04/01/27 Lab 3: LSU pedigree (HGAR1) continued
advertisement
 continued")
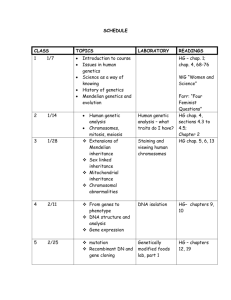
Biomathematics 207B/ Biostatistics 237/ Human Genetics 207B January 27, 2004 Laboratory 3: LSU PEDIGREE (HGAR1) CONTINUED –Is there evidence for a major gene? In this laboratory, we will attempt to exclude alternative explanations to major gene segregation for the observed inheritance of triglyceride levels. We will also test for gene by environment interactions. We will use the AIC (Akaike's Information Criteria) to compare models and we will set the parameter penalty k to 2. In the previous laboratory, we ran our most general model with codominant penetrance function ln( yi ) ageagei' bmibmii' axbagei' bmii' AAGAAi AaGAai ei ei ~ N 0, 2 . GAAi = 1 if A/A and 0 otherwise and GAai = 1 if A/a and 0 otherwise. We assumed Hardy Weinberg Equilibrium among founders so the probability of the A/A group was qA2, the probability of the A/a group was 2qA(1-qA) and the probability of the a/a group was (1-qA)2. For offspring the probability of being in each of the three groups depends on their parents' groups through the transmission probabilities, aa = P(A/a or A/A child | a/a parent), Aa = P(A/a or A/A child | A/a parent), and AA = P(A/a or A/A child | A/A parent). We were then able to reject the sporadic model. If your results were similar to mine, you found that the best model (using the AIC) was the Mendelian model. Specifically, I found an AIC for the general model was -2(-75.6013)+2(11) = 173.2026. My AIC for the Mendelian model was -2(-78.2674)+2(8) = 172.5348. When I reconsidered the sex effect, I still found that the best model does not include sex as a covariate. We will now consider some environmental inheritance models. (1) We first examine a simple environmental model. You will need to add a new "gene" to the model by selecting MODEL, GENES, and ADD TRAIT GENES. Name the model (for example, environmental) and pick an initial q value. This model has three groups A/a, A/A, a/a and the probability of being in a particular group does not depend on ones parents' groups. You may want to use the final values from the general model to help you select initial values. Select ENVIRONMENTAL transmission probabilities. What constraints have you placed on the ’s by specifying the environmental model? AA =_________________, Aa =________________, aa =__________________ (a) Now add the environmental "gene" to the penetrance model. Select MODEL, COVARIATES, ADD UNMEASURED COVARIATES. Select the CODOMINANT model for the inheritance mode. This means that you will be 1 fitting a model with three possible groups, A/A, A/a, and a/a. Choose initial values for the coefficients. There can only be one gene in the model at a time when using the maximum likelihood option so delete the covariate associated with the general model of transmission from the PHENOTYPE and add this newly defined covariate. Again you may have to run this model with several choices of starting parameters. lnL_env=________________________________________ Nparameters_env=___________________________ AICenv = -2lnL_env+2Nenv_________________ (c) What is the best model thus far? (2) Now we want to test a slightly more complicated environmental model. This model allows for difference in trait values between founders and offspring. (a) We now examine the founder, non-founder model. You will need to add a "gene" model by selecting MODEL, GENES, and ADD TRAIT GENES. Name the model (for example, founder) select initial values for qA. Select FOUDER / NON-FOUNDER transmission probabilities. What constraints have you placed on the ’s by specifying the founder/non-founder model? AA=__________________, Aa=_________________, aa=__________________ (b) Now add the founder "gene" to the penetrance model. Select MODEL, COVARIATES, ADD UNMEASURED COVARIATES. Select co-dominant model for the inheritance mode. Choose initial values for the coefficients. Add the specified gene to the phenotype model. Again you may have to run this model with several choices of starting parameters. lnL_fnf=________________________________________ Nparameters_fnf=___________________________ AICfnf = -2lnL_fnf+2Nfnf_________________ (c) What is the best model thus far? (3) Now try a restricted general model. This model has AA=1, Aa (unrestricted), and aa=0. This model is consistent with the presence of more than one gene influencing the trait, or genes and unmeasured environmental influences. 2 (a) Define a trait gene with these transmission probabilities. Go to MODEL, GENES, and ADD TRAIT GENE. You should specify the starting values for Aa and the allele frequency qA. The other transmission probabilities are fixed by the program (AA to 1 and aa to 0). The Mendelian model values provide reasonable starting points. (b) Add the unmeasured covariate associated with this restricted general gene using MODEL, COVARIATE, and ADD UNMEASURED COVARIATE. Select the CODOMINANT option. Again, the values you found under the Mendelian model provide reasonable starting values. (c) Once you have maximized the likelihood, determine the AIC. lnL_restr=________________________________________ Nparameters-restr=___________________________ AICrestr = -2lnL_restr+2Nrestr_________________ Decision: Which model is the best by AIC? (a) If your results are like mine you will find that the restricted general model is very slightly favored. Note that if the parameter penalty k were smaller (say 1.5), we would have found the general model favored and if k were larger (say 2.5), we would have found the Mendelian model favored. Another problem with the AIC is that it doesn't give us any significance - we don't know how confident we should be that the restricted general is the best model. Also note that in this case the restricted general model transmission parameters can vary by quite a bit and still give the same loglikelihood - this may mean that the likelihood is over parameterized. (b) Conduct a likelihood ratio test: (i) Test whether the Mendelian model can be rejected in favor of the restricted general model. Let the significance level be 0.05. In this case the Mendelian model is nested within the restricted general and the additional constraint on the Mendelian model, Aa = 1/2 is not a boundary. Therefore the LRT is asymptotically 2 with 1 degree of freedom. LRT = 2(lnL_restr-lnL_mend)=_____________________________ p_value (use excel for example to calculate)__________________ (ii) We can also test whether the Mendelian model can be rejected in favor of the general model. Again the Mendelian model is nested in the general model In this case two of the parameters are constrained to boundary values so the LRT has a distribution that is a mixture of 2 with 1, 2 and 3 degrees of freedom 3 with mixing parameters 1/4, 1/2, 1/4. If you got the same results as me, then you will find that the p value is ~0.077. All in all, it seems reasonable to accept the Mendelian model for now. We can retest whether the restricted general model improves the fit once we consider gene by environment interactions and alternative forms of inheritance to codominant inheritance. (4) Is there evidence of a genexbmi effect? (a) Define a genexbmi coefficient. Go to MODEL, COVARIATES, ADD MEASURED COVARIATES. Name the coefficient genexbmi, then click on the radio button, INTERACTION. Define a two-way interaction between bmi and the Mendelian codominant gene. Set the initial value range to 0 and 0. (b) Redefine the penetrance model by clicking on MODEL, PHENOTYPE, selecting TG, and EDIT PHENOTYPE. Add genexbmi to the penetrance model. (c) Run the model and compare the log likelihood to the log likelihood for the model without a genexbmi term using a Likelihood ratio test. (5) Is there evidence of a genexage effect? (a) Define a genexage coefficient. Go to MODEL, COVARIATES, ADD MEASURED COVARIATES. Name the coefficient genexage, then click on the radio button, INTERACTION. Define a two-way interaction between age and the Mendelian codominant gene. Set the initial value range to 0 and 0. (b) Redefine the penetrance model by clicking on MODEL, PHENOTYPE, selecting TG, and EDIT PHENOTYPE. Add genexage to the penetrance model. (c) Run the model and compare the log likelihood to the log likelihood for the model without a genexage term using a Likelihood ratio test. (6) If you found evidence for genexage and genexbmi effects then test for a genexbmixage effect. (a) Define a genexbmixage coefficient. Go to MODEL, COVARIATES, ADD MEASURED COVARIATES. Name the coefficient gxaxb, then click on the radio button, INTERACTION. Define a three-way interaction between age, bmi and the Mendelian codominant gene. Set the initial value range to 0 and 0. (b) Redefine the penetrance model by clicking on MODEL, PHENOTYPE, selecting TG, and EDIT PHENOTYPE. Add gxaxb to the penetrance model. (c) Run the model and compare the log likelihood to the log likelihood for the model without a gxaxb term using a Likelihood ratio test. (7) If you found evidence for any interactions then retest for a sex effect. 4 HOMEWORK (Due 2/5/2003): (A) Make a table that summarizes your results thus far. Write a few sentences that describe your conclusions. (B) Test whether HWE holds under the Mendelian model that fits best so far. What are the null and alternative hypotheses in this case? 5