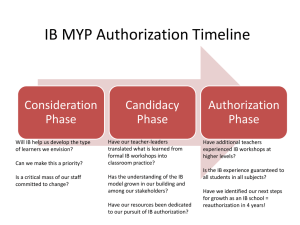
Breathing Life into the August 30th Agreement
advertisement

Breathing Life into the August 30th Agreement By Geoff Blackie INTRODUCTION & APPROACH I was recently giving a talk at a local area high school with two other law students on various human rights law issues. I was addressing HIV and AIDS in Africa, and thought to begin with some questions followed by statistics to shock. However, their answers regarding the number of infected in Africa, national infection rates and the number of orphans as a result of HIV/AIDS were higher than current statistics. For many, the scale of the problem is well-known (or even slightly exaggerated). Still the problem persists, infection rates are on the rise and deaths continue at an unprecedented rate with disastrous effects on the infrastructure of those societies.1 The battle against HIV/AIDS is fought by a number of dedicated individuals on many fronts. HIV/AIDS is an equality issue, a prevention issue, a debt relief issue, a poverty issue and an intellectual property issue. In 2003, the August 30th Agreement (the Agreement) brought changes to the World Trade Organization’s (WTO) Agreement on Trade Related Intellectual Property Rights (TRIPs). The Agreement allowed for, inter alia, importing nations to issue compulsory licenses for essential HIV/AIDS medications. Canada was the first developed country to announce that it would implement domestic legislation to allow for the issuance of compulsory licenses. Last year, on May 14th, the Jean Chrétien Pledge to Africa Act (Bill C-9) received Royal Assent.2 Despite these important developments on the intellectual property front, all is quiet; not a single pill has reached a developing nation using the August 30th Agreement. This paper’s central concern is how to utilize the August 30th Agreement; thus, the focus is on compulsory licenses for export. I shall first describe the concerns and actions that precipitated the Agreement. In the second section, I consider the TRIPs members that have signified intent to implement domestic legislation and are at different stages of the process. In the third section, I critically examine the Canadian legislation with reference to others models to answer the following questions: (1) How were central issues addressed and could the legislation have been better constructed to ensure success? (2) What steps are currently underway to gain a compulsory licence? (3) Can compulsory licensing systems in developed countries, specifically Canada, work? I. CONCERNS & ACTIONS RESULTING IN THE AUGUST 30TH AGREEMENT The African Pandemic and Access to Essential Medicines: 1 In Botswana and Swaziland the infection rate is nearly 40% (see the Clinton Foundation available at http://www.clintonfoundation.org/aids-globalcrisis.htm (last visited April 13, 2005). UNICEF estimates the number of orphans die to HIV/AIDS at 14 million see http://unicef.org/aids/index.html (last visited April 13, 2005). See Appendix A for the amendments. 2 Despite have received Royal Assent the amendments are not in force until all the companion legislation is in place. This has not yet been done. The amendments can be found at http://laws.justice.gc.ca/en/p4/notinforce.html. Note, the legislation available on Hansard is not the final version of the legislation Unfortunately, the crisis in Africa requires little introduction but much action. In Africa only 12%, or 700,000 of those infected, are receiving required treatment. Moreover, infections are rising more quickly than number receiving treatment.3 On World AIDS Day in 2003, the WHO released a plan to treat 3 million people in developing and transitional countries by the end of 2005 (the “3 by 5 initiative”). According to a Medicine Sans Frontier (MSF) press release, between July 2004 and January 2005 only 264,000 new patients were able to benefit from ARV treatment.4 The WHO recognized the importance of access to essential medicines in 1991 and was again a central concern within the WHO at the 2000 International AIDS Conference in Durban. At that time, few expected that ARVs could be made available to the poor in developing nations. Drug prices were simply too high; costs exceeded $10,000 per patient per year for first line treatments.5 Even in 2002 no one in the developing world had received ARVs through official donor support.6 As Kevin Outterson asserted, “precious years were lost because the drugs were too expensive for the developing world, and they were too expensive because of patent protection and fears of arbitrage.”7 However, in February 2001 an Indian generic company, Cipla, announced “the price heard around the world.”8 Cipla offered a standard package of ARVs for $350/year to NGOs and $600/year to governments in Africa. As additional Indian generic producers began producing the same drugs, prices continued to fall. By 2004 fixed dose combination medicine (FDCs)9 were available for less that $140/year and were mostly provided by four generic producers – three in India and one in South Africa.10 MSF is able to treat 25,000 patients only because the prices are a fraction of the prices in developed countries.11 Accordingly, the number of patients treated has risen dramatically, but remains drastically inadequate. The Debates: Life, Property and Innovation: See Medicine Sans Frontier (MSF) Press Release, “Global AIDS treatment efforts not on track” January 28, 2005 at http://www.msf.org/content/page.cfm?articleid=62168E79-1C22-4E97-8734B6F41C624CEA. 4 ibid. 5 When a patient is treated with ARVs the virus eventually develops immunity to the treatment. At this point there is a shift to second line ARVs that attack the virus in a different way. 6 Kevin Outterson Pharmaceutical Arbitrage: Balancing Access and Innovation in International Prescription Drug Markets 5 Yale J. Health Pol, L. & Ethics 193 t 255-256. 7 Ibid at 256-257. 8 Brook Baker, “Arthritic Flexibilities for Accessing Medicines: Analysis of WTO Action Regarding Paragraph 6 of the Doha Declaration on the TRIPS Agreement and Public Health” 14 Ind. Int'l & Comp. L. Rev. 613 at 615. 9 Essentially, FDCs combine various treatments into one pill. The most popular combination for first line treatment is a combination of Zidovudine (AZT), Lamivudine, and Nevirapine. FDCs minimize shipment and transport costs and, more importantly, greatly increase chances of compliance. It is much easier to remember to take one pill twice/day than a number of different pills at various points throughout the day. If the medication is not taken correctly there is a greater chance the virus will mutate more quickly. 10 The quality of the generic productions was assured through the WHO’s new pre-qualification program. See Baker supra note 7 at 615. 11 MSF supra note 2. 3 Professor Bryan Mercurio asserts that the debate between access to essential medicines and patent rights received global attention in 2000 when several pharmaceutical companies challenged the legality of South African legislation designed to allow for compulsory licenses of patented medicines. Simultaneously the US initiated WTO procedures challenging the legality of Brazil’s compulsory license system. Both the patent holders and the US, who supported the pharmaceutical companies, where bombarded with negative publicity. Largely as a result, the claims were dropped.12 Generally, increasing access to essential medicines is dependant on their prices. With finite funding, more ARVs can be bought and more money spent on developing the infrastructures to distribute the drugs. A number of methods have been used to reduce costs including: voluntary licensing, compulsory licensing, differential pricing, and bulk procurement.13 Domestic legislation that allows generic production of pharmaceuticals has played a central role in reducing costs. First, once generic companies are able to produce the drugs, the patent holder no longer holds a monopoly and prices necessarily drop closer to the manufacturing costs (see table 1 below). Second, a legitimate threat by a government to issue a compulsory license and allow generic production often provides the patent holder with an incentive to significantly reduce prices or allow a voluntary license on more favourable terms. While this happened in Brazil and South Africa, it is not unique to the developing world. When fears of an anthrax attack mounted in the United States government, it used the threat of a compulsory license to entice Bayer to provide Cipro at a greatly reduced cost.14 Bryan C. Mercurio, “Trips, Patents, and Access to Life-Saving Drugs in the Developing World” 8 Marq. Intell. Prop. L. Rev. 211 at 224. 13 The paper will focus on the attempts to make exporting under compulsory licenses viable. For more see Outterson supra note 5 who argues that differential pricing can allow for greatly reduced prices while still providing patent holders to make substantial gains. 14 Mercurio supra note 11 at 224. 12 TABLE 1: Effects of Generic Competition on ARV Prices15 Third, generic producers are able to provide essential medicines in forms that allow improved compliance with the required treatment regiment (e.g. FDCs). This is not an option in developed countries when different parties hold the patents to the drugs in the FDCs and empirically have demonstrated a lack of sufficient interest to cross-patent. The TRIPS Agreement is the international standard for protecting intellectual property rights and has drawn scorn from many developing countries.16 It became a central issue at the Doha during the Fourth WTO Ministerial Conference in 2001. To this extent, paragraph 4 of the Doha Declaration (the Declaration) asserted: We agree that the TRIPS Agreement does not and should not prevent members from taking measures to protect public health. Accordingly, while reiterating our commitment to the TRIPS Agreement, we affirm that the Agreement can and should be interpreted and implemented in a manner supportive of WTO members' right to protect public health and, in particular, to promote access to medicines for all.17 Paragraph 5(b) of the Declaration asserts the right of members to issue compulsory licenses for medicines to protect public health, “and the freedom to determine the This table was presented to the European Parliament in an MSF presentation by Ellen ‘t Hoen. It is available at msf.org. 16 See for example Michael Trebilcock who criticizes TRIPS for failing to recognize differences in comparative advantage vis a vis innovation and imitation. Michael Trebilcock and Robert Howse, The Regulation of International Trade (2d ed) (New York: Routledge, 1998) at 308. 17 Doha Declaration on the TRIPS Agreement and Public Health WT/MIN(01)/DEC/2, 20 November 2001, paragraph 4 at http://www.wto.org/english/thewto_e/minist_e/min01_e/mindecl_trips_e.htm 15 grounds upon which such licences are granted.”18 Paragraph 6 then recognizes the inadequacy of this solution for members who do not possess a manufacturing capacity in the pharmaceutical sector. It also instructed the Council on TRIPS to report a solution to the General Council by the end of 2002.19 The August 30th Agreement – The floor for compulsory licenses A solution to the paragraph 6 dilemma was not reached until August 30th, 2003. The responses have been mixed. Baker asserts that the current international regime should be “a floor and a ceiling.”20 He argues that developing countries should abandon the August 30th Agreement and return to a simplified process under Article 30 of the TRIPS Agreement. While an important consideration, this paper will focus on the potential within the legal framework created by the August 30th Agreement. The August 30th Agreement21 generated a list of eleven possible steps that an importing country must initiate for the issuance of a compulsory licence, this includes22: (1) The importing country must seek a voluntary license on commercially reasonable terms for a reasonable period of time. (2) If the importing country is not successful in obtaining a voluntary license, it must to apply to the WTO for a compulsory license. (3) If the compulsory license is for import, the importing country must assess its generic industry's capacity to produce the medicine locally. (4) If capacity insufficient, it must notify and explain to the TRIPS Council its decision to import. (5) The importing country must notify a potential exporter. (6) The exporter must also attempt to obtain a voluntary license on commercially reasonable terms for a reasonable period of time. 18 ibid at paragraph 5. ibid at paragraph 6. For a more detailed discussion of the Doha Declaration and the approach to the August 30th Agreement see Baker supra note 7 at 623-625. For a further discussion the role of IP laws in low income countries see Jean Lanjouw and Alan Sykes who support the enactment of IP laws in low income countries to encourage development in local markets for treating neglected diseases. A further concern is how to develop local markets for treating neglected diseases, addressed in Outterson supra note 5. 20 Baker supra note 7 at 618. 21 Implementation of Paragraph 6 of the Doha Declaration on the TRIPS and public health, Decision of the General Council of 30 August 2003, WT/L/540 1 September 2003 available at http://www.wto.org/english/tratop_e/trips_e/implem_para6_e.htm; for brief description see Yolanda Taylor (ed.), Battling HIV/AIDS: A Decision Maker’s Guide to Procurement of Medicines and Related Supplies (New York: The World Bank, 2004) at 121-125. 22 For similar lists see Correa infra note 95 at 401 and Anthony P. Valach, Jr. “Trips: Protecting the Rights of Patent Holders and Addressing Public Health Issues in Developing Countries” 4 Chi.-Kent J. Intell. Prop 156 at 167-169. 19 (7) The exporter must then seek a compulsory license from its own government on a single-supply basis. (8) The exporter will need to achieve product registration and prove bio-equivalence (as required by domestic law). This requires studies regarding toxicity and efficacy (unless access to the initial data from the original studies is granted with the compulsory license) (9) The exporter must determine the adequate royalty based on standards of reasonableness in the importing country. (10) The exporter must investigate pill size, shape, colour, labelling, and packaging of the patent-holder's product in the importing country and differentiate its new product in all respects, provided the measures taken are not too costly (11) Before shipment a website must be created, posting information about the quantities supplied and the distinguishing features of the product. In addition to the above specifics, the motivation behind the whole process must be non-commercial in nature. The General Council Chairperson’s Statement released with the decision asserted that the “system that will be established by the Decision should be used in good faith to protect public health and, without prejudice to paragraph 6 of the Decision, not be an instrument to pursue industrial or commercial policy objectives”.23 Questions remain about how this restriction impinges upon the ability of generic producers to make profits. Originally, the US had tried to limit the motivations to “humanitarian” only. However, preceding public outcries, the Chairperson’s statement was left more ambiguous. Some of the above processes do not create substantial burdens; others do.24 Fellmeth sees the central problem being that “developers must undertake the laborious and time-consuming task of obtaining marketing approval from the government agencies charged with protecting public health.”25 Essentially, the largest hurdle will not be imposed by the international system, but by the rigorous hurdles in domestic exporter legislation. As such, the domestic government has a crucial role to ensure that a legislative scheme allowing for the issuance of compulsory licenses for export works. II. LEGAL INITIATIVES TO IMPLEMENT THE AUGUST 30TH AGREEMENT The General Council Chairperson’s Statement available at http://www.wto.org/english/news_e/news03_e/trips_stat_28aug03_e.htm 24 The above list, and numerous variations of this list, all seem to assume that the motivation for a compulsory license will be initiated in the importing country. This need not be the case as will be addressed below. 25 Aaron Xavier Fellmeth Secrecy, Monopoly, and Access to Pharmaceuticals in International Trade Law: Protection of Marketing Approval Data Under the TRIPS Agreement 45 Harv. Int'l L.J. 443 at 445. 23 Since August 30th, 2003 a number of developed countries have taken steps to implement the Agreement. Presently, only Canada, Norway, the Netherlands and, just recently, India have passed legislation. Others are currently pursuing and evaluating potential legislation. This section of the paper will provide an analysis of what legislation does exist and what other frameworks are being proposed. Canada: Canada was one of the first to initiate legislation to take advantage of the August 30th agreement. Proposed legislation was introduced as Bill-56 in early November 2003, and was well received by many NGOs and generic pharmaceutical producers in Canada.26 Following extensive debate and consultation with various parties, the Jean Chrétien Pledge to Africa Act (Bill C-9) received Royal Assent on May 14, 2004.27 The legislation will take effect once all the companion legislation is passed. It was rumoured this would occur in January 2005; this was not the case. As of yet, there is little word as to when the regulations will be in place and the legislation will be in effect. The Canadian legislation is a product of compromise between the various stakeholders. Bill C-9, unfortunately, uses the August 30th Agreement as a floor and builds upwards. Its purpose is clear; section 21.01 lays out the goal of the legislation: “to facilitate access to pharmaceutical products to address public health problems afflicting many developing, and least developed countries, especially those resulting from HIV/AIDS, tuberculosis, malaria and other epidemics.”28 The legislation has a series of schedules. Schedule 1 is a list of patented drugs that can be produced under compulsory licenses. For the most part, the list is the WHO’s list of essential medicines that are patented in Canada. Schedules 2-4, respectively, lists the least developed countries, the developing countries that have chosen not to notify the WTO that they do not intend to import drugs under compulsory licenses, and the member countries that have stated they will issue compulsory licenses only in situations of emergency.29 The drugs do not have to be sold directly to government of the importing state. However, the applicant must provide the name of the governmental entity or “the person or entity permitted by the government of the importing country”.30 As such, an NGO31 could act as the purchaser but would have to be permitted by the importing state to do so. In addition, the NGO would have to inform the Canadian regulators what country wants which particular products and in what amount. It also appears that separate approval must be obtained for each state that the drugs will be imported to. Apotex press release, “Canadian-Owned Generic Company Prepared To Provide HIV/AIDS Drug to Developing Nations” November 7, 2003 available at http://www.apotex.com/PressReleases/2003110701.asp. (last visited March 29, 2005). 27 See www.hansard.ca 28 Section 21.01. 29 s.21.03. 30 s.21.04(2)(f). 31 For the purposes of this paper NGO is seen to include nongovernmental organization and intergovernmental organization (e.g. MSF, UNAIDS, The Global Fund, The World Bank, etc.) 26 When a party wishes to apply for a compulsory license they must file a notice of intent to apply to the Commissioner of Patents and attach a series of additional documents.32 They must provide notice of intent to identify the product, prescribing information, the quantity to be manufactured, information about the patentee, the importing country, and the terms of the contract that establishes how the product will be sold.33 Additionally, documentation must be provided: a copy of the WTO notification of intent to import (if they are a member state), the importers notification to the Canadian government and the patent status of the product in the importing country.34 The applicant must also provide the Commissioner with a solemn and statutory declaration stating that at least 30 days prior to filling the application the applicant sought form the patentee(s) a license to manufacture and sell the product to the importing country on reasonable terms and conditions and provided the patentee(s) with the information provided to the Commissioner with the application.35 The Commissioner will grant the compulsory licence once the applicant has paid the prescribed fee, and the below conditions are met:36 All the requirements in the regulations have been met (these regulations have not yet been published); The Minister of Health has informed the Commission that the product meets all relevant Food and Drugs Act requirements; The applicant has provided notice to the patent holders as required under s. 21.04; and Provides a certified copy of the notice given to the WTO specifying the product and quantities required by the WTO member (if they are a member) Before export, the licensee is responsible for publishing on a website the importing country, name and quantity of the product sold and other information required by the Food and Drug Regulations.37 In addition, a royalty must be paid to the patent holder. The Governor in Council who “must consider the humanitarian and noncommercial reasons underlying the issuance of authorizations” determines the royalty. The royalty is tied to the import countries U.N. Human Development Index (HDI). With the lower HDIs of developing countries, a generic firm is unlikely to have to pay royalty rates higher than 3%, and for many importing countries the rate will be below 1% of the 32 see generally s.21.04. s. 21.04(2). 34 s. 21.04(3). 35 ss.21.04(3)(c)(i)(ii) 36 ss.21.04(3) 37 s. 21.07 33 total value of the product being exported.38 If there is more than one patent holder, the royalty is divided equally among them.39 The compulsory licence is issue for a period of 2 years, unless another period is prescribed as per the regulations.40 This can be renewed for an additional 2 years by the Commissioner if the licensee applies, pays the fee and certifies that the quantity intended to have been exported was not exported before the licence expired. It can be renewed only once.41 The licensee’s authorization is non-transferable unless it is permitted by paragraph 31(e) of the TRIPS Agreement, which provides that the use of compulsory licensed product be non-assignable, “except with that part of the enterprise or goodwill which enjoys such use”.42 The compulsory licence expires when the two year licence ends, the Minister of Health no longer believes that the product meets the requirements of the Food and Drugs Act, the day the last of the product provided for in the licence is exported, 30 days after the product or importing country is removed from the schedules under the Act or any other date established in the regulations.43 The patentee can file a challenge to the Federal Court of Canada, challenging the accuracy of the information given by the licensee, its failure to meet obligations under the bill, or that the product is being diverted from the importing country. If this is demonstrated, the Federal Court can terminate the licence under section 21.14. The amendments also limit the scope of any commercial enterprising. If the price of the product is equal to or greater than 25% of the average price of the Canadian equivalent sold by, of with the consent of the patentee, the patentee can apply to the Federal Court for a ruling that the agreement is commercial in nature. The Court is instructed to consider the exporter’s need to make a reasonably sufficient return to sustain continued participation in humanitarian initiatives as well as the ordinary levels of profitability of commercial pharmaceutical agreements in Canada. Moreover, the Court is instructed to consider the international trends in prices of the products sold for humanitarian purposes. Richard Elliot “Generics for the developing world: a comparison of three approaches to implementing the WTO (World Trade Organisation) decision (F)” available at http://www.aidslaw.ca/Maincontent/issues/cts/Scrip-article-RElliot-241104.pdf (last visited April 27, 2005) 39 s. 21.08 40 s. 21.09 41 s. 21.12. 42 s. 21.11. 43 s. 21.13. 38 Norway:44 Norway also implemented legislation in May of 2004. It differs from the Canadian legislation. The Norwegian legislation does not contain a schedule of pharmaceutical products. Instead, a licensee is able to export “pharmaceutical products” as defined with reasonable breadth in the WTO decision. Whereas importing countries that are not WTO members must declare a national emergency to be considered under the Canadian legislation, the Norwegian legislation does not impose this burden, which was flatly rejected in the WTO negotiations. The Norwegian legislation is vague and uncertain about the nature of the duties of the generic firms to obtain a voluntary license. In addition to these uncertainties, the legislation is also unclear regarding what constitutes “adequate remuneration” for the patent holder.45 The European Commission: The European Commission has but forward a proposal to allow for uniform implementation of the conditions for granting compulsory licenses. EU members that have the capacity to issue compulsory licenses under domestic law are able to export.46 The EU draft regulation still requires approval by EU institutions. The EU proposal only allows the export of generic products to WTO members.47 Like the Norwegian legislation, the EC’s draft legislation is also vague, requiring that a generic firm attempt to obtain a voluntary license for a “reasonable period of time” which takes into account whether the importing member has declared a national emergency.48 Evidence of these attempts must be provided with the application.49 The legislation asserts that the “product(s) shall only be exported if those countries have issued a compulsory licence for the import and sale of the products”.50 Moreover, the EU draft legislation does not provide any material guidance on royalty rates51; however, this can be done in the domestic implementation by member states. There appears to be no time limit on the compulsory license. It can be terminated only if the licence conditions are not respected or the circumstances that lead to the licence no longer exist.52 Netherlands: 44 For jurisdictions outside Canada the analysis shall focus on the differences between the varying legislation, taking the Canadian as a base point, and not go through the specific provisions. Moreover, aside from India, none of the countries addressed below have a generic industry equal to the size of the Canadian industry. 45 Elliot supra note 37 46 Article 3. 47 Article 4. 48 Article 7. 49 Article 5(3)(f). 50 Article 8. 51 Article 8(9) requires: licensee shall be responsible for the payment of adequate remuneration to the right holder as determined by the competent authority taking into account the economic value of the use that has been authorised under the licence to the importing WTO member(s) concerned. 52 Article 14. On December 21, 2004, the State Gazzette published the “Policy rules on issuing compulsory licences pursuant to WTO Decision WT/L/540 on the implementation of paragraph 6 of the Doha Declaration on the TRIPS Agreement and public health, under section 57, subsection 1 of the Kingdom Act on Patents of 1995”.53 The Dutch legislation does not create an independent list of products, but relies on the WHO list.54 The importing state does not have to be a WTO member. The Minister is under a duty to ensure that the patent holder is unwilling to issue a voluntary license before issuing a compulsory license. However, in urgent cases the Minister is not required to investigate if the patentee was willing to issue a voluntary license.55 The legislation does not specifically lay out how royalty levels will be determined, but does state the economic value of the products in the importing state must be taken into consideration.56 The licensee must provide information on how diversion will be avoided.57 Importantly, the legislation specifically contemplates NGOs acting for a group of states: “The application shall be accompanied by an order addressed to the pharmaceutical manufacturer from an importing state, a group of states, or non-governmental organisation acting for one or more importing states”.58 India: India recently passed the Indian Patents (Amendment) Bill, 2005 and addresses the issuing of compulsory licences for export as provided for by the August 30th Agreement.59 Based on a reading of the provision dealing with the compulsory licenses, the new patent act is cursory in explaining the requirements. It only demands that a licence be issued in the importing country without manufacturing capacity and used to address public health concerns. Moreover, these requirements are without prejudice to the extent to which pharmaceutical products produced under a compulsory license can be exported under any other provision of the act.60 Regardless of how the legislation is crafted, there is a real and more general concern the India will lack the simple administrative process for issuing compulsory licenses. The questions surrounding the new Indian regime necessitates new and viable options in other regimes.61 Other Developments: Other states have noted intentions to move forward with legislation to implement the August 30th Agreement. According to France’s Minister of Health, Philippe Douste- 53 The legislation was obtained online through the CPTech at http://www.cptech.org/ip/health/cl/netherlands-export-rules.html (last visited April 27, 2005). 54 Article 1. 55 Article 2. 56 Article 5. 57 Article 3. 58 Article 3(2). 59 See Appendix B for the relevant section of the Indian Patent Amendment Act (2005). 60 Section 92A, Indian Patents Amendment Bill 2005 available at http://lawmin.nic.in/Patents%20Amendment%20Ordinance%202004.pdf. 61 See Amit Gupta “Patent Rights on Pharmaceutical Products and Affordable Drugs: Can TRIPS Provide a Solution” 2 Buff. Intell. Prop. L.J. 127 Blazy, “The bill is ready and we will be able to submit it to parliament in January.”62 In addition, Switzerland is in the process of proposing amendments to their Patent Act and on November 26, 2004 15 Congressmen introduced a draft Bill for Amending Korean Patent Act to the National Assembly.63 III. WHAT COMPULSORY LICENSE LEGISLATION SHOULD AND SHOULD NOT DO: THE CANADIAN EXAMPLE The end goal of the various implementing legislation is simple – to bring cheap but essential medication to those who need it but cannot produce it themselves. The means are more difficult. For better or worse, the August 30th Agreement laid out the loose framework for issuing compulsory licenses. Member states have implemented the General Council decision, often complicated the procedure and narrowed the scope of who can help, who can be helped and what can be provided. 1. The Key Issues: There are a number of differences among the legislations described above. In addition, there were approaches discussed but not taken (as demonstrated by the legislative history of Canada’s Bill C-9). None of the domestic implementing legislations are perfect, and they present various strengths and weaknesses. For legislation to be effective it must support commercial incentives that allow generic companies to participate. This means legislation that creates processes that are flexible, efficient and certain. If the process is inflexible, it will have trouble adapting to the varying needs of the parties. If the process is inefficient and burdened with various bureaucratic processes, it will increase the costs for generic companies and provide more opportunities for the patent holder to oppose the issuing of compulsory licences. If the process is uncertain and the likelihood of commercial success unclear, few generic manufacturers will risk the opportunity costs for what is likely to be a small return on their investment. The central issues that will affect these concerns include, but are not limited to: the right of first refusal, the listing of exportable products and importers, the remuneration for the patent holder, the role of NGOs, how the risk of diversion is addressed, the domestic requirements for registering the generic product, the non-commercial restrictions and the procurement role of NGOs. Each of these is discussed below. Common Issues Responsibility to seek a voluntary licence and the Right of first refusal The August 30th Agreement requires that applicants for a compulsory license first seek a voluntary licence on “reasonable commercial terms and conditions”.64 A “France to apply WTO generic drug pact by 2005” available at http://www.expatica.com/source/site_article.asp?subchannel_id=58&story_id=14233&name=France+to+a pply+WTO+generic+drug+pact+by+2005 63 See CPTech at http://www.cptech.org/ip/health/cl/ (last visited April 27, 2005) 64 August 30th Agreement supra note 20. 62 compulsory licence can only be issued if these efforts do not succeed within a “reasonable period of time”.65 This resulted in a right of first refusal under the original Canadian legislation and considerable criticism.66 Since then, the right of first refusal has been abandoned by the legislative scheme, and was not incorporated into the domestic legislation of other jurisdictions. The original Canadian legislation created a duty for the Commissioner of Patents to provide the patent holder with a copy of the application. This provided all the details of the contract.67 This is a significant disincentive for generic manufacturers.68 Upon viewing the application, the patentee could then undercut the contract and provide the drugs at a slightly lower cost. The motivation would be two-fold. First, the patent holder would be able to produce the drugs more cheaply since the machinery is already geared towards such production and there is no need to demonstrate bioequivalence. Moreover, it would be a strong warning to other generic companies not to invest time and money establishing a contract for generic production. Providing a right of first refusal would likely scuttle any commercial incentive for generic companies and therefore seriously undermine the legislative attempt to implement the August 30th Agreement.69 Richard Elliot, of the Canadian HIV/AIDS Legal Network, complements the Canadian legislative for keeping the “reasonable period of time” short (30 days).70 In addition, the duty to negotiate a voluntary licence is left with the applicant; therefore, the patent holder will not be provided with the details of the possible contract. Nonetheless, Elliot remains critical. The Canadian law could adopt the greater flexibility allowed for under TRIPS which allows for the issuance of a compulsory licence without seeking a voluntary licence. It can be waived in cases of emergency or public non-commercial use of the patent, or where the compulsory licence is issued to remedy an anticompetitive practice. This would eliminate the 30-day time period and added possibility of the patent holder hijacking the process.71 The CGPA notes that the patent holder already has the de 65 Ibid. See “Getting the Balance Right” a report by World Vision to the Standing Committee on Industry, Science and Technology regarding Bill C-9 (March 5, 2004) which commented, “the greatest concern about Bill C-9 relates to the provisions for the second ‘right of first refusal’.” Available at http://www.worldvision.ca/home/media/GettingtheBalanceRight-WorldVisionBriefonBillC-9.pdf 67 See the legislative summary of Bill C-9 available through Hansard at www.hansard.ca. 68 World Vision Report supra note 65, see also CGPA Report to the Standing Committee on Standing Committee on Industry, Science and Technology regarding Bill C-9, February 2004 available at www.hansard.ca. 69 In their submissions to the Standing Committee on Industry, Science and Technology reviewing the proposed legislation the CGPA asserted, “Canadian generic pharmaceutical companies will not seek out international contracts if they are required to pass any business they find to the brand-name company that holds the Canadian patent (and that company’s generic subsidiaries or licensees). Our member companies will be unlikely to participate if they do not have the ability to recoup their investments.” Supra note 67. 70 Elliot supra note 37. 71 It should be noted that having the patent holder “hijack” the agreement to provide medicines at low costs is beneficial on a one-off analysis. However, this would greatly reduce incentive for the generic manufacturer to initiate an application for a compulsory licence in the future. The lack of potential competition removes one of the chief incentives for the patent holder to provide essential medicines at reduced prices. 66 facto right of first refusal, “as they are the current patent holders of the products covered under this legislation”.72 This being said, the Canadian legislation remains a strong model. It provides a certain and relatively risk-free process for meeting the requirements of the Agreement. The European, Norwegian and Dutch legislation are vaguer on the demands for how a voluntary licence are met, creating an opportunity for slow down and legal challenge. Listing Products: The Canadian legislation has added an unnecessary limitation by creating a separate schedule of products for which a compulsory licence may be issued. TRIPS does not require Canada to create an independent list of products that can be exported. While a list was proposed at the WTO negotiations, it was rejected by WTO members.73 The various European implementations allow for exportation of “pharmaceutical products” as defined in the WTO decisions. The Agreement defines “pharmaceutical product” as including “any [patented product] needed to address the public health problems as recognized in paragraph 1 of the [Doha] Declaration.”74 International mechanisms already exist to ascertain what products are needed by developing countries.75 Canada’s decision to create a specific list of products, despite providing for the addition of other products, creates an unnecessary delay, further inflexibility and an additional opportunity for political lobbying by patent holders. In Canada new medicines were excluded from the list after drug industry lobbying. As an example, Bayer was able to keep moxifloxacin, a pneumonia therapy, off the list of medicines.76 As the original list was passing through the Canadian Parliament there was political lobbying regarding the inclusion of certain pharmaceutical products. Julie Tam of the CGPA explained many of the situations in which generic companies would seek compulsory licenses could arise unexpectedly.77 Clarithromycin is an anti-biotic often used to treat conditions such as pneumonia. Generic manufacturers were gearing up for production of Clarithromycin in anticipation of the patent expiring. However, the patent holder initiated judicial procedures pushing back the expiry date. At the time, if the amendments were in effect and Clarithromycin was on the list, the costs to the generic company of providing the drug under a compulsory licence would have been minimal. They could have supplied the drug under a two-year license while waiting to provide the drug domestically. However, Clarithromycin was not on the schedule of drugs available for export. Not surprisingly, Clarithromycin was one of the drugs that was removed from the list following lobbying by the research-based pharmaceutical industry. 72 Listing the Import Countries: CGPA Report supra note 67. Ellen ‘t Hoen supra note 14 74 Doha Declaration supra note 20. 75 e.g. the WHO’s list of essential medicines 76 t’ Hoen supra note 14; more generally see Elliot supra note 37 and CGPA Report supra note 67 77 This information was obtained through a telephone interview with Julie Tam in March, 2005 73 Listing potential importing countries creates unnecessary limitations. The Canadian legislation does not require that the importing country be a WTO member. Instead, Bill C-9 includes all least-developed countries and most developing countries. The proposed EU legislation does require the importing country be a WTO member and creates a distinction that does not reflect the nature of problem. Perhaps the Canadian law creates an unnecessary hurdle in requiring the developing nation to declare a national emergency as a precondition to import. WTO members do not face this same requirement, but only have to identify the quantity required, demonstrate an inability to domestically produce the drug and to issue a compulsory license.78 Appropriately, the Norwegian legislation only considers the need of the importing country rather than membership in a trade organization, and does not place the responsibility on developing nations to declare an emergency when evidence of the emergency is patently clear. Adequate Remuneration and Royalties for Patent Owners: Paragraph 3 of the Agreement requires that “adequate remuneration pursuant to Article 31(h) of the TRIPS Agreement shall be paid.”79 Royalty payments are used to fulfil this requirement. Originally the Canadian legislation imposed a royalty rate of 2%. However, generic manufactures contested this, asserting that this increased cost would reduce the incentive and what is already a slim profit margin.80 The current legislation recognized these concerns and reduced the royalties by incorporating a calculation that includes the UN’s Human Development Index. As a result, many of the least developed countries, that need the drugs most desperately, will only require a royalty payment of less than 1%. The Canadian process is good in that it provides cost certainty for the generic manufacturers and it is not overly burdensome. Conversely, the uncertainty present in the EU and Norwegian systems is a major flaw. Various concerns must be balanced, and the remuneration is meant to recognize the commercial loss of the patent holder.81 Using the HDI to ascertain the loss to the patent holder approaches a more accurate reflection of that loss. Trade Diversion: Concerns for trade diversion and parallel importing attract a great deal of attention from research-based pharmaceutical companies and government legislatures. In theory, the concern is clear and logical. A first line treatment, Cipla Triomune, is available for US$154 in developing countries and the bioequivalent sells for US$8773 in Australia.82 It would be a profitable business to purchase the drugs in Sub-Saharan Africa and then re-export them to Australia. Kevin Outterson notes that the market discrepancies are roughly similar to cocaine. In Columbia, the street value of one gram of cocaine is about US$3 to US$5; in the North America it is about US$100. Outterson then promptly asserts, “reality appears to depart from the neo-classical economic model, for there is 78 see Elliot supra note 37 supra note 20 80 See CGPA Report supra note 67 81 Mercurio supra note 11 at 242-244. 82 MSF paper supra note 3 79 quite limited evidence of dysfunctional arbitrage”.83 India has produced generic drugs for decades, yet they have not made there way into the western market. Much of the concern appears to be a white elephant. In 2002 GlaxoSmithKline claimed that 36 thousand packages of HIV/AIDS drugs (US$18 million) were diverted from West Africa to the EU. They brought a suit against various participants include Dowellhurst, a legal parallel trader. However, during the course of the trial it became apparent that 99% of the packages were not Glaxo’s charitable donations, but were ordinary commercial sales to Africa being sold at prices similar to those in Europe. Moreover, 99% of the packages were sold by Glaxo to addresses in France and likely never made it to Africa to be diverted, and Glaxo made no attempts to differentiate the drugs destined for charitable donation.84 While there have been other problems of local corruption in Indonesia, Chile and Lebanon, these do not provide evidence of diversion into western markets.85 That being said, legislation should not ignore the potential. Moreover, lowincome markets should not bear the administrative burden of anti-diversion measures. As such, the state issuing the compulsory licence should establish a regime ensuring that the products destined for developing countries are marked for easy differentiation. The WTO decision requires that, “Suppliers should distinguish such products through special packaging and/or special colouring/shaping of the products themselves, provided that such distinction is feasible and does not have a significant impact on price.”86 Provided that the requirements for product differentiation to not impose too burdensome a cost, it is important that they can be distinguished. There are already a number of industry practices regarding how to differentiate products. This requirement will likely not create a problem. Perhaps of greater concern than how exactly the products are to be distinguished is who is handling the products. Outterson notes, “the manufacturer also has the responsibility to deliver the essential medicines to a reputable supply chain”.87 This concern will be addressed below in considering who should be able to purchase drugs through compulsory licenses. “Commercial in nature” provision: The patent holder can challenge the application of a generic in the Canadian Federal Court if the price of the generic product in the contract is 25% or more of what the brand company charges for the product in Canada. This requirement derives from the General Council’s statements following the August 30th Agreement that the motivation behind issuing a compulsory license not be commercial in nature. This is well below the 63% of the brand price At which Canadian generic products typically enter the market. 83 Outterson supra note 5 at 262. Outterson supra note 5 at 263-265. 85 ibid at 265. 86 paragraph 2(b). 87 Outterson supra note 5 at 266. 84 However, this restriction clearly makes sense. The legislation is not designed to slowly introduce a competitive market based on current prices in the developed world. The goal is to drastically reduce the cost of medicines. If a drug were more than 25% of the price in developed countries, this would do little to resolve price-related access issues. National drug registration: A further impediment/cost in successfully acquiring a compulsory licence is the requirement to gain approval to produce the drugs under national drug registration authorities (NDRAs). This requires first reverse-engineering a patented medicine. Essentially, there are requirements that drugs made for export in Canada must meet Canadian safety standards. For generic medicines, this means demonstrating that the copy has the same effect on the body as the original product. This can become difficult in the HIV/AIDS context. First, there are limitations on access to data on the original studies of the patent holder. Without that data the generic companies must perform their own costly studies. Also, constructing FDCs will require a new set of studies.88 There are problems with the transnational administration of drug regulation. Many reputable systems duplicate themselves. The WHO pre-qualification system is not trusted by many developed countries, especially the United States. While any nation has the right to question the approval processes of others, one may question its rational when the drugs are only being made for export to a country that has accepted the WHO prequalification process. In addition, forcing generics to reverse-engineer the patent, or reproduce studies through extended data exclusivity (the US is currently negotiating TRIPS Plus agreements that extend data exclusivity for 5 or 10 years) is merely a means of artificially extending the patent. Depending on how states choose to recognise the balance between more affordable products and new innovation; these requirements may be acceptable. However, when legislation is designed to subvert patent laws under situations of (inter)national emergency it makes little sense to increase the cost on generic manufacturers and significantly delay the export of the essential medicines. 88 Jack Kay, COO of Apotex explained that it will entail demonstrating that the drugs, when combined, have the same effect on the effect on the blood as they do when administered separately and at different times. Uniquely Canadian and Not to be Repeated: Term of licence: The two-year limitation of the compulsory licence creates a further disincentive for generic companies. Generic manufacturers are expected to have small profit margins on a drug they are able to produce for two years. The license can only be renewed once, if the contract is not yet complete. A large percentage of the costs in producing the medicines are in proving bioequivalence and gearing up the plants for production of the particular product. Moreover, this complicates other uncertainties. Many of the drugs that are need are FDCs. These products will have multiple patent holders. Obtaining voluntary license for the products may not be simultaneous. As such, the narrow window of opportunity is a major disincentive for the generic industry (which may be made shorter depending on how the legislation plays out with various patents). This also creates a problem for importing countries that are not able to develop long-term health care solutions. The various European legislations do not have this limitation. Termination for “inaccurate information” Section 21.14 of Bill C-9 allows for patent holders to challenge the issuing of a compulsory licence if the application contained “material information” that is “inaccurate”. Lawyers who represent generic manufacturers are necessarily wary of such language.89 While the veracity of an application is important, there are always a number of uncertainties and gaps in information when working with least developed and developing countries. Fears that the accuracy of the information will be judged to a Canadian standard create yet another uncertainty for generic manufacturers. In essence, it provides a further opportunity for costly litigation. The EC legislation does not specifically arm the patent holder with this opportunity to challenge the licence. It goes more to what should be the central concern – that the conditions for the licence are met and the circumstances that precipitated the licence continue to exist. Procurement and the Role of Non Governmental Organizations: The role NGOs may play in procuring medication has been a hotly contested issue. In Canada, states remain the central player as the importer. An NGO can act as a purchaser but must be permitted by the importing state. Moreover, it appears separate approval must be obtained for each state that the drugs will be imported to. In their submissions to the Senate Foreign Affairs committee the research based companies supported the central role of the importing state: It is countries who will be held accountable at TRIPS Council or to Canada through diplomatic channels should aspects of this legislation not be followed. Therefore, their involvement is important. Experience on the ground also tells us The possibility of litigation and challenges under 21.14 was described by Tim Gilbert of Gilbert’s LLP as one of the “hidden barriers to access”. Gilbert’s LLP is a Toronto law firm that specializes in intellectual property law and represents a number of generic manufacturers. 89 that the best success happens when the local governments are involved and help take responsibility for improved health care regulations.90 This is not necessarily true. First, importing countries may not be members of the WTO, and are therefore not accountable to the TRIPS council. Second, the “best success” does not always involve the government. Involving the “government” of Somalia or the Democratic Republic of the Congo will do very little in ensuring access throughout the country. As a best-case scenario, the importing government can and should be involved, but the legislation should not require this. The EC legislation is vague on the issue, only requiring that the “product(s) shall only be exported if those countries have issued a compulsory licence for the import and sale of the products” (as is required by the August 30th Agreement).91 The Dutch legislation is more specific regarding the possibility of NGO involvement. “The application shall be accompanied by an order addressed to the pharmaceutical manufacturer from an importing state, a group of states, or non-governmental organisation acting for one or more importing states”.92 Certain NGOs have the potential to play a central role in getting essential medicines to developing countries. Many NGOs are well structured and financed. They have strong connections in possible importing countries that generic pharmaceutical companies do not. Moreover, generic companies may be more comfortable dealing with well-established NGOs. First, they may provide a buyer that is better known to the generic company. Second, a research-based company will fear that negative publicity that would arise if they were to challenge the compulsory licence of a generic manufacturer that is acting in concert with an NGO. However, most legislation leaves rules governing their involvement vague and peripheral. This should be changed. Current Concerns: To issue a compulsory license there are at least three players whose motivations (whether commercial or moral) must be aligned. The importing country has to develop the procedures and will have to notify the WTO (if it is a member) or the exporting state (if it is a non-member) of its need for a particular medicine. Moreover, when it is the central actor on the importing side, it must be able to make concrete the need and demand for the generic producer so that they can create a contract able to specify particular amounts. The exporting country must implement domestic legislation to allow for the issuing of compulsory licenses. In so doing, it must minimize the barriers by keeping the legislation flexible, efficient and certain. Third, and central, the commercial incentives must motivate generic producers to apply for compulsory licenses. The more uncertain and costly the process, the less likely generic firms will be to apply for a license. a. Generic Incentives: On November 6, 2003 then Prime Minister Jean Chrétien introduced legislation to implement the August 30th Agreement. On November 7, 2003 the Apotex Group of Remarks by Canada’s Research-Based Pharmaceutical Companies (May 12, 2004) available at www.hansard.ca. 91 Article 8. 92 Article 3(2). 90 Companies announced that it was prepared to produce a key HIV/AIDS drug under the legislation. Apotex is Canadian-owned pharmaceutical company with worldwide sales totalling over $800 million (Canadian) per year.93 In Canada it employs 5,500 Canadians, investing $190 million in research and development. Apotex asserted that it could manufacture Apo-Zidovidine, a generic equivalent of Retrovir-AZTÒ. Jack Kay, President and COO explained, “With our expertise and experience in producing the generic equivalent of AZTÒ, we can gear up fairly quickly…We are ready to assist in the fight against the HIV/AIDS disaster in Africa and other parts of the developing world.”94 The Canadian Generic Pharmaceutical Association (CGPA) has been involved throughout the legislative process. The CGPA was highly critical of the initial legislation tabled by the government and succeeded in pushing for changes regarding the right of first refusal and the high royalty rates. Its continued involvement demonstrates the prima facia interest of the generic industry. Generic incentives must be measured through a corporate perspective. While a desire to be good corporate citizens can grease the wheels, one cannot expect a pharmaceutical company to take massive and uncertain risks for small and temporary gains. While risk will always be there, and the reward is to be found in an impoverished market, a workable balance is possible because of the sheer volume of the pandemic. Creating Markets of Scale: For generic companies to begin the long process of seeking an application there must be a commercial reward. This does not need to be the fabled pot of gold, however, producing 200,000 tablets for a medical clinic in Harare does not justify the bureaucratic and legal costs of seeking an application, or the production and opportunity costs in research and product development. Those in the industry suspect that between 5 and 10 million dosage units would be required to justify the above cost and risks.95 As such, if an FDC is taken twice daily and the licence is limited to two years this would require (under perfect situations) between 3500 and 7000 patients all taking the medication for two years. Carlos Correa notes that the August 30th Agreement, “recognizes that the viability of the ‘solution’ largely depends on the existence of economies of scale that justify production”.96 Keith Maskus recognises the immense needs of poorer nations but "because the eligible import markets in really small countries will not be large, generic 93 Apotex press release available at http://www.apotex.com/CorporateInformation/Default.asp (last visited April 5, 2005). 94 Apotex press release available at http://www.apotex.com/PressReleases/20031107-01.asp (last visited April 5, 2005). 95 Conversations with Tim Gilbert, Julie Tam and Jack Kay. 96 Carlos M. Correa “The Nexus Symposium: An Interdisciplinary Forum on the Impact of International Patent and Trade Agreements in the Fight Against HIV and AIDS Introduction: TRIPS and Access to Drugs: Toward a Solution for Developing Countries Without Manufacturing Capacity (2003) 17 Emory Int'l L. Rev. 389 at 403. producers may not be interested in producing such small volumes and foregoing chances for economies of scale.”97 Combining markets can ameliorate this problem, and this establishes a potentially crucial role for NGOs. Presently, 700,000 Africans are receiving treatment. This is a massive market, even if profits per patient are minute; the possibility of gaining a foothold into this market could provide the necessary motivation. NGOs such as the Clinton Foundation, MSF and the Global Fund straddle borders and can access tens of thousands of patients.98 While a small fraction of the total population in need, this can still provide a sufficient market for a generic company to begin production and undergo the application process, especially with the potential of other markets. However, for this to be possible under the Canadian legislation MSF must involve all importing countries concerned and the generic producer would likely have to seek approval for each country MSF of another NGO proposes to use the drugs. The Dutch legislation specifically allows for a NGO to purchase for “a state or group of states”.99 This direct recognition is essential to creating the flexibilities and options necessary to make compulsory licences work. Ideally, the Canadian legislation would have made this clearer. As a baseline, the August 30th Agreement only requires that importing governments demonstrate an inability for domestic production and prevent diversion. The role of NGOs as a central procurement agency would likely not violate the Agreement. Moreover, current implementing legislations should design their requirements with this potential in mind. Briefly, a list of reputable NGOs can be established, the compulsory licensing procedures should specifically allow for single applications with multiple import states. In short, the legislative vision of a functioning compulsory licensing system should not be restricted to a generic manufacturer in a developed country contracting with a developing country for the provision of essential medicines. It should be expanded to clearly provide for the buyers to be responsible nongovernmental organizations who provide treatment in multiple countries in concert with those governments. Providing Market Certainty: The process is plagued with uncertainty. A pharmaceutical company considering a compulsory licence is unsure of what the legislation requires (but perhaps certain that 97 Keith Maskus, TRIPS, Drug Patents and Access to Medicines-Balancing Incentives for R&D with Public Health Concerns, Sept. 4, 2003, at 1, available at http://www.developmentgateway.org/download/206719/Maskus_on_TRIPS,_Drug_ Patents at 2 Quoted in Correa supra note 95. 98 Currently MSF treats 25,000 patients supra note 3. Also, the Clinton Foundation recently announced plans to treat 10,000 children in developing nations by the end of 2005 and is working with UNICEF to expand this number to 60,000 by the end of 2006. Moreover, the Clinton Foundation works with more than 30 countries. See “President Clinton Announces HIV/AIDS Initiative to Provide Treatment to 10,000 Children in Developing Countries” (April 11, 2005). The Global Fund hopes to provide the resources to treat 1.6 million people within the next five years. See Global Fund website available http://www.theglobalfund.org/en/about/aids/default.asp. Last visited April 13, 2005. 99 Article 3(2). there will be litigation resulting from these uncertainties), uncertain about real support from the government, unsure about market sizes and the prices it can charge. They may also be unsure about receiving the money. Many of the generic industry’s concerns would be mitigated if a group came forward, provided the money for 10 million doses, said ship the product here, and if this contract is successful, there is a larger market that will be opened. However, this is not the case. Aside from navigating the legislation, the market is uncertain and payment remains a question. Well-funded NGOs and foundations may provide a surer business partner. In the absence their involvement, government programs, such as Export Development Canada (EDC) or CIDA should make every effort to insure the business ventures undertaken. Current Developments and Potential for success: As the implementing regulations waiver in limbo, NGOs, academic institutions, a foreign government, a laws firm and a generic manufacturer are testing the workability of the Canadian legislation. Analysis and critique aside, the real test of how well the legislation works will only be answered by actually using it. To the author’s knowledge, there are two projects currently underway. b) Apotex and Medicines Sans Frontier: Apotex has expressed and demonstrated continued interest to provide generic medicines to developing nations. It is currently working in concert with MSF to gain regulatory approval of a FDC drug and have undertaken discussions with regulatory bodies toward this end. Apotex has also begun research on how to produce a FDC that includes Lamivudine, Nevirapine and Zidovudine. According to Jack Kay100, discussions with the Therapeutic Products Directorate of Health Canada101 in Ottawa have demonstrated support for obtaining an approval for generic products that will be produced under a compulsory licence. Nonetheless, Mr. Kay suspects that the process will require 12-18 months before approval is granted. Apotex has discussed the issue with the research-based pharmaceutical company that holds 2 of the 3 patents in Canada. Glaxo has indicated that there is a possibility that they will grant a voluntary licence for the products. Presently, it is hoped that MSF will be the purchaser of the medicines.102 Jack Kay recognizes that this FDC is currently being produced in India and it is unlikely that they will be able to compete with Indian prices. However, if they are able to approach the costs of the Indian products, the higher Canadian safety standards and product quality will hopefully justify the slightly higher price. Moreover, there is significant uncertainty about what India will be able to provide under their new patent regime and Brazil remains unwilling to export their generic products. As such, there is a risk (from a patient’s point of view) that Apotex generics will be required. 100 All information provided from Jack Kay was obtained through two telephone interviews in March and April 2005. 101 TPD is responsible for reviewing and approving new drugs and drugs already being sold to ensure safety and effectiveness. 102 MSF is likely interested in finding an alternative source for reliable generic products in light of the current uncertainties in India. c) Ghana, Gilberts LLP and the Access to Drugs Initiative In March of 2005 representatives from the law firm Gilberts LLP and the Access to Drugs Initiative (ADI)103 travelled to Ghana. The Ghanaian Ministry of Health, which indicated an interest in using Bill C-9 to acquire affordable generic ARVs, prompted the initiative. Ghana is not at the epicentre of the pandemic and only has a prevalence rate of roughly 3.1%104 thanks to an extensive prevention campaign that has kept infection rates down. Nonetheless, 72,000 persons in Ghana are living with HIV/AIDS; only 2000 currently have access to ARVs, which remain too expensive to permit an extensive treatment campaign. Medicines accessed through Bill C-9 would assist in the treatment of the 72,000 persons living with HIV/AIDS in Ghana. The various parties attempted to assess the viability of accessing Bill C-9. The dialogues have led to a Memorandum of Understanding (MOU) between ADI and the Ghanaian Ministry of Health. The MOU outlines how Bill C-9 can be exercised with the goal of providing sustainable treatment to Ghanaians living with HIV/AIDS. These measures include: 103 Reviewing Ghanaian patent legislation to ensure it conforms with the TRIPS Agreement and is amenable to the provisions of Bill C-9; Stimulating the supply of generic ARVs in Canada by liaising with Apotex Inc., the project’s generic drug producing partner; Fulfilling all requirements of the application procedure outlined in Bill C-9; Obtaining the necessary funding to achieve the WHO ‘3 by 5’ target in Ghana; Assisting in the training of Ghanaians to ensure the sustainability of ARV treatment sites; and Encouraging the bulk procurement of bioequivalent ARV treatment from generic producers to developing countries through organizations such as MSF. ADI is a collaborative project between the International Human Rights Program at the Faculty of Law and the Leslie Dan Faculty of Pharmacy, both at the University of Toronto. 104 UNAIDS.org. IV CONCLUDING REMARKS: A Piece of the Puzzle The AIDS pandemic continues to spread and exact a massive toll on nations struggling to develop. Providing these countries with affordable medicines is essential, not only for the millions of lives it will save, but to prevent the corrosion of the social infrastructure in nations hit most hardly. As the disease continues to spread in Eastern and Central Europe as well as Asia, the need will only grow. Procuring the drugs is only the first step, adequate health institutions are required to test for the virus, distribute the drugs, and educate patients about the importance of complying with the drug regiment to reduce the likelihood of mutation. Moreover, action will still be required to remedy deep scars to the family and social structures and the lost human capital in areas most affected by the pandemic.105 Nonetheless, none of this is possible if the first step is not made; the drugs must get to the patients. Several NGOs criticized the WTO for seeking a solution within Article 31 of TRIPS as being too bureaucratic and slow. They viewed an Article 30 solution as more practical due to the political nature of the world trading system: "Unlike the Article 31(f) amendment, an Article 30 exception [would have] allow[ed] the producer country to manufacture and export without issuing a compulsory license."106 This may very well be the case. However, the approach taken may provide an important piece of the puzzle. The concern is that affordable medication is provided to those suffering from HIV/AIDS. Whether the medication reaches Africa, Asia or South America through compulsory licences, voluntary licenses, negotiating reduced prices, donations or bulk purchasing is not important. However, if all these pathways are viable options there is a synergistic effect. The threat of a successful application motivates the patent holder to consider the other options more seriously. As explained by Outterson, “Sovereign threats of such compulsory licenses, public pressure from NGOs, and actual competition from generic companies persuaded PhRMA companies and the United States to embrace differential pricing of antiretroviral (ARV) medications for a number of poor countries combating HIV/AIDS.”107 While the Canadian legislation is far from perfect, the potential is there. Improving the legislation by increasing the time period for a compulsory license, providing a greater role for NGOs and keeping drug approval barriers to a minimum would improve the legislation and increase the likelihood and number of compulsory licenses sought. Nonetheless, the benefit of a viable threat is already visible. A generic company is beginning the process and the major patent holder has not responded with the normal hostility that exists between research based and generic pharmaceutical companies, but with an unofficial assertion that they will not stand in the way. Moreover, while Canada is one the international leader in the generic industry the drugs need not come from Canada for the legislation to be a success. If export continues in India, Brazil 105 Taylor supra note 20 at 3-5. Mercurio supra note 11 at 231; see also Gupta supra note 60. 107 Outterson supra note 5 at 224. 106 and China decide to enter the international market, or other developed countries initiate better legislation, the potential of competition from Canada will increase quality and push prices down. Again, only time will tell if this can work, and whether this will have an effect, but as MSF worker, Rachel Kiddle-Monroe, recently expressed during a presentation at the University of Toronto, “we have to hope that it can”108 and, as a number of current players are demonstrating, we have to see that it does. 108 This assertion was made at a recent lunch discussing the implementation of Bill C9. The talk was sponsored by the University of Toronto Faculty of Law’s International Human Rights Program. Appendix A: Amendments to the Canadian Patent Act 1. The Patent Act is amended by adding the following after section 21: USE OF PATENTS FOR INTERNATIONAL HUMANITARIAN PURPOSES TO ADDRESS PUBLIC HEALTH PROBLEMS Purpose 21.01 The purpose of sections 21.02 to 21.2 is to give effect to Canada's and Jean Chrétien's pledge to Africa by facilitating access to pharmaceutical products to address public health problems afflicting many developing and least-developed countries, especially those resulting from HIV/AIDS, tuberculosis, malaria and other epidemics. Definitions 21.02 The definitions in this section apply in this section and in sections 21.03 to 21.19. "authorization" « autorisation » "authorization" means an authorization granted under subsection 21.04(1), and includes an authorization renewed under subsection 21.12(1). "General Council" « Conseil général » "General Council" means the General Council of the WTO established by paragraph 2 of Article IV of the Agreement Establishing the World Trade Organization, signed at Marrakesh on April 15, 1994. "General Council Decision" « décision du Conseil général » "General Council Decision" means the decision of the General Council of August 30, 2003 respecting Article 31 of the TRIPS Agreement, including the interpretation of that decision in the General Council Chairperson's statement of that date. "patented product" « produit breveté » "patented product" means a product the making, constructing, using or selling of which in Canada would infringe a patent in the absence of the consent of the patentee. "pharmaceutical product" « produit pharmaceutique » "pharmaceutical product" means any patented product listed in Schedule 1 in, if applicable, the dosage form, the strength and the route of administration specified in that Schedule in relation to the product. "TRIPS Agreement" « Accord sur les ADPIC » "TRIPS Agreement" means the Agreement on Trade-Related Aspects of Intellectual Property Rights, being Annex 1C of the Agreement Establishing the World Trade Organization, signed at Marrakesh on April 15, 1994. "TRIPS Council" « Conseil des ADPIC » "TRIPS Council" means the council referred to in the TRIPS Agreement. "WTO" « OMC » "WTO" means the World Trade Organization established by Article I of the Agreement Establishing the World Trade Organization, signed at Marrakesh on April 15, 1994. Amending Schedules 21.03 (1) The Governor in Council may, by order, (a) on the recommendation of the Minister and the Minister of Health, amend Schedule 1 (i) by adding the name of any patented product that may be used to address public health problems afflicting many developing and least-developed countries, especially those resulting from HIV/AIDS, tuberculosis, malaria and other epidemics and, if the Governor in Council considers it appropriate to do so, by adding one or more of the following in respect of the patented product, namely, a dosage form, a strength and a route of administration, and (ii) by removing any entry listed in it; (b) on the recommendation of the Minister of Foreign Affairs, the Minister for International Trade and the Minister for International Cooperation, amend Schedule 2 by adding the name of any country recognized by the United Nations as being a least-developed country that has, (i) if it is a WTO Member, provided the TRIPS Council with a notice in writing stating that the country intends to import, in accordance with the General Council Decision, pharmaceutical products, as defined in paragraph 1(a) of that decision, and (ii) if it is not a WTO Member, provided the Government of Canada with a notice in writing through diplomatic channels stating that the country intends to import pharmaceutical products, as defined in paragraph 1(a) of the General Council Decision, that it agrees that those products will not be used for commercial purposes and that it undertakes to adopt the measures referred to in Article 4 of that decision; (c) on the recommendation of the Minister of Foreign Affairs, the Minister for International Trade and the Minister for International Cooperation, amend Schedule 3 by adding the name of any WTO Member not listed in Schedule 2 that has provided the TRIPS Council with a notice in writing stating that the WTO Member intends to import, in accordance with the General Council Decision, pharmaceutical products, as defined in paragraph 1(a) of that decision; and (d) on the recommendation of the Minister of Foreign Affairs, the Minister for International Trade and the Minister for International Cooperation, amend Schedule 4 by adding the name of (i) any WTO Member not listed in Schedule 2 or 3 that has provided the TRIPS Council with a notice in writing stating that the WTO Member intends to import, in accordance with the General Council Decision, pharmaceutical products, as defined in paragraph 1(a) of that decision, or (ii) any country that is not a WTO Member and that is named on the Organization for Economic Cooperation and Development's list of countries that are eligible for official development assistance and that has provided the Government of Canada with a notice in writing through diplomatic channels (A) stating that it is faced with a national emergency or other circumstances of extreme urgency, (B) specifying the name of the pharmaceutical product, as defined in paragraph 1(a) of the General Council Decision, and the quantity of that product, needed by the country to deal with the emergency or other urgency, (C) stating that it has no, or insufficient, pharmaceutical capacity to manufacture that product, and (D) stating that it agrees that that product will not be used for commercial purposes and that it undertakes to adopt the measures referred to in Article 4 of the General Council Decision. Restriction -- Schedule 3 (2) The Governor in Council may not add to Schedule 3 the name of any WTO Member that has notified the TRIPS Council that it will import, in accordance with the General Council Decision, pharmaceutical products, as defined in paragraph 1(a) of that decision, only if faced with a national emergency or other circumstances of extreme urgency. Removal from Schedules 2 to 4 (3) The Governor in Council may, by order, on the recommendation of the Minister of Foreign Affairs, the Minister for International Trade and the Minister for International Cooperation, amend any of Schedules 2 to 4 to remove the name of any country or WTO Member if (a) in the case of a country or WTO Member listed in Schedule 2, the country or WTO Member has ceased to be recognized by the United Nations as being a least-developed country or, in the case of a country that is not a WTO Member, the country has permitted any product imported into that country under an authorization to be used for commercial purposes or has failed to adopt the measures referred to in Article 4 of the General Council Decision; (b) in the case of a WTO Member listed in Schedule 3, the WTO Member has notified the TRIPS Council that it will import, in accordance with the General Council Decision, pharmaceutical products, as defined in paragraph 1(a) of that decision, only if faced with a national emergency or other circumstances of extreme urgency; (c) in the case of a WTO Member listed in Schedule 4, the WTO Member has revoked any notification it has given to the TRIPS Council that it will import pharmaceutical products, as defined in paragraph 1(a) of the General Council Decision, only if faced with a national emergency or other circumstances of extreme urgency; (d) in the case of a country listed in Schedule 4 that is not a WTO Member, (i) the name of the country is no longer on the Organization for Economic Co-operation and Development's list of countries that are eligible for official development assistance, (ii) the country no longer faces a national emergency or other circumstances of extreme urgency, (iii) the country has permitted any product imported into that country under an authorization to be used for commercial purposes, or (iv) the country has failed to adopt the measures referred to in Article 4 of the General Council Decision; (e) in the case of any country or WTO Member listed in Schedule 3 or 4, the country or WTO Member has become recognized by the United Nations as a least-developed country; and (f) in the case of any country or WTO Member listed in any of Schedules 2 to 4, the country has notified the Government of Canada, or the WTO Member has notified the TRIPS Council, that it will not import pharmaceutical products, as defined in paragraph 1(a) of the General Council Decision. Timeliness of orders (4) An order under this section shall be made in a timely manner. Authorization 21.04 (1) Subject to subsection (3), the Commissioner shall, on the application of any person and on the payment of the prescribed fee, authorize the person to make, construct and use a patented invention solely for purposes directly related to the manufacture of the pharmaceutical product named in the application and to sell it for export to a country or WTO Member that is listed in any of Schedules 2 to 4 and that is named in the application. Contents of application (2) The application must be in the prescribed form and set out (a) the name of the pharmaceutical product to be manufactured and sold for export under the authorization; (b) prescribed information in respect of the version of the pharmaceutical product to be manufactured and sold for export under the authorization; (c) the maximum quantity of the pharmaceutical product to be manufactured and sold for export under the authorization; (d) for each patented invention to which the application relates, the name of the patentee of the invention and the number, as recorded in the Patent Office, of the patent issued in respect of that invention; (e) the name of the country or WTO Member to which the pharmaceutical product is to be exported; (f) the name of the governmental person or entity, or the person or entity permitted by the government of the importing country, to which the product is to be sold, and prescribed information, if any, concerning that person or entity; and (g) any other information that may be prescribed. Conditions for granting of authorization (3) The Commissioner shall authorize the use of the patented invention only if (a) the applicant has complied with the prescribed requirements, if any; (b) the Minister of Health has notified the Commissioner that the version of the pharmaceutical product that is named in the application meets the requirements of the Food and Drugs Act and its regulations, including the requirements under those regulations relating to the marking, embossing, labelling and packaging that identify that version of the product as having been manufactured (i) in Canada as permitted by the General Council Decision, and (ii) in a manner that distinguishes it from the version of the pharmaceutical product sold in Canada by, or with the consent of, the patentee or patentees, as the case may be; (c) the applicant provides the Commissioner with a solemn or statutory declaration in the prescribed form stating that the applicant had, at least thirty days before filing the application, (i) sought from the patentee or, if there is more than one, from each of the patentees, by certified or registered mail, a licence to manufacture and sell the pharmaceutical product for export to the country or WTO Member named in the application on reasonable terms and conditions and that such efforts have not been successful, and (ii) provided the patentee, or each of the patentees, as the case may be, by certified or registered mail, in the written request for a licence, with the information that is in all material respects identical to the information referred to in paragraphs (2)(a) to (g); and (d) the applicant also provides the Commissioner with (i) if the application relates to a WTO Member listed in Schedule 2, a certified copy of the notice in writing that the WTO Member has provided to the TRIPS Council specifying the name of the pharmaceutical product, as defined in paragraph 1(a) of the General Council Decision, and the quantity of that product, needed by the WTO Member, and (A) a solemn or statutory declaration in the prescribed form by the person filing the application stating that the product to which the application relates is the product specified in the notice and that the product is not patented in that WTO Member, or (B) a solemn or statutory declaration in the prescribed form by the person filing the application stating that the product to which the application relates is the product specified in the notice and a certified copy of the notice in writing that the WTO Member has provided to the TRIPS Council confirming that the WTO Member has, in accordance with Article 31 of the TRIPS Agreement and the provisions of the General Council Decision, granted or intends to grant a compulsory licence to use the invention pertaining to the product, (ii) if the application relates to a country listed in Schedule 2 that is not a WTO Member, a certified copy of the notice in writing that the country has provided to the Government of Canada through diplomatic channels specifying the name of the pharmaceutical product, as defined in paragraph 1(a) of the General Council Decision, and the quantity of that product, needed by the country, and (A) a solemn or statutory declaration in the prescribed form by the person filing the application stating that the product to which the application relates is the product specified in the notice and that the product is not patented in that country, or (B) a solemn or statutory declaration in the prescribed form by the person filing the application stating that the product to which the application relates is the product specified in the notice and a certified copy of the notice in writing that the country has provided to the Government of Canada through diplomatic channels confirming that the country has granted or intends to grant a compulsory licence to use the invention pertaining to the product, (iii) if the application relates to a WTO Member listed in Schedule 3, a certified copy of the notice in writing that the WTO Member has provided to the TRIPS Council specifying the name of the pharmaceutical product, as defined in paragraph 1(a) of the General Council Decision, and the quantity of that product, needed by the WTO Member, and stating that the WTO Member has insufficient or no pharmaceutical manufacturing capacity for the production of the product to which the application relates, and (A) a solemn or statutory declaration in the prescribed form by the person filing the application stating that the product to which the application relates is not patented in that WTO Member, or (B) a certified copy of the notice in writing that the WTO Member has provided to the TRIPS Council confirming that the WTO Member has, in accordance with Article 31 of the TRIPS Agreement and the provisions of the General Council Decision, granted or intends to grant a compulsory licence to use the invention pertaining to the product, (iv) if the application relates to a WTO Member listed in Schedule 4, a certified copy of the notice in writing that the WTO Member has provided to the TRIPS Council specifying the name of the pharmaceutical product, as defined in paragraph 1(a) of the General Council Decision, and the quantity of that product, needed by the WTO Member, and stating that the WTO Member is faced with a national emergency or other circumstances of extreme urgency and that it has insufficient or no pharmaceutical manufacturing capacity for the production of the product to which the application relates, and (A) a solemn or statutory declaration in the prescribed form by the person filing the application stating that the product to which the application relates is not patented in that WTO Member, or (B) a certified copy of the notice in writing that the WTO Member has provided to the TRIPS Council confirming that the WTO Member has, in accordance with Article 31 of the TRIPS Agreement and the provisions of the General Council Decision, granted or intends to grant a compulsory licence to use the invention pertaining to the product, or (v) if the application relates to a country listed in Schedule 4 that is not a WTO Member, a certified copy of the notice in writing that the country has provided to the Government of Canada through diplomatic channels specifying the name of the pharmaceutical product, as defined in paragraph 1(a) of the General Council Decision, and the quantity of that product, needed by the country, and stating that it is faced with a national emergency or other circumstances of extreme urgency, that it has insufficient or no pharmaceutical manufacturing capacity for the production of the product to which the application relates, that it agrees that product will not be used for commercial purposes and that it undertakes to adopt the measures referred to in Article 4 of the General Council Decision, and (A) a solemn or statutory declaration in the prescribed form by the person filing the application stating that the product to which the application relates is not patented in that country, or (B) a certified copy of the notice in writing that the country has provided to the Government of Canada through diplomatic channels confirming that the country has granted or intends to grant a compulsory licence to use the invention pertaining to the product. Form and content of authorization 21.05 (1) The authorization must be in the prescribed form and, subject to subsection (2), contain the prescribed information. Quantity (2) The quantity of the product authorized to be manufactured by an authorization may not be more than the lesser of (a) the maximum quantity set out in the application for the authorization, and (b) the quantity set out in the notice referred to in any of subparagraphs 21.04(3)(d)(i) to (v), whichever is applicable. Disclosure of information on website 21.06 (1) Before exporting a product manufactured under an authorization, the holder of the authorization must establish a website on which is disclosed the prescribed information respecting the name of the product, the name of the country or WTO Member to which it is to be exported, the quantity that is authorized to be manufactured and sold for export and the distinguishing features of the product, and of its label and packaging, as required by regulations made under the Food and Drugs Act, as well as information identifying every known party that will be handling the product while it is in transit from Canada to the country or WTO Member to which it is to be exported. Obligation to maintain (2) The holder must maintain the website during the entire period during which the authorization is valid. Links to other websites (3) The Commissioner shall post and maintain on the website of the Canadian Intellectual Property Office a link to each website required to be maintained by the holder of an authorization under subsection (1). Posting on the website (4) The Commissioner shall, within seven days of receipt, post on the website of the Canadian Intellectual Property Office each application for authorization filed under subsection 21.04(1). Export notice 21.07 Before each shipment of any quantity of a product manufactured under an authorization, the holder of the authorization must, within fifteen days before the product is exported, provide to each of the following a notice, by certified or registered mail, specifying the quantity to be exported, as well as every known party that will be handling the product while it is in transit from Canada to the country or WTO Member to which it is to be exported: (a) the patentee or each of the patentees, as the case may be; (b) the country or WTO Member named in the authorization; and (c) the person or entity that purchased the product to which the authorization relates. Royalty 21.08 (1) Subject to subsections (3) and (4), on the occurrence of a prescribed event, the holder of an authorization is required to pay to the patentee or each patentee, as the case may be, a royalty determined in the prescribed manner. Factors to consider when making regulations (2) In making regulations for the purposes of subsection (1), the Governor in Council must consider the humanitarian and non-commercial reasons underlying the issuance of authorizations under subsection 21.04(1). Time for payment (3) The royalties payable under this section must be paid within the prescribed time. Federal Court may determine royalty (4) The Federal Court may, in relation to any authorization, make an order providing for the payment of a royalty that is greater than the royalty that would otherwise be required to be paid under subsection (1). Application and notice (5) An order may be made only on the application of the patentee, or one of the patentees, as the case may be, and on notice of the application being given by the applicant to the holder of the authorization. Contents of order (6) An order may provide for a royalty of a fixed amount or for a royalty to be determined as specified in the order, and the order may be subject to any terms that the Federal Court considers appropriate. Conditions for making of order (7) The Federal Court may make an order only if it is satisfied that the royalty otherwise required to be paid is not adequate remuneration for the use of the invention or inventions to which the authorization relates, taking into account (a) the humanitarian and non-commercial reasons underlying the issuance of the authorization; and (b) the economic value of the use of the invention or inventions to the country or WTO Member. Duration 21.09 An authorization granted under subsection 21.04(1) is valid for a period of two years beginning on the day on which the authorization is granted. Use is non-exclusive 21.1 The use of a patented invention under an authorization is non-exclusive. Authorization is non-transferable 21.11 An authorization is non-transferable, other than where the authorization is an asset of a corporation or enterprise and the part of the corporation or enterprise that enjoys the use of the authorization is sold, assigned or otherwise transferred. Renewal 21.12 (1) The Commissioner shall, on the application of the person to whom an authorization was granted and on the payment of the prescribed fee, renew the authorization if the person certifies under oath in the renewal application that the quantities of the pharmaceutical product authorized to be exported were not exported before the authorization ceases to be valid and that the person has complied with the terms of the authorization and the requirements of sections 21.06 to 21.08. One renewal (2) An authorization may be renewed only once. When application must be made (3) The application for renewal must be made within the 30 days immediately before the authorization ceases to be valid. Duration (4) An authorization that is renewed is valid for a period of two years beginning on the day immediately following the day of the expiry of the period referred to in section 21.09 in respect of the authorization. Prescribed form (5) Applications for renewal and renewed authorizations issued under subsection (1) must be in the prescribed form. Termination 21.13 Subject to section 21.14, an authorization ceases to be valid on the earliest of (a) the expiry of the period referred to in section 21.09 in respect of the authorization, or the expiry of the period referred to in subsection 21.12(4) if the authorization has been renewed, as the case may be, (b) the day on which the Commissioner sends, by registered mail, to the holder of the authorization a copy of a notice sent by the Minister of Health notifying the Commissioner that the Minister of Health is of the opinion that the pharmaceutical product referred to in paragraph 21.04(3)(b) has ceased to meet the requirements of the Food and Drugs Act and its regulations, (c) the day on which the last of the pharmaceutical product authorized by the authorization to be exported is actually exported, (d) thirty days after the day on which (i) the name of the pharmaceutical product authorized to be exported by the authorization is removed from Schedule 1, or (ii) the name of the country or WTO Member to which the pharmaceutical product was, or is to be, exported is removed from Schedule 2, 3 or 4, as the case may be, and not added to any other of those Schedules, and (e) on any other day that is prescribed. Termination by Federal Court 21.14 On the application of a patentee, and on notice given by the patentee to the person to whom an authorization was granted, the Federal Court may make an order, on any terms that it considers appropriate, terminating the authorization if the patentee establishes that (a) the application for the authorization or any of the documents provided to the Commissioner in relation to the application contained any material information that is inaccurate; (b) the holder of the authorization has failed to establish a website as required by section 21.06, has failed to disclose on that website the information required to be disclosed by that section or has failed to maintain the website as required by that section; (c) the holder of the authorization has failed to provide a notice required to be given under section 21.07; (d) the holder of the authorization has failed to pay, within the required time, any royalty required to be paid as a result of the authorization; (e) the holder of the authorization has failed to comply with subsection 21.16(2); (f) the product exported to the country or WTO Member, as the case may be, under the authorization has been, with the knowledge of the holder of the authorization, re-exported in a manner that is contrary to the General Council Decision; (g) the product was exported, other than in the normal course of transit, to a country or WTO Member other than the country or WTO Member named in the authorization; (h) the product was exported in a quantity greater than the quantity authorized to be manufactured; or (i) if the product was exported to a country that is not a WTO Member, the country has permitted the product to be used for commercial purposes or has failed to adopt the measures referred to in Article 4 of the General Council Decision. Notice to patentee 21.15 The Commissioner shall, without delay, notify the patentee, or each of the patentees, as the case may be, in writing of any authorization granted in respect of the patentee's invention. Obligation to provide copy of agreement 21.16 (1) Within fifteen days after the later of the day on which the authorization was granted and the day on which the agreement for the sale of the product to which the authorization relates was entered into, the holder of an authorization must provide by certified or registered mail, the Commissioner and the patentee, or each patentee, as the case may be, with (a) a copy of the agreement it has reached with the person or entity referred to in paragraph 21.04(2)(f) for the supply of the product authorized to be manufactured and sold, which agreement must incorporate information that is in all material respects identical to the information referred to in paragraphs 21.04(2)(a), (b), (e) and (f); and (b) a solemn or statutory declaration in the prescribed form setting out (i) the total monetary value of the agreement as it relates to the product authorized to be manufactured and sold, expressed in Canadian currency, and (ii) the number of units of the product to be sold under the terms of the agreement. Prohibition (2) The holder of an authorization may not export any product to which the authorization relates until after the holder has complied with subsection (1). Application when agreement is commercial in nature 21.17 (1) If the average price of the product to be manufactured under an authorization is equal to or greater than 25 per cent of the average price in Canada of the equivalent product sold by or with the consent of the patentee, the patentee may, on notice given by the patentee to the person to whom an authorization was granted, apply to the Federal Court for an order under subsection (3) on the grounds that the essence of the agreement under which the product is to be sold is commercial in nature. Factors for determining whether agreement is commercial in nature (2) In determining whether the agreement is commercial in nature, the Federal Court must take into account (a) the need for the holder of the authorization to make a reasonable return sufficient to sustain a continued participation in humanitarian initiatives; (b) the ordinary levels of profitability, in Canada, of commercial agreements involving pharmaceutical products, as defined in paragraph 1(a) of the General Council Decision; and (c) international trends in prices as reported by the United Nations for the supply of such products for humanitarian purposes. Order (3) If the Federal Court determines that the agreement is commercial in nature, it may make an order, on any terms that it considers appropriate, (a) terminating the authorization; or (b) requiring the holder to pay, in addition to the royalty otherwise required to be paid, an amount that the Federal Court considers adequate to compensate the patentee for the commercial use of the patent. Additional order (4) If the Federal Court makes an order terminating the authorization, the Federal Court may also, if it considers it appropriate to do so, make an order, on any terms that it considers appropriate, (a) requiring the holder to deliver to the patentee any of the product to which the authorization relates remaining in the holder's possession as though the holder had been determined to have been infringing a patent; or (b) with the consent of the patentee, requiring the holder to export any of the product to which the authorization relates remaining in the holder's possession to the country or WTO Member named in the authorization. Restriction (5) The Federal Court may not make an order under subsection (3) if, under the protection of a confidentiality order made by the Court, the holder of the authorization submits to a Court-supervised audit and that audit establishes that the average price of the product manufactured under the authorization does not exceed an amount equal to the direct supply cost of the product plus 15 per cent of that direct supply cost. Definitions (6) The following definitions apply in this section. "average price" « prix moyen » "average price" means (a) in relation to a product to be manufactured under an authorization, the total monetary value of the agreement under which the product is to be sold, expressed in Canadian currency, divided by the number of units of the product to be sold under the terms of the agreement; and (b) in relation to an equivalent product sold by or with the consent of the patentee, the average of the prices in Canada of that product as those prices are reported in prescribed publications on the day on which the application for the authorization was filed. "direct supply cost" « coût direct de fourniture » "direct supply cost", in relation to a product to be manufactured under an authorization, means the cost of the materials and of the labour, and any other manufacturing costs, directly related to the production of the quantity of the product that is to be manufactured under the authorization. "unit" « unité » "unit", in relation to any product, means a single tablet, capsule or other individual dosage form of the product, and if applicable, in a particular strength. Advisory committee 21.18 (1) The Minister and the Minister of Health shall establish, within three years after the day this section comes into force, an advisory committee to advise them on the recommendations that they may make to the Governor in Council respecting the amendment of Schedule 1. Functions of standing committee (2) The standing committee of the House of Commons that normally considers matters related to industry shall assess all candidates for appointment to the advisory committee and make recommendations to the Minister on the eligibility and qualifications of those candidates. Website for notices to Canada 21.19 The person designated by the Governor in Council for the purpose of this section must maintain a website on which is set out a copy of every notice referred to in subparagraphs 21.04(3)(d)(ii) and (v) that is provided to the Government of Canada through diplomatic channels by a country that is not a WTO Member. The copy must be added to the website as soon as possible after the notice has been provided to the Government of Canada. Review 21.2 (1) A review of sections 21.01 to 21.19 and their application must be completed by the Minister two years after this section comes into force. Tabling of report (2) The Minister must cause a report of the results of the review to be laid before each House of Parliament on any of the first fifteen days on which that House is sitting after the report has been completed. -- 2004, c. 23, Schs. 1 to 4: Appendix B: India’s Patent Amendment Bill 2005 This is the relevant section on the issuance for compulsory licences under India’s Patent (Amendment) Bill 2005 as passed by the Lok Sabha on 22/3/05 109: “92A. (1) Compulsory licence shall be available for manufacture and export of patented pharmaceutical products to any country having insufficient or no manufacturing capacity in the pharmaceutical sector for the concerned product to address public health problems, provided compulsory licence has been granted by such country. (2) The Controller shall, on receipt of an application in the prescribed manner, grant a compulsory licence solely for manufacture and export of the concerned pharmaceutical product to such country under such terms and conditions as may be specified and published by him. (3) The provisions of sub-sections (1) and (2) shall be without prejudice to the extent to which pharmaceutical products produced under a compulsory license can be exported under any other provision of this Act. Explanation.–For the purposes of this section, ‘pharmaceutical products’ means any patented product, or product manufactured through a patented process, of the pharmaceutical sector needed to address public health problems and shall be inclusive of ingredients necessary for their manufacture and diagnostic kits required for their use.”. 109 Available at http://lawmin.nic.in/Patents%20Amendment%20Ordinance%202004.pdf.