Introduction to Pediatric Hematology - Pedia 2 (1)
advertisement
")
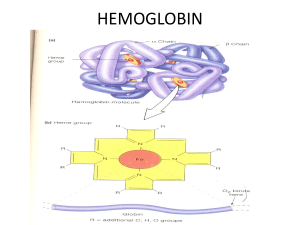
Introduction to Pediatric Hematology F Domiline C. Arca, MD, DPPS, DPSHBT, DPSPH Head, Section of Hematology Department of Pediatrics University of Perpetual Help DALTA Medical Center Goals: Learn approach to anemia and RBC disorders Be able to fully evaluate a complete blood count Understand the etiology of common hematologic conditions Know when to refer Definition: Hematology is the study of the normal and pathologic aspects of blood and blood elements Hematopoietic Stem Cells (HSC) Progenitor cells in the blood Derived from hemangioblast in the aorta-mesonephros-gonad (AGM) region o Give rise to the blood system that seeds the liver and then the bone marrow The Myeloid System From medullary cavity Consists of o RBCs (erythrocytes) o WBCs (neutrophils, monocyte, eosinophils, basophils) o Platelets (thrombocytes) Anemia Extremely common Worldwide, 1/3 of the population is anemic Definition of anemia: Hemoglobin (Hb) concentration more than two standard deviations below the normal for age and gender Anemia: Important clues: Patient’s history Physical examination Laboratory study Peripheral blood smear Symptoms: Hypotension, Orthostasis, Syncope, Shock → Hypovolemia 2° to acute hemorrhage Fatigue, Dyspnea, Light-headedness, Cognitive abnormalities, Ischemic pain→ Tissue hypoxia 2° to ↓ O2-carrying capacity of blood Tachycardia, Palpitations , Congestive heart failure→ Cardiovascular manifestations Note: Chronic anemia of gradual onset is generally well tolerated until the Hb declines to very low levels (≤8g/dl) Adaptive physiologic responses to anemia ↑ cardiac output due to ↑ heart rate (mild to moderate anemia) ↑ ventricular stroke volume (Severe anemia) Reflex vasoconstriction → Triggered by acute blood loss and hypovolemia Decrease vascular resistance →In chronic anemia Cerebral blood flow is preserved Decreased renal blood flow Expansion of intravascular volume (Kidneys retain salt & water) Unloading of O2 in tissues (↑levels of 2,3DPG) ↑ erythropoietin (Leads to ↑ RBC production) Laboratory Diagnostics CBC, platelet count Red cell indices – MCV, MCH, MCHC Peripheral Blood Smear reading: RBC morphology: Hypochromia, microcytosis, anisocytosis, poikilocytosis, target cells, polychromasia, inclusion bpdies WBC – estimated count, differential count including abnormal cells, toxic granulations, septic picture, ANC Platelets – estimated count: singly, in clumps, giant forms Laboratory Diagnosis of Iron Deficiency Anemia *increased RDW Decreased Hb, Hct, RBC count, retic count RBC morphology: hypochromia, microcytosis Decreased MCHC Decreased Serum Iron Increased TIBC Decreased serum ferritin Diagnostic Work-Up of Hemolytic Anemia Peripheral smear Reticulocyte stain Blood typing Coombs’ test Osmotic Fragility Test - (autohemolysis test) Hemoglobin electrophoresis - (Kleihauer method for fetal Hb) G6PD assay Bleeding Profile: CBC, platelet count Peripheral smear Bleeding time Prothrombin Time Partial Thromboplastin Time Thrombin Time Fibrinogen Fibrin Degradation Products Fibrin Monomers Bone marrow aspiration/biopsy When to do it – when 2 cell lines are down Helpful in: Very severe aplastic anemia, Acute leukemia, Lymphoma and solid tumors Bone marrow aspiration/biopsy: Very severe aplastic anemia - Pancytopenia , Bone marrow biopsy: empty marrow spaces, many fat spaces Acute leukemia: Bone marrow aspiration: (+) blasts Lymphoma and solid tumors: Bone marrow aspiration/biopsy: (+) metastatic cells in the marrow Etiologic Classification of Anemia Impaired Red Cell Formation: Deficiency, Bone marrow failure Blood Loss Hemolytic Anemia Nutritional Anemia: Iron Deficiency Anemia Still the most common cause of anemia in infants, children, and adolescents in our country Associated with cognitive and psychomotor abnormalities in children Most common cause: Inadequate dietary intake Iron Deficiency Anemia: Recommendations Source: 7th Edition of Preventive Pediatric Health Care Handbook 2014 DOH →- Recommends oral iron supplementation for infants, children, adolescents PPS and PSHBT; 6th Nat’l Nutrition Survey, Philippines 2003→ Recommends CBC monitoring at least once between 6-24 months, at least once bet 2-6 years, at least once bet 10-19 years for those at risk - Philippine Society of Adolescent Medicine Specialists - Recommend CBC at each stage of adolescence Iron Deficiency Anemia Diagnosis: Poor nutritional history Signs/symptoms of anemia Laboratory tests Positive response to oral iron supports the diagnosis Treatment of IDA Oral iron therapy o Elemental iron (ferrous sulfate, ferrous gluconate, ferrous fumarate) o Dose: 4.5-6.0 mg/kg/day in divided doses between meals in infants; 100-200 mg/day in older children Dietotherapy - Iron-fortified formulas, cereals; iron-rich foods Causes of Failure of Iron Therapy 1. Noncompliance 2. Timing of administration * empty stomach *Enhancers – low gastric pH, ascorbic acid, iron deficiency state 3. Deterrents – phytates, antacids, gluten diet 4. Iron given in a form poorly absorbed 5. Continuing unrecognized blood loss 6. Insufficient duration of therapy – at least 2 weeks 7. Inhibitors of iron absorption/utilization - lead, aluminum intoxication, chronic inflammation, neoplasms 8. Incorrect diagnosis - therapeutic failure of iron medication Note: * If causes 1 – 7 are ruled out, and the patient was given oral iron for about a month already and still NO response, consider another diagnosis like thalassemia. Refer to the Hematologist. Other nutritional anemia Megaloblastic anemia Cobalamin (B12) Deficiency Folic Acid deficiency Differential Diagnoses of Microcytic Anemias Iron deficiency Thalassemias Hemoglobinopathies Vitamin B6 deficiency Lead poisoning Sideroblastic anemias Hemolytic Anemia Hemoglobin defects Membrane defects Enzymatic defect Immune Clinical Triad of Hemolytic Anemia Pallor or anemia Jaundice Splenomegaly (may be absent in the newborn) Diagnostic Work-up of Hemolytic Anemia Clinical Triad of HA Presence of Hemolysis Blood typing Coombs’ Test (Direct & Indirect) ___________________________________ Positive Negative AIHA Osmotic Fragility Test _____________________________ Positive Negative Hereditary Spherocytosis Hemoglobin Electrophoresis _______________________________ Positive Negative Thalassemias Hemoglobinopathies G6PD assay ________________ Positive Negative G6PD deficiency Other causes (Infection) Long term care of children/adolescents with hemolytic anemias: 1. Monitor the growth curves, Hb, absolute reticulocyte counts, and size of the spleen. - Ultrasound of hepatobiliary tree for stones (bilirubin) 2. Transfuse PRBC only for symptomatic anemia to avoid iron overloading and toxicity. 3. Monitor patients for possible crises: hemolytic, aplastic, megaloblastic. 4. Give folic acid. DO NOT give oral iron. 5. When to do a splenectomy Hemolytic Anemia: Hemoglobin defects Thalassemia – a quantitative defect; reduced synthesis of globin chains, alpha or beta Hemoglobinopathy – a qualitative defect; substitution in amino acid sequence in globin chains May see combinations Ex. HbE-Beta thalassemia, which behaves like a thal major Thalassemias 2nd most common microcytic anemia A cause of hemolytic anemia A hemoglobin defect – decreased synthesis of either alpha or beta globin chains in the Hemoglobin molecule 2 types: Alpha thalassemia & Beta thalassemia Diagnostic test: Hemoglobin electrophoresis Pathophysiology: Reduced or absent synthesis of globin chains in the Hb molecule, alpha or beta (quantitative defect). Caused by mutations in the genes for the alpha and beta globin chains, resulting in severe ineffective erythropoiesis and extraordinary marrow expansion. Classification: Alpha Thalassemia Syndromes Silent carrier Thalassemia trait Hemoglobin H disease (4) Hydrops fetalis Beta Thalassemia Syndromes Silent carrier Thalassemia trait Thalassemia intermedia Thalassemia major Thalassemia Major “Cooley’s anemia” in 1925 Mode of inheritance: Usually autosomal recessive Usually diagnosed between 6 months and 2 years of age Pallor, jaundice, splenomegaly (clinical triad); also with hepatomegaly Craniofacial changes (classic thalassemic facies) Stunting of growth Diagnostic procedure: Hemoglobin electrophoresis Hb A1 (2 2) NV 96-98% Hb F (22) NV 1-2% Hb A2 (22) NV 2-3% Other laboratory tests: RBC morphology, tests for fetal Hb, skull x-ray (“hair-on-end” appearance), serum iron, serum ferritin Anticipatory Care: Hypertransfusion, supertransfusion to suppress erythropoiesis Folic acid supplementation Genetic counseling Hypertransfusion vs Supertransfusion Hypertransfusion hemoglobin level at >10 g/dL, mean of 12 g/dL if initiated in the first year of life: promotes normal initial growth and development limits the development of hepatosplenomegaly prevents disfiguring bone abnormalities reduces intestinal iron absorption decreases cardiac workload Supertransfusion pretransfusion hgb level >12 g/dL, mean of 14 g/dL more effectively eliminate chronic tissue hypoxia increase the quantity of transfused blood, increase iron loading no longer recommended Hypertransfusion, supertransfusion: Transfusion management and the optimal maintenance level of hemoglobin have evolved. Wolman and associates first recommended a pretransfusion hemoglobin level of 8.5 g/dL. This approach improved survival, but chronic illness, bone disease, and anemic cardiomyopathy persisted. To enhance quality of life, Piomelli and colleagues suggested maintaining the hemoglobin level at greater than 10 g/dL with a mean of 12 g/dL. Such “hypertransfusion,” if initiated in the first year of life, promotes normal initial growth and development, limits the development of hepatosplenomegaly, prevents disfiguring bone abnormalities, reduces intestinal iron absorption, and decreases cardiac workload. In 1980, Propper and colleagues proposed a “supertransfusion” program with a pretransfusion hemoglobin level greater than 12 g/dL and a mean of 14 g/dL to more effectively eliminate chronic tissue hypoxia. Initial studies reported that the quantity of blood required to maintain a higher hemoglobin level was no greater than that required for maintenance of a lower level because of a decrease in intravascular volume. Further data, however, suggested that increasing the pretransfusion hemoglobin level may simply increase the quantity of transfused blood and thus increase iron loading, and this regimen is no longer recommended Treatment: PRBC transfusion for symptomatic anemia Iron chelation (desferrioxamine,deferasirox, deferiprone), Vitamin C Splenectomy – hypersplenism, transfusion requirement(>250 ml/kg per year) Allogeneic bone marrow transplantation Gene therapy Other Causes of Hemolytic Anemia: G6PD deficiency Other Causes of Hemolytic Anemia Hereditary Spherocytosis - A red cell membrane defect - spectrin, ankyrin - Autosomal dominant in inheritance - Generally mild, well-compensated hemolysis - Splenectomy is curative Cause: RBC membrane defects Pathophysiology: - Osmotically fragile, spherical cells selectively trapped in the spleen. - Splenic sequestration and conditioning metabolic depletion, spheroidicity, hastened demise. Hereditary Spherocytosis Mode of inheritance: 75 % autosomal dominant w/ variable penetrance; (+) family history 25 % de novo mutation or recessive Hemolysis can be further triggered by infections – viral, bacterial Hereditary Spherocytosis Symptoms: Pallor, jaundice, splenomegaly → clinical triad of HA Formation of pigment gallstones Due to high turnover of heme Leg ulcers → In severe anemia Clinical classification: silent carrier mild moderate moderately severe severe * May be a cause of neonatal jaundice Hereditary Spherocytosis Diagnostic procedure: (+) Osmotic Fragility Test: incubated and unincubated phases Others: (+) spherocytes on peripheral smear, MCHC (+) autohemolysis test Hereditary Spherocytosis Anticipatory Guidance: Increase host resistance to prevent viral infections oral folic acid genetic counseling Other Causes of Hemolytic Anemia G6PD deficiency- A red cell enzyme defect Mode of inheritance: X-linked; random inactivation of the X chromosome Pathophysiology: Deficiency of the G6PD enzyme causes oxidant damage to the red cell, resulting in hemolysis Several mutants or variants of the G6PD enzyme G6PD Deficiency Symptomatology: Pallor, jaundice, splenomegaly Splenomegaly may be absent in the newborn Prolonged jaundice in the newborn period should make one suspect G6PD deficiency – exposure to naphthalene balls G6PD Deficiency Diagnostic procedure: G6PD assay, included in the Newborn Screening Tests Heinz bodies on reticulocyte stain Prevention: Avoidance of agents/conditions, Genetic counseling Agents/conditions commonly associated with hemolysis in G6PD-deficient individuals Drugs – Sulfonamides Antibacterials – Chloramphenicol Antimalarials Chemicals – Benzene, Naphthalene, Phyenylhydrazine, toluidine blue, TNT Infections – Viral hepatitis, pneumococcus Diabetic acidosis G6PD Deficiency Treatment: Removal of causative agent In the newborn period, naphthalene balls. Exchange transfusion in severe cases in the newborn. Transfusion of PRBC for symptomatic anemia Vitamin E? G6PD Deficiency Rare complication: Acute renal failure Prognosis: Good → compatible with a long life → avoidance of agents/conditions is very important long term care of children/adolescents with hemolytic anemias: 1. Monitor the growth curves, Hb, absolute reticulocyte counts, and size of the spleen. - Ultrasound of hepatobiliary tree for stones (bilirubin) 2. Transfuse PRBC only for symptomatic anemia to avoid iron overloading and toxicity. 3. Monitor patients for possible crises: hemolytic, aplastic, megaloblastic. 4. Give folic acid. DO NOT give oral iron. 5. When to do a splenectomy Approach to Bleeding BLEEDING DISORDERS • Factor deficiency • Von Willebrand Disease • Disorders of Fibrinogen • Congenital • Acquired • • • • • Anti-coagulation defects Disseminated Intravascular Coagulation Liver disease Vitamin K deficiency Massive transfusion Clinical Evaluation of a Bleeding patient Get a detailed History What is abnormal bleeding? Epistaxis unrelieved by 15 minutes of pressure Menstrual periods > 7 days associated with clots, saturation of pads or frequently stained clothing Detailed History What is abnormal bleeding? Bleeding from dental procedures Lasting beyond day of procedure Requiring a blood transfusion Ecchymoses size or character inconsistent with trauma What is abnormal bleeding? CLUE: + previous surgery or dental procedure without bleeding complications unlikely to have congenital bleeding disorder Family History - Important because most children have not encountered severe hemostatic challenges Ask about: Previous surgical procedures/dental extractions Transfusions Menstrual and obstetric histories 20% of girls with menorrhagia from menarche have congenital bleeding disorder Type of Bleeding Time of onset TIME OF ONSET TYPE OF BLEEDING Coagulation factor deficiencies (e.g. hemophilia) Platelet Disorder (or VWD) Mucosal membrane bleeding Gingival hemorrhage Epistaxis Menorrhagia Petechiae Bruising Acute * Deep muscle bleeding o Joint bleeding Acquired disorder Chronic Congenital disorder ITP Vitamin K deficiency VWD Coagulation factor deficiencies * over days to weeks Time of Onset Congenital Coagulation disorders At birth Birth process During circumcision In the first months of life With immunizations When they become mobile Experience mild trauma * Factor XIII deficiency Good initial hemostasis followed by persistent oozing (failure to form a firm clot) DETAILED HISTORY Liver disease Decreased production of coagulation factors Thrombocytopenia 2 splenic sequestration Malabsorption Renal disease Abnormal platelet function Impaired Vit K absorption Otherwise well ITP Congenital bleeding disorders PHYSICAL EXAMINATION Petechiae < 2mm Ecchymoses > 1 cm Purpura 2mm – 1 cm PHYSICAL EXAMINATION HEMARTHROSIS Hematoma PHYSICAL EXAMINATION HEMARTHROSIS PHYSICAL EXAMINATION INTRAMUSCULAR BLEED, PSOAS Laboratory Evaluation Initial lab tests CBC with platelet PTT (intrinsic) PT (extrinsic) PT Prothrombin time 7.5,10.2,1 Tests Extrinsic pathway: Factors VII, V, X, II, I aPTT activated Partial Thromboplastin Time Measures intrinsic pathway: Factors XII, XI, IX, VIII, X, V, II, I Bleeding Profile CBC, platelet count Thrombin Time Peripheral smear Fibrinogen Bleeding time Fibrin Degradation Products Prothrombin Time Fibrin Monomers Partial Thromboplastin Time DIFFERENTIAL DIAGNOSIS BASED ON INITIAL SCREEN ↑PT Normal plt, PTT ↑ PTT Normal plt, PT ↑ PT,PTT Normal plt Early Liver Dx Vit K Def F VIII def Vit K Def Liver Disease F VII Def Warfarin PT inhibitor (hemophilia or VWD) F IX, XI F XII def PTT inhibitor Massive Transfusion Oral anticoagulant (F II, V, X or fibrinogen def) DIFFERENTIAL DIAGNOSIS BASED ON INITIAL SCREEN ↑ PTT, PT All normal Platelet dec VWD ITP Platelet fxn d/o Mild factor def (VIII, IX, XI, XIII ) Fibrinolytic defect Collagen Disorder Vitamin C def Infection CVD Early BM failure and low plt DIC Liver Dysfxn Inherited platelet disorder (CAMT, TAR,BSS WAS, GPS) CAMT = Congenital Amegakaryocytic Thrombocytopenia TAR – Thrombocytopenia with Absent Radius WAS = Wiskott Aldrich Syndrome GPS = Gray Platelet Syndrome Von Willebrand Disease Most common inherited bleeding disorder Prevalence: 1% - by lab def’n only 10% symptomatic Quantitative or Qualitative deficiency of vWF Symptoms: Easy bruising Epistaxis from childhood Menorrhagia The Von Willebrand Factor Protein in plasma Function - Glues platelets to damaged endothelium - Binds and protects Factor VIII vWD- Classification Type 1 Classic ; 80% of patients Partial quantitative deficiency Type 2 Dysfunctional VWF Type 3 Nearly COMPLETE deficiency vWD-Inheritance Mostly Autosomal Dominant; can be Autosomal Recessive Theoretically, equal males and females But more females diagnosed (menorrhagia) Can be acquired rare Hypothyroidism, Wilms tumor, Cardiac disease, Renal disease or SLE / Valproic acid Most often caused by Ab to VWF vWD- S/Sx Increased bruising and excessive epistaxis Prolonged bleeding with trauma or surgery Menorrhagia Significant menorrhagia from menarche should prompt investigation for congenital bleeding disorders (commonly VWD) vWD-Labs Initial screen: - PT normal - PTT sometimes prolonged > in type 3 (factor VIII dec) - Platelet sometimes dec > in types 2 and 3 Most of the time: PT, PTT, platelet --- all NORMAL Blood type ‘O’ – normally lower vWF Bleeding time – prolonged Platelet function analyzer – prolonged closure time vWF assay - Definitive test TREATMENT Intermediate purity F VIII concentrates Cryoprecipitate Adjunctive Antifibrinolytic agents (Tranexamic acid / E-aminocaproic acid ) Prevents plasminogen plasmin For mucosal bleeding Topical thrombin and fibrin glue Estrogen containing contraceptive tx For menorrhagia HEMOPHILIA Essentials Factor VIII (or IX ) deficiency X-linked or spontaneous mutation (1/3) Bruising, soft tissue bleeding, hemarthrosis Prolonged PTT + dec factor VIII (or IX) levels Most common severe congenital bleeding disorder Prevalence Hemophilia A (Factor VIII) 1 / 10,000 males Hemophilia B (Factor IX) 1 / 50,000 males HEMOPHILIA – severity classification Factor VIII – reported in units / ml ( 1 unit/ml = 100% factor activity) - Normal range: 0.5 – 1.5 U/ml (50 – 150%) Classification - Severe (60% of cases) : < 1% factor VIII (spontaneous bleeding) - Moderate : 1 - < 5% - Mild : 5 – 40 % ( bleeding only with trauma and surgery) HEMOPHILIA- S/Sx Severe Hemophiliacs Usually initial presentation in 1st 2 years of life ( severe bruising and joint bleeds) 40 – 50% present in the 1st month of life 1-4% present in the neonatal period (birth trauma) Mild or Moderate hemophiliacs Boys Trauma related bruising or bleeding Excessive bleeding following surgery or dental extraction Girls ( carriers ) ~ Often with Factor VIII < normal Mild bruising or bleeding Heavy menstrual periods HEMOPHILIA complications Hemarthroses If recurrent joint destruction Intracranial hemorrhage Leading cause of death among hemophilliacs Intramuscular hematomas Compartment syndrome muscle and nerve death Acquired bleeding disorders Vitamin K deficiency/warfarin overdose Liver disease DIC ITP Bone Marrow Failure MCV inc because of production of stress rbc’s Examples of bone marrow failure o Aplastic Anemia o Fanconi’s Anemia o o Diamond-Blackfan anemia (Congenital hypoplastic anemia) Transient Erythroblastopenia of Childhood (TEC) Bone Marrow Failure Acquired or congenital Characterized by a reduction in the effective production of mature erythrocytes, granulocytes, and platelets by the bone marrow Bone marrow failure leads to various peripheral blood cytopenias o In some conditions, only one or two cell lines may be affected o In others such as aplastic anemia the result is pancytopenia Acquired Aplastic Anemia Pathogenesis: - Defects in hematopoietic stem cells - Failure of stromal microenvironment of the marrow - Presence of stem cell antibodies **Signs/symptoms due to pancytopenia Classification of Aplastic Anemia Very severe aplastic anemia Moderately severe aplastic anemia Mild aplastic anemia Treatment of Aplastic Anemia Supportive therapy - Blood component therapy - PRBC, platelets - Antibiotic therapy Definitive treatment - Stem cell transplantation - expensive - Antithymocyte globulin, antilymphocyte globulin - expensive - Ciclosporin – oral, may have good results - 25 mg, 100 mg caps - Corticosteroids, testosterone – least effective Reminders… When to Transfuse Red Cells We transfuse patients for severely symptomatic or pathologic anemia, not the numerical value of the Hemoglobin. Aim: to restore or maintain the oxygen-carrying capacity of the blood with minimal expansion of the blood volume. Patients with Hemolytic Anemia usually tolerate very low levels of Hemoglobin. Ex. Beta thalassemia major Slow transfusion of RBC’s for very severe anemia to avoid pulmonary congestion. < 10 ml/kg/transfusion Thank You! dlca 2019 Goodluck! goodluck! =) https://www.youtube.com/watch?v=O3raY5EH9Gw&list=UUTBeUxj7npf3gAOJqk-pmkw&index=8 please subscribe ;)