Template for Electronic Submission to ACS Journals
advertisement

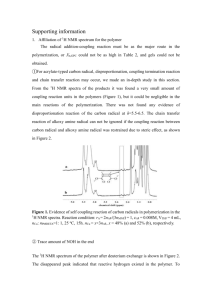
Solid Phase Modular Synthesis of Polymacromer Brushes and Their Isolation as Molecular Products by Photocleavage from the Substrate AUTHOR NAMES Benjamin I. Dach †, ‡, Xia Li †, Nicholas J. Turro †, ‡, and Jeffrey T. Koberstein ‡ ,* AUTHOR ADDRESS † Department of Chemistry and ‡ Department of Chemical Engineering, Columbia University, 3000 Broadway, New York, New York 10027 CORRESPONDING AUTHOR FOOTNOTE E-mail: jk1191@columbia.edu. Tel.: (212) 854-3120. Fax: (212) 854-3054 KEYWORDS Polymacromer, Solid Phase Synthesis, Polymer Brushes, Click chemistry, Photocleavage, Macromonomer mopolymacromers with integral molecular weights can be prepared by multiple addition cycles using a single macromonomer while segmented block copolymacromers of any desired sequence can be prepared using different macromonomers in each cycle. Azide-alkyne click chemistry, via the Hüisgen 1,3-dipolar cycloaddition mechanism, is employed as the coupling chemistry because the alkyne group can be readily protected and subsequently deprotected using relatively mild conditions. The click reaction can be initiated at room temperature with a copper catalyst (CuAAC) or thermally at temperatures as low as 70 °C without catalyst. The high chemoselectivity of the reaction enables the coupling of virtually any macromonomer, regardless of its chemical nature.14-16 The required heterobifunctional macromonomers, comprising one azide end group and a second protected alkyne end group, are prepared by atom transfer radical polymerization (ATRP).17 A limitation of the SPS method in this application is that the harsh reagents required for release of the final polymer product from the surface can potentially degrade the polymer. To circumvent this problem, a photochemical cleavage method is developed for release of the PM product from the substrate under extremely mild and universal conditions. The process for solid phase synthesis of a PM on a silicon wafer (i.e., SiO2 surface) is illustrated in Scheme 1 for four addition cycles of an -azido, -trimethylsilane-protected-alkyne poly(tert-butyl acrylate) [PtBA] macromonomer. The process consists of: ABSTRACT. We report a novel solid phase method for the sequential coupling of heterobifunctional macromonomers to form a new class of polymeric materials that we refer to as polymacromers. Starting from an azide functional substrate, azido, -protected-alkyne macromonomers are added step-by step by thermally initiated click reactions. After each addition step, the terminal alkyne group is deprotected to allow addition of another macromonomer. The use of highly chemoselective click chemistry for the coupling reactions allows virtually any macromonomer to be employed, regardless of its chemical nature. The polymacromers may be left as polymer brushes on the substrate, or can be isolated as molecular products by photocleavage of an ortho-nitrobenzyloxycarbonyl (NBOC) linkage incorporated at the substrate interface. The method is illustrated by forming homopolymacromers by sequential coupling of poly(tert-butyl acrylate) macromonomers. The results of characterization of the polymacromer bushes by ellipsometry, contact angle analysis and x-ray photoelectron spectroscopy, and direct measurements of the molecular weights of isolated products by gel permeation chromatography demonstrate that polymacromers can be prepared with a coupling efficiency approaching 100%. TEXT “Solid phase synthesis” (SPS) is a method by which molecules are bound to a solid surface (commonly a bead) and then synthesized step-by-step in a reactant solution. Compared to conventional solution synthesis, it is easier to remove excess reactant and by-products because the desired product is covalently bound to the solid surface. SPS is a modular method that generally employs building blocks with two complementary functional groups, one of which is protected. The synthetic pathway is controlled by the order of addition (and subsequent deprotection) of these building blocks to a solid substrate that is modified to present unprotected surface functional groups. SPS has become an important method for the synthesis of peptides,1-3 DNA4-8 and other biopolymers9,10 for which a certain asymmetric predetermined sequence is desirable, and has also been employed recently in combinatorial chemistry.11-13 We describe herein a new SPS method through which a specific, asymmetric sequence of macromonomers can be joined to produce a new class of polymers, polymacromers (PM). Ho- 1. attachment of an amine terminated primer (1, Chart 1) to the SiO2 surface; 2. reaction of the amine terminus with a nitrobenzyloxycarbonyl (NBOC) photo-cleavable linker possessing an alkyne terminus (2, Chart 1); 3. click reaction of the alkyne terminus with an -azide, -TMS-protected-alkyne macromonomer (M1, 3, Chart 1); 4. removal of the alkyne protecting group and repetition of step 3 to add a second polymer (M2, Chart 1); 5. repetition of the cycle to add two more polymer units (M3 and M4, Chart 1); 6. photocleavage and characterization of the final product PM1-4 (4, M1-M2-M3-M4, Chart 1). The chemistry involved in Scheme 1 is shown explicitly in Scheme 2. It is evident that SPS of PMs produces first a polymer 1 brush tethered by its end to the substrate. If desirable, the PM can subsequently be released from the substrate and isolated as a molecular polymer product by applying the photocleavage step. Primer mer synthesis and synthesis of the photocleavable linker are described in the supporting information. NBOC: photocleavable linker to M1 Scheme 2. Reaction scheme for solid phase synthesis of a PtBA PM on a SiO2 surface followed by photochemical cleavage of the product from the surface. Photocleaved PtBA Polymacromer PtBA (M1-M2-M3-M4) Macromonomer Chart 1. Materials used in the step-by-step synthesis of a PM. To evaluate the efficiency of the SPS method, four azido, -TMS-protected PtBA macromonomers were coupled sequentially to silicon wafers as described above. After each cycle, the resultant polymer brushes and the product isolated by photocleavage were both characterized by a battery of methods: X-Ray photoelectron spectroscopy (XPS), water contact angle (WCA), ellipsometry (ELP) and Gel Permeation Chromatography (GPC). The results of all of these independent methods confirm successful step-by-step covalent attachment of PtBA macromonomers to form the final PM product (4, M1-M2-M3-M4, Chart 1). The XPS analysis (PHI 5500 spectrometer, Al Kα monochromator X-ray source at 15 kV and 23.3 mA, 45° take off angle, penetration depth of 4.6 nm) of the surface was performed (Figure 1) at each addition step in the overall process, M 1, M1-M2, M1-M2-M3 and M1-M2-M3-M4 and after the photocleavage. Controls of the bare SiO2 surface and the surface primed with AHAMTES were also characterized. The results are summarized in Figure 1. The qualitative features of the data are: (1) compared to the bare SiO2 surface, the carbon signal (C 1s, 285 eV signal) increases as the primer and subsequent four layers of PtBA are attached to the surface; (2) upon photocleavage, the signals are similar to that of the surface with the primer. These results are consistent with the successful sequential step-by-step additions of four PtBA macromonomers and the complete removal of the final tetramacromer by photochemical cleavage. The results of the WCA measurements are shown in Figure 2. The hydrophilic SiO2 surface yields a low water contact angle of ~ 5°. Upon adding the less hydrophilic layers of primer, NBOC and macromonomers, the CA monotonically increases and approaches a limiting value of 90°, consistent with the value for pure PtBA.21 Upon photocleavage of the polymer film, the contact angle returns to a value close to that found for the primer. ELP measurements (ALPHA-SE® J.A. Woollam Co., Inc., USA. 70° fixed angle of incidence, Cauchy model) of the film thicknesses provide a quantitative means to evaluate the efficiency of the overall SPS process (Figure 3). The film thickness increases upon addition of the primer, the NBOC and the four PtBA macromonomers as expected. green = primer. yellow = NBOC photocleavable linker. Scheme 1. Schematic description of the solid phase synthesis of a four-step polymacromer on a silicon wafer and isolation of the product by subsequent photocleavage. Silicon wafers (prime grade N-doped [100] purchased from Addison Engineering) were cleaned with piranha solution, washed with Millipore distilled water and cut into 1 in x 1 in substrates. The primer layer, (aminohexyl)aminomethyltriethoxysilane (AHAMTES) was deposited by dipping the substrates into a toluene solution of 1 (Chart 1) and the surface was functionalized with the photocleavable alkyne functionality according to Scheme 2. -azido, -TMS-protected-alkyne PtBA macromonomers (MW = 10,000), prepared by atom transfer radical polymerization18 were spin coated onto the alkyne functional substrates from toluene and were subsequently heated to 110 °C for 18 hrs to effect a thermal click reaction.19 The terminal TMS groups were then removed by treatment with a solution of MeOH:DCM (1:10) with excess K2CO3 for 2 hr.20 A total of four spin coating/deprotection/thermal click cycles were used to prepare the four step PtBA polymacromer, which was photocleaved from the surface using UV light at a wavelength of 360 nm. Details regarding the solid phase synthesis procedure, macromono- 2 for the surface with just the primer. Film thicknesses determined by X-Ray reflectivity measurements, to be reported in a paper to follow, agree well with the ellipsometric thicknesses. The efficiency of coupling can be probed in more depth by analyzing polymacromers that have been isolated by cleavage from the substrate. GPC analyses (Knauer GPC system with a Knauer K-2301 refractive index detector, three Polymer Laboratories 5 μm particle size PLgel columns, linear polystyrene calibration standards ranging in molecular weight from 580-377,400 Da, THF eluant with a flow rate of 1.0 mL/min at room temperature) of isolated products are shown in (Figure 4). The signal quality is low because each silicon substrate yields only a limited amount of product however it is evident that the molecular weight increases in integral multiples of the number of macromonomers added, consistent with the linear behavior found in the ellipsometry data. Figure 1. XPS spectra of bare silica, silica-amine, silica-NBOC, silica-PtBA4, and silica substrate after photocleavage. Figure 4. GPC traces of the PtBA homopolymacromers produced by photocleavage after SPS: a) M1, b) M1-M2, c) M1-M2-M3 and d) M1-M2-M3-M4. The dotted line is the GPC trace of the original PtBA macromonomer. Figure 2. WCA measurements on bare, amine, NBOC, 1 PtBA macromonomer, 2 PtBA macromonomers, 3 PtBA macromonomers, 4 PtBA macromonomers, and photocleaved substrates (point on far right). The GPC traces do not evidence substantial signals for residual products from preceding cycles indicating that the conversion of the click reactions for each cycle is nearly 100%, consistent with the findings from the ellipsometric measurements. These results, which are completely independent of the previously described XPS, WCA and ELP measurements, confirm that the step-by-step SPS process proceeds according to the mechanism depicted in Schemes 1 and 2 to form the anticipated homopolymacromers: M1, M1-M2, M1-M2-M3 and M1-M2-M3-M4. In conclusion, we have developed a robust solid phase method for the step-by-step synthesis of a new class of materials, polymacromers. These interesting new materials are prepared by sequential coupling of -azido, -TMS-protected-alkyne PtBA macromonomers using thermally initiated azide-alkyne click chemistry. The method is illustrated for the preparation of homopolymacromers using the same macromonomer in each addition cycle. The preparation of block copolymacromers will be addressed in a paper to follow. The materials prepared on the substrates are unique polymer brushes, while molecular products may be isolated as linear polymacromers by application of a photocleavage technique. The brushes are unique in that they can be characterized as both “grafting to” and “grafting from”, in the latter case, if a macromonomer is considered equivalent to a monomer. The brush behavior that can be achieved with the new materials spans the entire range of properties possible between traditional “grafting to” and “grafting from” brushes. Figure 3. Measured (filled squares) and theoretical (open circles) film thicknesses (nm) from ellipsometric measurements for bare silicon oxide, AHAMTES, NBOC, PtBA1, PtBA2, PtBA3, PtBA4, and photocleaved substrates (point on far right). The ellipsometric thickness scales linearly with the number of macromonomer addition cycles and compares well with the theoretical thickness calculated assuming 100% conversion for the click reaction for each cycle. The value of the thickness for the photocleaved surface returns to a value close to that ACKNOWLEDGMENTS The authors thank the NSF for generous support of this research through grants IGERT-02-21589, 3 DMR-07-04054 and CHE-07-17518. The project was also sponsored by the Department of the Army, U.S. Army Research Office (JTK, grant 56531 CH). (11) Panicker, R. C.; Huang, X.; Yao, S. Q. Comb. Chem. 2004, 7, 547–556. (12) Mei, Y.; Beers, K. L.; Byrd, H. C. M.; VanderHart, D. L.; Washburn, N. R. J. Am. Chem. Soc. 2004, 126, 3472-6. (13) Hawker, C. J.; Wooley, K. L. Science 2005, 309, 1200-5. (14) Laibinis, P. E.; Whitesides, G. M.; Allara, D. L.; Tao, Y. T.; Parikh, A. N.; Nuzzo, R. G. J. Am. Chem. Soc. 1991, 113, 7152. (15) Senaratne, W.; Andruzzi, L.;,Ober, C. K. Biomacromolecules 2005, 6, 2427. (16) Wang, J. S.; Matyjaszewski, K. Macromolecules 1995, 28, 7901. (17) White, M. A.; Johnson, J. A.; Koberstein, J. T.; Turro, N. J. J. Am. Chem. Soc. 2006, 128, 11356. (18) Johnson, J. A.; Finn, M. G.; Koberstein, J. T.; Turro, N. J. Macromolecules, 2007, 40, 3589–3598. (19) Rengifo, H. R.; Chen, L.; Grigoras, C.; Ju, J.; Koberstein, J. T. Langmuir, 2008, 24, 7450–7456. (20) Chen, L.; Rengifo, H. R.; Grigoras, C.; Li, X.; Li, Z. Ju, J. Koberstein, J. T. Biomacromolecules 2008, 9, 2345. (21) Husemann, M.; Morrison, M.; Benoit, D.; Frommer, J.; Mate, C. M.; Hinsberg, W. D.; Hedrick, J. L.; Hawker, C. J. J. Am. Chem. Soc., 2000, 122, 1844-1845. SUPPORTING INFORMATION PARAGRAPH Experimental details, AFM images, and XPS multiplex spectra are included. This material is available free of charge via the Internet at http://pubs.acs.org. REFERENCES (1) Merrifield, R. B. J. Am. Chem. Soc. 1963, 85, 2149. (2) Orth, R.; Sieber, S. J. Org. Chem. 2009, 74, 8476-9. (3) Falsey, J. R.; Renil, M.; Park, S.; Li, S.; Lam, K. S. Bioconjugate Chem. 2001, 12, 346–353. (4) Yang, L.; Tang, X.; Weisbrod, C. R.; Munske, G. R.; Eng, J. K.; Haller, P. D. von; Kaiser, N. K.; Bruce, J. E. Anal. Chem. 2010, 82, 3556-66. (5) Fodor, S. P. A.; Read, J. L.; Pirrung, M. C.; Stryer, L.; Lu, A. T.; Solas, D. Science 1991, 251, 767–773. (6) Pease, A. C.; Solas, D.; Sullivan, E. J.; Cronin, M. T.; Holmes, C. P.; Fodor, S. P. A. Proc. Natl. Acad. Sci. 1994, 91, 5022–5026. (7) Singh-Gasson, S.; Green, R. D.; Yue, Y.; Nelson, C.; Blattner, F.; Sussman, M. R.; Cerrina, F. Nat. Biotechnol. 1999, 17, 974–978. (8) Hartmann, L.; Krause, E.; Antonietti, M.; Börner, H. G. Biomacromolecules. 2006, 7, 1239-44. (9) MacBeath, G.; Schreiber, S. L. Science 2000, 289, 1760– 1763. (10) Rusmini, F.; Zhong, Z.; Feijen, J. Biomacromolecules 2007, 8, 1775–1789. SYNOPSIS TOC 4