bip22580-sup-0004
advertisement

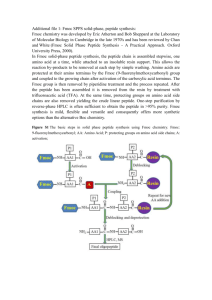
SUPPLEMENTARY MATERIAL CORRELATION BETWEEN MEMBRANE TRANSLOCATION AND ANALGESIC EFFICACY IN KYOTORPHIN DERIVATIVES Isa D. Serranoa‡, Vasanthakumar G. Ramub‡, Antónia R.T. Pintoa, João M. Freirea, Isaura Tavaresc,d, Montserrat Herasb, Eduard R. Bardajib, Miguel A. R. B. Castanhoa* a Instituto de Medicina Molecular, Faculdade de Medicina da Universidade de Lisboa, Av. Professor Egas Moniz, 1649-028 Lisboa, Portugal, isaserrano@gmail.com , arpinto@fm.ul.pt, joaofreire@fm.ul.pt, macastanho@fm.ul.pt, bLaboratori d'Innovació en Processos i Productes de Síntesi Orgànica (LIPPSO), Departament de Química, Universitat de Girona, Campus Montilivi, 17071 Girona, Spain. gangaramuvk@gmail.com, montserrat.heras@udg.edu, eduard.bardaji@udg.edu. cInstituto de Histologia e Embriologia, Faculdade de Medicina da Universidade do Porto, Alameda Professor Hernâni Monteiro, 4200319 Porto, Portugal; dInstituto de Biologia Molecular e Celular, Rua Campo Alegre, 4150-180 Porto, Portugal, isatav@med.up.pt. 1. SYNTHETIC PROTOCOLS General Synthetic Procedures. All chemicals used for organic synthesis were purchased from Sigma-Aldrich, Fluka, Bachem or Iris-Biotech and used without further purification. All compounds were analyzed for purity on high performance liquid chromatography (HPLC) and characterized by mass spectra under electrospray ionization (ESI-MS) (Figure s1). HPLC analyses were carried out using an Agilent Technologies 1200 Series HPLC instrument. Separations were achieved on an analytical 100-C18 Kromasil reversed phase column (4.6 mm x 40 mm; 3.5 μm particle size). The compounds were eluted using a linear gradient of 0–100% CH3CN in 0.1% TFA at flow rate of 1.0 mL∙min–1. The absorbance was measured at 220 nm. The HPLC retention time of each compound was determined when the peak was at its maximum height. ESI-MS analyses were performed with an Esquire 6000 ESI ion Trap LC/MS (Bruker Daltonics) instrument equipped with an electrospray ion source. The instrument was operated in the positive ESI(+) ion mode. Samples (5 μL) were introduced into the mass spectrometer ion source directly through an HPLC autosampler. The mobile phase (80:20 CH 3CN/H2O at a flow rate of 100 μL∙min–1) was delivered by a 1100 Series HPLC pump (Agilent). Nitrogen was employed as both the drying and nebulising gas. a) Solid phase synthesis of peptides GABA-KTP-NH2, AC-KTP-NH2, PRO-KTP-NH2, BU-KTP-NH2. General procedure. Peptides were prepared manually by the solid-phase method using Fmoc-type chemistry, Fmoc-Rink-MBHA resin (0.56 mmol∙g-1) as solid support, tert-butyl side chain protection for tyrosine and 2,2,5,7,8-pentamethylchroman-6-sulfonyl (pmc) for arginine. N-terminus derivatization was performed using NBoc amino butyric acid, acetic anhydride, propionic acid and butyric acid to obtain GABA-KTP-NH2, AC-KTPNH2, PRO-KTP-NH2 and BU-KTP-NH2 respectively. Couplings of the amino acids were mediated by O-(1Hbenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (3.8 equiv), Oxyma pure (4 equiv), and diisopropylethylamine (DIEA) (3 equiv) in N,N-dimethylformamide (DMF) at room temperature for 1 hour. S1 The completion of the reactions was monitored by the Kaiser test. Fmoc group was removed by treating the resin with 30 % piperidine in DMF (2 + 8 minutes). After each coupling and deprotection step, the resin was washed with DMF (6 x 1 minute) and air-dried. A mixture of trifluoroacetic acid (TFA), water and triisopropylsilane (TIS) was used as cleavage cocktail. Synthesis of GABA-KTP-NH2. The Fmoc-Rink-MBHA resin (450 mg) was placed into a plastic syringe fitted with a polypropylene frit to remove the Fmoc group and subsequently couple with arginine amino acid (4 equiv). After Fmoc group removal the resin was treated with tyrosine derivative (4 equiv). The resulting resin was deprotected with piperidine and derivatized at the N-terminus by coupling with N-Boc amino butyric acid (4 equiv). Next the peptide was cleaved from the solid support with TFA/H 2O/TIS (95:2.5:2.5). Following TFA evaporation and diethyl ether extraction, the resulting solid was dissolved in 0.1M aqueous HCl and lyophilized. This process was repeated three times to afford GABA-KTP-NH2 as a hydrochloride salt. HPLC: tR = 4.68 min, 95% pure. Synthesis of AC-KTP-NH2. The Fmoc-Rink-MBHA resin (450 mg) was placed into a plastic syringe fitted with a polypropylene frit to remove the Fmoc group and subsequently couple with arginine amino acid (4 equiv). After Fmoc group removal the resin was treated with tyrosine derivative (4 equiv). The resulting resin was deprotected with piperidine and derivatized at the N-terminus by treatment with acetic anhydride–pyridine– CH2Cl2 (1:1:1). Next the peptide was cleaved from the solid support with TFA/H 2O/TIS (95:2.5:2.5). Following TFA evaporation and diethyl ether extraction, the resulting solid was dissolved in 0.1M aqueous HCl and lyophilized. This process was repeated three times to afford AC-KTP-NH2 as a hydrochloride salt. HPLC: tR = 4.92 min, 94% pure. Synthesis of PRO-KTP-NH2. The Fmoc-Rink-MBHA resin (450 mg) was placed into a plastic syringe fitted with a polypropylene frit to remove the Fmoc group and subsequently couple with arginine amino acid (4 equiv). After Fmoc group removal the resin was treated with tyrosine derivative (4 equiv). The resulting resin was deprotected with piperidine and derivatized at the N-terminus by coupling with propionic acid (4 equiv). Next the peptide was cleaved from the solid support with TFA/H2O/TIS (95:2.5:2.5). Following TFA evaporation and diethyl ether extraction, the resulting solid was dissolved in 0.1M aqueous HCl and lyophilized. This process was repeated three times to afford PRO-KTP-NH2 as a hydrochloride salt. HPLC: tR = 5.18 min, 93% pure. Synthesis of BU-KTP-NH2. The Fmoc-Rink-MBHA resin (450 mg) was placed into a plastic syringe fitted with a polypropylene frit to remove the Fmoc group and subsequently couple with arginine amino acid (4 equiv). After Fmoc group removal the resin was treated with tyrosine derivative (4 equiv). The resulting resin was deprotected with piperidine and derivatized at the N-terminus by coupling with propionic acid (4 equiv). Next the peptide was cleaved from the solid support with TFA/H2O/TIS (95:2.5:2.5). Following TFA evaporation and diethyl ether extraction, the resulting solid was dissolved in 0.1M aqueous HCl and lyophilized. This process was repeated three times to afford BU-KTP-NH2 as a hydrochloride salt. HPLC: tR = 5.44 min, 96% pure. b) Solid phase synthesis of peptides IND-KTP-NH2 and IND-KTP. General procedure. Peptides were prepared manually by the solid-phase method using Fmoc-type chemistry, Fmoc-Rink-MBHA resin (0.56 mmol∙g-1) or Wang resin (0.80 mmol∙g-1) as solid supports and 2,2,5,7,8-pentamethylchroman-6-sulfonyl (pmc) S2 side chain protection for arginine. Couplings of the amino acids were mediated by O-(1H-benzotriazol-1-yl)1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (3.8 equiv), Oxyma pure (4 equiv), and diisopropylethylamine (DIEA) (3 equiv) in N,N-dimethylformamide (DMF) at room temperature for 1 hour. The completion of the reactions was monitored by the Kaiser test. Fmoc group was removed by treating the resin with 30% piperidine in DMF (2 + 8 minutes). After each coupling and deprotection step, the resin was washed with DMF (6 x 1 minute) and air-dried. A mixture of trifluoroacetic acid (TFA), water and triisopropylsilane (TIS) was used as cleavage cocktail. Synthesis of IND-KTP-NH2. The Fmoc-Rink-MBHA resin (450 mg) was placed into a plastic syringe fitted with a polypropylene frit to remove the Fmoc group and subsequently couple with arginine amino acid (4 equiv). After Fmoc group removal the resin was treated with N-methyl-1H-indole-3-carboxylic acid (4 equiv). The resulting compound was cleaved from the solid support with TFA/H 2O/TIS (95:2.5:2.5). Following TFA evaporation and diethyl ether extraction, the resulting solid was dissolved in 0.1M aqueous HCl and lyophilized. This process was repeated three times to afford IND-KTP-NH2 as a hydrochloride salt. HPLC: tR = 5.83 min, 97% pure. Synthesis of IND-KTP. The Wang resin (450 mg) was placed into a plastic syringe fitted with a polypropylene frit to couple with arginine amino acid (4 equiv). After Fmoc group removal the resin was treated with Nmethyl-1H-indole-3-carboxylic acid (4 equiv). The resulting compound was cleaved from the solid support with TFA/H2O/TIS (95:2.5:2.5). Following TFA evaporation and diethyl ether extraction, the resulting solid was dissolved in 0.1M aqueous HCl and lyophilized. This process was repeated three times to afford IND-KTP as a hydrochloride salt. HPLC: tR = 5.96 min, 99% pure. c) Solution phase synthesis of urea analogues uKTP-NH2 To a cooled solution (0 ºC) of Nα-Boc-Tyr(t-Bu)-OH (1 equiv) in THF (4 mL∙mmol-1), ethyl chloroformate (1 equiv) and N-methylmorpholine (1.2 equiv) were added successively. The mixture was stirred at 0 ºC for 30 minutes. After this time, NaN3 (2.0 equiv) was added, as an aqueous solution (1M), and the resulting mixture was continued stirring at 0 ºC for another 30 minutes. Next, the reaction mixture was diluted with CH2Cl2/toluene (1:1) and washed with saturated NaHCO3 solution and brine. The organic layer was dried over MgSO4, filtered and concentrated under vacuum to about one half its original volume. The resulting mixture was heated under reflux for 2 h. After the solution was cooled down to room temperature Arg-NH2x2HCl (1 equiv) and N-methylmorpholine (3 equiv) were added successively and stirred at room temperature for another 2 h. Next the resulting mixture was washed with HCl (1M), citric acid (1M) and brine. The organic layer was dried over MgSO4, filtered and evaporated under vacuum. The resulting residue was purified by crystallization to afford protected urea kyotorphin analogue Boc-uKTP-NH2 (40% overall yield) as a colorless solid (Figure s2). Protected peptide Boc-uKTP-NH2 was dissolved in HCl 4M in 1,4-dioxane and stirred at room temperature for 5 minutes. After this time, the solvent was evaporated under vacuum and the resulting residue dissolved in water and lyophilized to afford the deprotected urea analogue uKTP-NH2 (70% yield) as a hydrochloride salt (Figure s2). FIGURE s1 S3 FIGURE s2 2. PERMEABILITY OF KYOTORPHIN DERIVATIVES FIGURE s3 S4 FIGURE s1 ESI-MS of kyotorphin derivatives. FIGURE s2 Synthesis of urea analogues of kyotorphin. FIGURE s3 Fluorescence emission kinetics of KTP, KTP-NH2, Boc-uKTP-NH2, GABA-KTP-NH2, AC-KTPNH2, uKTP-NH2, IND-KTP-NH2, BU-KTP-NH2, PRO-KTP-NH2 and IND-KTP at apical (squares) and basal (circles) compartments at both POPC-covered (filled symbols) and blank (empty symbols) filters. Each group is an average of three independent measures. N=3, data shown as mean ± SEM. Lines represent guide to the eyes. During the first 10 h of the assay steady-state conditions are noticeable. S5