Metagenomics - Stanford AI Lab
advertisement

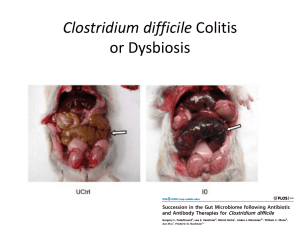
Metagenomics and the microbiome What is metagenomics? Looking at microorganisms via genomic sequencing rather than culturing Environmental use case: ag, biofuels, pollution monitoring Health use case: The human microbiome You = 1013 your cells + 1014 Why care about microbiome? bacterial cells More actionable genomics Source: http://www.med-health.net/Best-Time-To-Take-Probiotics.html http://www.mayo.edu/research/labs/gut-microbiome/projects/fecal-microbiota-transplant-c-diff-colitis Why care about microbiome? Diagnostic or modulatory implications in: Obesity, Diabetes, Fatigue, Pain disorders Anxiety, Depression, Autism Antibiotic resistant bacteria IBD and other gut disorders Cardiac function, cancer Diseases and the microbiome Source: The human microbiome: at the interface of health and disease. Nature reviews genetics Why care about microbiome? Publications containing ‘microbiome’ by date on Science Direct Goal 1: Composition Source: The human microbiome: at the interface of health and disease, Nature Reviews Genetics http://huttenhower.sph.harvard.edu/metaphlan Diversity measures Alpha diversity: how diverse is this population? Simpson’s index, Shannon’s index, etc Difference in alpha diversity before and after antibiotics Beta diversity: Taxonomical similarity between 2 samples Finding compositional associations between disease cohort and microbial makeup Sequencing for diversity Pyrosequencing the 16s ribosomal RNA subunit < 10 taxa appear in > 95% of people in HMP Recall the implicated diseases. Looks like GWAS common disease, small effect size + common disease, rare variant Goal 2: Functional profiling Source: The human microbiome: at the interface of health and disease. Nature reviews genetics Functional profiling Current: Which genes are present and are being transcribed In development: proteomics, metabolomics Sequencing for function Whole microbiome sequencing Avoids primer biases and is more kingdom agnostic Assembly is hard, especially where reference genomes don’t exist Two big problems Can’t understand the body without understanding the microbiome Can’t understand the microbiome by only looking at bacteria Read fragment assembly is very very hard in metagenomics Kingdom-Agnostic Metagenomics The players in your body Your cells Metabolites Bacteria Bacteriophages Other viruses Fungi That’s not complexity Source: A comprehensive map of the toll‐like receptor signaling network. Molecular Systems Biology Prokaryotic virome: bacteriophages Infect prokaryotic bacteria Transfer genetic material among prokaryotic bacteria Rapidly evolving Put constant selection pressure on bacterial microbiome Bacteriophages: deep sequencing results 60% of sequences dissimilar from all sequence databases More than 80% come from 3 families Little intrapersonal variation Large interpersonal variation, even among relatives Diet affects community structure Antibiotic resistance genes found in viral material Bacteriophages and function Cross the intestinal barrier possibly affecting systemic immune response Adhere to mucin glycoproteins potentially causing immune response in gut epithelium IBD/Chron’s: relative increase in Caudovirales bacteriophages Affect bacterial composition and/or host directly Eukaryotic virome Fecal samples from healthy children shows complex community of typically pathogenic viruses Includes plant RNA viruses from food Anelloviruses and circoviruses present in nearly 100% by age 5, likely from industrial ag Eukaryotic viruses and function Simian immunodeficient experiment showed enteric virome expansion Increased gut permeability and caused intestinal lining inflammation Acute diarrhea subjects showed novel viruses and highly divergent viruses with less than 35% similarity to catalogued viruses at amino acid level Meiofauna Fungi, protazoa, and helminths (worms) No experiments conducted with sampling to saturation, much more work to be done 18S sequencing showed 66 genera of fungi in gut and fungi were found in 100% of samples Most subjects had less than 10 genera But high fungal diversity is bad: increases in IBD, increases with antibiotic usage But it’s very hard Amplicon-based don’t work well for viruses Heterogeneous sample-prep is required Large differences in genome sizes from a few kb in viruses to 100+Mb in fungi Small genomes+divergence require lots of coverage to get contigs Getting the whole picture Source: Meta'omic Analytic Techniques for Studying the Intestinal Microbiome. Gastroenterology. The assembly problem Isn’t assembly easy? Recall: 500-1000 species of bacteria in the gut, but about 30 of them make up 99% of composition 33% of bacterial microbiome not well-represented in reference databases, > 60% for bacteriophages Coverage Coverage: mean number of reads per base L=read length, N=number of reads, G=genome size Problem, with 2nd gen WMS technologies, L is low and G is astronomical or unknown Thus, “full or sometimes even adequate coverage may be unattainable” Source: A primer on metagenomics Sequence length and discovery Source: A primer on metagenomics All is not lost Can use rarefaction curves to estimate our coverage All is not lost For composition analysis the phylogenetic marker regions (18S, 16S) work pretty well For functional analysis: can still find ORFs fairly reliably and can be aligned to homologs in databases Barring this, clustering and motif-finding yield some information Different sequencing approaches? Single-cell microfluidics in the future Now: hybrid long/short read approaches. “finishing” with Sanger sequencing Pacific biosciences SMRT approach SMRT errors are random, unbiased De novo assembly is 99.999% concordant with reference genomes HGAP: the SMRT assembly algorithm 1) Select longest reads as seeds 2) Use seed reads to recruit short reads 3) Assemble using off the shelf assembly tools 4) Refine assembly using sequencer metadata Source: Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods Seed selection Order reads according to length Considering reads above length L ~ 6kb Rough end-pair align reads until ~20x coverage is reached 17.7k seed reads, averaging 7.2kb in length, already at 86.9% accuracy compared to reference Recruiting short reads Align all reads to the seed reads Each read can be mapped to multiple seed reads, controlled by –bestn parameter -bestn must be chosen so that the coverage of seeds + short aligned reads is about equal to the expected coverage of the sequenced genome Use MSA and consensus to error correct long reads Result is 17.2k reads of length 5.7kb with 99.9% accuracy Overlap layout consensus assembly Source: Overview of Genome Assembly Algorithms. Ntino Krampis. http://www.slideshare.net/agbiotec/overview-of-genome-assembly-algorithms Refinement Use Quiver algorithm which looks at raw physical data from sequencer Uses an HMM and observed data to tell classify base calls as genuine or spurious Do a final consensus alignment, conditioned on Quiver’s probabilities Final result: 17.2k reads, length of 5.7kb, accuracy of 99.999506% Summary Most of the cells in your body aren’t yours But looking at bacteria alone is insufficient Expanding our view causes us to look for needles in haystacks which is beyond most conventional approaches Motif-finding and hybrid approaches will work until 3rd gen sequencing arrives References Cho, Ilseung, and Martin J. Blaser. "The human microbiome: at the interface of health and disease." Nature Reviews Genetics 13.4 (2012): 260-270. Wooley, John C., Adam Godzik, and Iddo Friedberg. "A primer on metagenomics." PLoS computational biology 6.2 (2010): e1000667. Chin, Chen-Shan, et al. "Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data." Nature methods 10.6 (2013): 563-569. Human Microbiome Project Consortium. "Structure, function and diversity of the healthy human microbiome." Nature 486.7402 (2012): 207-214. Norman, Jason M., Scott A. Handley, and Herbert W. Virgin. "Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities." Gastroenterology 146.6 (2014): 1459-1469. Morgan, X. C., and C. Huttenhower. "Meta'omic Analytic Techniques for Studying the Intestinal Microbiome." Gastroenterology (2014).