HLA Match Worksheet
advertisement

HLA and Matching
Learning Objectives
After completion of this module, the student will be able to
describe the role of HLA in matching
determine the likelihood of full and haplo matches through simulations and calculations for
related and unrelated donors
calculate genetic distances between two populations based on gene frequencies
Concepts
Simulating inheritance
Binomial and geometric distribution
Genetic distance
Knowledge and Skills
Excel functions
Simulating with Excel
Prerequisites
Basic familiarity with Excel
Citation: Neuhauser, C. HLA and Matching
Created: June 8, 2013 Revisions:
Copyright: © 2013 Neuhauser. This is an open-access article distributed under the terms of the Creative Commons Attribution
Non-Commercial Share Alike License, which permits unrestricted use, distribution, and reproduction in any medium, and allows
others to translate, make remixes, and produce new stories based on this work, provided the original author and source are
credited and the new work will carry the same license.
Page 1
The Story of Xilan Part 1: Related Donor
At age 13, Xilan was diagnosed with acute myeloid leukemia (AML), which was treated with
chemotherapy. Xilan was in remission for five years. At age 18, she was diagnosed with a recurrence of
leukemia after she reported feeling tired and running a fever. Although she tolerated the first treatment
well, her doctor recommends a hematopoietic cell transplant. She has four older siblings who
immediately volunteer to be tested for being a donor.
Task 1: Go to http://www.stanford.edu/dept/HPS/transplant/html/hla.html and learn about the
matching process.
Task 2: Below is Xilan’s family tree. Draw genotypes of the four children so that one is a full match, one
is a haplo match but not a full match, and two are no matches.
1,8,10
2,7,11
3,14,17
10,16,8
3,14,17
10,16,8
Figure 1: Xilan’s and her parents’ genotypes.
Task 3: Investigate through simulations how likely it is that at least one of Xilan’s siblings is a full or
haplo match.
To investigate the likelihood of a match, we simulate the genotypes of the four siblings. The genotypes
of the parents and of Xilan are given. We arbitrarily designate one of the haplotypes of each parent as
“0” and the other as “1” and assume that Xilan is of type 1-1. Since haplotypes are typically inherited as
blocks (i.e., we assume no recombination), we can code the haplotypes with “0” and “1”. The genotype
of each sibling is created randomly: each sibling inherits one of the two haplotypes of each parent at
random.
Citation: Neuhauser, C. HLA and Matching
Created: June 8, 2013 Revisions:
Copyright: © 2013 Neuhauser. This is an open-access article distributed under the terms of the Creative Commons Attribution
Non-Commercial Share Alike License, which permits unrestricted use, distribution, and reproduction in any medium, and allows
others to translate, make remixes, and produce new stories based on this work, provided the original author and source are
credited and the new work will carry the same license.
Page 2
Given
A
1
2
3
4
5
6
7
8
9
B
C
D
Type
Parent 1 1 8 10
Parent 2 2 7 11
0
Sibling 1
Sibling 2
Sibling 3
Sibling 4
0
1
1
0
=IF(RAND()<0.5,0,1)
E
F
G
1
3 14 17
10 16 8
Xilan is of type 1-1
1
1
0
0
1 8 10
3 14 17
3 14 17
1 8 10
10 16 8
10 16 8
2 7 11
2 7 11
=IF(C9=0,$C$3,$D$3)
=IF(D9=0,$C$4,$D$4)
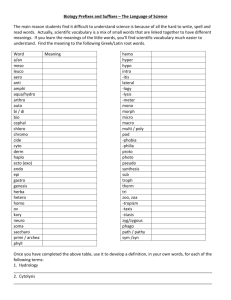
Figure 2: Screenshot
The screenshot in Figure 2 shows the Excel formulas to create the genotype of each sibling, and then
map it to the haplotypes. Columns F and G list the inherited haplotypes. Since Xilan is of type [{3, 14,
17), (10, 16, 8)], we see that Sibling 1 and Xilan are a haplo match, that is, they share one of the
haplotypes. Sibling 2 and Xilan are a full match; sibling 3 and Xilan are a haplo match, and Sibling 4 and
Xilan are no match.
The RAND function in Excel generates a uniformly distributed random variable between 0 and 1. Thus,
the command “IF(RAND()<0.5, 0,1)” results in a 0 with probability 0.5 and in a 1 with probability 0.5.
Citation: Neuhauser, C. HLA and Matching
Created: June 8, 2013 Revisions:
Copyright: © 2013 Neuhauser. This is an open-access article distributed under the terms of the Creative Commons Attribution
Non-Commercial Share Alike License, which permits unrestricted use, distribution, and reproduction in any medium, and allows
others to translate, make remixes, and produce new stories based on this work, provided the original author and source are
credited and the new work will carry the same license.
Page 3
The F9 key on a PC recalculates the sheet, and so, every time you hit the F9 key, you will see a new
realization. We want to keep track of how many siblings can serve as full or haplo matches. Figure 3
shows the screenshot of the spreadsheet
A
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
B
C
D
Type
Parent 1 1 8 10
Parent 2 2 7 11
0
Sibling 1
Sibling 2
Sibling 3
Sibling 4
0
1
1
0
Sibling 1
Sibling 2
Sibling 3
Sibling 4
Full
Match
FALSE
TRUE
FALSE
FALSE
E
F
G
1
3 14 17
10 16 8
Xilan is of type 1-1
1
1
0
0
Haplo
Match
TRUE
TRUE
TRUE
FALSE
1 8 10
3 14 17
3 14 17
1 8 10
10 16 8
10 16 8
2 7 11
2 7 11
Full
Match
Haplo
Match
Total
0
1
0
0
1
=IF(D12,1,0)
1
1
1
0
3
=SUM(G12:G15)
=AND(C9,D9)
=OR(C9,D9)
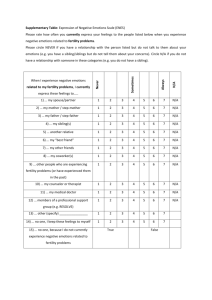
Figure 3: Counting the number of full and haplo matches.
A full match means that both haplotypes have to match. Since Xilan is of type 1-1, this means that the
sibling has to be of type 1-1 as well. The Excel command “AND” results in TRUE if both haplotypes are of
type 1. This is listed in Column C and Rows 12-15 for the four siblings. For a haplo match, we only
require that one of the two matches. This can be checked with the “OR” function, as shown in Column
D. Columns F and G translate the TRUE and FALSE into numbers, so that we can count the number of
times a “TRUE” occurs in Columns C and D, respectively. This translation is accomplished with the “IF”
function. The command “=IF(D12,1,0)” results in a “1” if the entry in D12 is “TRUE” and in a “0” if the
Citation: Neuhauser, C. HLA and Matching
Created: June 8, 2013 Revisions:
Copyright: © 2013 Neuhauser. This is an open-access article distributed under the terms of the Creative Commons Attribution
Non-Commercial Share Alike License, which permits unrestricted use, distribution, and reproduction in any medium, and allows
others to translate, make remixes, and produce new stories based on this work, provided the original author and source are
credited and the new work will carry the same license.
Page 4
entry in D12 is “FALSE”. The Cells F16 and G16 count the number of 1s in Cells F12:15 and G12:15,
respectively.
Create a set of new columns where you copy the counts for full and haplo match. To copy the entries in
Cells F16 and G16, highlight both cells, copy, and then paste the entries as values into two new cells.
Repeat this 500 times. We can either do this manually or write a Macro. Questions we can investigate
are, for instance, how many families have no siblings that are full matches or haplo matches.
Further Exploration
We can increase the family size and ask questions, such as how many siblings are needed until the first
full or haplo match occurs.
The probability of a match (full or haplo) can be calculated using the binomial distribution. The binomial
distribution models the number of successes in n independent trials where each trial has probability p of
success. The formula is
n k
P( ksuccesses in ntrials) 1
( p ) p
k
n k
Excel calculates this probability as well. For instance, to calculate the probability of 3 successes in 4 trials
where each trial has probability 0.25 of success would be
=BINOM.DIST(3,4,0.25,FALSE)
The “FALSE” tells Excel to calculate the probability mass function. If, instead, we wrote “TRUE”, Excel
would calculate the cumulative distribution function.
Each sibling can have one of four genotypes. A full match occurs in one out of the four possibilities, and
a haplo match in three out of the four possibilities. Use this information to compare the simulations to
the theory.
We can also ask how long we need to wait for the first success. This is described by the geometric
distribution. Again, we assume that the trials are independent and the probability of a success in each
trial is p. Then
P(first success in k thtrial) 1
( p ) p
k 1
To calculate the probability that the first success occurs in the 4th trial, the Excel command is
=NEGBINOM.DIST(3,1,0.25,FALSE)
Citation: Neuhauser, C. HLA and Matching
Created: June 8, 2013 Revisions:
Copyright: © 2013 Neuhauser. This is an open-access article distributed under the terms of the Creative Commons Attribution
Non-Commercial Share Alike License, which permits unrestricted use, distribution, and reproduction in any medium, and allows
others to translate, make remixes, and produce new stories based on this work, provided the original author and source are
credited and the new work will carry the same license.
Page 5
The syntax is
=NEGBINOM.DIST(number_f, number_s, probability_s, CUMULATIVE)
where number_f is the number of failures before the number_s-th success, and probability_s is the
probability of success. CUMULATIVE is a logical value. If CUMULATIVE is FALSE, the function returns the
probability mass function. If CUMULATIVE is TRUE, the function returns the cumulative distribution.
[Note that the geometric distribution is a special case of the negative binomial distribution when
number_s is equal to 1. In general, the negative binomial distribution calculates the probability that the
kth success (number_s) occurs on the nth trial (number_f+ number_s) when the trials are independent
and the probability of success is p (probability_s).] See http://office.microsoft.com/en-us/excelhelp/negbinom-dist-function-HP010335688.aspx for more information on this function.
The Story of Xilan Part 2: Unrelated Donor
It turns out that none of Xilan’s siblings is a suitable donor. Xilan is attending a local college, and when
her friends in college learn about the recurrence, they immediately volunteer to help. They quickly learn
about the National Marrow Donor Program (NMDP)
(http://www.youtube.com/watch?v=uXwUzEkrWf0) and decide to organize a Marrow Donor Registry
Drive to find a donor in the community (http://www.youtube.com/watch?v=3L8p_rhiPuw). Her friends
wonder how many donors they would need to recruit to find a match. They focus on the county where
the college is located.
Census data is available to learn about the ethnic/racial distribution in any county in the U.S.
(http://www.census.gov/2010census/). The NMDP uses the following four categories: EUR (Caucasian),
AFA (African American), API (Asian/Pacific Islander), and HIS (Hispanic).
Task 1: Pick a county. Find the number of people in the four ethnic/racial groups (EUR, AFA, API, and
HIS) in the county of your choice.
NMDP publishes data on haplotypes according to ethnic/racial groups. The data are available on their
website (http://bioinformatics.nmdp.org/HLA/Haplotype_Frequencies/Haplotype_Frequencies.aspx).
Task 2: Data from NMDP about the frequencies of the various haplotypes in each of the four
ethnic/racial groups can be downloaded
(http://bioinformatics.nmdp.org/HLA/Haplotype_Frequencies/Haplotype_Frequencies.aspx). To
illustrate the type of calculations, we will use serotype instead of genotype—the data for HLA-A
serotypes is in the spreadsheet.
Citation: Neuhauser, C. HLA and Matching
Created: June 8, 2013 Revisions:
Copyright: © 2013 Neuhauser. This is an open-access article distributed under the terms of the Creative Commons Attribution
Non-Commercial Share Alike License, which permits unrestricted use, distribution, and reproduction in any medium, and allows
others to translate, make remixes, and produce new stories based on this work, provided the original author and source are
credited and the new work will carry the same license.
Page 6
Task 3: Find the number of people in each ethnic/racial group in the county of your choice who are of
type HLA-A1, and thus find the likelihood of finding a match in the county for that serotype.
Task 4: The reciprocal of the likelihood you calculated in Task 3 is the expected number of individuals
required to find a match. You can also use the binomial distribution to find the number of individuals
required to find at least one match with probability, say, 0.95, if the probability of a successful match is
p:
P(at least one match in ntrials) 1- (noPmat
ch in n trials)
n
1 1
( p) 0.95
We can solve this for n:
1 (1 p) n0.95
0.05 (1 p) n
log(0.05 ) log(
n1
) p
log(0.05 )
n
log(1 p)
The probability for a successful match depends on the ethnic/racial composition of the county and the
frequencies of the serotype for each of the ethnic/racial groups. This is the likelihood you calculated in
Task 3.
The Story of Xilan Part 3: Genetic Distances
Xilan’s family immigrated from China. They belong to an ethnic minority, the Tujia
(http://en.wikipedia.org/wiki/Tujia_people). We use the serotype HLA-A to calculate the genetic
distance between the Tujia people and the four ethnic/racial groups in the NMDP database.
Cavalli-Sforza and Edwards1 used the following formula to calculate the genetic distance α between two
populations with gene frequencies [ p1, p 2 , , pn ] and [q1,q 2 , ,q n ] , respectively.
cos p1q1 p 2q 2
pn q n
The spreadsheet “Tujia” has the data for the HLA-A frequencies from NMDP and the Tujia population.
The data from the Tujia population is from the paper by Zhang et al.2
1
Cavalli-Sforza, L. L., & Edwards, A. W. (1967). Phylogenetic analysis. Models and estimation
procedures. American journal of human genetics, 19(3 Pt 1), 233.
Citation: Neuhauser, C. HLA and Matching
Created: June 8, 2013 Revisions:
Copyright: © 2013 Neuhauser. This is an open-access article distributed under the terms of the Creative Commons Attribution
Non-Commercial Share Alike License, which permits unrestricted use, distribution, and reproduction in any medium, and allows
others to translate, make remixes, and produce new stories based on this work, provided the original author and source are
credited and the new work will carry the same license.
Page 7
Task 1: Calculate the genetic distances between the Tujia population and the ethnic/racial groups based
on HLA-A. (Note that the Excel function ACOS calculates the angle α if cos(α) is given.) Which
ethnic/racial groups are closest to the Tujia population?
Further Exploration
We can use the HLA-A frequencies to calculate all pairwise genetic distances, and use the distances to
construct a tree that reflects the genetic distances.
2
Zhang, L., Cheng, D., Tao, N., Zhao, M., Zhang, F., Yuan, Y., & Qiu, X. (2012). Distribution of HLA-A,-B
and-DRB1 Genes and Haplotypes in the Tujia Population Living in the Wufeng Region of Hubei Province,
China. PloS one,7(6), e38774.
Citation: Neuhauser, C. HLA and Matching
Created: June 8, 2013 Revisions:
Copyright: © 2013 Neuhauser. This is an open-access article distributed under the terms of the Creative Commons Attribution
Non-Commercial Share Alike License, which permits unrestricted use, distribution, and reproduction in any medium, and allows
others to translate, make remixes, and produce new stories based on this work, provided the original author and source are
credited and the new work will carry the same license.
Page 8