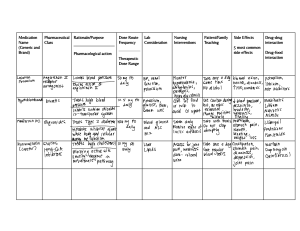
Clinical pharmacokinetics Applied pharmacokinetics is a branch of pharmacology that focuses on the study of how drugs move through the body. Pharmacokinetics deals with the processes of absorption, distribution, metabolism, and excretion (often referred to as ADME) of drugs. This field is crucial for understanding how drugs work in the body, how they are processed, and how their concentrations change over time. Here are the key components of applied pharmacokinetics: 1. Absorption: This phase involves the drug's entry into the bloodstream, typically through routes like oral ingestion, intravenous injection, inhalation, or topical application. The rate and extent of absorption influence the onset and intensity of drug effects. 2. Distribution: Once a drug is absorbed into the bloodstream, it is distributed throughout the body. Factors such as the drug's chemical properties, body composition, and blood flow to different tissues play a role in this process. The distribution can affect the drug's effectiveness and potential side effects. 3. Metabolism: Drugs are often metabolized (biotransformed) by the liver and other organs to become more water-soluble and easier for the body to eliminate. The liver's enzymes play a critical role in this phase. Metabolism can impact the drug's potency and duration of action. 4. Excretion: The final stage involves the removal of drug and its metabolites from the body. The kidneys are the primary organs responsible for excretion, but other routes such as feces, breath, and sweat can also be involved. Understanding the pharmacokinetics of a drug is essential for several reasons: • Dosing: It helps in determining the appropriate dosage and dosing intervals for a drug to achieve and maintain therapeutic levels in the body. • Individual Variation: Different individuals may have variations in their pharmacokinetic parameters, which can impact the drug's effectiveness and potential for adverse effects. Pharmacokinetic studies can guide personalized medicine. • Drug-Drug Interactions: Knowledge of a drug's pharmacokinetics is vital in predicting and managing potential interactions with other medications that a patient may be taking. • Toxicology: Understanding how drugs are processed and eliminated can provide insights into potential toxicity and help in designing safe dosing regimens. • Therapeutic Drug Monitoring: For certain drugs with narrow therapeutic windows or significant variability in individual responses, pharmacokinetic monitoring can be essential to adjust doses as needed. Applied pharmacokinetics is especially important in clinical practice, drug development, and research, as it provides a scientific basis for optimizing drug therapy and patient care. Pharmacokinetic parameters like half-life, clearance, volume of distribution, and bioavailability are commonly used to characterize how drugs behave in the body. 1 Absorption Clinical pharmacokinetics in drug absorption focuses on understanding how drugs are absorbed in the body in a clinical setting. This field is crucial in optimizing drug therapy and ensuring that patients receive the right dose of medication to achieve the desired therapeutic effect. Several factors influence drug absorption in a clinical context: 1. Route of Administration: The method by which a drug is administered (e.g., oral, intravenous, intramuscular, subcutaneous, topical, etc.) significantly affects its absorption. Different routes have varying rates and degrees of absorption. 2. Bioavailability: Bioavailability refers to the fraction of the administered drug that reaches the systemic circulation in its active form. For orally administered drugs, the extent of absorption can be influenced by factors such as first-pass metabolism in the liver and gastrointestinal factors. 3. Gastrointestinal Factors: In oral drug administration, the drug must pass through the gastrointestinal tract. Factors such as gastric emptying time, pH of the stomach, and the presence of food in the stomach can affect the rate and extent of absorption. 4. Drug Formulation: The formulation of the drug product (e.g., tablets, capsules, solutions) can impact its dissolution and absorption characteristics. Some drugs are formulated to have extended-release properties, while others are designed for rapid absorption. 5. Drug Interactions: Drug interactions can affect drug absorption. For example, some drugs may inhibit or enhance the absorption of others by affecting transporters or enzymes responsible for drug absorption. 6. Patient Factors: Patient-specific factors, including age, genetics, and individual variations in gastrointestinal physiology, can influence drug absorption. For example, differences in gastric pH or the activity of drug-metabolizing enzymes can affect drug absorption. 7. Disease States: Certain medical conditions can alter drug absorption. Patients with gastrointestinal disorders, liver disease, or conditions affecting blood flow to the absorption site may experience changes in drug absorption. 8. Dosage Forms: Different dosage forms, such as immediate-release, sustained-release, and controlled-release formulations, can have varying absorption profiles. Clinical pharmacokinetics in drug absorption involves monitoring and assessing these factors to optimize drug therapy. Key aspects of clinical pharmacokinetics include: • Dose Adjustment: Based on pharmacokinetic parameters like bioavailability, clinicians may adjust drug doses to ensure therapeutic drug levels are achieved. • Therapeutic Drug Monitoring: In cases where a drug has a narrow therapeutic window or significant inter-patient variability, therapeutic drug monitoring may be employed to ensure the drug is within the desired concentration range. • Personalized Medicine: By considering individual patient factors, including genetics, clinicians can tailor drug regimens to the specific needs and characteristics of each patient. 2 • Patient Education: Clinicians should educate patients on the importance of taking medications as directed, with or without food, and other relevant instructions to optimize drug absorption. Overall, clinical pharmacokinetics in drug absorption plays a vital role in ensuring the safety and efficacy of drug therapy in a clinical setting, promoting the rational use of medications, and minimizing adverse effects. Drug distribution Clinical pharmacokinetics in drug distribution focuses on understanding how drugs are distributed within the body once they have been absorbed into the bloodstream. This is an important aspect of pharmacokinetics as it helps clinicians and researchers determine the effective distribution of a drug throughout the body, which impacts its therapeutic effects and potential side effects. Here are some key considerations in clinical pharmacokinetics related to drug distribution: 1. Plasma Protein Binding: Many drugs bind to proteins in the bloodstream, particularly albumin. The degree of protein binding affects the amount of drug available for distribution to tissues. Drugs that are highly bound to plasma proteins have a lower free fraction available for distribution and may have a longer duration of action. 2. Tissue Perfusion: The blood flow to different tissues and organs can significantly impact drug distribution. Tissues with high blood flow, such as the heart, liver, and kidneys, often receive drugs more readily than those with lower blood flow, like fat or bone. 3. Tissue Composition: The composition of tissues, including factors like lipid solubility, can influence a drug's distribution. Lipid-soluble drugs tend to penetrate cell membranes more easily and may distribute more extensively in fatty tissues. 4. Blood-Brain Barrier: The blood-brain barrier is a highly selective membrane that separates the bloodstream from the brain and cerebrospinal fluid. Some drugs can penetrate this barrier, while others cannot. The ability to cross the blood-brain barrier is essential for drugs intended to affect the central nervous system. 5. Placental Barrier: In pregnant individuals, the placenta acts as a barrier that can influence drug distribution from the maternal bloodstream to the fetal circulation. Some drugs may cross the placenta readily, potentially affecting the developing fetus. 6. Organ Function: The function of various organs, particularly the liver and kidneys, can influence drug distribution. Impaired organ function may lead to altered drug distribution, potentially increasing the risk of toxicity. 7. Drug Accumulation: Some drugs may accumulate in specific tissues or organs due to factors like active transport mechanisms, tissue binding, or drug metabolism. This can affect the drug's distribution and elimination. Clinical pharmacokinetics in drug distribution involves assessing these factors to optimize drug therapy and ensure that the drug reaches its target site of action at the appropriate concentration. Key aspects of clinical pharmacokinetics related to drug distribution include: • Dose Adjustment: If a drug has a specific target organ or site of action, clinicians may need to adjust the dose to achieve therapeutic levels at that site while minimizing side effects in other tissues. 3 • Monitoring Drug Levels: In certain cases, therapeutic drug monitoring may be employed to measure drug concentrations in blood or specific tissues to ensure they are within the desired range. • Accounting for Disease States: Understanding how disease states affect tissue perfusion and organ function is crucial for managing drug distribution in patients with various medical conditions. • Consideration of Special Populations: For pregnant individuals, neonates, or individuals with altered physiology (e.g., elderly patients), clinicians need to consider the unique aspects of drug distribution. In summary, clinical pharmacokinetics in drug distribution is essential for optimizing drug therapy, ensuring that drugs reach their target sites, and minimizing the risk of adverse effects or suboptimal therapeutic outcomes. Drug Metabolism Clinical pharmacokinetics in drug metabolism involves studying how drugs are metabolized within the body once they have been absorbed. This branch of pharmacokinetics is crucial for understanding how drugs are transformed by the body's metabolic processes, as it greatly influences a drug's effectiveness, duration of action, and potential for toxicity. Here are some key aspects of clinical pharmacokinetics related to drug metabolism: 1. Liver Metabolism: The liver is the primary organ responsible for drug metabolism. It contains enzymes that chemically modify drugs to make them more water-soluble, allowing for easier elimination from the body. This phase is often called Phase I metabolism. 2. Cytochrome P450 Enzymes: A group of enzymes known as cytochrome P450 (CYP) enzymes play a significant role in drug metabolism. They are responsible for metabolizing a wide range of drugs and can influence how quickly or slowly a drug is eliminated from the body. 3. Drug Metabolites: After metabolism, drugs are often transformed into metabolites, some of which may be pharmacologically active, while others are inactive. The balance between active and inactive metabolites can affect the drug's efficacy and safety. 4. Enzyme Induction and Inhibition: Certain drugs can induce or inhibit the activity of drug-metabolizing enzymes. Enzyme induction can lead to faster drug metabolism, potentially reducing the effectiveness of co-administered drugs. Enzyme inhibition can lead to slower metabolism and increased drug concentrations, which can increase the risk of adverse effects. 5. Genetic Variability: Genetic factors can influence an individual's ability to metabolize specific drugs. Genetic polymorphisms can result in variations in enzyme activity, which, in turn, affects drug metabolism. Understanding a patient's genetic makeup can be important for tailoring drug therapy. 6. Drug-Drug Interactions: Knowledge of drug metabolism is essential for predicting and managing drug-drug interactions. Co-administered drugs can compete for the same metabolic pathways or affect the activity of drug-metabolizing enzymes. 4 7. Metabolic Pathways: Different drugs are metabolized by different metabolic pathways, and the metabolites produced may have different properties. Knowledge of these pathways is essential for predicting the fate of a drug in the body. Clinical pharmacokinetics in drug metabolism involves the following: • Dose Adjustment: Based on the metabolic rate of a drug, clinicians may need to adjust the dose to achieve and maintain therapeutic levels. • Therapeutic Drug Monitoring: In some cases, therapeutic drug monitoring may be used to measure drug and metabolite concentrations in the bloodstream to ensure they are within the desired range. • Genetic Testing: For certain drugs with known genetic variations affecting metabolism, genetic testing may be employed to tailor drug therapy to an individual's genetic profile. • Drug Safety: Understanding how a drug is metabolized is crucial for assessing its potential for toxicity, as some metabolites may be more toxic than the parent drug. Overall, clinical pharmacokinetics in drug metabolism is essential for optimizing drug therapy, understanding inter-individual variability in drug response, and managing drug interactions in a clinical setting. It plays a critical role in personalizing drug regimens to ensure safe and effective treatment. Drug elimination/excretion Clinical pharmacokinetics in drug elimination focuses on understanding how drugs are removed from the body once they have been absorbed and metabolized. Elimination processes are essential because they determine the duration of a drug's action, the potential for accumulation, and the risk of toxicity. Several key aspects are associated with clinical pharmacokinetics in drug elimination: 1. Renal Excretion: The kidneys play a central role in drug elimination. Many drugs and their metabolites are excreted in the urine. Renal excretion depends on factors such as glomerular filtration, tubular secretion, and tubular reabsorption. Impaired renal function can significantly affect drug elimination. 2. Hepatic Excretion: Some drugs and their metabolites are eliminated through the biliary system, which includes excretion into bile and subsequent elimination via the feces. The liver is responsible for this hepatic excretion, and the process can be influenced by factors like active transport mechanisms and enterohepatic recycling. 3. Non-Renal and Non-Hepatic Routes: Some drugs are eliminated through other routes, such as in sweat, tears, saliva, or breast milk. These routes may be significant for certain drugs, especially when considering their use in special populations like nursing mothers. 4. Drug Accumulation: In cases where a drug or its metabolites accumulate in the body due to slow elimination, dose adjustment may be necessary to avoid toxicity. This is particularly important for drugs with a narrow therapeutic index. 5. Drug-Drug Interactions: Understanding drug elimination processes is critical for assessing drug-drug interactions. Co-administered drugs can compete for renal excretion pathways, alter hepatic metabolism, or affect the enterohepatic circulation. 5 6. Individual Variability: There can be considerable inter-individual variability in drug elimination rates, particularly in renal excretion. Factors like age, renal function, and genetics can influence the rate of drug elimination. Clinical pharmacokinetics in drug elimination involves several considerations and actions: • Dose Adjustment: Depending on the elimination rate of a drug and the patient's characteristics (e.g., renal function), clinicians may need to adjust the dose to achieve therapeutic drug levels and avoid accumulation. • Therapeutic Drug Monitoring: In some cases, therapeutic drug monitoring may be used to measure drug and metabolite concentrations in the bloodstream to ensure they are within the desired range. • Renal Function Assessment: In patients with renal impairment, clinicians need to consider the impact on drug elimination and adjust drug regimens accordingly. • Monitoring for Adverse Effects: For drugs with potential for accumulation or toxicity, patients may need to be closely monitored for signs of adverse effects. Understanding clinical pharmacokinetics in drug elimination is essential for optimizing drug therapy, ensuring that drugs are cleared from the body at the appropriate rate, and preventing drug-related adverse events. It plays a critical role in personalized medicine and tailoring drug regimens to individual patient needs. Therapeutic drug monitoring Therapeutic Drug Monitoring (TDM) is a clinical practice that involves measuring and analyzing drug concentrations in a patient's bloodstream to ensure that a drug is administered at the right dose and is within the therapeutic range. TDM is used to optimize drug therapy, enhance efficacy, minimize adverse effects, and prevent toxicity. It is particularly important for drugs with a narrow therapeutic index (the range between the minimum effective dose and the minimum toxic dose) or drugs that exhibit significant variability in pharmacokinetics among different individuals. Key aspects of therapeutic drug monitoring include: 1. Measurement of Drug Levels: TDM involves the measurement of the concentration of a drug and, in some cases, its active metabolites in the patient's blood or other biological samples (e.g., urine or saliva). 2. Timing of Sampling: The timing of sample collection is critical. Samples are typically collected at specific times relative to the drug's administration to capture the drug's peak and trough concentrations. This timing may vary depending on the drug's pharmacokinetics. 3. Interpretation of Results: Clinicians interpret the drug concentration data in the context of therapeutic goals, patient characteristics, and the drug's pharmacokinetics. The goal is to maintain drug levels within a therapeutic range that optimizes therapeutic effect while avoiding toxicity. 4. Dose Adjustment: Based on the drug concentration data, clinicians may adjust the patient's dose. If drug levels are too low, the dose may be increased to achieve therapeutic levels. If levels are too high, the dose may be decreased to prevent toxicity. 6 5. Individualized Therapy: TDM allows for individualized drug therapy, taking into account a patient's unique pharmacokinetic and pharmacodynamic characteristics. This can be especially important when treating patients with renal or hepatic impairment or those on multiple medications. 6. Preventing Toxicity: For drugs with a narrow therapeutic index, TDM is vital in preventing drug toxicity. Regular monitoring ensures that drug levels remain within the safe and effective range. 7. Monitoring Compliance: TDM can also be used to assess patient compliance with medication regimens. Low drug levels may indicate non-compliance. 8. Special Populations: TDM is valuable for special patient populations, such as pregnant women, children, and the elderly, where pharmacokinetics can differ significantly from the general population. Common examples of drugs that are frequently monitored using TDM include antiepileptic drugs, certain antibiotics, antipsychotic medications, and immunosuppressants used in organ transplant recipients. TDM is especially important in clinical settings where precise dosing and therapeutic efficacy are critical. It helps to minimize the risk of underdosing (which may lead to treatment failure) and overdosing (which can cause adverse effects or toxicity). By providing a data-driven approach to drug therapy, TDM contributes to the safe and effective use of medications in a wide range of clinical applications. Drug clearance Drug clearance, often denoted by the symbol "CL," is a pharmacokinetic parameter that quantifies the rate at which a drug is removed from the body, typically through the process of elimination. It is an important parameter used to understand and predict drug concentrations in the bloodstream over time. Drug clearance is expressed in volume per unit of time (e.g., liters per hour) and can be used to determine the appropriate dosage and dosing interval for a drug. There are several types of clearance related to the various routes of drug elimination: 1. Renal Clearance: This represents the rate at which a drug is excreted by the kidneys. It is a major contributor to total drug clearance for many medications. Renal clearance depends on factors such as glomerular filtration, tubular secretion, and tubular reabsorption. Impaired renal function can lead to reduced drug clearance. 2. Hepatic Clearance: This refers to the rate at which the liver metabolizes and eliminates a drug. It encompasses the activity of hepatic enzymes responsible for drug metabolism and biliary excretion. Drugs that are hepatically cleared are typically transformed into metabolites that are then excreted in bile or urine. 3. Total Clearance: Total clearance is the sum of renal clearance and hepatic clearance (if applicable), as well as any other routes of elimination (e.g., lung or skin clearance). It represents the overall rate at which a drug is eliminated from the body. The formula for calculating total clearance is often expressed as follows: Total Clearance (CL_total) = Renal Clearance (CL_r) + Hepatic Clearance (CL_h) + Other Clearances 7 Understanding a drug's clearance is crucial for various reasons: • Dosing Regimens: Knowing the clearance of a drug helps determine the appropriate dosage and dosing frequency to maintain therapeutic levels in the body. For drugs with high clearance, more frequent dosing may be needed. • Individual Variability: Clearance can vary significantly among individuals, particularly in the case of renal clearance. Patients with impaired kidney or liver function may have reduced clearance, requiring dosage adjustments. • Drug-Drug Interactions: Knowledge of drug clearance is vital for predicting and managing drug interactions. Co-administered drugs can compete for elimination pathways or affect the activity of drug-metabolizing enzymes. • Therapeutic Drug Monitoring: Clearance can be used in conjunction with drug concentration measurements to assess and adjust drug therapy in specific patients, especially those with variability in pharmacokinetics. • Renal and Hepatic Disease: In patients with renal or hepatic disease, clearance may be significantly altered. Understanding these changes is essential for drug dosing and avoiding potential toxicities. In clinical practice, drug clearance is often used to calculate the maintenance dose of a drug, considering the desired steady-state concentration and the dosing interval. By adjusting drug dosages based on clearance, healthcare providers can tailor drug therapy to individual patients, optimize therapeutic outcomes, and minimize the risk of adverse effects. Volume of distribution The volume of distribution (Vd) is a pharmacokinetic parameter used to describe the apparent distribution of a drug within the body. It quantifies the relationship between the amount of drug in the body and the concentration of the drug in the bloodstream. The Vd provides insights into how a drug is distributed in bodily tissues and fluids and helps pharmacologists and clinicians understand the drug's pharmacokinetic behavior. The concept of the volume of distribution is essential for various pharmacokinetic calculations and dosing regimens. The Vd is typically expressed in liters (L) or liters per kilogram (L/kg) of body weight. It can be used to classify drugs into three main categories based on their distribution characteristics: 1. Low Vd: A low Vd (e.g., less than 0.6 L/kg) indicates that the drug is primarily confined to the bloodstream and does not distribute extensively into tissues. These drugs are often water-soluble and remain in the vascular compartment. 2. Moderate Vd: Drugs with a moderate Vd (e.g., 0.6 to 0.7 L/kg) have a relatively balanced distribution between the bloodstream and extracellular fluids and tissues. This category includes many commonly used medications. 3. High Vd: A high Vd (e.g., greater than 0.7 L/kg) suggests that the drug has extensive tissue distribution and may accumulate in tissues, such as fat, muscle, or organs. These drugs may exhibit a longer duration of action. The formula for calculating the volume of distribution is as follows: Vd = Amount of Drug in the Body (Dose) / Drug Concentration in Plasma (C) 8 The Vd can be influenced by several factors, including the drug's physicochemical properties, its ability to bind to plasma proteins or tissues, and the permeability of biological membranes. The Vd provides the basis for estimating the loading dose (the initial dose to achieve a desired drug level) and maintenance doses for a specific drug, taking into account factors such as the desired plasma concentration and dosing interval. Understanding the Vd is crucial in clinical pharmacology for various reasons: • Dosing Calculations: The Vd is used to determine the initial loading dose required to achieve a specific plasma concentration and subsequently the maintenance dose to maintain therapeutic levels. • Drug Monitoring: It helps in assessing and adjusting drug therapy, especially in patients with altered distribution characteristics due to factors like age, disease, or genetics. • Pharmacokinetic Properties: It provides insights into a drug's distribution behavior, which is important for selecting the appropriate drug therapy and predicting drug effects. • Drug Interactions: The Vd can help predict and understand potential drug interactions that affect a drug's distribution. In summary, the volume of distribution is a critical pharmacokinetic parameter that helps clinicians and pharmacologists understand how a drug distributes throughout the body and is essential for optimizing drug dosing regimens and therapeutic outcomes. Drug half life Drug half-life, often denoted as t½, is a fundamental pharmacokinetic parameter that measures the time it takes for the concentration of a drug in the body to decrease by half. It is a key parameter used to understand the rate at which a drug is eliminated from the body and how frequently doses need to be administered to maintain therapeutic drug levels. The concept of half-life is used to describe the pharmacokinetics of a drug and is important for various clinical and dosing considerations. Here's how it works: 1. First-Order Kinetics: Most drugs exhibit first-order kinetics, meaning that the rate of elimination is proportional to the drug's concentration in the body. As a result, the amount of drug eliminated in a given time is a fixed fraction of the total drug in the body. This leads to an exponential decrease in drug concentration over time. 2. Calculation: The half-life of a drug is calculated by determining the time it takes for the drug's concentration to drop to 50% of its initial value. Mathematically, it can be expressed as: t½ = (0.693 * Vd) / CL Where: • t½ is the half-life of the drug. • Vd is the volume of distribution of the drug. • CL is the clearance of the drug. 9 3. Clinical Implications: • A short half-life indicates that the drug is eliminated relatively quickly, and frequent dosing is often required to maintain therapeutic levels. • A long half-life suggests that the drug is eliminated slowly, and less frequent dosing may be sufficient to maintain therapeutic levels. Understanding a drug's half-life is crucial for various aspects of pharmacotherapy: • Dosing Regimens: The half-life helps determine the dosing frequency required to maintain consistent therapeutic drug levels. For drugs with short half-lives, more frequent dosing is necessary, while drugs with long half-lives may require less frequent dosing. • Steady-State: It takes approximately five half-lives for a drug to reach steady-state concentrations, where the rate of drug administration matches the rate of elimination. This is important for determining how long it takes to achieve a stable drug effect. • Drug Accumulation: A long half-life can lead to drug accumulation in the body, which may necessitate dose adjustments to prevent toxicity. Conversely, a short half-life may require more frequent dosing to avoid suboptimal therapy. • Understanding Pharmacokinetics: Half-life is one of the key parameters used to characterize a drug's pharmacokinetics and helps in predicting drug behavior in the body. • Drug Interactions: Knowledge of a drug's half-life is important for understanding and predicting potential drug interactions that may affect its clearance. In summary, the half-life of a drug is a critical pharmacokinetic parameter that provides insights into the rate of drug elimination from the body. It has practical implications for dosing regimens, steady-state attainment, and avoiding drug accumulation or suboptimal therapy. Drug accumulation Drug accumulation refers to the gradual increase in the concentration of a drug in the body over time, especially when multiple doses of the drug are administered. Accumulation occurs because the rate of drug administration exceeds the rate of drug elimination, resulting in higher drug concentrations in the body. This can have important implications for drug therapy, as excessive accumulation may lead to toxicity, while insufficient accumulation may result in suboptimal therapeutic effects. Here are some key points to understand about drug accumulation: 1. Dosing Frequency: Drug accumulation is more likely to occur with drugs that have a long half-life (the time it takes for the drug concentration to decrease by half) and are administered at relatively short intervals. When the dosing frequency is high, each subsequent dose adds to the concentration of the previous dose, leading to accumulation. 2. Steady-State: It typically takes around five half-lives for a drug to reach a steady-state concentration. At steady-state, the rate of drug administration equals the rate of drug elimination, resulting in a stable and consistent drug concentration in the body. 10 3. Maintenance Dosing: To achieve and maintain a steady-state drug concentration within the therapeutic range, clinicians often calculate maintenance doses. These are administered at intervals that correspond to the drug's half-life and its desired steadystate concentration. 4. Tolerance and Toxicity: Accumulation can lead to both therapeutic tolerance and potential toxicity. If drug levels accumulate to toxic concentrations, the patient may experience adverse effects. On the other hand, if the drug concentration remains subtherapeutic, the patient may not experience the desired therapeutic effect. 5. Dose Adjustments: To manage drug accumulation, clinicians may need to adjust the dosing regimen. For drugs with a long half-life, the dose or dosing interval may need to be reduced to prevent excessive accumulation and potential toxicity. Conversely, for drugs with a short half-life, doses or dosing intervals may need to be increased to maintain therapeutic levels. 6. Individual Variation: Patients with altered pharmacokinetics, such as those with impaired kidney or liver function, may be more susceptible to drug accumulation. In such cases, careful monitoring and dose adjustments are important. 7. Clinical Considerations: In certain situations, such as critical care or anesthesia, clinicians closely monitor drug levels and adjust dosing to rapidly achieve and maintain therapeutic drug concentrations. 8. Therapeutic Drug Monitoring: For drugs prone to accumulation, therapeutic drug monitoring (TDM) is often used. TDM involves regular measurement of drug concentrations in the patient's blood to ensure that they are within the therapeutic range. In summary, drug accumulation is a pharmacokinetic phenomenon that occurs when the rate of drug administration exceeds the rate of elimination, leading to an increase in drug concentration in the body. Clinicians need to consider factors like the drug's half-life, dosing frequency, and patient-specific characteristics when managing drug accumulation to achieve safe and effective drug therapy. Bioavailability Drug bioavailability refers to the fraction of an administered drug that reaches the systemic circulation in its active form, allowing it to have a therapeutic effect. It is a critical pharmacokinetic parameter that measures how efficiently a drug is absorbed and becomes available to exert its intended pharmacological action. Bioavailability is typically expressed as a percentage. Factors affecting drug bioavailability include the route of administration, the drug's chemical properties, and various physiological factors. Understanding and optimizing bioavailability is crucial for designing effective drug formulations and dosing regimens. Here are key aspects related to drug bioavailability: 1. Routes of Administration: Different routes of drug administration affect bioavailability. For example: • Oral administration (by mouth) often results in variable bioavailability due to factors like gastrointestinal transit time, first-pass metabolism in the liver, and interactions with food and other drugs. 11 • Intravenous administration provides complete bioavailability because the drug is directly introduced into the bloodstream. • Other routes, such as intramuscular, subcutaneous, transdermal, and inhaled, have varying levels of bioavailability based on drug characteristics and local factors. 2. First-Pass Effect: For drugs administered orally, the first-pass effect occurs when a drug is metabolized or partially eliminated by the liver before reaching the systemic circulation. This process can significantly reduce oral bioavailability. 3. Chemical Properties: A drug's chemical properties, including solubility and stability, influence its ability to be absorbed and achieve bioavailability. 4. Formulation: The formulation of a drug product, such as tablets, capsules, and injections, can affect its bioavailability. Factors like the rate of drug release and dissolution play a role. 5. Physiological Factors: Individual variations in gastrointestinal physiology, genetics, and health status can affect drug absorption and bioavailability. Conditions like gastrointestinal disorders or impaired liver function may alter bioavailability. 6. Food Interactions: The presence of food in the gastrointestinal tract can influence drug absorption. Some drugs should be taken with food to improve their bioavailability, while others should be taken on an empty stomach to avoid interactions. 7. Drug Interactions: Concurrent use of multiple drugs can lead to drug interactions that affect bioavailability. Some drugs may inhibit or enhance the absorption of others. 8. Dosage Form: Different dosage forms, such as immediate-release, sustained-release, and controlled-release formulations, can have varying bioavailability profiles. Optimizing bioavailability is crucial for ensuring that a drug achieves the desired therapeutic effect while minimizing the risk of underdosing or overdosing. This often involves considerations such as selecting the appropriate route of administration, designing drug formulations that enhance absorption, and considering individual patient factors that may impact bioavailability. Measuring and understanding drug bioavailability is a key step in drug development and in clinical practice, as it helps ensure that the intended drug effects are achieved while minimizing the potential for adverse effects or suboptimal treatment outcomes. Designing a dosage regimen Designing a dosage regimen involves creating a structured plan for administering a medication to achieve the desired therapeutic effect while minimizing adverse effects and ensuring patient compliance. Dosage regimens are tailored to the specific drug, patient population, and medical condition. Here are the key steps involved in designing a dosage regimen: 1. Define the Treatment Goals: • Determine the therapeutic goals, such as symptom relief, disease management, or prevention. • Specify the target drug concentration or desired clinical effect. 12 2. Understand the Drug Properties: • Analyze the pharmacokinetic properties of the drug, including its half-life, bioavailability, volume of distribution, and clearance. • Consider the drug's mechanism of action, pharmacodynamics, and potential adverse effects. 3. Select the Route of Administration: • Choose the most appropriate route of drug administration (e.g., oral, intravenous, subcutaneous, topical) based on factors like the drug's properties, patient preferences, and medical condition. 4. Determine the Dosage Form: • Select the appropriate dosage form (e.g., tablets, capsules, injections, suspensions) that ensures drug stability, proper drug release, and patient acceptability. 5. Calculate the Dosing Frequency: • Determine how often the drug should be administered based on the drug's halflife, desired steady-state concentration, and therapeutic goals. • Consider the convenience and feasibility of the dosing schedule for the patient. 6. Establish the Initial Dose: • Calculate the initial loading dose (if needed) to quickly achieve therapeutic levels or bypass loading with maintenance dosing. • Use principles such as the loading dose = (Vd × Desired concentration) / F, where Vd is the volume of distribution and F is bioavailability. 7. Determine Maintenance Dose: • Calculate the maintenance dose to maintain steady-state therapeutic levels. • Maintenance dose = (Desired concentration × CL) / F, where CL is clearance. 8. Consider Patient Factors: • Account for individual patient characteristics, such as age, weight, renal or hepatic function, genetics, and comorbidities. • Adjust the dosage regimen based on these factors, especially for pediatrics, geriatrics, and special populations. 9. Monitor and Adjust: • Implement therapeutic drug monitoring (TDM) when appropriate. Regularly measure drug concentrations in the patient's blood or other relevant tissues. • Adjust the dosage regimen based on TDM results and clinical response, particularly for drugs with a narrow therapeutic index or significant interindividual variability. 13 10. Educate the Patient: • Provide clear instructions to the patient regarding proper dosing, timing, and any special considerations, such as taking with or without food. • Address potential adverse effects, interactions, and the importance of adherence. 11. Plan for Follow-Up: • Schedule follow-up visits or monitoring to assess the patient's response, make necessary dosage adjustments, and evaluate the therapy's effectiveness and safety. 12. Evaluate and Revise as Needed: • Continuously assess the patient's condition and response to the drug and be prepared to modify the dosage regimen as necessary. Dosage regimen design is an iterative process that may require adjustments over time to optimize therapy based on a patient's individual needs and changing clinical circumstances. Collaboration between healthcare providers, pharmacists, and patients is essential for successful dosage regimen design and management. Target concentration A target drug concentration, also known as the target therapeutic concentration or target therapeutic range, is the specific concentration or range of concentrations of a drug in the body that is associated with the desired therapeutic effect for a particular medical condition. Target drug concentrations are used as a reference point in drug therapy to guide dosing regimens, ensuring that a patient receives the appropriate amount of medication to achieve therapeutic benefits while minimizing the risk of adverse effects. Key points related to target drug concentrations include: 1. Individual Variation: The target concentration may vary from one patient to another due to factors such as age, weight, genetics, organ function, and the severity of the medical condition. A target concentration may need to be adjusted to account for these individual differences. 2. Therapeutic Window: Drugs typically have a therapeutic window, which is the range of concentrations between the minimum effective concentration (MEC) and the minimum toxic concentration (MTC). The target concentration is usually within this therapeutic window. 3. Efficacy and Safety: The target concentration is chosen to provide the desired therapeutic efficacy while minimizing the risk of adverse effects or toxicity. It's a delicate balance between achieving the intended treatment goals and avoiding harm. 4. Therapeutic Drug Monitoring (TDM): TDM is a process that involves measuring the drug's concentration in the patient's blood or other relevant tissues to assess whether it falls within the target range. TDM helps ensure that the patient is receiving the right dose of medication. 5. Dosing Adjustments: Based on TDM results, clinicians may adjust the drug dosage to achieve or maintain the target concentration. Dosage adjustments are made to keep drug levels within the desired range and optimize treatment. 14 6. Examples: • In the case of some antibiotics, like vancomycin, target concentrations are established to ensure adequate bacterial killing while preventing kidney damage. • In antiepileptic therapy, the target concentration of an antiepileptic drug is chosen to minimize seizures while avoiding toxic effects. 7. Continuous Monitoring: For certain medications, particularly those with a narrow therapeutic index or drugs with significant inter-individual variability, continuous monitoring and adjustment of the dosage regimen are crucial. It's important to note that target drug concentrations are specific to each drug and the medical condition it is intended to treat. These values are determined through clinical trials and research and are often based on factors such as efficacy, safety, and patient outcomes. Healthcare providers use target drug concentrations as a guide when prescribing and monitoring drug therapy, with the goal of achieving the best possible therapeutic outcomes for each patient. Maintenance dose A maintenance dose is a regular, ongoing dose of a medication administered to maintain the concentration of the drug within the therapeutic range. It is part of a dosing regimen designed to ensure that a patient consistently receives the right amount of medication to achieve and sustain the desired therapeutic effect while preventing underdosing or overdosing. Key points about maintenance doses include: 1. Steady-State: Maintenance doses are used to maintain drug levels in the body within a steady-state concentration. In steady state, the rate of drug administration matches the rate of drug elimination, resulting in a stable and consistent drug concentration. 2. Dosing Frequency: The dosing frequency of a maintenance dose depends on factors such as the drug's half-life, desired therapeutic concentration, and dosing interval. It may be administered once daily, multiple times a day, weekly, or according to a different schedule. 3. Loading Dose vs. Maintenance Dose: In some cases, a loading dose is given initially to rapidly achieve therapeutic drug levels, followed by maintenance doses to sustain those levels. The loading dose is often higher than the maintenance dose. 4. Individualization: Maintenance doses can be adjusted based on individual patient characteristics, such as age, weight, renal or hepatic function, genetics, and comorbidities. These factors can affect the rate of drug clearance and distribution. 5. Therapeutic Drug Monitoring (TDM): Maintenance doses are often used in conjunction with TDM, a process that involves measuring drug concentrations in the patient's blood. TDM helps clinicians assess whether the drug is within the target therapeutic range and make necessary dosage adjustments. 6. Examples: • In the case of an antihypertensive medication, the maintenance dose is administered daily to maintain normal blood pressure. 15 • For an antiepileptic drug, the maintenance dose is given to prevent seizures on an ongoing basis. 7. Titration: The process of finding the appropriate maintenance dose may involve dose titration, which is gradual dose adjustment based on clinical response and, when applicable, drug concentration measurements. 8. Duration of Therapy: The duration of maintenance therapy can vary widely, depending on the patient's medical condition. Some conditions require long-term or even lifelong therapy, while others may require a shorter duration of treatment. The design of a maintenance dose considers the drug's pharmacokinetics, the patient's characteristics, and the therapeutic goals. Clinicians aim to establish a dosing regimen that ensures consistent, effective therapy while minimizing the risk of side effects or adverse reactions. Regular follow-up and monitoring are important to assess the patient's response to the medication and make any necessary dose adjustments. Loading dose A drug loading dose, often referred to simply as a "loading dose," is an initial, higher dose of a medication given at the beginning of a drug therapy to quickly achieve the desired therapeutic concentration in the body. Loading doses are used for drugs that have a long half-life, drugs with a delayed onset of action, or when a rapid therapeutic effect is needed. The loading dose is typically followed by maintenance doses to sustain the desired drug concentration. Key points about loading doses include: 1. Rapid Attainment of Therapeutic Levels: The primary purpose of a loading dose is to rapidly achieve therapeutic drug levels in the body, often bypassing the time it would typically take for the drug to accumulate to the desired concentration through maintenance doses alone. 2. Higher Than Maintenance Dose: The loading dose is usually higher than the subsequent maintenance doses. It is calculated based on the volume of distribution (Vd) and the desired therapeutic concentration, considering the pharmacokinetics of the drug. 3. Particularly Relevant for Drugs with Long Half-Lives: Loading doses are frequently used for drugs with long half-lives because it may take an extended period to reach steady-state concentrations using maintenance doses alone. 4. Examples: • In the case of an anticoagulant like warfarin, a loading dose may be given to rapidly achieve an anticoagulant effect. • For antibiotics like vancomycin, a loading dose can help achieve effective bacterial killing. 5. Transition to Maintenance Doses: After the loading dose, patients are often switched to lower, regular maintenance doses to sustain therapeutic drug levels while minimizing the risk of adverse effects or toxicity. 6. TDM and Dose Adjustment: Therapeutic drug monitoring (TDM) may be used to ensure that the desired drug concentrations have been achieved with the loading dose. TDM can also help guide subsequent dose adjustments. 16 7. Timing and Frequency: The timing and frequency of loading doses depend on the specific drug and its pharmacokinetics. In some cases, loading doses are administered as a single bolus, while in others, they may be given in divided doses over a short period. 8. Clinical Context: Loading doses are administered in various clinical contexts, including inpatient and outpatient settings, emergency medicine, and critical care. 9. Safety Considerations: Due to the higher dose, careful monitoring is essential to prevent the risk of adverse effects or toxicity associated with loading doses. Dosage calculations should be accurate. Loading doses are a valuable tool in drug therapy, allowing clinicians to rapidly achieve therapeutic drug levels when time is of the essence. They are especially important for drugs with slow onset of action, when immediate clinical benefits are needed, or for achieving a target concentration rapidly. Proper dosing and monitoring are critical to ensure the safety and efficacy of loading dose regimens. 17