(solid line) eluted with solvent A = H 2 O + 0.1% TFA and
advertisement
 eluted with solvent A = H 2 O + 0.1% TFA and")
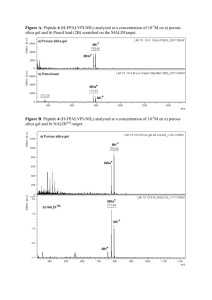
Supporting Information for Azide-Rich Peptides via an On-Resin Diazotransfer Reaction Jeannette E. Marine, Xiaoli Liang, Shuang Song and Jonathan G. Rudick Department of Chemistry, Stony Brook University, Stony Brook, NY 11794-3400, United States ADDITIONAL SYNTHESIS SCHEMES .............................................................................................................. S1 EXPERIMENTAL SECTION ................................................................................................................................. S2 CHROMATOGRAMS AND MASS SPECTRA .................................................................................................... S6 REFERENCES ....................................................................................................................................................... S10 1 H AND 13C NMR SPECTRA..................................................................SERROR! BOOKMARK NOT DEFINED. Additional Synthesis Schemes Scheme S1. Synthesis of alkyne 6. Scheme S2. Chemical structure of conjugate 7. S1 Experimental Section Assembly of Ac-YLKKLLKLLKKLLK-NH2 (3). Peptide 1a was assembled on ChemMatrix Rink amide resin (0.50 mmol, 0.9628 g) in a glass-fritted peptide reaction vessel and cleaved from the resin to yield peptide 3. The sequence of operations is detailed in the table below: Table S1. Protocol for the synthesis of Ac-YLKKLLKLLKKLLK-NH2 (3). Mass of Reagents (g) Cycle Fmoc-AA-OH HBTU 1 Single Coupling: Fmoc-Lys(Boc)-OH 2.3219 1.8593 Capping N-Fmoc Deprotection 2 Single Coupling: Fmoc-Leu-OH 1.7894 1.8655 Capping N-Fmoc Deprotection 3 Single Coupling: Fmoc-Leu-OH 1.7750 1.8643 Capping N-Fmoc Deprotection 4 Single Coupling: Fmoc-Lys(Boc)-OH 2.3715 1.8803 Capping N-Fmoc Deprotection 5 Single Coupling: Fmoc-Lys(Boc)-OH 2.3225 1.8678 Capping N-Fmoc Deprotection 6 Double Coupling: Fmoc-Leu-OH 1.7639 1.8588 1.7748 1.8564 Capping N-Fmoc Deprotection 7 Single Coupling: Fmoc-Leu-OH 1.17690 1.8614 Capping N-Fmoc Deprotection 8 Single Coupling: Fmoc-Lys(Boc)-OH 2.3292 1.8778 Capping N-Fmoc Deprotection 9 Single Coupling: Fmoc-Leu-OH 1.7471 1.8877 Capping N-Fmoc Deprotection 10 Single Coupling: Fmoc-Leu-OH 1.7781 1.8548 Capping N-Fmoc Deprotection 11 Single Coupling: Fmoc-Lys(Boc)-OH 2.3251 1.8457 Capping N-Fmoc Deprotection 12 Single Coupling: Fmoc-Lys(Boc)-OH 2.3099 1.8425 Capping N-Fmoc Deprotection 13 Single Coupling: Fmoc-Leu-OH 1.7792 1.8679 Capping N-Fmoc Deprotection 14 Single Coupling: Fmoc-Tyr(tBu)-OH 2.2917 1.8649 N -Fmoc Deprotection Acetylation of the N-terminus Cleavage and Side-Chain Deprotection S2 HOBt 0.7898 0.7696 0.7660 0.7732 0.7898 0.7863 0.7522 0.7586 0.77700 0.7810 0.7963 0.7631 0.7760 0.7799 0.7877 Assembly of Ac-Y(tBu)LK(Boc)K(Alloc)LLK(Alloc)LLK(Boc)K(Alloc)LLK(Alloc)-NH-Resin (1b). Resinbound peptide 1b was assembled on ChemMatrix Rink amide resin (0.25 mmol, 0.5510 g) in a glass-fritted peptide reaction vessel. The sequence of operations is detailed in the table below: Table S2. Protocol for the synthesis of Ac-Y(tBu)LK(Boc)K(Alloc)LLK(Alloc)LLK(Boc)K(Alloc)LLK(Alloc)NH-resin (1b). Cycle 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Mass of Reagents (g) Fmoc-AA-OH HBTU 1.1272 0.9184 Single Coupling: Fmoc-Lys(Alloc)-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Leu-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Leu-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Lys(Alloc)-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Lys(Boc)-OH Capping N-Fmoc Deprotection Double Coupling: Fmoc-Leu-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Leu-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Lys(Alloc)-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Leu-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Leu-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Lys(Alloc)-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Lys(Boc)-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Leu-OH Capping N-Fmoc Deprotection Single Coupling: Fmoc-Tyr(tBu)-OH N-Fmoc Deprotection Acetylation of the N-terminus S3 HOBt 0.3845 0.8831 0.9281 0.3840 0.8853 0.9263 0.3827 1.1290 0.9239 0.3868 1.1739 0.9286 0.3876 0.8890 0.8904 0.9198 0.9174 0.3767 0.3862 0.8906 0.9201 0.3835 1.1157 0.9211 0.3878 0.8857 0.9207 0.3831 0.8835 0.9188 0.3879 1.1220 0.9287 0.3802 1.1192 0.9278 0.3811 0.8883 0.9250 0.3824 1.1482 0.9315 0.3844 (2,2-Dibutyl-5-methyl-1,3-dioxan-5-yl)methanol (S1). Compound S1 was prepared according to a previously reported procedure.1 The reaction was performed in a 250-mL, one-neck flask equipped with a magnetic stir bar and Soxhlet extractor containing molecular sieves. To an ice-water bath-cooled suspension of 1,1,1tris(hydroxymethyl)ethane (12.08 g, 0.1005 mol) in anhydrous C 6H6 (100 mL) and 5-nonanone (19.0 mL, 0.110 mol), BF3·Et2O (0.5 mL) was added slowly. The suspension was stirred for 30 min, and then heated at reflux for 7 d. The reaction was cooled to room temperature, the residual solids were removed by filtration, and the crude product was obtained by rotary evaporation of volatiles from the filtrate. The product was purified by flash column chromatography (SiO2, hex/EtOAc 4:1 to 3:2) to give S1 as slightly yellow oil (16.16 g, 66%). TLC (SiO2, 7:3 hex/EtOAc): Rf = 0.37. 1H NMR (400 MHz, CDCl3, δ, ppm): 3.70 (d, J = 5.7 Hz, 2H), 3.64 (d, 2J = 11.7 Hz, 1H), 3.62 (d, 2J = 11.0 Hz, 1H), 3.58 (d, 2J = 11.7 Hz, 1H), 1.76 (m, 2H), 1.60 (m, 3H), 1.33 (m, 8H), 0.92 (t, J = 7.1 Hz, 3H), 0.90 (t, J = 7.0 Hz, 3H), 0.82 (s, 3H). 13C NMR (100 MHz, CDCl3, δ, ppm): 100.9, 66.6, 66.0, 36.9, 34.8, 30.1, 26.0, 25.3, 23.3, 23.2, 17.9. ESI–MS (m/z): [M + H]+ calcd for C14H28O3, 245.2; found 245.1. GPC: Mn = 190, Mw/Mn = 1.06. The spectral data agree with literature.1 2,2-Bis[(2,2-dibutyl-5-methyl-1,3-dioxan-5-yl)methoxymethyl]ethene (S2). Compound S2 was prepared according to a literature procedure.1 A solution of S1 (33.55 g, 137.3 mmol) in anhydrous THF (140 mL) was added dropwise to an ice-water bath-cooled suspension of dry NaH (4.00 g, 166 mmol) in anhydrous THF (20 mL), methallyl dichloride (6.3 mL, 54 mmol) and 15-crown-5 (2.7 mL, 14 mmol). The reaction mixture was heated at reflux for 16 h under a N2 atmosphere while stirring. The reaction mixture was quenched with deionized water. The product was extracted with EtOAc. The organic washings were combined, washed once with saturated NaCl (aq) solution and dried over anhydrous MgSO4. The solids were removed by filtration. The volatiles were removed by rotary evaporation. The resulting product was purified by flash chromatography (SiO 2, hex to 7:3 hex/EtOAc) to yield S2 as a colorless liquid (28.41 g, 96%). TLC (SiO2, 7:3 hex/EtOAc): Rf = 0.67. 1H NMR (500 MHz, CDCl3, δ, ppm): 5.13 (s, 2H), 3.97 (s, 4H), 3.66 (d, 2J = 11.7 Hz, 4H), 3.50 (d, 2J = 11.7 Hz, 4H), 3.39 (s, 4H), 1.72 (m, 2H), 1.60 (m, 2H), 1.32 (m, 16H), 0.90 (t, J = 6.9 Hz, 6H), 0.89 (t, J = 7.0 Hz, 6H), 0.85 (s, 6H). 13C NMR (125 MHz, CDCl3, δ): 143.2, 113.3, 100.7, 73.4, 72.3, 66.1, 36.0, 34.3, 31.1, 25.8, 25.3, 23.3, 23.2, 18.6, 14.3. ESI–MS (m/z): [M + H]+ calcd for C32H61O6, 541.4; found, 541.5. GPC: Mn = 480, Mw/Mn = 1.05. The spectral data agree with literature.1 2,2-Bis[(2,2-dibutyl-5-methyl-1,3-dioxan-5-yl)methoxymethyl]ethanol (S3). Compound S3 was prepared according to a literature procedure.1 To a solution of S2 (12.14 g, 22.45 mmol) in anhydrous THF (9 mL), a 0.5 M 9-BBN in THF solution (67 mL, 34 mmol) was added dropwise under a N 2 atmosphere. The reaction mixture was stirred under a N2 atmosphere for 18 h. The reaction was cooled in an ice-water bath while 3 M NaOH (aq) solution (30 mL, 90 mmol) was added dropwise to the reaction mixture. After the addition was complete, the reaction mixture was stirred under a N2 atmosphere at room temperature for 15 min. The reaction vessel was cooled in an ice-water bath and 30 wt% H2O2 (aq) solution (30 mL) was added dropwise to the reaction mixture. After the addition was complete, the reaction mixture was stirred under a N 2 atmosphere at room temperature for 3 h. The reaction mixture was saturated with K2CO3. The product was extracted with EtOAc. The organic washings were combined and dried over anhydrous MgSO4. The solids were removed by filtration. The volatiles were removed from the filtrate by rotary evaporation. The resulting product was purified by flash chromatography (SiO2, hex to 7:3 hex/EtOAc) to yield S3 as a colorless liquid (10.74 g, 86%). TLC (SiO 2, hex/EtOAc = 7:3): Rf = 0.42. 1H NMR (500 MHz, CDCl3, δ): 3.75 (dd, J = 5.4 Hz, J = 5.5 Hz, 2H), 3.62 (dd, 2J = 11.7 Hz, 4J = 2.6 Hz, 4H), 3.57 (dd, 2J = 11.8 Hz, 3J = 6.3 Hz, 1H), 3.56 (dd, 2J = 9.3 Hz, 3J = 5.4 Hz, 1H), 3.54 (dd, 2J = 11.1 Hz, 3J = 5.4 Hz, 1H), 3.53 (dd, 2 J = 9.2 Hz, 3J = 6.2 Hz, 1H), 3.51 (d, 2J = 10.0 Hz, 4H), 3.44 (d, 2J = 8.9 Hz, 1H), 3.42 (d, 2J = 9.2 Hz, 2H), 3.40 (d, 2 J = 9.0 Hz, 1H), 2.57 (t, J = 5.7 Hz, 1H), 2.15 (m, 1H), 1.72 (m, 4H), 1.60 (m, 4H), 1.32 (m, 16H), 0.91 (t, J = 6.8 Hz, 6H), 0.90 (t, J = 7.0 Hz, 6H), 0.88 (s, 6H). 13C NMR (125 MHz, CDCl3, δ): 100.9, 74.7, 71.7, 66.1, 64.5, 41.6, 36.2, 34.4, 30.8, 25.9, 25.3, 23.3, 23.2, 18.5, 14.3. ESI–MS (m/z): [M + H]+ calcd for C32H63O7, 559.5; found, 559.5. GPC: Mn = 620, Mw/Mn = 1.04. The spectral data agree with literature.1 2,2-Bis[(2,2-dibutyl-5-methyl-1,3-dioxan-5-yl)methoxy]ethyl propargyl ether (S4). To a solution of S3 (5.00 g, 8.95 mmol) in anhydrous THF (15 mL), 80 wt% propargyl bromide in toluene solution (1.5 mL, 13 mmol) and NaH (0.30 g, 12 mmol) were added. The reaction was stirred at room temperature under N2 for 48 h. The reaction mixture was quenched with deionized water. The product was extracted with EtOAc. The organic washings were combined, washed once with saturated NaCl (aq) solution and dried over anhydrous MgSO4. The solids were removed by filtration. The EtOAc was removed from the filtrate by rotary evaporation. The resulting product was purified by flash column chromatography (SiO2, hex to 4:1 hex/EtOAc) to yield S4 as a colorless liquid (4.30 g, 81%). TLC (SiO2, 4:1 hexanes/EtOAc): Rf = 0.63. 1H NMR (400 MHz, CDCl3, δ, ppm): 4.10 (d, J = 2.4 Hz, 2H), 3.66 (d, J S4 =11.7 Hz 4H), 3.56 (d, J = 5.9 Hz, 2H), 3.48 (d, J = 14.4 Hz, 4H), 3.46 (d, J = 8.7 Hz, 4H), 3.33 (dd, J1 = 8.0 Hz, J2 = 8.9 Hz, 4H), 2.39 (t, J = 2.4 Hz, 1H), 2.19 (m, 1H), 1.72 (m, 4H), 1.62 (m, 4H), 1.32 (m, 16H), 0.92 (t, J = 6.7 Hz, 6H), 0.90 (t, J = 6.9 Hz, 6H), 0.86 (s, 6H). 13C NMR (100 MHz, CDCl3, δ): 100.7, 80.2, 77.5, 74.3, 69.9, 68.9, 66.2, 58.6, 40.4, 35.6, 34.5, 31.5, 25.8, 25.4, 23.27, 23.25, 18.7, 14.3. HRMS–ESI (m/z): [M + H]+ calcd for C35H65O7, 597.4730; found, 597.4728. GPC: Mn = 620, Mw/Mn = 1.06. 2,2-Bis[2,2-di(hydroxymethyl)propyloxymethyl]ethyl propargyl ether (6). To a solution of S4 (0.50 g, 0.84 mmol) in a 1:1 v/v mixture of THF/MeOH (8 mL), Dowex 50WX2 (8.50 g) was added. The mixture was stirred at room temperature under N2 for 2 d. The solids were removed by filtration. The resulting product was purified by flash column chromatography (SiO2, CH2Cl2 to 19:1 CH2Cl2/MeOH) to yield S5 as colorless oil (0.2350 g, 81%). TLC (SiO2, 19:1 CH2Cl2/MeOH): Rf = 0.18. 1H NMR (400 MHz, DMSO-d6, δ): 4.25 (t, J = 5.3 Hz, 4H), 4.09 (d, J = 2.4 Hz, 2H), 3.44 (d, J = 5.9 Hz, 2H), 3.37 (t, J = 2.4 Hz, 2H), 3.33 (d, J = 5.9 Hz, 4H), 3.24 (d, J = 5.3 Hz, 8H), 3.16 (t, J = 9.0 Hz, 4H), 2.03 (m, 1H), 0.76 (s, 6H). 13C NMR (100 MHz, DMSO-d6, δ): 80.4, 77.0, 73.4, 69.2, 68.0, 64.0, 57.7, 40.0, 35.6, 16.8. HRMS–ESI (m/z): [M + H]+ calcd for C17H33O7, 349.2226; found, 349.2218. GPC: Mn = 300, Mw/Mn = 1.08. S5 Chromatograms and Mass Spectra Figure S1. Chromatograms of peptide 3 (solid line) eluted with solvent A = H2O + 0.1% TFA and a) solvent B = MeCN/H2O (9:1 v/v) + 0.1% TFA and b) solvent B = MeOH/H2O (9:1 v/v) + 0.1% TFA. The dashed line indicates the solvent composition during elution of the peptide at 1 mL/min from a C18 column. S6 Figure S2. Chromatogram of peptide 3 (solid line). The dashed line indicates the solvent composition during elution of the peptide at 1 mL/min from a C4 column (solvent A = H 2O + 0.1% TFA; solvent B = iPrOH/MeCN/H2O (6:3:1 v/v/v) + 0.1% TFA). Figure S3. MALDI-TOF Mass spectrum of peptide 3. S7 Figure S4. Chromatograms of peptide 2a (solid line) eluted with solvent A = H2O + 0.1% TFA and a) solvent B = i PrOH/MeCN/H2O (6:3:1 v/v/v) + 0.1% TFA, and b) solvent B = iPrOH/MeCN/H2O (3:6:1 v/v/v) + 0.1% TFA. The dashed line indicates the solvent composition during elution of the peptide at 1 mL/min from a C18 column. Figure S5. MALDI-TOF Mass spectrum of peptide 2a. S8 Figure S6. MALDI-TOF Mass spectrum of peptide 4. Figure S7. Chromatograms of peptide 2b (solid line) eluted with solvent A = H2O + 0.1% TFA and a) solvent B = MeCN/H2O (9:1 v/v) + 0.1% TFA, and b) solvent B = iPrOH/MeCN/H2O (3:6:1 v/v/v) + 0.1% TFA. The dashed line indicates the solvent composition during elution of the peptide at 1 mL/min from a C18 column. S9 Figure S8. MALDI-TOF Mass spectrum of peptide 2b. Figure S9. Chromatogram of conjugate 7 (solid line) eluted with solvent A = H2O + 0.1% TFA and solvent B = i PrOH/MeCN/H2O (3:6:1 v/v/v) + 0.1% TFA. The dashed line indicates the solvent composition during elution of the peptide at 1 mL/min from a C18 column. References (1) Grayson, S. M.; Fréchet, J. M. J. J. Am. Chem. Soc. 2000, 122, 10335-1034 S10