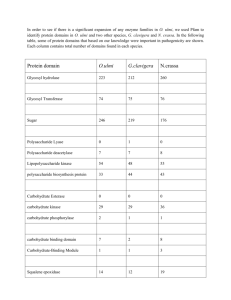
Structure of coenzyme F420H2 oxidase (FprA), a di-iron flavoprotein from methanogenic Archaea catalyzing the reduction of O2 to H2O Henning Seedorf1, Christoph H. Hagemeier1, Seigo Shima1, Rudolf K. Thauer1, Eberhard Warkentin2 and Ulrich Ermler2 1 Max Planck Institute for Terrestrial Microbiology, Marburg, Germany 2 Max Planck Institute for Biophysics, Frankfurt am Main, Germany Keywords coenzyme F420; crystal structure; di-iron center; F420H2 oxidase; O2 detoxification Correspondence U. Ermler, Max Planck Institute for Biophysics, Max-von-Laue-Str. 3, D-60438 Frankfurt am Main, Germany Fax: +49 69 63031002 Tel: +49 69 63031054 E-mail: ulrich.ermler@mpibp-frankfurt.mpg.de (Received 14 November 2006, revised 11 January 2007, accepted 17 January 2007) doi:10.1111/j.1742-4658.2007.05706.x The di-iron flavoprotein F420H2 oxidase found in methanogenic Archaea catalyzes the four-electron reduction of O2 to 2H2O with 2 mol of reduced coenzyme F420(7,8-dimethyl-8-hydroxy-5-deazariboflavin). We report here on crystal structures of the homotetrameric F420H2 oxidase from Methanothermobacter marburgensis at resolutions of 2.25 Å, 2.25 Å and 1.7 Å, respectively, from which an active reduced state, an inactive oxidized state and an active oxidized state could be extracted. As found in structurally related A-type flavoproteins, the active site is formed at the dimer interface, where the di-iron center of one monomer is juxtaposed to FMN of the other. In the active reduced state [Fe(II)Fe(II)FMNH2], the two irons are surrounded by four histidines, one aspartate, one glutamate and one bridging aspartate. The so-called switch loop is in a closed conformation, thus preventing F420 binding. In the inactive oxidized state [Fe(III)FMN], the iron nearest to FMN has moved to two remote binding sites, and the switch loop is changed to an open conformation. In the active oxidized state [Fe(III)Fe(III)FMN], both irons are positioned as in the reduced state but the switch loop is found in the open conformation as in the inactive oxidized state. It is proposed that the redox-dependent conformational change of the switch loop ensures alternate complete four-electron O2 reduction and redox center re-reduction. On the basis of the known Si–Si stereospecific hydride transfer, F420H2 was modeled into the solvent-accessible pocket in front of FMN. The inactive oxidized state might provide the molecular basis for enzyme inactivation by long-term O2 exposure observed in some members of the FprA family. Oxidases catalyze oxidation reactions with O2 as electron acceptor, which is reduced to either H2O2 [E¢(O2 ⁄ H2O2) ¼ + 0.28 V] or H2O [E¢(O2 ⁄ H2O) ¼ + 0.81 V]. The four-electron reduction of O2 to H2O generally proceeds without involving O2–, H2O2 or OH as free intermediates. This is essential, as the superoxide anion radical O2– [E¢(O2– ⁄ H2O2) ¼ + 0.89 V], H2O2 [E¢(O2 ⁄ H2O2) ¼ + 1.35 V] and the OH radical [E¢(OH ⁄ H2O) ¼ + 2.3 V] are very strong one-electron oxidants that are highly toxic to living cells, as shown by the finding that some eukaryotic organisms deliberately produce these reactive oxygen species via oxidases to defend themselves against intruding bacteria [1,2]. We have recently discovered in methanogenic Archaea a coenzyme F420H2 oxidase that catalyzes a fourelectron reduction of O2 to H2O, and have provided evidence that the enzyme is involved in O2 detoxification in these strictly anaerobic microorganisms [3]. In cell extracts of Methanothermobacter thermoautotrophicus, Abbreviation F420, 7,8-dimethyl-8-hydroxy-5-deazariboflavin, coenzyme F420. 1588 FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS F420H2 oxidase is one of the most prominent proteins [4]. The tetrameric cytoplasmic enzyme is composed of only one type of subunit, of molecular mass 45 kDa, and contains, per subunit, one FMN and a di-iron center. It is specific for coenzyme F420 (7,8-dimethyl-8hydroxy-5-deazariboflavin) as electron donor (apparent Km ¼ 30 lm) and O2 as electron acceptor (apparent Km ¼ 2 lm), with an apparent Vmax of the purified enzyme of 180 s)1 [3]. Coenzyme F420 is a 5-deazaflavin derivative, and as such transfers hydride anions rather than single electrons. Upon reduction, 1,5-dihydrocoenzyme F420 is formed, with a prochiral center at C5 (Fig. 1). The F420H2 oxidase has been shown to be Siface stereospecific with respect to C5 of the deazaflavin [5]. Coenzyme F420 is found in high concentrations only in methanogenic and sulfate-reducing Archaea. F420H2 oxidase is not related to other H2O-forming oxidases such as heme–copper oxidases [6–10], cytochrome bd quinol oxidases [11–14], the multicopper oxidases [15–17], or the apparently only FAD-containing NADH oxidases from anaerobic bacteria [18–21]. F420H2 oxidase is, however, phylogenetically related to the A-type flavoprotein family (FprA) [22]. One functionally and structurally characterized member of this family is the bacterial cytoplasmic NO reductase, which also contains FMN and a nonheme nonsulfur di-iron center as prosthetic groups. This enzyme catalyzes the two-electron reduction of 2NO to N2O and H2O with reduced rubredoxin, but also efficiently catalyzes the four-electron reduction of O2 to 2H2O with the same one-electron donor [23–28]. Interestingly, the cytoplasmic NO reductase from Escherichia coli (X-ray structure unknown) has an extra module at the C-terminus containing a rubredoxin-like center, FMN and an NADH-binding site [23,29]. In comparison, F420H2 oxidase catalyzes neither the reduction of O2 with reduced rubredoxin nor the reduction of NO with F420H2 [3]. This difference in reductant specificity is surprising for homologous enzymes, considering that F420 is a deazaflavin (771 Da) that transfers hydride Fig. 1. Structures of F420H2 and of FMNH2, both viewed from the Si face. The Re and Si faces of the flavin isoalloxazine ring are defined relative to C5 of the oxidized deazaflavin F420 [52]. anions at a redox potential (E¢) of ) 360 mV [30], whereas rubredoxins are iron–sulfur proteins (6000 Da) that transfer single electrons at redox potentials around 0 ± 100 mV [31]. We report here on the crystal structures of F420H2 oxidase from Methanothermobacter marburgensis in a reduced state (2.25 Å) and two oxidized states (1.7 Å and 2.25 Å), and compare them with the 2.5 Å resolution structure of the rubredoxin:NO ⁄ O2 oxidoreductase from Desulfovibrio gigas (31% sequence identity with F420H2 oxidase) [27] and with the 2.8 Å structure of the rubredoxin:NO ⁄ O2 oxidoreductase from Moorella thermoacetica (41% sequence identity) [28]. Of particular interest is the redox state-dependent position and coordination of the iron atoms and the structural basis for the specificity of F420H2 oxidase for coenzyme F420H2 in comparison to that of the two paralogous enzymes for reduced rubredoxin. Results and Discussion Structural basis F420H2 oxidase from M. marburgensis heterologously produced in E. coli was isolated and crystallized anaerobically and in the presence of dithiothreitol. Therefore, the isolated enzyme should be in a completely reduced state with respect to both FMN and the di-iron center. This assumption is corroborated by the UV ⁄ visible spectrum of the enzyme, which was typical for a fully reduced flavoprotein, and by the absence of an EPR signal, which is consistent with a diferrous or a diferric center, in which the two irons are antiferromagnetically coupled [24]. The first structure determined at 2.25 Å resolution (Table 1) was based on a crystal in a monoclinic form (grown in the presence of F420H2) frozen in liquid nitrogen within the anaerobic tent. A second and third structure at 2.25 Å and 1.7 Å resolution (Table 1) were derived from crystals of a tetragonal and monoclinic crystal form, respectively, that were frozen in a nitrogen gas stream outside the anaerobic tent and thus, before freezing, air-exposed for several minutes at 18 C. We assume that the first crystal structure reflects an active, predominantly reduced enzyme state [Fe(II)Fe(II)FMNH2], the second an inactive oxidized enzyme state [Fe(III)FMN] and the third an active oxidized [Fe(III)Fe(III)FMN] and active reduced [Fe(II)Fe(II)FMNH2] state superimposed. Despite considerable efforts, crystals of the enzyme were not obtained under aerobic conditions. F420H2 oxidase from M. marburgensis was found in the crystals ) according to packing considerations ) as a homotetrameric oligomer (Fig. 2A), which FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 1589 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Structure of di-iron flavoenzyme F420H2 oxidase H. Seedorf et al. H. Seedorf et al. Table 1. Data collection and refinement statistics Crystallization Crystal properties Space group Cell constants (Å), () No. of monomers in the asymmetric unit Data collection Wavelength (Å) Resolution (Å) Multiplicity Completeness (%) Rsym (%)a I ⁄ rI (last shell) Refinement Rcryst (%)b Rfree (%)c No. of reflections No. of protein atoms Average B-factor (Å2) Protein, di-iron, FMN Bond length rms (Å) Bond angle rms () F420H2 oxidase (anaerobic) F420H2 oxidase (air-exposed) F420H2 oxidase (air-exposed) 0.2 M (NH4)2SO4, 0.1 M Mes ⁄ KOH (pH 6.5), 16–22% poly(ethylene glycol) MME 5000 0.2 M (NH4)2SO4, 0.1 M Mes ⁄ KOH (pH 6.5), 16–22% poly(ethylene glycol) MME 5000 0.2 M (NH4)2SO4, 0.1 M Mes ⁄ KOH (pH 6.5), 8–16% poly(ethylene glycol) MME 5000, 15% glycerol P21 97.8, 123.1, 135.9, 103.4 8 P21 73.7, 120.9, 92.7, 110.4 4 P43212 88.7, 450.4 4 SLS-X10SA 1.000 2.25 (2.3–2.25) 2.6 (2.5) 97.6 (97.8) 6.6 (36.4) 13.7 (1.8) SLS-X10SA 0.979 1.7 (1.77–1.7) 4.6 (2.1) 99.2 (98.9) 7.8 (41.8) 9.9 (2.3) SLS-X10SA 0.992 2.25 (2.32–2.25) 4.5 (2.4) 97.2 (74.0) 6.3 (13.7) 16.7 (7.1) 20.6 27.0 103 861 25 349 44.8, 39.4, 27.4 18.6 21.8 156 441 12 652 33.3, 28.9, 20.6 18.8 23.4 80 298 12 652 24.8, 20.6, 15.8 0.011 1.36 0.018 1.87 0.011 1.30 P P Rsym¼ |Ii)Ælæ|/ Ii , where Ii is the observed intensity and Ælæ is the averaged intensity obtained from multiple observations of symmetryP P related reflections. b Rcryst¼ hkl(|Fobs|)|Fcalc|)/ hkl|Fobs| . c Rcryst where 5% of the observed reflections (randomly selected) are not used for refinement. a is in agreement with previous results (m ¼ 170 kDa) based on gel filtration experiments [32]. The tetramer is composed of a loose dimer of two dimers documented by an intradimeric and interdimeric buried surface of 12% (five ion pairs) and 9.5% (16 ion pairs), respectively, relative to the entire monomer and dimer surface areas. Compared to F420H2 oxidase, the interdimer contact areas found in the crystal structures of rubredoxin:NO ⁄ O2 oxidoreductase from D. gigas (7.5%; six ion pairs), and of rubredoxin:NO ⁄ O2 oxidoreductase from Mo. thermoacetica (1.2%; no ion pairs), are smaller, which is in line with their presence as a dimer in solution [25]. As the catalytically productive oligomeric state is the homodimer (see below), the differences in quaternary structure may reflect differences in thermoadaptation rather than differences in function. The homodimers of the FprA family members reveal a highly similar architecture, reflected by the rmsd of about 1.5 Å between the Ca atoms of the monomers, and by the analogous arrangements of the two mono1590 mers (Fig. 2B). Briefly, each (F420H2 oxidase) monomer is built up of two modules, an N-terminal b-lactamase-like domain (residues 1–252) harboring a di-iron center, and a C-terminal flavodoxin-like domain (residues 253–404) containing FMN. Two monomers assemble via a head-to-tail arrangement, such that the b-lactamase and the flavodoxin domains face each other, thereby forming two separated and presumably independent active sites (Fig. 2B). Thus, at the intradimer interface, the di-iron site of one monomer is positioned close to the FMN of the other and vice versa. Whereas the pyrimidine portion of FMN is directed to the protein surface, its dimethylbenzyl group points to the di-iron center. The iron closer to FMN is, in the following, referred to as proximal iron, and the other as distal iron. The distance between N5 of FMN and the proximal Fe of about 9 Å is within a suitable range to allow rapid electron transfer [33]. In contrast, the di-iron center and the FMN in one monomer are about 40 Å apart, which is too far for electron transfer at significant rates (Fig. 2). FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Structure of di-iron flavoenzyme F420H2 oxidase Fig. 2. Overall structure of F420H2 oxidase. (A) Molecular surface representation of the tetramer. The tetramer is composed of two functional dimers, each formed by a head-to-tail arrangement of two monomers, colored blue ⁄ green and dark gray ⁄ light gray). (B) Ribbon diagram of the dimer. The monomer is composed of a flavodoxin-like domain (light green ⁄ light blue) harboring FMN (stick model) and a b-lactamase-like domain (green ⁄ blue), with the di-iron center depicted as orange spheres. The active sites are located at the interfaces between two monomers of the functional dimers. N5 of FMN and the proximal iron (closest to FMN) are sufficiently close for rapid electron transfer. Binding of the di-iron center The di-iron center differs dramatically between the active reduced, inactive oxidized and active oxidized F420H2 oxidase states (Fig. 3), but also within each structure, as reflected by differences between the monomers in the asymmetric unit and by alternative conformations within one monomer. In the active reduced enzyme state (present in the monoclinic crystals frozen in the anaerobic tent and partly in the air-exposed monoclinic crystals), each iron ion is tetracoordinated by two imidazole nitrogens (proximal Fe, His83 and His151; distal Fe, His88 and His233), one carboxylate (proximal Fe, Glu85; distal Fe, Asp87), and one bridging carboxylate (Asp170) (Fig. 3A). Each iron ion contains, approximately trans to His83 and His88, a fifth coordination site. Both sites are oriented towards each other and constitute the dioxygen-binding site (see below). The two Fe(II) ions are in van der Waals contact with each other, their distances being 3.5 ± 0.2 Å. The described primary ligation shell essentially corresponds to that found in the rubredoxin-dependent enzymes. In contrast to the latter enzymes, the average site occupancy of the proximal iron in F420H2 oxidase is reduced to approximately 0.4, based on a refinement with equal temperature factors of the two irons. This finding is in line with biochemical data that indicate one iron to be more loosely bound to the enzyme than the other [34]. The low occupancy of the proximal iron leads to an increase of the temperature factor of its surroundings but not to a significant alteration of its structure. In the inactive oxidized state (present in the airexposed tetragonal crystals), the proximal iron is completely absent, and the ligands to iron in the reduced state have dramatically changed their position, such that the enzyme is definitively inactive (Fig. 3B). The side chain of Glu85 is rotated away from the proximal iron-binding site and constitutes, together with His26 and His267, a new remote metal (iron)-binding site. Its nature as a metal is compatible with the distance between the metal and the three ligands of 2.0 Å, 2.1 Å and 2.5 Å, as well as with the height of the electron density peak. Tyr25 evades the new metal-binding site and becomes hydrogen-bonded to Asp87, which itself is slightly shifted away from the distal iron. In other respects, the distal iron-binding site corresponds to that found in the reduced state. The imidazole group of His151 ligated to the proximal iron in the reduced state is shifted by more than 10 Å, and this is paralleled by a large conformational rearrangement of the loop between Pro148 and Pro153, referred to in the following as the switch loop (Fig. 3B). Whereas in the reduced state this loop is conformationally closed and directed to the di-iron center and to FMN, in the oxidized state it flips and creates an open conformation with respect to the accessibility of the redox centers from bulk solvent. Interestingly, the unusual nonprolyl cis peptide bond formed by Leu150 and His151 in the reduced state is thereby converted to a trans peptide bond (Fig. 3B). A nonprolyl cis peptide bond at this position, which is necessary to project the imidazolyl ring towards the proximal iron, was also found in the rubredoxin:NO ⁄ O2 oxidoreductase from D. gigas but not in the 2.8 Å crystal structure of rubredoxin:NO ⁄ O2 oxidoreductase from Mo. thermoacetica, possibly due to their low resolution. Unexpectedly, in the inactive oxidized state, His151, Asp330 and a water molecule (or a hydroxyl ion) that is hydrogen-bonded to Arg340 and Lys337 build up another new metal-binding FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 1591 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Structure of di-iron flavoenzyme F420H2 oxidase H. Seedorf et al. H. Seedorf et al. Fig. 3. Structures of the di-iron-binding site of F420H2 oxidase. The active site is formed at the homodimer interface, where the di-iron center of one monomer (green) is juxtaposed to FMN of the other monomer (blue). Active site amino acid residues and FMN are shown as stick models, and the two irons as orange spheres. (A) In the active reduced state, each of the irons is ligated to two histidines (His83, His88, His151 and His233), one aspartate or glutamate, and one bridging aspartate. The switch loop (red) (a-chain between Pro148 and Pro153) (the residues are not shown) is in a closed conformation. Note that His151 projects from the switch loop towards the proximal iron (closest to FMN), due to a cis peptide bond between Leu150 (not shown) and His151. Trp152 shields the completely buried di-iron center from bulk solvent (monoclinic crystal resolved to 2.25 Å). (B) In the inactive oxidized state, the proximal iron is absent but, alternatively, two new remote metals are found. The switch loop (black) is in an open conformation. The proximal iron-ligating residues Glu85, His83 and His151 dramatically change their conformation; in particular, the last of these moves more than 10 Å as part of the switch loop (tetragonal crystal resolved to 2.25 Å). (C) In the active oxidized state, both the proximal and the distal irons are present as in the active reduced state, but the switch loop adopts an open conformation (black). The active oxidized state is found superimposed with the active reduced state, such that the closed conformation (red) is also visible in the electron density map (monoclinic crystal resolved to 1.7 Å). site located at the protein surface. His83, another ligand of the proximal iron in the reduced state, is rotated by about 90 around the Ca–Cb bond, and is 1592 now hydrogen-bonded to the hydroxyl group of Ser232, which has also changed its conformation (Fig. 3B). Notably, a conformational change of a histidine ligated to the distal iron was detected in rubredoxin:NO ⁄ O2 oxidoreductase from D. gigas, in contrast to the rubredoxin:NO ⁄ O2 oxidoreductase from Mo. thermoacetica [28] and F420H2 oxidase. A third enzyme state was tentatively extracted from the electron density of the air-exposed monoclinic crystal, which contains both irons in a similar position and an occupancy as found in the reduced state. Additionally, Glu85 and Asp87 adopt the conformation of the reduced state, and the remote metal-binding site is either not occupied or very little occupied (depending on the considered monomer in the asymmetric unit). However, the switch loop reveals electron density not only for the closed conformation of the reduced state but also for the open conformation of the inactive oxidized state, the ratio being 60% to 40%. Consequently, the air-exposed monoclinic crystals includes, besides the active reduced state, a new superimposed state referred to as the active oxidized state (Fig. 3C). The active oxidized state is characterized by a di-iron center and a switch loop in the open conformation, the rearrangement from the closed conformation being presumably triggered by iron oxidation upon air exposure of the crystals. Therefore, we consider the active oxidized state as an intermediate of the catalytic cycle after O2 reduction. Note that the proximal iron FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Structure of di-iron flavoenzyme F420H2 oxidase is only ligated to Glu85 and Asp170 but not additionally to His83 and His151, as found in the reduced state. Redox-dependent changes of the ligation in di-iron proteins were previously reported for methane monooxygenase reductase hydroxylase [35] and ribonucleotide reductase [36], where, however, only carboxylate groups of glutamates and aspartates are subject to conformational alterations. O2-binding site The ligand geometry of the di-iron center in the reduced state offers an attractive O2-binding site within a pocket coated by the iron-ligating residues Asp87, Glu85, His151, and His233, as well as by Tyr25, His26, His175, Phe198 and Leu202 (Fig. 4). In the eight monomers of the asymmetric unit, the O2-binding pocket is either empty or occupied by a solvent molecule loosely bound to the distal iron. Whereas in Fig. 4. The O2-binding site of F420H2 oxidase. Active site amino acids, FMN and sulfate are shown as stick models. The dioxygenbinding site is surrounded by a pocket coated by residues His233, Tyr25, His26, His175, Phe198, Asp87, Glu85, His151 and Leu202 (the last four amino acids are not shown). His26 and His175 are candidates for transferring protons to the peroxo and oxo intermediates (see text). Tyr25 and Phe198 are exchanged in the structurally closely related NO reductases by phenylalanine and tyrosine (pink). Therefore, Tyr25 and Phe198 are probably responsible for the finding that F420H2 oxidase does not show NO reductase activity. In the active oxidized state (monomer C), the distal iron is ligated to a tentatively identified sulfate ion. The two irons are shown as as orange spheres, and a water molecule as a blue sphere. the inactive oxidized state the O2-binding site is destroyed, the electron density map derived from the airexposed monoclinic crystals reveals partial occupation. In monomers A and B, the extra electron density is most compatible with a diatomic molecule positioned slightly closer to the distal than to the proximal iron and perpendicular to the connection line between the two irons. In this conformation, one atom ligates to the proximal and distal irons and the other interacts with Tyr25 and Asp87. In monomer C, extra electron density linked to the distal iron is tentatively interpreted as a sulfate ion (Fig. 4). A sulfate anion is plausible, due to the shape and height of the electron density peak, the favorable hydrogen bond interactions with His27 and His175, and the presence of 0.2 m (NH4)2SO4 in the crystallization buffer. Moreover, an additional water molecule could be identified between the two irons and opposite to Asp170. Interestingly, extra electron density around the distal iron atom suggests an alternative iron position closer to the putative sulfate ligand due to ligand binding or due to the altered redox state. Covalent Fe(III)–ligand complexes are also observed in toluene and methane monooxygenase hydroxylase with acetate, formate and azide as anion ligands, thereby also corroborating the presence of the Fe(III) oxidation state [37]. In monomer D, the water molecule opposite to Asp170 is again visible, but the electron density connected with the distal iron could not be reasonably interpreted. The undefined iron adduct contacts a solvent molecule that is hydrogen-bonded to His26 and His175. The shape of the O2-binding pocket is approximately conserved in the structures of rubredoxin:O2 ⁄ NO oxidoreductases and of F420H2 oxidase, which has no NO reductase activity. However, the side chains protruding into the pocket partly vary, and might account for the different specificity. Phe198 in F420H2 oxidase (Fig. 4) is replaced by tyrosine in the rubredoxindependent enzymes, and the importance of this has been proven by the decrease of the NO reductase activity of the Tyr fi Phe mutant in rubredoxin:NO ⁄ O2 reductase [28]. Phe198 in F420H2 oxidase from M. marburgensis is strictly conserved in other FprA enzymes from methanogenic Archaea (supplementary Fig. S1), most of which contain at least one FprA with F420H2 oxidase activity (an exception is Methanopyrus kandleri). Another crucial residue is Tyr25 (Fig. 4), which is invariant in methanogenic Archaea and replaced by a phenylalanine in the rubredoxin-dependent enzymes. It protrudes from a loop variable within the FprA family, and its hydroxyl group interacts with the Fe-ligating carboxylate group of Glu85 and Asp87. The side chain of Tyr25 is in van FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 1593 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Structure of di-iron flavoenzyme F420H2 oxidase H. Seedorf et al. H. Seedorf et al. der Waals contact with the putative ligand in the O2-binding site, and it might be speculated that its hydroxyl group interferes with the bulky N2O, thus preventing its formation. Binding of FMN and modeling of F420H2 The conformation and binding characteristics of FMN are nearly identical in all of the analyzed structures of F420H2 oxidase but also in comparison to those of other members of the FprA family. However, the specific FMN–polypeptide interactions can be most accurately described in F420H2 oxidase, due to the higher resolution. FMN has an essentially planar isoalloxazine ring (Fig. 5), which is compatible with FMN being in either the reduced or the oxidized state [38]. A large number of polar contacts are formed between the peptide nitrogens of Met266, His267, Gly268, Ser269, Thr270, Tyr319, Asp320, Gly353, and Gly354, as well as Gly356 and the pyrimidine and phosphate components of FMN, indicating a rigid binding mode. Whereas the Re face of the ring is attached to residues Thr317, Ile318, Tyr319 and Met266 of the flavodoxinlike domain, the Si face is solvent-accessible, and a water-filled pocket is placed between the isalloxazine ring and the opposite monomer (Fig. 5). This pocket can be reliably considered as the F420H2-binding site, although the experimental verification by structure determination of an enzyme–F420 complex was not Fig. 5. The F420H2-binding site of F420H2 oxidase in the active oxidized state. F420H2 (yellow stick model) is modeled into its binding pocket with its Si face oriented towards the Si face of FMN (blue stick model). C5 of F420H2 and N5 of FMN, between which the hydride is transferred, are positioned within the van der Waals distance (approximately 3 Å). In this conformation, the Re face of F420H2 is attached to the switch loop in the open conformation (black), and the pyrimidine group of F420 reaches the di-iron center. 1594 feasible. Remarkably, solely in the oxidized state, the available space in front of the Si face of the FMN ring is sufficient to accommodate the bulky deazaisoalloxazine ring of F420H2 (open conformation), whereas in the reduced state (closed conformation) the switch loop is directed towards the prosthetic groups, and the bulky side chains of His151 and Trp152 block F420H2 binding. Model building of F420H2 was governed by the experimentally determined Si-face stereospecificity of the hydride transfer to and from F420 [5], which defines the orientation of the deazaflavin relative to the FMN face, by the assumed aromatic stacking interactions between the two ring systems observed in various systems [39,40], and by the required proximity between C5 of F420H2 and N5 of FMN (Fig. 5), implying that the generated complex is competent for hydride transfer [40]. Thus positioned, the tricyclic F420 ring is sandwiched between the isoalloxazine ring of FMN and the segment between His151 and Pro153 of the switch loop, whereby the imidazole group of His151 interacts with the bottom of F420H2 and the side chain of Trp152 with its face (Fig. 5). The crucial residue Trp152 is kept in place by a hydrogen bond between its indole nitrogen atom and the hydroxyl group of Tyr319. The l-lactyl-l-glutamyl-l-glutamic acid phosphodiester portion of F420 (see Fig. 1) was placed at the interface between the subunits such that its phosphate group is anchored by His117 and His267, which are both strictly conserved, and its first carboxylate group by Lys272. In this conformation, the mentioned F420H2 portion replaces a water chain that extends from the Si side of FMN to the bulk solvent, and therefore requires only minor displacements of the polypeptide (Fig. 5). In the crystal structures of rubredoxin:NO ⁄ O2 oxidoreductases from D. gigas and of rubredoxin: NO ⁄ O2 oxidoreductase from Mo. thermoacetica, the pocket is filled up from the entrance side by the side chains of Trp347 and Met146, which are both conserved in the rubredoxin-dependent enzymes but replaced by an asparagine and a leucine in F420H2 oxidase (supplementary Figs S1 and S2). F420H2 cannot enter the pocket, and this effectively precludes direct interaction of this electron donor with the FMN of the active site. On the other hand, where and how rubredoxin with a molecular mass of approximately 6 kDa binds to the two rubredoxin-dependent enzymes and not to F420H2 oxidase is not yet known. The mentioned Trp347 would be a candidate for shuttling electrons from rubredoxin to FMN. The structure-based analysis of the substrate binding in F420H2 oxidase teaches us once again that, on the FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Structure of di-iron flavoenzyme F420H2 oxidase Structure of di-iron flavoenzyme F420H2 oxidase basis of sequence homology, the function of proteins cannot be inferred even if their crystal structures are known in detail. In the FprA family, the electron donor and acceptor specificity and the accompanied redox mechanisms are totally different, although the structural framework, the binding mode of FMN and the di-iron center, as well as the electron transfer process, are strictly conserved. As discussed in detail, only a few side chain exchanges are sufficient to prevent or allow NO versus O2 as electron donor and to block or favor F420H2 binding over FMN. The catalytic reaction The F420H2 oxidase reaction represents a ping-pong process where, in a first reaction, four electrons from the diferrous di-iron and FMNH2 are transferred to the dioxygen, thereby forming two water molecules without the release of reactive oxygen species, and in a second reaction, the two redox centers are re-reduced by two hydride transfer reactions between F420H2 and FMN. The first half-cycle is assumed to begin with the FMN and the di-iron center of the enzyme both in the fully reduced state [Fe(II)Fe(II)FMNH2], for which the structure has been established. As a first step, the enzyme binds one molecule of O2 transiently, forming a peroxo intermediate bridging the two iron atoms, as suggested by mechanistic studies with di-iron(II) complexes [41,42]. Then, a first water molecule is released, leaving behind the enzyme in the diferric l-O(H) FMNH2 state (reaction in Scheme 1). Fe(II)Fe(II)FMNH2 þ O2 þ 2 Hþ ! Fe(III)OFe(III)FMNH2 þ H2 O ðScheme 1Þ Then, two electrons are transferred from the reduced FMN to the l-O(H) bridge between the two irons in the diferric state, with the release of the second water molecule (reaction in Scheme 2). Fe(III)OFe(III)FMNH2 ! Fe(III)Fe(III)FMN þ H2 O ðScheme 2Þ We assume that the generated Fe(III)Fe(III)FMN state is reflected in the active oxidized structure. The second half-cycle proceeds with binding of the first F420H2 and subsequent reduction of FMN, from which the electrons are shuttled one by one to the irons. After release of F420, a second F420H2 binds, reduces FMN and leaves the active site (reactions in Schemes 3 and 4). Fe(III)Fe(III)FMN þ F420 H2 ! Fe(II)Fe(II)FMN þ F420 þ 2 Hþ ðScheme 3Þ Fe(II)Fe(II)FMN þ F420 H2 ! Fe(II)Fe(II)FMNH2 þ F420 ðScheme 4Þ The enzyme is now back in the reduced FMN and diferrous state. Electron transfer between the reduced FMN and the proximal iron across the homodimeric subunit interface is most likely mediated via the dimethylbenzyl group of FMN and His151 or Asp85 (Fig. 3A). Both residues have a minimal distance to C8 of the flavin ring of 3.7 Å. Trp152 and Tyr319, flanking the mentioned residues, might additionally support a rapid electron transfer process between the reactions in Schemes 1 and 2. Proton transfer to the peroxo and oxo intermediates generated during oxygen reduction might be directly or indirectly accomplished by the strictly conserved residues His26 and His175, which are both accessible to bulk solvent (Fig. 4). In the reduced and active oxidized state, the two pronounced histidines are too far away (4.0–4.5 Å) from the O2-binding site, and a water molecule visible in the electron density map between their side chains (in monomer D) might be used as mediator. However, His26 can be positioned in hydrogen bond contact with a tentatively modeled O2 upon minor structural rearrangements, as seen in the inactive oxidized state (Fig. 3B). Experimental evidence is provided that the FprA oxidase reaction avoids the release of reactive oxygen species [29], which requires a direct and controlled four-electron reduction of O2. Structural data suggest that the sequential course of the complete O2 reduction and the complete prosthetic group re-reduction are ensured by the redox-dependent conformation of the switch loop (Fig. 3C). In the case that the di-iron center and FMN are reduced, the side chains of the key residues His151 and Trp152, protruding from the switch loop, complete the di-iron center for O2 activation and block the access of F420H2 (which is compatible with the unsuccessful cocrystallization experiments with F420H2 oxidase in the reduced state and F420H2). When the prosthetic group becomes oxidized upon O2 reduction, the switch loop is rearranged, thereby abolishing the catalytic competence of the di-iron center and allowing the binding of F420H2 and the subsequent hydride transfer. A hypothetical scenario might be that iron oxidation weakens the interactions between the proximal iron and His151, leading to an energetically favorable cis–trans isomerization of the peptide bond between Leu150 and His151, thereby inducing the structural rearrangement of the switch loop. For comparison, a stepwise O2 reduction is realized in a related iron–sulfur and flavin-containing ferredoxin oxidase found in methanogenic Archaea, but FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 1595 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License H. Seedorf et al. H. Seedorf et al. also in other anaerobic prokaryotes, that catalyze the reduction of O2 to H2O with H2O2 as free intermediate [43]. Interestingly, despite its completely different O2 activation mechanism, several architectural features are common to those described for the FprA family, such as its homotetrameric organization, its head-totail arrangement of two monomers juxtaposing FMN and the [4Fe)4S] cluster from two different monomers, and the similar fold of the FMN-binding domain [44]. The outlined mechanism provides no functional role for the inactive oxidized state structurally characterized for F420H2 oxidase. However, it is conceivable that the displacement of the proximal iron to the remote metalbinding sites over a distance of about 6 Å and 15 Å (Fig. 3B) is related to the inactivation of rubredoxindependent NO reductases after multiple O2 reduction cycles. A shift of the proximal iron would be energetically plausible, as its fixation by ligands is reduced in the active oxidized state, and because it can move concomitantly with the swinging side chain of Glu85 to constitute, with His26 and His267, an efficient metalbinding site. Inactivation of FprAs in the presence of large amounts of O2 might be biologically useful, as the cell would lose reducing power without eventually getting rid of the oxygen. Experimental procedures Purification and crystallization The fprA gene from M. marburgensis (DSMZ2133) was overexpressed in E. coli as described, except that the cells were grown in 2 L of trypton ⁄ phosphate medium rather than in LB medium [3,5]. Purification was performed under exclusion of oxygen in an anaerobic chamber (Coy) filled with 95% N2 ⁄ 5% H2 (v ⁄ v) and containing a palladium catalyst for O2 reduction with H2. Initial trials to crystallize F420H2 oxidase were performed with the hanging ⁄ sittingdrop, vapor-diffusion method using Basic and Extension crystallization kits from Sigma-Aldrich (Sigma-Aldrich, St Louis, USA). For the screens, 2 lL of the enzyme solution (containing 20 mgÆmL)1 of F420H2 oxidase) and 2 lL of reservoir solutions were mixed and incubated at 4 C. Under aerobic conditions, crystals of FprA were not observed. However, under anaerobic conditions and in the presence of 1 mm dithiothreitol, crystals were obtained at 10 C using 0.2 m (NH4)2SO4, 0.1 m Mes ⁄ KOH (pH 6.5) and 30% poly(ethylene glycol) [30% poly(ethylene glycol) monomethylether 5000] (MME) 5000 or 0.2 m Mgformate. Optimization of crystal quality, mainly varying the drop size (20 lL), precipitant concentrations and additional agents, resulted in three different crystal forms (see Table 1). 1596 Data collection, structure determination and refinement Data were collected at the beam line X10SA of the SwissLight-Source (Villigen, Switzerland) from anaerobically grown crystals, the first kept in an oxygen-free atmosphere and the second exposed to air. Processing and scaling were performed with the hkl [45] and xds [46] packages. The quality of the data and crystallographic parameters are summarized in Table 1. The structure of the enzyme based on the air-exposed monoclinic crystals was solved by molecular replacement using epmr [47] based on the 2.8 Å structure of rubredoxin:NO ⁄ O2 oxidoreductase from Mo. thermoacetica [28]. Using the 2.5 Å structure of rubredoxin:NO ⁄ O2 oxidoreductase from D. gigas [27] gave less reliable results, although the structures of the two rubredoxin-dependent enzymes are very similar, with respect to both the primary structure (42% sequence identity) and the quaternary structure (rmsd 1.3 Å for the Ca atoms of the two models). The phases for the other two crystals were obtained by molecular replacement using the model from the air-exposed monoclinic crystals [47]. Refinement of the structures based on crystals were performed using o [48] and cns [49], applying the four-fold noncrystallographic symmetry (NCS) relationship for the lower resolution data. Refinement was completed with the program refmac5 [50], using the TLS option (each monomer was treated as a separate TLS group), maximum likelihood minimization and isotropic B-value refinement. The refinement statistics are given in Table 1. Except for the C-terminal arginine, the entire polypeptide chain is visible in the electron density map. The stereochemical quality of the model was checked with the program procheck [51]. Figures 2–5 were generated with pymol (http://www.pymol.org). The coordinates of the structures based on anaerobically treated crystals, on air-exposed monoclinic crystals and tetragonal crystals are deposited in the Protein Data Bank (http://www.rcsb.org) with accession numbers 2OHI, 2OHH and 2OHJ, respectively. Acknowledgements This work was supported by the Max Planck Society and by the Fonds der Chemischen Industrie. We thank Hartmut Michel for continuous support, and the staff of the X10SA beamline at the Swiss-Light-Source, Villigen for assistance during data collection. References 1 Fang FC (2004) Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol 2, 820–832. FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Structure of di-iron flavoenzyme F420H2 oxidase 2 El-Benna J, Dang PMC, Gougerot-Pocidalo MA & Elbim C (2005) Phagocyte NADPH oxidase: a multicomponent enzyme essential for host defenses. Arch Immunol Ther Exp 53, 199–206. 3 Seedorf H, Dreisbach A, Hedderich R, Shima S & Thauer RK (2004) F420H2 oxidase (FprA) from Methanobrevibacter arboriphilus, a coenzyme F420-dependent enzyme involved in O2 detoxification. Arch Microbiol 182, 126–137. 4 Farhoud MH, Wessels HJ, Steenbakkers PJ, Mattijssen S, Wevers RA, van Engelen BG, Jetten MS, Smeitink JA, van den Heuvel LP & Keltjens JT (2005) Protein complexes in the archaeon Methanothermobacter thermautotrophicus analyzed by blue native ⁄ SDS-PAGE and mass spectrometry. Mol Cell Proteomics 4, 1653–1663. 5 Seedorf H, Kahnt J, Pierik AJ & Thauer RK (2005) Si-face stereospecificity at C5 of coenzyme F420 for F420H2 oxidase from methanogenic Archaea as determined by mass spectrometry. FEBS J 272, 5337–5342. 6 Michel H, Behr J, Harrenga A & Kannt A (1998) Cytochrome C oxidase: structure and spectroscopy. Annu Rev Biophys Biomed 27, 329–356. 7 Giuffre A, Stubauer G, Sarti P, Brunori M, Zumft WG, Buse G & Soulimane T (1999) The heme-copper oxidases of Thermus thermophilus catalyze the reduction of nitric oxide: evolutionary implications. Proc Natl Acad Sci USA 96, 14718–14723. 8 Brunori M, Giuffre A & Sarti P (2005) Cytochrome c oxidase, ligands and electrons. J Inorg Biochem 99, 324– 336. 9 Zumft WG (2005) Nitric oxide reductases of prokaryotes with emphasis on the respiratory, heme-copper oxidase type. J Inorg Biochem 99, 194–215. 10 Branden G, Pawate AS, Gennis RB & Brzezinski P (2006) Controlled uncoupling and recoupling of proton pumping in cytochrome c oxidase. Proc Natl Acad Sci USA 103, 317–322. 11 Hill BC, Hill JJ & Gennis RB (1994) The room-temperature reaction of carbon-monoxide and oxygen with the cytochrome bd quinol oxidase from Escherichia coli. Biochemistry 33, 15110–15115. 12 Dmello R, Hill S & Poole RK (1996) The cytochrome bd quinol oxidase in Escherichia coli has an extremely high oxygen affinity and two oxygen-binding haems: implications for regulation of activity in vivo by oxygen inhibition. Microbiol UK 142, 755–763. 13 Zhang J, Hellwig P, Osborne JP, Huang HW, MoenneLoccoz P, Konstantinov AA & Gennis RB (2001) Sitedirected mutation of the highly conserved region near the Q-loop of the cytochrome bd quinol oxidase from Escherichia coli specifically perturbs heme b(595). Biochemistry 40, 8548–8556. 14 Zhang J, Barquera B & Gennis RB (2004) Gene fusions with beta-lactamase show that subunit I of the cytochrome bd quinol oxidase from E. coli has nine Structure of di-iron flavoenzyme F420H2 oxidase 15 16 17 18 19 20 21 22 23 24 25 26 transmembrane helices with the O2 reactive site near the periplasmic surface. FEBS Lett 561, 58–62. Lee SK, George SD, Antholine WE, Hedman B, Hodgson KO & Solomon EI (2002) Nature of the intermediate formed in the reduction of O2 to H2O at the trinuclear copper active site in native laccase. J Am Chem Soc 124, 6180–6193. Johnson DL, Thompson JL, Brinkmann SM, Schuller KA & Martin LL (2003) Electrochemical characterization of purified Rhus vernicifera laccase: voltammetric evidence for a sequential four-electron transfer. Biochemistry 42, 10229–10237. Riva S (2006) Laccases: blue enzymes for green chemistry. Trends Biotechnol 24, 219–226. Ross RP & Claiborne A (1992) Molecular-cloning and analysis of the gene encoding the NADH oxidase from Streptococcus faecalis 10c1 ) comparison with NADH peroxidase and the flavoprotein disulfide reductases. J Mol Biol 227, 658–671. Matsumoto J, Higuchi M, Shimada M, Yamamoto Y & Kamio Y (1996) Molecular cloning and sequence analysis of the gene encoding the H2O-forming NADH oxidase from Streptococcus mutans. Biosci Biotechnol Biochem 60, 39–43. Brown DM, Upcroft JA & Upcroft P (1996) A H2Oproducing NADH oxidase from the protozoan parasite Giardia duodenalis. Eur J Biochem 241, 155–161. Kawasaki S, Ishikura J, Chiba D, Nishino T & Niimura Y (2004) Purification and characterization of an H2Oforming NADH oxidase from Clostridium aminovalericum: existence of an oxygen-detoxifying enzyme in an obligate anaerobic bacteria. Arch Microbiol 181, 324– 330. Wasserfallen A, Ragettli S, Jouanneau Y & Leisinger T (1998) A family of flavoproteins in the domains Archaea and Bacteria. Eur J Biochem 254, 325–332. Gomes CM, Giuffre A, Forte E, Vicente JB, Saraiva LM, Brunori M & Teixeira M (2002) A novel type of nitric-oxide reductase. Escherichia coli flavorubredoxin. J Biol Chem 277, 25273–25276. Silaghi-Dumitrescu R, Coulter ED, Das A, Ljungdahl LG, Jameson GNL, Huynh BH & Kurtz DM (2003) A flavodiiron protein and high molecular weight rubredoxin from Moorella thermoacetica with nitric oxide reductase activity. Biochemistry 42, 2806– 2815. Silaghi-Dumitrescu R, Kurtz DM, Ljungdahl LG & Lanzilotta WN (2005a) X-ray crystal structures of Moorella thermoacetica FprA. Novel diiron site structure and mechanistic insights into a scavenging nitric oxide reductase. Biochemistry 44, 6492–6501. Rodrigues R, Vicente JB, Felix R, Oliveira S, Teixeira M & Rodrigues-Pousada C (2006) Desulfovibrio gigas flavodiiron protein affords protection against nitrosative stress in vivo. J Bacteriol 188, 2745–2751. FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 1597 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License H. Seedorf et al. H. Seedorf et al. 27 Frazao C, Silva G, Gomes CM, Matias P, Coelho R, Sieker L, Macedo S, Liu MY, Oliveira S, Teixeira M et al. (2000) Structure of a dioxygen reduction enzyme from Desulfovibrio gigas. Nat Struct Biol 7, 1041–1045. 28 Silaghi-Dumitrescu R, Ng KY, Viswanathan R & Kurtz DM (2005b) A flavo-diiron protein from Desulfovibrio vulgaris with oxidase and nitric oxide reductase activities. Evidence for an in vivo nitric oxide scavenging function. Biochemistry 44, 3572–3579. 29 Gomes CM, Vicente JB, Wasserfallen A & Teixeira M (2000) Spectroscopic studies and characterization of a novel electron-transfer chain from Escherichia coli involving a flavorubredoxin and its flavoprotein reductase partner. Biochemistry 39, 16230–16237. 30 Thauer RK (1998) Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiology 144, 2377– 2406. 31 Lin IJ, Gebel EB, Machonkin TE, Westler WM & Markley JL (2005) Changes in hydrogen-bond strengths explain reduction potentials in 10 rubredoxin variants. Proc Natl Acad Sci USA 102, 14581–14586. 32 Wasserfallen A, Huber K & Leisinger T (1995) Purification and structural characterization of a flavoprotein induced by iron limitation in Methanobacterium thermoautotrophicum Marburg. J Bacteriol 177, 2436–2441. 33 Page CC, Moser CC, Chen XX & Dutton PL (1999) Natural engineering principles of electron tunnelling in biological oxidation–reduction. Nature 402, 47–52. 34 Seedorf H (2003) F420H2-Oxidase, ein neuartiges vier Elektronen umsetzendes Flavoprotein in methanogenen Archaea. Diploma Thesis, Philipps-University, Marburg. 35 Rudd DJ, Sazinsky MH, Merkx M, Lippard SJ, Hedman B & Hodgson KO (2004) Determination by X-ray absorption spectroscopy of the Fe–Fe separation in the oxidized form of the hydroxylase of methane monooxygenase alone and in the presence of MMOD. Inorg Chem 43, 4579–4589. 36 Sintchak MD, Arjara G, Kellogg BA, Stubbe J & Drennan CL (2002) The crystal structure of class II ribonucleotide reductase reveals how an allosterically regulated monomer mimics a dimer. Nat Struct Biol 9, 293–300. 37 Sazinsky MH & Lippard SJ (2006) Correlating structure with function in bacterial multicomponent monooxygenases and related diiron proteins. Acc Chem Res 39, 558–566. 38 Moonen CTW, Vervoort J & Muller F (1984) Reinvestigation of the structure of oxidized and reduced flavin: carbon-13 and nitrogen-15 nuclear magnetic resonance study. Biochemistry 23, 4859–4867. 39 Warkentin E, Mamat B, Sordel-Klippert M, Wicke M, Thauer RK, Iwata M, Iwata S, Ermler U & Shima S (2001) Structures of F420H2:NADP+ oxidoreductase 1598 40 41 42 43 44 45 46 47 48 49 50 51 52 with and without its substrates bound. EMBO J 20, 6561–6569. Pejchal R, Sargeant R & Ludwig ML (2005) Structures of NADH and CH3-H4 folate complexes of Escherichia coli methylenetetrahydrofolate reductase reveal a Spartan strategy for a ping-pong reaction. Biochemistry 44, 11447–11457. Kryatov SV, Rybak-Akimova EV, MacMurdo VL & Que L Jr (2001) A mechanistic study of the reaction between a diiron(II) complex [FeII(2)(mu-OH)2(6-Me3TPA)2](2+) and O2 to form a diiron(III) peroxo complex. Inorg Chem 40, 2220–2228. Costas M, Cady CW, Kryatov SV, Ray M, Ryan MJ, Rybak-Akimova EV & Que L Jr (2003) Role of carboxylate bridges in modulating nonheme diiron(II) ⁄ O(2) reactivity. Inorg Chem 42, 7519–7530. Cruz F & Ferry JG (2006) Interaction of iron–sulfur flavoprotein with oxygen and hydrogen peroxide. BBA Gen Subjects 1760, 858–864. Andrade SL, Cruz F, Drennan CL, Ramakrishnan V, Rees DC, Ferry JG & Einsle O (2005) Structures of the iron–sulfur flavoproteins from Methanosarcina thermophila and Archaeoglobus fulgidus. J Bacteriol 187, 3848– 3854. Otwinowski Z & Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26, 795–800. Kissinger CR, Gehlhaar DK & Fogel DB (1999) Rapid automated molecular replacement by evolutionary search. Acta Crystallogr D 55, 484–491. Jones TA, Zou JY, Cowan SW & Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47(2), 110–119. Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS et al. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54, 905–921. Murshudov GN, Vagin AA & Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53, 240– 255. Laskowski RA, MacArthur MW, Moss DS & Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26, 283–291. Sumner JS & Matthews RG (1992) Stereochemistry and mechanism of hydrogen transfer between NADPH and FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Structure of di-iron flavoenzyme F420H2 oxidase methylenetetrahydrofolate in the reaction catalyzed by methylenetetrahydrofolate reductase from pig liver. J Am Chem Soc 114, 6949–6959. Supplementary material The following supplementary material is available online: Fig. S1. Sequence alignment of F420H2 oxidase from Methanothermobacter marburgensis, rubredoxin:NO ⁄ O2 oxidoreductase from Desulfovibrio gigas, rubredoxin: NO ⁄ O2 oxidoreductase from Moorella thermoacetica, F420H2 oxidase from Methanobrevibacter arboriphilus, and other FprAs from methanogenic archaea assumed to have F420H2 oxidase activity. The amino acids involved in FMN binding are highlighted in yellow, and those involved in iron coordination are highlighted in red. The amino acids lining the cavity above the di-iron center are given in blue. The prominent trypto- Structure of di-iron flavoenzyme F420H2 oxidase phan between FMN and the di-iron site is in green. The two amino acids linked via a cis peptide bond are indicated by asterisks. Other amino acids conserved in all sequences are highlighted in gray. Fig. S2. Structures of the F420H2 pocket at the interface of two subunits in the functional dimer of (A) F420H2 oxidase from Methanothermobacter marburgensis, (B) rubredoxin:NO ⁄ O2 oxidoreductase from Desulfovibrio gigas, and (C) rubredoxin:NO ⁄ O2 oxidoreductase from Moorella thermoacetica. This material is available as part of the online article from http://www.blackwell-synergy.com Please note: Blackwell Publishing is not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article. FEBS Journal 274 (2007) 1588–1599 ª 2007 The Authors Journal compilation ª 2007 FEBS 1599 17424658, 2007, 6, Downloaded from https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2007.05706.x by European Molecular Biology Laboratory EMBL, Wiley Online Library on [19/02/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License H. Seedorf et al.