YG practical 2015 Project 1
advertisement

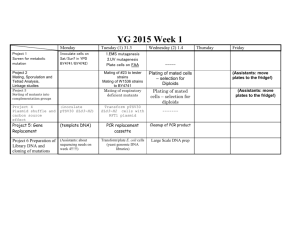
YG 2015 P1 Project 1: Selection for metabolic mutations The very early work done in yeast was concerned with the characterization of metabolic pathways and the characterization of the enzymes involved. We will carry out a mutagenesis that is geared towards identifying mutations in the tryptophane biosynthesis pathway. During this practice, you will learn how to perform mutagenesis by UV-irradiation and chemical mutagenesis with ethylmethane sulfonate and perform a selection for a biosynthetic pathway mutation A selection will only allow the mutants we are interested in to grow (in contrast to a screen, in which you have to look at the phenotype of individual colonies, which is more work-intensive).In general, if you can think of a good selection to obtain mutants you desire, it is always preferable to a screen. Unfortunately, selections are not always possible. In this experiment, you will be using plates containing 5-fluoroanthranilic acid (5-FAA). The 5-FAA selection is specific for mutations in the yeast tryptophan synthesis pathway. Mutations in TRP1, TRP3, TRP4 and TRP5 will lead to resistance of yeast to 5-FAA. The enzymes encoded by these genes cannot distinguish between their natural substrate and 5-FAA and convert it into a highly toxic end product (5-fluorotryptophan), and cells that are wild type for the tryptophane synthesis pathway (with exception of the anthranilate synthase step) die on 5-FAA. Cells that carry a lossof-function mutation in TRP1, TRP3, TRP4 or TRP5 cannot make tryptophan, but will survive on media supplemented with this amino acid. 5-FAA media are often used by yeast geneticists to force loss of a TRP1containing plasmid in plasmid shuffle procedures (see project 4 for plasmid shuffle – we will use a different approach, though) 1 YG 2015 P1 Day 1: Mutagenesis The spontaneous rate of mutation in most organisms is very low (10-7 per division per gene in yeast), and to screen for spontaneous yeast mutants would require a large amount of plates and a long time to screen. We can, however, increase the mutation rate (to a range of 10-3 to 10-5 per gene) by using mutagens (UV light and a chemical mutagen), and save ourselves a lot of material and work. UV-light is a powerful mutagen (remember that the next time you go out to get a tan next summer – put on sunscreen!) and causes the formation of thymidine dimers in DNA, which will be removed by the DNA repair machinery, but not always replaced correctly. The range of mutations that can be obtained by UV-irradiation is somewhat limited by this specific lesion it causes, but it is good enough for our purposed and fairly safe. One or two groups will also be using an alkylating agent called EMS (ethylmethane sulfonate), which is toxic, so people handling this agent need to be careful and to be working in the hood. UV light is quantified by units of energy, which is expressed in Joules. We will use the department’s Stratalinker apparatus, which is normally used to crosslink DNA to a solid support for Northern/Western reports. The experiments will be more reproducible this way, because we can program the amount of energy used during the mutagenesis. In principle, you could do the same procedure on a UV-box for ethidium bromide stained gels, and exposing for varying amounts of time. We are using strains BY4741 (MATa; his3 Δ 1; leu2 Δ 0; met15 Δ 0; ura3Δ0) and BY4742 (MATa ; his3Δ1; leu2Δ0; lys2Δ0; ura3Δ0) for this project. We inoculated cultures yesterday in YPD. EMS mutagenesis (First thing in the morning after introduction!) Ethylmethane sulfonate (EMS) is a fairly toxic alkylating agent. Avoid contact or inhalation. Wear gloves all the time while you are handling materials that contain active EMS. EVERY item that has come in contact with EMS must be rinsed with/submerged in 6 % sodium thiosulfate solution for inactivation. Transfer 1ml of overnight culture of BY4741 (Group 1) or BY4742 (Group 2) into sterile microfuge tubes and spin down for 10 seconds. Resuspend cells in 1.5 ml of sterile 0.1 M NaHPO4 buffer (pH 7) transfer 2x 0.7 ml of resuspended cells to two white capped (Sarsted) tubes containing 1 ml of 0.1 M NaHPO4 buffer (pH 7) add 50 ul of EMS to one of the tubes and mix well (vortex briefly) (the other tube is the unmutagenized control) incubate both tubes for 1 hour, occasionally inverting the tubes gently transfer 0.2 ml of each of the cell suspensions to a separate screw cap pointy-end Falcon tubes containing 8ml of sterile sodium thiosulfate and mix gently and incubate for 5 minutes at room temperature. 2 YG 2015 P1 Spin down cells at maximum speed in pointy end blue cap tubes for 10 min (not in round ended white cap tubes, as you may not get a good enough pellet), gently decant the supernatant. Add 2 ml of YPD, and resuspend cells incubate in shaker for 3 hours make 1/500 , 1/5000 and 1/50000 dilutions of each culture and spread 200 ul of each dilution on YPD plates (growth control to determine killing rate) spread the rest on 10 5-FAA plates (about 200 ul each) incubate at 30oC for several days UV mutagenesis Spread 200ul of 1:5 diluted overnight culture of BY4741 (Group 3) or BY4742 (Group 4) on each 5-FAA plates (10 plates per GROUP!) – 20 plates total. UV-irradiate plates with 2400 microjoules (BY4741) or 3400 microjoules (BY4742) in the Stratalinker (the agar has to face the UV-bulbs; remember to take off the lids!). Do not irradiate more than six plates at a time. make a 1:5000 and 1:50000 dilution of the original culture and plate 200 l of each of these dilutions on two separate YPD plates (4 plates total per group). Expose one of each of these dilution plates to 2400 microjoules (BY4741) or 3400 microjoules (BY4742) microjoules UV light, and leave one of each dilution plates unexposed. These are our controls for cell titer and killing rate. Day 2: Waiting….. Day 3 (Tuesday 2nd week): Determine killing rates The following formula is used to determine the killing rate: Number of colonies on irradiated plate 1- Number of colonies on non-irradiated plate x 100 Killing rate (%) if there are survivors on the 5-FAA plates, pick those and re-streak on YPD plates (pick at least 12 colonies per group; it is not necessary to streak for single colonies; patches are fine). - Transfer plates with mutant patches into incubator and grow at 30oC o/n 3 YG 2015 P1 Day 4 (Wednesday 2nd week) 4: Confirm 5-FAA-resistant/Trpphenotype, mating tests Confirmation of phenotype Streak cells from mutant patches on YPD you picked yesterday (5-FAAresistant mutants) on 5-FAA plates, as well as SC – Trp plates for single colonies. You can make 6-8 sectors per plate. Make sure you have numbered your candidates so yu can identify them later on! Include a trp1 mutant (Strain W1536) and TRP1 wild type controls (BY4741 or BY4742 . These are positive (=growing) and negative (=not growing) controls on the 5-FAA plate. Whenever possible, controls should be grown on the same plate as your samples, and every plate should have its own set of controls!! !!On the SC-Trp plate, the trp1 mutant strain is the negative control (=not growing), and the TRP1 strain is the positive (=growing) strain!! Day 5 (Friday 2nd week): Verify Trp- phenotype of 5-FAA mutants, Select Diploids of 5-FAA mutants - Check the 5-FAA resistant mutant colonies that we streaked on the SC-Trp plates/5-FAA plates on Wednesday. If they grow on 5-FAA, but not on SC-Trp, we can be confident that they are real mutants; isolates that fail this test will be discarded and not used for the further analysis Matings: mate 5-FAA resistant, Trp- mutants against wild type of opposite mating type (this is again to determine the dominant or recessive nature of the mutations) mate mutants of one mating type against mutants of the opposite mating type (the groups with mutants of opposite mating type will exchange four or five of their mutants for this – we do this to be able to sort the mutants into complementation groups). How many complementation groups do you expect from the 5-FAA resistance mutant screen? Transfer all the plates into the 30oC incubator and let grow until next week 4 YG 2015 P1 Day 6 /Tuesday 3rd week): Diploid selection of 5-FAA resistant mutant mating with wild type or mutant of opposite mating type: - BY4741 and BY4742 carry different marker mutation. Neither of the haploid strains are able to grow on SC –Met, -Lys plates, but the diploids will be streak diploids for single colonies on SC-Met-Lys plates make sure to include the parental controls (like in Project 2) - grow over the weekend in 30oC incubator Transfer all plates to the 30oC incubator and grow until Friday (assistants take out plates) - Inoculate several of your Trp-/5-FAA resistant mutants (5 per group?) in 2 ml YPD for transformation tomorrow (incubate o/n in 30oC shaker) Day 7 (Wednesday 3rd week): Transformation of selected FAA resistant mutant with TRP1 plasmid Transform 5-FAA resistant/Trp- mutants you inoculated yesterday with plasmid YEplac112 (one-step transformation; see day 1 project 4); streak on SC-Trp plates Day 8 (Tuesday 4th week): Test of phenotype of diploids with 5-FAA mutants Pick two colonies of each diploid type, plate for single colonies of SC-Trp and 5FAA plates. Make sure to use Trp+ and Trp- as well as original mutant controls (see Day 4) Incubate for two days in 30oC incubator Score transformations of 5-FAA mutants with TRP1 plasmid - Were any the mutants obtained by selection for 5-FAA resistance complemented by the YEplac112 plasmid containing the TRP1 marker? 5 YG 2015 P1 Day 9 (Wednesday 4th week): Waiting Day 10 (Friday 4th week): Score diploid growth - score for dominance or recessiveness of mutations - how many complementation groups do you find? Review of results 6