AM Module 1. Genome assembly: Overview and Experimental design
advertisement

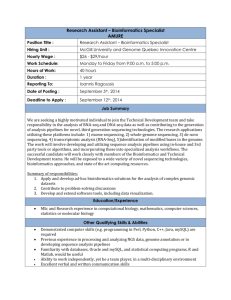
GCAT-SEEK Eukaryotic Genomics Workshop Eukaryotic Genomics Breakout Session Outline Tues, Day 2 AM Module 1. Genome assembly: Overview and Experimental design Module 2. Linux tutorial: Some basics and software installation Module 3. Genome assembly: Data quality and genome size estimation using Jellyfish Module 4. Genome assembly: Data quality and error correction Module 5. Genome assembly: Assembly algorithms PM Module 6. Genome annotation: Overview and manual annotation of a eukaryotic gene Module 7. Genome annotation with Maker: Introduction and repeat finding Module 8. Genome annotation with Maker: Gene finder training Module 9. Genome annotation with Maker: Putting it all together Weds, Day 3 AM Module 10. Genome annotation with Maker: Processing results Module 11. Genome annotation: Using a genome browser to observe evolutionary patterns PM TBA Thurs, Day 4 AM TBA 1 Module 1. Genome assembly: Overview & experimental design Background The ultimate goal of genome assembly is to completely construct intact chromosomes. Rapidly decreasing cost per bp has made it possible to randomly sequence even entire large mammalian genomes like that of human ( >3GBp) by densely covering the genome with 100X coverage in short sequence reads. This is analogous to snow falling down on your yard, randomly coating all exposed surfaces. The fundamental problem of genome assembly is sequencing through repetitive DNA. One cannot sequence through a repeat with reads that are shorter or the same size as the length of the repeat. Here, using four 5 bp reads one can not determine whether the correct path is (1) or (2) above. However, using paired end data one can determine a unique sequence because the insert size (15bp) exceeds the length of the repeat (5bp). Note that given the data above, connecting the two contigs into a single scaffold resulted in placement of two Ns. Some repeats are long and abundant in eukaryotic genomes. Long interspersed nuclear elements (Lines) are about 6Kbp long and comprise over 15% of the genome in humans. Short nuclear elements (e.g. such as Alu elements) are just as abundant in humans, but are only about 350bp long. In practice, in a genome sequencing project one aims for the attaining the largest and fewest DNA pieces (known as contigs) with as few errors as possible. Errors in sequencing and assembly may result in misjoins (or chimeras), where disparate parts of the genome are connected erroneously. Contigs may be connected by paired read data to form scaffolds. Often there is a closely estimated distance between 2 paired reads, but without information to completely fill in the gaps, resulting in stretches of Ns, or unknown nucleotide sequence. Recent genomes are considered “good” having N50 scores in the millions of bp. An N50 is the size of the smallest scaffold such that 50% of the genome is contained in scaffolds of size N50 or larger (Salzberg et al. 2012). Assemblies should have scaffolds with N50 sizes big enough to contain entire genes for the organism of interest that can then be subject to comprehensive annotation. Gene size is proportional to genome size as shown in the figure below from Yandell and Ence (2012). The figure shows that for a species with a 1Gbp genome, gene size will be about 5Kbp. One would expect an assembly with an N90 of 5Kbp to contain about 90% of genes on single scaffolds in this case. 3 Genome projects are of great usefulness in providing a centralized location for the research community for that organism to map information. The following view of the UCSC genome browser (http://genome.ucsc.edu) shows a slice of the human genome project. Many different tracks of data have been mapped to this location, the beta globin gene, by the research community. In Module 11 you will go to the UCSC genome browser and investigate evolutionary patterns at the Beta globin gene. Draft genomes are more appropriate for some uses than others. Draft genomes are appropriately used for determining gene sequences, including promoters, exons, introns, and other regulatory sequences. They are also appropriate for determining variable sites such as single nucleotide polymorphisms and insertion/deletions. They are less appropriate for determining copy number of genes because the assembled sequence may not be complete, and repeats tend to “collapse” into one sequence given that different sequences look the same to an assembler. Duplicated areas of genomes can be underrepresented. For this reason the amount of assembled DNA may underestimate true genome length. Because draft genomes come in many small pieces, determining gene order is difficult. When planning a genome sequencing project some considerations regarding the genomes itself are important in determining sequencing strategy. Planning for adequate sequencing coverage of the genome in raw data is essential (>100x). Genome size is such an important consideration in designing a sequencing project because of the cost of obtaining that much sheer data. Because the location of any given sequence read is random, 100X coverage is necessary to ensure coverage of all regions of the genome, except for difficult to sequence regions. To make matters worse, because organisms’ genes get bigger as the genome gets bigger, better assemblies are needed in organisms with larger genomes to achieve comprehensive gene annotation. 4 Understanding %GC content is helpful to determine necessary coverage because some commercial sequencing platforms, such as Illumina, have biases against certain nucleotides, (Illumina is somewhat biased against AT coverage; Dohm et al. 2008). Different sequencing platforms have different biases in error rates. One study showed that Illumina has highest A to C error rates, which was partly explained because both are called by a red detection laser; Dohm et al. 2008). Illumina has low overall error rates of about 0.1%, but error rates of 3% were estimated at the 3’ end of reads (Dohm et al. 2008). Imbalance in GC content in genome content or in sequencing coverage will result in the need for higher overall coverage (Kenney et al. 2010). Species vary remarkably in percent of repetitive DNA, with some plants having greater than 80% of DNA in repeats, leading to very large, difficult-to-sequence genomes. Species with larger genomes with more repetitive DNA will require special library construction involving “jumping” or “mate-pair” libraries that sequence the ends of large fragments, in addition to shorter paired-end libraries. Mate-pair sequencing is important in connecting relatively distant contigs and making them into scaffolds. This is an essential step to produce long scaffolds (millions of bp) in genomes with a large fraction of repetitive DNA. The Mate-Paired sequencing strategy connects contigs across repetitive regions. The figure below shows the difference between paired-end and mate-pair genomic DNA libraries. Sequencing of large mammalian and plant genomes typically involves high (>40x) coverage using a series of fragment and mate pair libraries of different sizes. For example for Giant Panda, the first large mammalian genome, fragment sizes of 150bp, 500bp, 2kbp, 5kbp and 10kbp were used to produce 96x coverage of a 2.4Gbp genome 5 (Li et al 2010; see Schatz et al. 2010 for a review of early shotgun assemblies). Fragment library paired-end sequencing DEF ABC 180bp Produces the sequence reads Forward: ABC Reverse: FED Orientation: “Innie” or “Normal Forward/Reverse” Mate paired sequencing ABC DEF 3500 bp Produces the sequence reads Forward: DEF Reverse: CBA Orientation: Outie or “Reverse” Circularize DEF ABC DEF ABC Cut DEF ABC Sequence ABC DEF Orientation: Outie F IGURE 1. F ORMATION OF PAIRED - END AND MATE - PAIR LIBRARIES . P AIRED - END LIBRARIES PRODUC E FRAGMENTS IN NORMAL FORWARD - REVERSE OR “ INNIE ” ORIENTATION AND MATE - PAIR LIBRARIES PRODUCE FRAGMENTS IN AN ORIENTATION THAT IS REVERSED COMPARED TO PAIRED - END LIBRARIES , CALLED “ OUTIE .” Other genome sequencing experimental design considerations include cost per bp, error rates, error types, single end, paired end, or mate pair sequencing sequencing, and read length. For deNovo genome assembly mate pairs are essential to get past repeats. Long reads (from Pac Bio) or jumping libraries are often combined with short read data sets to aid in increasing assembly. For bacterial-sized genomes, decent assemblies can be performed from 2X250bp MiSeq Runs (Magoc, Pabinger, Canzar et al. 2013). A summary of important characteristics for different sequencing platforms is summarized at the following web page: http://www.molecularecologist.com/next-gen-fieldguide-2013/. So far it has been updated every year. Go there now. As of 2013, Tables 1a and 1b show that for plant and animal genome sequencing and resequencing, the Illumina HiSeq and Miseq get the highest grades. This is due 6 to low cost per bp, large throughput, and low error rates. The long reads of the MiSeq would make it the favored choice for small eukaryotic genomes due to reduced cost concerns and ability to sequence through small repeats like SINEs. Table 2 shows that Illumina platforms have the lowest cost per bp. Table 3 shows that Illumina data has among the lowest error rates of current machines, but not the absolute lowest. Goals Choose and justify the appropriate methods for whole genome sequencing using Next Gen sequencing technology Apply NextGen sequencing methodologies to solve their own research questions V&C core competencies addressed Apply process of science: Design of genome sequencing approach GCAT-SEEK sequencing requirements None Computer/program requirements for data analysis Web browser Protocols None Assessment See essays below. Time line of module One hour of lecture. Discussion topics for class Q. Does a nucleotide of sequence using the Illumina platform cost closer to 1 cent per bp or 1 cent per million bp? Q. Which sequencing platform has the absolute lowest sequencing error rates? Why do you think it is not winning the sequencing battle to Illumina? Q. Design a genome sequencing project for a mammal for 100 X coverage. Include as many details about platform and cost as possible. 7 Q. Design a sequencing project for a bacteria for 100 X coverage. Include as many details about platform and cost as possible. Q. Look up repeat content for several bacteria, plants, animals, and fungi. How would this affect a genome sequencing approach? Q. Why is it important to sequence genomes? References Literature cited Dohm JC, Lottaz C, Borodina T, Himmelbauer H. 2008. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res, 36:e105+. Kenney DR, Schatz MC, Salzberg SL. 2010. Quake:quality-aware detection and correction of sequencing errors. Genome Biology 11:R116 Li et al. 2010. The sequence and de novo assembly of the giant panda genome. Nature. 463: 311-317 Magoc T, Pabinger S, Canzar S, et al. 2013. An evaluation of genome assemblers for bacterial organisms. Bioinformatics online advanced access. [The GAGE-B project] Salzberg SL, Phillippy AM, Zimin A, et al. 2012. GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res 22: 557-567. [important online supplements!] Shatz MC, Delcher AL, Salzberg SL. 2010. Assembly of large genomes using second-generation sequencing. Genome Res 20: 1165-1173. Yandell M, Ence D. 2012. A beginner's guide to eukaryotic genome annotation. Nature Reviews Genetics 13:329-342. Further reading ASSEMBLATHON The Assemblathon (http://assemblathon.org) Earl D, Bradnam K, St John J, et al. 2011. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res 21:2224-2241 Bradnam KR, Fass JN, Alexandrov A, et al. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. arXiv preprint arXiv:1301.5406 Other Assembly Reviews Alkan C, Sajjadian S, Eichler E Limitations of next-generation genome sequence assembly. 2011. Nat Methods 8:61-65 8 Birney E. 2011. Assemblies: The good, the bad, the ugly. Nature Methods. 8:59-60. Compeau PEC, Pevzner PA, Tesler G. 2011. How to apply de Bruijn graphs to genome assembly. Nature Biotechnology. 29:987-991. Miller JR, Koren S, Sutton G. 2010. Assembly algorithms for next-generation sequencing data. Genomics 95: 315-327. Paszkiewicz K, Studholme DJ. 2010. De novo assembly of short sequence reads. Briefings in Bioinformatics. 11: 457-472. Pop M. 2009. Genome assembly reborn: recent computational challenges. Briefings in Bioinformatics. 10:354-366. Zhang et al. 2011. A practical comparison of De Novo genome assembly software tools for nextgeneration sequencing technologies. PLos ONE 6: e17915. Sequencing Technology Updates http://www.molecularecologist.com/next-gen-fieldguide-2013/ 9