or insect resistance; traits that are often controlled by a single gene. In recent years, more complex traits, such as dough functionality in wheat and nutritional quality of rice have been improved by the use of biotechnology. The current challenges for genetic engineering of plants will be to understand and control factors causing transgene silencing, instability and rearrangement, which are often seen in transgenic plants and highly undesirable in lines to be used for crop development. Further improvement of current cereal cultivars is expected to benefit greatly from information emerging from the areas of genomics, proteomics and bioinformatics. 5.3.1 Transfer of Whole Genome - Examples from Wheat, Archis & Brassica In recent years, with advent of the development of efficient plant regeneration systems in cereal crops, the field of recombinant DNA technology has opened up new avenues for genetic transformation of crop plants. Monocots particularly cereals were initially considered difficult to genetically engineer, primarily due to their recalcitrance to in vitro regeneration and their resistance to Agrobacterium. Continuous efforts and systematic screening of cultivars and tissues for regeneration potential, development of various DNA delivery methods and optimization of gene expression cassettes have led the development of reliable transformation protocols for the major cereals including wheat. Since the production of first fertile transgenic wheat plants in 1992s, microprojectile-mediated gene transfer has proved the most successful method for genetic transformation of wheat not only for the introduction of marker genes but also agronomically important genes for improving quality of wheat flour, transposons tagging, building resistance against fungal [67] pathogen and insects, engineering male sterility, and resistance to drought stress. Despite tremendous successes in producing fertile transgenic wheat plants using various methods and approaches, elite cultivars of wheat still remain recalcitrant to transformation. Moreover, in comparison with other major cereals like rice and maize, the development of a high throughput wheat transformation system has been slowed and severely affected by genotype effects on plant regeneration, low transformation efficiencies and problems with transgene inheritance and stability of expression. Majority of the researcher worldwide have used genetic engineering to tailor wheat for specific end-use by using immature embryos as the primary target tissue for the delivery of desired foreign genes. Hence, we have focused our attention to the work done on stable gene expression and transformation of wheat by employing microprojectile bombardment and Agrobacterium as a source of foreign DNA delivery into immature embryos. Recent advances in wheat transformation especially successes in genetic transformation of wheat with agronomically important genes and novel and innovative approaches for wheat transformation based on different selection schemes are also discussed. Wide hybrids in the Triticeae tribe have been attempted and studied for over a 100 years. The first such hybrid was between wheat and rye (Wilson, 1876). Rimpau (1891) described 12 plants recovered from seed of a wheat-rye hybrid that represented the first Figures 5.37 : 3-D anaglyphs of pollen grains stained with acridine orange. Bar = 10μm. (3) a single Hoheria sp. pollen grain; (4) Myosotis monroi pollen grains on the surface of a stigma; (5) Agathis australis (kauri) pollen grain obtained from a swamp profile, estimated age 10,000 years. (6) and (7) 3-D anaglyphs of dividing somatic cells of Vicia faba, broad bean, root-tips stained with Feulgen. (6) mostly interphase cells, note the bright heterochromatic knobs. Also note the early prophase at left, late prophase in middle, and metaphase top right. Bar = 5μm; (7) a prominent anaphase in the center and a prophase cell at top left. Bar =3μm. (8) 3-D anaglyph of germinating yeast spores stained with acridine orange. Bar = 5μm. Color images of these anaglyphs are found on the cover of this issue. Anaglyph 3-D glasses are included with this issue. View the color image using the glasses: Left eye = blue, right eye = red. triticale. Farrer (1904) reported studies on wheat-barley [68] hybridization; however, Shepherd and Islam (1981) considered it improbable that these were true intergeneric hybrids. Several perennial grasses were hybridized with wheat in the early 1930s with the objectives of transferring disease resistance and perenniality into annual crops. Many hybrids involving Triticum and several Aegilops species were also made during the 1920s and 1930s from which the genomic relationships of the two genera were derived. The large-scale practical use of the hybrids, however, was delayed until the advent of colchicine treatment in the late 1930s. The ability to double the chromosome number of hybrids using colchicine had both practical and theoretical consequences. The production of fertile amphiploids provided the way to develop X Triticosecale Wittmack as a new cereal crop. Also advanced were evolutionary studies when McFadden and Sears (1946) resynthesized T. aestivum, discovering that Ae. tauschii (syn. T. tauschii) was the D-genome donor to bread wheat. With the advancement of hybridization techniques (Kruse, 1973) and embryo culture (Murashige, 1974), wide hybridization became a more common practice involving more perennial species. In reviews of the progress of wide hybridization, intense interest was expressed among breeding programmes in utilizing the genetic resources available in the perennial Triticeae for cereal improvement (Dewey, 1984; Sharma and Gill, 1983a; Mujeeb-Kazi and Kimber, 1985; Sharma, 1995; Wang, 1989). Of the approximately 325 species in the tribe Triticeae, about 250 are perennials and 75 are annuals (Dewey, 1984). Relatively few perennials have been hybridized with wheat essentially because of the complexity of doing so and due to embryo rescue/regeneration constraints. The perennials, which include many important forage grasses, have the potential to serve as a vital genetic reservoir for the improvement of annual grasses. These include the major cereals: bread wheat, durum wheat, triticale, barley and rye. Perennials successfully utilized for improving wheat are predominantly in the Thinopyrum group. This section is focus on achieving agricultural production targets with emphasis on bread and durum wheat. Such production targets are to be achieved by enforcing crop improvement protocols based upon the utilization of genetic diversity, crucial for durability of stress resistances and tolerances and for ensuring sustainability. Major emphasis is devoted to consideration of the exploitation of ‘alien’ genetic diversity, encompassing interspecific and intergeneric hybridization categories. 5.3.2 Transfer of Individual Chromosomes and Chromosome Segments Chromosome 5B mechanism - There seems to be no parallel to the chromosome 5B-like manipulative approach that encompasses mono-5B, phph or nullitetrasomic stocks as the maternal wheat sources in wide-crosses. These stocks enhance wheat/alien recombinations in the F1 hybrids and all involve the ph system (Sharma and Gill, 1983a, 1983b, 1983c; Darvey, 1984; Mujeeb-Kazi et al., 1984; Forster and Miller, 1985; Sharma and Bäenziger, 1986). The resultant F1 hybrids exhibit a high meiotic chromosome pairing frequency, but obtaining back-cross derivatives was considered to be a major problem. Sharma and Gill (1986) encountered similar constraints when T. aestivum x Aegilops species hybrids were produced. Subsequently, Ter-Kuile et al. (1988) reported success with the ph maternal system using T. aestivum x Ae. variabilis as [69] the test cross. Since then, numerous ph manipulative high pairing F1 hybrids have been routinely produced and advanced to BCI or BCII (Rosas et al., 1988). However, as an alternative, since a general constraint prevails, it may be appropriate to produce the F1 hybrid with a highly crossable wheat (PhPh) and either back- or top-cross it with the phph stock (Sharma and Gill, 1986). Additional options for influencing the PhPh locus are associated with this locus being suppressed by Ae. Mutica or Ae. speltoides; a procedure that could be incorporated at the F1 stage with a low recombination hybrid or on desired alien disomic addition lines. Achieving high recombination is emphasized primarily because the T. aestivum crop species with its phenomenal cytogenetic flexibility via Ph manipulation offers remarkable opportunities for alien gene transfers and incorporation of homoeologous segments introduced in the best location in the recipient wheat chromosomes. Some other novel systems for genetic manipulation in intergeneric hybridization have experimental priority and are targetted to yield a tremendous agricultural impact via germplasm developed by this procedure. The following are strategies that the authors feel must be pursued vigorously in the quest to keep product development at a pace ahead of the population surge anticipated during the next two decades. Exploiting these strategies is anticipated to render available diverse genes from alien sources that have been insufficiently utilized and that the authors feel shall provide a durable and sustainable practical focus through gene pyramiding. The need to match or exceed the impact of the T1BL.1RS spontaneous translocation (Rajaram et al., 1983) and the induced T1AL.1RS germplasm (Islam-Faridi and Mujeeb-Kazi, 1995; Villareal et al., 1996) ranks high. With the relative ease of hybridization (Sharma, 1995) and the range of hybrids and alien chromatin exchange stocks available (Jiang et al., 1994; Friebe et al., 1996), some diversification seems appropriate as the next century begins. Can intergeneric hybridization protocols be modified to yield faster returns of quality end products? Though closely related genomes hold a priority for wheat improvement, additional genes from ‘diverse’ gene pools also offer unique resistance durability and are anticipated to contribute to sustainable cropping systems. The various gene sources contributing to C. sativus resistance elucidates this concept. These genes, when pyramided, have the potential to ensure resistance durability across several locations where C. sativus is a wheat production constraint. From the earlier BH1146 cultivar resistance, the improvement present in the Th. curvifolium plus Chinese wheat cultivars has been significantly dramatic (cultivars Chirya and Mayoor) and has remained durable across several countries for approximately 12 years. However, complacency in not introgressing more diverse genes must not prevail for C. sativus or for any other biotic stress. In essence, the progress from BH1146 to Th. curvifolium and/or usage of Chinese wheat cultivar derivatives (Chirya and Mayoor), coupled with the extensive variation identified in the A-, B- and D-genome accessions, is seen as a guarantee for stability over years to come. It is a path being followed for C. sativus and can be extended to address other stress objectives. The above approach, as the next century begins, is totally removed from some wide hybridization views. Fedak et al. (1994) expressed that wide-crossing for purposes of gene transfer be done as a last resort when the variability for a particular trait is exhausted or is non-existent in the primary gene pool. The authors, however, view wide-crossing as a complementary approach, conducted simultaneously and integrated within conventional breeding programmes. This approach contributes multiple diverse [70] novel genes from all gene pools by pyramiding them with the conventional genetic resource present in the primary pool commonly used by breeders. Even if these are ‘major’ alien genes, their multiplicity and diversity shall provide an advantage when pyramided with the conventionally available ‘minor’ genes that recognizably contribute to durable resistance. Distantly related species (e.g. tertiary gene pool) are complex to exploit, but their potential use in crop improvement is very high. To exemplify, the contributions of S. cereal in wheat improvement cannot be overlooked (Islam-Faridi and Mujeeb-Kazi, 1995; Mujeeb-Kazi et al., 1996), and a major role of Thinopyrum species for BYDV resistance further attests to the use of this distant diversity (Henry et al., 1996). Involvement of other tertiary gene pool species in wheat germplasm has been the subject of a few recent reviews (Jiang et al., 1994; Sharma, 1995; Friebe et al., 1996). A modified tertiary gene pool transfer strategy, which should receive greater emphasis in the future, is aimed at providing maximum recombination between wheat and alien species chromosomes in the early hybrid generation stages. The enhanced recombination will be a consequence of cytogenetic Ph locus manipulation, irradiation, callus induction, etc. Ph manipulations will involve the use of chromosome 5B genetic stocks and use of the relatively newer PhI germplasm option (Chen et al., 1994). The latter warrants more exploitation. The sooner the wheat/alien chromosomal exchanges occur in an intergeneric hybridization programme involving tertiary gene pool species, the sooner appropriate breeding protocols will be incorporated, stress screening coupled with homozygosity will find its place and, with current molecular diagnostic strength, alien introgression(s) identification will occur and be exploited. Researchers' vision is to shorten the tertiary gene pool conventional genetic transfer protocols to a short-term productoriented programme akin to the Figure 5.38 : Molecular marker - assisted genetic interspecific approach that capitalizes manipulation involving the ph locus on chromosome upon the primary and secondary gene 5B mediated by maize induced doubled haploidy. pool species. The vision projected above will better address the incorporation of polygenically controlled traits, hopefully embolic, into wheat of which the alien Th. elongatum chromosomal control (3E, 4E and 7E) for salinity tolerance is one example (Dvorak et al., 1988). [71] Immediate priority has been assigned to a doubled haploid wheat/maize-based manipulation protocol that utilizes the Ph F1 wheat/alien hybrids maintained at the International Maize and Wheat Improvement Center (CIMMYT) as a living herbarium. The protocol is applicable to amphiploids and fertile BCI combinations. The double haploidy role in salvaging Ph-based F1 hybrids has become an option to enable phmediated alien introgression(s) without having to remake complex F1 hybrids using the ph genetic stock (Sears, 1977) as the maternal parent. Because of CIMMYT’s living F1 herbarium involving wheat and several alien species (Ph locus present), BCI derivatives can be produced by pollinating these Ph F1 wheat/alien hybrids with the Chinese Spring Ph ph wheat genetic stock. The BCI progenies (Ph ph) are crossed with maize to yield poly-haploids that possess the Ph or ph locus. The entire wheat and alien chromosomal complement is represented. The haploids derived from the Ph BCII=back-cross II ph BCI derivatives possessing the ph recessive gene get identified at Figure 5.39 : Schematic showing the crossing scheme the seedling stage by a polymerase of Triticum aestivum /Aegilops chain reaction (PCR) based tauschii direct hybridization diagnostic analysis (Gill and Gill, and back-cross advance 1996; Qu et al., 1998), which enhances programme efficiency (Figure 5.38) and sets a crop improvement programme in place using an integration of the breeding methodologies. The ph locus facilitates wheat/alien chromosomal exchanges and generates translocation stocks that are between homoeologous and nonhomoeologous chromosomes. They are Robertsonian translocations generally, but smaller alien exchanges are also produced. The diagnostic protocols are FISH followed by Giemsa C-banding. Derivatives of interest possess the critical translocation chromosome(s), but also have entire alien chromosomes that are eliminated via backcrossing (Mujeeb-Kazi, 2001b). [72] Current emphasis has shifted for delivering farm crop products at a fast pace. Such impacts shall be a consequence of gene transfers from the closely related diploid species and their accessional diversity. In some cases, even swifter results can be obtained as exemplified by the Dgenome direct crossing procedures (Figure 5.39) (Alonso and Kimber, 1984; Gill and Raupp, 1987). Greater efficiency emerges by mediating direct transfers with sexually induced double haploidy (DH) for achieving rapid homozygosity (Mujeeb-Kazi and RieraLizarazu, 1996).Identifying genes in different resistant accessions and pyramiding these prior to crossing with wheat also appears challenging (Figure 5.40, Figure 5.41). One approach is to intercross resistant synthetic hexaploids for a stress, advance the F1, select resistant F2/F3 derivatives that combine the effect of the two divergent Ae. tauschii accessions, incorporate double haploidy and end up with a homozygous SH=synthetic hexaploid stock. Figure 5.39 : Schematic showing steps involved in pyramiding accessional diversity of Aegilops tauschii Figure 5.40 : Schematic indicating a novel gene pyramiding strategy associating stressresistant diverse genomes (AA of Triticum monococcum and DD of Aegilops tauschii) SH = Synthetic Hexaploid *Seven such double haploids result Use of this stock in wheat improvement will simultaneously permit incorporating two different genes from the Ae. tauschii accessions (Figure 5.40). Another approach is intercrossing two genomically divergent resistant diploid accessions and developing tetraploid stocks. Use of resistant A and D-genome accessions is an example (Figure 5.41). In order to hasten such gene identifications, the- DH approach has provided further usage and is now being advantageously utilized for modified complete or partial monosomic analyses. The partial analysis is conducted when [73] resistance is associated with the D genome of SH wheats (Figure 5.42). The F1 monosomics of 1D to 7D chromosomes (2n=6x=40 + 1D to 40 + 7D) when crossed with maize yield 21 chromosome polyhaploids with the 1D to 7D contributions coming from resistant SH wheats. Doubling these n=3x=21 polyhaploid plants with colchicines results in stable 42 chromosome double haploids. Each DH now possesses the homozygous 1D to 7D chromosomes of the resistant SH parent being analysed for the chromosomal location(s) of the resistant gene(s). Upon screening, the non-segregating resistant DHs are attributed with having the gene(s) in them. The stable monosomic derived DH germplasm, apart from simplifying the conventional monosomic analyses, also facilitates global distribution of the developed germplasm. The germplasm enables experimental repetition without have necessary when the conventional monosomic analytical procedure is followed. Alien Transfers for Durum Wheat Improvement - Progress in durum wheat/alien species hybridization has not been as exhaustive as bread wheat. However, the need to diversify the durum genetic base is crucial and can be achieved by incorporating the diversity of the primary, secondary and tertiary gene pools. The intergeneric recombination constraints can presumably be overcome by using the ph1c Capelli genetic stock that requires greater investigating to fit agricultural goals. For interspecific durum wheat improvement, the A and the B genome diversity through their AAAABB/AABBBB amphiploid routes allows for cross combinations to be made between the resistant amphiploids and elite durum cultivars. This facilitates introgression and exploitation of resistant traits in breeding programmes by utilizing appropriate breeding protocols. This alien diversity based durum improvement programme is currently in its infancy, but the authors do anticipate contributions for resistant transfers to be achieved for durums. Challenging is the exploitation of D-genome resistances for durum wheat improvement, and at a high priority would be the transfer of scab (F. graminearum) resistance genes. Additionally, it must be mentioned the potential been assigned to these diseases of Dgenome resistance transfers to durum wheat of genes associated with salinity tolerance, Figure 5.42 : Steps drought tolerance, S. tritici, C. sativus and associated in conducting a BYDV resistance, with quality being an partial monosomic integral part in all A-, B- and D-genome analysis, where resistance accessional transfers. These genomic transfers is located within the hexaploid would be a consequence of recombinational synthetic (2n=6x=42) D-genome events due to the preferential A- and D- chromosomes [74] genomic chromosome pairing represented as seven bivalents in the presence of the ph locus. The bivalents are generally of the A- and D-genome chromosomes and univalents of the B genome, as inferred separately from meiotic C-banding data (unpublished). The authors’ current tester system to demonstrate the D- to A-genome genetic exchange efficacy is for C. sativus and S. tritici from some D-genome resistant accessions. Durum wheat cultivars are highly susceptible for both of these biotic stresses, and since CIMMYT has ideal screening protocols with reliable screening locations in Mexico, priority has from the secondary gene pool, the potential of using Ae. speltoides resistance diversity for several stresses does also exist for durum wheat improvement. Whole-Arm Translocation - One possibility of introducing into a cultivated background less than an entire alien chromosome is to substitute one arm of the latter for a corresponding wheat arm. In, wheat, such transfers are made possible by the strong tendency of univalent chromosomes to misdivide at meiosis and give rise to one-armed (telocentric) chromosomes. If both an alien chromosome and its wheat homoeologs are present as univalents and misdivide in the same sporocyte, the resulting telocentrics can occasionally fuse at the centromere and produce a bibrachial chromosome, with one alien and one wheat arm. One major limit of such a method for introducing alien variation resides in the low frequency with which the correct, balanced combination can be recovered, whether from spontaneous or from radiation-induced events. Moreover, a whole arm often possesses too much unwanted alien chromatin along with the target gene(s). however, before the extensive use of induced homoeologous pairing as the method of choice and, in some instances, to make possible use of desired introgressions involving A or B genome chromosomes already obtained at the 6s level, a number of alien transfers had been attempted into durum wheat involving entire chromosomal arms. Among the few that met with relatively good success in a tetraploid background is the 1BL.1RS translocation. This translocation, together with the corresponding one involving wheat chromosome 1A (1A.1RS), represents the most successful wheat-alien transfer effectively employed in common wheat breeding worldwide. The short arm of Rye chromosome 1R is known to carry many important genes for resistance to wheat pathogens, including the yellow rust resistance gene Yr9, the leaf rust resistance gene Lr26, the stem rust resistance gene Sr31, the powdery mildew resistance gene Pm8 and Pm17 and the green bug resistance genes Gb2 and Gb6. Transfer of Chromosomal Segments - The general outcome of the research described above demonstrates that a whole alien arm, particularly if it originates from relatively distant alien species, finds little acceptance by durum wheat. Among the possible strategies of chromosome engineering that enable controlled introductions of chromosome segments from related Triticeae into cultivated wheat, the one based on manipulations of the wheat chromosome pairing control system, particularly the use of mutations of Ph1, is by far the most effective. Radiation-induced transfers have the inherent drawback of producing essentially random translocation. Although many such translocations in wheat tend to involve homoeologous chromosomal segments, these are greatly outnumbered by those involving non homoeologous. Moreover, even for compensating transfers, attainment of intercalary translocations remains an extremely rare event. The only documented case of an [75] intercalary wheat-alien transfer produced by radiation treatment appears to be that of a non compensating translocation of a relatively short 6RL segment containing the H25 gene for resistance to Hessian Fly proximally inserted into the wheat 4AL arm. This introduction, originally obtained in common whe3at, was recently incorporated via homologous recombination into durum. Gametic transmission of translocated chromosome, showing disturbance on the male side in the BC1 generation, appeared normal in BC2 and BC3 derivatives. Moreover, the resultant translocation stock was vigorous and had a seed set similar to the durum parent Cando thus demonst6rating its usefulness in breeding. Apart from this exceptional case, the past and more recent experience on wheat chromosome engineering clearly highlights that the Ph1-mediated approach offers the greatest promise for obtaining alien transfers. This is because disturbance of the recipient chromosome and genotype balance, if any, is minimal, and the benefits of the alien genetic contribution(s) are more readily available for practical utilization. One reason for this is the cytogenetic affinity of the recipient chromosome to the donor chromosome, because homoeologous are almost exclusively involved in Ph1-promoted pairing. When the critical alien chromosome or chromosome arm is as an addition or substitution into wheat genome, the potential for pairing between the alien and the wheat homeologs is the highest if the two are present as univalents. Another important advantage of the Ph1mediated approach resides in the possibility to progressively shorten the amount of alien chromatin flanking the desired gene(s) during successive steps of the transfer procedure. This can be accomplished in different ways, depending on the material available as well as on the position of the target gene(s) on the chromosome. If the alien products with short, terminal alien segments. If, however, the alien gene has a median or more proximal location, additional manipulations are needed to further shorten the alien segment due to the expected low frequency of double crossover, especially between homoeologous chromosomes. One possibility consists of allowing the donor and recipient chromosomes to undergo repeated rounds of Ph1-induced homoeologous recombination. This, however, can cause an excessive accumulation of unwanted background translocations, leading to considerable gametic and zygotic instability and eventually to loss of potentially desirable types. An alternative and perhaps better strategy was originally suggested and successfully applied by Sears. By combining two complementary transfer chromosomes, which resulted from single exchanges on the proximal and distal sides of the target gene, respectively, crossing-over can occur in the homologous region shared by them. This will give rise to a product equivalent be determined by applying FISH with a highly repeated (pSc119.2) and low-copy RFLP sequence (PSR907) as probe. These physical markers allowed the alien segment to be precisely located distal to the 3BS Xprs907 locus, in the adjacent subtelomic interval separating the two most distal pSC119.2 sites of 3BS, is known to be genetically positioned at less than 25 centimorgans (cM) from the 3B centromere. The above example highlights how molecular cytogenetic techniques, such as FISH using specific DNA sequences or total genomic DNA (fl-GISH) of the alien species as probe, allow a precise assessment of the physical amount of exchanged material. This represents a critical parameter to estimate the potential impact of an alien transfer on the recipient genotype, particularly when operating at the less tolerant 4x ploidy level. [76] 5.3.3 Methods for Detecting Alien Chromatin Hybrid Validation: Diagnostics which also Include Detection of Alien Chromatin - Initial hybrid identification is based upon mitotic counts in root tips collected at various hybrid development stages (Mujeeb-Kazi and Miranda, 1985). A Figure 5.43 : Figure no. 1, 2 and 3 showing somatic chromosomes of Triticale “Badger”, a pair of 1B/1R chromosomes and an interphase cell in “Ning 8026” respectively probed with labelled rye DNA and blocked with unlabelled wheat DNA, the rye chromosomes and chromatin (brown) could be followed. Figure 4, 5 showing the somatic chromosomes of Triticale Badger and the 1R(1D) substitution line “84056-1-36-1” respectively probed with PTA 71, six signals in Badger and eight in “840561-36-1” could be observed (arrows). [77] normal intergeneric F1 hybrid possesses half the chromosome number of each parent involved in the combination. For hybrids of different polyploidy levels, a mere number count is adequate initial verification. There are, however, cases where the alien species are hexaploid, such as wheat, and hybrids would then have 42 chromosomes. These may be difficult to classify categorically as hybrids, but with superb primary and secondary constriction resolution of wheat 1B, 6B and 5D chromosomes (Mujeeb-Kazi and Miranda, 1985), identification of hexaploid hybrids is simplistic. Additional identification can be made by employing chromosome-banding techniques. Karyotypic differences play a part, but positive claim to hybridity must be accompanied by clear meiotic analyses. In some situations, the alien genome may be totally or partially eliminated, resulting in the production of polyhaploid/haploid or aneuploid F1 hybrids. The two aspects are classified under: (i) genome elimination; and (ii) aneuploid F1 hybrids. A modified hybrid phenotype is also good proof of hybridity, but often a hybrid may not express the features of one parent as in the case of wheat x barley or its reciprocal combination (Islam et al., 1978). In general, however, a majority of the F1 wheat x alien (reciprocal also) hybrids involving all the A-B- and D-genome diploids, annual and perennial, Triticeae genera and species exhibit a co-dominant phenotype (Mujeeb-Kazi et al., 1987, 1989). Similar is the phenotype modification of their amphiploids. More recent diagnostic procedures utilized for the detection of hybrids and/or their advanced derivatives are in situ hybridization (FISH, GISH, etc.), electrophoretic analyses (biochemical) and molecular detection of the presence of alien chromatin. Some examples are as follows:- (1) Fluorescence In-Situ Hybridization Techniques for Brassica: Methodological Development and Practical Applications Fluorescence In Situ Hybridization (FISH) methods were developed for Brassica which allow the physical localization of labeled DNA sequences at chromosomal sub-arm level. In addition, Genomic In Situ Hybridization (GISH) enabled all Brassica diploid genome components apart from the highly similar A and C genomes to be distinguished, and R genome chromatin was reliably detected in Brassica napus x Raphanus sativus hybrids. Hybrids among the Brassicaceae can be relatively easily produced and are an ideal method for generating new lines containing agronomically important pest and disease resistance genes. Efficient monitoring of alien chromatin is however critical for successful transfer of alien chromosomes or chromosome segments containing useful genes. The small size of chromosomes in Brassica and its close relatives is particularly problematic for the identification of alien chromosomes in Brassica hybrids, and translocations cannot be reliably identified by conventional cytogenetic methods. GISH was shown to be an effective practical method for detecting amounts and locations of foreign chromatin in Brassica hybrids, while other FISH applications were useful for chromosome identification and investigations of Brassica genome evolution. (2) Identification of Alien Chromatin and Ribosomal DNA in Wheat by In-Situ Hybridization - In situ hybridization has used to detect somatic cells of hexaploid Triticale “Badger” 1B/1R translocation line “Nine 8026” and IR (1D) substitution line “84056-1-36” using biotin labelled total rye genomic DNA and wheat rDNA as probe (Figure 5.43). [78] (3) FISH and RFLP Marker-Assisted Introgression of Festuca mairei Chromosomes into Lolium perenne - Plant breeders and geneticists have attempted for several decades to combine perennial ryegrass (Lolium Figure 5.44 : Southern hybridization of the HindIII - or EcoRI-digested genomic DNA of Lolium perenne (Lp), Festuca mairei (Fm), and their hybrid derivatives with (A) TF109, identifying the presence of F. mairei genome in F1, F2, and BC1 plants; (B) TF515, a Festuca genomespecific probe, showing strong hybridization only in F1, BC1, and some BC1G1 plants (M = labeled DNA/HindIII fragments) perenne L.) with fescue (Festuca spp.) to create novel forage grasses containing both high forage quality and good drought tolerance (Figure 5.44). Difficulty in selecting true hybrids with alien chromatin or chromosome addition–substitution has been a major barrier in Festuca x Lolium breeding programs. In this investigation, fluorescence in situ hybridization (FISH) and restriction fragment length polymorphism (RFLP) markers were used to monitor transfer of Festuca mairei, St. Yves chromosomes into L. perenne through intergeneric hybrids. Among 64 hybrid plants of the BC1G1 generation (intercrossed progeny of first backcross to L. perenne), chromosome addition and substitution of F. mairei were identified by FISH using total genomic DNA of F. mairei as a probe (Figure 5.45). Forty-two clones from a PstIgenomic DNA library of F. arundinacea Schreb, were used to screen for the presence of F. mairei DNA in the hybrid plants. These RFLP probes rapidly identified presence of the F. mairei genome in F1, F2, BC1, but not in BC2 plants. In contrast, genomic FISH on meiotic cells effectively detected any F. mairei chromosomes as well as chromosomal pairing relationships in any hybrid. By Figure 5.45 : Genomic FISH using labeled F. mairei DNA as a probe effectively detected substitution of a pair of Festuca mairei chromosomes in a Lolium-like hybrid plant [79] monitoring and selectively introducing F. mairei chromosomes into ryegrass, these molecular markers may accelerate the Festuca x Lolium breeding for improvement of ryegrass. Figure 5.46 : Detection of rye chromatin using genomic in situ hybridization. Yellow-orange to yellow fluorescence shows hybridization of the total genomic DNA rye probe. A: Metaphase chromosome spread of the disomic wheat-rye addition line 1R showing two rye chromosomes identified by yelloworange arms and bright yellow telomeres (see arrows). B: Metaphase chromosome spread of Atlas, showing the absence of rye DNA probe hybridization. C: Metaphase spread of Century, showing the detection of two rye chromosome arms by the yellow-orange fluorescence and bright yellow telomeres (see arrows). D: The number of alien chromosome arms can also be counted in interphase nuclei of Century where they form two distinct fluorescent yellow domains (see arrows). [Normal View 101K | Magnified View 298K] [80] (4) Use of fluorescence genomic in situ hybridization (GISH) to detect the presence of alien chromatin in wheat lines differing in nuclear DNA content - Genomic in situ hybridization was used to identify the number and size of alien chromosomes or chromosome arms. Probe hybridization sites were detected by FITC-linked antibodies that fluoresced yellow under blue light excitation and allowed visualization of rye chromatin material. TA3601, a disomic wheat-rye addition line involving the addition of two complete rye chromosomes, is pictured in Figure 5.46A. Forty-four chromosomes were observed with two chromosomes appearing yellow-orange with each end of the chromosome stained bright yellow. The bright yellow-stained chromosome telomeric ends represent heterochromatic regions of the chromosome. In addition, nuclei were also seen to have four distinct signals representing the heterochromatic regions of genetic material. These results were observed in all four of the disomic wheat-rye addition line. Figure 5.46B is a metaphase spread of Atlas, with an absence of the FITC signal, indicating the absence of rye chromatin. Century (Figure 5.46C) had 42 chromosomes with two chromosomes that exhibited one red arm and one yellow-orange arm with a bright yellow signal on the telomere. The nuclei of Century (Figure 5.46D) exhibited two labeled domains in the nuclei at interphase. The near-isolines of Century also displayed two FITC-stained chromosome arms and two yellow domains in the nucleus. All of the Century-derived plants analyzed were observed to have the positive signal. Chisholm metaphase spreads had an absence of FITC signal. Nuclei were absent of any labeled domains. Near isolines derived from Chisholm did not exhibit FITC signals in either metaphase chromosome spreads or nuclei. At no time was a positive signal observed in the Atlas or Chisholm lines or the isolines of Chisholm. 5.3.4 Produciton, Characterization and Utility of Alien Addition & Subtraction Lines RNA Splicing Production of alien chromosome addition / substitutions and their utility in plant genetics - Plant geneticists and breeders have gained great interest in extending genetic variation of crop plant using exotic germplasm from related species. In a long term crossing programme, known as introgressive hybridization, economically or otherwise important genes were incorporated into the recipient parent by sexual or somatic hybridization between the crop and a related species or genera, followed by consecutive backcrossing with the recipient parent while selecting for the favourable trait(s). In the offspring families aneuploidy individuals could thus be isolated containing only a single alien chromosome added to the full cell complement of the recipient parent. Monosomic addition were first described by Leighty and Taylor (1924) but use and potential were better demonstrated in the comprehensive study of O’Mara (1940). Khus (1973), Gupta (1995) and Sybega (1992) gave a full overviews of alien addition (The commonly used term monosomic addition lines is incorrect because lines assume homozygosity for a particular gene or chromosome variant, and so will not segregate in the next generation) in relation to other aneuploids in plant genetics. Although the number of papers on alien addition are almost countless, reports on full sets with all alien chromosomes appeared only for monosomic additions with chromosomes [81] of Beta webbiana, B. patellaris and B. procumbens to beet, Solanum lycoperiscoides to tomato, tomato to potato, Oryza officinalis to rice, a number of Triticeae species, including rye and barley, to Triticum aestivum, maize to oat, and onion to Allium fistulosm. Table 5.5 present an overview of complete sets, together with their parental species, marker selections, cytogenetics and references. Production of additions and substitutions - Three major barriers can be distinguished in introgressive hybridization : i) incompatibility or incongruence between the parental species, and hence the difficulties in producing viable interspecific or intergeneric hybrids; ii) sterility of the F1 and sometimes also of the BC1, thus hampering transfer of alien chromosomes through backcrossing and ii) rare or negligible meiotic recombination between the alien chromosomes and one of its homoeologous counterparts, preventing incorporation of alien chromatin into the recipient genome. Methods have been developed to overcome these natural crossing barriers. One of them makes use of chromosome doubling to produce allopolyploids from which fertile allotriploids can be derived. A second method makes use of pollen mixes, containing pollen for syngamy and “mentor” pollen from the maternal species to facilitate fertilization by foreign pollen. Alternatively, desirable traits can be transferred indirectly using bridge cross with a related (wild) species or cultivar that is compatible with either parental species. Although time consuming, such interim crosses have successfully been applied to various crop species including alfalfa, beet, lettuce and onion species. Melchers et al. (1978) was the first to make somatic hybrids, later followed by many others. Table 5.5: The development of cell culture technology for protoplast fusion and regeneration, for the production of somatic hybrids from fused protoplasts of related species that are incompatible in a sexual cross was a further step to bridge the gap between non-crossable A highly advanced example of somatic hybridization technology was developed by Sree Ramulu et al. who isolated micronuclei containing one or few chromosome s from the donor species fused them with complete euploid protoplasts from the recipient crop. This microprotoplast fusion technique directly produced asymmetric somatic hybrids with the cell complement of a monosomic addition, thus skipping the problems of interspecific incongruity and BC1 sterility. Although potentially very promising, this advanced strategy never received large-scale practical application. Hybrid embryo abortion after fertilization is an important problem generally occurring in (somatic) hybrids from wide crosses and reflects disharmony between the genomes of the parental species that result into embryo mortality, endosperm break down, seed inviability and hybrid sterility. Different in vitro embryo rescue techniques were developed to overcome this problem of hybrid embryo degeneration and have been applied to a large variety of crops and ornamental species. Once the backcross progenies could be recovered, aneuploid individuals with alien chromosomes, including monosomic additions could be isolated. As maintenance of monosomic additions was difficult due to sterility, inferior viability and low chromosome transmission through the germline , additions of potato, onion and beet background were often kept in vitro or retained by [82] vegetative propagation. Transmission of the alien chromosome sand viability of the additions can difficult to obtain. Production of disomics was only possible when the alien chromosome was transmitted also through the male germ line, or they can be obtained by meiotic non-disjunction of the alien chromosome in the monosomic addition parent. Use Figure 5.47 : Genomic painting (genomic in situ hybridization) shows the number and behaviour of alien chromosomes in backcross individuals. Here we show the monosomic addition carrying chromosome 3 of tomato added to tetraploid potato (2n=4x=+1=49) hybridized with total genomic tomato DNA as probe and 50x unlabeled potato DNA as competitor. (a) Mitotic metabphase complement. (b) Pachytene complement. Bars equal 10 μm. of derived ditelosomic additions, in which single individual alien chromosomes were, replaced by their telocentrics, often improved chromosomal transmission considerably. The final step in introgressive hybridization programmes involves meiotic recombination between alien chromosome and one of its homoeologous counterparts in order to stably incorporate its chromatin into the recipient parental genome. Generally, additions derived from wide interspecific or intergeneric hybrids display little or no homoeologous recombination at all, and hence leave the alien chromosome s as univalents at metaphaseI. In the corresponding substitution lines, association of homoeologous chromosomes may be far higher due to the absence of homologous partners. Transfer of desired chromosome regions to wheat is generally most successful in the Ph1 background of wheat allowing high levels of homoeologous recombination. Other means of alien chromosome introgression can be achieved by breakage and fusion of chromosome fragments, either spontaneously by radiation-induction or by gametocidal factor in Aegilops cylindrical, which have especially been applied in the cereal species. Characterization of the alien chromosome(s) - Monosomic additions can be selected on the basis of specific alien traits, like disease resistance, aberrant plant phenotype, species-specific molecular markers and karyotype analysis to demonstrate the presence of an extra chromosome. Morphological traits can be qualitative, like the [83] characteristic liguleless leaves of the maize chromosome 3 monosomic addition in oat and the monogenic dominant resistance genes, or inherit quantitatively, such as plant size and spike morphology. The morphological traits in the monosomic addition set of Beta vulgaris carrying an extra chromosome from B. procumbens or B. patellaris appear only partly chromosome-specific, and hence are not adequate for the identification of all alien chromosomes without additional markers. Monosomic additions derived from hybridization between genetically related parental species can have phenotypes often resembling its corresponding primary trisomics. Alien chromosomes in the additions may sometimes be morphologically distinguishable, as was shown in a karyotype analysis of four nematode resistant monosomic addition of beet containing different alien chromosomes from Beta procumbens or B. patellaris. Chromosome morphology was for a long time described in terms of centromere position, arm lengths, C- and N-banding profiles, and heterochromatin pattern in cell complements at pachytene. Flow cytometry can be helpful when chromosome size and GC/AT ratio are sufficient to identify the alien chromosome in the flow karyogram and can even be applied to isolate large numbers of alien chromosomes, as was demonstrated for a monosomic addition with maize chromosome 9 to oat. The by far most important tool to visualize alien chromosomes in genomic in situ hybridization (GISH) or genomic painting, a fluorescence in situ hybridization protocol using total genomic donor DNA as probe and non-labelled DNA from the recipient parent as competitor. An example of GISH of a monosomic addition of tomato chromosome 3 in potato is shown in the Figures 5.47a and 5.47b. Reports on establishing the number of alien chromosomes in intergeneric backcross families are numerous, like tomato to potato, maize to oat, Beta corolliflora in beet, Solanum brevidens to potato and Sgenome chromosomes in wheat. The technique is also considered effective in painting and studying individual chromosomes in plants. GISH has also the advantage of demonstrating interspecific and intercenteric translocations, and shows recombinant chromosomes resulting from homoeologous recombination. For the identification of the alien chromosome in the Figure 5.48 : RFLP molecular markers for the addition stocks, additional characterization and selection of monosomic additions with a specific alien chromosome. Southern analysis is required using hybridization of Dral/HindIII markers, T=tomato, chromosome specific “wild” P=potato and 1-11, a series of offspring plants from a monosomic addition 3 backcross. [84] morphological traits, isozyme markers including RFLPs, RAPDs, microsatellites and repetitive sequence DNA fingerprints (Figure 5.48). The integrity and identification of the alien chromosome in a monosomic addition carrying a Solanum brevidens chromosome in potato background were obtained by sequential genomic painting and FISH with BACs diagnostic for each chromosome. The advantage of this 2-step procedure is that both genomic origin and genetic identity of the alien chromosome can be established in a single experiment. Properties of additions - The potential of alien additions for breeding programmes largely depends on the genetic distance of the parental species and hence, on the possibility of the alien chromosome to recombine with one of its homoeologous counterparts by crossing over. When parent can be combined in a sexual cross, such as Wheat and Rye, Maize and Pennisetum, Festuca and Lolium, crossovers between homoeologous chromosomes are not rare, which may reveal recombinant chromosomes, even in the first backcross generations. In the case of distant parental species when somatic hybridization and embryo rescue are required for the production of monosomic additions, the alien chromosome generally fails to synapse and / or recombine at meiotic prophase I, remains lagged behind in the equatorial plane at anaphase-I and may get lost at later stages. Its univalence may result in centromere breakage and thus producing monotelosomic and isotelosomic additions in the progeny. In the derived disomic additions, the two alien chromosomes will pair and form chiasmata, resulting in the formation of univalents and hence, in unbalanced gametes. The extent of crossover recombination between the homoeologous in the monosomic additions depends primarily on the genetic relation between the parental species, but can also vary between the different alien chromosomes in the monosomic additions and in different genetic backgrounds. When homoeologous recombination in the interspecific hybrids is rare, it will be even more seldom or entirely absent in the derived backcross generations due to high preferential pairing. Additional causes for suppression of crossover and recombination are the heterochromatic pericentromeric chromosome regions and heterozygosity for (small) chromosomal rearrangements like duplications, inversions and translocations or pericnetromeric regions. Geneticists have undertaken several strategies to improve homoeologous recombination. When genotypes lacking the Ph1 locus that controls suppression of homoeologous recombination are often used in breeding programmes for transferring desirable genes from rye, and other related cereal species to wheat. More recent studies on the mode of Ph1 have shown that the locus not directly control homoeologous recombination suggesting the need for an entirely new approach to the introgression of alien genetic variation into wheat. Very little is known about other genetic systems controlling synapsis and recombination between homoeologous chromosomes in plant polyploids. In Lolium amphidiploids diploidizing genes carried by A-chromosomes and supernumerary B-chromosomes controlled bivalent formation. This system has so far not been applied to introgressive hybridization programmes. Diploid plants with one chromosome replaced by its homoeologous can be easily obtained in backcross offspring families of interspecific hybrids and monosomic additions. The heteromorphic (homoeologous) bivalents in such monosomic substitutions generally demonstrate higher levels of crossover recombination between the alien [85] chromosomes and its homoeologous counterpart than in the corresponding monosomic addition, and are therefore more appropriate for producing recombination chromosomes. A large series of substitution lines have been produced from Festuca pratensis x Lolium perenne hybrids and used GISH to establish the sites of homoeologous crossover events in the recombinant chromosomes. The range of substitution lines each with different recombinant chromosomes provided excellent material for physical mapping the introgressed F. pratensis chromosomes segments and comparing genetic and physical maps for the molecular markers on these chromosomes. Gamma-ray irradiation of monosomic oat-maize additions was used by produce radiation hybrid lines containing centric maize chromosome fragments or oat-maize translocation chromosome. These lines, which lack one or more parts of the original maize chromosome, allow mapping of molecular markers on a sub-chromosome level of the maize genome. Transmission rates are generally far higher through the female than the male line, and greatly vary between and among the addition sets, as was found for Oryza australiensis to rice, Solanum lycopersicoides to tomato and onion to A. fistulosum. In the monosomic additions of Solanum lycopersicoides chromosomes to tomato, transmission for chromosome 10 was 24%, whereas for chromosome 6 no transmission at all could be recorded. In an extensive study transmission values amounted 0-32% for chromosome 9, whereas that of chromosome 6 varied from 14-88% between the different families. Transmission rate can vary considerable for the different alien chromosomes and is generally higher for the larger chromosomes. A recent study encompassing three consecutive generations in the Brassica rapa monosomic additions in Raphanus sativs, revealed average transmission rates ranging from 26% to 44%. Applications and Scientific impact of additions and substitutions These are as follows : 1. Introgressive hybridization 2. Gene/markers localization 3. Construction of chromosome specific libraries 4. Heterologous gene expression 5. Organization of the alien chromosomes 6. Studies on chromosome pairing of alien chromosome pairs using GISH 7. Use in genomics and physical mapping 5.3.5 Genetic Basis of Inbreeding and Heterosis Heterosis is a term used in genetics and selective breeding. The term heterosis, also known as hybrid vigour or outbreeding enhancement, describes the increased strength of different characteristics in hybrids; the possibility to obtain a genetically superior individual by combining the virtues of its parents. Heterosis is the opposite of inbreeding depression, which occurs with increasing homozygosity. The term often causes controversy, particularly in terms of the selective breeding of domestic animals, because it is sometimes believed that all crossbred plants or animals are genetically superior to their parents; this is true only in certain circumstances: when a hybrid is seen to be superior to its parents, this is known as hybrid [86] vigor. When the opposite happens, and a hybrid inherits traits from their parents that makes them unfit for survival, the result is referred to as outbreeding depression. Typical examples of this are crosses between wild and hatchery fish that have incompatible adaptations. Genetic basis of heterosis - When a population is small or inbred, it tends to lose genetic diversity. Inbreeding depression is the loss of fitness due to loss of genetic diversity. Inbred strains tend to be homozygous for recessive alleles that are mildly harmful (or produce a trait that is undesirable from the standpoint of the breeder). Heterosis or hybrid vigor is the tendency of outbreed strains to exceed both inbred parents in fitness. Selective breeding of plants and animals, including hybridization, began long before there was an understanding of underlying scientific principles. In the early 20th century, after Mendel's laws came to be understood and accepted, geneticists undertook to explain the superior vigor of many plant hybrids. Two competing hypotheses, which are not mutually exclusive, were developed: Dominance hypothesis. The dominance hypothesis attributes the superiority of hybrids to the suppression of undesirable recessive alleles from one parent by dominant alleles from the other. It attributes the poor performance of inbred strains to loss of genetic diversity, with the strains becoming purely homozygous at many loci. The dominance hypothesis was first expressed in 1908 by the geneticist Charles Davenport (Figure 5.49). Overdominance hypothesis. Certain combinations of alleles that can be obtained by crossing two inbred strains are advantageous in the heterozygote. The overdominance hypothesis attributes to heterozygote advantage the survival of many alleles that are recessive and harmful in homozygotes. It attributes the poor performance of inbred strains to a high percentage of these harmful recessives. The overdominance hypothesis was developed independently by Edward M. East (1908) and George Shull (1908). Figure 5.49 Figure- Genetic basis of heterosis. Dominance hypothesis. (Scenario A) - Fewer genes are under-expressed in the homozygous individual. Gene expression in the offspring is equal to the expression of the fittest parent. Overdominance hypothesis (Scenario B) - Overexpression of certain genes in the heterozygous offspring. (The size of the circle depicts the expression level of gene A) Dominance and overdominance have different consequences for the gene expression profile of the individuals. If over-dominance is the main cause for the fitness advantages of heterosis, then there should be an over-expression of certain genes in the [87] heterozygous offspring compared to the homozygous parents. On the other hand, if dominance is the cause, fewer genes should be under-expressed in the heterozygous offspring compared to the parents. Furthermore, for any given gene, the expression should be comparable to the one observed in the fittest of the two parents. Historical retrospective - Population geneticist James Crow, who in his younger days believed that overdominance was a major contributor to hybrid vigor, has undertaken a retrospective review of the developing science. According to Crow, the demonstration of several cases of heterozygote advantage in Drosophila and other organisms first caused great enthusiasm for the overdominance theory among scientists studying plant hybridization. But overdominance implies that yields on an inbred strain should decrease as inbred strains are selected for the performance of their hybrid crosses, as the proportion of harmful recessives in the inbred population rises. Over the years, experimentation in plant genetics has proven that the reverse occurs, that yields increase in both the inbred strains and the hybrids, suggesting that dominance alone may be adequate to explain the superior yield of hybrids. Only a few conclusive cases of overdominance have been reported in all of genetics. Since the 1980s, as experimental evidence has mounted, the dominance theory has made a comeback. Crow writes, "The current view ... is that the dominance hypothesis is the major explanation of inbreeding decline and the high yield of hybrids. There is little statistical evidence for contributions from overdominance and epistasis. But whether the best hybrids are getting an extra boost from overdominance or favorable epistatic contributions remains an open question." Inbreeding depression is usually defined as the lowered fitness or vigour of inbred individuals compared with their non-inbred counterparts, observed in many (but by no means all) organisms. Its converse is heterosis, the 'hybrid vigour' manifested in increased size, growth rate or other parameters resulting from the increase in heterozygosity in F1 generation crosses between inbred lines. Problems arise in defining fitness and vigour. So long as we are discussing populations of wild plants, we have the concept of Darwinian fitness: if individuals of one genotype survive to breed more than others, then that genotype confers greater fitness. Fitness is an observed quantity that integrates the effect of all characters that influence the ability of the organism to live and reproduce. As natural selection can only act to increase the frequency of an allele in proportion to the extent to which that allele increases fitness, it was predicted (Falconer, 1989) that the amount of heritable variance for a trait would be inversely proportional to its effect on the organism's fitness. In other words, there would be more inheritable variance in relatively 'neutral' characters than in ones that increased the organism's viability or fecundity. Studies on three very different animal species tended to confirm this prediction regarding individual characters (Kruuk et al., 2000), but the measurement of overall fitness in populations remains problematical. Vigour is another vague concept: but to most growers it is synonymous with rate of growth or biomass accumulation. In an annual, this is well correlated with production of grain, flowers or fruit since these plants maximise their investment in seed production. But a perennial may increase its growth rate only to reinvest these resources in further vegetative reproduction; examples are bulbil watsonia and crow garlic that have become weeds by the strategy of vegetative reproduction with little or no seeding. [88] Vigour has sometimes been used as a measure (or even a near-synonym) of fitness in discussions of heterosis. But a plant can be too vigorous for its own good: it can become structurally unstable and vulnerable to destruction by mechanical stress or dependent on a higher and more reliable supply of water and nutrients than the environment can guarantee. And an individual plant whose accelerated growth rate causes it to flower long before the rest of its population may - if it can self-pollinate - have increased its fitness. But its fitness will be reduced to zero if it's a self-incompatible annual. The relation between fitness and vigour in a wild population is not a straight line, but a curve with a maximum: In the language of games theory, natural selection might be said to favour strategies that lead to a minimax outcome - the optimum attainable in the real world rather than the theoretical maximum. Darwinian fitness is not a single character, but the outcome of every trait in an individual that influences the contribution it makes to the next generation. In populations of wild plants, heterosis for a range of quantities presumed to have a strong influence on fitness is more meaningful than heterosis for vigour alone. For example, Buza et al. (2000) measured germination rate, seedling survivorship and vegetative growth rate in their attempt to quantify inbreeding depression in relict Swainsona populations. Balanced polymorphisms in populations are often due to the heterozygotes having a higher fitness than either homozygote (Ford, 1964). Possible explanations of this heterosis for overall fitness include: 1. The formation of homozygotes in which alleles with more or less recessive deleterious effects become fully expressed, but had been masked by the dominants in heterozygotes. Recessive and mildly deleterious mutations are common, and if located close to a beneficial allele they will increase in frequency as it does, being "sheltered" from selection in the heterozygotes. By the time a beneficial mutant is common enough for its homozygotes to occur in significant numbers in the population, it may be linked with enough harmful recessives to make these homozygotes less fit than the heterozygote (Fisher, 1927; Ford, 1965). In this case, heterosis is said to be caused by dominance complementation. This may also be described loosely as epistasis of the harmful genes over the beneficial ones - but only if that term is used in the broadest sense. 2. Overdominance - the fitness of the heterozygote per se is higher than either homozygote. This could arise because genes normally have pleiotropic effects, contributing too many measurable traits of the plant. If a mutation is selected for a positive effect on fitness or some desired trait, its other effects are likely to be deleterious. Because a mutant allele at first spreads through a population via [89] heterozygotes, selection will tend to modify and optimise its action by making its beneficial effects dominant and its deleterious effects recessive (Gustafsson, 1950; Sheppard, 1953). This hypothesis has been used to explain balanced polymorphisms, for example in the case of Dactylis glomerata (Apirion & Zohary, 1961). In both explanations, a beneficial dominant effect is associated with a deleterious recessive effect. Whether these correlated effects are due to the same gene or to closely linked but separate genes may be a difficult point to decide. Both overdominance and dominance complementation may contribute in varying degrees to observed cases of heterosis. But evidence for actual instances of overdominance remains scarce. Three predictions of the dominance complementation hypothesis suggest how it might be tested: 1. Inbreeding depression due to genes with deleterious recessive effects that are linked to genes with beneficial dominant effects may be removed by selection; but if it is due to overdominance it cannot. 2. A period of inbreeding can 'purge' deleterious recessive alleles from a population since these will have an increased chance of appearing in homozygotes than can be eliminated by selection (Barrett & Charlesworth, 1991). 3. If two strains of a plant are each selected for heterosis in the F1 produced by crossing them with a third strain, heterosis will also increase in the F1 between these selected strains because they are being purged of deleterious alleles. But if heterosis was due to overdominance, the two strains would converge, since each would be selected for alleles that differed from those of the tester strain at each locus, and their hybrid will show inbreeding depression instead of heterosis. Wild plants that are normally inbreeders, or have a mixed inbreeder/outbreeder strategy, might be expected to have a genetic architecture that minimises inbreeding depression. In one experiment confirming this, the inbred seeds from cleistogamous flowers of Viola species had only slightly lower fitness than the outcrossed seeds from chasmogamous flowers on the same plants (Berg & Redbo-Torstensson, 1999). Inbreeding depression is caused mainly by deleterious recessive alleles in both Mimulus guttatus (an outbreeder) and the closely related inbreeder M. micranthus (Dudash & Carr, 1998). Natural selection in experimentally inbred lines of M. guttatus purged the few alleles with major deleterious effects, but numerous other alleles, each with a small effect, accounted for almost all the depression (Willis, 1999). Cultivated plants - But most of the practical interest in heterosis centers on cultivated plants, and when plant breeders say 'fitness' they really mean 'crop yields'. It is more useful to speak of heterosis for a particular statistic of the crop: for example, leaf area or seed size. Heterosis is also modified by the interactions between genotype and environment in cultivation. Hybrid sorghum can show heterosis for yield; but this effect varies widely between trials conducted at sites differing in seasonal water supply (Chapman et al., 2000), so that it is more meaningful to characterise a particular hybrid line as showing heterosis for yield at a specific locality or under certain environmental conditions. In practice, yield and other measurable traits desired by breeders are often correlated with traits that increase the fitness of the plant: increased efficiency of metabolism, [90] photosynthesis etc. As a convenient approximation, we can speak loosely of 'beneficial' and 'deleterious' effects. Heterosis for size, vigour or yield is most evident in outbreeding crops. The classic case is maize; in which heterosis has long been exploited in the production of uniformly highyielding F1 seed in commercial quantities. The definitive experiment by Sprague (1983) showed that this instance of heterosis is due wholly or mainly to dominance complementation. Over many years, two maize populations were each selected for increased yield in the hybrids produced by crossing them with another inbred 'tester' strain. F1 hybrids between the two selected strains showed increased yield, as predicted by the dominance complementation hypothesis. They also produced increased yields in hybrids with other testers. If dominance complementation is the only factor involved in heterosis for yield, the high productivity of hybrid maize may have been matched by open-pollinated cultivars if enough work had been invested in their development. However, Fu & Dooner (2002) have reported the surprising discovery that maize cultivars differ in the set of genes they carry: some loci in one cultivar lack corresponding alleles in another line. In this case, any component of the heterosis effect due to complementation between different loci could never be fixed in an open-pollinated line, since these loci would remain on separate chromosomes. Maize may be a special case among crop plants - both because its genome has been doubled in size by retrotransposon activity that produced multiple paralogs of some genes, and also because it has been more radically changed by the domestication process than have other crops such as rice. Xiao et al. (1995) demonstrated that overdominance is not a major cause of heterosis for yield in a cross between the two subspecies of rice, because there was no correlation between most traits and overall genome heterozygosity, heterozygotes were never superior to both homozygotes in analysis of quantitative trait loci, and some F8 inbred lines were actually superior to the F1 for all traits evaluated. Trials support the hypothesis that heterosis in wheat is due to dominance complementation, with linkage and interaction of alleles (Pickett & Galwey, 1997) - to the loss of those who would like to exploit hybrid wheat as they have done hybrid maize. In some other outbreeding crop plants, such as cucumbers (Cramer & Wehner, 1999), crossing inbred lines rarely leads to heterosis. The hypothesis of dominance complementation is also supported by evidence that polyploids are to some extent buffered against inbreeding depression because they have additional copies of each gene (Bingham et al., 1994; Husband & Schemske, 1997). A third explanation of heterosis - An alternative theory was proposed by Milborrow (1998). He suggested that growth of a plant may be limited by the genes that regulate certain metabolic pathways down to lower levels than the maximum possible. Heterozygotes may partially escape this regulation because they have two slightly different alleles for these genes, allowing greater flow on these pathways. This is not overdominance; but, like the overdominance hypothesis, it predicts that heterozygotes have an inherent advantage in vigour that cannot be duplicated by any amount of selection in open-pollinated homozygous lines. The most interesting part of Milborrow's theory is the implication as to why there are so many "weak" alleles in a population to start with. These are not sublethal mutants that [91] have accumulated under the protection of closely linked genes that are strongly selected, but a necessary part of the genetic adaptation of the population. Natural selection will maintain weak alleles because an individual with all the strong alleles will be vigorous but of lower fitness. Therefore, selection maintains the average individual in the population at that level of vigour which maximises fitness. This may explain observations that the level of heterosis in wild populations depends on their habitat, sometimes termed habitat-dependent heterosis (Lloyd, 1980). The fitness advantage of heterozygotes is often greater under more severe conditions (e.g.- Dudash, 1990). Genetic variation allows more vigorous and less vigorous individuals to exist and take advantage of changing conditions under which they may have the advantage. This is a mixed strategy; and as Von Neumann & Morgenstern (1953) showed, mixed strategies are necessary to achieve minimax outcomes. But artificial selection of plants in cultivation aims to maximise desired parameters yield, size etc - within an artificial, optimum environment. It may therefore be possible to purge the alleles that cause inbreeding depression from cultivated lines. (1) Genetic basis of heterosis and inbreeding depression in rice (Oryza sativa L.) - The genetic basis of heterosis was studied through mid-parent, standard variety and better parent for 11 quantitative traits in 17 parental lines and their 10 selected hybrids in rice (Oryza sativa L.). The characters were plant height. days to flag leaf initiation, days to first panicle initiation, days to 100% flowering, panicle length, flag leaf length, days to maturity, number of fertile spikelet/panicle, number of effective tillers/hill, grain yield/10-hill, and 1000-grain weight. In general the hybrids performed significantly better than the respective parents. Significant heterosis was observed for most of the studied characters. Among the 10 hybrids, four hybrids viz., 17A×45R, 25A×37R, 27A×39R, 31A×47R, and 35A×47R showed highest heterosis in 10-hill grain yield/10-hill. Inbreeding depression of F2 progeny was also studied for 11 characters of 10 hybrids. Both positive and negative inbreeding depressions were found in many crosses for the studied characters, but none was found significant. Selection of good parents was found to be the most important for developing high yielding hybrid rice varieties. (2) Overdominant epistatic loci are the primary genetic basis of inbreeding depression and heterosis in rice. I. Biomass and grain yield - To understand the genetic basis of inbreeding depression and heterosis in rice, main-effect and epistatic QTL associated with inbreeding depression and heterosis for grain yield and biomass in five related rice mapping populations were investigated using a complete RFLP linkage map of 182 markers, replicated phenotyping experiments, and the mixed model approach. The mapping populations included 254 F(10) recombinant inbred lines derived from a cross between Lemont (japonica) and Teqing (indica) and two BC and two testcross hybrid populations derived from crosses between the RILs and their parents plus two testers (Zhong 413 and IR64). For both BY and GY, there was significant inbreeding depression detected in the RI population and a high level of heterosis in each of the BC and testcross hybrid populations. The mean performance of the BC or testcross hybrids was largely determined by their heterosis [92] measurements. The hybrid breakdown (part of inbreeding depression) values of individual RILs were negatively associated with the heterosis measurements of their BC or testcross hybrids, indicating the partial genetic overlap of genes causing hybrid breakdown and heterosis in rice. A large number of epistatic QTL pairs and a few maineffect QTL were identified, which were responsible for >65% of the phenotypic variation of BY and GY in each of the populations with the former explaining a much greater portion of the variation. Two conclusions concerning the loci associated with inbreeding depression and heterosis in rice were reached from our results. First, most QTL associated with inbreeding depression and heterosis in rice appeared to be involved in epistasis. Second, most (approximately 90%) QTL contributing to heterosis appeared to be overdominant. These observations tend to implicate epistasis and overdominance, rather than dominance, as the major genetic basis of heterosis in rice. 5.3.6 Exploitation of Hybrid Vigour Hybrid vigour, which overcomes “inbreeding depression”, and which is a way to increase genetic diversity and stress resilience in a crop, is an important breeding concept. It is especially relevant to canola in Australia. Theoretical and experimental understanding of hybrid vigour in several crops is an extremely sound reason to be confident that GM canola hybrids are more profitable than conventional varieties. There are many silly untrue assumptions floating around the community about commercial seed hybrid varieties - eg that commercial seeds are sterile or do go grow without fertilizer, that it is worth going back to explain what hybrids are. The story starts with maize (Indian corn) in America. In 1908 the U.S. plant breeder G. H. Shull reported the results of crossing maize plants from two different inbred lines. The cross between two highly homozygous lines produced heterozygous offspring, because the lines were homozygous for different sets of genes. The result was astonishing. The two inbred lines had each produced about 20 bushels of maize per acre in the last crop. Their outbreed offspring quadrupled this yield, to 80 bushels per acre! This unanticipated strength in the heterozygous outcross was called hybrid vigor; and such hybrids have played an important role in increasing maize yield. Hybrid vigour can be fully exploited when plants are cross pollinators rather than selfpollinators. Thus exploitation of hybrid vigour is not possible with self-pollinating wheat and barley, but is possible with maize, sunflower and sorghum. Although the commercial breeding of hybrid maize has focussed on yield improvements, there are interesting complexities in the actual traits that allow this achievement. Modern hybrids are highly stress tolerant plants, and they can be seeded at high plant densities per acre. Scientific publications of Duvick and Cassman (1999) and Tollenaar et al. (1994) show that modern maize hybrids have increased stress tolerance rather than high yield potential. Because of the spectacular potential of hybrid vigour demonstrated with maize, plant breeders have tried to develop artificial methods to allow practical cross-breeding with other crops. The Chinese have developed non-GM artificial methods that generate rice hybrids, and Bayer market GM canola hybrids. The phenomenon of heterosis and its importance in crop improvement have been known since decades. Recognizing that commercial exploitation of hybrid vigour would, [93] however, depend on economic viability of hybrid seed production. Varied genetic and non-genetic approaches for selective emasculation of one of the parents have been evolved and used. Cytoplasmic-genetic male sterility is the worldwide used system among them Yet, its cumbersome nature, labour expansive seed production method and restircted parental choice warranting an alternative has led to the discovery in some of the cereal crops. Physiological characterization of various sources of photoperiod and temperature sensitive genic male sterility (TGMS/PGMS) in a model crop like rice has helped group them into 3-4 categories on the basis of their critical sterility and fertility points, while genetic analysis has revealed it to follow simple mendelian mode of inheritance and 2-3 putative genes (pms1, pms2, tms1, tms2, tms3) to govern them. Development of stable and agronomically superior PGMS and TGMS lines using promising gene sources and identification of ideal locations/seasons for hybrid seed production and multiplication of parental TGMS or PGMS line has facilitated evolution of two-line hybrids of rice for commercial planting. It is hoped that more precise understanding of the physiology, biochemistry, genetics and molecular bases of these environment sensitive male sterility sources would greatly help extent this innovative system of hybrid breeding to a wide range of crop plants in the coming year. Exploitation of hybrid vigour on a commercial scale involves extra expenditure in the production of hybrid seeds. In a study for developing the hybrid of brinjals an increase in yield of hybrids was in the order of 50 to 100 percent. Over the parental means and this compensate for the extra cost of production. This is a sufficient reason to advocate the use of hybrid seeds of brinjals for commercial growing in India. In Tamilandu region of India it is recently proof that exploitation of hybrid vigour is very successful which was done for developing rice hybrid (Oryza sativa L.) through green manure and leaf colour chart (LCC) based nitrogen application. 5.4 LET US SUM UP Type II restriction enzymes cut DNA at specific base sequences. Some restriction enzymes make staggered cuts, producing DNA fragments with cohesive ends; others cut both strands straight across, producing blunt-ended fragments. There are fewer long recognition sequences in DNA than short sequences. Eukaryotic DNA comprises three major classes: unique-sequence DNA, moderately repetitive DNA, and highly repetitive DNA. Unique-sequence DNA consists of sequences that exist in one or only a few copies; moderately repetitive DNA consists of sequences that may be several hundred base pairs in length and is present in thousands to hundreds of thousands of copies. Highly repetitive DNA consists of very short sequences repeated in tandem and present in hundreds of thousands to millions of copies. When double-stranded DNA is heated, it denatures, separating into singlestranded molecules. On cooling, these single-stranded molecules pair and re-form double-stranded DNA, a process called renaturation. A C0t curve is a plot of a renaturation reaction. [94] Physical-mapping methods determine the physical locations of genes on chromosomes and include deletion mapping, somatic-cell hybridization, in situ hybridization, and direct DNA sequencing. Additions can basically be subdivided into two classes, each with their own properties and applications: (i) Class 1 in which homoeologous recombination between the alien chromosome and one of its homoeologous counterparts occurs at reasonable frequency. These additions produce various recombinants and so are particularly suitable for introgressive hybridization, for comparing physical and genetic maps, and for studying the effect of individual chromosomes on crossover (chiasma) distribution and frequencies. More knowledge is required especially about the genes controlling homoeologous recombination, chiasma distribution and unreduced gamete formation, and for genetic programmes aiming at producing monosomic substituions in order to force homoeologous chromosomes to pair and synapse. (ii) In class 2 homoeologous recombination does not occur in a normal genetic background, i.e. not disturb in genes controlling meiotic recombination, homoeologous recombination or unreduced forms of meiosis (like first and second division restitution). This group of interspecific aneuploids are less attractive3 for breeders as long as introgressive hybridization cannot be achieved by homoeologous recombination, but can provide significant information for various cytogenetic and genomic studies including chromosome disposition, molecular organization of repetitive and single copy sequences, heterologous gene expression and starting material for flow sorted or microdissected alien chromosome samples for chromosome specific DNA sequences. By virtue of its several advantages over the traditional cytoplasmic-genetic male sterility system, the environment sensitive genic male sterility would be an ideal alternative for effective exploitation of hybrid vigour in rice and other crops. Nevertheless, sustainable use of this system on a commercial scale would require further refinement of the system, keeping in view the impending global warming, the effect of temperature-light interaction and the influence of not very well understood Fd and other minor gene complexes on the stability of fertility-sterility reversion. The following are some of the areas, warranting more in-depth study: o Streamlining of breeding methodology for development of PGMS and TGMS lines. o Precise characterization of all the usable PGMS and TGMS lines derived from different sources for the benefit of users in the seed industry. Development and use of molecular markers for improving breeding/selection efficiency. o Development of EGMS system for developing more productive intersubspecific hybrids. [95] 5.5 CHECK YOUR PROGRESS NOTE: (1) Write your answer in the space given below. (2) Compare your answer with the one given at the end of this unit. ( א1) Fill in the blanks : (a) The DNA of the nucleus with its associated proteins is called …………….. (b) The cells of some …………… may contain 40 times more DNA than those of humans. (c) ………. restriction enzymes recognize specific sequences and cut the DNA at nearby sites, usually about 25 bp away. (d) …………………… technique was developed using highly radioactive DNA probes, which were detected by radiography. (e) HAT selection, which stands for ……………., ……………, and …………., three chemicals that are used to select for hybrid cells. (f) Scientists who study chromosomes are known as ……………………. ( א2) Write the answer of following questions : (a) Describe the multigene family in bacteria and insect. (b) Explain how production of alien addition/substitution is useful. 5.6 CHECK YOUR PROGRESS : THE KEY ( א1) (a) Chromatin (b) Salamanders (c) Type III (d) In situ hybridization (e) Hypoxanthine, aminopterin and thymidine (f) Cytogeneticists ( א2) (a) see section 5.2.5 (b) see section 5.3.4 [96] 5.7 ASSIGNMENT Collect the information about Indian plants in which alien gene transfer methods have been applied successfully. 5.8 REFERENCES Our courteous thanks to following two authors/publishers for preparing the various section of this chapter:B. Alberts et al., ‘Molecular Biology of the Cell’: 4th Ed. (2002). Garland. Benzamin A. Pierce, ‘Genetics : A Coneptual Approach’ Other helping resources are as follows:Scalenghe F, Turco E, Edstrom JE, Pirrotta V, Melli M. 1981. “Microdissection and cloning of DNA from a specific region of Drosophila melanogaster polytene chromosomes”. In Chromosoma. 82(2):205-16. Mapping Genomes - From Genomes by T. A. Brown, 2002. Linkage disequilibrium maps and association mapping - J. Clin. Invest., 2005. Linkage Disequilibrium in Humans: Models and Data - Am. J. Hum. Genet., 2001. Cytogenetic and Molecular Studies on Tomato Chromosomes using Diploid Tomato and Tomato Monosomi Additions in Tetraploid Potato From Song-Bin Chang. Linkage Disequilibrium and the Search for Complex Disease Genes - Genome Research, 2000. Gray JW, Cram LS. 1990. Flow karyotyping and chromosome sorting. In: Flow Cytometry and Sorting. New York: Wiley Liss, 503-529. Heslop-Harrison JS, Schwarzacher T. 1995. Flow cytometry and chromosome sorting. In: Fukui K, ed. Plant Chromosomes: Laboratory Methods. CRC, Baton Rouge. In press. Dewey, D.R. 1984. The genomic system of classification as a guide to intergeneric hybridization with the perennial Triticeae. In J.P. Gustafson, ed. Gene manipulation in plant improvement, p. 209-279. New York, NY, USA, Plenum Press. Allen Klinger, member, IEEE, Arnold kochman, and Nikitas Alexandridis. 1971. “Computer Analysis of Chromosome Patterns: Feature Encoding for Flexible Decision Making”. In IEEE Transactions on Computers, Vol. C-20, No. 9 Pawley JB (editor) (2006). Handbook of Biological Confocal Microscopy, 3rd ed., Berlin: Springer. ISBN 038725921x. Hybrid Vigor inn Plants and its Relationship to Insect Pollination - a section from Insect Pollination Of Cultivated Crop Plants by S.E. McGregor, USDA ****** [97]