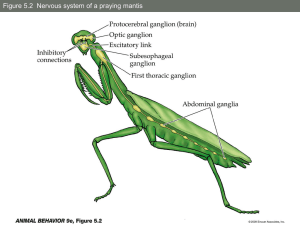
Genetics of Human Sexual
Development
Warning! If you are legally required to get parental
permission before viewing sex-related material, please do so
before proceeding with this presentation.
Levels of Sexual Development
• There are 3 levels to sexual development:
– chromosomal sex: presence or absence of the Y chromosome
– gonadal sex (primary sex determination): whether the gonads
develop as testes or ovaries depends on the presence or absence of
the SRY gene, usually found on the Y chromsome
– phenotypic sex (secondary sex determination): all of the internal
and external structures develop along male or female lines
depending on which hormones are secreted by the gonads.
• Phenotypic sex also has a couple of distinct systems: the
internal ducts, and the external genitalia
• Two important times: pre-natal development and puberty
Chromosomal Sex Determination
•
•
We have 46 chromosomes: 23 pairs,
one set from each parent.
One pair of chromosomes is the sex
chromosomes, X and Y.
– the other chromosomes just have
numbers: 1-22.
•
A person with 2 X chromosomes
(46,XX) is female, and a person with
an X and a Y (46,XY) is male.
Human karyotype: chromsomes
stained to show bands, from a male
Meiosis
•
Reproduction involves putting one copy of each chromosome into each sperm cell
or egg cell: the process of meiosis. So, meiosis starts with 46 chromosomes,
chooses one from each pair, and puts 23 chromosomes in each sperm or egg.
– For males, 1/2 the sperm get an X chromosome, and the other 1/2 get a Y chromosome.
•
Sometimes meiosis goes wrong (non-disjunction), and a sperm or egg might get 2
sex chromosomes, or 0 sex chromosomes, leading people with 47 or 45
chromosomes. More on this later...
Fertilization
• Fertilization means
the sperm joins the
egg, creating the
zygote, which is the
first cell of the new
individual person.
– 23 chromosomes
from sperm plus
23 from the egg
restores the total
of 46
chromosomes.
– And, the new
individual is now
either 46,XX
(female) or
46,XY (male)
The SRY Gene
• How the Y chromosome determines sex.
• The SRY gene, located on the Y chromosome, is the
primary determinant of sexual development.
– That is, if a developing embryo has a functional SRY
gene in its cells, it will develop as a male. And, if
there is no functional SRY, the embryo develops as
female.
• Although the SRY gene is usually on the Y
chromosome, it occasionally gets transferred to the
X.
– this leads to 46,XX males
• Also, sometimes the SRY gene is inactivated by
mutation.
– Leading to 46,XY females (Swyer syndrome)
– it is also possible to have a partially inactive SRY
gene, leading to ambiguous genitalia
Early Gonad Development
• Before 6-7 weeks of
development, the gonad is
indifferent: neither male
nor female.
• It develops from the same
tissue as the kidneys and
adrenal glands.
• Also developing by this
time: 2 sets of ducts that
will eventually lead to the
outside world.
– Wolffian ducts =
male
– Mullerian ducts =
female
Gonad Differentiation
• If SRY is present in the
indifferent gonad at 6
weeks, it gets activated.
This in turn activates other
genes, and the indifferent
gonad is converted to a
testes.
• In the absence of SRY, a
different set of genes is
activated, and the
indifferent gonad becomes
an ovary.
• The germ cells, which
actually become sperm or
eggs, migrate into the
gonad about this time.
Development of Phenotypic Sex
• The cells of the newly formed testes start secreting the
hormone testosterone.
– Testosterone secretion peaks about week 16, with levels similar to
those found in adult males. After this, the testosterone level drops
to about the same level as female fetuses.
– The testes also secrete another hormone: Mullerian inhibiting
substance (MIS) (aka anti-Mullerian hormone, AMH).
• Another important process in the developing male: during
the last trimester of pre-natal life, the testes migrate
(“descend”) from the kidney region into the scrotum.
– Under the control of a third testes hormone: “insulin-like
hormone 3”
• The developing ovary secretes estrogen, which is
important after birth, but estrogen from the mother
completely swamps it out before birth.
•
•
In the early embryo, two duct
systems form. After the gonad
differentiates into a testis or ovary,
one set of ducts develops further
while the other set degenerates.
Testosterone causes the Wolffian
ducts to develop into male
structures: epididymus, vas
deferens, seminal vesicles.
– In the absence of testosterone, the
Wolffian ducts disappear (except
a bit becomes the adrenal glands
in both sexes)
•
Mullerian inhibiting substance
causes the Mulerian ducts to
disappear.
– In the absence of MIS, the
Mullerian ducts develop into
the Fallopian tubes, uterus,
and upper vagina.
Internal Ducts
Another Duct Picture
Development of the External Genitalia
• This process is controlled by the presence
or absence of dihydrotestosterone (DHT).
• Testosterone gets converted into DHT by the
enzyme 5-alpha reductase, which is found in the
testes and the skin.
• Both sexes start out with the same structures, which
develop along different lines under the influence of
testosterone and DHT.
• The default condition in female: in the absence of
DHT, the external genital structures develop along
female lines.
• DHT also causes hair loss: male pattern baldness.
Testosterone is converted to DHT locally. Rogaine
works by blocking 5-alpha reductase
•
•
In the absence of DHT, the
genital swellings form the
labia majora; the genital
folds remain unfused and
form the labia minora; the
genital tubercle forms the
clitoris and the urogenital
sinus forms the lower part
of the vagina.
With DHT present, the
genital swellings migrate
and become the scrotum;
the urogenital folds enlarge
and enclose the penile
urethra and become the
shaft of the penis; the
genital tubercle becomes
the glans penis; and the
urogenital sinus forms the
prostate gland
External Development
Side View
Childhood and Puberty
•
During childhood, sex hormone levels are very low in both sexes.
– There is a surge of sex hormones in both boys and girls for a few weeks just after
birth. Significance is unknown.
• Puberty begins when the brain and hypothalamus start producing the
neurohormone GnRH (gonadotropin releasing hormone). This
hormone then stimulates production of LH and FSH by the pituitary
gland.
• LH and FSH stimulate the testes and ovaries to start producing large
amounts of testosterone and estradiol (a form of estrogen).
– In boys, some of the testosterone is converted to estradiol, which causes a growth
spurt, and sometimes leads to temporary breast development.
• The adrenal glands also secrete male sex hormones, in both boys and
girls, starting in late childhood.
– After puberty starts, the ovaries also produce androgens.
Review of Normal Sexual Development
• The default condition is female. Male development occurs only when the
SRY gene (on the Y chromosome) is present.
– The primary sex organ, the gonad, becomes an ovary in the absence of SRY, and
a testis in the presence of SRY.
• The testes secrete two hormones: testosterone and Mullerian-inhibiting
substance. These control the development of the internal reproductive ducts.
• Two duct systems are present in the embryo: Wolffian and Mullerian
– In males, testosterone causes the Wolffian ducts to develop into the vas deferens,
seminal vesicles, and epididymus. Also in males, MIS causes the Mullerian ducts
to degenerate.
– In females, the absence of MIS causes the Mullerian ducts to develop into the
fallopian tubes, uterus, and upper vagina. The absence of testosterone causes the
Wolffian ducts to degenerate.
• The external genitalia develop in the male pattern if dihydrotestosterone
(DHT) is present. Female genitalia develop in the absence of DHT.
• Very little change occurs in childhood, but puberty brings a big surge in sex
hormones, which modify the structures formed before birth.
Variant Conditions
•
The large majority of people develop as either completely male or
completely female. However, 1% or more of the population has
some variant condition.
–
–
–
•
Chromosomal variations
Gene mutations
External conditions
A few important terms:
– Gynecomastia: development of breasts in a male
– Hypospadias: the urethra exits the male body at the base of the penis
instead of at the tip, due to failure of the urethra to become enclosed
by the urogenital folds.
– Hermaphrodite: a person exhibiting both male and female
characteristics
• a true hermaphrodite has both testicular and ovarian tissue,
sometimes as separate organs but more frequently as an
ovotestis: a single organ with different parts. Very rare.
• Male pseudo-hermaphrodite has testes and no ovaries; a female
pseudo-hermaphrodite has ovaries and no testes.
– Worth noting: the Intersex Society of North America finds the term
“hermaphrodite” offensive, and prefers “intersex”.
Chromosomal Variants
•
•
Meiosis, the form of cell division that generates the
sperm and eggs, carefully puts exactly 1 copy of
each chromosome pair into each cell.
Sometimes meiosis goes wrong and puts 0 or 2
copies of some chromosome into a sperm or egg
cell.
– the best example of this: Down syndrome, which
starts with a sperm or egg with 2 copies of
chromosome 21.
– Maternal age effect: more frequent in older mothers
•
•
•
•
The sex chromosomes are quite tolerant of variants.
Most common types involve 45 or 47 chromosomes
There are many other, rarer types, with 48 or even
49 chromosomes, such as 49,XXXXY. Such
conditions almost always lead to serious mental
deficiencies.
The general rule: if the Y is present, the person is
internally and externally male.
Klinefelter Syndrome: 47,XXY
•
•
•
Occurs about 1 per 500 male births. It is the most
common type of sex chromosome variant.
The presence of the Y chromosome causes a
47,XXY person to be male, both externally and
internally, because the testes are formed.
Root symptom: small testes, leading to low
testosterone levels. Most, but not all, are sterile.
•
At puberty, reduced facial and body hair, broader
hips, breast development.
•
47,XXY children tend to be taller, less physically
strong and coordinated, and more quiet and shyer
than their peers. Some language and learning
problems are common: often slow to learn to speak
and read.
•
Testosterone replacement therapy helps with some
of the physical symptoms. Speech therapy and
educational services also help.
46,XX males, with the SRY
gene on the X, have the
Klinefelter appearance.
Turner Syndrome: 45,X
•
•
Only one X chromosome, sometimes called XO.
Since there is no Y chromosome, the primary gonad
is the ovary, and 45,X people are female.
About 1 in 2500 live female births.
– 10% of all spontaneous abortions (miscarriages) are
due to Turner syndrome; about 98% of all Turner’s
embryos die before birth
•
•
Ovaries completely non-functional, so 45,X women
are sterile, with no production of sex hormones and
development of secondary sexual characteristics at
puberty.
Some characteristic physical abnormalities: short
stature, low hairline, webbed skin at neck. Kidney
and circulatory system problems
•
Often have problems with spatial reasoning and
mathematics. Also social difficulties: inability to
understand others’ emotions.
•
Can be treated with growth hormone and estrogen.
You need 2 X chromosomes for
proper ovarian development.
46,XY females (non-functional
SRYgene) resemble Turner’s
47,XYY
•
•
•
•
•
•
About 1 in 1000 live male births. Most XYY’s are never
detected: a very mild condition.
since 1960, newly discovered chromosome variants aren’t given
the discoverer’s name
It was once thought to create hyper-aggressive males with a
tendency towards criminal behavior.
– Richard Speck, the killer of eight student nurses in 1966,
pretended (falsely) to be an XYY to obtain leniency.
– A 1968 letter to the Lancet claimed that XYY men were in
prison at a rate "25-60 times as high as the prevalence in
the general population”, based on finding 2 XYY’s.
– the plot of Aliens 3 involves a prison planet for XYY’s.
XYY’s are generally normal in appearance, but with average
height about 7 cm above expected and normal build. Perhaps
acne is more common than average, but this is disputed.
They are often more physically active, somewhat delayed in
emotional maturity, and have a slight increase in learning and
speech problems.
Fertile, normal sex drive, very rarely pass 2 Y’s to sons.
1970’s British TV series:
He had an extra Y, which
made him a macho criminal!
47,XXX
• About 1 in 1000 live female births. So mild
as to be only rarely detected. Also called
triplo-X.
• Originally called “superfemale” (early
1960’s). <rolls eyes>
• Widely varying symptoms, including none
at all.
• Slightly more passive and quiet as babies,
less assertive, delayed motor and linguistic
skills. Delayed emotional maturity and
social skills. Some have slightly decreased
intelligence and learning difficulties.
•
Lower back problems are common. Fertility
normal, don’t generally pass 2 X’s to
children.
•
•
•
•
Both terms refer to people who have 2 different chromosome
sets in different cells. For example, a 46,XX/47,XXY person
has some cells with 46 chromosomes and other cells with 47.
A mosaic starts out with a single fertilized egg. During an early
cell division in the embryo, one cell gained or lost a
chromosome. (This is non-disjunction, the same event that
happens in meiosis to generate Klinefelter’s, etc.)
A chimera starts out with two separate fertilized eggs, fraternal
twins. The two embryos fuse together to form a single
individual.
– It is not uncommon to have fraternal twins sharing some
blood cells, a “blood chimera”
– fused embryo chimeras are very rare: there are about 30-40
known XX/XY chimeras (and undoubtedly an equal
number same sex chimeras). “tetragametic chimera”
– Chimerism is probably the way most true hermaphrodites,
who have both ovarian and testicular tissue, are formed.
However, actual XX/XY chimeras have been everything
from normal male, through various degrees of ambiguous
genitalia, to normal female.
Sexual development can be quite variable in such people,
because the characteristics depend on which cells have which
chromosome complement.
Mosaics and
Chimeras
Gene Mutations
•
•
•
•
The variants up to now all involve whole chromosomes, which have lots of
genes on them. The effects of changing the dosage of many genes tend to be
widespread but mild. (or completely lethal, as with most non-sex
chromosomes).
Now we are going to look at several gene mutations. In these cases, only one
gene is affected, but it is completely knocked out. This can lead to large
effects, but limited to a few subsystems in the body.
Rates are different: for chromosome changes, about 1 in 1000 births is a
typical frequency. For gene mutations, each parent needs to contribute a
mutated copy of the gene, so rates are usually 1 in 10,000 births or less.
Inheritance is also a factor here: most chromosomal variants are spontaneous
events and don’t run in families. Gene mutations are usually inherited
variants: there is often a family/community history of the variant type.
– New mutations do occur spontaneously, but it’s rare. Most gene variants are
inherited from the parents.
5-alpha Reductase Deficiency (5-ARD)
•
•
•
•
5-alpha reductase is the enzyme that converts testosterone into
DHT. If both copies of the gene that makes this enzyme are
defective, the person has 5-ARD.
– Recall that DHT is responsible for the development of male
external genitalia
At birth, people with 5-ARD have undescended testes and male
ducts (with no female ducts), but genitalia that appear
somewhere between female and ambiguous, including a a very
small penis with hypospadias (which appears to be an enlarged
clitoris), and a short vagina. Often raised as girls
At puberty, the increase in testosterone is large enough that some
DHT gets made, and they develop a male appearance: the testes
descend, the penis enlarges, facial hair appears, the voice
deepens, muscles develop.
Large group in the Dominican Republic: maybe 1 in 90 men.
Called Guevedoces, a corruption of “huevos a los doce” (eggs-testicles- at age 12). Raised as girls, they easily switch to the
male role.
– Other groups found in Malta, Jordan, Pakistan, New Guinea
Also, a character on the TV show
Nip and Tuck has this condition
Guevodoces Case
Androgen Insensitivity
•
Incidence about 1 in 20,000 births
•
Used to be called “testicular feminization”. 46,XY with normal
(undescended) testes. The testes secrete testosterone, but the cells lack
a receptor for it. No receptor = no response to the hormone. Complete
androgen insensitivity, CAIS.
•
As a result, the male ducts (vas deferens, epididymus, seminal vesicles)
are not present. However, the testes secrete MIS, which causes the
female ducts (uterus, fallopian tubes, upper vagina) to degenerate.
•
External genitalia develop as male if DHT is present, but
testosterone and DHT use the same receptor. So, female
external genitalia, including the lower 2/3 of the vagina.
•
At puberty, the testes again secrete testosterone. The enzyme
aromatase converts it into estradiol. Thus, female secondary sexual
characteristics develop. Often “voluptuously feminine”. No
menstruation of course: no ovaries and no uterus. Pubic and armpit hair
is usually scant or absent.
–
Occasionally, the undescended testes can become cancerous, so they are
often surgically removed after puberty is complete (so as to get normal
female development).
Partial Androgen Insensitivity
•
Sometimes, the testosterone receptors work
inefficiently, due to less drastic mutations than in
CAIS. In these cases, the body cells respond in a
variable manner to testosterone, leading a a wide
variety of ambiguous genitalia. PAIS = partial
androgen insensitivity. Also called Reifenstein
syndrome.
–
•
•
•
there is also mild androgen insensitivity (MAIS), which
leads to completely male appearance internally and
externally, but with some impairment of masculinization
at puberty.
Variable symptoms: can be predominantly male (with
hypospadia, abnormal scrotum, small penis),
predominantly female (with enlarged clitoris, fused
labia, separate vaginal and urethral openings), or
ambiguous genitalia (microphallus--less than 1 cm
long), labia-like scrotum, hypospadia, gynecomastia.
Similar variability in male internal ducts; females
ducts are usually absent due to MIS secretion.
Sometimes people with PAIS change gender identity
after puberty, in either direction.
Congenital Adrenal Hyperplasia
•
•
•
•
•
The adrenal glands sit on top of the kidneys and secrete a variety
of steroid hormones, including cortisone (stress response),
aldosterone (salt balance) and androgens (male sex hormones).
Steroid hormones are made from cholesterol through a series of
biochemical steps. Any one of these steps can be inactivated by
mutation. However, about 95% of CAH cases involve defects in
the enzyme 21-hydroxylase.
21-hydroxylase is needed to make cortisol and aldosterone (but
not androgens). Cortisol is secreted in response to the pituitary
hormone ACTH, in a feedback loop. So, if there isn’t enough
cortisol being made, more ACTH is made, and this causes the
adrenal gland to grow larger (hyperplasia).
And, all of those steroid molecules that were destined to become
cortisol and aldosterone get diverted into male sex hormones
(androstendione and testosterone), which don’t need the 21hydroxylase.
Very little effect on male fetus, which is already making
testosterone, except that after birth the lack of salt regulation can
lead to death from excess salt secretion (salt-wasting).
•
Female fetuses with 21-hydroxlase deficiency have some
problems due to the flood of androgens released by the
adrenal gland. The ovaries are normal, and the female
(Mullerian) ducts are also normal (since no MIS is made).
•
Main effects are on the external genitalia: enlarged clitoris,
sometimes with an enclosed urethra (i.e. like the penis),
labia can fuse and become scrotum-like, vaginal opening
can be partly or completely closed.
•
Appearance at birth varies a lot. Some appear to be normal
male with undescended (because non-existent) testes.
However, the chromosomes are XX, the gonads are
ovaries, and the uterus and fallopian tubes are usually
intact.
•
Normally, very little androgen is made in childhood. CAH
causes excess androgens throughout life, leading to rapid
growth, but an early closure of the bone growth plates: a
very short adult. Also: early puberty, with menstrual
problems (and poor sperm production in males).
•
The other hormones, aldosterone and cortisol, need to be
replaced. The cortisol replacement calms the ACTH
activity, leading to less androgen production.
CAH
CAH is the most frequent cause
of non-standard genitals in
genetically female (XX) children.
Some Environmental Causes
•
Progestin-induced virilization. Progestin was used to prevent miscarriages
in the 1950’s and 60’s. Related to this is the use of androgens to treat
endometriosis during that time period, and occasional accidental use of
androgens. 160 known cases.
– XX fetuses develop as normal females with functioning ovaries, but they may
develop some male secondary characteristics and often have enlarged clitorises.
Effects are very similar to CAH.
•
Freemartin: usually seen in cattle: female and male twins, with testosterone
from male leaking over to the female due to a shared placenta. Normal female
appearance, but undeveloped ovaries and masculinized behavior. Rare or
unknown in humans.
– Aldous Huxley’s book Brave New World has human freemartins created
by hormone treatment of fetuses.
And Lots More…
• We have covered the main causes of variation in
human biological sex. However, there are many
other, rarer conditions that also affect this.
• As always with biology, there are exceptions to
every rule, exceptions to everything I said here
today.
• What any individual feels about their body, and
how society reacts to these variations, is more a
matter for psychology and sociology than for
biology.