hernandez_heidi_processprion
advertisement

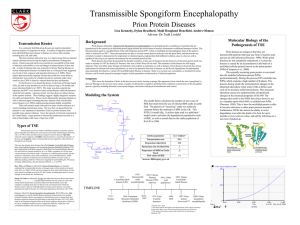
Hernandez 1 Heidi Hernandez Dr. Dockter Cluster 7/ Biomedical Sciences The Process of Prions The “protein only” theory states that the infectious agent causing transmissible spongiform encephalopathies is a conformational isomer of PrP, a mass protein primarily articulated in brain, and is powerfully maintained by many streaks of evidence. Prion diseases are so far unique between conformational diseases in that they are infectious, not only experimentally but also by normal routes, mainly by consumption. A remarkable feature of prions is their astonishing resistance to conventional decontamination actions, and their ability to bind to exteriors of metal and plastic without losing ability of a pathogen to establish an infection. This property, primarily detected in a clinical scenery, is today being examined in investigational settings, mutually in animals and in cell culture. The term used by countless researchers to define the pathogen that causes transmissible spongiform encephalopathies (TSE) which are characterized by degeneration of the nervous system diseases in mammals is called prions. Prions are tremendously tiny, smaller than the small infectious agent that replicates only inside the living cells of other organisms, and even through an electron microscope only aggregations (collection, or the gathering of things together), not singular prions, can be seen. Prions are unique microorganisms in that they give the impression to have no nucleic acid and in so doing contrast from viruses, bacteria, fungi and other pathogens. Prions refuse to accept actions that break down nucleic acid and destroy biological Hernandez 2 arrangements of pathogens. Additionally, prions vary from other pathogens in that they are liable for genetic, sporadic and acquired forms of neurodegenerative infection. Likewise, for the reason that prions are an irregular form of a normal protein that is genetically encoded, they do not produce an immune response in the host as would a foreign infectious agent (BSE info). The typical cellular arrangement of the prion protein is present in most or all the specialized cells transmitting nerve impulses, and it can be concluded from the protein’s position within the cell. Prion molecules are established at the cell surface, where they are secured to the nerve cell membrane by short fatty acid molecules called glycosylphosphatidylinositols (GPIs), which are attached to the prions following synthesis of the proteins. Prions are, consequently, most likely to play a roles in some characteristic of the cell-to-cell signaling process. Prion proteins are uninterruptedly recycled amongst the membrane and the endosomes, which are membrane-bounded compartment inside eukaryotic cells. (Mo). Prion diseases are categorized by the removal of PrPSc, a misfolded and aggregated isoform of the host-encoded cellular prion protein (PrPC), inside the central nervous system (CNS) and other organs (A. Adrianno). When prions are existing in the brain, they duplicate by making normal prion protein (PrPC) molecules to refold into the irregular, pathogenic shape. The normal prion protein assembly is understood to consist Hernandez 3 of a number of flexible coils called alpha helices. In the abnormal form of the protein, some of these helices are stretched out into flat structures called beta sheets. The ordinary protein is broken down by cellular enzymes called proteases, but the unusual protein shape is resistant to these enzymes. As a result, as prions replicate, they are not broken down by proteases nonetheless, they accumulate in brain tissue. The growth of prions in the brain causes neuronal cells to die and a type of protein called amyloid, accumulates in plaques, or flat areas, and causes degeneration of brain tissue. Prions disturb the normal cell process of protein recycling which causes an accumulation of defective proteins and the death of the cell. The damage of neural cells produce small holes in the brain tissue and a sponge-like appearance under the microscope, hence giving rise to the term spongiform disease (BSE info). Transmissible spongiform encephalopathies (TSEs), or prion diseases, are disorders that show evidence of decline of the central nervous system (CNS) that lead to motor dysfunction, disorder of the mental process, and death. Prion diseases consist of scrapie of sheep, bovine spongiform encephalopathy in cattle, and such human diseases as Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome, and fatal familial insomnia. Neither humoral nor cellular immunologic responses have been identified in prion diseases (C.Weissmenn). “The conversion of Hernandez 4 normal prion protein (PrPC) into abnormal prion protein (PrPSc) and replication of prions in the brain causes degeneration of neural tissue and, ultimately, death. In TSE linked to consuming TSE-infected material, it is theorized that once prions are ingested, they are taken up by the lymphoid tissue that drains the gastrointestinal tract including Peyer's Patches and other nodes” (BSE info). Prions also have been found in tonsil, spleen and appendix. From the lymphatic system, research proposes that prions replicate access and move through the autonomic nervous system to the central nervous system. Once in the brain, the higher absorption of cellular prion protein speeds up the replication process. Prions also may enter lesions or wounds in the oral cavity, and access the vagus nerve as a passageway to the brain (BSE info). Recent laboratory investigation using luminous dye to "brand" scrapie proteins has pursued prions as they raid nerve cells and then travel along wire-like circuits to points of contact with other cells. This gives the idea to be the way the prions that cause TSE invade nerve cells and are transported along neural circuits all the way through the nervous system (BSE info). Prions are exceedingly unaffected to disinfectants, heat, ultraviolet radiation, ionizing radiation and formalin. However, prions can be deactivated by heat, by chemicals and by an arrangement of heat, chemicals, pressure and time. Prions can be Hernandez 5 demolished through incineration, providing the incinerator can maintain a temperature of 900 F for four hours. “In an autoclave, prions can be deactivated by using a temperature of 270 F at 21 psi for 90 minutes. If the infectious material is in a solution of sodium hydroxide, deactivation will occur after one hour at 250 F and 21 psi . A commercial disinfectant called Environ LpH also has been shown to be effective at deactivating prions. Prion disinfection occurs with a 1 percent solution of LpH for 10 hours or with a 10 percent LpH solution for one hour”(BSE info). Dead bodies of contaminated animals can be deactivated into a sterile alkaline solution using an alkaline hydrolysis digester. This comprises of an insulated steam-jacketed stainless steel vessel which functions at up to 70 psi and 300 F into which sodium hydroxide and water is added, heated and continuously circulated. This process reduces proteins into salts of free amino acids, and the temperature and alkali absorptions deactivate prions by ending their peptide bonds (BSE info). Prions are unknown contagious pathogens that cause a cluster of invariably fatal neurodegenerative diseases by an entirely novel mechanism. Prion diseases may be present as genetic, infectious, or sporadic disorders, all of which involve modification of the prion protein (PrP). Bovine spongiform encephalopathy (BSE), scrapie of sheep, and Creutzfeldt–Jakob disease (CJD) of humans are among the most notable prion Hernandez 6 diseases. Prions are transmissible particles that are deprived of nucleic acid and seem to be composed entirely of a modified protein (PrPSc). The species of a specific prion is encoded by the sequence of the chromosomal PrP gene of the mammals in which it last replicated. In contrast, to pathogens carrying a nucleic acid genome, prions appear to convert strain-specific properties in the tertiary structure of PrPSc. Hernandez 7 Works Cited Aguzzi, Adriano, and Caihong Zhu. "Five Questions on Prion Diseases." PLoS Pathogens. Public Library of Science, 8 May 2012. Web. 25 July 2015. "BSE Info - Prions." BSE Info - Prions. BSE, n.d. Web. 25 July 2015. C. Weissmann, M. Enaria, P-C Klohn, D. Rossia, and E. Flechsiga. "Transmissions of Prions." The Journal of Infectious Diseases. Oxford Journals, n.d. Web. 25 July 2015. Mo. "Prion Protein Infection Mechanism Identified." Neurophilosophy. Science Blogs, 7 Oct. 2008. Web. 25 July 2015. Hernandez 8