General Requirements for the Submission of IDE Application
advertisement

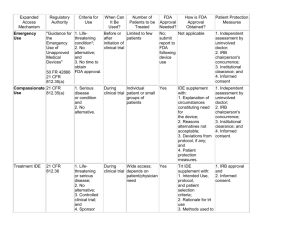
General Requirements for the Submission of IDE Applications for Clinical Research Studies I. General Applicability of the FDA Regulations Governing the Submission of IDE Applications Clinical investigations involving the use of a FDA-approved or unapproved device are generally exempt from the requirement for submission of an IDE application if the research is not being conducted for the purpose of evaluating the safety and effectiveness of the device for a specific clinical indication1 II. Clinical Investigations to Evaluate the Safety and/or Effectiveness of Devices Not Currently Approved for Commercial Marketing by the U.S. Food and Drug Administration (FDA)2 A. Significant risk device studies. The submission of an IDE application is required if the reviewing institutional review board (IRB) determines that the non-approved device, or its proposed use in the research study, constitutes a “significant risk” to the research subjects. 1. A “significant risk device study” is defined by FDA regulations3 as “a study of a device that presents a potential for serious risk to the health, safety, or welfare of a subject and (1) is an implant4; or (2) is used in supporting or sustaining human life; or (3) is of substantial importance in diagnosing, curing, mitigating or treating disease, or otherwise prevents impairment of human health; or (4) otherwise presents a potential for serious risk to the health, safety, or welfare of a subject.” 2. A “non-significant risk device study” is a study of a device that does not meet the FDA’s definition for a “significant risk device study.” B. Studies of diagnostic devices. 21 CFR 812.2 Applicability – “General. The IDE regulations apply to all clinical investigations of devices to determine safety and effectiveness.” 2 I.e., devices for which the FDA has not previously granted Pre-Market Approval (PMA) or 510k approval. 3 21 CFR 812.3(m) 4 An “implant” is defined by the FDA as “a device that is placed into a surgically or naturally formed cavity of the human body and is intended to remain there for a period of 30 days or more”. The FDA may determine that devices placed into human subjects for shorter periods of time are also implants. 1 1 Clinical investigations of diagnostic devices are exempt from the requirement for the submission of an IDE application if the testing: 1. Is non-invasive5; 2. Does not require an invasive sampling procedure that presents significant risk; 3. Does not by design or intention introduce energy into a subject; and 4. Is not used as a diagnostic procedure without confirmation by another medically established diagnostic product or procedure. III. Clinical Investigations to Evaluate the Safety and/or Effectiveness of Devices Currently Approved for General Marketing by the U.S. Food and Drug Administration (FDA) A. Clinical investigations to evaluate (compare) the safety and/or effectiveness of a FDA-approved device for the clinical indication(s) currently specified in the FDA-approved product labeling The submission of an IDE application is not required for clinical investigations (e.g., product comparison studies) directed at evaluating the safety and/or effectiveness of an approved device for a clinical indication that appears currently in the FDA-approved product labeling for that device. B. Clinical investigations to evaluate the safety and/or effectiveness of a FDA-approved device for a clinical indication that is not currently specified in the FDA-approved product labeling (i.e., an “off-label” indication) The submission of an IDE application is required if the reviewing IRB determines that the device, or its proposed use in the research study, constitutes a “significant risk” to the research subjects. 1. A “significant risk device study” is defined by FDA regulations6 as “a study of a device that presents a potential for serious risk to the health, 21 CFR 812.3 (k) “Noninvasive” means a device or procedure “that does not by design or intention: (1) penetrate or pierce the skin or mucous membranes of the body, the ocular cavity, or the urethra; or (2) enter the ear beyond the external auditory canal, the nose beyond the nares, the mouth beyond the pharynx, the anal canal beyond the rectum, or the vagina beyond the cervical os.” Blood sampling that involves simple venipuncture is considered noninvasive, and the use of surplus samples of body fluids or tissues that are left over from samples taken for noninvestigational purposes are also considered noninvasive. 6 21 CFR 812.3(m) 5 2 safety, or welfare of a subject and (1) is an implant7; or (2) is used in supporting or sustaining human life; or (3) is of substantial importance in diagnosing, curing, mitigating or treating disease, or otherwise prevents impairment of human health; or (4) otherwise presents a potential for serious risk to the health, safety, or welfare of a subject.” 2. A “non-significant risk device study” is a study of a device that does not meet the FDA’s definition for a “significant risk device study.” An “implant” is defined by the FDA as “a device that is placed into a surgically or naturally formed cavity of the human body and is intended to remain there for a period of 30 days or more”. The FDA may determine that devices placed into human subjects for shorter periods of time are also implants. 7 3