Introduction to Microarray Analysis
advertisement

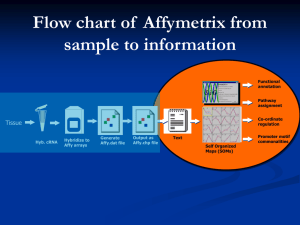
Introduction to Microarray Analysis Uma Chandran PhD, MSIS Department of Biomedical Informatics chandran@pitt.du 412-648-9326 10/17/12 What is a microarray Probes on surface Arrays can detect Glass beads, chips, slides mRNA microRNA Methylation SNP High throughput 10000s of specific probes Measure global gene expression, SNP calls, LOH, amplification, methylation etc Questions that can be asked Can measure global changes Which mRNAs are high in disease versus normal, i.e, out of the 1000s of mRNAs expressed in the cell at any time Are there single nucleotide polymorphism that are markers for a disease – many studies on for example, autism, schizophrenia Are there methylation changes in disease versus normal ARRAY DESIGN Affymetrix Insert oligo slide Probes are synthesized on a chip Probes are oligonculeotides of a specified length Generally 25 mers At each x, y location a particular oligonucleotide is synthesized in 1000s of copies at that location Affymetrix • • • Feature: a location on the array with a particular oligonucleotide sequence Oligonucleotides are synthesized using a photolithographic manufacturing • process The oligo on the chip is called the probe and RNA (or DNA) that it hybridizes to is called the target Affy array design Probe set Affymetrix Probe design Multiple probe sets/gene Probe sets are selected based on GenBank dbEST RefSeq Bioinformatics approaches Design at the time of chip design However, this may be incorrect as genome builds update Affymetrix data Annotation The probe set id and sequence are contained in reference files This id never changes However, annotations change with genome builds Many software tools to annotate Some involve new BLAST of the sequences Mask out probe sets Affymetrix Chips for Human HGU95, HGU133A, B, HGU133 set Very low ~ 10 units 20K + Cannot compare genes within chips Mouse Rat Chimpanzee Plants Many other species Dynamic range 54K probe sets on the HGU133, 30+ to known genes and ESTs Control probes like GAPDH Spike in bacterial probes For example, a transcript that is expressed at 500 units may not be more abundant than one that is expressed at 200 units This is due to probe binding affinities etc However, can compare the same probe across multiple chips Difficulty in probe design makes it difficult to compare from one version to another Affymetrix workflow from: http://wwwnmr.cabm.rutgers.edu/academics/biochem694/reading/DalmaWeiszhau sz_2006.pdf Illumina Illumina Each bead has one type of oligo and thousands of these oligos/bead Bead is deposited on wells in glass slides. The beads are decoded by a step by proprietary technology Microarray analysis objectives Data Preprocessing Data Analysis Analysis questions Treatment Class Comparison Class Discovery Expression - Which genes/miRs are up or down in tumors v normal, untreated v treated SNP – Which regions are amplified or deleted Within the tumor samples, are there subgroups that have a specific expression profile? SNP – amplification or deletion common to subgroups? Class prediction, pathway analysis etc Integrative analysis Proteomic and genomic SNP and expression Methylation and expression Normal Challenges in microarray analysis Different platforms Ilumina, Affymetrix, Agilent…. Many file types, many data formats Need to learn platform dependent methods and software required Analysis How to get started? Which methods? Which software? Many freely available tools. Some commercial Analysis software and methods will depend on platform. SNP analysis is different from expression Software used may be very specific to SNP For example, Excel cannot open large SNP files How to interpret results Public databases Many sources for public data – labs, consortia, government Publications require that data files including raw files be made public GEO – http://www.ncbi.nlm.nih.gov/ geo/ Array Express http://www.ebi.ac.uk/arraye xpress/#ae-main[0] Hands on #1 Look at GEO Search Data Set with the term Exercise Exercise Heart Human Identify Platform by clicking on GSE record Try restricting by platform such as Affymetrix or Illumina Affy data Normalization method Signal value Probe set Id Total probesets Raw files Data pre-processing Affy produces many files - .dat, .cel, .chp etc Process these to produce data that can be opened in excel or .txt Illumina produces different file types Data Preprocessing Objective Convert image of thousands of signals to a a signal value for each gene or probe set Multiple step Image analysis Background and noise subtraction Normalization Summarized expression value for a probe set or gene Gene 1 Gene 2 Gene 3 . Gene10000 100 150 75 500 Data Pre-processing Go from .DAT file to feature quantification The first step where .DAT file is aligned to a grid and the features are quantified is usually performed by Affy’s proprietary algorithm .DAT .CEL file .CEL file contains the feature quantifications .CEL file still has probes spread over the chip Values still need to be summarized to probe set level; for example 90525_at = 250 units 250 Data Pre-processing – Step 1 Image processing Usually done using proprietary software Affy: convert .dat file to .cel file May perform noise subtraction, background Illumina: Bead Studio software to convert bead level data to next level of data Data Preprocessing – Step 2 Normalization Bring all the experiments up to the same scale Multi-step process depending on technology Summarized expression value for a probe set or gene Affy: .cel to .chp; need .cdf file which describes the file layout Ilumina: normalization option and background subtraction option using Bead Studio Gene 1 Gene 2 Gene 3 . Gene10000 100 150 75 500 .CEL +.CDF to .CHP In going from .CEL to .CHP file to generate signal values, the multiple probes within a probe set are “averaged” to produce a single value for that gene/transcript Normalization Corrects for variation in hybridization etc Important for all high throughput platforms Assumption that no global change in gene expression Without normalization Treated Intensity value for gene will Gene 1 100 be lower on Chip B Gene 2 150 Many genes will appear to Gene 3 75 be downregulated when in . reality they are not Gene10000 500 Control 50 75 32 250 How to normalize? Many methods – Affy MAS5.0 Median scaling – median intensity for all chips should be the same Known genes, house keeping, invariant genes Quantile - RMA Normalization method may differ depending on platform Illumina – cubic spline Affymetrix Which method to choose? Choose method .cel to .chp file Know the biology After normalization from .cel .chp file .txt file A Before After 100 200 B 50 (down) 200 (no change) Normalization Affy data Normalization method Signal value Probe set Id Total probesets Raw files Workflows normalization Affy .dat file > .cel file > .chp file > .txt file cdf file Affy software needed for .dat > cel The rest of the steps can be carried out by other tools Illumina Through Bead Studio Bkg subtraction > normalization with various options > background normalization > .txt file Need bead studio to carry out these steps and raw files not necessarily given Illumina Does not have .DAT, .CEL, .CDF and .CHP files There is no chip definition or chip layout as in Affy However, the identity of each bead has to be decoded vial proprietary software Illumina Data preprocessing Signal normalization Raw files are .txt files Probe id Affy v Illumina Affy 25mer Probe synthesized on chips Multiple probes/probeset May have multiple probes/transcript .dat, .cel, .cdf, .chp file types Normalization methods such as quantile Txt output can be used for downstream data analysis Annotations can be updated Illumina Longer oligo Bead technology Single probe May have multiple probes/transcript Image file processed by Bead Studio Several normalization methods Txt output can be used for downstream data analysis Annotations can be updated Hands on #2 -Data analysis Import data into BRB Which files to import .cel file if performing normalization through BRB Or mport already normalized file as .txt file for further analysis Steps in analysis - Import Affy Import all files into Affy tools such as Expression console Normalize and generate signal values using Affy MAS5.0 Assess QC using GAPDH, B-actin and control probes for spike in and hybridization Then, import into other tools such as BRB for analysis Illumina Depending on background subtraction/normalization, may have generated negative values Check QC metrics, such as did chip pass? Remove negative values Import into tools such as BRB Step in Data analysis – Normalization Import raw data into a tool Has data been normalized? After normalization, check distribution If not, which method to use? What is available for a particular platform If not available in tools, is R code or package available Are there any batch effects? Is the data log transformed? If not, should you log transform? When? After or before normalization? Are there missing or negative values in data? What should be done? Impute? Remove rows Steps in Data analysis – update Annotations Very important step Annotations updated Annotations provided may often be incorrect Multiple probe sets for each gene BRB – Array tools Website Excel plug in; R and fortran Import, choose correct format For Affy: .cel files Or directly from processed files Process using GCRMA or MAS5.0 Attaches annotation Create experiment labels Class Discovery Objective? Can data tell us which classes are similar? Are there subgroups? Do T-ALL, T-LL, B-ALL fall into distinct groups? Methods Hierarchical clustering K-means, SOM etc These are Unsupervised Methods Class Ids are not known to the algorithm For example, does not know which one is cancer or non cancer Do the expression values differentiate, does it discover new classes Multidimensional scaling MDS Class comparison – differential expression analysis What genes are up regulated between control and test or multiple test conditions Normal v tumor Treated v untreated Fold change Not sufficient, need statistics Statistics t test, non-parametric, fdr, Class comparison Many analysis methods May produce different results Different underlying statistics and methods t test t test with permutations SAM Emperical bayesian Depends on underlying assumptions about data High throughput data with many rows and few samples What is the distribution Variance from gene to gene Save raw data files to try different methods and compare results Fold change does not take variation into account low variability medium variability high variability Modified from madB http://nciarray.nci.nih.gov/ Differentially expressed gene Differentially expressed gene. A low-reliable estimate Differentially expressed gene. Powerful and exact statistical tests must be used Hypothesis Testing Normal Tumor d mean1 mean2 Null hypothesis Alternative hypotheses Statistical power t test Test hypothesis that the two means are not statistically different Adding “confidence” to the fold change value Mean Standard deviation Sample size Calculates statistic You choose cutoff or threshold Give me gene list at a cutoff of p <0.05 95% confidence that the mean for that gene between control are treated are different Experimental Design – Very important!!! Sample size How many samples in test and control Will depend on many factors such as whether tissue culture or tissue sample Power analysis Replicates Technical v biological Biological replicates is more important for more heterogenous samples Need replicates for statistical analysis To pool or not to pool Sample acquistion or extraction Depends on objective Laser captered or gross dissected All experimental steps from sample acquisition to hybridization Microarray experiments are very expensive. So, plan experiments carefully t tests Results might look like At a p<0.05, there are 300 genes up and 200 genes downregulated 95% confidence that the means of these genes in the two groups is different At a p < 0.05, x genes up and y genes down with a fold change of at least 3.0 Multiple comparison Microarrays have multiple comparison problem p <= 0.05 says that 95% confidence means are different; therefore 5% due to chance 5% of 10000 is 500 500 genes are picked up by chance Suppose t tests selects 1000 genes at a p of 0.05 500/1000 ;Approximately 50% of the genes will be false Very high false discovery rate; need more confidence How to correct? Correction for multiple comparison p value and a corrected p value Corrections for multiple comparisons Involve corrections to the p value so that the actual p value is higher Bonferroni Benjamin-Hochberg Significance Analysis of Microarrays Tusher et al. at Stanford Hands on BRB Class comparison Choose comparison Which tests are available? P value cutoff How is multiple correction testing being done? Stringent p value, fdr How is the output reported? Can you figure out how many genes are regulated at different p values and different cutoffs How to interpret results Look at gene lists generated by our analysis v those generated in the paper BRB – Class Comparison Output folder Check the .html file Look at results P value Fold change Annotation Click on annotation Cut and paste save into Excel Issues Annotation Multiple probe sets for a gene Annotation files will get updated Which one is correct? Where does it map? How to report the genes? How to compare between platforms Different chips within same platform Biological annotation Difficult to interpret experimental results 350 4500 201120_s_at progesterone receptor membrane component 1 PGRMC1 4000 300 204253_s_at vitamin D (1,25dihydroxyvitamin D3) receptor VDR 250 200 204254_s_at vitamin D (1,25dihydroxyvitamin D3) receptor VDR 150 204255_s_at vitamin D (1,25dihydroxyvitamin D3) receptor VDR 213692_s_at Vitamin D (1,25dihydroxyvitamin D3) receptor VDR 100 50 201121_s_at progesterone receptor membrane component 1 PGRMC1 3500 3000 201701_s_at progesterone receptor membrane component 2 PGRMC2 2500 208305_at progesterone receptor PGR 2000 1500 213227_at progesterone receptor membrane component 2 PGRMC2 1000 228554_at progesterone receptor PGR 500 0 0 1 10 19 28 37 46 55 64 73 82 91 100 109 118 127 136 1 10 19 28 37 46 55 64 73 82 91 100 109 118 127 136 200 100 0 Unlogged Expression value 300 Which probe/probe set is correctly aligned to the gene? 205225_at 211233_x_at 211234_x_at 211235_s_at 211627_x_at Affymetrix probeset 215551_at 215552_s_at 217163_at 217190_x_at Probe set errors Types of Probe Error Cross Hybridization Mismatched Probe Intron Probe SNPs ESR1 probes in UCSC genome browser How to manipulate Gene lists Create gene lists Venn Diagram Can be done even though study done on different platforms Compare MAS and RMA Venn Diagram Compare B-ALL v T-LL and T-LL v B-ALL Venn Diagram http://www.pangloss.com/seidel/Protocols/venn.cgi http://ncrr.pnl.gov/software/VennDiagramPlotter.stm Conclusion Other analysis Class prediction Gene list from class comparison can be used in pathway analysis HSLS pathway workshops on Ingenuity, DAVID, Pathway Architect Future: Integrate expression data with other data such as snp or microRNA GEO has some data analysis features ESR1 probes in UCSC genome browser Next Gen Sequencing Directly sequence DNA to determine SNP CN Expression, mRNA, microRNA Protein binding sites Methylation Initial steps depend not on hybridization but also on base pairing or complementarity and DNA synthesis Data analysis extremely challenging Next Gen Sequencing Applications Sequence varation – WGS, Exome Seq Structural rearrangements – WGS, Exome Seq Copy number – WGS, Exome Seq Epigenetic changes such as methylation – Methyl Seq DNA – protein binding – CHIP Seq mRNA expression – RNA Seq Next Gen Sequencing Read mapping Alignment Denovo assembly Mapping to reference genome Based on complementarity of a given 35 nucleotide to the entire genome Computationally intensive Million of 35 bp reads has to search for alignment against the reference and align spefically to a given regions Large file sizes Sequence files in the TB Aligned file BAM files Several hundred GB Reference genome Sequence variation Analysis pipeline- CHIP-Seq