BL 415 Genetics
advertisement

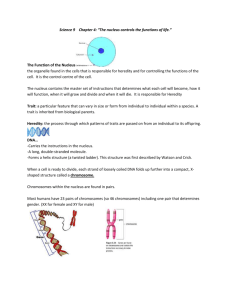
BL 414 Genetics Spring 2006 Study Guide for Test 3 Chapter 8: Chromosomes karyotype: representation of the chromosomes in the metaphase stage of mitosis, by arranging the pairs of homologous chromosomes in order of size, with the sex chromosomes placed at the bottom right of the page Chromosomes stained with Giesma exhibit characteristic banding patterns, dark bands are called G bands, contain heterochromatin (highly repetitive DNA) and are gene poor. In between the dark bands are the interbands, called R-bands – these contain euchromatin and are gene-rich. -the dark and light segments due to differential staining are used to identify specific locations on each chromosome A more recent technique for labeling chromosomes is “chromosome painting” which is done by hybridization of fluorescently labeled chromosome specific DNA with metaphase chromosomes – this results in a karyotype that is color-coded for each homologous chromosome pair The human karyotype contains 22 pairs of autosomes and 2 sex chromosomes, with a total of 46 individual chromosomes. Human chromosomes are identified by their karyotype number 1-22 or as the X or Y chromosome. The chromosomes are also placed in groups A-G according to their size. Smaller regions within chromosomes are identified by location on the arm on either side of the centromere and the labeling of bands. The shorter arm is called the p arm and the longer arm is the q arm. More specific location is given by the number for the region, subregion, band and interband. See Figure 8.3 for these designations. For example, the gene for Rh blood group is located near 1p36.2, which is on the p arm of chromosome 1, the second band in subregion 36. Nomenclature used in describing chromosomes and chromosomal abnormalities, particularly alterations in the structure of the chromosome – which could be seen in the karyotype as a change from normal banding patterns on chromosomes: ter: the terminal portion of a chromosome, pter: terminal portion of p arm, qter: terminal portion of q arm – could be used to indicate a deletion, inversion or duplication +: indicates an extra chromosome or part of a chromosome – for example +21 means an extra copy of chromosome 21, also known as trisomy 21 or Down syndrome -: indicates a missing chromosome or part of a chromosome mos: mosaic – an individual with more than one genetic makeup in different tissues or cells of that individual dup: duplication – a region of the chromosome is present twice dirdup: direct duplication – the duplicated region is in the same direction as the original copy, also called tandem duplication if they are next to each other in the same direction tandem duplications lead to further multiplication of copies through uneven crossing over, in which chromosomes line up in a slightly shifted alignment and crossover takes places between identical copies at different positions on the chromosome – see Figure 8.16 invdup: inverted duplication – the second copy of a gene is in the reverse direction of the first copy del: deletion – a segment of the chromosome is missing – one or more genes may be missing inv: inversion – the linear order of a group of genes is the reverse of the normal order t: translocation – a interchange of segments of chromosomes that are not homologous rcp: reciprocal translocation – the parts are reciprocally exchanged – if reciprocal translocation occurs in only one pair of chromosomes and the other chromosomes are normal, the organism will be semisterile, that is only half of its gametes will have the full complement of genes in the genome: see Figure 8.24 and 8.25 rob: Robertsonian translocation – the centromeres of two nonhomologous acrocentric chromosomes become fuse to from a single centromere of a single chromosome – this can occur with chromosome 21, which could lead to Down syndrome in the children of parents with a Robertsonian translocation (about 3% of Down syndrome patients have been found to have a parent with a Robertsonian translocation r: Ring chromosome – a chromosome whose ends are joined, lacks telomere, rare i: Isochromosome – a chromosome with two identical arms containing homologous genes Classification of chromosomes by the relative position of the centromere: Metacentric chromosome: centromere in the middle of the two arms – appears as a Vshape during anaphase Submetacentric chromosome: centromere is off-center appears as a J-shape during anaphase Acrocentric chromosome: centromere is very close to one end appears as an I-shape during anaphase Acentric chromosome: lacking a centromere: lost during cell division because it does not attach to a spindle Dicentric chromosome: has two centromeres: often lost during cell division because it will have problems separating into one or the other daughter cell – it may create a bridge between the daughter cells, not reach the nucleus of either daughter cell, or break and the daughter cells will have broken copies of the chromosome. Dosage compensation: Human females have two copies of X and males have one copy. So if there is no compensation for the difference in amount of X genes, males will express half of much from genes on X as females do, or looking at it the other way, females will express twice as much as males. This would not work well in an organism, because the level of gene expression is very important to normal function. This is why dosage compensation is necessary - there needs to be something to equalize the expression of X genes in males and females. X-inactivation: In humans this is accomplished by inactivating one of the X chromosomes in every cell of females. During early embryonic development one X is randomly inactivated. All descendant cells of this embryonic cells keep the same X inactivated. The X-inactivation initiates at the XIC “X-inactivation center” near the centromere – the chromosome condenses, is coated with an RNA transcript called Xist that is essential for X-inactivation, and heavy methylation occurs along the chromosome In some cells, the inactivated X chromosome is visible as a densely staining Barr body in the cell nucleus For this reason, human females are mosaic for X-linked genes because in different cells and tissues of a human individual, the maternal or paternal X chromosome may be the active copy and therefore different alleles of X-linked genes will be expressed throughout her cells There are a few genes in X that are not silenced by gene inactivation Some of these have homologs in the Y chromosome – and they can undergo crossover during meiosis – therefore they do not behave as X-linked genes but behave as autosomal genes and are said to show pseudoautosomal inheritance – the regions of the X and Y chromosomes on which they reside, PARp and PARq, are called pseudoautosomal regions Polyploidy in plants Many plants species have genomes composed of multiple complete sets of chromosomes The number of single chromosomes of each types is called the monoploid chromosome set. A diploid species has two copies of the monoploid set, triploid has three copies, tetraploid has four copies of the monoploid set, hexaploid – 6 copies, octoploid – 8 copies, decaploid – 10 copies. The chromosome number present in the gametes of a species is the haploid number and is one-half of the number of chromosomes in the somatic cells. Autopolyploidy refers to the case where all of the chromosomes in the species derive from a single ancestral species. Allopolyploidy is the case where complete sets of chromosomes have come from two or more different ancestral species. Polyploidy can arise sexually from unreduced gametes or asexually by means of endoreduplication in which a cell undergoes chromosome replication but not chromosome separation or cytoplasmic division. See Figure 8.32 Ch. 11 – see chapter outline Ch. 14 DNA mutations and repair – see Types of Mutations chart Sources of mutations Replication slippage – responsible for triplet expansion, which are large tracts of trinucleotide repeats – see Figure 14.8 Triplet expansion diseases: genetic diseases caused by a disruption in the gene expression due to a long triplet repeat region – e.g. Fragile X syndrome, which causes mental retardation. In Fragile X, the protein FMR1 is not produced in adequate amounts, causing problems in nervous system development and learning Anticipation: because the number of repeats increases from generation to generation, there is in an increase in the severity of the disease in each successive generation Transposable elements, or transposons: large sequences of DNA move throughout the genome – their insertion into sites within chromosomes may disrupt the activity or regulation of genes in the insertion site Cut-and-paste elements: transposable elements that insert by a nicking the target DNA, leaving overhanging DNA, and subsequent insertion and relegation LTR retrotransposons: use an RNA transcript as an intermediate, and have long terminal repeats at both ends Non-LTR retrotranposons: use an RNA transcript as an intermediate, but do not have long terminal repeats at both ends – LINE and SINE elements are this type of transposon and they are the most abundant types of transposons in mammals There are many copies of transposons in the human genome but they do not seem to be transpositionally active. They may have function during organismic stress (such as heat or starvation) because SINE elements are known to be transcribed when an organism is under stress, and they promote protein production under stressful conditions. Studies of sequence similarities and changes in the transposon repeats can indicate the activity of transposition and it appears that only 1 in 600 new mutations in humans are due to transpositions, whereas the number is 1 in 10 for mice. Therefore, transposition does not appear to be a major source of mutation in humans. Hot spots of mutation: certain sequences are more likely to undergo mutation: Mutagens: Oxidizing agents: deaminates cytosine or adenine – if adenine is deaminated this can cause an AG transition, if cytosine is deaminated, uracil is made, but there is a cellular repair mechanism to remove uracils from DNA, involving DNA uracil glycosylase, if methyl-cytosine is deaminated, thymine is made causing a CT transition, e.g. nitrous acid Base analog: increases the rate of base mispairs – can cause transitions, e.g. 5-Bromodeoxyuridine alkylating agents: add methyl or larger alkyl group to DNA bases, causes mispairing and transitions e.g. EMS or nitrogen mustard Intercalating agents: inserting between the base pairs in DNA – distort the DNA and cause insertion or deletion of one or a few bases – this could cause a frameshift mutation, e.g. acridine orange, proflavin UV radiation: energy absorption from UV light causes chemical reaction linking pyrimidines together, especially thymines which form covalently bound thymine dimers, which lead to blockage of DNA replication and RNA transcription, there are cell repair mechanisms for pyrimidine dimers but they may be overwhelmed in the case of excessive exposure to UV light Ionizing radiation e.g. X rays, radioactive material emitting and radiation, Create free radicals which damage tissue and DNA Ch.15 Cell Cycle and Cancer – refer to powerpoint slides, notes on slides Cancer is not one disease, but a collection of diseases united by certain characteristics: – Uncontrolled cell proliferation – Caused by mutations in several genes – Occurs in somatic cells – About 99% of cancer is caused by sporadic mutations, and 1% is caused by inherited mutation Cancer cells Normal cells No contact inhibition – pile up on top Stop growing and dividing upon of each other in petri dish contact with others cells – single sheet on petri dish No apoptosis Apoptosis (programmed cell death) occurs when cell is damaged or starving Immortal – cells never cease dividing, Cell senescence occurs - cells stop generation after generation – this dividing after several generations property is necessary for cell lines Clonal – cells are descendants of a single ancestral cell Cells differentiate according to normal development The genetics of cancer: Cancer results from mutations in somatic cells, so in most cases it is not hereditary When sporadic mutations in several genes governing the cell growth and cell death occur cell can become cancerous In the small number of cancers that are familial, one mutation is inherited and others sporadically occur Mutations of genes involved in the cell cycle cause cancer: G1 S transition Apoptosis (programmed cell death) Tumor-suppressor genes, the most important of which is p53 Oncogenes and proto-oncogenes What is the cell cycle? It’s a division of the lifetime of a cell (between its formation and cell division) into 4 phases including DNA synthesis and mitosis: many genes and proteins, discovered in yeast studies, are involved in the maintenance and regulation of the cell cycle Cyclins are proteins involved in cell-cycle control – their levels of expression go through regular cycles Timing of synthesis and destruction of Cyclin + Cyclin-dependent protein kinase (Cdk) complexes during cell cycle If something is amiss in the cell, it does not proceed through the cell cycle – growth is arrested at cell cycle checkpoints Tumor suppressor protein p53 is involved in the control of DNA damage The balance of Bax and Bcl2 proteins determines the fate of a cell (normal, apoptosis or immortalization) Cell cycle regulatory genes affected in tumors: tumor-suppressor genes: Tumor-suppressor genes Alteration Consequence p53 p16/p19ARF Loss of G1/S, S, and G2/M checkpoint functions Mutation p21 Mutation Loss of G1/S, and S checkpoint functions RB Mutation Promotes proliferation Bax Mutation Failure to promote apoptosis Bub1p Mutation Loss of spindle assembly checkpoint function Cell cycle regulatory genes (proto-oncogenes/oncogenes) affected in tumors: Proto-oncogenes Alteration Consequence Cyclin D` Cdk4 Amplification or overexpression Amplification Cdk4 Mutation EGFR (epidermal Amplification growth factor) FGFR (fibroblast growth-factor receptor) Ras Amplification Ras Mutation Bcl2 Overexpression by translocation next to strong enhancer Amplification Mdm2 Promotes entry into S phase Proliferation b/c of Constitutive activation of growth factor pathway Proliferation from constitutive growth signal Inactivates GTPase – constitutive activation of growth factor pathway Blocks apoptosis Mimics loss of p53 About 1% of cancer is from familial cancer syndromes – these involve the inheritance of a mutation in a cancer gene. The incidence and type of cancer depends on the particular mutation.