Abstract template for the IDS 2006
advertisement

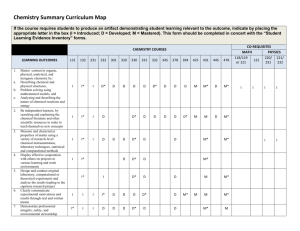
1:a NT-Forskningskonferensen Karlstad 5 maj 2015 ON THE USEFULNESS OF COMPUTATIONAL CHEMISTRY K. Andersson Institutionen för Matematik och Datavetenskap. Tel.:054-7001873, E-mail: kerstin.andersson@kau.se Abstract: Computational chemistry and physics can be considered as bridging the gap between theory and experiment. Computations can give detailed explanations of processes that are not so easily captured in experiments. In the presentation an introduction of computational chemistry will be given together with some example problems, where computational chemistry can be useful for the research conducted at Karlstad University. INTRODUCTION Computational chemistry attempts to calculate, using computers, the predictions of quantum theory for molecular systems and solids. Central to the calculations is the many-body problem, which usually requires enormous computer resources for accurate solutions. By various approximations (and/or the inclusion of experimental data) the application domain can be enlarged. In the presentation only accurate ab initio methods will be considered and therefore the application domain necessarily is restricted to fairly small model systems. The calculations have been performed using the MOLCAS quantum chemistry software package (www.molcas.org). The central ingredient in this package is the multi-configurational wave function (in the complete active space (CAS) self-consistent field (SCF) framework). This allows the calculation of qualitatively correct wave functions (beyond the HartreeFock level) for both ground and excited electronic states. By incorporating dynamical electron correlation effects through second-order perturbation theory (in energy) also quantitatively correct descriptions are given, important for accurate calculations of energy differences. In the example problems discussed below the calculation of energy differences is a common factor. EXAMPLE MODEL SYSTEMS In order to give a flavor of the kind of calculations that can be performed, two problem areas will be covered, both present in the research conducted at Karlstad University. Firstly, a solid-state problem (steel) will be discussed and secondly an organic chemical problem (organic solar cells) will be considered. Steel In order to understand the behavior of steel alloys, molecular dynamics (MD) simulations can be performed (Holleboom and Alasqalani, 2015). The forces between particles can be estimated through pair potentials. The pair potential between one pair of atoms in steel (vanadium and carbon) has been calculated using the MOLCAS software (see Fig. 1). The dissociation energy (an energy difference) of 0.170 H obtained using secondorder perturbation theory (CASPT2) compares well with the experimental value of 0.160 ± 0.009 H (Gupta and Gingerich, 1981). However, even though the pair potential can be considered accurate it is still an approximation in MD simulations. Fig. 1. Pair potential for vanadium and carbon Organic solar cells The active layer of an organic solar cell typically consists of a mixture of electron donor molecules (polymers), electron acceptor molecules (polymers or fullerene derivatives) and solvent molecules. By an applied electric field a direction is enforced on the solar cell bulk material which drives the photo-excited electrons and their corresponding holes (see step 1 in Fig. 2) to different electrodes and thereby generates a photocurrent. The holes are transported along the donor polymer chain and the electrons by hopping between acceptor fullerene molecules. The central process in the working of a solar cell is the formation of a charge transfer (CT) complex at the interface between donor and acceptor material (see step 3 in Fig. 2)l. A proper understanding of the mechanisms around the CT complex, which involve non-equilibrium conditions, excited states, recombination (radiative/non-radiative) etc., may be achieved using computational chemistry. Here it is essential that energy differences are properly described since it is the relative positions of energy levels that are critical (see the black bars in Fig. 2). As a concluding remark the following quotation from the pedagogical and visionary article in Science by Sariciftci et al. (1992) is given: “The wide range of possible conducting polymers and conjugated oligomers available as donors and C60 derivatives (for example in polymeric form) as acceptors implies an important scientific opportunity.” ACKNOWLEDGEMENTS The author is grateful to L. J. Holleboom for generating the graphs in Fig. 1 and to A. Alasqalani for providing a nice problem (VC). REFERENCES Gupta S. K. and K. A. Gingerich (1981), Mass spectrometric study of the stabilities of gaseous carbides of vanadium, niobium, and molybdenum, J. Chem. Phys., Vol. 74, pp. 3584-3590. Holleboom L. J. and A. Alasqalani (2015), Oral communication. Ma, Z. (2013), Studies of morphology and chargetransfer in bulk-heterojunction polymer solar cells, LiU-Tryck, Linköping, Sweden. Sariciftci N. S., L. Smilowitz, A. J. Heeger and F. Wudl (1992), Photoinduced electron transfer from a conducting polymer to buckminsterfullerene, Science, Vol. 258, pp. 1474-1476. Fig. 2. The working principle of a solar cell (Ma, 2013) According to Sariciftci et al. (1992) the electron transfer (step 3 in Fig. 2) describes the formation of an ion radical pair and they give criteria for this to occur, which involves ionization potentials, electron affinities and electronic energies of ground and excited states of the participating donor and acceptor molecules. Again energy differences are of importance and it is critical to choose quantum chemical methods that describe these well. In some preliminary studies, using the MOLCAS software, the lowest electronic states (both ground and excited), ionization potentials and electron affinities have been calculated for some simple conjugated cycle compounds. All these properties could be calculated with an error compared to experiment of less than 0.2 eV for benzene (C6H6) and indenyl (C9H7). In the presentation more results and details of the calculations will be given. These initial studies will continue by including more and larger conjugated cycle compounds (models of acceptor molecules) and linear conjugated compounds (models of donor molecules). Further, by combining a linear conjugated compound and a cyclic conjugated compound, charge-transfer states will be calculated. The gole is, by this systematic approach, to find patterns and rules that may be useful in the design of organic solar cells.