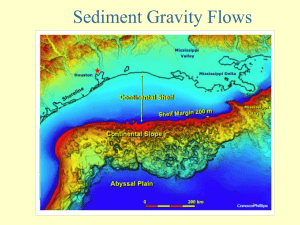
Sediment sampling stations
advertisement

Book of Protocols ECASA Project 1 Table of Contents page Table of Contents Selection of sampling stations Sampling Field Log Hydrography “Large-scale” description of the seafloor at the study site, by visual & other means Sediment sampling Grab sampling Benthos Macrobenthos procedures Meiofauna procedure Phytobenthos procedure Sediment chemistry Sediment redox potential, Eh Sediment grain size analysis (granulometry) Sediment organic matter Sediment organic carbon Total nitrogen in sediment Inorganic and organic phosphorus in sediment Water column (water quality) sampling Secchi disk transparency Dissolved nutrients in seawater Nitrate/Nitrite in seawater Orthophosphate in seawater Ammonium in seawater Total N & total P in seawater Particulate matter in seawater Particulate organic carbon and nitrogen in seawater Particulate organic phosphorus in seawater Chlorophyll a determination Bioassay studies to assess nutrient levels around aquaculture 1 2 2 3 4 6 6 9 9 16 18 20 20 22 23 24 26 27 28 29 31 34 34 35 35 38 40 42 43 46 2 Selection of sampling stations Station positions should reflect gradients of impact. The principle is that some rationale must be used to maximise the chance that one of the stations is selected in the area of maximum impact. This can be done by modelling (using an appropriate current record) or by a pre-survey which could be done by sediment sampling for e.g. redox, by acoustic methods or by video, etc. We want to maximise the chance of detecting a gradient and we also would like to consider the distance from the farm that effects of the farm can be detected. This will probably take a minimum of 5 stations per site but could require several more. A reference station should be at a similar depth and sediment type but not be influenced by the farm. If this is not possible, an additional station added to the transect may reveal background conditions by comparing the trend away from the farm i.e. if stations become more similar with distance. Sampling Field Log One of the key elements necessary to ensure successful fieldwork is a good and reliable system for data collection and handling. It is essential that careful records are kept of all details related to the sampling and measurement of field data. A typical field log should include: – date of sampling; – person responsible for sampling; – project or contract identification code; – sketch of sampling station transect in relation to cage groups – cage group numbering system (so that biomass for cages can be matched to husbandry sheets) – geographical co-ordinates for each sampling station (for each replicate sample in case of boat drift during sampling), including time of day, whether the ship was anchored or not, weather conditions during sampling; – water depth (m) at each sampling station; – sampling programme for each sampling station (number of samples, sampling of background parameters etc.); – sampling device used for each station and each replicate sample – other comments (such as rejected samples, delays and any problems experienced and the causes); – quality of sample i.e. disturbed/not disturbed. 3 Hydrography Source: MERAMED project field handbook Hydrography is one of the key features used in site selection, as it indicates: a) flux of good quality water via the finfish or shellfish gear (water quality, animal welfare); b) spatial dispersal of effluents, which may spread the load over sufficiently large area to reduce benthic impact on seafloor (environmental health), c) supply of planktonic food to shellfish (enhanced growth). Hydrographic characterization needs to be done before the intensive WP5 site study as it will serve as the basis for pre-selecting sampling stations (see below). For microtidal sites (e.g. Mediterranean) current meter records should be as long as possible; at the very least 15 days, but preferably months of data. For macrotidal sites (e.g. France, Atlantic coast), 15 days may be sufficient to capture the tidal cycle. The most common types of instruments used for hydrographic measurements around fish farms are rotary, electromagnetic and acoustic doppler current profilers (ADCP). The first two instrument types measure current speed and direction at a discrete depth, the third type profiles the water column with some limitations at surface and bed. A sampling interval of 10 minutes is recommended for long-term deployments (e.g. up to 1 month). Short-term deployments (i.e. of a few days) should use a 5-minute sampling interval. Where acoustic profilers are being used, care should be taken to ensure that the standard deviation of the measurements is sufficiently small. Total battery life and memory capacity should be taken into account when setting up the instrument. Select a mooring type depending on local site conditions (i.e. level of exposure of site, local fishing activities), intended mooring location, instrument type and length of deployment. The mooring should be located outside the shadowing effect of the cages. Ushaped mooring arrangement is recommended for long-term deployments as it allows three methods for recovery in the event of the surface buoys being lost. Where a surface marker is lost, recovery can take place from the opposite end. Where both surface markers are lost, recovery can take place by snagging the groundline with a grapnel. A conductivity, temperature and depth (CTD) profile is useful at the deployment and recovery stages of the hydrographic instruments. This gives information on the depth of any water column features in relation to instrument depth. The depth and length of deployment of each instrument depends on the objectives of the current meter study. It also relates to any modelling intended for the survey site, in which case the modelling guidelines can be consulted. The following recommendations are made with respect to current meter deployment depth: - if three instruments are available – deploy surface, mid-water and near-bed - if one instrument is available – deploy mid-water or below the depth of the cage bottom 4 - if two instruments are available, then consideration should be given to the level of resuspension expected at the site before deciding on instrument deployment depths. Near-bed current speed exceeding 10 cm s-1 causes resupension of fish farm material and so near-bed measurements are crucial at sites where these conditions exist. Therefore, deploy surface and near-bed if greater than 5 % of near-bed current speeds are expected to exceed 10 cm s-1, otherwise deploy surface and mid–water. If profiler is available – deploy either ‘looking down’ or ‘looking up’ but consideration must be given to the effect of surrounding fish farm equipment and the limitations of current measurement at the surface and near-bed Actual deployment depths will depend on the total depth of the water column, but measurement of surface current and current below the bottom of the net pen is desirable. In the case where a counter current to the surface flows exists at depth, this should be measured if possible. A deployment of 1 month is recommended for studies aiming at measuring the general hydrodynamic features of a site. The effect of season must also be taken into account where possible, as seasonal wind patterns are likely to change hydrographic patterns. Measurement of current in the same season as any intensive benthic sampling is ideal, but this may not be practical. Need to select one sampling station which should avoid shadowing effect of cages but should be sufficiently close to cages to sample general flow conditions in the area of expected deposition. “Large-scale” description of the seafloor at the study site, by visual & other means Source: Davies et al. (2001) Video or still photography of the seafloor, along a transect (from the fish/shellfish farm toward the reference station) is extremely desirable as it will provide large-scale information on macrophytobenthos, epibenthic macrofauna, seafloor status, etc. which core or grab sampling cannot do. Video can be used for interpreting and ground truthing data from an acoustic survey or as a primary survey technique for habitat mapping. If video is limited by turbidity good timing of the survey may improve the quality of a video survey considerably, e.g. in tidal areas water clearance is often best around slack tide. Good synchronization between video and GPS data is a prerequisite for mapping habitats. Video cameras can be towed above the seafloor. In areas where obstacles (rocks, wrecks etc.) can be expected, the camera system should be protected by a frame. The frames can also be used for close-up inspections when placed on the seafloor. The resolution of still pictures from photo cameras is much better than the resolution of video footage. Thus, an additional photo camera facilitates the interpretation of videos considerably. The framecamera should be towed at a constant distance above the ground. A weighted rope of known length within the view of the camera is a simple but very helpful way to achieve 5 this goal. Alternatively, the video camera and accessory equipment (lamps, still photo) can be mounted on a sledge. Comprehensive guidelines for identifying biotopes using video techniques and in situ survey of sublittoral epibiota using towed sledge video and still photography have been published in the Marine Monitoring Handbook (Procedural Guideline No. 3-5, pages 241-251, and No. 3-14, pages 331-337). If a frame or sledge based camera system is not available and depth does not exceed ca. 30 m and only a small area needs to be surveyed for ground-truthing a hand-held video- or photo camera can be employed (see Marine Monitoring Handbook, Procedural Guideline No. 3-13, 327-330) or habitat classifications can be conducted by scientific divers who are familiar with habitats and species typical for the region (Marine Monitoring Handbook, Procedural Guideline No. 3-3, pages 233-239). Transect Photography. For monitoring the impact of an existing mariculture facility a photographic documentation along a transect line has been shown to be a valuable tool. Necessary equipment are standard diving equipment, a compass, a good visible transect line (e.g. yellow, minimum length 50 m, diameter ca. 3 mm) on a seawater-resistant reel, an underwater photo-camera (focal length should be not longer than 28 mm for 35 mm film) with an external strobe. A lead weight (1 to 2 kg) is attached to the beginning of the line and the line marked with tape every 5 m, for distances longer than 50 m every 10 m and the distance written on the tape. Even if the number is not always legible on the photos, it is helpful for evaluation and orientation of the divers. The weight is placed at the point of maximum impact, e.g. in the centre under a fish cage. From there, the diver swims towards the desired direction, preferably with the current, thereby unrolling the transect line. While swimming back, the diver exposes a horizontal photo at each mark (with the mark visible in the picture) for qualitative overview pictures. If quantitative pictures are desired (e.g. for counting the number of feed pellet) vertical pictures should be taken with a ruler within the picture or a photo camera with a frame should be used. Acoustic ground discrimination systems (seabed classification systems) allow for mapping bathymetry, sediment type and some types of habitats (vegetation, mussel beds) in a range of a few kilometres around the planned location of aquaculture facility. For a proper sampling design, the desired spatial resolution of the map should be defined in advance, e.g. an area of a certain seabed type and a maximum diameter of 10 m should be identified with a probability of at least 90 %. This can be achieved by choosing the distance between tracks scanned by the echosounder appropriately. Depending on the system in use a more or less extensive ground truthing (diver observations, photo, video or grab samples) is necessary to link sea bed types with habitat types. Standard Operational Procedures vary with the technique, hard- and software in use, thus a thorough training of staff is necessary. Systems based on multibeam technology or sidescan sonar allow for a faster and more accurate mapping but are more expensive and thus less widely available. Comprehensive guidelines on seabed mapping using acoustic ground discrimination interpreted with ground truthing have been published in the Marine Monitoring Handbook (Procedural Guideline No. 1-3, pages 183-197, No. 1-4, pages 199-209). 6 References Davies J, Baxter J, Bradley M, Connor D, Khan J, Murray E, Sanderson W, Turnbull C and Vincent M (2001) (eds.) Marine Monitoring Handbook. http://www.jncc.gov.uk/page-2430, ISBN 1 86107 5243. Rumohr H (1995) Monitoring the marine environment. Sci. Mar. 59 (Suppl.1) 129-138. Smith C, Rumohr H (2003) Imaging Methods. In: AD McIntyre, A Eleftheriou (eds) Methods for Study of Marine Benthos. Blackwell. Sediment sampling Timing of sampling is crucial - sampling should be done when impact is likely to be high. Number of sediment samples. At each station, at least 4 grab replicates should be taken for macrofauna, and triplicate cores for sediment chemistry. If an analysis is very expensive or complicated to perform, duplicates may be sufficient. Diver sampled cores. Sediment cores should be taken by divers where practical since divers can apply quality control (i.e. discard bad samples and re-sample in situ) to the sampling process. Core diameters should be 5cm for sediment chemistry and 10cm for macrofauna. Core depth should be at least 12cm for sediment chemistry and at least 6cm for macrofauna. 1. The corer should be inserted slowly and smoothly into the sediment in a vertical position. 2. If necessary, facilitate penetration with the help of a rubber head mallet 3. The depth of penetration into the sediment must exceed 10 cm1 4. When the desired depth is reached, close the corer’s upper end with a stopper 5. Remove the corer slowly. Care should be taken to retain the core while removing the corer from the sediment. If needed, remove the sediment around the corer by digging for facilitating core removal. 6. Close the lower end of the corer with a stopper and place it in a carrying basket Caution must be exercised to ensure that the cores are not be turned up-side down during transport, as this will mix the enclosed sediments! Grab sampling Source: Stubbs et al. 1987 Where it is not possible to collect by diving, samples will generally be taken by van Veen grab with a sampling area of 0.1 m2. Grab sampling is suboptimal for sediment geochemistry or for meiofauna sampling because the stratified structure of the sediments is usually compromised by the sediment collecting mechanism of the grabs. However, 1 Required sample’s depth is 10 cm, but due to possible sediment loss that might occur while removing the corer from the sediment, the corer should be pushed deeper in the sediment 7 because van Veen grabs are affordable and are easy to use from a small boat, these will most likely be used by farmers and regulators monitoring sediments below fish farms. Whereas box corers yield undisturbed sediments, they are not recommended in this project because most farmers/regulators will not have access to box corers (expensive) or to the large boats (with large crane/winch) needed to use these. If possible, samples could be taken with a Craib corer as this yields undisturbed sediment profiles. Gravity corers and grabs tend to fluidize the samples and destroy the vertical stratification of the sediments, thereby making chemical or meiobenthos data extracted from them meaningless. Benthic sampling using a Van Veen grab A 0.1 m2 modified Van Veen grab may be used to collect sediment samples for physical, chemical, and infaunal analysis (Figure 2) (Stubbs et al. 1987). The grab may be galvanized, stainless steel, or Teflon-coated. All surfaces of the grab must be clean and free of rust. Either single or tandem Van Veen grabs are acceptable. Grabs will be used if: * a large (surface area and volume) sample is required, e.g. for macrofauna * it is not possible to sample with cores by divers or by using a corer However - grabs are not suitable for surface layer sampling (e.g. for meiofauna or some other analyses) because they disturb and "blow off" the surface layer Grab sampling procedures Prior to deployment, the grab is cocked with the safety key in place. The grab is then hoisted over the side, the safety key is removed, and the grab is lowered at 2 m/sec until it is 5 m above the bottom. From this point, it is lowered at 1 m/sec to minimize the effects of bow wave disturbance. After bottom contact has been made (indicated by slack in the lowering wire), the tension on the wire is slowly increased, causing the lever arms to close the grab. Once the grab is back on board, the top doors are opened for inspection. Criteria for acceptable grab samples Upon retrieval of the grab the acceptability of the sample must be determined. Acceptability is based upon two characteristics of the sample: sample condition and depth of penetration. Sample condition is judged using criteria for surface disturbance, leakage, canting, and washing (Figure 3). Acceptable sample condition is characterized by an even surface, with minimal surface disturbance, and little or no leakage of the overlying water. Heavily canted samples are unacceptable. Samples with a large amount of "humping" along the midline of the grab indicating washing of the sample during retrieval are also unacceptable. While some humping will be evident in samples from firm bottoms where penetration has been poor, this is due to the closing action of the grab and is not evidence of unacceptable washing. 8 Figure 2. Modified Van Veen grab sampler recommended for marine recieiving-water monitoring programs in Southern California: a) cocked position; b) tripped position (modified from Stubbs et al. 1987) Figure 3. Examples of acceptable and unacceptable grab sample condition If the sample condition is acceptable, the overlying water is drained off and the depth of penetration determined. The overlying water in grabs intended for infaunal samples may be drained but all drained water must be captured for screening with the sediments (see Sample Processing below). Extra caution should be taken to drain the overlying water from the grabs for chemistry and toxicity samples. It is recommended that siphoning or decanting be employed for these grabs to avoid disturbance and loss of the surface sediments. It is important to get the best sample possible. For infaunal samples, sediment penetration depth must be at least 5 cm; however, penetration depths of 7-10 cm should be obtainable in silt (fine sand to clay). The depth of penetration is determined by insertion of a plastic ruler vertically along the grab midline and measurement of the depth of sediment to the nearest 0.5 cm. 9 Sediment Description The field description of sediments is required following measurement of penetration depth. The sediment description should encompass the following: at minimum the sediment should be characterized as being shell hash, gravel, sand, or mud (silt and/or clay), but if possible, the following should be done: surface colour and colour change with depth as a possible indicator of redox state; smell: sulfide (the odor of H2S or rotten eggs), oily (the odor of petroleum tar), or humic (a musty, organic odor). Typically, sediments will have no particular odor. General sediment colors (e.g., black, green, brown, red, yellow) description of sediment types, including important notes, e.g., the occurrence of concretions, loose algae, etc. Precise position fixing during sampling is essential. The position and the depth should be controlled and documented during station work. References Stubbs, H.H., D.W. Diehl, and G.P. Hershelman. 1987. A Van Veen grab sampling method. So. Calif. Coastal Water Res. Proj., Long Beach, CA., Tech. Rep. No. 204. Benthos Benthos samples will consist of the entire grab (i.e. no a priori subsampling) or the entire core. Macrofauna samples must be sieved sequentially at 1mm and 0.5mm and macrofauna will be identified to lowest practical taxon. Meiofauna will be taken from all possible sites by cores. If necessary, meiofauna may be subsampled from van Veen grab, by using sub-sampling cores. Where possible, meiofauna should be taken from cores collected by a Craib corer. Macrobenthos Procedures Assorted Sources Separation of Fauna from the Sediment The transfer of the sample to the sieve, the sieving procedure, and the transfer of the animals to the fixation jar are the steps during sample treatment most likely to introduce sources of error. To reduce the magnitude of these errors, the number of steps in the sampling and sieving procedures should be kept as small as possible and attention should be paid to the following procedures. Sieving can be conducted either aboard the survey vessel as samples are collected or onshore after a sampling excursion has been completed. In the first case, sieving usually precedes fixation and is conducted primarily on live organisms. In the second case, sieving generally occurs after fixation and is therefore conducted on dead organisms. Comparability between the results of these two techniques may be influenced by at least two factors. First, because fixation may cause some taxa to distort their shape or autotomize (i.e., cast off body parts), the sieving characteristics of those taxa may change 10 following fixation. Second, sieving characteristics of live organisms may differ from those of dead individuals. This bias occurs primarily for soft-bodied organisms (e.g., polychaetes) that can crawl through mesh openings or entangle themselves on the screen when they are sieved live. A major problem that may be encountered when organisms are fixed in sediment before being sieved is that the fixative either will not reach all buried organisms or will not reach them in time or in sufficient concentration to prevent some deterioration. Because deteriorated individuals may decompose completely or fragment upon sieving, their sieving characteristics can be modified substantially by inadequate fixation. Therefore, if samples are fixed in sediment, extra care should be taken to ensure that organisms are fixed adequately. For example, the sample container can be rotated gently immediately after fixation and again after 12-24 h to ensure adequate fixative penetration. From a logistical standpoint, sieving of samples in the field is generally preferred for surveys in which a large number of samples are collected during each cruise. Field sieving results in a considerable reduction in the volume of material that must be stored on the vessel (i.e., where space is often limiting) and later transported to the laboratory. Use of Relaxants Relaxants are often used when processing benthic macroinvertebrate samples for at least two major reasons. First, relaxants facilitate taxonomic identifications (and morphometric measurements) by reducing the tendency of organisms to distort then shape or autotomize when exposed to a fixative (Gosner 1971). Complete organisms having a natural appearance are easier to identify correctly than are fragmented and/or distorted specimens. For some taxonomic groups (e.g., Maldanidae), complete organisms are required for species-level identification. A second reason for using a relaxant is to ensure that animals are sieved whole, if sieving follows fixation. The tendency for some taxa (especially polychaetes) to autotomize if not relaxed can influence sieving by reducing the size of individuals. Because relaxation can influence taxonomic identification and sieving, data comparability between studies that use a relaxant and those that do not use one may be affected. The magnitude of these effects is unknown, but probably is greatest for softbodied taxa that are difficult to identify (e.g., some polychaetes) and smallest for taxa encased in a hard enclosure such as a calcareous shell (e.g., most molluscs) or an exoskeleton (e.g., crustaceans), particularly if the hard parts are the primary taxonomic characters used for identification. Recommended treatments for main marine groups: Sponges: Fix and preserve in 5% formaldehyde. Calcareous sponges should be preserved in 75% ethanol as formaldehyde can decalcify the specimens. Hydroids: Relax in 8% MgCl2 (or 15% MgSO4 or Menthol crystals). Fix in 5% formaldehyde for at least 24 hours; transfer to 75% ethanol for preservation *Actinians: Allow to relax in seawater then narcotise by replacing slowly with either 8% MgCl2 or Soda water to 50% (or 10% MgSO4 plus 1 or 2 drops of formaldehyde every 15 minutes) Nemerteans: Relax in 8% MgCl2 or add Menthol crystals to water 11 *Polychaetes: Relax in 8% MgCl2 (or gradual addition of 70% ethanol, or 20% MgSO4, or 0.15% propylene phenoxetol to water). Fix in 5% formaldehyde for 24 hours then transfer to 1.5% propylene phenoxetol (this preserves colour, but if unavailable 75% ethanol will do). Ideally, don't fix in ethanol and don't leave in formaldehyde. *Priapulids, sipunculans, echiurans: Relax using menthol crystals with a few drops of alcohol added after an hour (or put straight into 8% MgCl2) Small Crustaceans: Relax in soda water (or add a few drops of 70% ethanol to water or use 0.15% propylene phenoxetol). *Opisthobranchs: Relax in 8% MgCl2, fix and preserve in 5% formaldehyde or transfer to propylene phenoxetol after fixation. Bryozoans: Calcified bryozoans fix and preserve in 75% ethanol, fleshy or membranous ctenostomes fix in 5% formaldehyde for 24 hours then transfer to propylene phenoxetol for preservation Echinoderms: Fix in excess 75% alcohol, replace after a few days due to dilution from body fluids. Do not preserve long term in formaldehyde as the acid can dissolve the calcareous ossicles and plates, which are essential for identification, particularly of holothurians. *Ascidians: Relax using Menthol crystals (or immerse in 8% MgCl2). Fix in 5% formaldehyde. They can be preserved in propylene phenoxetol, or left in formaldehyde. *IMPORTANT to relax these groups BEFORE fixation if need to identify later Sieving options There are new designs of sieving tables with hand-controlled water sprinklers, which help to reduce the physical stress on the people involved while at the same time retaining the quality of the sampled specimens (Figure 3). Also, tilting devices for the full sample container, providing the option to fix the container at a certain angle over the sieve, are of use to reduce spilling and to avoid destructive tools. One example of a smaller sieve holder is shown in Figure 4. With this stand, the sieve residue can be transferred to the sample container with only the help of a sprinkler bottle, thereby avoiding the need for spoons or other scraping tools. For descriptive surveys, sieves used for extraction of the macrofauna from sediments should have a mesh size of 1.0 mm. The use of a finer sieve of mesh size 0.5 mm, or even finer, is recommended for special purposes. The sieve mesh should be checked from time to time for damage and wear. If a finer sieve is also used, the sieve fractions should be treated separately, and the results should be given for the single and the summed fractions. If re-sieving of samples is carried out, a mesh size finer than that of the initial sieve should always be used. Small sieves may be cleaned with an ultrasonic bath. The use of brushes should be avoided to prevent possible alterations of the mesh size. Distortion of woven mesh sieves occurs with increasing frequency of use. This can introduce considerable errors in the collection of small organisms. Moreover, the use of a square mesh introduces additional inaccuracies in collecting organisms in the size range of approximately the mesh size since the mesh diagonal width is greater than the nominal mesh width. The use of larger sieves is encouraged because the risk of clogging is reduced, for example, sandy samples may rapidly fill or even overfill smaller sieves. Larger sieves also reduce the risk of 12 spilling when transferring samples from containers/buckets to the sieve. This risk can also be kept low by using integrated sieve tables, as shown in Figure 3. Figure 3. Cross-section of an integrated sieving table where the sample is first emptied onto a coarse sieve (~5 mm) from where it is washed with a hand sprinkler douche onto the final 1 mm (0.5 mm) sieve (Design provided by G. Fallesen, Aarhus, Denmark). A growing number of institutes are changing to round mesh sieves, owing partly to a perceived improvement in the condition of the animals retained and partly to the theoretical improvement in mesh selectivity. Further work is required to establish a basis for using either type of sieve. Errors associated with the use of different sieves are like to be small in relation to other sources of sampling error. Figure 4. Sieve holder to provide a careful transfer of the sieve residue to the sample vessel (no tools needed-only a funnel and a wash bottle)(Design provided by G. Fallesen, Aarhus, Denmark). 13 Sieving Procedure Sieving should be conducted according to the following procedure: Each grab and box core sample should be sieved, stored, and documented separately. The grab or box core should be emptied into a container or washing table, and then the sample should be transferred portion by portion onto the sieves, as a sediment-water suspension. The use of sprinklers or hand-operated douches to suspend the sample is recommended. Very stiff clay can be gently fragmented by hand in the water of the container. The sieve must be cleaned after each portion has been sieved to avoid clogging and to ensure an equal mesh size throughout the entire sieving procedure. In order to avoid damaging fragile animals, the most gentle way to sieve a sample is to gently agitate the sieve surface under the water surface of a water-filled container until all sediment that can pass the sieve is washed through. On no account should water jets (i.e., deck hose) be used against the sieve surface. Fragile animals, such as some polychaetes, should be picked out by hand during the sieving, to minimize damage. Also, stones and large shells should be picked out, to avoid a grinding effect on the organisms and the sieve. All material retained on the sieve should be carefully flushed off the sieve, with water from below, into an appropriate recipient and fixed. The use of spoons or other scraping tools should be avoided. When the 0.5 mm sieve is used, the 0.5 mm and the 1 mm fractions must be kept separate throughout all further processing. Fixation Fixation and conservation (preservation) are two different steps in the treatment of a sample. The former procedure is employed to coagulate and harden the tissue of the organisms, while the latter prevents them from rotting and decaying. Improperly fixed specimens may create problems during further treatment, i.e., through fragmentation of specimens or loss of appendages. Some zoological museums will only accept properly (formalin-) fixed specimens for further analysis and curation. All the material retained on the sieves should be fixed in a buffered 4 % formaldehyde solution (1 part 40 % formaldehyde solution and 9 parts filtered sea water). For buffering, 100 g of hexamethylene tetramine (= Hexamine, = Urotropine) can be used per 1 litre of concentrated formaldehyde (36-40 %). Sodium tetraborate (= Borax) in excess may also be used. Sponges are best preserved by putting them directly into absolute ethyl alcohol so as to prevent fragmentation. Formaldehyde is regarded as a toxic compound, and probably also carcinogenic, and should, therefore, be handled with great care. Appropriate means of laboratory air suction or ventilation should be provided for all procedures. For animal sorting, the samples should first be thoroughly washed with tap water and left to soak over night so that sorters are not exposed to formalin vapour. Other fixation fluids that do not release formalin gas have been tested, such as formaldehyde depot chemicals (Dowicil 75 and Kohrsolin) used in clinics for sterilization purposes. The effects of these fluids on dry weight and ash-free dry weight are marked and the effects on long-term storage are unclear, so that no unequivocal recommendation can be given (Brey, 1986). 14 Staining To facilitate sorting and to increase sorting accuracy, especially for small animals, staining the sample with, e.g., Rose Bengal, is recommended. However, in some cases, staining may cause problems with species identification and the time gained during sorting will therefore be more than offset. Zoological museums will not accept stained material for taxonomic purposes. The following procedure has been shown to give good results: Wash the sample free from the preservation fluid by using a sieve with a mesh size smaller than 0.5 mm x 0.5 mm. Allow the sieve to stand in Rose Bengal stain (1 g dm-3 of tap water plus 5 g of phenol for adjustment to pH 4-5) for 20 minutes with the sample well covered. Wash the sample until the tap water is no longer coloured, As an alternative, Rose Bengal (4 g dm-3 of 40 % formaldehyde) may be added to the fixation fluid. Overstained specimens may be destained in alkaline (pH 9) fluids. Sieving of Fixed Material Samples may be sieved 'alive', as is the usual practice, or preserved. If they are preserved, it must be realized that the sorting characteristics are different from those for live fauna and result in apparently higher abundance and biomass figures. Intercalibrations of both procedures should be performed. In publications, it should always be stated whether the sieved material was fresh (alive) or fixed. Sorting Sorting must be done using some magnification aid (magnification lamp, stereomicroscope). Any finer fraction (< 1 mm) should always be sorted under a stereomicroscope. When taxa occur in great numbers (e.g., Polydora, Phoronids, Capitellids), it may be advisable to split the samples to reduce the counting time. Different types of sample splitters can be used. Rare species should be counted from whole samples. The accuracy of the sample-splitting device should be adequately assessed. To reduce sorting time, a sorting aid (such as the one described by Pauly (1973) or a 'fluidized sand bath' (after P. Barnett, see Holme and McIntyre, 1984)) may be used, provided that its efficiency has been satisfactorily checked for the particular bottom material studied. The Ludox method (see Higgins and Thiel, 1988) has successfully been applied to meiobenthos work and may also prove useful for the extraction of soft-bodied macrofauna. In coarse sand, the following procedure may be recommended: the sediment is fixed and placed on a PVC trough 5 m long, 20 cm wide, and 20 cm high (an ordinary gutter of the same length may also be used). Water is poured over the sediment from one closed side and the extracted fauna caught on a sieve on the other (open) side (Vanosmael et al., 1982). If samples are sorted alive, care should be taken to avoid predation within the sample. Biomass Determination The following measures of biomass determination can be used: wet weight, dry weight, and/or ash-free dry weight, either from fresh or fixed material. Furthermore, energy content (J) and / or matter equivalents (C, N, P) may be determined, using fresh material 15 only. Fresh wet weight is to be preferred to formalin wet weight, but if the latter has to be used, weighing should not be done until at least three months after fixation (Brey, 1986). The wet weight is obtained by weighing after the external fluid has been removed on filter paper. The animals are left on the filter paper until no more distinct wet traces can be seen. Animals with shells are generally weighed with their shells; the water should be drained off bivalves before weighing. When shell-free weights are given, the shell weight should be included in the data list. Echinoids should be punctured to drain off the water before blotting on filter paper. As soon as the non-tissue water has been removed, the organisms are weighed with the accuracy required (for adult macrofauna: 0.1 mg). In case tube-building animals have to be weighed together with their tubes, appropriate correction factors should be established. The dry weight should be estimated after drying the fresh material at 60 oC, or by freeze drying, until constant weight is reached (at least 12-24 hours, depending on the thickness of the material; large bivalves may need up to 96 hours). Dry weights obtained by lyophilization (freeze drying) are slightly higher than those obtained by oven drying. For Mytilus, lyophilized tissues weighed 10.9 % more than oven-dried tissues (Gaffney and Diehl, 1986). The use of ash-free dry weight is recommended in routine programmes, because it is the most accurate measure of biomass (Rumohr et al., 1987; Duineveld and Witte, 1987). However, it destroys specimens, and the consequences of this should be carefully considered. Ash-free dry weight should be estimated after measuring dry weight. It is determined after incineration at 500 oC in an oven until weight constancy is reached (about 6 hours, depending on sample and object size). The temperature of the oven should be checked with a calibrated thermometer because there may be considerable temperature gradients (up to 50 oC) in a muffle furnace. Caution is advised to avoid exceeding a certain temperature (> 550 oC), at which a sudden loss of weight may occur owing to the formation of CaO from the skeletal material of many invertebrates (CaC03). This can reduce the weight of the mineral fraction by 44 %. Such decomposition occurs very abruptly and within a small temperature interval (Winberg, 1971). Before weighing, the samples must be kept in a desiccator while cooling down to room temperature after oven drying or removal from the muffle furnace. To estimate biomass from length or size measurements, conversion factors may also be used (Rumohr et al., 1987; Brey et al., 1988). References Brey, T. 1986. Estimation of annual P/B-ratio and production of marine benthic invertebrates from length-frequency data. Ophelia Supplement, 4: 45-54. Callaway, R., Robinson, L. & Simon P.R. 2003. Methods Manual, Managing Fisheries to Conserve Groundfish and Benthic Invertebrate Species Diversity (MAFCONS Project) Duineveld, G.C.A., and Witte, H.J. 1987. Report on an intercalibration exercise on methods for determining ash-free dry weight of macrozoobenthos. ICES CM 1987/L:39. Gaffney, P.M., and Diehl, W.J. 1986. Growth, condition and specific dynamic action in the mussel Mytilus edulis recovering from starvation. Marine Biology, 93: 401-409. 16 Gosner, KL 1971. Guide to identification of marine and estuarine invertebrates. John Wiley, New York, New York, USA. Higgins, R.P., and Thiel, H. 1988. Introduction to the study of meiofauna. Smithsonian Institute Press, Washington, D.C. 470 pp. Holme, N.A., and McIntyre, A. 1984. Methods for the study of marine benthos. IBP Handbook, 16. Second edition. Oxford. 387 pp. Pauly, D. 1973. Über ein Gerät zur Vorsortierungg von Benthosproben. Berichte der Deutschen Wissenshaftlichen Kommission für Meeresforschung, 22 (4): 458-460. Rumohr, H., Brey, T., and Ankar, S. 1987. A compilation of biometric conversion factors for benthic invertebrates of the Baltic Sea. Baltic Marine Biologists, Publication No. 9. 56 pp. Vanosmael, C., Willems, K.A., Claeys, D., Vincx, M., and Heip, C. 1982. Macrobenthos of a sublittoral sandbank in the Southern Bight of the North Sea. Journal of the Marine Biological Association of the UK, 62: 521-534. Winberg, G.G. 1971. Methods for the estimation of the production of aquatic animals. Academic Press, London, New York. 175 pp. Meiofauna procedure Source: University of Crete Required Corers Rubber stoppers Core plunger Corers basket Rubber mallet Sample containers Sample labels Syringe 7% MgCl2 10% buffered formalin (4% formaldehyde) Washbottles Spatulas Cylindrical, transparent, plastic tubes (proposed inner diameter 4.4 cm) with smooth internal surface and beveled lower end to facilitate sediment penetration and core removal. The length should be at least 15 cm (Fig. 1a) Appropriate diameter for tight-fitting to the coring tubes (Fig. 1) Same diameter with the corers for extruding the sediment core (Fig. 1b) Will ensure the upright position of the core samples ~ 500 ml volume Indicate area, station, replicate and date ~ 20 ml volume 7.5 g MgCl2 6H2O dissolved in 100 ml distilled water. Used as a narcotic agent Filtered seawater (through a 45 um sieve) should be used as dilutant to prevent contamination with planktonic species. The formalin should be buffered with 200 g Borax per liter Used for distilled/filtered water, MgCl2 Helpful for slicing the cores 17 Sampling In sediments, coring is the best quantitative sampling technique for meiobenthos, because when corers are used with care they collect a known area or volume of sediment with all depths equally represented and all animals present before sampling are captured. a b Figure 1. a) Corers, rubber stoppers and b) core plunger for sampling meiobenthos. If possible, subtidal samples should be taken by SCUBA divers (Fleeger et al. 1988). Divers usually obtain superior samples because they are able to position the samplers with care and insert the corer slowly (McIntyre 1971). In addition, the presence of the investigator will often yield important insights about the ecology of the site or practical aspects of the sampling (Fleeger et al. 1988). However, if diving is not possible, a Craib type corer (Fig. 2a) may be used instead. Subsampling from a sample collected with a multiple SMBA type corer, grab or box corer is also an alternative (Fig 2b,c). In the above case, caution should be taken to avoid pseudoreplication by taking each subsample from different deployments. a b c Figure 2. Different samplers used on research vessels for meiobenthic studies. a) Craib corer, b) box corer, c) multiple corer. Processing cores 18 It is easiest to process the cores by working with another person. The bottom stopper is removed and replaced by the core plunger, to avoid loss of the sample. Following this, the upper stopper is removed and the overlying water is transferred, with a syringe, to a sample jar (because meiofauna may have swum out of the sediments to the water). Next, the sediment is pushed out of the corer and the top 10cm are transferred to the sample jar. A 7% MgCl2 solution is added to the jar until the sample is fully covered. Stir gently and allow 10 minutes to react. Next, the sample is fixed by adding buffered formalin to achieve a final concentration of 10% (take into consideration the volume of sediment, water and MgCl2). Invert the jar several times to mix the fixative and sediment and store at room temperature until the samples may be processed further. References Elmgren R (1973) Methods of sampling sublittoral soft bottom meiofauna. Oikos Supplement 15: 112-120 Fleeger JW, Thistle D, Thiel H (1988) Sampling equipment. In: Higgins RP, Thiel H (eds) Introduction to the study of meiofauna. Smithsonian Institution Press, Washington DC, London, p 115-125 Pfannkuche O, Thiel H (1988) Sample processing. In: Higgins RP, Thiel H (eds) Introduction to the study of meiofauna. Smithsonian Institution Press, Washington DC, London, p 134-145 Giere O (1993) Meiobenthology. The microscopic fauna in aquatic sediments. SpringerVerlag, Berlin McIntyre AD (1971) Deficiency of gravity corers for sampling meiobenthos and sediments. Nature 231:260 Phytobenthos procedure Source: HIMOM Background Phytobenthos refers to the algae that may be found on/in the seafloor sediments. Sediments are sampled and the algal pigments are quantified by spectrophotometer and by HPLC. The sampling strategy is a key issue in these studies due to non-uniform vertical and horizontal distribution of the phytobenthos. In muddy sediments, phytobenthos is found mostly in the top 1.5 cm of sediment whereas in sandy sediments it is possible to find phytobenthos as deep as 15 cm depth (Cartaxana et al., 2006). Phytobenthos distribution may be very patchy and the sampling strategy should take this into account. No extraction procedure exists that will provide 100% extraction efficiency. Efficiency varies with species and acts differently for the various pigments. The 90% acetone as recommended in this protocol generally provides ca. 90% efficiency (Wiltshire et al. 2000). Moreover, acetone extraction is especially efficient for polar pigments, like chlorophyll a, and thus can be used for the analytical methods described below. Unfortunately, some extraction-resistant species such as cyanobacteria and Zostera exist on the tidal flats, which require mechanical (e.g. grinding) and aggressive chemical (e.g. toxic dimethylformamide and methanol) means to extract these pigments. However, due 19 to other problems associated with these extraction methods, they will not be described here. Required - Freezer - Freeze-Dryer - balance - 90% buffered acetone - ultrasound bath - centrifuge - ultrasound bath Sampling: A small petri dish is used to take surface sediment samples at intertidal sites. Ideally, the petri dish should have 20 ml volume and a height of 2 cm. The Petri dish is placed on the sediment and collected by means of a plastic card that is slid under the plate to break the contact with underlying sediment. This method works very well with mud, however, for sampling sand, may want to sample a deeper depth, based on previous (preliminary) experience regarding vertical distribution of the phytobenthos. If sampling in a subtidal area, sediments may be taken by divers, by Craib corer or by box corer (sediment surface must NOT be disturbed) and then subsampled using the Petri dish method. Lab work: Extraction should always be carried out under dim light, keeping the samples and solvent cold/on ice. 1. Sediment samples should be frozen as soon as possible and then freeze-dried 2. Weigh out an amount (about 15g) of freeze-dried sediment and transfer to 50ml centrifuge tube 3. Add about 10% (w/w) quartz sand (MERCK- particle size 10-30µm) to the tube 4. Add 15ml acetone (10% saturated sodium carbonate : 90% pure acetone) to the tube. 5. Place the tubes in an ultrasound bath containing -4ºC water for 90 minutes. 6. Place the sealed tube with sample in a freezer (-20ºC) to extract for 24 hours (vortex after that). 7. At end of 24 h, vortex the tubes and centrifuge 10 min at 3000 rpm. 8. Take the extract for pigment analysis, using HPLC or spectrophotometer. References Bowles, N. D., Paerl, H. W. & Tucker, J. 1985. Effective solvents and extraction periods employed in phytoplankton carotenoid and chlorophyll determinations. Can. J. Fish. Aquat. Sci. 42: 1127-31 Cartaxana, P., Mendes, C.R., van Leeuwe, M.A & Brotas, V., 2006. Comparative study on microphytobenthic pigments of muddy and sandy intertidal sediments of the Tagus estuary. Est. Coast. Shelf Sci. 66: 225-230. 20 Wiltshire, K.H., M. Boersma, A. Molle & H. Buhtz, 2000. Extraction of pigments and fatty acids from the green alga Scenedesmus obliquus (Chlorophyceae). Aquatic Ecol. 34: 119-126. Wright, S. W., S. W. Jeffrey & R. F. C. Mantoura, 1997. Evaluation of methods and solvents for pigment extraction. In: Jeffrey, S. W., Mantoura, R. F. C., and Wright, S. W. [Eds], Phytoplankton pigments in oceanography: guidelines to modern methods. UNESCO, Paris, pp. 261-82. Sediment chemistry At each station, triplicate cores will be taken for sediment chemistry. If an analysis is very expensive or complicated to perform, duplicates may be sufficient. Sediment chemistry will be conducted almost exclusively on sediment cores. The first thing that will be measured in the sediments is the vertical profile of redox potential. Cores (or sub-cores taken from grabs) will be sliced at 1 cm intervals from sediment surface to 12cm depth for sediment geochemistry. If the redox discontinuity layer is deeper than 12 cm, should slice down to the redox discontinuity depth. Chemical variables will be measured from cores sampled by SCUBA divers or in subcores taken from grab samples. It is essential that grabs are of good quality for chemical analysis, with as little disturbance as possible. If the samples have been disturbed, best thing to do is toss the sample and sample again to obtain a good, undisturbed sediment sample. If this is not possible, should note this and the reason additional cores could not be taken and analyze only the top 2cm layer. The sediment slices will be taken for granulometry, loss on ignition, total organic carbon, total nitrogen, total phosphorus. Sediment redox potential, Eh Source: ICRAM Organic enrichment of sediments usually leads to reduced conditions which equate to “bad” sediment quality, wherein natural benthic communities undergo substantial changes. The oxidation-reduction (redox) conditions in surface sediments depend on the degree of organic enrichment and can be assessed by measuring the vertical redox potential profile (expressed in mV) in the top 15 cm (Zobell, 1946). The redox state of sediment is the result of the combined effect of biological and chemical processes of reversible and/or irreversible nature and is therefore difficult to define. It has been pointed out that the concentrations of the components reacting reversibly are, in most cases, too small to get reliable results from redox measurements. Thus, what is really measured is a mixed potential which is not useful for chemical equilibrium calculations (Bågander, 1978). However, Eh profiles still provide useful information, since the decrease in Eh with the depth is related to the decrease in the dissolved oxygen 21 concentration in the pore water. Negative Eh values are therefore associated with anoxic conditions, in which the degradation of the organic matter is performed by anaerobic bacteria. In marine sediments, these bacteria mainly use sulphate as the electron acceptor, which is reduced to hydrogen sulphide (Porrello et al., 2005; Chamberlain, 2002; Danovaro et al., 2004). Redox potential is measured by profiling an electrode down a sediment core to as deep as is necessary to detect the redox discontinuity layer (normally 10 or 15cm), i.e. the point at which redox values change abruptly from highly negative values to either less negative, or to positive values. Redox potential will be measured in duplicate cores at each station and only in undisturbed cores. Required: Surface sediment in cores, at least 0-20 cm depth Ruler (to measure sediment height) Portable pH/Eh meter Redox electrode, with shaft >15cm long, preferably as thin as possible, with Platinum ring indicator Double junction silver/silver chloride reference electrode Redox electrode calibration: The ZoBell solution is used as a reference; this solution (0.003 M potassium ferricyanide, 0.003 M potassium ferrocyanide, and 0.1 M potassium chloride) has an Eh value of +430 mV at 25°C. Measurement: The redox potential should be measured by carefully pressing the redox electrode into the intact sediment core (in the tube) as soon as possible after collection (the longer the sediment sits in the collection tube, the more it changes due to biogeochemical processes that directly affect redox values), at depth intervals of 1cm from the surface to 15cm depth. Measuring redox by inserting electrodes into holes in the side of the cores leads to artifacts (see Hionchey and Schaffner 2005) and is therefore not recommended. Moreover, slicing the sediments to measure redox in these is not recommended as exposure to the air will rapidly and drastically alter the redox potential values. It is essential that the measurements be done using either a combination redox electrode or a redox electrode that is connected to a reference electrode that is in contact with the sediment porewater. The measurements will be recorded in mV when the meter readings stabilize at each depth. In order to clarify the effect of sampling method on the data, sediments should be sampled (several replicates) at same site by both van Veen grab and by Craib corer or by SCUBA diver sampled corers. Redox profiles should be run directly on the sediments in the grab and subsequently, subsamples should be taken from the grab for redox profiles and these should be compared to redox profiles performed on cores taken either by SCUBA divers or by Craib corer. References 22 Bågander, L.E. 1978. An evaluation of the use of redox measurements for characterizing recent sediments. Estuarine and coastal Marine Science, 6: 127-134. Chamberlain, J., 2002. Modelling the environmental Impacts of Suspended Mussel (Mytilus edulis L.) Farming. Ph-D Thesis, Napier Univeristy, Edimburgh. Danovaro, R., Gambi, C., Luna, G.M., Mirto, S., 2004. Sustainable impact of mussel farming in the Adriatic Sea (Mediterranean Sea): evidence from biochemical, microbial and meiofaunal indicators. Marine Pollution Bulletin, 49: 325-333. Hinchey, E.K. and L. C. Schaffner. 2005. An evaluation of electrode insertion techniques for measurement of sediment redox potential in estuarine sediments. Chemosphere 59:703-710. Porrello, S., Tomassetti, P., Manzueto, L., Finoia, M.G., Persia, E., Mercatali, I., Stipa, P., in press. The influence of marine cages on the sediment chemistry in the Western Mediterranean Sea. Aquac. Zobell, C. E., 1946. Studies on redox potential of marine sediments. Bulletin of the American Association of Petroleum Geologists 30, 477-511. Sediment grain size analysis (granulometry) Source: HIMOM Introduction The objective of grain size analysis is to determine the grain size characters of sediment samples and subsequently classify samples based upon their constituent parts. Particle size analysis is performed using laser diffraction techniques (<2mm fraction) and sieving techniques (>2mm fraction). Sediments containing a mixture of both fractions are analysed by both methods and the results integrated. The results from the particle size analysis are used to generate size distribution plots, to calculate statistical operators including mean and sorting, and to obtain %clay, %silt, %mud, %sand and %gravel in order to classify samples into sediment types. The laser diffraction technique is described in the HIMOM manual. The sieving technique is executed as follows: 1. Put the sediment sample in an oven at 60-80.C to calculate total dry weight (it’s advisable to take a sample of ca. 100g dry weight). 2. Remove the fraction <63um, by sieving the sample through a 63 um sieve under a water flow. 3. Dry the remaining fraction in an oven at 60-80.C 4. Put this fraction in the analytical sieve shakers to separate the different fractions (>1000um; 1000-500; 500-250; 250-125; 125-63um). 5. Weigh the residuals in each sieve, and express as a percentage of total weight, including the weight of the fraction <63um, which is calculated by subtraction. Go to HIMOM Book of Protocols, p. 45 for further details on granulometry. 23 Sediment organic matter Ratio of refractory to labile organic matter using Loss on Ignition (LOI) measurements Source: Loh 2005 (SAMS) Introduction There are some data in the literature on LOI using this method, but generally little for fish farms. It might be expected to work very well in fish farms owing to the high flux of labile organic matter to sediments, and it should be possible to use this to evaluate recovery status. This indicator is less complex and time-consuming than total organic carbon (TOC) measurements, which are routinely carried out in EIA studies, yet correlate very poorly with impact, as TOC includes both labile and refractory material. Method Loss on ignition is carried out in a temperature-monitored muffle furnace. Approximately 0.5 g of dried, ground and sieved (200um) sediment sample is weighed precisely into a crucible. Crucibles with sediment are then ashed (250°C for 16 hours). When cooled, the crucibles are reweighed. Sediments are then heated to 500°C (Kristensen and Andersen, 1987) for 16 hours (Sutherland, 1998). When cool, they are weighed again. According to Kristensen (1990), the weight loss in the lower temperature range (130-280°C) is due to evaporation (i.e. dehydrogenation of hydroxylated aliphatic structures, decarboxylation of acid groups, and generation of low molecular weight volatile compounds); oxidative degradative degradation of aliphatic carbohydrates; random chain-scission of weak bonds; cross-linking and peroxide formation; formation of compounds of less-ordered structure; cyclization; and formation of carbonaceous char. Plant materials rich in carbohydrates are combusted at the lower temperatures. The weight loss in the high temperature region (280-520°C) is due to oxidation of aromatic groups (polyphenolic compounds such as lignin, humic substances and kerogens) and char. The percentage weight reduction after both 250°C and 500°C are measures of the sediment labile and refractory organic matter, respectively. Mook and Hoskin (1982) found significant (p<0.05) weight losses between 200°C and 300°C; hence the mean 250°C is chosen to be used here. Kristensen and Andersen (1987) found that most calcite was combusted between 500-800°C, hence the 500°C is used in this study, as most organic carbon would be completely separated with this temperature. Hirota and Szyper (1975) also separated organic and inorganic compounds by heating at 500°C. Rp index was introduced by Kristensen (1990) to characterize the composition of various biogenic organic materials at different decomposition stages. It is defined as, Rp = PII/(PI+PII) where PI is the weight loss in the temperature range 0 °C and 250 °C and PII is the weight loss in the temperature range 250 °C and 500 °C. Calculations of percentage organic matter and Rp values are as follows: 24 Let Ws = original weight of sediment sample (approximately 0.5 g) W0 = weight of crucible and sediment before combustion W250= weight of crucible and sediment alter combustion at 250°C W500= weight of crucible and sediment after combustion at 500°C PI = weight loss in temperature range 0°C and 250°C = WO - W250 PII = weight loss in temperature range 250°C and 500°C = W250 - W500 and, Rp= PII/(PI+PII) % labile OM = (PI/Ws) x 100% % refractory OM = (PII/Ws) x 100% % total OM = W0-W500 Ws X 100% or, % total OM = % labile OM + % refractory OM References Hirota, J. & J. P. Szyper, 1975. Separation of total particulate carbon into inorganic and organic components. Limnol. Oceanogr. 20: 896–900. Kristensen, E. 1990. Characterization of biogenic organic matter by stepwise thermogravimetry (STG). Biogeochemistry 9:135–159 Kristensen, E. and F.Ø. Andersen. 1987. Determination of organic carbon in marine sediments: a comparison of two CHN-analyzer methods. J. Exp. Mar. Biol. Ecol. 109: 15-23. Loh, P.S., 2005. An assessment of the contribution of terrestrial organic matter to total organic matter in sediments in Scottish sea lochs. PhD thesis, UHI Millenium Institute, 350 pp. Mook, DH and Hoskin CM. 1982. Organic determinations by ignition: caution advised. Est. Coast. Shelf Sci. 15: 697-699. Sutherland, R. A., 1998. Loss-on-ignition estimates of organic matter and relationships to organic carbon in fluvial bed sediments. Hydrobiologia 389: 153–167. Sediment organic carbon Source: ICRAM Introduction Carbon in marine sediments occurs as organic carbon (OC) intimately linked to the metabolic processes and remains of plants and animals and as carbon contained within biogenic and abiogenic carbonate minerals. Successful determination of sediment organic carbon relies upon the separation of organic from inorganic carbon (IC). Several methods of analysis have been employed for the separation of organic from inorganic forms of 25 carbon (reviewed in Verardo et al.,1990). Although the determination of total carbon (TC) content in marine sediments is straightforward by a carbon-hydrogen-nitrogen (CHN) elemental analyzer, that of organic carbon presents difficulties. This is because organic material is a complex mixture, and some of these components may be lost from the sediment at the temperatures both above and below those for the loss of other materials, such as structural water and carbonates. The determination of OC is usually a lengthy procedure, involving weighing of the sediment sample, careful pretreatment of the sample by a suitable acid to remove IC, drying of the sediment, reweighing of sediment and analysis by CHN analyzer (Tung & Tanner, 2003). Required Falcon test-tube Broad knife Ruler (to measure sediment height) Centrifuge (to extract interstitial water from the sediments) Freeze-dryer (to lyophilize the sediments) Mortar (to homogenize sediments) Sieve with 200 µm mesh Micro-analytical balance Procedure Sampling: Collect the sediment sample, by means of a corer device; Remove the upper 1 cm layer of the sediment, homogenize in a clean vessel, transfer to a centrifuge tube and store on ice during transit to the laboratory Sample processing: Samples are centrifuged to remove interstitial water and lyophilized (freeze-dried) Freeze-dried samples are gently pulverized using clean mortar and pestle and subsequently sieved at 200 µm; 6-10 mg of homogenized sediment are transferred to a special soft Silver cup (for elemental analysis) Treat each sample by adding 20 µl of 1N HCl, to oxidize inorganic carbonate. If effervescence continues, this step of the acidification procedure must be repeated; Wait for 10 minutes; Dry sample in 60°C oven for 10 minutes, Addition of 20 µl of HCl 1:1. If effervescence continues, this step of the acidification procedure must be repeated; Wait for 10 minutes; Dry sample in 60°C oven for 10 minutes Addition of 20 µl of HCl 25%. If effervescence continues, this step of the acidification procedure must be repeated; Wait for some minutes; Desiccate samples in oven, drying overnight at 60°C 26 Analysis: Silver cups with samples are folded and carbon content is determined using a calibrated carbon-hydrogen-nitrogen (CHN) Elemental Analyzer. References Tung, J. W. T. and Tanner, P. A., 2003. Instrumental determination of organic carbon in marine sediments, Marine Chemistry, 80 (2-3): 161-170. Verardo, D.J. 1990. Determination of organic carbon and nitrogen in marine sediments using the Carlo Erba NA-1500 Analyzer. Deep-Sea Research 37 (1): 157-165. Total nitrogen in sediment Source: ICRAM Introduction The production of biodeposits (faeces and pseudofaeces) due to mussel cultivation, can cause an increase in the total nitrogen concentrations in the sediment underneath mussel lines (Chamberlain, 2002; Aleffi et al., in press). In a recent study (Christensen et al., 2003) observed higher C/N ratios in surface sediments underneath mussel lines as compared to a reference site. The same is true for sediments underlying intensive fish farms, since the major fish species reared are carnivores that require high protein (= high nitrogen content) diets. Procedure Sampling, sample processing and analysis are identical to those described for organic carbon determination (above), but without the acidification steps. References Aleffi, I.F., Bettoso, N., Solis-Weiss, V., Tamberlich, F., Predonzani, S., Fonda-Umani, S., submitted to ICES – Journal of Marine Science. Effects of suspended mussel culture on the macrozoobenthos in the Gulf of Trieste (Northern Adriatic Sea, Italy). Chamberlain, J., 2002. Modelling the environmental Impacts of Suspended Mussel (Mytilus edulis L.) Farming. Ph-D Thesis, Napier Univeristy, Edimburgh. Christensen, P.B., Glud, R.N., Dalsgaard, T., Gillespie, P., 2003. Impacts of longline mussel farming on oxygen and nitrogen dynamics and biological communities of coastal sediments. Aquaculture 218: 567-588. 27 Inorganic and organic phosphorous in sediment Source: Strickland and Parsons (1972) with modification by Aspila (1976) Reagents Stock Ammonium Molybdate Solution Dissolve 30g of ammonium molybdate tetrahydrate in 1l of DI water Keep in a cool dark place Stock Antimony Potassium Tartrate Solution Dissolve 1.5g of antimony potassium tartrate in 1l of DI water Keep in a cool dark place 10% Sulphuric acid Ascorbic Acid Solution Dissolve 60g of ascorbic acid in 1l of DI water Keep in a cool dark place. Stable for about a month Working Colour Reagent Carefully mix 100mls of ammonium molybdate stock solution, 250mls of 10% sulphuric acid 100mls of ascorbic acid and 50mls of antimony potassium tartrate solution. Keep in a cool dark place. Stable for about 6 hours 1M Hydrochloric acid Potassium orthophosphate (KH2PO4) 10mM stock solution Accurately weigh 136mg into 100mls of 1M hydrochloric acid. Potassium orthophosphate standards Prepare 0, 50, 100, 200, 400 and 800uM solutions by pipetting 0.5, 01.0, 2, 4 and 8mls of potassium orthophosphate standard into 1M hydrochloric acid and making up to 100mls. This will give a calibration able to measure phosphorus between 0 and 0.1% in a 0.3g dried sample aliquot Procedure Inorganic phosphorus Weigh accurately between 0.2 and 0.3g of dried, homogenised and sieved (200 um) sediment into a centrifuge tube and add 12mls of 1M HCl. Place on a shaking table for 16 hours and then centrifuge or filter. To 10mls of extract add 1ml or working colour reagent and let stand for 20 min before measurement by spectrophotometer at 885nm. Prior to measurement, calibrate the spectrophotometer with standards and the blank. Calculate inorganic phosphorus content of sediment: 28 (u-molarity of extract) x (12/1000) x 31 1000000 x wt. of sample (g) x 100% Total phosphorus Weigh accurately between 0.2 and 0.3g of dried, homogenised and sieved (200 um) sediment into a crucible and ignite at 550 C for 1hour. After the crucible has cooled empty contents into a centrifuge tube and add 12mls of 1M HCl. Repeat extraction and measurement procedure of inorganic phosphorus determination. Organic phosphorus content can then be calculated from: Organic phosphorus = Total phosphorus – Inorganic phosphorus References Aspila, K.I. Agemain, H and Chau, A. 1976. A semi-automated method for the determination of inorganic, organic and total phosphorus in sediments. Analyst, 101 187-197 Strickland J. and Parsons T. 1972. A practical handbook of seawater analysis. Bulletin 167 Fisheries Research Board of Canada, Ottawa Water column (water quality) sampling Selection of sampling stations The same principles as described above, regarding sediment sampling, should dictate the selection of water column sampling stations. Station positions should reflect gradients of impact, normally aligned with the axis of predominant current and could consist of the following distances from the farm: 0, 25, 50, 100, 200m and 1000m. Unlike the footprint of fish farms that is often evident on the underlying sediments, the influence of aquaculture activities on surrounding water quality is generally quite small and measurable only within close proximity to the farm perimeter. It is noteworthy that in highly dispersive areas, with fast currents, we may limit water column sampling to 2 stations – one in the vicinity in the farm and one at reference site, outside zone B, because it is very difficult to detect a gradient in such highly dynamic environments. Sampling dates should correspond to peak production and warmest water conditions in order to capture maximal impact conditions. Samples should be taken along a diel or tidal cycle (minimal sampling should include one at low water and another at high water in tidal regimes) to include extreme conditions, e.g. DO sag at end of the night. Optimally, should establish diel measurement of all variables at a minimum of 2 stations (e.g. one at the farm site and second at anticipated intermediate effect station) within the area of anticipated (from hydrographical data) effect and another at a distant reference station. The water column will be sampled by CTD, and by bottle sampler at 3 depths; surface, mid-depth and near bottom and Secchi disk depth will be determined at all stations. In addition, the algal growth bioassay should be conducted at all possible study sites. 29 CTD vertical profiles at each station should include: salinity, temperature, sigma-t, dissolved oxygen, chlorophyll, PAR (photosynthetically active radiation), light transmission, turbidity (nephelometer). Water samples will be collected using Niskin (or similar) bottles, and taken for dissolved nutrients (including: ammonia, nitrate, nitrite, phosphate, silicate, total N, total P) and for suspended particulate matter (including: total suspended solids (including POC, PON, POP) and chlorophyll (for CTD fluorescence ground-truthing). For shellfish farm sites, the phytoplankton community composition is extremely important and should be characterized, where possible. Secchi disk transparency Source: Michigan Lake & Stream Associations, Inc Background The Secchi disk is used to measure how deep a person can see into the water. Even though the Secchi disk measurement of water clarity is an approximate evaluation of the transparency of water, it is used primarily for its simplicity. A more accurate measurement of underwater irradiance can be made by the use of a photometer. Vertical light penetration, as measured by Secchi disk, may also serve as a proxy for the phytoplankton biomass in a water body. The greatest value of the Secchi disk measurements occurs when readings at each station are recorded and compared from week to week, month to month and season to season. Several factors are involved in depth determination, including the eyesight of the viewer, the time of day the readings are taken (midday- between 10:00 and 14:00 is preferred), the reflectance of the disk, the color of the water, clay particles or other materials suspended in the water, etc. Equipment Secchi disk - 8 inch diameter metal disk painted in alternate black and white quadrants (see below) with waterproof tape or rope marked to 20cm resolution. Procedure The disk is lowered into the water by unwinding a waterproof tape or marked line (20cm resolution) to which it is attached and until the observer loses sight of it. The disk is then raised until it reappears. The depth of the water where the disk vanishes and reappears is the Secchi disk reading. The depth level reading on the tape at the surface level of the water is recorded. In order that the Secchi Disk measurement be done to provide the greatest accuracy, the following conditions should be met: 1. The same person should perform all readings since sharpness of vision varies from person to person. 30 2. The reading should be taken on the same day of the week, if taking weekly readings or at least not more than one day before or after the same day of the week. 3. It is preferable that the measurement be taken between 10:00 a.m. and 2:00 p.m. so that the light rays from the sky are at a similar angle each time the reading is taken. 4. Avoid taking the measurement when the water is choppy or rough. 5. Readings should be taken on the shady side of the boat. 6. The reading should be taken at the same location each week. Record: a. Cloud cover (see Cloud Cover Field Guide below). b. Depth of Secchi disk disappearance and reappearance, time of day, tide, current condition (strong, mild, or none), wind/wave condition (ripples, small waves, whitecaps), viewer’s initials and any other significant notes. Secchi disk depth procedure 31 Dissolved nutrients in seawater Source: ICES 2004 The commonly designated nutrients are inorganic nitrogen compounds (NO3 -, NO2 – ,NH4 +), phosphate (PO4 3-) and silicate (SiO4 3-). Total phosphorus (Ptot) and total nitrogen (Ntot) are also included because of their importance in relation to ecosystem analysis and budgets. Nutrients in sea water are considered trace compounds and their analysis is liable to various sources of contamination. Sea water for nutrient analysis is usually collected from research vessels or ships of opportunity (e.g., ferry boats, fishing boats, coast guard or navy vessels). The reference method for measuring nutrients in the following (including storage and pre-treatment) is Grasshoff (1976) “Methods of Seawater Analysis”. Sample handling Special attention must be paid to possible nutrient sample contamination generated by the boat or ship. Wastewater discharged from wash basins, showers, and toilets contains significant amounts of phosphorus and nitrogen compounds and, therefore, can contaminate the surface waters to be sampled. For this reason, the water sampler must be deployed far from wastewater outlets, even if no sewage is discharged at the time of sampling. Although most modern ships are equipped with special sewage tanks, they are often emptied at sea owing to a lack of appropriate reception facilities in ports. In addition, there are potential problems with kitchen garbage. Mixing by the ship’s propeller can disturb the natural distribution of the nutrients in the surface layer, particularly as regards oxygen. These problems, including the exact location of the ship, should be considered along with the natural variability. Phosphorus and nitrogen compounds are secreted from human skin. However, touching of the sampler and the sample bottles by hands does not cause problems unless the sample comes into contact with the outer surface of the sampler or sample bottle. This is something that should never happen since the outer surfaces cannot be kept free of contamination on-board a ship. In view of the potential for contamination, the analyst should preferably supervise the collection of samples. The attaching of bottles to a hydrowire or the preparation of a rosette and the subsequent removal and transport of samples to the ship’s laboratory should be done by trained personnel. The written instructions for the collection of samples should include the precautions to be taken when a sub-sample is transferred to the storage container. The instructions must include the details of the essential record of the sample: station location, station code, depth of sampling, date, time, etc., and the identity of the person responsible for sampling. Storage of samples The stability of nutrients in seawater samples depends strongly on the season and the location from which the samples were taken. Nutrients in seawater samples are generally unstable. Grasshoff (1976) recommends that ammonia and nitrite are measured no later than one hour after sampling. Samples for nitrate, phosphate, and silicate should preferably be analysed within six hours after sampling, and no later than ten hours. If for practical reasons samples cannot be analysed within these time limits, the corresponding 32 data should be flagged if stored in databases, unless the storage method has been validated. Samples should be stored protected from light and refrigerated. Plastic bottles must be used if silicate is measured. New sample bottles sometimes adsorb nutrients onto their walls. The new bottles, if necessary, should be cleaned with phosphate-free detergent, rinsed generously with distilled/deionized water, and left filled with sea water containing nutrients for a few days. Then checks for adsorption of nutrients onto the walls or losses due to transformation to another chemical form should be carried out. Sample bottles should always be rinsed with the seawater sample from the sampler before they are filled. As regards ammonia determination, glassware for ammonia should always be cleaned with dilute hydrochloric acid. If samples cannot be analysed within the abovementioned time limits, the following methods of storage can be recommended. Silicate - 0–4° C protected from light. Do not freeze (polymerization may occur). Nitrite - Freezing or 0–4° C protected from light. Do not acidify (rapid decomposition). Ammonia - No known preservation methods are applicable. Nitrate - Freezing. Total nitrogen - Freezing or 0–4° C protected from light. Do not acidify (enhanced risk of contamination). Phosphate - Freezing or acidification. Total phosphorus - Freezing or acidification with sulphuric acid, store at 0–4° C, protected from light. The addition of mercury or chloroform is an alternative preservation method for all nutrients except ammonia. However, these chemicals can affect the reaction kinetics, especially with automated methods, and this effect should be evaluated by the laboratory. The same chemical preservation of calibrants and quality controls can compensate for this effect. The use of mercury should be minimized and optimum disposal procedures should be ensured. These preservation methods are all second choice to immediate analysis. They should, as mentioned, be validated by each laboratory, taking into account the concentration levels, storage time and environment, differences in sample matrices, and the analytical method of the laboratory. Since no preservation method for nutrients can, at present, be recommended for general use, each laboratory must validate its storage methods for each nutrient before they are used routinely. Sample pre-treatment Sea water contains microorganisms and other suspended matter of different composition. In some cases, these particles bias the measurement of the nutrient in the soluble phase. The suspended matter can be removed either by filtration or centrifugation. Unnecessary manipulation of the sample should be avoided, but in particle-rich waters (e.g., coastal waters, during plankton blooms), filtration or centrifugation may become necessary. It is important that the procedure used for filtration/centrifugation has been validated. For removing algae from the water sample, a GF/C filter is adequate. For work in open oceans with low concentrations of suspended matter, GF/F filters are considered suitable for suspended matter separation from open sea water. Filtration in closed systems with a neutral gas is recommended. Centrifugation is especially advisable for samples destined for ammonia determination. If a sample containing particles is not filtered, the turbidity causes light scattering which can bias a colorimetric measurement. In this case, a 33 turbidity blank should be carried out by measuring light absorption of the sample before adding the colour-forming reagents. Appropriate chemical analytical methods The choice of an analytical method should be based on the following criteria: the method should measure the desired constituent, i.e., be adequately specific, with accuracy sufficient to meet the data needs in the presence of interferences normally encountered in natural samples; the method should be sufficiently simple and rapid to permit routine use for the examination of large numbers of samples. The reference methods used for manual nutrient measurements are described by Grasshoff (1976). Any changes to the reference methodology should be validated before use for routine work. Apart from manual methods, various automated methods are in use, including different types of continuous flow analysis (CFA, steady-state mode, and peak mode) or flow injection analysis (FIA or Reverse Flow Injection). The analyst has to be aware of the effects of the different analytical conditions in automated analysis which might affect accuracy. Calibration and the blank Stock standard solutions should be prepared separately for each nutrient using analytical grade reagents that can be pre-treated to a precise stochiometric composition, e.g., by drying excess moisture. Reagents containing crystal water should be dried at a sufficiently low temperature in order not to remove the crystal water (the drying temperature is compound dependent). Stock standard solutions containing more than 1 mM are stable for long periods (up to one year refrigerated), but working calibration solutions must be prepared daily and used within hours of preparation. Blank sea water may be prepared from a bulk sample of offshore surface sea water collected in summer, when the nutrients are at low or below-detection concentrations (Kirkwood, 1994). Blank sea water and reagents totally devoid of nutrients are, however, difficult to achieve, especially regarding the content of ammonia. Optimum handling precautions should be taken to minimize the content of nutrients to below approximately 10% of the measuring range. The concentrations of nutrients in the blank and reagents can be assessed by the standard addition method. For ammonia analysis, the salinity of the samples affects the reaction kinetics, mainly due to the buffer effect of marine water which results in a sub-optimum end pH. This effect can give biased results, especially with kinetically dependent automated methods. In the Baltic Sea, the salinity ranges from approximately 0 to 30, and therefore the size of this bias will be variable. This kinetic effect should be checked by standard addition, or by checking the pH of the reagent-sample mixture, which should be in the range between 10.5 and 11. Whenever compensation for this bias is deemed necessary, one of the following methods is suggested: a) If all samples have the same salinity, calibrate using the addition of calibrants to one of the samples. In some situations, low-nutrient sea water can be prepared by aging and filtering natural sea water (as mentioned above). b) Empirical correction in accordance with the measured sample salinity or pH value. For all photometric nutrient measurements, differences in light refraction, caused by 34 differences in the salt concentration, can give rise to shifts in blank/baseline values, especially in light-measuring cells with round windows. This can be compensated by using blanks and calibrants of the same salt concentration as the samples. Particles can give rise to light-scattering effects that result in interferences in all photometric nutrient analyses. This bias can be avoided by measuring the sample before addition of the colour reagent, or by filtration or centrifugation where this does not cause contamination. References Grasshoff, K. 1976. Methods of seawater analysis. Verlag Chemie, Weinheim, New York. ICES. 2004. Chemical measurements in the Baltic Sea: Guidelines on quality assurance. Ed. E. Lysiak-Pastuszak and M. Krysell. ICES Techniques in Marine Environmental Sciences, No. 35. 149 pp. Kirkwood, D. 1994. Nutrients: Practical notes on their determination in seawater. In ICES/HELCOM Workshop on Quality Assurance of Chemical Analytical Procedures for the Baltic Monitoring Programme. Ed. by G. Topping and U. Harms. Baltic Sea Environment Proceedings No. 58: 23–47. Nitrate/Nitrite in seawater (Cadmium reduction method) (Lachat QuickChem method #31-107-04-1-C; http://www.lachatinstruments.com/) Principle Nitrate is quantitatively reduced to nitrite by passage of the sample through a copperized cadmium column. The nitrite (reduced nitrate plus original nitrite) is then determined by diazotizing with sulfanilamide followed by coupling with N-(1-naphthyl) ethylenediamine dihydrochloride. The resulting solution has a magenta color, which is detectable at 520 nm. Procedure Nitrite/nitrate will be determined on autoanalyser/flow injection analyser following the standard colorimetric methods (Grasshoff et al. 1983). Samples containing high concentrations of iron, copper or other metals may give low results. EDTA is added to the buffer to reduce this interference. OrthoPhosphate in seawater (Lachat QuickChem method #31-115-01-1-G; http://www.lachatinstruments.com/) Principle Ammonium molybdate and antimony potassium tartrate react in an acid medium with phosphate to form an antimony-phospho-molybdate complex. This complex is reduced to an intensely blue-colored complex by ascorbic acid. The color produced is proportional to the phosphate concentration in the sample. Though there is a density difference between seawater and reagent water the bias is less than 2%. 35 Though the method is written for seawater and brackish water it is also applicable to nonsaline sample matrixes. The method is calibrated using standards prepared in deionized water. Once calibrated, samples of varying salinities (0 to 35 ppt) may be analyzed. The determination of background absorbance is necessary only for samples, which have color absorbing at 880 nm. Procedure Orthophosphate will be determined manually or on autoanalyser/flow injection analyser following standard colorimetric methods (Grasshoff et al. 1983). Ammonium (Phenolate method) in seawater (Lachat QuickChem Method #10-107-06-1-C; http://www.lachatinstruments.com/) Principle This method is based on the Berthelot reaction. Ammonia reacts with alkaline phenol, then with sodium hypochlorite to form indophenol blue. Sodium nitroprusside is added to enhance sensitivity. The absorbance of the reaction product is measured at 630 nm and is directly proportional to the original ammonia concentration. Note: EDTA is added to the sample in-line in order to prevent precipitation of calcium and magnesium ions. This method is often adapted for use on an autoanalyzer, to allow for automated analysis of multiple samples and replicates. Total N & total P (persulfate digestion) in seawater Source: Qualls (1989) Introduction This digest is applied to water samples to sweep the nitrogen from all N compartments into nitrate and the phosphorus from all P compartments into orthophosphate. The resulting digests are analyzed by manual or automated colorimetry (e.g. Technicon autoanalyzer) for nitrate-N and orthophosphate-P. This method is both safer and more effective than traditional Kjeldahl techniques. It is based on the method of Koroleff (1983) as modified by Qualls (1989). Equipment 1) Chemical autoclave or pressure cooker 2) 13x100 mm glass screw-cap culture tubes with teflon-lined caps. (Tubes are acid-washed in 20% HCl and muffled at 500 degrees C for two hours. Caps are acid-washed in 50% HCl) 3) autoclave-safe test tube racks 4) 100 ml, 200 ml or 500 ml acid-washed volumetric flask for oxidizing reagent (depending on how much reagent is needed) 5) 500 ml acid-washed volumetric flask for 3.75M NaOH stock 6) 1000 ul and 5000 ul automatic pipetters 36 7) weigh boats and clean chemical spatula Reagents 1) fresh deionized H2O 2) low-N potassium peroxydisulfate (e.g. Fisher P282-100) 3) boric acid (e.g. Baker 0084-01) 4) low-N NaOH if stock is needed 5) EPA-certified Nutrient 2 quality control digest standard NaOH stock: Place ~350 ml diH2O in 500 ml volumetric on stir plate. Add 75.0 g NaOH. Stir to dissolve; remove stir bar and bring to volume. Cap with parafilm and invert to mix. Allow to stand ~30 minutes and recheck volume. Sample tube handling: Wash all sample test tubes (70 mL etched glass tubes) with HOT phosphate free soapy water and triplicate flush with dH2O. Acid wash the tubes and triplicate rinse with DI H2O. After drying, store sample tubes capped until day of sample collection. Oxidizing reagent: Place clean volumetric of appropriate size on stir plate; into it rinse in the appropriate amounts of reagent from the table below with diH2O. Bring to about 80% of flask volume with diH2O and stir to dissolve; takes ~15 minutes on stir plate (gentle warming may help.) When dissolved, remove stir bar and bring to final volume with diH2O. Cap with parafilm and invert to mix. Allow to stand ~30 minutes and recheck volume. 100 ml 200 ml 250 ml 500 ml persulfate 5.2 g 10.4 g 13 g 26 g boric acid 3.12 g 6.24 g 7.8 g 15.6 g NaOH stock 10 ml 20 ml 25 ml 50 ml (This reagent may be stored 7 days at room temperature. Crystalizes when refrigerated.) EPA Nutrient-2: 10 ml concentrate from ampoule in 1000 ml diH2O (or 5 ml concentrate in 500 ml.) yields 5.00 mg/liter total nitrogen and 1.50 mg/liter total phosphorus. (Digests for total nitrogen and total phosphorus are checked by digesting and analyzing E.P.A. Nutrient-2 QC solutions formulated to challenge digestion techniques alongside every batch of unknowns) Procedure 1) bring samples to room temperature if chilled or frozen 37 2) make up fresh digest reagent, and NaOH stock if needed 3) obtain acid-washed, muffled digest tubes; label them. 4) for field samples (unknowns) and EPA2 (standard) samples: on first pass through the sample set, pipette 5 ml sample into each labeled digest tube. Cap loosely to exclude dust. on second pass, pipette 1 ml oxidizing reagent into each digest tube. Cap tightly and mix well (invert several times.) 5) For reagent blanks, pipette only 1 ml oxidizing reagent into tube and cap tightly. (take care! Qualls (1989) states: "For low level samples the variability in the reagent blanks determines the limit of detection, not the error associated with the NO3 and PO4 analyses themselves.") 6) Place capped tubes in autoclave, 30 minutes on liquid cycle. (= 30 minutes on "sterilize" in addition to all other cycle segments. If using pressure-cooker field method, time 30 minutes after coming to canning temperature in addition to warmup and cooldown times.) 7) After tubes are cool, add 5 ml diH2O to all reagent blank tubes so that the total volume of liquid in these tubes is the same as in the others. (note, Qualls (1989): "Since distilled or deionized water contains significant N, the dilution water [for the blanks] is added after the digestion.") 8) Analyze digest-tube contents manually or with autoanalyzer using the nitratenitrite and orthophosphate manifolds. Post-analysis calculations 1) Take the mean of the reagent blank determined values. Throw out any that are >2 std. deviations above the mean (for nitrogen in particular this indicates that the tube cap has cracked during the autoclave step and admitted atmospheric N to the tube.) 2) To compensate for color absorption by the digest reagents, subtract the mean reagent blank N and P values from the autoanalyzer determined values for each unknown or EPA2 sample. 3) The effect of diluting the samples by the addition of digest reagents must be reversed, using a dilution factor (df): sample volume + reagent volume df = -------------------------------------initial sample volume In the case of the above procedure, where initial sample volume is 5 ml and reagent volume is 1 ml, 5 ml sample + 1 ml reagent df = ---------------------------------5 ml sample = 1.2 Find the actual value of the undiluted sample by multiplying the determined value (after reagent blank subtraction) by the df, as follows: True analyte concentration = (raw determined value - rblank value)*(df) 38 Comments 1) Successful digests have pH in the range 5 to 8; incomplete digests are ~2. This can be checked with wide-range pH paper. There is no reliable correlation between final digest pH and the yellow color developed in some digests, so the color cannot be used to spot incomplete digests. 2) Instead of using reagent blanks, it is possible to digest the calibration standards (including water blanks, i.e. calibration standards of content zero), the D3 (recalibrant) and ref3 (reference check) and the W (baseline drift correction) cups. Thus with digest reagent in both samples and calibants, the reagent's contribution to total absorbance will be compensated for automatically. (This strategy is of course useable only if all samples in the run are using the same reagent/diluent ratio.) It may be necessary to have different dilutions of the EPA2 QC standards for nitrate and phosphate to get both into the optimum manifold range (e.g. if the PO4 manifold range is 0.2-1 ppm while the expected TP content of EPA2 is 1.5 ppm PO4, the QC digests will be offscale for phosphate unless diluted.) Perform these dilutions before digestion and then use the same reagent/diluent ratio for everything. References D'Elia, C. F., P. A. Steudler, and N. Corwin. 1977. Determination of total nitrogen in aqueous samples using persulfate digestion. Limnol. Oceanogr. 22, 760-764. Koroleff, F. 1983. Simultaneous oxidation of nitrogen and phosphorus compounds by persulfate. p.168-169. In K. Grasshoff, M. Eberhardt, and K. Kremling, eds., Methods of Seawater Analysis. 2nd ed., Verlag Chemie, Weinheimer, FRG. Koroleff, F.1983. Appendix A pp. 131-138. In The biogeochemical properties of dissolved organic matter in a hardwood forest ecosystem: their influence on the retention of nitrogen, phosphorus, and carbon. Ph.D. dissertation, University of Georgia Institute of Ecology, Athens, Georgia, USA. Langner, C. L. and P. F. Hendrix. 1982. Evaluation of a persulfate digestion method for particulate nitrogen and phosphorus. Water Res. 16, 1451-1454. Qualls, R. G. 1989. Determination of total nitrogen and phosphorus in water using persulfate oxidation: a modification for small sample volumes using the method of Particulate matter in seawater Source: Plymouth Marine Laboratory and ICES (2004) Introduction This procedure addresses determination of Total Suspended Solids (TSS; which is also known as Suspended Particulate Matter; SPM = total particulate matter, TPM), Particulate Inorganic Matter (PIM) and Particulate Organic Matter (POM). The particle size of organically bound carbon of particles (POC) generally ranges between 0.45 µm and 300 µm. This includes both living organisms, such as phytoplankton, yeasts, bacteria, and microzooplankton, and detrital particles and 39 aggregates. The production and decomposition of biogenic particles as well as their fractional removal to the deep sea control the distribution of most trace elements in the oceans. Microbial decomposition, desorption, and dissolution of suspended or sinking marine particles can release elements associated with labile (e.g., organic) fractions back to the sea water. On the other hand, particles can scavenge trace elements from the dissolved phase and thereby transport them to sediments. Analysis of the composition and distribution of the particulate fractions in the oceans is therefore required to understand the behaviour and geochemical cycling of both major and trace elements. Required pre-washed, ashed and weighed GF/F 47mm filters, prepared as below, stored in clean petri plates clean forceps 100% acetone in wash bottle for washing off all surfaces that come in contact with filters Freshly distilled water in wash bottle Filtration manifold with filter holders for 47mm filters 0.5M Ammonium formate (31.5 g/l) Dessicator Drying oven Muffle furnace Concentrated HCl in Erlenmeyer or beaker for acid-fuming filters Long (>20cm) forceps for holding filter over acid fumes Fume hood for acid-fuming process 50mm (diameter) Petri plates Filter-preparation: a. To remove fine loose particles of filter, separate and soak in distilled water for > 1h; agitate and rinse 3-4 times in distilled water. b. Partially dry each filter on suction head to remove excess water (this prevents sticking to foil in the next step). c. Place filters individually into foil envelope/fan and oven dry overnight. d. Carefully number each filter on the exposed margin (soft lead pencil or pre-tested pen) and lay out (slightly overlapping) on foil tray, fit a lid and ash in muffle furnace at 450°C for >4h. e. Cool in dessicator; all handling of filters, from this point on, using clean (acetone) forceps only to avoid contamination. f. Remove individually and weigh to 5 places, standardising the time it takes to weigh (filters increase in weight as they take up atmopheric moisture), and store place in numbered petri-slides. Particulate Organic Matter and Total Particulate Matter determination : a. Filter the required volume of homogenised material (see below) b. Rinse filter twice with 10 ml of 0.5M Ammonium formate solution to remove salt and then rinse with distilled water around margin of the filtration cup, having removed filter head (do all of the above with pump running), and when dry, return filter to petrislide. c. Oven dry filters (60°C for 2 days, 40°C for 1 week) and store in dessicator. 40 d. Weigh (from dessicator, to 5 places, as above, preferably with the same balance) for total particulate matter (TPM). e. Ash at 450°C in muffle furnace for > 4h f. Weigh (from dessicator to 5 places, as above, preferably with the same balance) for inorganic particulates (PIM). g. Do all of the above using at least 10 blank filters (prepared and processed as above, but without sample) for each experimental day (changes in weight before and after experimentation are used to correct for changes in balance calibration and/or filter water content). Absolute care in the preparation and processing of these filters as described is essential, for small errors in weight at these stages will significantly bias ratios and other results calculated later. Many experiments have been ruined by lack of attention to the above details! Reference ICES. 2004. Chemical measurements in the Baltic Sea: Guidelines on quality assurance. Ed. E. Lysiak-Pastuszak and M. Krysell. ICES Techniques in Marine Environmental Sciences, No. 35. 149 pp. Particulate organic carbon and nitrogen in seawater Source: Plymouth Marine Laboratory and ICES (2004) Required: 25mm filters prepared as below, stored in acid-washed Petri plates. Access to CHN analyzer Note: it is necessary to remove all organics and keep free from contamination using gloves and covers, etc. The sample should be handled and transferred between containers as little as possible to avoid contamination during the steps between sampling and analysis (see Grasshoff et al., 1999 and ISO, 1999). It is important to obtain a representative sample, which under certain circumstances, e.g., during heavy algal blooms, can be achieved by shaking the water sampler immediately before taking the sub-sample. The homogeneity of the sample may be verified, for example, by separately analysing sub-samples from the upper and lower layers of the bottle. Filter-preparation is as follows: a. Ash at 450°C for >4h, laid out in foil tray with foil cover. b. Cool in dessicator. c. Store in box until required. d. When using: filter material through, then wave filter in HCl vapour for 15 secs to destroy inorganic material (do not rinse filter), then place in numbered acid-washed petri-slide and oven dry at < 40 oC. e. Upon removal from oven, seal petri-slide with cover. 41 Storage of samples Filters containing particulate matter collected for POC analysis should be dried under vacuum for at least one day and stored dry in a desiccator with silica gel or, preferably, stored in a freezer and later dried in a drying oven at 60 °C for 30 min, cooled in a dessicator and weighed prior to analysis. Sample processing The amount of sample (volume of water) required will depend on water quality. Measures of chlorophyll a are very sensitive, so that one only needs enough sample on the filter such that one can see a change in colour. Measures of CHN require about 4 times more sample volume on filters of the same size. However, the measure of POM and TPM requires as much sample as is reasonable on the filter, and certainly more than 2 mg TPM. Especially when filtering natural seawater, this means filtering until the filter is almost blocked. This may only require 500 ml in times of algal bloom or resuspension, but when seston levels are low, may need to filter up to 3 liters per sample (as has been done in eutrophic UK waters). All filtering must be quantitative (i.e. we need to know the initial total volume of each sample, and the separate volumes of that sample filtered for separate determinations of Chl a, CHN and POM/TPM; thus allowing us to calculate the total of each within the sample as a whole). Examples, for mesotrophic waters (e.g. ~ 8 ug/l chl) for TPM, PIM, POM 300ml on 47mm GF/F (rinse with NH4 formate) 2 duplicate filters for CHN 50ml on 25mm GF/F (acid fumes) 2 duplicate filters Therefore, for each sample, require: 2 2 2 for Chl a 10ml on 25mm GF/F (no rinse) 2 duplicate filters 47mm GFFs (washed, ashed, weighed) 25mm GFFs (ashed only) 25mm GFFs (no pretreatment) After filtering, place all CHN and POM/TPM filters in the low temperature (< 40 oC) oven to dry (more than 40 oC will result in loss of lipids!!). Chl a samples (see below) must not be dried; instead, fold in half with material on inner side, and stored in freezer. References Grasshoff, K., Kremling, K., and Ehrhardt, M. (eds.) 1999. Methods of seawater analysis. VCH, Weinheim, New York ICES. 2004. Chemical measurements in the Baltic Sea: Guidelines on quality assurance. Ed. E. Lysiak-Pastuszak and M. Krysell. ICES Techniques in Marine Environmental Sciences, No. 35. 149 pp. 42 ISO. 1999. Water quality – Guidelines for the determination of total organic carbon (TOC) and dissolved organic carbon (DOC). ISO 8245. International Organization for Standardization, Geneva. Particulate organic phosphorus in seawater Source: Marine Biogeochemistry - Practical Course in Biological Oceanography, University of Kiel, Germany, 2005. Organic phosphorus compounds are converted to orthophosphate (i.e. inorganic dissolved phosphate) by cooking with potassium peroxydisulphate. The inorganic dissolved phosphate is then measured colorimetrically, following Grasshoff (1983). During cooking with peroxydisulphate, some chloride is oxidised to chlorine. Because it may interfere with the measurement, this chlorine is then reduced by ascorbic acid (added in surplus, before the addition of the mixed reagent; see below). Interferences The corresponding reaction with silicic acid (up to 200 μm l-1 Si) (see 4.5) does not occur at a pH below 1.0. The reagents are designed for seawater in such a way, that the pH adjusts itself around 1.0. If the samples, already mixed with the reagents, are allowed to stand for more than 30 minutes AsO43- reacts slowly and a heteropoly acid forms. However, the arsenate content in seawater amounts only to about 0.03 μm l-1 As. Hydrogen sulphide in concentrations above 60 μmol l-1 H2S-S (= 2 mg S2- (sulphide)) disturbs the analysis. In such samples the sulphide has to be oxidized with bromine water and the surplus bromine is driven out by airflow prior to analysis. Calibration From the stock solution (10 μmol ml-1 PO43--P) a calibration series of 0, 1 and 2 μmol l-1 PO43- is set up. With a pipette 0.1 or 0.2ml are transfered into a 1 litre measuring flask and filled up to 1 litre with deionized water. For the 0 μmol l-1 PO43- standard only deionized water is used. The calibration series is treated as described in the chapter “Execution of the Determination”. Range: 0 – 10 μmol l-1 PO43--P (corresponding to 0 – 0,35 μmol/35 ml) Precision: ± 0.02 μmol l-1 PO43--P (corresponding ± 0,0007 μmol/35 ml) Procedure A GF/F filter (blank or sample) is transferred to a 60ml Duran glass bottle and 35 ml of deionized water are added to this. Next, 2ml potassium peroxidisulphate solution are added, the bottle is closed and shaken. Samples are placed in an autoclave, or a household pressure cooker filled 2 cm high with deionized water, and cooked for 30 min at 121 C (at 15 psi), then cooled to room temperature. Next, 1 ml of ascorbic acid solution is added to each sample. Shake and wait for 1 min before adding 1 ml of mixed reagent II. 12 ml of this blue colored solution are transferred into centrifuge tubes and centrifuged at high 43 speed for 10 min. Following centrifugation, a sample of the cleared solution is transferred to a cuvette and the absorption is measured at 882 nm against deionized water. Reagents 4.5 M Sulphuric acid: Slowly add 250 ml concentrated H2SO4 (98%) to about 750 ml deionized water and make up to a volume of 1000 ml. (the mixture must be cooled – wear safety glasses!!!!) Ammonium heptamolybdate: Dissolve 9.5 g (NH4)6Mo7O24*4H2O in about 50 ml deionized water and fill up to 100 ml. Store in polyethylene bottles. Potassium antimonyltartrate: Dissolve 3.25 g of potassium antimonyltartrate K(SbO)C4H4O6*0.5H2O in 100 ml deionized water. Mixed-reagent II: Slowly add 125 ml 4.5 M H2SO4 to 47 ml ammonium molybdate solution, then 5.2 ml potassium antimonyltartrate solution and 73 ml deionized water (total volume of 250ml) Potassium peroxydisulphate: 30ml of 4.5 M sulphuric acid are given to 200 ml deionized water. Then add 10 g of potassium peroxydisulphate (reagent remains stable during about 1 week) Ascorbic acid: Dissolve 14 g ascorbic acid (C6H8O6) in 200 ml deionized water (at 4°C durable for 30 days) Stock solution: Dissolve 1.361 g dried potassium hydrogen phosphate (KH2PO4) in deionized water and fill up to 1 000 ml (= 10 μmol ml-1) Calculation of concentrations with a calibration series Calibration series 0, 1 and 2 μmol l-1 (= 0,035 und 0,070 μmol/35 ml) 3–fold determination Examples Ø EBl ~ 0.015 absorption of reagent blank Ø ESt1 ~ 0.115 absorption of Standard 0,035 μmol / 35ml PO43—P Ø ESt2 ~ 0.215 absorption of standard 0,070 μmol / 35 ml PO43—P Chlorophyll a determination Source: Plymouth Marine Laboratory Required: Clean water sampler, e.g. Niskin bottle Filtration system with 25mm or 47mm filter holders 50mm Petri plates 47mm or 25mm GF/F filters Filter forceps (flat) Dispensor for 90% buffered acetone solution Homogenizer for GF/F filters 90% buffered (MgCO3) acetone Wash bottle with 90% buffered acetone Glass bottle for acetone waste Centrifuge tubes 44 Spatula Lint-free tissues (e.g. Kimwipes) for cleaning cuvettes HCl 1 N Sampling 1. Filter water sample (e.g. 1000 ml) through 47mm (or 25mm) GF/F filter at relatively low vacuum pressure (<250 mm Hg). Smaller volumes may be adequate, e.g. 10 to 100ml, depending upon predicted chlorophyll concentration. 2. Fold filter in half (sample side inwards) place in labeled petri-dish and freeze until required. Chlorophyll extraction 1. For natural seawater and more delicate species (e.g Phaeocystis; Isochrysis ) : place filter in bottom of centrifuge tube, add 10 ml 90% acetone then refrigerate (4°C) for at least 16, but not more than 24 hours 2. Gently invert tube several times before decanting off into fluorometer tube or spectrophotometer cuvette (see further on for fluorometer or spectrophotometer protocols). 2. For more robust species: place filter at the bottom of homogeniser tube, add 2 - 4 ml taken from a measured volume of 10 ml of 90% Acetone. Homogenise at low speed for approximately 20 seconds. a) Pour into centrifuge tube. Scrape out any remaining bits of filter with spatula and add these to centrifuge tube. Rinse homogeniser tube with remaining 6-8 ml 90% acetone and add this to the centrifuge tube,. Cap centrifuge tube and store in refrigerator for between 16 and 24 hours. Clean off homogeniser head and tube between samples. b) Remove samples from fridge and centrifuge for 5 mins at 3,000 to 4,000 rpm (if centrifuge has cooling facility then set this at 5 - 10 °C). Note: Throughout storage and analysis of samples, exposure to light (especially strong sunlight) should be avoided or at least minimised. Fluorometric determination3 If possible, use a fluorometer (e.g. Turner Designs TD700) that does not require the acid addition to. If not possible, measure sample fluorescence before and after addition of 2 drops (~100 ul) of 1N HCl to the cuvette. Chlorophyll a (µg litre) = (FA (RB - RA)*Ve (ml))/Vf (ml) FA = calibration factor of fluorometer (calculated as µg litre) RB = fluorescence reading before addition of 1N HCL RA = fluorescence reading after addition 1N HCL 2 Note that time filters left in fridge MUST be standardised; assumption is that up to 95% of the chlorophyll is released in the first 4 hours, but the remainder is released very slowly, expect c. 100% recovery after 24 hours. If experiment involves making comparisons between robust/non-robust species then ALL filters must be homogenised. 3 See Strickland J.D.Hand Parsons T.R. A Practical Handbook of Seawater Analysis. Fisheries Research Board of Canada (Ottawa 1968) Bulletin 167. 45 Ve = Vol.of acetone extract (ml) Vf = Vol. of sample filtered (ml) Spectrophotometric determination Use wavelength 750nm to measure turbidity and 663nm for chl a, using bandwidth of 1 nm. Ensure digital display knob is set on absorbance! Measuring sequence: read absorbance at 750nm (E750), 663nm (E663), 750nm; add 2 drops HCl (leave 30 sec), read at 663nm (E663a) Rinse cuvette with 90% acetone between samples. Chlorophyll a (µg litre) =( 26.7((E663 - E750) - (E663a - E750)) x vol.extract (ml)) / (vol filtered (litres) x path length (cm) ) E663 = abs. prior to HCL E663a = abs. after addition of HCL Quality Assurance Sampling QA • Keep the samples cool and in the dark. • For chlorophyll a it is recommended that the sample is filtered immediately after sampling or, at least, as soon as possible thereafter to avoid deposition of cells. If storage is unavoidable, the filters should be deep frozen (< -20 oC). Spectrophotometric or fluorometric chlorophyll a analysis QA • The analysis should follow ISO 10260; departure from this has to be documented, and evidence of comparability of the data provided. • The samples/filters and the chlorophyll a extracts should be handled in subdued light. • Avoid evaporation of the extraction solvent during extraction and measurement procedures. • The measurements should be done immediately after clearing the extracts; the preference is for equipment for measuring the whole spectrum (800–350 nm) for easier checking of shifting of the chlorophyll peaks. • Validate the spectrophotometer and the fluorometer at least once a year, or when changes of the equipment are required. • Calibrate the equipment with a certified reference material, if possible; use control charts. Reference Strickland J.D.Hand Parsons T.R. 1968. A Practical Handbook of Seawater Analysis. Fisheries Research Board of Canada (Ottawa 1968) Bulletin 167. 46 Bioassay studies to assess nutrient levels around aquaculture Source: University of Crete (modification of Dalsgaard (2006) protocol) Background Release of nutrients from fish farms is traditionally monitored by analyzing dissolved nutrients in the waters around the fish cages. Two major drawbacks appear with this approach: the release of nutrients varies diurnally suggesting a sampling around the clock for documenting this release, which would lead to a large number of samples and a high cost for monitoring; the nutrients lost from fish cages are diluted in large volumes of water rendering the documenting of their small increase in concentration difficult by standard analytical techniques. The use of bioassays can overcome the above mentioned problems as it integrates the effects of the aquaculture over time and responds to all bio available nutrients, organic or inorganic. The approach of bioassay is simply to expose phytoplankton or macroalgae to the waters next to the aquaculture facility for a period of 3-6 days and measure the growth of these primary producers as a function of distance from the facility and thus describe the horizontal extent of the effects of nutrient release. Required Surface water 25 m mesh sieve/plankton net Spectra/por 1 dialysis membrane Collected from the control station of the site regenerated cellulose with a molecular weight cut-off of 6-8 kilo Dalton. The flat width of the membrane is 10 cm Plastic coated metal wire Nylon mesh bags to protect dialysis bags Metal rod/plate Rope Weights (100g) Buoys Anchors Bioassay setup 1. Cut the dialysis membrane into pieces of 30 cm length 2. Soak the pieces of the membrane in distilled water until they become soft (1-2h) 3. Close one end of them with a plastic coated metal wire 4. Filter surface water from the control station of the site through a 25 m mesh sieve to remove larger grazers 5. Dispense the filtered water into the dialysis bags (ca 600 ml/dialysis bag) 6. Close the dialysis bags with plastic coated metal wire 7. Five replicate bags will be needed for each station (ca 6.4 cm diameter, ca 20 cm long) 8. Place each bag in a nylon mesh bag for hanging and protecting the dialysis bags 9. Using a rope and a metal plate hold together 5 replicate bags (Fig. 1) 10. A buoy on top and an anchor at the bottom will be needed for holding each bioassay setup ca 1.5 m below the surface at each site (Fig. 1) 11. At each station one bioassay setup should be incubated for 5 days. 47 buoy metal plate dialysis bags weight (optional) mooring weight Figure 1. Bioassay setup. Designed by the IMBC team participating in MedVeg project (based on Dalsgaard 2006). Analysis 1. Biomass of phytoplankton is measured as chlorophyll-a concentrations according to standard protocols used for water column measurements 2. Before the analysis the volume of each dialysis bag must be recorded References Dalsgaard, T. and Krause-Jensen, D. 2006. Monitoring nutrient release from fish farms with macroalgal and phytoplankton bioassays. Aquaculture 256:302-310. Mura MP, Agusti S (1996) Growth rates of diatoms from coastal Antarctic waters estimated by in situ dialysis incubation. Marine Ecology Progress Series 144: 237245 Mura MP, Agusti S, delGiorgio PA, Gasol JM, Vaque D, Duarte CM (1996) Losscontrolled phytoplankton production in nutrient-poor littoral waters of the NW Mediterranean: In situ experimental evidence. Marine Ecology Progress Series 130: 213-219 48