pathophysiology in hyperammonemic syndromes
advertisement

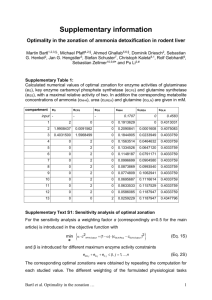
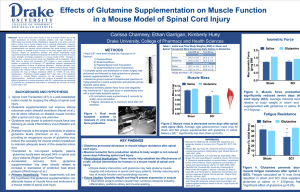
PATHOPHYSIOLOGY IN HYPERAMMONEMIC SYNDROMES: THE MANY FACES OF GLUTAMINE Roger F Butterworth PhD DSc. Dept. of Medicine, University of Montreal and Neuroscience Research Unit, Hopital St-Luc (CHUM), 1058 St-Denis, Montreal, Qc, Canada H2X 3J4 The absence of effective hepatic clearance of excess ammonia in the form of urea, as occurs in urea cycle enzymopathies (UCDs) and in several types of liver failure leads to increases in circulating and tissue concentrations of glutamine. Studies using classical biochemical approaches or, more recently, 1H-NMR spectroscopy demonstrate a positive correlation between brain glutamine increases and the severity of neurological symptoms in these disorders. These findings have led to the assumption that increased brain glutamine synthesis rates are predictive of the severity of neurological complications as a result of the osmotic effects of the amino acid on astrocyte cell volume regulation. Studies using 1H/13C NMR spectroscopy provide evidence of increased de novo synthesis of glutamine in brain in acute hyperammonemia ; but increases of synthesis do not correlate with either the severity of encephalopathy or with the presence of brain edema (Chatauret et al., Gastroenterol., 125, 815, 2003). These findings led to the suggestion that the capacity of the brain to synthesize glutamine is limited in hyperammonemic syndromes, and studies show that the mechanism responsible for this limitation involves tyrosine nitration of the glutamine sythetase (GS) gene and protein involving NMDA receptoractivated nitric oxide production (Schliess et al., FASEB J., 16, 739, 2002). In UCDs and in liver failure, skeletal muscle becomes the organ primarily responsible for removal of excess ammonia and this adaptation results from post-translational induction of the GS gene and protein in muscle (Chatauret et al., J Hepatol., 44, 1083, 2006). 13C-NMR studies confirm that the increase in muscle glutamine synthesis involves both the pyruvate carboxylase and dehydrogenase metabolic pathways. These alterations of inter-organ trafficking of ammonia and the particular importance of muscle in its removal in conditions of urea cycle deficiency accounts for the current resurgence of interest in (1) maintaining adequate dietary protein and (2) the use of L-ornithine Laspartate whose ammonia-lowering action involves primarily the stimulation of muscle glutamine synthesis (Rose et al., Metab Brain Dis., 13, 24, 1998). It is important to bear in mind that other metabolic and regulatory pathways may impact on ammonia and/or glutamine homeostasis, particularly in the brain. These pathways include (1) glutamine deamination by glutaminase, an enzyme with a predominantly neuronal localization, (2) glutamine transaminase leading to the production of the neurotoxic metabolite, alpha-ketoglutaramate (AKGM), and (3) recently cloned and characterized neuronal and astrocytic high affinity glutamate transporters (SNATs). Surprisingly little attention has been given to date to the possibility that increases in circulating and/or brain glutamine in hyperammonemic disorders could result from inhibition of glutaminase by its product despite the fact that it has been demonstrated that neomycin lowers circulating ammonia levels by inhibition of glutaminase rather than an action at the GI tract. One proposed new theory related to the glutaminase pathway suggests that the accumulation of glutamine (“A Trojan Horse”) by the astrocyte could afford an indirect pathway whereby ammonia is generated via the action of glutaminase in the astrocyte (Albrecht and Norenberg, Hepatol., 44, 788, 2006). However, the theory has been challenged on multiple grounds. The transaminase pathway has not been further investigated even though concentrations of AKGM are increased in UCDs and in liver failure. On the other hand, findings of a selective reduction in expression of the glutamine transporter SNAT-5 (responsible for glutamine exit from the astrocyte) in a liver failure model has raised the issue of “glutamine trapping “within these cells in hyperammonemia (Desjardins et al., ref). Such a trapping mechanism could explain the phenomenon of cytotoxic brain edema and, given the key role that glutamine from the astrocyte plays as precursor of neurotransmitter amino acids, could also contribute to the imbalance between excitatory and inhibitory neurotransmission that is considered to occur in both UCDs and in liver failure. Funded by the Canadian Institutes of Health Research