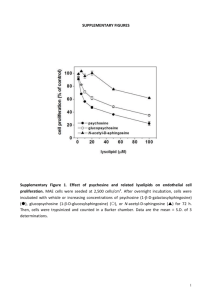
Supplementary Data

SUPPLEMENTARY MATERIALS AND METHODS
Reagents. Psychosine (1-
-D-galactosylsphingosine) from bovine brain with a length of sphingoid base of C18 carbon atoms (mw: 461.63, purity ≥98%), glucopsychosine (1-
-Dglucosylsphingosine) from glucocerebrosides from human Gaucher′s spleen and N -acetyl-Dsphingosine (mw: 341.53, purity ≥98%) were purchased from Sigma Chemical Co. (St.
Louis, MO). Stock solutions were prepared in dimethyl sulfoxide (DMSO) and diluted directly in cell culture medium to a final DMSO concentration equal to or lower than 0.5%
(vol:vol); control cells were treated in parallel with the same doses of vehicle that were ineffective in all the assays. VEGF was from Calbiochem Biochemicals. Recombinant FGF2 was expressed and purified from E. coli (Bastaki et al.
, 1997).
Cell cultures. Immortalized Balb/c murine aortic endothelial (MAE) cells and murine brain microvascular endothelial (MBE) cells were obtained from R. Auerbach (University of
Wisconsin, Madison, WI) and grown in DMEM (Gibco Life Technologies, Rockville, MD) added with 10% fetal calf serum (FCS) (Invitrogen, Carlsbad, CA). FGF2T -MAE cells, an highly tumorigenic subclone of FGF2-overexpressing MAE cells (Gualandris et al.
, 1996), were grown in DMEM supplemented with 4 mM glutamine (Gibco) and 10% FCS. 1G11 cells, a microvascular endothelial cell line cloned from the lung of C57BL/6NCrLBR mice
(Dong et al.
, 1997), and microvascular endothelial cells isolated from subcutaneous sponges implanted in mice of the same strain (SIE cells) were provided by A. Vecchi (Istituto Mario
Negri, Milan, Italy) and grown in DMEM supplemented with 1 mM glutamine, 1% nonessential amino acids, 1 mM sodium pyruvate and 20% FCS in the presence of 10 ng/ml
FGF2 and 10
g/ml heparin on gelatin-coated dishes. Bovine aortic endothelial (BAE) cells
(provided by A. Vecchi, Istituto Mario Negri, Milan, Italy) were cultured in DMEM supplemented with 10% heat-inactivated donor calf serum. Human umbilical vein endothelial
(HUVE) cells were used at early (I-IV) passages and grown on plastic surface coated with porcine gelatin (Sigma) in M199 medium (Invitrogen) supplemented with 20% fetal calf serum (FCS, Invitrogen), endothelial cell growth factor (100
g/ml) (Sigma) and porcine heparin (100
g/ml, Sigma).
Cell proliferation assays.
MAE, FGF2T -MAE and BAE cells were seeded at 20,000 cells/cm
2
whereas 1G11 cells were seeded at 50,000 cells/cm
2
. After overnight incubation,
1
cells were incubated with vehicle or increasing concentrations of psychosine for 24 h. Then, cells were trypsinized and counted in a Burker chamber. When indicated, MAE cells were seeded at 20,000 or 2,500 cells/cm
2
and counted after 72 h or 120 h of incubation, respectively.
Wounding of endothelial cell monolayers.
Wounds were created in 1G11 or MAE cell monolayers with a 1.0-mm-wide rubber policeman. Then, cells were incubated in fresh medium added with 10% FCS and increasing concentrations of psychosine. At different time points, the percentage of cells at the edge of the wound showing cell membrane ruffles were counted under a inverted microscope at x 400 magnification (Olympus IX51 microscope with
Camedia C-404 digital camera; Olympus Biosystem, Munich Germany). After 16 h, wounds were photographed and endothelial cells invading the wound were quantified by computerized analysis of the digitalized images. When indicated, cell monolayers on glass coverslips were wounded and analyzed for MTOC repositioning.
MTOC reorientation. Repositioning of the microtubule organization center (MTOC) was evaluated by immunostaining with an anti-pericentrin antibody (BabCO, Richmond, CA) as described (Belleri et al.
, 2005). Briefly, MAE cell monolayers were fixed 3 h after wounding in ice-cold methanol for 10 min at –20°C. After saturation with goat serum in PBS, cells were incubated with the primary antibody, followed by biotinylated anti-rabbit IgG (Molecular
Probes, Eugene, OR) and Texas-red avidin (Vector Laboratories, Burlingame, CA). Finally, cells were stained with 0.5
g/ml DAPI to visualize nucleus position. To determine MTOC position, 70-80 cells in the first row of cells adjacent to the wound and in the inner monolayer were then selected per experimental point. Each cell was visually divided by drawing two perpendicular lines across the cell nucleus, thus defining a front quadrant covering 25% of the cell surface and containing the leading edge (see Fig. 1b ). The percentage of migrating and quiescent cells with MTOC located inside the front quadrant was then calculated.
Statistically, a random distribution of the MTOC around the nucleus, as observed in the quiescent monolayer, will result in 25% of cells with MTOC located in the front quadrant.
GALC fluorometric enzyme assay. LRh-6-GalCer ( N -lissamine rhodaminyl-6aminohexanoylgalactosyl ceramide) was synthesized as described (Marchesini et al.
, 1990).
Then, 5 nmoles of LRh-6-GalCer in 2:1 chloroform/methanol were concentrated and
2
dissolved in 5 µl of dimethyl sulfoxide (DMSO) and 25 µl of 0.2 M citrate phosphate buffer, pH 4.4. The enzyme source (50 µg of cell pellets) and water were added to a final volume of
100 µl and incubated for 3 h at 37°C. The reaction was extracted with 1.9 ml of 2:1 v/v chloroform/methanol and 0.4 ml of water. The lower phase was collected and evaporated under nitrogen. Samples were spotted on glass-coated silica gel plates and developed in
25:25:25:9:16 volumes chloroform/ethyl acetate/ n -propanol/0.25 M KCl/methanol. The fluorescent ceramide spots (LRh-6-Cer) were visualized under an ultraviolet lamp, scraped, extracted with methanol, and evaporated under nitrogen. Samples were suspended in 1.0 ml of 6:4 v/v chloroform/methanol and read in a spectrofluorometer (excitation and emission wavelength at 565 and 575 nm).
3D fibrin gel sprouting assay. FGF2T -MAE cell aggregates were prepared on agarosecoated plates and embedded in fibrin gel as described (Gualandris et al.
, 1996). Then, culture medium containing increasing concentrations of psychosine were added on the top of the gel in the presence of 10
g/ml aprotinin to prevent the dissolution of the substrate.
Formation of radially growing cell sprouts was observed during the next 24 h. Sprouts were photographed at x 40 magnification (Olympus IX51 inverted microscope with Camedia C-4040 digital camera) and quantified by computerized analysis of the digitalized images.
Preparation of three-dimensional collagen gels. Three-dimensional gels of reconstituted rat tail tendon type I collagen fibrils (Boehringer Mannheim Italia, Milan, Italy) were prepared as described (Bastaki et al.
, 1997). Then, BAE cells were seeded on the top of collagen gel
(80,000 cells/well) and allowed to reach confluence. Cell cultures were then treated with fresh medium containing FGF2 plus VEGF (both at 30 ng/ml) and added with 10% FCS in the absence or in the presence of psychosine. After 24 h, cells were photographed at x 100 magnification (Olympus IX51 inverted microscope) and endothelial cells invading the gel, in a plane of focus beneath the cell monolayer surface, were quantified by computerized analysis of the digitalized images.
Immunocytochemistry.
BAE cells were seeded on gelatin-coated glass coverslips in DMEM added with 10% FCS. After overnight incubation, cells were treated with 50
M psychosine.
At different time points cells were washed, fixed in 3% paraformaldehyde/2% sucrose in
PBS, permeabilized with 0.5% Triton-X100, and saturated with goat serum in PBS. Then,
3
cells were incubated with rodaminate-phalloidin, photographed at x 63 magnification
(Axiovert S100 epifluorescence microscope, Zeiss, with Camedia C-4040 digital camera) and the percentage of cells with actin disassembly was then calculated.
Endothelial cell adhesion and spreading. Glass coverslips were coated with 10
g/ml of bovine fibronectin (FN) (Sigma) dissolved in 100 mM NaHCO
3
, pH 9.6. After 16 h of incubation at 4°C the solution was removed and coverslips were washed three times with
PBS. Then, BAE cells were seeded at 25,000/cm
2
on FN-coated coverslips in the presence of vehicle or 50
M psychosine. At different time points, adherent cells were stained with rhodamine-phalloidin and the percentage of adherent cells and of cells showing a spread morphology (Yoshimura et al.
, 1995, Yu et al.
, 1998) were counted in three random fields under an inverted microscope at x 200 magnification .
Chick embryo chorioallantoic membrane assay. Gelatine sponges containing vehicle or
500 ng of FGF2 with or without psychosine (1.6
moles) were prepared as described (Ribatti et al.
, 2006) and placed on top of the chicken embryo chorioallantoic membrane (CAM) of fertilized White Leghorn chicken eggs at day 8 of incubation (8 eggs per experimental group).
At day 12, blood vessels converging toward the implant were counted by two observers in a double-blind fashion under a stereomicroscope (STEMI-SR, x2/0.12; Zeiss).
Mice. Breeder twitcher heterozygous mice (C57BL/6J, twi/+; Jackson Laboratories, ME,
USA) were maintained under standard housing conditions. Animal handling protocols were in accordance with Italian institutional guidelines for animal care and use. Twitcher mutation was determined by polymerase chain reaction (PCR) on DNA extracted from clipped tails
(Sakai et al.
, 1996). In all the experiments, littermate wild type ( wt ), heterozygous carrier
( twi/+ ) and homozygous ( twi/twi ) animals were used.
Brain expression of angiogenic growth factors.
Brains from wild type ( wt ), heterozygous carrier ( twi/+ ) and homozygous ( twi/twi ) mice were analysed for the expression of different pro-angiogenic factors by quantitative RT-PCR and data were normalized for Gapdh and
Actin expression as described (Coltrini et al.
, 2012). To this purpose, total RNA was extracted from frozen samples and contaminating DNA was digested using DNAse, following indications reported in RNeasy Micro Handbook (Qiagen, Valencia, CA, USA ).
4
Two
g of total RNA were retrotranscribed with MMLV reverse transcriptase (Invitrogen) using random hexaprimers in a final 20
l volume. Quantitative PCR was performed with a
Biorad iCycler iQ TM Real-Time PCR Detection System using a iQ TM SYBR Green Supermix
(Biorad) according to manufacturer’s instructions. The primers are listed in Supplementary
Table 1 .
Morphometric evaluation of murine brain vascular density. Brain cortex and periventricular area samples obtained from wild type ( wt ), heterozygous carrier ( twi/+ ) and homozygous ( twi/twi ) mice were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer.
Eight
m histological sections collected on poly-L-lysine coated slides (Sigma) were deparaffinized and stained with a three-layer-avidin-biotin-immunoperoxidase system technique. Sections were rehydrated in a xylene-graded alcohol scale and then rinsed for 10 min in 0.1 M phosphate-buffered saline (PBS). Intrinsic peroxidase activity was blocked with
3% H
2
O
2
for 10 min in the dark. Then, sections were treated with 0.1% trypsin (Sigma) in
CaCl
2
0.01 M for 30 min at room temperature and then sequentially exposed to: (1) primary antibody anti-factor VIII-related antigen/von Willebrand’s factor (FVIII/vWF, Dako,
Glostrup, Denmark) diluted 1:100 in RPMI-1640 medium supplemented with 10% heatinactivated FCS overnight at 4°C, (2) biotinylated swine anti-rabbit Ig (Dako) diluted 1:300 in RPMI-1640 supplemented with 10% heat-inactivated FCS for 15 min at room temperature, and (3) streptavidin-peroxidase conjugate (Vector, Burlingame, Calif., USA) diluted 1:250 in
PBS for 15 min at room temperature. The immunodetection was performed in 0.05 M acetate buffer pH 5.1, 0.02% 3-amino-9-ethylcarbazole grade II (Sigma), and 0.05% H
2
O
2
for 20 min at room temperature. The sections were counterstained with Gill’s hematoxylin number 2
(Polysciences, Warrington, Pa., USA), and mounted in buffered glycerin. A preimmune rabbit serum (Dako) replacing the primary antibody served as a negative control. Then,
FVIII/vWF + vascular density was assessed on 30 sections per brain sample by two investigators with a double-headed light microscope (Axioplan II, Zeiss). Four to six 200x fields covering almost the whole of each of four sections per sample were examined with a square reticulum (0.78 mm
2
) inserted in the eyepiece. Means
1 standard deviation (SD) were determined for each section, sample and group of samples.
3D rendering of cortex microvasculature.
Brains from P36 wt and twi/twi mice were fixed overnight at 4°C in 4% paraformaldehyde and embedded in 6% agarose. Agarose-embedded
5
samples were cut at a thickness of 50 µm using a vibratome, and free-floating sections were incubated overnight at 4°C with an anti-CD31 antibody (BD Pharmingen, San Diego, CA) diluted 1:100 in PBS containing 0.2% BSA and 0.2% Triton X-100. After several rinses in
PBS, sections were incubated overnight at 4°C with secondary 594-conjugated antibody
(Invitrogen) diluted 1:250 in PBS containing 0.1% Triton X-100. After further rinsing in
PBS, the sections were mounted in a drop of anti-bleaching mounting medium (Vectashield,
Vector Laboratories, Burlingame, CA, USA) and examined under an Axiovert 200 M microscope (Zeiss) equipped with ApoTome optical sectioning device (Zeiss). Z-stacks images were acquired with a 20x objective at 0.32 pixels/µm resolution. In the vertical axis, slices were 1 µm apart. Image processing was carried out with AxioVision 4.8 software
(Zeiss) and 3D renderings were obtained using ImageJ software.
Confocal immunofluorescence microscopy. Deparaffinized brain sections and cryosectioned samples 12 μm thick obtained from 3
twitcher and 3 control brains were utilized. Sections were incubated for 30 min in blocking buffer (1% bovine serum albumin,
2% FCS in PBS, pH 7.4) and exposed overnight at 4°C to mouse anti-GFAP monoclonal antibody (Novocastra Laboratories, Newcastle, UK) and rabbit anti-ZO-1 polyclonal antibody (Abcam, Cambridge, UK) diluted 1:50 and 1:5 in blocking buffer, respectively.
After washing in PBS, sections were incubated for 2 h with Alexa Fluor 488 goat anti-mouse antibody and Alexa Fluor 555 goat anti-rabbit antibody (Invitrogen) diluted 1:200 in normal goat serum. All samples were incubated for 5 min with 0.01% TO-PRO-3 (Invitrogen) for nuclear staining and mounted in Vectashield (Vector Laboratories). Negative controls, obtained by replacing primaries antibodies with normal mouse serum and normal rabbit serum (Santa Cruz Biotechnology) showed no staining of the sections. The sections were examined under a Leica TCS SP2 (Leica, Wetzlar, Germany) confocal laser scanning microscope using 40x and 63x objective lenses with either 1x or 2x zoom factors. A sequential scan procedure was applied during image acquisition of the two fluorophores.
Confocal images were taken at 200 nm intervals through the z-axis of the section covering a total depth of 10 μm. Images from individual optical planes and multiple serial optical sections were analyzed, digitally recorded and stored as TIFF files using Adobe Photoshop software (Adobe Systems Inc., San Jose, CA, USA).
Transmission electron microscopy. Small pieces of brain cortex samples were fixed by immersion in a 0.1 M phosphate buffered mixture of 3% glutaraldehyde for 3 h and washed
6
in the same buffer for 12 h. The specimens were then post-fixed with 1% osmium tetroxide for 1 h, dehydrated in an ascending ethanol series and embedded in Epon 812. Semithin sections were cut for orientation purposes and stained with toluidine blue. Thereafter, ultrathin sections of 60 nm were cut with a diamond knife on an LKB-V ultratome, stained with uranyl acetate and lead citrate and examined with a Zeiss EM109 electron microscope.
Evaluation of brain vascular permeability.
Vessel walls were biotinylated in vivo by intracardiac injection of sulfosuccinimidyl-6-(biotinamido) hexanoate (sulfo-NHS-LC-biotin,
Thermo Fisher Scientific, Rockford, IL, USA) as described (Rybak et al.
, 2005). Five min after injection, biotinylated brains were then excised, fixed in 4% paraformaldehyde overnight at 4°C and embedded in 6% agarose. Agarose-embedded samples were cut at a thickness of 100 µm using a vibratome, and frontal cortex sections were stained with Alexa
Fluor 488-conjugated streptavidin (Invitrogen). Z-stacks images were acquired with a 10x objective at 0.65 pixels/µm resolution using a Axiovert 200 M microscope (Zeiss) equipped with ApoTome optical sectioning device (Zeiss). In the vertical axis, slices were 2 µm apart.
Image processing was carried out with AxioVision 4.8 software (Zeiss).
GALC immunofluorescence analysis. Seven µm thick OCT-embedded frozen sections of brain cortex from wild type mice were fixed for 10 min in cooled acetone, air dried, and washed three times with Tris-buffered saline (TBS). After blocking with 1% BSA in TBS plus 0.1% Triton-X100, sections were incubated overnight at 4°C with an anti-GALC rabbit polyclonal IgG antibody (1:100 dilution, Protein Tech Group, Inc, Chicago, IL), followed by
1 h incubation at room temperature with Alexa Fluor 488 anti-rabbit IgG antibody (1: 500 dilution, Invitrogen). Sections were then incubated for 2 h at room temperature with biotinconjugated Bandeiraea Simplicifolia BSI-B4 lectin (1:500 dilution, Sigma), followed by 1 h incubation at room temperature with streptavidin Alexa Fluor 594 (1:500 dilution,
Invitrogen). Images were taken using a Zeiss LSM 510 META confocal laser scanning microscope equipped with a Plan-Neofluor 40x/1.3NA oil objective.
Matrigel plug angiogenesis assay . In a first set of experiments, C57BL mice were injected s.c. with 400
l of Matrigel (Trevigen, Gaithersburg, MD) containing PBS or 300 ng of FGF2 in the absence or in the presence of 200
M psychosine. In a second set of experiments, wild type, twi/+ and twi/twi mice were injected s.c. with 400
l of Matrigel containing PBS or 500 ng of FGF2 plus 100 ng of VEGF. After 7 d, the vascular response was quantified by anti-
7
CD31 immunostaining of frozen sections of the plugs (5 sections/plug) followed by computerized image analysis of CD31
+
areas in 6 microscopic fields per section. To this purpose, sections were incubated with a rat IgG2a anti-mouse CD31 monoclonal antibody
(1:200 dilution, 1 h at room temperature; BD Pharmingen), followed by incubation with biotinylated goat anti-rat IgG antibody (at 1:200 dilution, Santa Cruz). Sections were then exposed to avidin-biotin-peroxidase complex Vectastain ABC Kit (Vector, Laboratories) and peroxidase color reaction was developed with 3-amino 9-ethyl carbazole AEC Peroxidase substrate kit (Vector Laboratories). Also, the levels of expression of the endothelial markers
CD31 in Matrigel plugs was evaluated by quantitative RT-PCR. To this purpose, total RNA was extracted from Matrigel plugs using TRIzol Reagent according to manufacturer’s instructions (Invitrogen). Purified total RNA was dissolved in RNase free water at 1:1 (w:v) ratio and contaminating DNA was digested using DNase, following the protocol reported in
DNase 1 Amplification Grade kit (Sigma). Four μl of total RNA were retrotranscribed with
MMLV reverse transcriptase (Invitrogen) using random hexaprimers in a final 20 μl volume.
Quantitative PCR was performed with a Biorad iCycler iQ TM Real-Time PCR Detection
System using a iQ TM SYBR Green Supermix (Biorad) according to manufacturer’s instructions. Each PCR reaction was performed in triplicate on one plate and fluorescence data were recorded using iCycler software (BioRad). Relative expression levels were normalized in respect to murine Gapdh mRNA levels. The primers are listed in
Supplementary Table 1 .
Murine aorta ring assay. This was performed as described (Ronca et al.
, 2010). Briefly, aortic rings were obtained by cross-sectioning the mouse thoracic aorta at 1 mm intervals.
Rings were placed individually on the bottom of 24 well-plates with the luminal axis lying parallel to the bottom of the plate. Next, 30
l of polymerizing fibrin solution (prepared as described above) were applied onto each ring. After 5 min, wells were added with 600
l of serum-free endothelial cell basal medium (Clonetics) plus 10
g/ml aprotinin in the presence of 30 ng/ml of VEGF. When indicated, aorta rings were incubated in serum-free medium with 8 µg/ml polybrene and lentiviral particles harboring the murine
GALC cDNA or control
EGFP cDNA (kindly provided by A. Biffi, San Raffele Scientific Institute, Milan). After 24 h, rings were embedded in fibrin gel and incubated with VEGF in the presence of 10% FCS.
Then, vessel sprouts were counted under a stereomicroscope at 5 d.
8
In some experiments, histological sections of the aorta rings were stained with hematoxylin and eosin or were decorated with anti-CD31 antibodies. Also, samples were analyzed for the levels of Vegfr2/Kdr mRNA expression by quantitative RT-PCR as described above and data were normalized for CD31 expression. The primers are listed in Supplementary Table 1 . siRNA GALC knockdown in HUVE cells. HUVE cells silencing was carried out with a pool of concentrated lentiviral particles containing three shRNA target-specific constructs designed to knockdown the human GALC gene expression (sc-60669-V; Santa Cruz
Biotechnology) while shRNA lentiviral particles encoding scrambled shRNA sequence (sc-
108080; Santa Cruz Biotechnology) were used as control. Cells were seeded in 24 well plates
(10.000 cells/cm
2 ) and infected for 7 h in medium containing 8 μg/ml of polybrene with
5x10
4
lentiviral particles. Puromycin (0.8 μg/ml) was added 24 h later as selection agent.
Psychosine extraction and quantification. Lyophilized HUVE cells (4 x 10
6 cells/sample) were homogenized on ice with a Polytron homogenizer (IKA T8.10, Werke). Total lipids were extracted with chloroform-methanol (2:1), filtered and evaporated to dryness followed by solid phase extraction with LC-SCX SPE columns (Supelco, PA). Lipids were isolated as described (Galbiati et al.
, 2007). The psychosine fraction, containing lyso-lactosylceramide
(Matreya, PA) added as internal reference, was eluted with 7.5 ml of chloroform-methanol
1:2. Eluates were dried and resuspended in 5 mM ammonium formate/methanol. We analyzed psychosine using a HPLC tandem mass spectrometry (LC/MS/MS) system consisting of a Shimadzu Shimpak VP-ODS-C18 reverse phase 4.5x150 mm for HPLC and a
Shimadzu LC-MS-8030 triple quadrupole mass spectrometer. Psychosine concentration on each sample was normalized to protein concentration determined by the Bradford Assay.
HUVE cell motility assay. HUVE cell motility was assessed by time lapse videomicroscopy.
Cells were detached from culture plates and resuspended in M199 containing 5% FCS, allowed to adhere on fibronectin-coated dishes and stimulated with 30 ng/ml of VEGF.
Constant temperature (37°C) and pCO
2
(5%) were maintained throughout the experimental period by means of an heatable stage and climate chamber. Cells were observed under an inverted photomicroscope (Axiovert 200M, Zeiss, Oberkochen, Germany) and phase-contrast snap photographs (one frame every 10 min) were digitally recorded for 8 h. Cell paths (20-50 cells per experimental point) were generated from centroid positions and migration
9
parameters were analyzed with the “Chemotaxis and Migration Tool” of ImageJ software
(http://rsbweb.nih.gov/ij).
Histopathology of human GLD biopsy.
Representative formalin-fixed paraffin embedded tissue sections from autopsy brain specimens of one GLD patient (age 2.5 y) and a control brain obtained anonymously from one patient with no neurological abnormalities who died for respiratory complications were submitted to hematoxylin and eosin (H&E) and immunohistochemical stainings for evaluation of histological diagnosis and vessels morphology and distribution. Samples were obtained from the Eunice Kennedy Shriver
National Institute of Child Health and Human Development, at the University of Maryland
School of Medicine in Baltimore, Maryland. Use of this material was approved by the
Institutional Review Board of College of Medicine, University of Illinois, (Chicago, IL).
Briefly, sections were de-waxed, rehydrated and endogenous peroxidase activity blocked in
0.3% H
2
O
2
/methanol for 15 min. Heat-induced antigen retrieval, when needed, was obtained using a microwave-oven in 1.0 mM EDTA buffer (pH 8.0). Sections were then washed and pre-incubated in 5% normal goat serum in Tris-HCl before incubation for 2 h with primary antibodies. Single immunostains were revealed by ChemMATE Envision Rabbit/Mouse-HRP
(DAKO Cytomation, Glostrup, Denmark) followed by diaminobenzidine/hydrogen peroxide as chromogen and hematoxylin as counterstain. For double immunostains, sections were again incubated with a specific primary antibody for which appropriate biotinylated secondary antibodies were applied (DAKO) followed by incubation with streptavidinconjugated alkaline phosphatase (DAKO) and Ferangi Blue chromogen kit (Biocare Medical,
Concord, CA) and methyl green counterstaining. The following primary antibody and dilutions were used: polyclonal anti-GFAP (DAKO; 1:200), monoclonal anti-CD68 (DAKO; clone KP1, 1:300), monoclonal anti-SMA (DAKO; clone HHF35, 1:100), monoclonal anti-
CD31 (Novocastra; clone 4C9, 1:50), polyclonal anti-Factor VIII-related antigen
(Novocastra; 1:100). Images were acquired by Cell F imaging software (Soft Imaging System
GmbH) with an Olympus DP70 camera mounted on a BX60 Olympus microscope.
Statistical analysis.
Comparisons among multiple groups were performed by ANOVA.
Comparisons between two groups were achieved with Student’s unpaired t -test.
10
REFERENCES
Bastaki M, Nelli EE, Dell'Era P, Rusnati M, Molinari-Tosatti MP, Parolini S, et al. Basic fibroblast growth factor-induced angiogenic phenotype in mouse endothelium. A study of aortic and microvascular endothelial cell lines. Arterioscler Thromb Vasc Biol 1997;
17: 454-64.
Belleri M, Ribatti D, Nicoli S, Cotelli F, Forti L, Vannini V, et al. Antiangiogenic and vascular-targeting activity of the microtubule-destabilizing trans-resveratrol derivative
3,5,4'-trimethoxystilbene. Mol Pharmacol 2005; 67: 1451-9.
Coltrini D, Di Salle E, Ronca R, Belleri M, Testini C, Presta M. Matrigel plug assay: evaluation of the angiogenic response by reverse transcription-quantitative PCR.
Angiogenesis 2013; 16: 469-77.
Dong QG, Bernasconi S, Lostaglio S, De Calmanovici RW, Martin-Padura I, Breviario F, et al. A general strategy for isolation of endothelial cells from murine tissues.
Characterization of two endothelial cell lines from the murine lung and subcutaneous sponge implants. Arterioscler Thromb Vasc Biol 1997; 17: 1599-604.
Galbiati F, Basso V, Cantuti L, Givogri MI, Lopez-Rosas A, Perez N, et al. Autonomic denervation of lymphoid organs leads to epigenetic immune atrophy in a mouse model of Krabbe disease. J Neurosci 2007; 27: 13730-8.
Gualandris A, Rusnati M, Belleri M, Nelli EE, Bastaki M, Molinari-Tosatti MP, et al. Basic fibroblast growth factor overexpression in endothelial cells: an autocrine mechanism for angiogenesis and angioproliferative diseases. Cell Growth & Differentiation 1996;
7: 147-60.
Marchesini S, Preti A, Aleo MF, Casella A, Dagan A, Gatt S. Synthesis, spectral properties and enzymatic hydrolysis of fluorescent derivatives of cerebroside sulfate containing long-wavelength-emission probes. Chem Phys Lipids 1990; 53: 165-75.
Ribatti D, Nico B, Vacca A, Presta M. The gelatin sponge-chorioallantoic membrane assay.
Nat Protoc 2006; 1: 85-91.
Ronca R, Benzoni P, Leali D, Urbinati C, Belleri M, Corsini M, et al. Antiangiogenic activity of a neutralizing human single-chain antibody fragment against fibroblast growth factor receptor 1. Mol Cancer Ther 2010; 9: 3244-53.
Rybak JN, Ettorre A, Kaissling B, Giavazzi R, Neri D, Elia G. In vivo protein biotinylation for identification of organ-specific antigens accessible from the vasculature. Nature methods 2005; 2: 291-8.
11
Sakai N, Inui K, Tatsumi N, Fukushima H, Nishigaki T, Taniike M, et al. Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe's disease. J Neurochem 1996; 66: 1118-24.
Yoshimura M, Nishikawa A, Nishiura T, Ihara Y, Kanayama Y, Matsuzawa Y, et al. Cell spreading in Colo 201 by staurosporin is alpha 3 beta 1 integrin-mediated with tyrosine phosphorylation of Src and tensin. J Biol Chem 1995; 270: 2298-304.
Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. J Biol Chem 1998; 273: 21125-31.
12