Script - Chemistry
advertisement

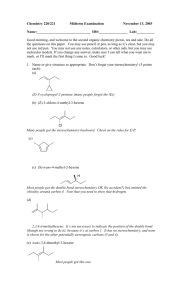
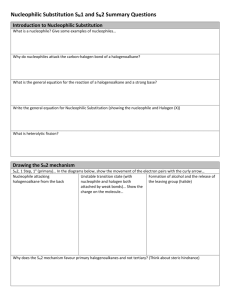
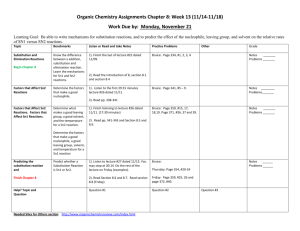
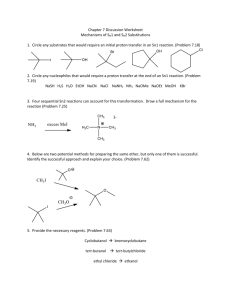
Script Basics Curly arrow mechanisms are used a lot in chemistry to show the mechanism of a reaction, but they can look scary and complicated. How do you know where to put the arrows and dots? Once you know the basics, curly arrow mechanisms are quite easy. Curly arrows show the transfer of electrons. Electrons move from electron rich areas to electron poor areas. A single headed arrow shows the movement of only one electron, and is needed to describe free radical reactions. A double headed arrow shows the movement of an electron pair, such as from a lone pair or a bond. We will mostly use the double headed curly arrow. The tail of the arrow begins at the original location of the electrons, which could be a lone pair, a negative charge on a nucleophile, a base, or the bond between two atoms. The head of the curly arrow always points to where the electrons are going. For example, when a carbonyl group is protonated, the electron pair on the oxygen grabs a positively charged hydrogen, a proton. This is described mechanistically by drawing a doubleheaded curly arrow from the oxygen lone pair to the proton. In the product the electron pair now forms a bond between these two atoms. Protonations are not only very fast, they are also reversible. In the reverse reaction, the bond between the oxygen and the hydrogen is broken, the electrons move from the bond back to the oxygen, leaving a proton behind. Again, this is illustrated by the double headed arrow moving from the bond back to the oxygen. Remember also, the charges on either side of the reaction should balance. Curly arrows are only used to show the movement of electrons, so the H+ does not attack the oxygen, the electrons on the oxygen attacks the proton H+. The H+ is an electrophile, as it is attracted to a negative centre, whereas the oxygen is a nucleophile, as it is attracted to a positive centre. SN2 The first reaction we will look at is the SN2 reaction; a nucleophilic substitution that follows second order kinetics, meaning the rate of the reaction relies on 2 molecules. This is a substitution reaction that is typical for many primary and secondary alkyl halides and related compounds with a good leaving group. The nucleophile always attacks from the back, on the opposite side from the leaving group, expelling the leaving group. In this reaction, the nucleophile, a hydroxide ion, attacks the electrophilic carbon centre, on the opposite side from the bromide leaving group. A new bond is formed with the carbon at the same time as the bond to the bromide ion breaks. This forms a transition state, where the carbon is partially bonded to both groups at the same time. The leaving group breaks away, and we get the product, an alcohol. SN2 reactions always go with inversion of stereochemistry, which will be important if the starting material is chiral. SN1 Another substitution reaction is the SN1 reaction, where the 1 indicates first order kinetics. The rate of this substitution depends only on one reaction participant. It occurs under acidic conditions and is incompatible with strong bases, such as hydroxide or alkoxide. The SN1 reaction proceeds through a carbocation intermediate. Tertiary halides favour SN1 substitution. The presence of three substituents blocks direct attack of the nucleophile from the back, and the substituents help to stabilise the intermediate carbocation. In this example, the bromide ion leaving group breaks away first from the rest of the molecule. This process is slow and reversible, and leaves a positively charged carbocation intermediate. The nucleophile then attacks the carbocation, and loss of a proton results in the neutral product. As the intermediate is a trigonal planar carbocation, the nucleophile can attack from either face, leading to a racemic product. E2 We will now consider elimination reactions. The first of these is the E2 reaction, which is a second order reaction. An E2 elimination requires that the leaving group, the 2 carbons forming the C=C double bond and the hydrogen are all in the same plane. We call this arrangement “antiperiplanar” and it is a key feature of the E2 mechanism. The base, ethoxide in this case, removes the proton β to the leaving group. The electron pair of the C-H bond moves to form the carbon-carbon π-bond, and the carbon-chlorine bond is broken, with the electrons moving with the chlorine. All of this happens at the same time and produces the alkene product, ethanol and a chloride ion. Depending on the arrangement of the groups on the carbons before elimination, either a cis or a trans product can sometimes be formed. E1 Elimination can also occur by an E1 mechanism. Like an SN1 substitution, the E1 elimination is a two-step process which is favoured by tertiary compounds and involves a carbocation intermediate. Both SN1 and E1 occur under acid conditions. As with the SN1 mechanism, the chlorine leaving group separates first, and a carbocation is formed. This step is slow and reversible. In the absence of a good nucleophile, the carbocation then loses a proton to form the C=C double bond of the alkene product. Key features of substitution and elimination reactions SN1 and SN2 are both substitution reactions. A good leaving group, such as bromine or chlorine, is substituted for the attacking nucleophile. E1 and E2 are both elimination reactions, where groups are removed from the compound, forming alkene products. SN1 and E1 are similar in mechanism, as both proceed via a carbocation intermediate, and require an acid catalysis. Elimination by E1 is often a side reaction during an SN1 substitution and is particularly prone to happen at higher temperature. SN2 and E2 also have similarities in that they pass through a transition state, and no intermediate is involved. The E2 reaction will occur under basic conditions. Addition on C=C Let us now look at addition reactions. The carbon-carbon double bond of an alkene is electron rich, so most reactions with it involve electrophiles. For example, hydrogen halides are strong acids which can protonate an alkene, such as 2-methyl-2-propene. Protonation leads to a carbocation intermediate. This is then attacked by the bromide counter-anion to give the addition product. The addition of HBr to the alkene follows Markownikoff’s rule and the bromine ends up on the more substituted former alkene carbon. The reason for this is that protonation occurs first and will always give the more substituted and thus more stable carbocation intermediate. Addition on C=O Addition can also occur on the C=O double bond. The carbonyl group is polarised, with the electronegative oxygen bearing a delta negative charge and the carbonyl carbon a delta positive charge. Reactions of the carbonyl group are dominated on the addition of the nucleophile to the electron deficient carbon. The mechanism, and rate of reaction, varies with the nucleophile. A good nucleophile, such as a Grignard reagent, will attack the carbonyl carbon directly. The resulting alkoxide is then protonated during an acidic work up. The reaction of a ketone or aldehyde with a poor nucleophile doesn’t work unless it is catalysed by an acid. Protonation of the carbonyl group is essential before a bad nucleophile can attack. A typical example includes the formation of an oxime. In this example, the carbonyl group is protonated. This makes the carbonyl carbon much more delta positive. The nucleophile, hydroxylamine, now can attack the electron deficient carbon. The nitrogen then loses a proton, while the OH picks up another proton and is transformed in a good leaving group. The lone pair of the nitrogen comes in to form a double at the same time when water is eliminated. Loss of a proton finally gives the oxime product. Carboxylic acids and their derivatives (esters, amides, and acid chlorides) differ from ketones and aldehydes in that they have a leaving group attached to the carbonyl group. So, attack of a nucleophile at an ester gives initially a tetrahedral intermediate, but the reaction doesn’t stop there. The tetrahedral intermediate then loses the leaving group and reforms the C=O double bond. This is saponification of an ester, and the overall reaction is a “substitution” at an acyl carbon. Electrophilic Aromatic Substitution The final mechanism we will look at is that of an electrophilic aromatic substitution. Unlike alkenes which prefer addition reactions, the reaction of benzene with an electrophile involves the replacement, substitution, of a group, in most cases just a hydrogen atom, to give a substituted benzene. Many aromatic compounds are electron rich in nature, and prefer substitution by an electrophile. The electrophile is generally an electron deficient group, such as a cation; it could even be a proton although this won’t be very productive. A typical example of an electrophilic aromatic substitution is the Friedel-Crafts alkylation, where an alkyl halide is combined with anhydrous AlCl3 in situ. Aluminium trichloride is a Lewis acid which triggers the dissociation of the carbonchlorine bond, and the result is an alkyl cation. In truth, this is a rather simplistic view since primary alkyl cations in particular are so unfavourable that the carbon-chlorine bond is only weakened rather than broken, but it acts as a separate cation. Benzene now reacts with the alkyl cation. We indicate this by drawing a curly arrow from the benzene to the carbocation. Although the aromaticity of the benzene ring is destroyed, the intermediate carbocation (called the Wheland intermediate) is stabilised by resonance and the positive charge is shared around the ring. In the final step, the electrons from the carbon hydrogen bond move into the ring and regenerate the aromatic ring. At the same time, the hydrogen atom and the positive charge are lost in form of a proton. The overall result is that the hydrogen has been substituted by an alkyl group. There are many other types of aromatic substitution. There is the related Friedel-Crafts acylation which uses an acyl halide and involves a resonance-stabilised acyl cation as the electrophile. Another common example is halogenation where a halogen, such as chlorine, when combined with catalytic amounts of iron(III) chloride becomes polarised, so that the aromatic ring reacts with what is almost like a Cl+. Nitrations involve an NO2+ electrophile, the nitronium ion, which is generated in situ from a mixture of concentrated nitric acid and concentrated sulfuric acid. This is an important reaction as these can be reduced to aminobenzenes. These are very important in the pharmaceutical industry, in the production of paracetamol.