Inheritance patterns:
Monogenic (Mendelian) Inheritance
Polygenic and Multifactorial Inheritance
Mitochondrial Inheritance
Inheritance patterns
Inheritance patterns trace the transmission of
genetically encoded traits, conditions or diseases to
the offsprings.
There are several modes of inheritance:
Single Gene or Mendelian
Polygenic and Multifactorial
Mitochondrial
Single Gene Inheritance
Genetic conditions caused by a mutation in a single gene follow predictable patterns of
inheritance within families. Single gene inheritance is also referred to as Mendelian
inheritance as they follow transmission patterns he observed in his research on peas.
There are 3 types of Mendelian inheritance patterns:
1.
Autosomal: the gene responsible for the phenotype is located on one of the 22 pairs
of autosomes (non-sex determining chromosomes).
2.
X-linked: the gene that encodes for the trait is located on the X chromosome.
3.
Y-linked (holandric): the gene that encodes for the trait is located on the Y
chromosome
Dominant: conditions that are manifest in heterozygotes (individuals with just one
copy of the mutant allele).
Recessive: conditions are only manifest in individuals who have two copies of the
mutant allele (are homozygous).
Autosomal dominant (AD)
Dominant conditions are expressed in individuals
who have just one copy of the mutant allele.
The pedigree on the right illustrates the transmission
of an autosomal dominant trait.
Affected males and females have an equal probability
of passing on the trait to offspring.
Affected individual’s have one normal copy of the
gene and one mutant copy of the gene, thus each
offspring has a 50% chance on inheriting the mutant
allele.
As shown in this pedigree, approximately half of the
children of affected parents inherit the condition and
half do not.
AD – Incomplete penetrance
A typical pedigree from a
family with a mutation in the
BRCA1 gene.
Fathers can be carriers and
pass the mutation onto
offspring.
Not all people who inherit
the mutation develop the
disease, thus patterns of
transmission are not always
obvious.
Autosomal dominant (AD)
Huntington Disease
Myotonic muscular dystrophy
Achondroplasia (short-limbed dwarfism)
Polycystic kidney disease (ADPKD)
Brachydactyly
Polydactily
Syndactyly
Adactyly
Osteogenesis imperfecta
Gout
Familial hypercholesterolemia
Hypercalcemia (familial)
Marfan syndrome
Familial Polycystic ovary syndrome (PCOS)
Neurofibromatosis
Huntington Disease
Huntington's disease (HD) is a neurodegenerative genetic disorder that affects muscle
coordination and leads to cognitive decline and psychiatric problems. It typically
becomes noticeable in mid-adult life. HD is the most common genetic cause of abnormal
involuntary writhing movements called chorea, which is why the disease used to be
called Huntington's chorea.
The Huntingtin gene (HTT=HD=IT15) on 4p16.3 provides the genetic information for a
protein that is also called "huntingtin". Expansion of a CAG triplet repeat stretch
within the Huntingtin gene results in a different (mutant) form of the protein, which
gradually damages cells in the brain, through mechanisms that are not fully understood.
The genetic basis of HD was discovered in 1993 by an international collaborative effort
spearheaded by the Hereditary Disease Foundation.
Huntington Disease
Increases in the number of repeats (and hence earlier age of onset and severity of
disease) in successive generations is known as genetic anticipation. Instability is
greater in spermatogenesis than oogenesis;
Individuals with more than sixty repeats often develop the disease before age 20, while
those with fewer than 40 repeats may not ever develop noticeable symptoms;
Life expectancy in HD is generally around 20 years following the onset of visible
symptoms;
Most life-threatening complications result from muscle coordination and, to a lesser
extent, behavioral changes induced by declining cognitive function.
The largest risk is pneumonia, which causes death in one third of those with HD. As the
ability to synchronize movements deteriorates, difficulty clearing the lungs and an
increased risk of aspirating food or drink both increase the risk of contracting
pneumonia. The second greatest risk is heart disease, which causes almost a quarter of
fatalities of those with HD.[
Huntington Disease
Recommended (highly) to see what Huntington is all about
An excellent French documentary (subtitled in English) about a family carrying such a
genetic “burden”, including aspects of their life and expectancies
As a reminder, the disease has a complete penetrance (100%) make the disease, usually
after 35-40 years of age, and transmit it to their progenitors
http://www.youtube.com/watch?v=0qOdGvoOXI0 (it takes 1 hour and a half)
Other AD conditions
Myotonic muscular dystrophy (dystrophia myotonica, myotonia atrophica) is a
chronic, slowly progressing, highly variable, inherited multisystemic disease. It is
characterized by wasting of the muscles (muscular dystrophy), cataracts, heart
conduction defects, endocrine changes, and myotonia.
Achondroplasia is a common cause of dwarfism. It occurs as a sporadic mutation in
approximately 75% of cases (associated with advanced paternal age) or may be inherited
as an autosomal dominant genetic disorder. People with achondroplasia have short
stature, with an average adult height of 131 centimeters for males and 123 centimeters for
females. Achondroplastic adults are known to be as short as 62.8 cm.
Polycystic kidney disease (PKD or PCKD, also known as polycystic kidney syndrome)
is a cystic genetic disorder of the kidneys. There are two types of PKD: autosomal
dominant polycystic kidney disease (ADPKD) and the less-common autosomal recessive
polycystic kidney disease (ARPKD). Polycystic kidney disease is one of the most
common life-threatening genetic diseases, affecting an estimated 12.5 million people
worldwide.
Other AD conditions
Brachydactyly (short fingers/toes)
Polydactily (extra fingers/toes)
Syndactyly (two or more digits are fused together)
Adactyly (congenital absence of fingers and/or toes)
Osteogenesis imperfecta types I-V (OI and sometimes known as brittle
bone disease, or "Lobstein syndrome") is a congenital bone disorder. People
with OI are born with defective connective tissue, or without the ability to make
it, usually because of a deficiency of Type-I collagen. As a genetic disorder, OI
has historically been viewed as an autosomal dominant disorder of type I
collagen. In the past several years, there has been the identification of
autosomal recessive forms. Most people with OI receive it from a parent but in
35% of cases it is an individual (de novo or "sporadic") mutation. There are eight
different types of OI, Type I being the most common, though the symptoms
vary from person to person.
Osteogenesis imperfecta
Other AD conditions
Gout (also known as podagra when it involves the big toe). is a medical condition
usually characterized by recurrent attacks of acute inflammatory arthritis—a red,
tender, hot, swollen joint. The metatarsal-phalangeal joint at the base of the big toe is
the most commonly affected (approximately 50% of cases). However, it may also present
as tophi, kidney stones, or urate nephropathy. It is caused by elevated levels of uric acid
in the blood. The uric acid crystallizes, and the crystals deposit in joints, tendons, and
surrounding tissues. The occurrence of gout is partly genetic, contributing to about 60%
of variability in uric acid level.
Familial hypercholesterolemia (abbreviated FH) is a genetic disorder characterized
by high cholesterol levels, specifically very high levels of low-density lipoprotein (LDL,
"bad cholesterol"), in the blood and early cardiovascular disease. Many patients have
mutations in the LDLR gene that encodes the LDL receptor protein, which normally
removes LDL from the circulation, or apolipoprotein B (ApoB), which is the part of LDL
that binds with the receptor; mutations in other genes are rare. Patients who have one
abnormal copy (are heterozygous) of the LDLR gene may have premature cardiovascular
disease at the age of 30 to 40. Having two abnormal copies (being homozygous) may
cause severe cardiovascular disease in childhood. Heterozygous FH is a common genetic
disorder, inherited in an autosomal dominant pattern, occurring in 1:500 people in most
countries; homozygous FH is much rarer, occurring in 1 in a million births.
Other AD conditions
Hypercalcemia - Familial hypocalciuric hypercalcemia is a condition that
can cause hypercalcemia, a serum calcium level typically above 10.2 mg/dL. It is
also known as familial benign hypocalciuric hypercalcemia (FBHH) where
there is usually a family history of hypercalcemia which is mild, a urine calcium
to creatinine ratio <0.01, and urine calcium <200 mg/day.
Familial Polycystic ovary syndrome (PCOS) is one of the most common
female endocrine disorders. PCOS is a complex, heterogeneous disorder of
uncertain etiology, but there is strong evidence that it can to a large degree be
classified as a genetic disease. PCOS produces symptoms in approximately 5%
to 10% of women of reproductive age (12–45 years old). It is thought to be one of
the leading causes of female subfertility and the most frequent endocrine
problem in women of reproductive age. The genetic component appears to be
inherited in an autosomal dominant fashion with high genetic penetrance but
variable expressivity in females; this means that each child has a 50% chance of
inheriting the predisposing genetic variant(s) from a parent, and if a daughter
receives the variant(s), then the daughter will have the disease to some extent.
Other AD conditions
Marfan syndrome (also called Marfan's syndrome) is a genetic disorder of
the connective tissue. People with Marfan tend to be unusually tall, with long
limbs and long, thin fingers. The syndrome is inherited as a dominant trait,
carried by the gene FBN1, which encodes the connective protein fibrillin-1.
People have a pair of FBN1 genes. Because it is dominant, people who have
inherited one affected FBN1 gene from either parent will have Marfan
syndrome. Marfan syndrome has a range of expressions, from mild to severe.
The most serious complications are defects of the heart valves and aorta. It may
also affect the lungs, the eyes, the dural sac surrounding the spinal cord, the
skeleton and the hard palate.
Other AD conditions
Neurofibromatosis (commonly abbreviated NF; neurofibromatosis type 1 is
also known as von Recklinghausen disease) is a genetically-inherited
disorder in which the nerve tissue grows tumors (neurofibromas) that may be
benign and may cause serious damage by compressing nerves and other tissues.
Neurofibromatosis is an autosomal dominant disorder, which means only one
copy of the affected gene is needed for the disorder to develop. Therefore, if
only one parent has neurofibromatosis, his or her children have a 50% chance of
developing the condition as well. The severity in affected individuals can vary;
this may be due to variable expressivity. Approximately half of cases are due to
de novo mutations and no other affected family members are seen. It affects
males and females equally.
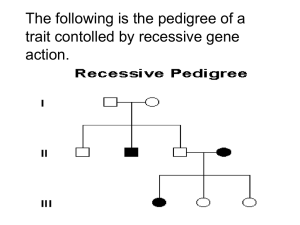
Autosomal Recessive (AR)
Recessive conditions are clinically manifest
only when an individual has two copies of the
mutant allele.
When just one copy of the mutant allele is
present, an individual is a carrier of the
mutation, but does not develop the
condition.
Females and males are affected equally by
traits transmitted by autosomal recessive
inheritance.
When two carriers mate, each child has a 25%
chance of being homozygous wild-type
(unaffected); a 25% chance of being
homozygous mutant (affected); or a 50%
chance of being heterozygous (unaffected
carrier).
Note: Affected individuals are
indicated by solid black symbols
and unaffected carriers are
indicated by the half black symbols.
Autosomal Recessive (AR)
Cystic fibrosis
Phenylketonuria (PKU)
Albinism
Galactosemia
Xeroderma pigmentosum
Fanconi anemia
Bloom syndrome
Tay-Sachs
Hemochromatosis
Cystic fibrosis (CF or mucoviscidosis)
Affects most critically the lungs, and also the pancreas, liver, and intestine. It is
characterized by abnormal transport of chloride and sodium across an epithelium,
leading to thick, viscous secretions.
The name cystic fibrosis refers to the characteristic scarring (fibrosis) and cyst formation
within the pancreas, first recognized in the 1930s.
Difficulty breathing is the most serious symptom and results from frequent lung
infections that are treated with antibiotics and other medications. Ultimately, lung
transplantation is often necessary as CF worsens.
Other symptoms, including sinus infections, poor growth, and infertility affect other
parts of the body.
GENETICS:
CF is caused by a mutation in the gene for the protein cystic fibrosis transmembrane
conductance regulator (CFTR). This protein is required to regulate the components of
sweat, digestive fluids, and mucus.
CF is most common among Caucasians; 4% of people of European descent carries
one allele for CF (by far the most common mutation is ΔF508, but there are >1000)
Individuals with cystic fibrosis can be diagnosed before birth by genetic testing, or
by a sweat test in early childhood.
Other AR conditions
Phenylketonuria (PKU) is a metabolic genetic disorder characterized by a mutation in
the gene for the hepatic enzyme phenylalanine hydroxylase (PAH), rendering it
nonfunctional. This enzyme is necessary to metabolize the amino acid phenylalanine
(Phe) to the amino acid tyrosine. When PAH activity is reduced, phenylalanine
accumulates and is converted into phenylpyruvate (also known as phenylketone), which
is detected in the urine. Untreated PKU can lead to mental retardation, seizures and
other serious medical problems. The mainstream treatment for classic PKU patients is a
strict PHE-restricted diet (requires severely restricting or eliminating foods high in
Phe, such as meat, chicken, fish, eggs, nuts, cheese, legumes, milk and other dairy
products) supplemented by a medical formula containing aminoacids and other
nutrients. The current recommendation is that the PKU diet should be maintained for
life. Patients who are diagnosed early and maintain a strict diet can have a normal
life span with normal mental development.
Albinism also called achromia, achromasia, or achromatosis) is a congenital
disorder characterized by the complete or partial absence of pigment in the skin, hair
and eyes. While an organism with complete absence of melanin is called an albino an
organism with only a diminished amount of melanin is described as albinoid.
Other AR conditions
Galactosemia is a rare genetic metabolic disorder that affects an individual's ability to
metabolize the sugar galactose properly. Although the sugar, lactose, metabolizes to
galactose, galactosemia is not related to and should not be confused with lactose
intolerance. The only treatment for classic galactosemia is eliminating lactose and
galactose from the diet. Even with an early diagnosis and a restricted diet, however,
some individuals with galactosemia experience long-term complications such as speech
difficulties, learning disabilities, neurological impairment (tremor).
Xeroderma pigmentosum (XP) is a disorder in which the ability to repair damage
caused by ultraviolet (UV) light is deficient. In extreme cases, all exposure to sunlight
must be forbidden, no matter how small; as such, individuals with the disease are often
colloquially referred to as Children of the Night. Patients with XP are at a high risk for
developing skin cancers, such as basal cell carcinoma.
Other AR conditions
Fanconi anemia is a genetic disease with an incidence of 1 per 350,000 births, with a higher
frequency in Ashkenazi Jews and Afrikaners in South Africa. FA is the result of a genetic defect in a
cluster of proteins responsible for DNA repair. As a result, the majority of FA patients develop
cancer, most often acute myelogenous leukemia, and 90% develop bone marrow failure (the
inability to produce blood cells) by age 40. About 60-75% of FA patients have congenital defects,
commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and
developmental disabilities. Around 75% of FA patients have some form of endocrine problem, with
varying degrees of severity. Median age of death was 30 years in 2000. Treatment with androgens
and hematopoietic (blood cell) growth factors can help bone marrow failure temporarily, but the
long-term treatment is bone marrow transplant if a donor is available. Because of the genetic defect
in DNA repair, cells from people with FA are sensitive to drugs that treat cancer by DNA
crosslinking, such as mitomycin C.
Bloom syndrome is characterized by short stature and predisposition to the development of
cancer. Cells from a person with Bloom syndrome exhibit a striking genomic instability that
includes excessive homologous recombination.
Tay-Sachs (I gave you a separate ppt for it)
Other AR conditions
Hemochromatosis (iron overload) indicates accumulation of iron in the body from any cause.
The most important causes are hereditary hemochromatosis (HHC), the most common
genetic disease in Europe (1:200-300). The gene responsible for HHC (known as HFE gene) is
located on chromosome 6; the majority of HHC patients have mutations in this HFE gene (other
genes involved C283Y and H63D). HHC is characterized by an accelerated rate of intestinal iron
absorption and progressive iron deposition in various tissues that typically begins to be expressed in
the 3rd to 5th decades of life, but may occur in children. Hemochromatosis can be asymptomatic
(75%) and be discovered by routine blood tests or may present with the following clinical features:
Cirrhosis of the liver
Diabetes due to pancreatic islet cell failure
Cardiomyopathy
Arthritis (iron deposition in joints)
Testicular failure
Tanning of the skin (bronze diabetes)
Joint pain and bone pain
Routine treatment consists of regularly scheduled bloodletting (500ml). For those unable to
tolerate routine blood draws, there is a chelating agent available for use (Deferoxamine).
A third of those untreated develop hepatocellular carcinoma.
X-linked Dominant
Because the gene is located on the X
chromosome, there is no transmission
from father to son, but there can be
transmission from father to daughter
(all daughters of an affected male will
be affected since the father has only one
X chromosome to transmit).
Children of an affected woman have a
50% chance of inheriting the X
chromosome with the mutant allele.
X-linked dominant disorders are
clinically manifest when only one copy
of the mutant allele is present.
X-linked Dominant
Some forms of Retinitis Pigmentosa
Chondrodysplasia Punctata
Hypophosphatemic rickets
= X-linked hypophosphatemia (XLH)
=Hypophosphatemic vitamin D-resistant rickets
(HPDR)
Amelogenesis imperfecta
X-linked Dominant
Some forms of Retinitis Pigmentosa (RP) is an
inherited, degenerative eye disease that causes severe
vision impairment and often blindness. The progress of
RP is not consistent. Some people will exhibit symptoms
from infancy, others may not notice symptoms until later
in life. Generally, the later the onset, the more rapid is
the deterioration in sight.
Fundus of patient with retinitis pigmentosa, mid
stage (Bone spicule-shaped pigment deposits are
present in the mid periphery along with retinal
atrophy, while the macula is preserved although
with a peripheral ring of depigmentation. Retinal
vessels are attenuated.)
From a review by Christian Hamel, 2006.
X-linked Dominant
Chondrodysplasia Punctata is a clinically and genetically diverse group of
rare diseases, first described by Conradi, that share the features of stippled
(presenting small dots) epiphyses and skeletal changes.
Amelogenesis imperfecta presents with abnormal formation of the enamel or
external layer of teeth. Enamel is composed mostly of mineral, that is formed
and regulated by the proteins in it. People afflicted with amelogenesis
imperfecta have teeth with abnormal color: yellow, brown or grey. The teeth
have a higher risk for dental cavities and are hypersensitive to temperature
changes. This disorder can afflict any number of teeth.
X-linked Dominant
Hypophosphatemic rickets = X-linked hypophosphatemia (XLH) =Hypophosphatemic
vitamin D-resistant rickets (HPDR) is an X-linked dominant form of rickets (or osteomalacia)
that differs from most cases of rickets in that ingestion of vitamin D is relatively ineffective. It can
cause bone deformity including short stature and genu varum (bow leggedness). It is associated
with a mutation in the PHEX gene sequence (Xp.22) and subsequent inactivity of the PHEX protein.
The prevalence of the disease is 1:20000
X-linked Recessive
X-linked recessive traits are not clinically
manifest when there is a normal copy of the
gene.
All X-linked recessive traits are fully evident in
males because they only have one copy of the X
chromosome, thus do not have a normal copy of
the gene to compensate for the mutant copy.
For that same reason, women are rarely affected
by X-linked recessive diseases, however they are
affected when they have two copies of the
mutant allele.
• Duchenne muscular dystrophy
(DMD)
Because the gene is on the X chromosome there
• Hemophilia A
is no father to son transmission, but there is
father to daughter and mother to daughter and
son transmission.
If a man is affected with an X-linked recessive
condition, all his daughter will inherit one copy
of the mutant allele from him.
• X-linked severe combined
immune disorder (SCID)
• Some forms of congenital
deafness
Duchenne muscular dystrophy (DMD)
Is affecting around 1 in 3,600 boys, which results in muscle
degeneration and eventual death. The disorder is caused by a
mutation in the dystrophin gene, located on the human X
chromosome, which codes for the protein dystrophin, an
important structural component within muscle tissue that
provides structural stability to the dystroglycan complex (DGC) of
the cell membrane. While both sexes can carry the mutation,
females rarely exhibit signs of the disease. Symptoms usually
appear in male children before age 6 and may be visible in
early infancy. Even though symptoms do not appear until early
infancy, laboratory testing can identify children who carry the
active mutation at birth. Progressive proximal muscle
weakness of the legs and pelvis associated with a loss of
muscle mass is observed first. Eventually this weakness spreads
to the arms, neck, and other areas. Early signs may include
pseudohypertrophy (enlargement of calf and deltoid muscles), low
endurance, and difficulties in standing unaided or inability to
ascend staircases. As the condition progresses, muscle tissue
experiences wasting and is eventually replaced by fat and fibrotic
tissue (fibrosis). By age 10, braces may be required to aid in
walking but most patients are wheelchair dependent by age
12. Later symptoms may include abnormal bone development that
lead to skeletal deformities, including curvature of the spine. Due
to progressive deterioration of muscle, loss of movement occurs,
eventually leading to paralysis. Intellectual impairment may or
may not be present but if present, does not progressively worsen as
the child ages. The average life expectancy for patients
afflicted with DMD is around 25.
X-linked Recessive
Hemophilia A is the most common type of hemophilia. It is also
known as factor VIII deficiency or classic hemophilia. It is largely an
inherited disorder in which one of the proteins needed to form blood
clots is missing or reduced. In about 30% of cases, there is no family
history of the disorder and the condition is the result of a spontaneous
gene mutation.
Approximately one in 5,000 males born in the United States has
hemophilia. All races and economic groups are affected equally.
When a person with hemophilia is injured, he does not bleed harder or
faster than a person without hemophilia, he bleeds longer. Small cuts
or surface bruises are usually not a problem, but more traumatic
injuries may result in serious problems and potential disability (called
"bleeding episodes").
Hemophilia A
Normal plasma levels of FVIII range from 50% to 150%. There are different levels of hemophilia:
mild, moderate, and severe, depending on the amount of clotting factor in the blood:
1.
People with mild hemophilia have 6% up to 49% of the normal clotting factor in their
blood. Most patients usually have problems with bleeding only after serious injury, trauma or
surgery. In many cases, mild hemophilia is not diagnosed until an injury, surgery or tooth
extraction results in prolonged bleeding. The first episode may not occur until adulthood.
Women with mild hemophilia often experience menorrhagia, heavy menstrual periods, and
can hemorrhage after childbirth.
2.
People with moderate hemophilia about, 15% of the hemophilia population, have 1% up to 5%
of the normal clotting factor in their blood. They tend to have bleeding episodes after injuries
and some without obvious cause. These are called spontaneous bleeding episodes.
3.
People with severe hemophilia about 60% of the hemophilia population, have <1% of the
normal clotting factor in their blood. They have bleeding following an injury and may have
frequent spontaneous bleeding episodes, often into their joints and muscles.
Hemophilia A
Everyone inherits two sex chromosomes, X and Y, from his or her parents. A female inherits one X
chromosome from her mother and one X chromosome from her father (XX). A male inherits one X
chromosome from his mother and one Y chromosome from his father (XY). The gene that causes
hemophilia is located on the X chromosome.
A woman who gives birth to a child with hemophilia often has other male relatives who also have
hemophilia. Sometimes, a baby will be born with hemophilia when there is no known family
history. This means either that the gene has been "hidden" (that is, passed down through several
generations of female carriers without affecting any male members of the family) or the change in
the X chromosome is new (a "spontaneous mutation").
There are four possible outcomes for the baby of a woman who is a carrier. These four possibilities
are repeated for each and every pregnancy:
1. A girl who is not a carrier
2. A girl who is a carrier
3. A boy without hemophilia
4. A boy with hemophilia
With each pregnancy, a woman who is a carrier has a 25% chance of having a son with hemophilia.
Since the father's X chromosome determines the baby will be a girl, all the daughters of a man with
hemophilia will be carriers. None of his sons, which is determined by the father through his Y
chromosome, will have hemophilia.
Hemophilia A
Hemophilia A
In general, small cuts and scrapes are treated with regular first-aid: clean the cut, then
apply pressure and a band-aid. Individuals with mild hemophilia can use a non-blood
product called desmopressin acetate (DDAVP) to treat small bleeds. Deep cuts or
internal bleeding, such as bleeding into the joints or muscles, require more complex
treatment. The clotting factor missing (VIII or IX) must be replaced so the child can
form a clot to stop the bleeding.
Some factor products are made from human blood products such as donated plasma.
Others, called "recombinant factor," are made in a laboratory and do not use human
blood products. The Medical and Scientific Advisory Council of the National
Hemophilia Foundation encourages the use of recombinant clotting factor products
because they are safer. Your doctor or your HTC will help you decide which is right for
you. All factor treatments are injected or infused directly into the veins.
In cases of severe hemophilia, doctors sometimes recommend giving a regimen of
regular factor replacement treatments (a therapy called prophylaxis) to prevent bleeding
episodes before they happen. The Medical and Scientific Advisory Council of the
National Hemophilia Foundation recommends prophylaxis as optimal therapy for
children with severe hemophilia A and B.
X-linked Recessive
X-linked severe combined immune disorder (X-SCID) is
an immunodeficiency disorder in which the body produces very little T cells
and NK cells. In the absence of T cell help, B cells become defective.
It is an x-linked recessive trait, stemming from a mutated (abnormal) version of the
IL2-RG gene located at xq13.1 on the X-chromosome, which is shared between
receptors for IL-2, IL-4, IL-7, and IL-15.
Persons afflicted with X-SCID often have infections very early in life, before three
months of age. This occurs due to the decreased amount of immunoglobulin G (IgG)
levels in the infant during the three-month stage.
This is followed by viral infections such as pneumonitis, an inflammation of the lung
which produces common symptoms such as cough, fever, chills, and shortness of
breath.
X-linked Recessive
Some forms of congenital deafness:
X-linked recessive inheritance causes hearing loss in only a small number (about 3%) of
people with hearing loss.
With X-linked recessive inheritance, only boys are affected.
Girls can be carriers of the gene. That means they could pass it on to their sons in the future.
If a mother is a carrier of the hearing loss gene, her sons will have a 50% chance of having hearing
loss.
If a mother is a carrier of the hearing loss gene, none of her daughters will have hearing loss. But
half of them will be carriers.
X-linked types of hearing loss can be a mix of conductive and sensorineural hearing loss.
If there is a recessive gene for hearing loss on only one of the mother’s X chromosomes, she will
have normal hearing. She would be called a “carrier.” Half of her children will get the hearing loss
gene. Her daughters will get the normal gene on the X chromosome from their father. Sons won’t
have a second X chromosome because they will have gotten the Y chromosome from their father. So
they will have hearing loss even though they have only one copy.
X-linked recessive congenital deafness
In this picture the mother is a carrier of an X-linked hearing loss gene called Xd.
Only her sons who get the Xd from her and the Y from their father will have
hearing loss.
Y-linked (holandric) traits
Hypertrichosis of the ears
Polygenic and Multifactorial
Inheritance
Most diseases have multifactorial inheritance patterns.
As the name implies, multifactorial conditions are not caused by a single
gene, but rather are a result of interplay between genetic factors and
environmental factors.
Diseases with multifactorial inheritance are not genetically determined,
but rather a genetic mutation may predispose an individual to a
disease. Other genetic and environmental factors contribute to whether
or not the disease develops.
Numerous genetic alterations may predispose individuals to the
same disease (genetic heterogeneity).
For instance coronary heart disease risk factors include high blood
pressure, diabetes, and hyperlipidemia. All of those risk factors have
their own genetic and environmental components. Thus multifactorial
inheritance is far more complex than Mendelian inheritance and is more
difficult to trace through pedigrees.
DISEASES:
• Alzheimers disease
• Heart disease
• Some cancers
• Neural tube defects
• Schizophrenia
• Insulin-dependent
Diabetes mellitus
CHARACTERS:
· Height, weight
• Intelligence
· Skin, eyes and hair color
· Dermatoglyphics
· Blood pressure
Mitochondrial Inheritance (1)
Mitochondria are organelles found in the cytoplasm of cells.
Mitochondria are only inherited from the mother’s egg, thus
only females can transmit the trait to offspring, however they
pass it on to all of their offspring.
The primary function of mitochondria is conversion of molecule
into usable energy.
Thus many diseases transmitted by mitochondrial inheritance
affect multiple organs with high-energy use such as the heart,
blood, skeletal muscle, liver, and kidneys, becoming a complex
texture of diseases, usually lethal in early childhood.
The difficulty arises when no mtDNA defect can be found or
when the clinical abnormalities are complex and not easily
matched to those of more common mitochondrial disorders.
Mitochondrial Inheritance (2)
Mitochondria are unique in that they have multiple
copies of a circular chromosome = mtDNA
Each human cell contains thousands of
copies of mtDNA. At birth these are
usually all identical (homoplasmy).
By contrast, individuals with
mitochondrial disorders resulting from
mtDNA mutations may harbor a mixture
of mutant and wild-type mtDNA within
each cell (heteroplasmy)
The percentage level of mutant mtDNA
may vary among individuals within the
same family, and also among organs and
tissues within the same individual. This is
one explanation for the varied clinical
phenotype seen in individuals with
pathogenic mtDNA disorders.