HPM/CGH REC Guidelines - Trinity College Dublin
advertisement

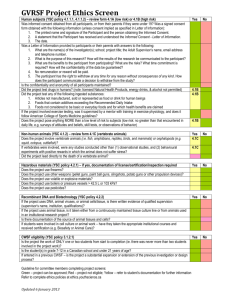
Version Redrafted 18 February 2013 TRINITY COLLEGE DUBLIN HEALTH POLICY AND MANAGEMENT / CENTRE FOR GLOBAL HEALTH RESEARCH ETHICS COMMITTEE GUIDELINES FOR THE SUBMISSION OF ETHICAL APPROVAL APPLICATIONS 1 Version Redrafted 18 February 2013 TABLE OF CONTENTS 1. ABOUT THE COMMITTEE ...................................................................................................................... 3 2. PURPOSE OF THE GUIDELINES .............................................................................................................. 3 3. RELEVANT RESEARCH ETHICS AND GOVERNANCE LITERATURE ............................................................ 4 4. THE ETHICAL REVIEW PROCESS ............................................................................................................ 6 4.1 COMMITTEE MEETINGS.............................................................................................................................. 6 4.2 OUTCOME OF ETHICAL REVIEW.................................................................................................................... 7 4.3 ANNUAL REVIEW AND PROGRESS REPORT...................................................................................................... 7 4.4 PROCEDURE FOR PROPOSING AMENDMENTS TO APPROVED PROTOCOLS ............................................................. 8 5. ETHICAL APPROVAL DOCUMENTATION................................................................................................ 9 5.1 SUBMISSION OF FULL PROTOCOLS ................................................................................................................ 9 5.2 THE CONSENT DOCUMENTATION ............................................................................................................... 10 5.3 GUIDELINES FOR STORAGE OF INFORMATION ................................................................................................ 12 5.4 CHECKLIST FOR THE REVIEW OF SUBMITTED PROTOCOLS ................................................................................ 13 5.5 (A) TEMPLATE FOR INFORMED CONSENT FORM ................................................................................ 16 5.5 (B) TEMPLATE FOR PARTICIPANT INFORMATION LEAFLET ................................................................................ 18 2 Version Redrafted 18 February 2013 1. About the Committee The Health Policy & Management / Centre for Global Health Research Ethics Committee (henceforth HPM/CGH REC) was established to promote high ethical standards in ensuring the protection of the rights and welfare of potential research participants and that researchers are guided in conducting ethical research. To this end the HPM/CGH REC has developed a set of guidelines and standard operating procedures to ensure that research conducted under the auspices of the two Academic Units is independently reviewed in a fair and consistent manner. In addition to the review of submitted research protocols, the HPM/CGH REC will work to ensure the ethical conduct of research through continuing development of guidelines for human research participant protection in keeping with established national and international principles of research ethics and governance, as well as periodically providing training on research ethics, so as to build capacity for researchers in conducting ethical research as well as to build capacity of the Committee members in ensuring appropriate research governance. It is mandatory that all research – whether undertaken by staff or students – conducted under the auspices of the two Academic Units be reviewed by the HPM/CGH REC. The only exception concerns studies that involve clinical and/or therapeutic interventions, in which case the researchers are strongly encouraged to submit for ethical approval by the TCD Faculty of Health Sciences Research Ethics Committee. 2. Purpose of the Guidelines This document provides guidelines for individuals preparing applications to the HPM/CGH REC for ethical review of research projects. Students Research Projects A student must at the planning stage discuss his or her project with their supervisor in preparing for the submission of an ethical approval application to the REC. The supervisor is considered to be in charge of overseeing the research project. The supervisor is responsible for reviewing the student’s application to ensure that it is complete and accurate before passing it to the REC for review. Once the REC has completed the review the results will be issued to both the supervisor and the student. Please contact the HPM/CGH REC Secretary for clarification of these guidelines, the application process or the application form. 3 Version Redrafted 18 February 2013 3. Relevant Research Ethics and Governance Literature Applicants should be familiar with the various national and international guidelines concerning the involvement of human subjects in research. Specifically, the HPM/CGH REC recommends the following literature on research ethics and governance as key reference documents. These guidelines are available online at the URL addresses outlined below, and copies of same are also available to students through their course intranets. Copies could also be made available from the Committee Secretary upon request. These recommended guidelines include, but are not limited to, the following: 1. Trinity College Dublin Good Research Practice http://www.tcd.ie/about/policies/assets/pdf/TCDGoodResearchPractice.pdf 2. Declaration of Helsinki http://www.wma.net/en/30publications/10policies/b3/index.html.pdf?print-media-type&footerright=[page]/[toPage] 3. The Belmont Report http://www.hhs.gov/ohrp/humansubjects/guidance/belmont.html 4. Annual Reports and Guidelines from the Office of the Data Protection Commissioner (Ireland), such as Guidelines to Data Controllers http://www.dataprotection.ie/documents/forms/NewAGuideForDataControllers.pdf 5. EU Legislation on Biomedical Ethics http://cordis.europa.eu/search/index.cfm?fuseaction=search.resultlist&#page=1@perPage=10@q=3F2F5 5FBF64446E2A0C57199C9668472@type=hom@showtype=lib@sortBy=RELEVANCE@sortOrder=DESC 6. Guidance Note on Healthcare Management Research and the Role of Gatekeeper within Such Studies. For guidelines using human participants in behavioural research see: US Department of Health and Human Services: Office for Human Research Protections: http://www.hhs.gov/ohrp/policy/index.html The American Anthropological Association's code of Ethics: http://www.aaanet.org/committees/ethics/ethcode.htm The British Sociological Association: http://www.britsoc.co.uk/equality/ 4 Version Redrafted 18 February 2013 When research involves children or other potentially vulnerable groups, rigorous adherence to the appropriate professional codes of ethical practice is required and particular attention must be paid to issues such as access, informed consent by both participants and carers, and duty of care. Detailed guidelines for such research are available: The (UK) National Children’s Bureau: http://www.ncb.org.uk/PDF/Guidelines%20for%20research%20Oct%202009.pdf 5 Version Redrafted 18 February 2013 4. The Ethical Review Process 4.1 Committee Meetings The HPM/CGH REC meets for the purpose of evaluating protocol submissions three times a year. Dates of these meetings will be confirmed at the operational meeting to be held at the beginning of each academic year. Full protocols and all attachments (please see Section 5 for more details) must be received by the application deadline date (usually two weeks before the Committee meeting). All ethical approval applications will require HPM/CGH REC review. There are two levels of review: Level 1: Full Committee review. Each application will be independently reviewed by three reviewers before being discussed by all members of the Committee at one of their monthly meetings. Applications can be awarded full approval or conditional approval by the Committee, or they could be rejected outright (please see 4.2 for more details). If an application is conditionally approved, the applicant must re-submit an application for review by the Chair (see Level 2) within the time-frame specified (usually three weeks). If an application is rejected, the applicant may re-submit an application for full Committee review at a later meeting. Level 2: Review by the Chair. Supplementary information and/or amendments to proposals that have been awarded conditional approval following full Committee review will be considered by the Chair, who has the authority (delegated from the Committee) to decide whether the amended application receives full approval or a further conditional approval. In rare cases whereby the Chair deems an amended application to warrant a rejection in spite of the initial conditional approval, this would be brought back for a full Committee review. Researchers who are unsure about whether their proposed research requires full Committee review should consult the Chair of the Committee through the Committee Secretary. 6 Version Redrafted 18 February 2013 4.2 Outcome of Ethical Review Written responses from the HPM/CGH REC will be sent by the Committee Secretary within 5 working days after the Committee meeting. The different outcomes from the review process are as follows: I. Full Approval: Research can begin immediately upon receipt of the ethical approval letter. II. Conditional Approval: Research cannot be initiated. The researcher is required to submit additional information or a revised proposal as outlined in the REC response letter. The Chair will review this information as soon as it is received. The Chair will give Full Approval or a further Conditional Approval, or rejection. III. Rejection: Research cannot be initiated. Full resubmission of protocol is advised. A formal letter of ethical approval will be issued to principal investigators whose protocols have received full approval. 4.3 Annual Review and Progress Report The HPM/CGH REC is committed to a partnership approach regarding research governance. Such partnership rests on open and honest communication between the investigator and the Committee as the project unfolds. When unforeseen circumstances arise, the Committee seeks to provide appropriate guidance in a manner that protects the research participants, the members of the research team as well as the College. All applicants with approved protocols are required to submit a one-page progress report one calendar year after the date of ethical approval. Those who are conducting longitudinal studies have to submit such reports annually. Those who are conducting studies that involve an iterative research design (such as action research, or certain types of sequential mixed methods studies) may need to submit a progress report earlier than a year as significant changes or departures from the initially approved protocols arise. Notices of closure/termination of research, e.g. operational research study evaluation should reach the Committee six months before the date of termination of research activities. Documents to be submitted include: i) Progress report (Please use the progress report form available from the Committee Secretary) 7 Version Redrafted 18 February 2013 ii) iii) Copies of exemption and/or approval by relevant local research ethics committees / institutional review boards. Original feedback documents from such local research governance bodies must also be included. Any further materials as requested by the Committee. 4.4 Procedure for Proposing Amendments to Approved Protocols Once protocols have been approved, investigators could propose further amendments if fieldwork conditions demand adjustments to the initially-approved protocols. Amendments to approved protocols must be reviewed and formally approved by the Committee before implementation. The following should be considered as changes to approved protocols that warrant further approval by the Committee: i) Changes in sample size ii) Changes in study sites iii) Review of payments to research participants iv) Additional study objectives v) Changes in the approved informed consent, PIL, invitation to participate in the research, etc. Advice should be sought in the first instance from the Committee Chairperson if the investigator is unsure about the need for further approval of amendments to their protocols. Change/Replacement of Principal Investigator The Committee should be notified of a change of the PI. The likely impact of such a change on the study should be outlined in writing to the Committee. Submission of Amendments i) Areas of amendments to approved protocols must be clearly stated. ii) There should be a point-by-point justification for each point amended. iii) Amended applications should include three hard copies and an electronic copy of all amended protocols (application form and appendices). All amendments must be highlighted via the use of track changes on the Word document. 8 Version Redrafted 18 February 2013 5. Ethical Approval Documentation 5.1 Submission of Full Protocols Investigators must submit full protocols at the first instance of applying for ethical approval by the HPM/CGH REC. Full protocols comprises of a completed application form as well as all relevant appendices: i) Completed application form – all sections must be answered (including the insertion of “not applicable” to those questions irrelevant to the proposed study); signature(s) from investigator(s) and, in the case of student applications, the signature of their principal supervisors, must be inserted. The checklist on the front-page of the application must be completed. ii) Appendices – these include the following: Informed consent form – please see Section 5.5 for more details. Participant information leaflet (PIL) – please see Section 5.5 for more details. Copy of research instruments – please indicate if the draft or final instruments are attached with the application. In the case of the former, applicants must supply the final instruments to the Research Ethics Committee as soon as they are ready. Letters regarding Gatekeeper Permissions – these could either be draft letters seeking gatekeeper permissions; or they could be actual letters received from gatekeepers confirming their permission for investigators to conduct the proposed study pending receipt of ethical approval. Letters confirming Exemptions or Approvals from Local Research Ethics Committees (if available at the time of application). Three hard copies of the complete application and all relevant appendices, including appropriate signatures from the investigator(s) as well as their supervisor(s) where relevant, plus one electronic copy of the same documentation, must be submitted to the Committee Secretary by 5pm of the stated application deadline date. Supporting documents (such as letters confirming gatekeeper’s permission to conduct the study) should also be provided as part of the documentation if they are available at the time of submission. Three hard copies of these documents must accompany the application as an appendix, and an electronic copy should also be provided to the Committee Secretary as far as practicable (hard copy letters could be scanned and emailed to the Committee Secretary). 9 Version Redrafted 18 February 2013 5.2 The Consent Documentation The consent documentation consists of the Participant Information Leaflet (PIL), and an Informed Consent Form. The consent documentation should be written in lay language. The consent document should be written so that the intended participants are likely to understand the important information. In situations where the research participants of the target population do not understand the English Language adequately, the consent document must be translated to the relevant language of the participants. The Informed Consent Form is a legal document in which the project must be comprehensible to a reader without reference to the Participant Information Leaflet. It must contain Project Title, names of the Principal Investigators and a summary of what will be involved for a participant. This summary should be written in the first person since the signatory is agreeing to undertake whatever procedures are involved. Participants must agree that they understand what is involved in participation in the project, consent to take part on a voluntary basis and understand that they may withdraw at any time at their request or be withdrawn by the investigator without any affect on access to services or legal rights. If appropriate, the investigator may also wish to obtain agreement to: o Use of data in other future studies without the need for additional consent. o Access to non-anonymised patient records. o Waiver of intellectual property rights. o Consent to possible publication of results. In an interview based study it is good practice to inform participants that copies of the transcript of their interview will be made available to them and, where appropriate, to provide them with the opportunity of deleting any wording that they may perceive as identifying them. Applications should indicate that these procedures will be followed. If a commercial sponsoring body is involved, the investigator will need to name the sponsor and obtain consent for a copy of the signed form to be sent to that body. The form needs to be signed by the actual research participant (or a parent or guardian in the case of the participant being unable to understand the scope, nature or significance of the study or in the case of the participant being under 18 years) and dated. The above section must be followed by a signed declaration by the researcher that s/he: 10 Version Redrafted 18 February 2013 Has explained the study. Has answered questions. Believes that the participant understands and is freely giving consent. The Informed Consent Form must be signed by each individual research participant and by the researcher. The researcher should retain the original of the signed form in a secure file. A copy of the signed consent form should be provided back to the participant for his/her own reference. Please follow the template given for the Participant Information Leaflet and the Informed Consent Form in Section 5.5(a) and 5.5(b). Photographic Consent The REC can only consider giving approval for the taking of photographs where this is an integral part of the research. Where photographs are being taken for other reasons (including for use in presentations or publications) this falls outside the remit of the committee. In cases where the REC is asked to approve photographs as part of the research process it will require a clear and explicit consent process. Role of the gatekeeper The role of a gatekeeper is to protect the interests of the participants and to ensure that they are not under any pressure to participate. They act as a neutral buffer between the researcher and the participant. For example if a lecturer wanted to give a questionnaire to their students, the students might feel pressurised to take part. Therefore a third party (the gatekeeper) should distribute the questionnaires, thus removing the direct link between the researcher and the participant, thereby eliminating the pressure to participate. Informed assent from minors It is accepted by the Irish courts and international guidelines that minors have independent rights. All minors regardless of age should therefore be informed as fully as is practicable about the research and agree to be involved. The child should be asked if they agree (assent) to be in the study and be asked to sign an assent form. The assent form should be a simpler version of the consent form that parents/guardians sign. If they do not wish to do so, then this must take precedence over any consent given by a responsible adult. 11 Version Redrafted 18 February 2013 5.3 Guidelines for storage of information The investigator must consider carefully the documents that will bear personal information such as participant’s name, address and telephone number. Often only one document, i.e. the consent form, will need to bear such information. An identity code should be generated for each participant, and any personal identifying information should be removed from raw data, such as interview transcripts. The identification key should be stored securely and separately from the main data. Only members of the research team as designated on the approved protocols could have access to raw data. Transcriptionists and other data-entry personnel who may have access to raw data must sign a confidentiality agreement that contain the same terms and conditions for maintaining data protection and participants’ confidentiality as stated in the approved protocols. The investigator must formally inform the Committee if s/he decides to employ the services of transcriptionists/data entry personnel if this is not already stated in the original approved protocols. Data should be handled in the following way: 1. Each researcher will store the document bearing personal information in a locked cabinet with access strictly restricted to personnel working on the study. 2. All computerised data/information must be encrypted and stored on passwordprotected computer terminals with access restricted to only designated members of the research team. This data is more prone to loss or theft and normal username/password access may not be sufficient to protect against unlawful access to the information. 3. Due care and caution must be exercised when transporting data from one computer terminal to another. The Committee advises against the use of USB memory sticks and other portable media to transport raw data containing personally identifiable information of the research participants. 4. Raw data will be stored for the duration of the study, i.e. until the work is fully reported and disseminated. Anonymised data will then be kept in a locked cabinet for the period as recommended by the College research best practice guidelines (typically five to ten years). Guidelines from the Data Protection Commissioner of Ireland must always be adhered to over and above the general guidance given above. 12 Version Redrafted 18 February 2013 5.4 Checklist for the Review of Submitted Protocols We advise that all applicants study the checklist below so as to meet the review requirement of the HPM/CGH REC. Study Objectives and Rationale Are the specific study objectives and questions clearly specified? Is there appropriate justification for the proposed study? Study Design Is the study design adequately described and justified? Is the study design appropriate to address the stated objectives? Is the study feasible within the stated study time-frame? Participant Selection Are inclusion and exclusion criteria clearly specified and appropriate? If members of vulnerable groups are included, is this justified? Are the selected participants able to contribute to the stated study objectives? Is the principle of distributive justice adequately incorporated into the inclusion and exclusion criteria for the research protocol? Is subject selection equitable? Participant Recruitment Are the methods for recruiting potential subjects well defined? Are the location and timing of the recruitment process acceptable? Is the individual performing the recruitment appropriate for the process? Are all recruitment materials submitted and appropriate? Where relevant, are there acceptable methods for screening participants prior to recruitment? Do the stated procedures for participant recruitment minimize risk for potential research coercion, especially for members of vulnerable populations? Data Collection Procedures Are the research procedures clearly described and justified? Are the sites for research participation appropriate from the perspectives of maintaining confidentiality and respecting the participants’ integrity and autonomy? 13 Version Redrafted 18 February 2013 Are there adequate plans to inform participants about specific research results if necessary? Data Analysis Are the plans for data analysis defined and justified, including the use of stopping rules and endpoints for randomized control trials? (Investigators who propose the use of RCTs as part of their study design are strongly encouraged to apply for ethical approval from the Trinity College Faculty of Health Sciences Research Ethics Committee). Compensation and Costs for Subjects Are there adequate provisions to avoid out-of-pocket expenses by the research subject, or is there sufficient justification to allow subjects to pay? Is the amount or type of compensation or reimbursement reasonable? If members of vulnerable groups are involved, who receives the compensation, and is this appropriate (i.e. does not constitute risk of research inducement)? Privacy and Confidentiality Are there adequate provisions to protect the privacy and ensure the confidentiality of the research participants? Are there adequate plans to store and code the data? Is the use of identifiers or links to identifiers necessary, and how is this information protected? Potential Risk, Discomforts, and Benefits for Participants Are the risks and benefits adequately identified, evaluated, and described? Are the potential risks minimised and likelihood of benefits maximised? Is the risk/benefit ratio acceptable for proceeding with the research? Are there appropriate procedures for addressing the potential risks of research participation for members of vulnerable populations? Are the principles of voluntary participation and informed consent upheld in all stages of the proposed research and manifested in all research protocols? Funding Are there any funding conflicts? Are there any concerns over the integrity of the research? Has the investigator addressed these concerns? 14 Version Redrafted 18 February 2013 Local Ethical Approval Has the investigator any plan for applying for local ethical approval? If not, is there an acceptable justification provided? Other Issues (Appendices) Are adequate references provided? 15 Version Redrafted 18 February 2013 5.5 (a) TEMPLATE FOR INFORMED CONSENT FORM Please use the exact wording below for the Participant’s Declaration of Agreement and the Statement of Investigator’s Responsibility: PROJECT TITLE: PRINCIPAL INVESTIGATORS: BACKGROUND (Provide short summary of what project involves for participants, including the procedures to be carried out and the assurance of confidentiality. If relevant it should be stated that participants have a right to their transcripts.) DECLARATION: I have read, or had read to me, the information leaflet for this project and I understand the contents. I have had the opportunity to ask questions and all my questions have been answered to my satisfaction. I freely and voluntarily agree to be part of this research study, though without prejudice to my legal and ethical rights. I understand that I may withdraw from the study at any time and I have received a copy of this agreement. PARTICIPANT'S NAME: ………………………………………………………….. CONTACT DETAILS: ………………………………..…………………………….. PARTICIPANT'S SIGNATURE: ……………..…………………………………….. Date:………………………….. Where the participant is incapable of comprehending the nature, significance and scope of the consent required or is under 18 years old, the form must be signed by a person legally competent to give consent. NAME OF CONSENTER, PARENT or GUARDIAN:……………………………… SIGNATURE:……………………………………………………………………….. RELATION TO PARTICIPANT:………………………………………………… Statement of investigator's responsibility: I have explained the nature and purpose of this research study, the procedures to be undertaken and any risks that may be 16 Version Redrafted 18 February 2013 involved. I have offered to answer any questions and fully answered such questions. I believe that the participant understands my explanation and has freely given informed consent. INVESTIGATOR’S SIGNATURE:………………………… Date:………………….. 17 Version Redrafted 18 February 2013 5.5 (b) Template for Participant Information Leaflet Please adapt as required. Study Title: Name of Investigator: Name of Supervisor: This study is conducted in partial fulfillment of the M.Sc. Health Services Management degree at Trinity College Dublin. 1. Research purpose and procedures: a statement that gives an explanation of the research, the expected duration of the participant’s involvement in the study, a description of the procedures to be followed, and identification of any procedures which are experimental; 2. Risks and discomforts: a description of any reasonably foreseeable risks or discomforts to the subject Research-related injury: for research involving more than minimal risk, an explanation as to whether any medical treatments are available if injury occurs and, if so, what they consist of, or where further information may be obtained; Unforeseeable risks: a statement that the particular treatment or procedure may involve risks to the subject (or to the embryo or foetus, if the subject is or may become pregnant) which are currently unforeseeable; 3. Potential benefits: a description of any benefits to the subject or to others that may reasonably be expected from the research; 4. Alternative procedures or treatments: a disclosure of appropriate alternative procedures or courses of treatments, if any, that might be advantageous to the participant; 5. Provisions for confidentiality: a statement describing the extent, if any, to which confidentiality of records identifying the participant will be maintained; 6. Voluntary participation and the right to discontinue participation without penalty: a statement that participation is voluntary, refusal to participate will involve no penalty or loss of benefits to which the subject is otherwise entitled, and the participant may discontinue participation at any time without having to give a reason, and without any penalty or loss of benefits to which the subject is otherwise entitled. A statement that explains that any significant new findings developed 18 Version Redrafted 18 February 2013 during the course of the research that may relate to the participant’s willingness to continue participation will be provided to the participant. 7. Contacts for additional information: an explanation of whom to contact for answers to pertinent questions about the research and research subject’s rights, and whom to contact in the event of a research-related injury to the subject. 8. Termination of participation by the investigator: anticipated circumstances under which the participant’s involvement may be terminated by the investigator. 9. Permissions: the investigator has obtained permission to conduct the study from [organisational gatekeeper]. The study has also obtained ethical approval from the HPM-CGH Research Ethics Committee at Trinity College Dublin, as well as from the [local research ethics committee]. 10. Access to transcripts: In an interview based study, there should be a statement informing participants that copies of the transcript of their interview will be made available to them and, where appropriate, provide them with the opportunity of deleting any wording that they may perceive as identifying them. 19