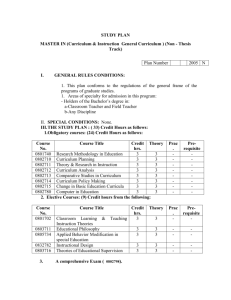
Table of contents - European Medicines Agency
advertisement

Rapporteurs day 180 joint CHMP and PRAC response
assessment report
Overview
<Invented name>
<(Active substance)>
EMEA/H/C/<xxx>
Applicant:
CMHP Rapporteur:
CHMP Co-rapporteur:
PRAC Rapporteur:
EMA EPL:
EMA PM:
Start of the procedure:
Date of this report:
Deadline for comments:
Table of contents
1. Recommendation ..................................................................................... 6
2. Executive summary ................................................................................. 7
2.1. Problem statement ............................................................................................... 7
2.2. About the product ................................................................................................ 7
2.3. The development programme/Compliance with CHMP Guidance/Scientific advice ......... 7
2.4. General comments on compliance with GMP, GLP, GCP ............................................. 7
2.5. Type of application and other comments on the submitted dossier.............................. 7
3. Scientific overview and discussion .......................................................... 7
3.1. Quality aspects .................................................................................................... 8
3.2. Non clinical aspects .............................................................................................. 8
3.3. Clinical aspects .................................................................................................... 9
3.4. Risk management plan ........................................................................................ 10
3.5. Pharmacovigilance system ................................................................................... 11
3.6. New active substance status ................................................................................ 11
4. Orphan medicinal products .................................................................... 11
5. Benefit risk assessment......................................................................... 11
5.1. Conclusions ....................................................................................................... 12
6. CHMP list of outstanding issues to be addressed in an oral explanation
and/or in writing ....................................................................................... 12
6.1. Quality aspects .................................................................................................. 12
6.2. Non clinical aspects ............................................................................................ 13
6.3. Clinical aspects .................................................................................................. 13
6.4. Pharmacovigilance system ................................................................................... 13
6.5. Risk management plan ........................................................................................ 13
6.6. New active substance status ................................................................................ 13
7. Proposed conditions for marketing authorisation and product
information ............................................................................................... 13
7.1. Proposed list of post-authorisation measures* ....................................................... 13
7.2. Other conditions ................................................................................................. 14
7.3. Summary of product characteristics (SmPC) .......................................................... 14
7.4. Labelling ........................................................................................................... 14
7.5. Package leaflet (PL) ............................................................................................ 14
8. Annex 1 (as appropriate) ..................................................................... 15
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 2/15
Administrative information
Invented name of the medicinal product:
INN (or common name) of the active
substance(s):
Applicant:
Applied Indication(s):
Pharmaco-therapeutic group
(ATC Code):
Pharmaceutical form(s) and strength(s):
CHMP Rapporteur contact person:
Name:
Tel:
Fax:
Email:
CHMP Co-Rapporteur contact person:
Name:
Tel:
Fax:
Email:
PRAC Rapporteur contact person:
Name:
Tel:
Fax:
Email:
EMA Product Lead:
Name:
Tel:
Fax:
Email:
Procedure Manager:
Name:
Tel:
Fax:
Email:
Quality:
Name(s)
Tel:
Fax:
Email:
Names of the Rapporteur assessors
(internal and external):
Non-clinical:
Name(s)
Tel:
Fax:
Email:
Names of the Co-Rapporteur assessors
(internal and external):
Clinical :
Name(s)
Tel:
Fax:
Email:
Quality:
Name(s)
Tel:
Fax:
Email:
Non-clinical:
Name(s)
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 3/15
Tel:
Fax:
Email:
Names of the PRAC Rapporteur assessors
(internal and external):
Clinical:
Name(s)
Tel:
Fax:
Email:
Name(s)
Tel:
Fax:
Email:
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 4/15
List of abbreviations
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 5/15
1. Recommendation
Based on the review of the data and the Applicant’s response to the CHMP LoOI on quality, safety,
efficacy and risk management plan, the Rapporteurs consider that the application for <product name>,
<an orphan medicinal product> in the treatment of <indication>,
<is approvable <provided that the applicant commits to perform a number of post authorisation
measures <and specific obligations > to be reported back to the CHMP within predefined timeframes. A
preliminary list of such post-authorisation measures <and specific obligations> are in section VI of this
report> and <provided that points related to the RMP are taken into account in the next update of the
RMP>.
<With reference to Part 4 G of the Annex I to Directive 2001/83/EC, the Rapporteurs consider that: a
marketing authorisation under exceptional circumstances can be recommended since <the
indication(s) for which the medicinal product in question is intended are encountered so rarely that the
applicant cannot reasonably be expected to provide comprehensive evidence/data on the <quality>,
<safety> and <efficacy> of the medicinal product.> <in the present state of scientific knowledge,
comprehensive information on the <quality>, <safety> and <efficacy> of the medicinal product
cannot be provided by the applicant.> <comprehensive information on the <quality>, <safety> and
<efficacy> of the product cannot be provided because it would be contrary to generally accepted
principles of medical ethics to collect such information.>>
<is not approvable since major objections still remain, which preclude a recommendation for
marketing authorisation at the present time. The details of these major objections can be summarised
as follows:
Quality aspects
Non-clinical aspects
Clinical aspects
Pharmacovigilance system
Risk management plan
Proposal for questions to be posed to additional experts
Inspection issues
<New active substance status>
Based on the review of the data and the Applicant’s response to the CHMP LoOI, the Rapporteurs
consider that the active substance <active substance> contained in the medicinal product <product
name>
<is to be qualified as a new active substance <in itself.> <in comparison to the known
<isomer/mixture of isomers/complex /derivative/salt of {INN (salt) approved} previously authorised in
the European Union as {name of the medicinal product approved} as it differs significantly in
properties with regard to safety and efficacy from the previously authorised substance.>
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 6/15
<is not to be qualified as a new active substance <in itself.> <in comparison to the known
<isomer/mixture of isomers/complex /derivative/salt of {INN (salt) approved} previously authorised in
the European Union as {name of the medicinal product approved} as it does not differ significantly in
properties with regard to safety and efficacy from the previously authorised substance.>
2. Executive summary
2.1. Problem statement
2.2. About the product
2.3. The development programme/Compliance with CHMP
Guidance/Scientific advice
2.4. General comments on compliance with GMP, GLP, GCP
2.5. Type of application and other comments on the submitted dossier
Legal basis
Conditional approval
Approval under exceptional circumstances
Accelerated procedure
Biosimilarity
1 year data exclusivity
Significance of paediatric studies
3. Scientific overview and discussion
The structure of this AR is in accordance with the Day 180 LoOI and shall be updated at
the different stages of the CHMP review (Day 180/CHMP AR/EPAR) so as to constitute a
self standing document. See also the Day 80 Overview Guidance.
It should therefore be sufficiently detailed to eventually be used for the
CHMP(Withdrawal)AR and (W)EPAR and give sufficient justifications for the LoQ/LoI as
appropriate.
Tables and graphs to display results are encouraged.
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 7/15
3.1. Quality aspects
3.1.1. Introduction
3.1.2. Active Substance
General Information
Manufacture, characterisation and process controls
Specification
Stability
Comparability exercise for Active Substance
3.1.3. Finished Medicinal Product
Description of the product and Pharmaceutical Development
Manufacture of the product and process controls
Product specification
Stability of the product
Comparability exercise for Finished Medicinal Drug Product
Adventitious agents
GMO
3.1.4. Discussion on chemical, pharmaceutical and biological aspects
3.1.5. Conclusions on the chemical, pharmaceutical and biological aspects
3.2. Non clinical aspects
3.2.1. Pharmacology
3.2.2. Pharmacokinetics
3.2.3. Toxicology
3.2.4. Ecotoxicity/environmental risk assessment
3.2.5. Discussion on non-clinical aspects
3.2.6. Conclusion on non-clinical aspects
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 8/15
3.3. Clinical aspects
Tabular overview of clinical studies
3.3.1. Pharmacokinetics
3.3.2. Pharmacodynamics
3.3.3. Discussion on clinical pharmacology
3.3.4. Conclusions on clinical pharmacology
3.3.5. Clinical efficacy
Dose-response studies and main clinical studies
Clinical studies in special populations
Analysis performed across trials (pooled analyses AND meta-analysis)
Supportive study(ies)
3.3.6. Discussion on clinical efficacy
Design and conduct of clinical studies
Efficacy data and additional analyses
<Additional expert consultation>
<Assessment of paediatric data on clinical efficacy>
3.3.7. Conclusions on clinical efficacy
3.3.8. Clinical safety
Patient exposure
Adverse events
Serious adverse events and deaths
Laboratory findings
Safety in special populations
Immunological events
Safety related to drug-drug interactions and other interactions
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 9/15
Discontinuation due to AES
Post marketing experience
3.3.9. Discussion on clinical safety
<Additional expert consultation>
<Assessment of paediatric data on clinical safety>
3.3.10. Conclusions on clinical safety
[Conclude on whether the responses to questions related to safety impact the safety
specification in the RMP.]
3.4. Risk management plan
Safety Specification [to be assessed by the CHMP rapporteur]
[Please copy table from RMP Part II Module SVIII]
The applicant identified the following safety concerns in the RMP:
Table XX: Summary of the Safety Concerns
Summary of safety concerns
Important identified risks
<List here>
Important potential risks
<List here>
Missing information
<List here>
[Please update the scientific discussion on the safety specification.]
<New safety specification that should be taken into account by the PRAC>
Pharmacovigilance Plan [to be assessed by the PRAC rapporteur]
[Please copy RMP table III.5.1 (only studies of categories 1-3)]
Table XX: Ongoing and planned studies in the PhV development plan
Activity/Study title
(type of activity,
Objectives
Safety concerns
Status
Date for
addressed
Planned,
submission of
started,
interim or final
study title [if
known] category
reports
1-3)*
(planned or
actual)
*Category 1 are imposed activities considered key to the benefit risk of the product.
Category 2 are specific obligations
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 10/15
Category 3 are required additional PhV activity (to address specific safety concerns or to measure effectiveness
of risk minimisation measures)
[Please update the scientific discussion on pharmacovigilance activities.]
Risk minimisation measures for <product name(s)> [to be assessed by the
PRAC rapporteur]
[The RMP may cover more than one medicinal product. In some circumstances risk
minimisation measures may be specified per product, or certain risks may not be
relevant to all products.]
[Please copy table from RMP section V.3.]
Table XX: Summary table of additional Risk Minimisation Measures [only for safety concerns
that require additional risk minimisation measures]
Safety concern
Routine risk minimisation
Additional risk minimisation
measures
measures
[Please update the scientific discussion on the additional risk minimisation measures.]
Public summary of the RMP [to be filled in by the PRAC rapporteur]
<The public summary of the RMP < may require><does not require> revision. >
<PRAC Outcome>
If applicable, the outcome of the PRAC plenary discussion should be added in
that section, the above sections should be updated at the time of the
circulation of the updated CHMP overview AR.
3.5. Pharmacovigilance system
3.6. New active substance status
4. Orphan medicinal products
<According to the conclusion of the COMP (Opinion dated 00/00/00) the prevalence of the “condition”
<state the condition> is <> per 10000 individuals in the EU.>
<N/A>
5. Benefit risk assessment
[Update this section at Day 180. See Day 80 template/guidance for instructions]
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 11/15
Benefits
Beneficial effects
Uncertainty in the knowledge about the beneficial effects
Risks
Unfavourable effects
Uncertainty in the knowledge about the unfavourable effects
Balance
Importance of favourable and unfavourable effects
Benefit-risk balance
Discussion on the benefit-risk assessment
5.1. Conclusions
The overall B/R of <name of product> <is> <positive> provided <general statement on conditions>;
is <negative>.
6. CHMP list of outstanding issues to be addressed in an oral
explanation and/or in writing
6.1. Quality aspects
Note: In case the ASMF procedure is used the following should be stated in case
potential serious risks to public health or other concerns are being raised on the
restricted part of the ASMF:
“For <potential serious risks to public health> <and> <Other concerns> on the restricted part of the
ASMF see separate LoOI on the ASMF”
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 12/15
6.2. Non clinical aspects
6.3. Clinical aspects
6.4. Pharmacovigilance system
6.5. Risk management plan
6.6. New active substance status
7. Proposed conditions for marketing authorisation and
product information
7.1. Proposed list of post-authorisation measures*
[Please also consider Appendix 1 of the CHMP D180 JAR Clinical AR to fill in
the below table. This table should be reserved to include conditions that are
part of the marketing authorisation, such as specific obligations, Annex II
conditions, additional PK, efficacy and safety studies that have arisen based
on the assessment of the data (non-clinical and clinical).]
Post-authorisation measure (s)
Motivation
Proposed post-authorisation measure 1
with proposed classification:
Motivation/Background information on
measure, including due date:
1.
Proposed post-authorisation measure 2
with proposed classification:
Motivation/Background information on
measure, including due date:
2.
Proposed post-authorisation measure 3
with proposed classification:
Motivation/Background information on
measure, including due date:
3.
Proposed post-authorisation measure X
with proposed classification:
Motivation/Background information on
measure, including due date:
X.
* Classification: category 1= Annex II D condition; category 2= Annex II E specific obligations;
category 3 = All other studies reflected only in the RMP (non-clinical, PK, PASS)
Proposed list of recommendations:
Description of post-authorisation measure(s)
1.
2.
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 13/15
7.2. Other conditions
[Please state in this section all additional risk minimisation measures.]
7.3. Summary of product characteristics (SmPC)
7.4. Labelling
7.5. Package leaflet (PL)
User consultation
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 14/15
8. Annex 1 (as appropriate)
CHMP questions on the ASM (active substance manufacturer) restricted
part of the ASMF
NOTE that this annex should not be sent to the MAH but only to the holder of the ASMF.
<Invented name>
Rapporteurs day 180 joint CHMP and PRAC response assessment report
Overview
Rev05.15
Page 15/15