Some practical notes about imputation
advertisement

Some practical notes about imputation
The most important precondition of imputation is that study and reference panel should
be as compatible as possible. This is because the whole process is based on the common
SNPs, so we have to make sure that no discrepancies are included. Specifically, study
and reference should be aligned in the same genome assembly, checked for inverted
SNPs alleles and corrected for minor allele frequency (MAF) and Hardy Weinberg
Equilibrium (HWE) differences. In case the reference and the study are aligned in
different genome assembly, we recommend the re-alignment of the study panel in the
assembly of the reference rather than the opposite. This is because the haplotype
structure of the reference panel that has been extracted through phasing will be
distorted if the position of the markers will be altered through the re-alignment process.
The process of re-alignment of a genotype dataset in a new genome assembly is called
liftovering. GATK [6] is one of the tools that perform liftovering.
Reference panels that have been pre-processed from imputation can be acquired from
the websites of imputation software:
Impute2:
http://mathgen.stats.ox.ac.uk/impute/impute_v2.html#download_reference_data
BEAGLE:
http://faculty.washington.edu/browning/beagle/beagle.html#reference_panels
Mach: http://www.sph.umich.edu/csg/abecasis/MACH/download/1000G-PhaseIInterim.html
Alternatively an imputation reference panel can be created from the raw data. For
example raw variant calls of the 1000 Genomes Project data in compressed Variant Call
Format [5] are available here (October 2011 release):
ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521
The data are in compressed Variant Call Format [5] and one of the most known tools to
manage VCF files is vcftools. After downloading we can extract the SNPs, disregard any
other variation and apply a basic quality filter of over 1% for MAF and over 10-4 for
HWE with vcftools. We strongly advise to scrutinize the reference panel for any HWE or
MAF discrepancy, as it is more likely that it will be transferred to the imputation results.
The qq-plot of the chi-square values of the HWE of the SNPs that passed the above filter
reveal that 1KG contains excessive SNPs with abnormal allele distributions. By applying
a more stringent filter we can correct the inflation but, of course, we will decrease the
variation in the reference panel.
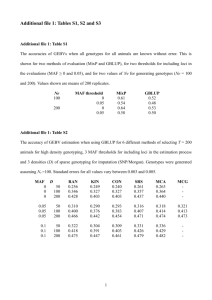
The next step contains the format conversions. In [table 1] we present the tools needed
to perform the appropriate conversions. To impute with BEAGLE we will have to
convert the study and the reference from VCF to BEAGLE. To impute with IMPUTE2 we
will have to convert the reference panel to hap and legend format and the study panel to
gen and samples format.
Origin Format
VCF
Target Format
PED / MAP
Tool
vcftools [8]
VCF
Hap, legend
vcftools
Notes
This can be very slow for
large datasets. We
recommend converting from
VCF to TPED/TFAM and then
use PLINK to convert to
PED/MAP
IMPUTE2 reference panel.
PED / MAP
PED / MAP
gen, samples
beagle
gtools [9]
linkage2beagle [10]
Does not support missing
genotypes. Only markers
with 100% calling rate will
be exported.
IMPUTE2 study panel
BEAGLE study and reference
panel
The HWE and MAF filtering should be applied to the study panel as well. The next step is
the correction for possible strand alignment issues. Impute2 and MACH contain options
to fix misaligned alleles between study and reference panel by inverting the alleles
whenever this is possible. Practically this corrects for AC/GT and AG/CT misalignments
whereas all the other combinations (for example AC / AG) are considered incompatible
and the respective SNPs are removed from the study panel. We do not recommend
relying in these corrections and always align the genotypes in study panel to the
assembly of the reference. IMPUTE2’s reference datasets are aligned in the ‘+’ strand, so
if the architecture of the study’s genotype platform is known, the strand correction can
be performed prior to the imputation. An extra quality step is to check for significant
differences in minor allele frequencies. An absolute difference of more than 0.25
indicates a major structural dissimilarity coming possibly from a genotyping error.
Here we demonstrate how we can apply the presented filters by using PLINK software
[7]. We assume that the study and the reference are in PED / MAP format.
Apply MAF and HWE filtering in the study
plink --study --maf 0.01 --hwe 0.0001 --recode --out study_1
Substitute the SNP identifier with the chromosomal position.
cat study_1.map | awk '{print $1, $1"_"$4, $3, $4}' > study_2.map
cat reference_1.map | awk '{print $1, $1"_"$4, $3, $4}' > reference_2.map
cp study_1.ped study_2.ped
cp reference_1.ped reference_2.ped
Create an alphanumerically sorted file of the positions:
cat study_2.map | awk '{print $4}' | sort > study.pos
cat reference_2.map | awk ‘{print $4}’ | sort > reference.pos
Get the common positions:
comm -12 study.pos reference.pos > common.pos
Extract the Allele Frequencies from common SNPs between study and reference:
plink --file study_2 --freq --extract common.pos --out study_2
plink --file reference_2 --freq --extract study.pos --out reference_2
Paste the two generated frequency files horizontally:
paste study_2.frq reference_2.frq > common.frq
The “normalize.py” python script in the appendix parses the common.frq file and
reports the SNPs that have incompatible alleles, inverted alleles and abnormal
frequencies:
python normalize.py
Remove the incompatible and abnormal SNPs:
plink --file study_2 --exclude abnormal.txt --recode --out study_3
plink --file study_3 --exclude incompatible.txt --recode --out study_4
We can also flip the inverted alleles:
plink --file study_4 --flip inverted.txt --recode --out study_5
Now we can convert the files study5.map and study5.ped to the suitable format of the
imputation tool of our choice according to table 1. The final step is the splitting of the
study panel to bins of samples and to chromosomal regions. The size of the splitting is
up to the existing computational environment and the parallelizing options.
Quality measures
Here we present the various metrics that assess the quality of an imputation
experiment. The metrics are divided into two categories whether real unimputed
genotypes are available or not.
The most common imputation metric is the R2 that represents the correlation between
the imputed and the real genotypes. Since real genotypes are unknown, various
statistics can be used to estimate it. Marchini et al. [1] presents a thorough review about
the R2 metrics used by MACH, BEAGLE, SNPTEST and IMPUTE v1 and v2 software. In
the appendix, we provide source code for the estimation of these metrics. Another R2
metric [2] is the ratio of the variance of the imputed allele dosage and the variance of the
true allele dosage. The variance of the true allele dosage is unknown, but it can be
estimated as 2p(1−p) under Hardy-Weinberg equilibrium, where p is the estimated
allele frequency.
Other metrics include also the allelic frequency error and the standardized allele
frequency error [3]. After assessing a suitable R2 value we can draw useful conclusion
from plotting the percentage of SNPs that exhibit R2 over than 0.8 for various minor
allele frequency bins (for an example see [11]). This graph will show how rare and
common SNPs where imputed and can be used to compare different imputation
reference panels.
In cases where real data are available the most useful metric is the R2 of the correlation
between allelic dosage from imputation and true genotyped dosage (code available in
appendix). We can also assess the performance of the imputation R2 value by estimating
the overconfident and under-confident genotypes. Overconfident are the wrongly
imputed genotypes with high R2 values and the opposite are the under-confident
genotypes. Another qualitative metric is the concordance between real and imputed
genotypes. The large number of concordant genotypes of the homozygous major alleles
can easily bias the concordance estimation. This is why if A and B are major and minor
alleles respectively, it is essential to provide an estimations for the non-reference
sensitivity that is the concordance of genotypes that are A/B or B/B in the “real” dataset.
GATK contains options to perform these estimations for VCF files. For comparing
different imputation methods we can assess the graph of the percentage discordance
versus percentage of missing genotypes for various thresholds of the genotype
probability [4]. To construct the graph, we measure the discordance between imputed
and real data versus the missing genotypes for an increasing threshold for the genotype
probabilities from 0.33 to 0.99.
References
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
[10]
[11]
Jonathan Marchini & Bryan Howie. Genotype imputation for genome-wide
association studies. Nature Reviews Genetics 11, 499-511. Supplementary
information S3.
P.I. de Bakker, M.A. Ferreira, X. Jia, B.M. Neale, S. Raychaudhuri, B.F. Voight
Practical aspects of imputation-driven meta-analysis of genome-wide
association studies
Brian L. Browning and Sharon R. Browning. A Unified Approach to Genotype
Imputation and Haplotype-Phase Inference for Large Data Sets of Trios and
Unrelated Individuals
Howie BN, Donnelly P, Marchini J, 2009 A Flexible and Accurate Genotype
Imputation Method for the Next Generation of Genome-Wide Association
Studies. PLoS Genet 5(6): e1000529. doi:10.1371/journal.pgen.1000529
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A.,
Handsaker, R. E., Lunter, G., Marth, G. T., Sherry, S. T., McVean, G., Durbin, R.,
1000 Genomes Project Analysis Group. The variant call format and VCFtools.
Bioinformatics (2011) 27 (15): 2156-2158.
Mark A DePristo, Eric Banks, Ryan Poplin, Kiran V Garimella, Jared R Maguire,
Christopher Hartl, Anthony A Philippakis, Guillermo del Angel, Manuel A Rivas,
Matt Hanna, Aaron McKenna, Tim J Fennell, Andrew M Kernytsky, Andrey Y
Sivachenko, Kristian Cibulskis, Stacey B Gabriel, David Altshuler & Mark J Daly. A
framework for variation discovery and genotyping using next-generation DNA
sequencing data. Nature Genetics 43, 491–498 (2011) doi:10.1038/ng.806
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J,
Sklar P, de Bakker PIW, Daly MJ & Sham PC (2007) PLINK: a toolset for wholegenome association and population-based linkage analysis. American Journal of
Human Genetics, 81.
http://vcftools.sourceforge.net/
http://www.well.ox.ac.uk/~cfreeman/software/gwas/gtool.html
http://faculty.washington.edu/browning/beagle_utilities/utilities.html
Zhaoming Wang, Kevin B Jacobs, Meredith Yeager, Amy Hutchinson, Joshua
Sampson, Nilanjan Chatterjee, Demetrius Albanes, Sonja I Berndt, Charles C
Chung, W Ryan Diver, Susan M Gapstur, Lauren R Teras, Christopher A Haiman,
Brian E Henderson, Daniel Stram, Xiang Deng, Ann W Hsing, Jarmo Virtamo,
Michael A Eberle, Jennifer L Stone, Mark P Purdue, Phil Taylor, Margaret
Tucker& Stephen J Chanock. Improved imputation of common and uncommon
SNPs with a new reference set. Nature Genetics 44, 6–7 (2012)
doi:10.1038/ng.1044.
http://www.nature.com/ng/journal/v44/n1/full/ng.1044.html
Appendix
Scripts:
normalize.py. Correct for inverted SNPs, Minor Allele Frequency differences and
incompatible SNPs between reference and study panel
http://www.bbmriwiki.nl/svn/Imputation/PaperMethodology/scripts/normali
ze.py
BEAGLE’s Allelic R2:
http://www.bbmriwiki.nl/svn/Imputation/PaperMethodology/scripts/beagle_r
2.py
Real Allelic R2:
http://www.bbmriwiki.nl/svn/Imputation/PaperMethodology/scripts/real_alle
lic_r2.py