Genomic Firsts
1976: RNA virus -- Phage MS2 (3 kbp)
1977: DNA virus -- Phage Φ-X174 (6 kbp)
1995: Bacteria -- Haemophilus influenzae (1.8 Mbp)
1995: Eukarya -- Saccharomyces cerevisiae (12 Mbp)
1996: Archaea -- Methanococcus jannaschii (1.6 Mbp)
2000: draft human genome -- J. Craig Venter (3 Gbp)
Genome Sequencing Explosion
Genome Sequencing Explosion
Three domains of life
16S rRNA sequences
Woese 1987
Global phylogeny
of 191 organisms
derived from 31
conserved protein
genes.
Tree is fairly well
resolved and
agrees mostly
with rRNA tree.
Ciccarelli et al
(2006) Science
Genomic streamlining in prokaryotes
Proteobacteria
(from Higgs & Attwood)
~1000 bp/gene
short intergenic regions
Efficiency in the Genome
Small organisms care about DNA
replication time.
No wasted space
High coding density (85-90%)
1 gene per 1000 bases in
prokaryotes
Haemophilus influenzae
1762 genes in 1.8 Mb
Human
23000 genes in 3080 Mb
Eukaryotic genomes have lots of
transposons and repetitive
sequences.
Hou and Lin – PLoS ONE 2009
The larger organelle genomes
also have a greater fraction of
non-coding sequence, but small
animal mitochondria fit the trend
of the bacteria.
large variation in genome size between bacteria
Sorangium cellulosum
(14000kb)
11599 codong sequences
Soil bacterium
Tremblaya princeps (140kb)
121 coding sequences
Endosymbiont in insect cells
McCutcheon and Moran
Nature Reviews (2012)
McCutcheon and Moran
Nature Reviews (2012)
Reduced size genomes
evolve independently in
different lineages. Usually
on long branches = fast
sequence evolution.
Subdivisions of proteobacteria identified using
16S rRNA originally
proteobacteria
- Agrobacterium tumefaciens - genetic
engineering
- Rickettsia conorii – ticks – spotted fever
- Rickettsia prowazeckii – lice – typhus
proteobacteria
-Neisseria meningitidis
- N. gonorrhoea
proteobacteria
-Escherichia coli – commensal – lab study
- Yersinia pestis – plague
- Haemophilus influenzae – respiratory pathogen.
(First bacterial genome)
- Xanthomonas / Xylella – plant pathogens
proteobacteria
- Helicobacter pylori – intestinal infections
Considerable change in GC content among related genomes.
Short genomes are derived from longer genomes – lots of deletions in
cases of intracellular parasites and endosymbionts.
Pathogens and intracellular bacteria have
low GC content –
May be a result of metabolic cost of
synthesis of G and C being higher (Rocha
and Danchin, 2002)
These genomes are also small
– use it or lose it!
This may explain correlation of GC content
with genome size
It has also been argued that there is a general
mutation bias towards AT, and that selection for GC
keeps this from going to very low GC in most
organisms. This stabilizing selection might be
weaker in smaller intracellular organisms. Therefore
smaller genomes have more AT.
...However, two extremely small genomes break the
trend. Maybe these have a mutation bias in the
other direction (towards GC) – this is not yet
measured.
Circular representation of the R. conorii genome (strain Malish 7). The outermost circle indicates
the nucleotide positions. The second and third circles locate the ORFs on the plus and minus
strands, respectively. Function categories are color-coded [see Web fig. 1 (10)]. The fourth and
fifth circles locate tRNAs. The locations of three rRNAs are indicated by black arrows. The sixth
and seventh circles indicate the locations of repeats. The eighth circle shows the G-C skew (GC/G+C) with a window size of 10 kb. The region locally breaking the genome colinearity with R.
prowazekii is indicated by a shaded sector. The four major genomic segments involved in this
rearrangement are colored in blue, yellow, green, and red. Ogata et al – Science (2001)
Illustration of the colinearity. Three distinct segments from the R. conorii genome aligned
with the homologous segments from the R. prowazekii genome are shown. These
segments were chosen to show three types of gene alteration: split genes in R.
prowazekii (top), a split gene in R. conorii (middle), and a gene remnant in R. prowazekii
(bottom).
Comparison of genomes of related organisms shows synteny –
but relatively rapid evolution of gene order
Mycoplasma genitalium and M.
pneumoniae
Each dot shows a high-scoring BLAST
match between a gene of one species
and a gene of the other species
Gene gain via Horizontal Gene Transfer
(mostly prokaryotes)
Gene gain via Gene Duplication
(mostly eukaryotes)
Genomic streamlining in symbionts and pathogens
McCutcheon & Moran (2012)
Free-living bacteria
• Selection to maintain reasonably
large set of functional genes.
• Gene acquisition balances gene loss
• HGT mediated by viruses and
plasmids gain of functions
• Some cells are competent for DNA
uptake (transformation)
• Homologous recombination can
eliminate some deleterious mutations
Host-restricted parasites and
endosymbionts
• Fewer essential genes because of
environment provided by host
• Smaller effective population size
(bottlenecks)
• Reduced selection against slightly
deleterious mutations & Reduced
opportunity for homologous
recombination faster sequence
evolution, reduced functionality and
stability of proteins (need for high level
of chaperones)
• Reduced selection against the deletion
of slightly beneficial genes, inherent
bias toward deletions, & reduced
opportunity to acquire genes
horizontally gene loss much faster
than gene gain.
Balance between selection and mutation in a large population
nk = number of individuals with k
deleterious mutations
N = total population size
U = number of deleterious mutations
per genome per generation
Assume no advantageous mutations.
Back-mutations are very rare.
Fitness w = (1-s)k
For a very large population, selection balances mutation.
There is a stationary state:
nk
N
(U / s )
k!
k
exp( U / s )
Muller’s Ratchet –
Acumulation of deleterious mutations in asexual species with small populations
If N is fairly small, then the
number of individuals in the fittest
class, n0, can be very small.
This fluctuates, and eventually
goes to zero.
If there are no back-mutations, the
fittest class is gone forever.
This is one click of the ratchet.
More and more deleterious mutations
with time until “mutational meltdown”
kills the species
fitness
Muller’s Ratchet is stopped by recombination
mutation
After one click of the ratchet, every
chromosome has at least one
deleterious mutation,
but they don’t all have the same one.
Initial population
recombination
Cross-over can recreate the
fittest class. This is much more
likely than back-mutation in
sexual species.
Muller’s Ratchet and the Evolution of Sex
• Two-fold cost of males in sexual species must be a big benefit of sex to
outweigh this cost
• A few parthenogenetic species are derive from sexual ancestors. These do
not do well in the long term.
• The ability of recombination to stop Muller’s ratchet is one large advantage of
sex, and is one possible reason for the prevalence of sexual species.
• Host-parasite co-evolution is probably another important reason.
• Maybe most free-living bacteria should be thought of as sexual, not asexual.
• Uptake of fragments of DNA from similar cells gives the possibility of
homologous recombination. This functions like sex in eukaryotes. It can
remove deleterious mutations.
• Uptake of DNA from distantly related organisms (Horizontal Gene Transfer)
can lead to the spread of beneficial genes
• When bacteria become obligate parasites or endosymbionts, they become
truly asexual.
• Consequences are gene loss and accumulation of deleterious mutations.
Global phylogeny
of 191 organisms
derived from 31
conserved protein
genes.
Tree is fairly well
resolved and
agrees mostly
with rRNA tree.
Ciccarelli et al
(2006) Science
Need to consider Eukaryotes separately for 2
reasons.
(i) Almost everyone believes there is a tree for
Eukaryotes.
(ii) Origin of Eukaryotes is a later unique event
that is very likely not tree-like.
Do prokaryotic taxa
mean anything?
-Proteobacteria?
Enterobacteriaceae?
E. coli?
Criticisms of the Prokaryotic Tree of Life (Bapteste et al. 2009)
“Belief in the universal tree of life is stronger than the evidence from genomes that
supports it.”
1. Circularity of tree methods – Phylogenetic methods always produce a tree of
some kind.
2. Statistical problems – weak signals from many individual genes. Failure to reject
the consensus tree is not necessarily support for it.
3. Systematic biases in phylogenetic methods.
4. Large-scale exclusion of conflicting data. Core genes not necessarily
representative of a species tree.
5. Closely related species may exchange genes more frequently.
6. Unrelated species in similar niches may exchange genes more frequently.
Convergent evolution?
This is an interesting paper but take it with a pinch of salt
Spectrum of Opinions
1. The tree of rRNA and translational genes is the species tree. Other
genes appear to give different trees just because of noise and
phylogenetic errors. HGT is unimportant.
2. The tree of rRNA and translational genes is the best information we
have about the tree of cell divisions and speciations. Most genes follow
this tree most of the time, even if most genes may have been
horizontally transferred at some point in their history.
3. The tree of rRNA and translational genes tells us only about the history
of these genes, and is therefore not particularly important. There are
other essential groups of genes that follow other evolutionary paths.
We need a network representation, not a single tree.
4. HGT is so frequent that all genes follow different histories. Therefore
tree-building is a waste of time. We only get results that look like trees
because our methods are designed to produce trees.
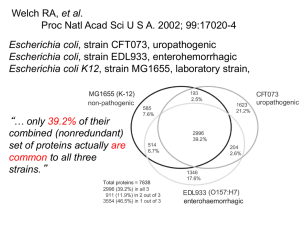
Gene Content Variation among E. coli genomes.
Evidence for horizontal transfer –
Welch et al (2002).
Core genome = intersection of sets
Pangenome = union of sets
Core and Pan-genome of E. coli
Core genome
Pan-genome
Rasko et al (2008) J. Bacteriol.
Rapid Gain and Loss of genes among closely related genomes of Bacillus
Hao and Golding (2006) Genome Research
• Assumes a tree to begin with (many conserved genes)
• Only two of the patterns shown require more than one character change
• Does not distinguish HGT from innovation
Tree of Archaea based on signature genes
Gao and Gupta (2007) BMC Genomics
• Signature genes are those that are shared by all members of a group and are not
posessed by any other speies.
• Can the tree be constructed from gene content alone?
• Does not show events that do not fit the hierarchical tree.
• What about transfers within niches? Groups of genes confer metabolic activity
Phylogeny of three domains of life based on shared gene content
SHOT – Korbel et al (2002)
S = fraction of genes that are orthologues between two species
d = -lnS
Input d to NJ method
Major domains and groups of bacteria are obtained the same as for rRNA
Does not work for very reduced genomes of parasites & symbionts
Always possible to explain a presence/absence pattern by
either multiple deletions or by horizontal transfer.
Examples from Dagan et al (2007)
(a) Loss only, (b) Single origin, (c) Origin + 1 HGT, (d) Orign + 2 HGTs
The problem is, we don’t know the ratio of HGT to deletions….
Reconstructing ancestral genomes using parsimony (Dagan et al 2007)
If HGT is disallowed or penalized too much, then ancestral genomes must
have been far larger than any current genomes.
If HGT is too frequent then ancestral genomes are apparently too small.
This helps to find a moderate value for the ratio of HGT to deletions.
Method of Collins & Higgs (2012)
Collect genomes
from NCBI
All-vs-All BLASTP
Single-linkage clustering
Identification of universal
single-copy clusters
Global amino acid alignment
Concatenation of alignments
Phylogenetic reconstruction using
Maximum Likelihood
Core and Pangenomes
Closed – means that pangenome size
tends to a maximum as number of
genomes increases
Open – means that pangenome keeps
increasing as you add new genomes
Fitting the data suggests that the
pangenome is open for most groups of
bacteria and that Gpan (n) increases in
proportion to ln(n).
This is expected on a tree like a
coalescent (a). On a star tree (b), it would
increase linearly with n.
Gene Frequency Spectra
9 Prochlorococcus genomes
Baumdicker et al (2009)
G(k) is the number of genes found in k
genomes from a group of n.
There is a U-shape: many genes found in
only 1 or 2 genomes, a certain number of
core genes in (almost) all n, and fewer
genes in between.
The U-shape applies at all scales from
species to the full bacerial domain.
293 Bacterial genomes
Lapierre and Gogarten (2009)
Collins and Higgs (2012)
Core, Shell and Cloud genes
(Koonin and Wolf – 2012)
The role of gene duplication:
Gene family size distributions
Collins and Higgs (2011)
Modelling duplication and deletion of genes
u
0
1
2
2
2
3
3
3
etc.
4
Origin of Mitochondria
Sequence similarity to Rickettsia – within proteobacteria
Also conserved gene order between Rickettsia and the
mitochondrial genome of the protist Reclinomonas (one of the
largest mitochondrial genomes).
Gene order and phylogeny for Hodgkinia
(very small endosymbiont – see assignment 3)
Shows it has evolved independently of the lineage
leading to Rickettsia and mitochondria
Derived change in Rickettsia not
shared with Hodgkinia
Hodgkinia placed within Rhizobiales –
raises questions of GC content bias and long
branch attraction
Long Branch Attraction - An artefact of phylogenetic methods that tends to put
unrelated species with rapid evolution together.
It can also draw long branch species closer to the root, because they are attracted
to the outgroup.
Rooting the
tree of life
using
ancient gene
duplications
Long Branch attraction and the tree of rRNA
(Gribaldo and Philippe 2002)
Typical tree in older papers shows many lineages on long
branches close to the roots of Bacteria and Eukarya
Are there any
eukaryotes that never
had mitochondria?
Were ancestral organisms
hyperthermophiles?
Root is usually inferred from
ancient gene duplications –
eg EFTu and EFG
After correcting for long branch attraction...
Microsporidia are now related to fungi.
They have small genomes with lots of gene
loss and rapid sequence evolution.
Current thought says there may never
have been eukaryotes without
mitochondria. Eukaryotes evolved by
fusion of an protobacterium with an
archaeon. The event that created the
mitochondria also created the nucleus.
Phylogeny of major bacterial groups is still
uncertain. Deduction of temperature at base of
tree is difficult. Most papers still argue for
hyperthermophiles at common ancestor of
archaea and bacteria.
Seems strange! This
would make prokaryotes
monophyletic after all
Root is still most likely here,
although this paper
questions it.
Growth temperature mapped onto
the rRNA tree
Or was there a mesophilic
origin after all?
Competing hypotheses for the origin of the eukaryotic host cell.
TA Williams, et al. Nature 504, 231-236 (2013) doi:10.1038/nature12779
Eocyte hypothesis:
Standard picture:
The root is on the bacterial branch
There is a common ancestor of
archaea amd eukaryotes
The root is (still) on the bacterial
branch
Eukaryotes fall within the archaea.
They have a common ancestor with
Eocytes/Crenarchaeota.
Only Two Domains!
Maybe Giant Viruses are a Fourth Domain?
RNA polymerase sequences
from Global Ocean SurveyGOS
Wu et al. (2011)