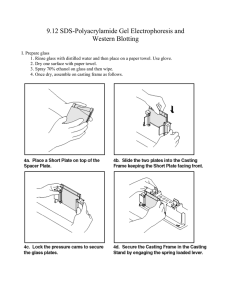
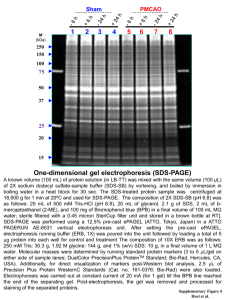
E Lab session 2: SDS-PolyAcrylamide Gel Electrophoresis (PAGE) of proteins INTRODUCTION Electrophoresis is used to separate complex mixtures of proteins, In polyacrylamide gel electrophoresis, proteins migrate in response to an electrical field through pores in the gel matrix; pore size decreases with higher acrylamide concentrations. The combination of gel pore size and protein charge, size, and shape determines the migration rate of the protein. Electrophoresis of protein samples in polyacrylamide gels is an indispensable analytical and, in some cases, preparative tool for the protein scientists. Electrophoresis can be used to separate and compare complex protein mixtures, evaluate purity of a protein during the course of its isolation, and provide estimates of physical characteristics such as subunit composition, isoelectric point, size, and charge. Each type of electrophoretic separation can be conducted in a variety of gel sizes ranging from microgels (e.g., Phast gels from Hoefer Pharmacia) only slightly larger than a postage stamp to giant gels much larger than this page (Garrels, 19791; Young et al., 19832). In general, the time required to electrophorese, stain, and destain small gels is very short, and a minimal amount of sample is consumed. In contrast, larger gels consume more reagents, sample, and time, but provide increased resolution. Small gels are therefore recommended for rapid screening, and larger gels are indicated when maximum resolution is required, as in analysis of complex mixtures or samples containing very similar components. The most common one-dimensional gel methods utilize the detergent sodium dodecyl sulfate (SDS) to solubilize, denature, and impart a strong negative charge to proteins. Although many onedimensional SDS gel methods have been published over the past 30 years, the single most widely used method is that initially described by Laemmli (1970)3. SDS-based gel separations are very robust as most, but not all, proteins are readily solubilized in SDS solutions. Most proteins bind a 1 Garrels, J.I. 1979. Two-dimensional gel electrophoresis and computer analysis of proteins synthesized by cloned cell lines. J. Biol. Chem. 54:7961-7977. 2 Young, D.A., Voris, B.P., Maytin, E.V., and Colbert, R.A. 1983. Very high resolution two-dimensional electrophoretic separation of proteins on giant gels. Methods Enzymol. 91:190-214. 3 Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. 1 Pr. Jaouad ANISSI uniform amount of SDS per microgram of protein, which imparts a uniform charge density per unit mass to provide a separation based on the mass of the polypeptide chain. ELECTRICITY AND ELECTROPHORESIS Many researchers are poorly informed concerning the electrical parameters of running a gel. It is important to note that the voltages and currents used during electrophoresis are dangerous and potentially lethal. Thus, safety should be an overriding concern. Safety Considerations 1. Never remove or insert high-voltage leads unless the power supply voltage is turned down to zero and the power supply is turned off. Always grasp high-voltage leads one at a time with one hand only. Never insert or remove high-voltage leads with both hands. This can shunt potentially lethal electricity through the chest and heart should electrical contact be made between a hand and a bare wire. On older or homemade instruments, the banana plugs may not be shielded and can still be connected to the power supply at the same time they make contact with a hand. Carefully inspect all cables and connections and replace frayed or exposed wires immediately. 2. Always start with the power supply turned off. Have the power supply controls turned all the way down to zero. Then hook up the gel apparatus: generally, connect the red highvoltage lead to the red outlet and the black high-voltage lead to the black outlet. Turn the power supply on with the controls set at zero and the high-voltage leads connected. Then, turn up the voltage, current, or power to the desired level. Reverse the process when the power supply is turned off: i.e., to disconnect the gel, turn the power supply down to zero, wait for the meters to read zero, turn off the power supply, and then disconnect the gel apparatus one lead at a time. CAUTION: If the gel is first disconnected and then the power supply turned off, a considerable amount of electrical charge is stored internally. The charge will stay in the power supply over a long time. This will discharge through the outlets even though the power supply is turned off and can deliver an electrical shock. Ohm’s Law and Electrophoresis Understanding how a gel apparatus is connected to the power supply requires a basic understanding of Ohm’s law: v𝑜𝑙𝑡𝑎𝑔𝑒 = 𝑐𝑢𝑟𝑟𝑒𝑛𝑡 × 𝑟𝑒𝑠𝑖𝑠𝑡𝑎𝑛𝑐𝑒, or 𝑉 = 𝐼𝑅. A gel can be viewed as a resistor and the power supply as the voltage and current source. Most power supplies deliver 2 Pr. Jaouad ANISSI constant current or constant voltage. Some will also deliver constant power: power = voltage × current, or 𝑉𝐼 = 𝐼 2 𝑅. The discussion below focuses on constant current because this is the most common mode in vertical SDS-PAGE. Most modern commercial equipment is color-coded so that the red or positive terminal of the power supply can simply be connected to the red lead of the gel apparatus, which goes to the lower buffer chamber. The black lead is connected to the black or negative terminal and goes to the upper buffer chamber. This configuration is designed to work with vertical slab gel electrophoreses in which negatively charged proteins or nucleic acids move to the positive electrode in the lower buffer chamber (an anionic system). When a single gel is attached to a power supply, the negative charges flow from the negative cathode (black) terminal into the upper buffer chamber, through the gel, and into the lower buffer chamber. The lower buffer chamber is connected to the positive anode (red) terminal to complete the circuit. Thus, negatively charged molecules, such as SDS-coated proteins and nucleic acids, move from the negative cathode attached to the upper buffer chamber toward the positive anode attached to the lower chamber. SDS-PAGE is an anionic system because of the negatively charged SDS. Most SDS-PAGE separations are performed under constant current (consult manufacturer’s instructions to set the power supply for constant current operation). The resistance of the gel will increase during SDS-PAGE in the standard Laemmli system. If the current is constant, then the voltage will increase during the run as the resistance goes up. Power supplies usually have more than one pair of outlets. The pairs are connected in parallel with one another internally. If more than one gel is connected directly to the outlets of a power supply, then these gels are connected in parallel. In a parallel circuit, the voltage is the same across each gel. In other words, if the power supply reads 100 V, then each gel has 100 V across its electrodes. The total current, however, is the sum of the individual currents going through each gel. Therefore, under constant current it is necessary to increase the current for each additional gel that is connected to the power supply. Two identical gels require double the current to achieve the same starting voltages and electrophoresis separation times. Multiple gel apparatuses can also be connected to one pair of outlets on a power supply. This is useful with older power supplies that have a limited number of outlets. When connecting several gel units to one outlet, make certain the connections between the units are shielded and protected from moisture. The gels can be connected in parallel or in series (Fig. 1). 3 Pr. Jaouad ANISSI Figure 1: Series and parallel connections of gel tanks to power supply. Gel thickness affects the above relationships. A 1.5-mm gel can be thought of as consisting of two 0.75-mm-thick gels run in parallel. Because currents are additive in parallel circuits, a 0.75-mm gel will require half the current of the 1.5-mm gel to achieve the same starting voltage and separation time. If a gel thickness is doubled, then the current must also be doubled. There are limits to the amount of current that can be applied. Thicker gels require more current, generating more heat that must be dissipated. Unless temperature control is available in the gel unit, a thick gel should be run more slowly than a thin gel. RAPID COOMASSIE BLUE STAINING OF PROTEIN GELS Coomassie brillant blue R250 (CBR-250) and silver staining are the most widely used methods for the routine visualization of proteins separated by SDS-PAGE. CBR-250 is an organic dye that complexes with basic amino acids, such as arginine, lysine, and histidine, as well as tyrosine. Conventional CBR250 staining is capable of detecting as little as 30-100 ng of protein, but sensitivity can be improved by performing the staining and destaining at elevated temperatures. NOTE: Milli-Q-purified water or equivalent should be used throughout the protocols. PRACTICE: DENATURING (SDS) DISCONTINUOUS GEL ELECTROPHORESIS OBJECTIVE: Separate and characterize a mixture of proteins along with standard proteins using SDS-PAGE. Introduction One-dimensional gel electrophoresis under denaturing conditions (i.e., in the presence of 0.1% SDS) separates proteins based on molecular size as they move through a polyacrylamide gel matrix 4 Pr. Jaouad ANISSI toward the anode. The polyacrylamide gel is cast as a separating gel (sometimes called resolving or running gel) topped by a stacking gel and secured in an electrophoresis apparatus. After sample proteins are solubilized by boiling in the presence of SDS, an aliquot of the protein solution is applied to a gel lane, and the individual proteins are separated electrophoretically. 2Mercaptoethanol (2-ME) or dithiothreitol (DTT) is added during solubilization to reduce disulfide bonds. Materials Chemicals - Acrylamide Solution, Ammonium Persulfate (APS) 10% Bis-Acrylamide BPB DL-Dithiothreitol (DTT) Glycerol Glycine PBS, 10X Solution, pH 7.4 Sodium Dodecyl Sulfate (SDS) Solution TEMED (Tetramethylethylenediamine) Thiourea (Thiocarbamide) Tricine Tris Tris-Glycine-SDS Buffer, 10X Solution Reagents Separating and stacking gel solutions (Table 1) H2O-saturated isobutyl alcohol orH2O 1.5 MTris⋅Cl/SDS, pH 8.8 (Table 1) 0.5 Tris⋅Cl/SDS, pH 6.8 Ammonium Persulfate (APS) 10% Protein samples to be analyzed 2× and 1× SDS sample buffer (see REAGENTS AND SOLUTIONS) Protein molecular-weight-standards mixture (Table 2) 2× SDS sample buffer (see REAGENTS AND SOLUTIONS) 1× SDS electrophoresis buffer (see REAGENTS AND SOLUTIONS) Electrophoresis apparatus: SE400 Air-Cooled Vertical Protein Electrophoresis Unit Glass Plates, 18 x 16 cm 0.45-μm filters (used in stock solution preparation) 25-ml Erlenmeyer side-arm flask Vacuum pump with cold trap 1.5-mm Teflon comb with 1, 3, 5, 10, 15, or 20 teeth Micropipette Constant-current power supply. 5 Pr. Jaouad ANISSI Table 1 : Recipes for Polyacrylamide Separating (resolving) and Stacking Gelsa Pour the separating gel 1. Assemble the glass-plate sandwich of the electrophoresis apparatus according to manufacturer’s instructions using two clean glass plates and two 1.0-mm spacers. If needed, clean the glass plates in liquid Alconox or RBS-35 (Pierce). These aqueous-based solutions are compatible with silver and Coomassie blue staining procedures. Figure 2 : Assembled SE400 Air-Cooled Vertical Protein Electrophoresis Unit 2. Lock the sandwich to the casting stand. 3. Prepare the separating gel solution as directed in Table 1, degassing using a rubber-stoppered 25-ml Erlenmeyer side-arm flask connected with vacuum tubing to a vacuum pump with a cold trap. After adding the specified amount of 10% ammonium persulfate and TEMED to the degassed solution, stir gently to mix. Table 1 was prepared as a convenient summary to aid in the preparation of separating and stacking gels. The stacking gel is the same regardless of the 6 Pr. Jaouad ANISSI separating gel used. The desired percentage of acrylamide in the separating gel depends on the molecular size of the protein being separated. Generally, use 5% gels for SDS-denatured proteins of 60 to 200 kDa, 10% gels for SDS-denatured proteins of 16 to 70 kDa, and 15% gels for SDS-denatured proteins of 12 to 45 kDa (Table 1). 4. Using a Pasteur pipet, apply separating gel solution to the sandwich along an edge of one of the spacers until the height of the solution between the glass plates is ∼3/4. Use the solution immediately; otherwise, it will polymerize in the flask. Sample volumes <10 μl do not require a stacking gel. In this case, cast the resolving gel as you normally would, but extend the resolving gel into the comb to form the well. The proteins are then separated under the same conditions as used when a stacking gel is present. Although this protocol works well with singleconcentration gels, a gradient gel is recommended for maximum resolution. 5. Using another Pasteur pipet, slowly cover the top of the gel with a layer (∼1.5 cm thick) of H2Osaturated isobutyl alcohol, by gently layering the isobutyl alcohol against the edge of one and then the other of the spacers. Be careful not to disturb the gel surface. The overlay provides a barrier to oxygen, which inhibits polymerization, and allows a flat interface to form during gel formation. The H2O-saturated isobutyl alcohol is prepared by shaking isobutyl alcohol and H2O in a separatory funnel. The aqueous (lower) phase is removed. This procedure is repeated several times. The final upper phase is H2O-saturated isobutyl alcohol. 6. Allow the gel to polymerize 30 to 60 min at room temperature. A sharp optical discontinuity at the overlay/gel interface will be visible on polymerization. Failure to form a firm gel usually indicates a problem with the ammonium persulfate, TEMED (N,N,N′,N′- tetramethylethylenediamine), or both. Ammonium persulfate solution should be made fresh before use. Ammonium persulfate should “crackle” when added to the water. If not, fresh ammonium persulfate should be purchased. Purchase TEMED in small bottles so, if necessary, a new previously unopened source can be tried. Pour the stacking gel 7. Pour off the layer of H2O-saturated isobutyl alcohol and rinse with 0.5M Tris⋅Cl/SDS, pH 8.8. Residual isobutyl alcohol can reduce resolution of the protein bands; therefore, it must be completely removed. The isobutyl alcohol overlay should not be left on the gel longer than 2 hr. 8. Prepare the stacking gel solution as directed in Table 1. Use the solution immediately to keep it from polymerizing in the flask. 7 Pr. Jaouad ANISSI 9. Using a Pasteur pipet, slowly allow the stacking gel solution to trickle into the center of the sandwich along an edge of one of the spacers until the height of the solution in the sandwich is ∼1 cm from the top of the plates. Be careful not to introduce air bubbles into the stacking gel. 10. Insert a 0.75-mm Teflon comb into the layer of stacking gel solution. If necessary, add additional stacking gel to fill the spaces in the comb completely. Again, be careful not to trap air bubbles in the tooth edges of the comb; they will cause small circular depressions in the well after polymerization that will lead to distortion in the protein bands during separation. 11. Allow the stacking gel solution to polymerize 30 to 45 min at room temperature. A sharp optical discontinuity will be visible around wells on polymerization. Prepare the sample and load the gel 12. Dilute a portion of the protein sample to be analyzed 1:1 (v/v) with 2× SDS sample buffer and heat 3 to 5 min at 100°C in a sealed screw-cap microcentrifuge tube. If the sample is a precipitated protein pellet, dissolve the protein in 50 to 100 μl of 1× SDS sample buffer and boil 3 to 5 min at 100°C. Dissolve protein-molecular-weight standards mixture in 1× SDS sample buffer according to supplier’s instructions as a control (Table 2). For dilute protein solutions, consider adding 5:1 protein solution/6× SDS sample buffer to increase the amount of protein loaded. Proteins can also be concentrated by precipitation in acetone, ethanol, or trichloroacetic acid (TCA), but losses will occur. For a 0.8-cm-wide well, 25 to 50 μg total protein in <20 μl is recommended for a complex mixture when staining with Coomassie blue, and 1 to 10 μg total protein is needed for samples containing one or a few proteins. If silver staining is used, 10- to 100-fold less protein can be applied (0.01 to 5 μg in <20 μl depending on sample complexity). To achieve the highest resolution possible, the following precautions are recommended. Prior to adding the sample buffer, keep samples at 0°C. Add the SDS sample buffer (room temperature) directly to the 0°C sample (still on ice) in a screw-top microcentrifuge tube. Cap the tube to prevent evaporation, vortex, and transfer directly to a 100°C water bath for 3 to 5 min. Let immunoprecipitates dissolve for 1 hr at 56°C in 1× SDS sample buffer prior to boiling. DO NOT leave the sample in SDS sample buffer at room temperature without first heating to a 100°C to inactivate proteases (see Critical Parameters and Troubleshooting). Endogenous proteases are very active in SDS sample buffer and will cause severe degradation of the sample proteins after even a few minutes at room temperature. To test 8 Pr. Jaouad ANISSI for possible proteases, mix the sample with SDS sample buffer without heating and leave at room temperature for 1 to 3 hr. A loss of high-molecular-weight bands and a general smearing of the banding pattern indicate a protease problem. Once heated, the samples can sit at room temperature for the time it takes to load samples. 13. Carefully remove the Teflon comb without tearing the edges of the polyacrylamide wells. After the comb is removed, rinse wells with 1× SDS electrophoresis buffer. The rinse removes unpolymerized monomer; otherwise, the monomer will continue to polymerize after the comb is removed, creating uneven wells that will interfere with sample loading and subsequent separation. 14. Using a Pasteur pipet, fill the wells with 1× SDS electrophoresis buffer. If well walls are not upright, they can be manipulated with a flat-tipped needle attached to a syringe. Table 2 : Molecular Weights of Protein Standards for Polyacrylamide Gel Electrophoresisa 15. Attach gel sandwich to upper buffer chamber using manufacturer’s instructions. 16. Fill lower buffer chamber with the recommended amount of 1× SDS electrophoresis buffer. 17. Place sandwich attached to upper buffer chamber into lower buffer chamber. 9 Pr. Jaouad ANISSI 18. Partially fill the upper buffer chamber with 1× SDS electrophoresis buffer so that the sample wells of the stacking gel are filled with buffer. Monitor the upper buffer chamber for leaks and if necessary, reassemble the unit. A slow leak in the upper buffer chamber may cause arcing around the upper electrode and damage the upper buffer chamber. 19. Using a 25- or 100-μl syringe with a flat-tipped needle, load the protein sample(s) into one or more wells by carefully applying the sample as a thin layer at the bottom of the wells. Load control wells with molecular weight standards. Add an equal volume of 1× SDS sample buffer to any empty wells to prevent spreading of adjoining lanes. Preparing the samples at approximately the same concentration and loading an equal volume to each well will ensure that all lanes are the same width and that the proteins run evenly. If unequal volumes of sample buffer are added to wells, the lane with the larger volume will spread during electrophoresis and constrict the adjacent lanes, causing distortions. The samples will layer on the bottom of the wells because the glycerol added to the sample buffer gives the solution a greater density than the electrophoresis buffer. The bromphenol blue in the sample buffer makes sample application easy to follow visually. 20. Fill the remainder of the upper buffer chamber with additional 1× SDS electrophoresis buffer so that the upper platinum electrode is completely covered. Do this slowly so that samples are not swept into adjacent wells. Run the gel 21. A. Stain gel in coomassie stain solution at room temperature overnight. Gels can also be stained rapidly by placing them at 55 °C in a shaking water bath for 30– 45 min. B. Place gel in Destain solution I at room temperature. Change the destain solution when it reaches a deep blue color until clear background results. C. Store the gel in Destain solution II. For a more sensitive method, silver stain protocol is recommended. REAGENTS AND SOLUTIONS 10 Pr. Jaouad ANISSI Use Milli-Q-purified water or equivalent for the preparation of all buffers. For common stock solutions, 30% acrylamide/0.8% bisacrylamide The Laemmli system is the most common electrophoresis protocol for SDS-denatured proteins. The leading ion in this discontinuous buffer system is chloride and the trailing ion is glycine. Accordingly, the resolving gel and the stacking gel contain Tris-Cl buffers (of different concentration and pH), and the electrophoresis buffer contains Tris-glycine. All buffers contain 0.1% SDS. Polyacrylamide gel composition is indicated by two different percentages: The total percent of acrylamide (%T) in the resolving gel, which can range from 4 to 20%, determines the pore size. Commonly, the amount of crosslinker used (%C) is 2.6%. - Mix 30.0 g acrylamide and 0.8 g N,N′-methylenebisacrylamide with H2O in a total volume of 100 ml. - Filter the solution through a 0.45-μm filter and store at 4°C in the dark. - Discard after 30 days, as acrylamide gradually hydrolyzes to acrylic acid and ammonia. CAUTION: Acrylamide monomer is neurotoxic. Mask should be worn when weighing acrylamide powder. Gloves should be worn while handling the solution, and the solution should not be pipetted by mouth. 4× Tris⋅Cl/SDS, pH 6.8 (0.5 M Tris⋅Cl containing 0.4% SDS) Dissolve 6.05 g Tris base in 40 ml H2O. Adjust to pH 6.8 with 1 N HCl. Add H2O to 100 ml total volume. Filter the solution through a 0.45-μm filter, add 0.4 g SDS, and store at 4°C up to 1 month. 4× Tris⋅Cl/SDS, pH 8.8 (1.5 M Tris⋅Cl containing 0.4% SDS) Dissolve 91 g Tris base in 300 ml H2O. Adjust to pH 8.8 with 1 N HCl. Add H2O to 500 ml total volume. Filter the solution through a 0.45-μm filter, add 2 g SDS, and store at 4°C up to 1 month. 11 Pr. Jaouad ANISSI a. The recipes produce 15 ml of separating gel and 5 ml of stacking gel, which are adequate for a gel of dimensions 0.75 mm × 14 cm × 14 cm. The recipes are based on the SDS (denaturing) discontinuous buffer system of Laemmli (1970). b. All reagents and solutions used in the protocol must be prepared with Milli-Q-purified water or equivalent. c. Units of numbers in table body are milliliters. The desired percentage of acrylamide in the separating gel depends on the molecular size of the protein being separated. See annotation to step 3, Basic Protocol 1. d. Best to prepare fresh. Preparation of separating gel In a 200-ml side-arm flask, mix reagents in table 1: - MilliQ-H2O. - 30% acrylamide/0.8% bisacrylamide solution, - 4× 1.5 M Tris⋅Cl/SDS, pH 8.8, - Degas under vacuum about 5 min. - Add 10% ammonium persulfate and TEMED. - Swirl gently to mix. - Use immediately. STACKING GEL (3.9% acrylamide) - In a 200-ml side-arm flask, mix reagents in table 1: - MilliQ-H2O. - 30% acrylamide/0.8% bisacrylamide solution, - 4× 1.5 M Tris⋅Cl/SDS, pH 8.8, - Degas under vacuum about 5 min. - Add 10% ammonium persulfate and TEMED. - Swirl gently to mix. - Use immediately. - Use immediately. Failure to form a firm gel usually indicates a problem with the persulfate, TEMED, or both. REAGENTS USED IN GELS 12 Pr. Jaouad ANISSI SDS electrophoresis buffer, - 5×15.1 g Tris base (0.125 M final) - 72.0 g glycine (0.96 M final) - 5.0 g SDS, 0.5% final; - H2O to 1000 ml - Dilute to 1× or 2× for working solution, as appropriate Do not adjust the pH of the stock solution, as the solution is pH 8.3 when diluted. Store at 0° to 4°C until use (up to 1 month). SDS sample buffer, 2× (for discontinuous systems) - 25 ml 4× Tris⋅Cl/SDS, pH 6.8 (Table 10.2A.1) - 20 ml glycerol (20% final) - 4 g SDS [4% final] - 2 ml 2-ME or 3.1 g DTT (0.2% 2-ME or 0.2 M DTT final) - 1 mg bromphenol blue (0.001% final) - Add H2O to 100 ml and mix - Store in 1-ml aliquots at −70°C Coomassie blue G-250 staining solution - 200 ml acetic acid - 1800 ml H2O - 0.5 g Coomassie blue G-250 - Mix 1 hr and filter (Whatman no. 1 paper) - Store at room temperature indefinitely - The final solution is 0.025% (w/v) Coomassie blue G-250 in 10% (v/v) acetic acid. Gel-fixing solution: Add 500mL of USP-grade 95% (v/v) ethanol to 300 mL of HPLC grade water. Add 100 mL of reagent grade acetic acid and adjust the total volume to 1000 mL with water. The final concentrations are 50% (v/v) ethanol in water with 10% (v/v) acetic acid. Gel-washing solution: Add 500mL of HPLC-grade methanol to 300 mL of HPLC grade water. Add 100mL of reagent grade acetic acid and adjust the total volume to 1000 mL with HPLC grade water. The final concentrations are 50% (v/v) methanol in water with 10% (v/v) acetic acid. 13 Pr. Jaouad ANISSI Stain solution: Dissolve 0.4g of Coomassie blue R250 in 200 mL of 40% (v/v) HPLC grade methanol in water with stirring as needed. Filter the solution to remove any insoluble material. Add 200mL of 20% (v/v) acetic acid in water. The final concentration is 0.1% (w/v) Coomassie blue R350, 20% (v/v) methanol, and 10% (v/v) acetic acid. Destaining solution: Add 500mL of HPLC- grade methanol to 300 mL of HPLC grade water. Add 100 mL of reagent grade acetic acid and, after mixing, adjust the total volume to 1000mL with water. The final concentrations are 50% (v/v) methanol in water with 10% (v/v) acetic acid. Storage solution: Add 25mL of reagent grade acetic acid to 400mL of HPLC grade water. After mixing, adjust the final volume to 500mL with water. The final concentration of acetic acid is 5% (v/v). 14 Pr. Jaouad ANISSI REPORT: 1. Calculate the missing concentrations of each reagent for a volume of 30 mL resolving gel solution, 5 ml stacking gel solution: Reagent Initial concentration pH Final concentration Milli Q-H2O Acrylamide-Bisacrylamide solution Tric-Cl Ammonium persulfate SDS TEMED (99% purity, density=775 kg/m³, MW 116.24 g/mol) ≈55.5 M? 30 % acrylamide, 0.8% Bis-acrylamide 1.5 M 10% 10% 8.8 - ≈55.5 M? Reagent Initial concentration pH Final concentration Milli Q-H2O Acrylamide-Bisacrylamide solution Tric-Cl SDS Ammonium persulfate TEMED (99% purity, density=775 kg/m³, MW 116.24 g/mol) ≈55.5 M? 30 % acrylamide, 0.8% Bis-acrylamide 0.5 M 10% 10% 6.8 - ≈55.5 M? 2. Calculate the table below for each sample loaded in each well: Sample Name Loaded amount (µg) Sucrase Volume loaded (µL) 100 100 100 100 100 100 100 BSA (Bovine Serum albumin) Control 3. Paste the photography of the stained gel 15 Pr. Jaouad ANISSI Rf Volume (mL) 9.60 12.50 7.50 0.15 0.30 0.01 Volume (mL) 6.00 1.34 1.25 0.10 0.05 0.01 3.1. Describe and interpret the obtained result …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… …………………………………………………………………………………………………………… 16 Pr. Jaouad ANISSI