Epilepsy Syndromes
advertisement

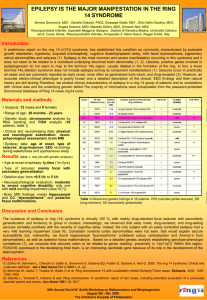
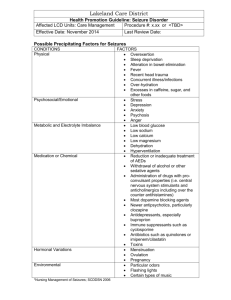
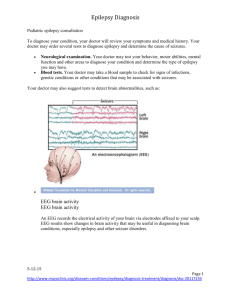
Epilepsy syndromes Name/Age Class Neonatal Benign Familial Neonatal Epilepsy (BFNE) Day 2-15 Focal familial (like BNC or fifth day fits) Early Myoclonic Encephalopathy (EME) first month, up to 3 months Otahara Syndrome (IEE with suppression bursts) 10 days to 3 months Mechanism AD KCNQ 2-3 Potassium Channel, Nicotinic Ach 20q13, 8q24 Epileptic Glycine encephalopathy encephalopathy, other metabolic syndromes, (csf and serum neurotransmitter, amino/organic acid) pyridoxine Epileptic Structural lesions, encephalopathy Aicardi syndrome, Porencephaly Semiology EEG Treatment Prognosis Clusters of focal clonic seizures, apnea, sec generalization, last 3-4 months Erratic progressive myoclonus, Apnea, tonic spasms, partial motor sz Normal interictal, theta pointu alternant, suppress to SW Suppressionburst EEG None, or phenobarb Spontaneous recovery, 10-15% have later epilepsy Many AEDs DPH, PB, Benzos, lignocaine (pyridox/biotin creatine PRN) Poor response to AEDs, developmental arrest, microcephaly, atrophy, 50% die by 1 year (severe) Tonic spasms Suppressionburst EEG ACTH, KD Phenobarb, Benzos, TPM, lignocaine, ZNM Developmental arrest, often go on to West Syndrome, can die (surgery can help) ACTH, KD Infantile West syndrome 4-7 months Epileptic Various mostly encephalopathy structural, or genetic or metabolic causes Clusters of infantile spasms, increasing in drowsiness, myoclonic-tonic Hypsarrythmia with electrodecreme ntal seizures ACTH, Vigabatrin (VGB in TS) Pyridoxine, ketogenic diet Developmental arrest, 85% have poor outcome cognitively, 630% die by age 3 Myoclonic epilepsy Idiopathic of Infancy (MEI) generalized 4-36 months epilepsy Family history of seizures or febrile seizures in 30%, male predominate Epilepsy of infancy with migrating focal seizures (MMPEI) malignant <6 months Benign Familial Infantile Epilepsy <12 months Infection, hypoxia, structural lesions Dravet Syndrome (SMEI –severe myoclonic epilepsy of infancy) <12 mo Myoclonic encephalopathy in nonprogressive disorders Symptomatic focal epilepsy Genetic idiopathic focal epilepsy SCN2A Sodium channel AD 2q23 (6p, 19q, 2q) assoc with paroxysmal chorea, ATPase Genetic SCN1A epileptic Sodium channel encephalopathy 2q24 sporadic Epileptic encepalopathy Myoclonic, or reflex visual seizures, febrile seizures, GTC in adolescence Clusters of focal polymorphic seizures, with frequent secondary generalization Clusters of brief partial seizures, often clonic, head and eye deviation Fast SW or poly SW with myoclonus, normal background IEA migrates to nearby areas with rhythmic theta becoming more continuous Seizures in wakefulness, IEA focal spikes or fast activity, otw normal VPA Benzodiazepin es Respond well to AEDs, mild cognitive impairment.. CBZ, VPA, PB Favorable response, normal development Long Febrile seizures, GTC, then myoclonic seizures yrs 1-4, then atypical absence or focal hemiclonic sz Normal, then gen SW, photosensitive, and heat sensititve Stiripental, VPA Clobazam, TPM, ZNM Ketogenic diet (avoid CBZ, DPH, Na channel drugs) Ataxia, psychomotor retardation, refractory seizures, poor outcome but sz stabilize Prolonged myoclonic status Continuous slow SW, asynchronous myoclonus Progressive cognitive decline 28% mortality Neuropsychologic impairment, poor prognosis Childhood Generalized Idiopathic epilepsy with generalized febrile seizures plus (GEFS+) 3 mo-6 years Paniotopoulos Idiopathic focal Syndrome (early benign childhood epi with occipital spikes) 5 years FH febrile sz SCN1A/B Sodium channel Or GABRG2, GabaA ?SCN1A GTC with fever after 6, or then GTC without fever, febrile myoclonus Pronounced autonomic features – EMESIS, LOC, atonia, head and eye deviation, salivation Epilepsy with myoclonic-astatic (atonic) seizures (Doose) 18 mo-5 yr, max at 3 years BECTs 2-13 years (9-10 max) Idiopathic generalized Some with SCNA1 Myoclonicatonic, massive jerk, then drop Idiopathic genetic focal AD Nocturnal Frontal Lobe Epilepsy (ADNFLE) 11 years Genetic focal AD 15q23 More males, elongator protein complex ELP4 11p AD Nicotinic Ach R CHRNA4 CHRNB2 Focal seizures hemifacial motor, oropharyngeal sensory, GTC Jerking in NREM sleep, automatisms, vocalizations, dystonia Normal Favorable Occipital spikes, (30% with other spikes or none), IEA is shifting or multifocal, high amplitude spikes Atypical 4-7 Hz SW or polySW, (or 24 Hz SW) None, or benzos, CBZ, DPH, LEV Remits after 1-2 years, favorable outcome, many have only 5-6 seizures VPA, ESM, TPM, ACTH, ZNM, LMT, ketogenic diet Variable, 50% remit Diphasic trains of centrotemporal spikes, often bilateral Ictally – frontally dominant slow discharges Often not needed, or GBP, CBZ, PB, DPH, VPA, OZC All remit Generally favorable Late-onset, childhood occipital onset epilepsy (Gastaut) 8-9 years Idiopathic focal 21-37% have FH Visual colors, hallucinations and temporary blindness, hemiclonic sz, automatisms Epilepsy with myoclonic/absence s (Tassinari) or like Jeavons eyelid myoclonia 7 years Lennox Gastaut Syndrome 3-10 years Idiopathic generalized Epileptic encephalopathy with continuous SW during sleep (CSWS) 4-7 yrs Landau Kleffner Syndrome (LKS) 3-8 years Epileptic 30% abnormal encephalopathy MRI Loss of consciousness with severe rhythmic myoclonus and absences Tonic, absence and atonic seizures (multiple types) Generalized or focal seizures, atypical absence in wakefulness More boys (70%), 30% have family history Epileptic 75% have prior encephalopathy insult, others idiopathic Epileptic Hypometabolism encephalopathy PET temporal lobe Acquired aphasia (verbal auditory agnosia, 80% seizures Occipital SW on Eye closure (attenuate with eye opening) “fixation off” sensitivity, ictal low ampl fast spikes, more in sleep 3 Hz SW associated with myoclonus, 30% photoparoxysm al Slow 1.5-2.5 SW, sleep GPFA at 10 Hz CSWS -- 85% SWS has SW CBZ, VPA, etc, wean after 2 yrs seizure free Favorable, remit in adolescence VPA, LMT, ESX, CZP, LEV, TPM, Diamox Some are resistant to medications, 50% with cognitive changes VPA, CBZ TPM, LMT, RUF, ketogenic diet Diazepam, Prednisone, LEV, VPA, ESM MR, developmental delay ?callosotomy Cognitive deterioration, but can improve, 25% near normal CSWS and also in wakefulness at times Diazepam, Prednisone, LEV, VPA, LMT, ESM, IvIG Cognitive changes, but resolution of EEG in adolescence 50% near normal Childhood Absence Epilepsy (CAE) 4-10 years Idiopathic generalized Juvenile Absence Epilepsy (JAE) 7-17 years Juvenile Myoclonic epilepsy (JME) 8-26 years Idiopathic generalized Epilepsy with Grand Mal seizures on awakening (EGMA) 16-19 yrs Idiopathic generalized Adult Mesial Temporal lobe Epilepsy MTLE Idiopathic generalized Structural focal AD lateral temporal Genetic focal lobe epilepsy with auditory features 15-25 Gaba R Chloride channel GABARG2, rh1 CLCN2 10% GLUT1, like PKDyskinesia EF-hand, EFHC1 CLCN2 Clustering absences, 50% with GTC 3 Hz SW 4-20 sec Ethosuximide, VPA, LMT, ketogenic diet Normal development, test refractory for GLUT 1 Sporadic absences, most have GTC AM myoclonus GTC >90% Absences 30% 3.5 Hz SW VPA, LMT, LEV Polyspikewave, Photosensitive in 30-35% VPA, LEV, TPM, Felbamate, Clobazam Favorable, but less likely to remit with GTC Lifelong seizures, Problems with semantic or verbal fluency GTC on awakening, 80% with myoclonis or absence >3 Hz SW, photosensitive VPA, LMT, TPM, LEV 75% remit MTS Epigastic rising, automatisms, refractory Anterior temporal epileptiform actiivty CBZ, OXC, ZNM, LEV, LMT, TPM,.. Refractory, responds to surgery AD 10q24 Leucine-rich repeat epilepsy protein Voltage gated potassium channels VGKC Auditory hallucinations GabaA R GABRA1 PPR1, (photic) EF-hand motif protein, EJM CLCN2 gene 40% genetic, 60% male Responds to AEDs Chromosomal defects with epilepsy Wolf-Hirshhorn Year 1 Glut 1 glucose transporter defect Angelman Syndrome 18-24 months Miller-Dieker Chr 4, greek warrior, beak microceph GLUT1 Low CSF glucose Chr 15 happy puppet Ring 20 14-3-3 17p13 LISI YW2AE lissencephalopath y 20 Trisomy 21 21 Rett Syndrome X chromosome MECP2 Methyl CPG binding protein 3-4 hz sw Sz 6-12 months, ataxia GTC, CPS, Absence Ketogenic diet 2 hz SW Low ear, upslanting eyes, high forehead Eyelid myoclonus, nonconvulsive absence status Generalized seizures more common with aging Hand wringing 66% GTC ACTH, benzos Microcephaly, atrophy Cognitive dysfunction, Ataxia, tremor, seizures improve May be refractory 2-3 Hz SWand frontal SW Mild-moderate learning problems and refractory seizures Late gradually progressive dementia Slow background, Central or temporal discharges Regression, microcephaly, ataxia, autism, sz Girls-only epilepsy <3 years Pyridoxine deficiency Birth to 3 months Biotinidase deficiency neonatal Progressive myoclonic epilepsies UnverrichtLundborg Syndrome 6-15 years Lafora Body Disease Neuronal ceroid lipofuscinosis: Jansky- X linked, 50% daughters, PCDH19 Familial sporadic AR 5q31 Lysine degradation, (antiquin deficiency) AR 3p25 AR Cystatin B 21q22 Clusters of febrile seizures 1-3 months, focal or generalized convulsive Severe seizures first few days of life Alopeicia, dermatitis, GTC GTC severe myoclonus, absences, ataxia AR 6q24 Lafora bodies in skin bx, PAS+ polyglucosan, EPM2A mutation AR, 11p15 Finnish (adult Ad form) Typical Treat with Pyridoxine GTC, blindness Myoclonic, atonic, GTC, MR in 2/3, psychiatric – ADHD, aggression, psychosis, variable outcome Jittery, with less sleep, MR leukodystrophy even with Rx Treat with biotin SW at 2-3 or 46 Hz, slow bkgd, photosensitive Starts at 3 Hz, then 6-12 Hz in late disease, Photosensitive at low frequencies Giant VEP, occipital spikes with low VPA, clobazam ?restrict carbs? Gene rx, vit e, c, methionine? Progressive, unfavorable, but slow little cognitive decline Severe, to death in 2-10 years Dementia, psychosis, ataxia, Death within 5-8 Bielshowski, SpiegelmeyerVogt, Battens, Kufs lipopigment in liposomes or mutation TPP1, CLN3, CLN5 absence frequency photic years Sialidosis AR NEU1 Alpha Neuraminidase deficiency in leukocytes, galactosidase Maternal more, Mitochondrial, t-RNA-lysine mutation, MTTK AD CAG repeat Ataxia, cherry red spot, GTC and myoclonus Low voltage fast activity 10-20 Hz vertex seizures Gradual decline MERRF DRPLA Neurocutaneous Syndromes NF1 NF2 Gen SW 2-5 Hz, sometimes focal IEA Chorea, ataxia ? AD AD TS complex Vigabatrin Ataxia Telangiectasia AR Incontinenti Pigmenti Xlinked Sturge Weber Sporadic Maple syrup urine disease DNPH Low glucose GTC vertex and wicket spikes hypotonia