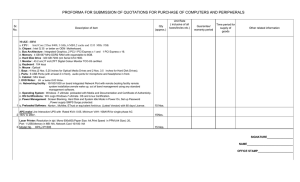
Nosology of genetic skeletal disorders - 2023 revision.pdf
advertisement

Revised: 13 January 2023 Accepted: 17 January 2023 DOI: 10.1002/ajmg.a.63132 ORIGINAL ARTICLE Nosology of genetic skeletal disorders: 2023 revision Sheila Unger 1 | Carlos R. Ferreira 2 | Geert R. Mortier 3 | Houda Ali 4 | Alistair Calder 7 | Daniel H. Cohn 8,9 | Débora R. Bertola 5,6 Valerie Cormier-Daire 10 | Katta M. Girisha 11 | Outi Makitie 14,15 | Deborah Krakow 13 | | Christine Hall 12 | Stefan Mundlos 16 | | Ravi Savarirayan 19 | Gen Nishimura 17 | Stephen P. Robertson 18 David Sillence 20 | Marleen Simon 21 | V. Reid Sutton 22 | Matthew L. Warman 23,24 | Andrea Superti-Furga 1 1 Division of Genetic Medicine, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland 2 Skeletal Genomics Unit, Metabolic Medicine Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland, USA 3 Center for Human Genetics, University Hospital Leuven and KU Leuven, Leuven, Belgium 4 INSERM, US14-Orphanet, Paris, France 5 Unidade de Genética, Instituto da Criança, Hospital das Clínicas da Faculdade de Medicina, Universidade de São Paulo, São Paulo, Brazil 6 Departamento de Genética e Biologia Evolutiva, Instituto de Biociências, Universidade de São Paulo, São Paulo, Brazil 7 Radiology Department, Great Ormond Street Hospital for Children, NHS Foundation Trust, London, UK 8 Department of Molecular, Cell and Developmental Biology, University of California, Los Angeles, Los Angeles, California, USA 9 Department of Orthopaedic Surgery, University of California, Los Angeles, Los Angeles, California, USA 10 Paris Cité University, Reference Center for Skeletal Dysplasia, INSERM UMR 1163, Imagine Institute, Necker Enfants Malades Hospital (AP-HP), Paris, France 11 Department of Medical Genetics, Kasturba Medical College, Manipal, Manipal Academy of Higher Education, Manipal, Karnataka, India 12 Emerita Consultant Paediatric Radiologist at Great Ormond Street Childrens' Hospital, London, UK 13 Departments of Obstetrics and Gynecology, Orthopaedic Surgery and Human Genetics, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, California, USA 14 Children's Hospital, University of Helsinki and Helsinki University Hospital, Helsinki, Finland 15 Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden 16 Institut für medizinische Genetik und Humangenetik, Charité - Universitätsmedizin Berlin, Berlin, Germany 17 Department of Radiology, Musashino-Yowakai Hospital, Tokyo, Japan 18 Department of Women's and Children's Health, Dunedin School of Medicine, University of Otago, Dunedin, New Zealand 19 Murdoch Children's Research Institute and University of Melbourne, Parkville, Victoria, Australia 20 Specialities of Genomic Medicine and Paediatrics and Adolescent Health, Sydney University Clinical School, Children's Hospital, Westmead, NSW, Australia 21 Department of Genetics, University Medical Center Utrecht, Utrecht, The Netherlands 22 Department of Molecular & Human Genetics, Baylor College of Medicine & Texas Children's Hospital, Houston, Texas, USA 23 Department of Orthopedic Surgery, Boston Children's Hospital, Harvard Medical School, Boston, Massachusetts, USA 24 Department of Genetics, Harvard Medical School, Boston, Massachusetts, USA Equal contributions from Sheila Unger and Carlos R. Ferreira. This is an open access article under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs License, which permits use and distribution in any medium, provided the original work is properly cited, the use is non-commercial and no modifications or adaptations are made. © 2023 The Authors. American Journal of Medical Genetics Part A published by Wiley Periodicals LLC. This article has been contributed to by U.S. Government employees and their work is in the public domain in the USA. Am J Med Genet. 2023;1–46. wileyonlinelibrary.com/journal/ajmga 1 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Received: 22 December 2022 UNGER ET AL. Correspondence Andrea Superti-Furga, Division of Genetic Medicine, Lausanne University Hospital, Lausanne, Switzerland. Email: asuperti@unil.ch Abstract The “Nosology of genetic skeletal disorders” has undergone its 11th revision and now contains 771 entries associated with 552 genes reflecting advances in molecular delineation of new disorders thanks to advances in DNA sequencing technology. Funding information The International Skeletal Dysplasia Society; University of Lausanne; DBT/Wellcome Trust; India Alliance; Sigrid Jusélius Foundation; CureKids New ZEaland; National Health and Medical Research Council of Australia, Grant/Award Number: #GNT2018081 The most significant change as compared to previous versions is the adoption of the dyadic naming system, systematically associating a phenotypic entity with the gene it arises from. We consider this a significant step forward as dyadic naming is more informative and less prone to errors than the traditional use of list numberings and eponyms. Despite the adoption of dyadic naming, efforts have been made to maintain strong ties to the MIM catalog and its historical data. As with the previous versions, the list of disorders and genes in the Nosology may be useful in considering the differential diagnosis in the clinic, directing bioinformatic analysis of nextgeneration sequencing results, and providing a basis for novel advances in biology and medicine. When the first “Nosology” was compiled and published in 1970, few [International nomenclature of constitutional bone diseases. Constitu- would have predicted that it would gain such an important role in tional bone diseases without known pathogenesis], 1971; [Interna- genetics clinics and research to motivate an 11th revision 52 years tional nomenclature of constitutional diseases of bone], 1970; later (Table 1). Yet, the reasons that stimulated the first Nosology are McKusick & Scott, 1971). At that time, however, the work was called the same that uphold the present new revision: coping with the a “Nomenclature” rather than a Nosology; the goal was to bring all wealth of novel information on the growing number and variety of scholars to use the same name for the same condition. skeletal phenotypes with a genetic basis and trying to assure a common naming system to facilitate diagnosis and communication. Molecular criteria began to inform the Nosology in the 1980s, first with osteogenesis imperfecta and the discovery of genetic vari- The recognition of chromosomal aneuploidies at the transition ants in collagen 1. The concept of “bone dysplasia families” originating between the 50s and the 60s provided a biological foundation to from different pathogenic variants in a single gene was proposed in genetics and ushered in a first golden era of clinical genetics. The the 1980s (Spranger, 1985) and confirmed in the 1990s, with the Birth Defects Conferences between 1969 and 1971 signaled the COL2A1 and FGFR3 disorders being prominent examples. Ever since, awareness and importance of clinical genetics throughout the sixties the Nosology has straddled the fence between defining disorders and initiated a prolific period in disease identification, delineation, based either on their clinical and radiographic features or on the and description. In this context, it became apparent that “chondro- responsible genes (Beighton et al., 1992; Bonafe et al., 2015; dysplasia” was not a single diagnosis, but that many distinct, true- Hall, 2002; International nomenclature and classification of the osteo- breeding forms existed; for example, diastrophic dysplasia, spondylo- chondrodysplasias (1997). International working group on constitu- epiphyseal dysplasia congenita, and the so-called “pseudo-Morquio” tional diseases of bone, 1998; International nomenclature of disorders were distinguished and clinically described. In addition, constitutional diseases of bone, 1979; International nomenclature of biochemistry allowed subtypes of clinically similar disorders to be constitutional diseases of bone. Revision, May 1983, 1983; distinguished, the most notable example at the time being the muco- Lachman, 1998; Mortier et al., 2019; Superti-Furga & Unger, 2007; polysaccharidoses. The contribution of radiologic features and radi- Warman et al., 2011). ologists to the delineation of skeletal dysplasias deserves explicit Traditionally, the Nosology has been organized into groups of recognition. In many instances, it was the radiographic features and disorders—initially based on radiographic criteria, then by biochemical their time dependent evolution (the “fourth dimension” emphasized criteria (metabolic pathways) and subsequently, more and more, by by the late Andres Giedion) that permitted not only to discriminate functional and molecular criteria. The organization into groups has between disorders that had outward similarities but also to recog- been maintained in the current revision as it helps in finding the disor- nize a gene's radiographic signature in phenotypically distinct disor- ders relevant for a particular patient of finding. On the other hand, ders (e.g., Achondrogenesis type 2 and Spondyloepiphyseal dysplasia Nature has more complexity than can be captured in the Nosology, congenita) and thus create the first gene families (Spranger, 1985) and our attempt at classification is necessarily both arbitrary and a (and see below). simplification, as many disorders might warrant classification in more In 1970, the first prototype of the “Nosology” was drafted (A nomenclature for constitutional (intrinsic) diseases of bone, 1971; than one group. Thus, we have elected to drop the term “classification” from the title; this is just a “Nosology”. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License 2 Thanatophoric dysplasia (type 1), FGFR3‐related Thanatophoric dysplasia (type 2), FGFR3‐related Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN), FGFR3‐related Achondroplasia, FGFR3‐related Hypochondroplasia, FGFR3‐related NOS 01‐0010 NOS 01‐0020 NOS 01‐0030 NOS 01‐0040 NOS 01‐0050 AD AD AD AD AD Inheritance FGFR3 FGFR3 FGFR3 FGFR3 FGFR3 Gene or locus 146000 100800 616482 187601 187600 MIM No. Radiographic differences between types 1 and 2 are correlated to specific FGFR3 variants Includes previous “platyspondylyic dysplasia type San Diego” Notes Achondrogenesis, COL2A1‐related (formerly type 2, type Langer‐Saldino) Hypochondrogenesis, COL2A1‐related Platyspondylic dysplasia, type Torrance, COL2A1‐related Spondyloepiphyseal dysplasia congenita (SEDC), COL2A1‐ related Spondyloepimetaphyseal dysplasia, COL2A1‐related Kniest dysplasia, COL2A1‐related Spondyloperipheral dysplasia, COL2A1‐related SED with metatarsal shortening, COL2A1‐related Stickler syndrome, COL2A1‐related Dysplasia of the proximal femoral epiphyses, COL2A1‐ related NOS 02‐0010 NOS 02‐0020 NOS 02‐0030 NOS 02‐0040 NOS 02‐0050 NOS 02‐0060 NOS 02‐0070 NOS 02‐0080 NOS 02‐0090 NOS 02‐0100 AD AD AD AD AD AD AD, AR* AD AD AD COL2A1 COL2A1 COL2A1 COL2A1 COL2A1 COL2A1 COL2A1 COL2A1 COL2A1 COL2A1 150600, 608805 108300 609162 271700 156550 184250, 184253, 184255, 616583 183900, 604864 151210 200610 200610 Heterogeneous condition, not all cases are due to COL2A1 variants (usually p.G393S; p.G717S; p. G1170S). The condition called “Meyer dysplasia of the hip” is not associated with COL2A1 variants Monoallelic loss‐of‐function variants; see also COL11A1, COL11A2, COL9A1, COL9A2, COL9A3 Often associated with the p.R275C variant; formerly “Czech dysplasia” Like Torrance dysplasia, often variants in the C‐propeptide of collagen 2 Also known as “SED with marked metaphyseal changes”. Includes SEMD type Strudwick, SMD type Algerian, SED type Stanescu, dysspondyloenchondromatosis, and some cases of SMD “corner fracture” type Includes mild SED with premature onset arthrosis, also known as osteoarthrosis with mild chondrodysplasia; includes Namaqualand type hip dysplasia. Mild SED cases may resemble MED (see note). AR*: very rare SED cases with biallelic COL2A1 variants have been reported Often variants in the C‐propeptide of collagen 2 Achondrogenesis type 2 and hypochondrogenesis form one phenotypic continuum Achondrogenesis type 2 and hypochondrogenesis form one phenotypic continuum 3 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License (Continues) See also the pseudoachondroplasia‐multiple epiphyseal dysplasia group for recessively inherited variants of Stickler syndrome as well as for overlapping phenotypes with normal stature and premature onset arthrosis; as well as spondylometaphyseal dysplasia Sutcliffe (or “corner fractures” type), FN1‐related Type 2 collagen disorders Group 2 See also group 33 for craniosynostosis syndromes linked to FGFR3 variants, as well as CATSHL in group 30 and LADD syndrome in group 40 for other FGFR3‐related phenotypes; rare FGFR3 missense variants have been reported in idiopathic short stature but a causal link is not yet established and their significance remains unclear Name of group/name of disorder FGFR3 chondrodysplasias Group 1 The nosology of genetic skeletal disorders: 2023 revision. Group number/number of disorder TABLE 1 UNGER ET AL. Stickler syndrome, COL11A1‐related Marshall syndrome, COL11A1‐related Stickler syndrome, COL11A2‐related (non‐ocular type) Fibrochondrogenesis, COL11A1‐related Fibrochondrogenesis, COL11A2‐related Otospondylomegaepiphyseal dysplasia (OSMED), recessive type, COL11A2‐related Otospondylomegaepiphyseal dysplasia (OSMED), dominant type, COL11A2‐related NOS 03‐0010 NOS 03‐0020 NOS 03‐0030 NOS 03‐0040 NOS 03‐0050 NOS 03‐0060 NOS 03‐0070 AD AR AR, AD AR, AD AD AD AD, MOS Inheritance COL11A2 COL11A2 COL11A2 COL11A1 COL11A2 COL11A1 COL11A1 Gene or locus 184840 215150 614524 228520 184840 154780 604841 MIM No. Formerly Weissenbacher‐Zweymüller syndrome and Stickler syndrome type 3 One report with homozygous p.G901E variant in two affected sibs (PMID 22499343) Can also result from somatic mosaicism for a COL11A1 variant Notes Sulfation disorders Achondrogenesis, SLC26A2‐related (formerly achondrogenesis type 1B, or type Fraccaro) Atelosteogenesis, SLC26A2‐related (formerly atelosteogenesis type 2) Diastrophic dysplasia, SLC26A2‐related Multiple epiphyseal dysplasia, SLC26A2‐related (autosomal recessive type, rMED) Spondylo‐epi‐metaphyseal dysplasia, PAPSS2‐related Chondrodysplasia with congenital joint dislocations, IMPAD1‐related Chondrodysplasia with congenital joint dislocations, CHST3‐related Chondrodysplasia with hypomyelinating leucodystrophy, SLC35B2‐related Ehlers‐Danlos syndrome, musculocontractural type, CHST14‐related Ehlers‐Danlos syndrome, musculocontractural type, DSE‐ related Osteochondrodysplasia, brachydactyly, and overlapping malformed digits (OCBMD), CHST11‐related NOS 04‐0010 NOS 04‐0020 NOS 04‐0030 NOS 04‐0040 NOS 04‐0050 NOS 04‐0060 NOS 04‐0070 NOS 04‐0080 NOS 04‐0090 NOS 04‐0100 NOS 04‐0110 AR AR AR AR AR AR AR CHST11 DSE CHST14 SLC35B2 CHST3 IMPAD1 PAPSS2 SLC26A2 SLC26A2 AR SLC26A2 SLC26A2 AR AR AR 618167 615539 601776 See 610788 143095 614078 612847 226900 222600 256050 600972 Includes adducted thumb‐clubfoot syndrome Includes recessive Larsen syndrome, humero‐spinal dysostosis, and SED type Omani Some features similar to Catel‐Manzke syndrome, TGDS‐ related, as well as to Desbuquois syndrome, CANT1‐ related Formerly “SEMD Pakistani type”; includes the former “recessive brachyolmia, recessive type” as well as the older entities “Toledo brachyolmia” and “Hobaek brachyolmia” See also multiple epiphyseal dysplasias and pseudoachondroplasia group (group 9) Includes former entities de la Chapelle dysplasia, McAlister dysplasia, and neonatal osseous dysplasia Formerly known as achondrogenesis, type Fraccaro UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 4 See also Stickler syndrome type 1 in collagen 2 group (Group 2) as well as recessive forms of Stickler syndrome in the pseudoachondroplasia‐multiple epiphyseal dysplasia group (Group 9) Name of group/name of disorder Type 11 collagen disorders Group 3 (Continued) Group number/number of disorder TABLE 1 4 (Continued) Inheritance AR Gene or locus HS2ST1 MIM No. 619194 Notes Baratela‐Scott syndrome, XYLT1‐related Desbuquois dysplasia (with accessory ossification center in digit 2), CANT1‐related Desbuquois dysplasia (with short metacarpals and elongated phalanges, Kim type), CANT1‐related SEMD with joint laxity (Hall type or leptodactylic type), KIF22‐related SEMD with joint laxity, EXOC6B‐related SEMD with joint laxity (Beighton type), B3GALT6‐related (Ehlers‐Danlos syndrome, spondylodysplastic type 2, EDSSPD2) Ehlers‐Danlos syndrome, spondylodysplastic type 1 (EDSSPD1), B4GALT7‐related Multiple joint dislocations, short stature, craniofacial dysmorphisms, and skeletal dysplasia, with or without heart defects, B3GAT3‐related Skeletal dysplasia with joint laxity and advanced bone age (SDJLABA), CSGALNACT1‐related Skeletal dysplasia with joint dislocations and amelogenesis imperfecta, SLC10A7‐related NOS 05‐0020 NOS 05‐0030 NOS 05‐0040 NOS 05‐0050 NOS 05‐0060 NOS 05‐0070 NOS 05‐0080 NOS 05‐0090 NOS 05‐0100 AR SLC10A7 CSGALNACT1 B3GAT3 AR AR B4GALT7 B3GALT6 AR AR EXOC6B KIF22 CANT1 CANT1 XYLT1 AR AD AR AR AR 618363 618870 245600 130070 271640 618395 603546 251450 251450 615777 The phenotype is very variable and has been reported also as “Larsen‐like” or as “pseudodiastrophic dysplasia”. Intellectual disability and severe osteopenia with fractures have been observed. The OMIM entry includes older descriptions that are probably unrelated. Formerly known as “EDS, progeroid form”; includes Larsen syndrome, La Reunion type; see also B3GALT6 deficiency above Includes MIM 609465—Al‐Gazali syndrome as neonatal form Phenotype resembles SEMD‐JL leptodactylic or type Hall (preceding line) May have intellectual disability; formerly Desbuquois dysplasia type 2 Filamins and related disorders Frontometaphyseal dysplasia, FLNA‐related Frontometaphyseal dysplasia, MAP3K7‐related Frontometaphyseal dysplasia, TAB2‐related NOS 06‐0010 NOS 06‐0020 NOS 06‐0030 AD AD XL TAB2 MAP3K7 FLNA 617137 305620 (Continues) No MIM entry yet; TAB2 gene also associated with MIM 614980—congenital heart defects, nonsyndromic, 2 FLNA gene also associated with MIM 300049, MIM 300321, MIM 314400, MIM 300048, MIM 300049 (see) and conditions below in this group 5 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 6 Note: remarkably, this group contains several disorders of glycosaminoglycan synthesis. In spite of this group being named after a clinical feature (dysplasias with joint dislocations), the phenotypes in this group are related to those of the preceding Group 4 (sulfation disorders) and of the following Group 6 (filamin disorders) justifying its placement here. See also: Temtamy type brachydactyly, CHSY1‐related, as well as SEMD with microcephaly, retinal dystrophy and hearing loss, PISD‐related (Liberfarb syndrome), for other conditions with congenital dislocations, as well as EDSSPD3, SLC39A13‐related, in the SEMD group. Dysplasias with multiple joint dislocations NOS 05‐0010 See also filamin disorders (Group 6) and dysplasias with multiple joint dislocations (Group 5) for other conditions with dislocations, as well as brachydactyly, CHSY1‐related, for phalangeal changes reminiscent of the sulfation disorders. Developmental delay with corpus callosum, skeletal, and renal abnormalities, HS2ST1‐related Name of group/name of disorder Group 5 NOS 04‐0120 Group number/number of disorder TABLE 1 UNGER ET AL. Cardiospondylocarpofacial syndrome, MAP3K7‐related Melnick‐Needles syndrome, FLNA‐related Otopalatodigital syndrome type 1 (OPD1), FLNA‐related Otopalatodigital syndrome type 2 (OPD2), FLNA‐related Terminal osseous dysplasia (TOD), FLNA‐related Larsen syndrome, FLNB‐related Atelosteogenesis type 1, FLNB‐related Atelosteogenesis type 3, FLNB‐related Spondylocarpotarsal synostosis syndrome, FLNB‐related Spondylocarpotarsal synostosis syndrome, RFLNA‐related Spondyocarpotarsal synostosis syndrome with contractures and pterygia, MYH3‐related Frank‐ter Haar syndrome, SH3PXD2B‐related NOS 06‐0050 NOS 06‐0060 NOS 06‐0070 NOS 06‐0080 NOS 06‐0090 NOS 06‐0100 NOS 06‐0110 NOS 06‐0120 NOS 06‐0130 NOS 06‐0140 NOS 06‐0150 Inheritance AR AD, AR AR AR AD AD AD XL XL XL XL AD Gene or locus SH3PXD2B MYH3 RFLNA FLNB FLNB FLNB FLNB FLNA FLNA FLNA FLNA MAP3K7 MIM No. 249420 178110, 618469 272460 108721 108720, 112310 150250 300244 304120 311300 309350 157800 Includes previous Borrone dermatocardioskeletal syndrome frequently biallelic loss of function variants; monoallelic missense variants in the MYH3 gene associated with MIM 193700‐Arthrogryposis 2A, and MIM 618436‐ Arthrogryposis 2B3 Entity proven, no MIM entry yet Includes Boomerang dysplasia, Piepkorn dysplasia, and spondylohumerofemoral (giant cell) dysplasia Includes digitocutaneous dysplasia Includes osteodysplasty Notes Proteoglycan core proteins disorders Dyssegmental dysplasia, HSPG2‐related Myotonic chondrodystrophy, HSPG2‐related (Schwartz‐ Jampel syndrome) Spondylo‐epiphyseal dysplasia, ACAN‐related (dominant, Kimberley type) Spondylo‐epi‐metaphyseal dysplasia, ACAN‐related (recessive, aggrecan type) Short stature with advanced bone age, ACAN‐related SEMD, BGN‐related (Camera type) TRPV4 disorders Metatropic dysplasia, TRPV4‐related NOS 07‐0010 NOS 07‐0020 NOS 07‐0030 NOS 07‐0040 NOS 07‐0050 NOS 07‐0060 Group 8 NOS 08‐0010 AD, MOS XL AD AR AD AR AR TRPV4 BGN ACAN ACAN ACAN HSPG2 HSPG2 156530 300106 165800 612813 608361 255800 224410, 224400 Includes so‐called “hyperplastic”, lethal, and non‐lethal forms. Can also result from somatic mosaicism for a TRPV4 variant. The BGN gene is also associated with a connective tissue‐ arterial aneurysms disorder (Meester‐Loeys syndrome, MIM300989) Sometimes with osteochondritis dissecans; other cases short stature with no skeletal features and normal bone age Variable severity; includes previous Burton dysplasia Variable severity; Includes both former Silverman‐ Handmaker and Rolland‐Desbuquois types UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 7 See also chondrodysplasia with congenital joint dislocations, CHST3‐related (“recessive Larsen syndrome”) and the group of dysplasias with multiple dislocations, above (Group 5) Name of group/name of disorder NOS 06‐0040 (Continued) Group number/number of disorder TABLE 1 6 (Continued) Spondyloepiphyseal dysplasia, Kozlowski type Brachyolmia, TRPV4‐related Familial digital arthropathy with brachydactyly, TRPV4‐ related NOS 08‐0030 NOS 08‐0040 NOS 08‐0050 Inheritance AD AD AD AD Gene or locus TRPV4 TRPV4 TRPV4 TRPV4 MIM No. 606835 113500 184252 184095 Notes Previously known as “pseudo‐Morquio syndrome type 2”. Includes the obsolete MIM 168400‐parastremmatic dwarfism entry, a phenotypic variant. Pseudoachondroplasia, COMP‐related Multiple epiphyseal dysplasia, COMP‐related Multiple epiphyseal dysplasia, MATN3‐related Multiple epiphyseal dysplasia, CANT1‐related Multiple epiphyseal dysplasia, COL9A1‐related Multiple epiphyseal dysplasia, COL9A2‐related Multiple epiphyseal dysplasia, COL9A3‐related Stickler syndrome, recessive type, COL9A1‐related Stickler syndrome, recessive type, COL9A2‐related Stickler syndrome, recessive type, COL9A3‐related Multiple epiphyseal dysplasia with microcephaly and nystagmus (Lowry‐Wood syndrome), RNU4ATAC‐ related NOS 09‐0010 NOS 09‐0020 NOS 09‐0030 NOS 09‐0040 NOS 09‐0050 NOS 09‐0060 NOS 09‐0070 NOS 09‐0080 NOS 09‐0090 NOS 09‐0100 NOS 09‐0110 RNU4ATAC 226960 120270 614284 614134 600969 600204 614135 617719 607078 132400 177170 See also Microcephalic osteodysplastic primordial dwarfism, RNU4ATAC‐related, as well as Roifman syndrome, RNU4ATAC‐related, both in the primordial dwarfism group (Group 21), for conditions with different severity from the RNU4ATAC gene See also Groups 2 and 3 AR AR Short rib–polydactyly syndrome (SRPS), DYNC2H1‐ related Short rib–polydactyly syndrome (SRPS), IFT80‐related Short rib–polydactyly syndrome (SRPS), IFT81‐related Short rib–polydactyly syndrome (SRPS), WDR34‐related Short rib–polydactyly syndrome (SRPS), WDR60‐related NOS 10‐0010 NOS 10‐0020 NOS 10‐0030 NOS 10‐0040 NOS 10‐0050 WDR60 WDR34 IFT81 IFT80 DYNC2H1 615503 615633 617895 611263 613091, 263520 (Continues) There is significant clinical and radiological overlap between SRP1/3 and ATD. Some forms of both remain unlinked to the known genes. This gene can also be responsible for chondroectodermal dysplasia (Ellis‐van Creveld), see below. 7 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License AR AR AR Skeletal disorders caused by abnormalities of cilia or ciliary signaling Group 10 See also multiple epiphyseal dysplasia, recessive type, SLC26A2‐related, as well as ASPED. Some COL2A1 variants can make a MED‐like phenotype. Some MED or MED‐like phenotypes remain genetically unclear. COL9A3 AR COL9A2 AR AR COL9A3 COL9A1 AD COL9A2 COL9A1 CANT1 MATN3 COMP COMP AR AD AD AR AD AD AD Pseudoachondroplasia and the multiple epiphyseal dysplasias Group 9 Missense variants in the TRPV4 gene can be responsible for different types of peripheral neuropathies (see under MIM 605427). The TRPV4 skeletal phenotypes can sometimes be associated with neuropathy. Spondyloepimetaphyseal dysplasia, TRPV4‐related (Maroteaux type) Name of group/name of disorder NOS 08‐0020 Group number/number of disorder TABLE 1 UNGER ET AL. (Continued) Short rib–polydactyly syndrome (SRPS), DYNC2LI1‐ related Short rib–polydactyly syndrome (SRPS), NEK1‐related Short rib–polydactyly syndrome (SRPS), IFT122‐related Short rib–polydactyly syndrome (SRPS), WDR19‐related Short rib‐polydactyly syndrome (SRPS), INTU‐related Short rib‐polydactyly syndrome (SRPS), TRAF3IP1‐related Endocrine‐cerebro‐osteo dysplasia (ECO), CILK1‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), DYNC2H1‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), DYNC2LI1‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), WDR34‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), TCTEX1D2‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), WDR60‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), WDR19‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), IFT140‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), TTC21B‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), IFT122‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), WDR35‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), IFT43‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), IFT80‐related NOS 10‐0070 NOS 10‐0080 NOS 10‐0090 NOS 10‐0100 NOS 10‐0110 NOS 10‐0120 NOS 10‐0130 NOS 10‐0140 NOS 10‐0150 NOS 10‐0160 NOS 10‐0170 NOS 10‐0180 NOS 10‐0190 NOS 10‐0200 NOS 10‐0210 NOS 10‐0220 NOS 10‐0230 NOS 10‐0240 Name of group/name of disorder Inheritance AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR Gene or locus IFT80 IFT43 WDR35 IFT122 TTC21B IFT140 WDR19 WDR60 TCTEX1D2 WDR34 DYNC2LI1 DYNC2H1 CILK1 TRAF3IP1 INTU WDR19 IFT122 NEK1 DYNC2LI1 MIM No. 611623 617866 614091 See 269860 613819 266920 614376 See 615503 617405 See 615633 See 617088 613091 612651 See 607380 617925 614091 269860 263520 617088 Subsumed under SRPS (MIM 269860) Gene also known for nephronophthisis (MIM 613820) WDR19 is associated with MIM 614091, 614376, 614378, 615633 as well as with nephronopthisis (MIM 614377), Senior‐Loken syndrome (MIM 616307) and Mainzer‐ Saldino syndrome (see below) TRAF3IP1 also known as IFT154 WDR19 is associated with MIM 614091, 614376, 614378, 615633 as well as with nephronopthisis (MIM 614377), Senior‐Loken syndrome (MIM 616307) and Mainzer‐ Saldino syndrome (see below) Possibly also digenic inheritance combining NEK1 with DYNC2H1 variants Notes UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License NOS 10‐0060 Group number/number of disorder TABLE 1 8 (Continued) Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), IFT172‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), IFT81‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), IFT52‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), CFAP410‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), CEP120‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), KIAA0586‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), GRK2‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), TRAF3IP1‐related Short‐rib thoracic dysplasia (formerly asphyxiating thoracic dysplasia—Jeune syndrome), KIAA0753‐related Axial spondylometaphyseal dysplasia, CFAP410‐related Axial spondylometaphyseal dysplasia, NEK1‐related Chondroectodermal dysplasia (Ellis‐van Creveld), EVC1‐ related Chondroectodermal dysplasia (Ellis‐van Creveld), EVC2‐ related Chondroectodermal dysplasia (Ellis‐van Creveld), WDR35‐ related Chondroectodermal dysplasia (Ellis‐van Creveld), DYNC2LI1‐related Chondroectodermal dysplasia (Ellis‐van Creveld), GLI1‐ related Chondroectodermal dysplasia (Ellis‐van Creveld), SMO‐ related Orofaciodigital syndrome type 4 (Mohr‐Majewski), TCTN3‐related Orofaciodigital syndrome type 2 (Mohr syndrome), NEK1‐ related NOS 10‐0260 NOS 10‐0270 NOS 10‐0280 NOS 10‐0290 NOS 10‐0300 NOS 10‐0310 NOS 10‐0320 NOS 10‐0330 NOS 10‐0340 NOS 10‐0350 NOS 10‐0360 NOS 10‐0370 NOS 10‐0380 NOS 10‐0390 NOS 10‐0400 NOS 10‐0410 NOS 10‐0420 NOS 10‐0430 Name of group/name of disorder Inheritance AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR Gene or locus NEK1 TCTN3 SMO GLI1 DYNC2LI1 WDR35 EVC2 EVC1 NEK1 CFAP410 KIAA0753 TRAF3IP1 GRK2 KIAA0586 CEP120 CFAP410 IFT52 IFT81 IFT172 MIM No. 252100 258860 See See 165220 See 617088 225500 See 252100 602271 619479 See 607380 See 109635 616546 616300 602271 617102 617895 615630 (Continues) A single case with compound heterozygosity missense variants reported See also Weyers acrofacial (acrodental) dysostosis (MIM 193530) KIAA0753 variants also associated with orofaciodigital syndrome (MIM 617127) and with Joubert syndrome (MIM 619476) TRAF3IP1 also known as IFT154 Gene also associated with Joubert syndrome (MIM 616490) Described in severe cases resembling SRPS; the CEP120 gene is also associated with Joubert syndrome (MIM 617761) Notes 9 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License NOS 10‐0250 Group number/number of disorder TABLE 1 UNGER ET AL. (Continued) Cranioectodermal dysplasia (Levin‐Sensenbrenner), WDR35‐related Cranioectodermal dysplasia (Levin‐Sensenbrenner), WDR19‐related Cranioectodermal dysplasia (Levin‐Sensenbrenner), IFT40‐ related Cranioectodermal dysplasia (Levin‐Sensenbrenner), IFT43‐ related Joubert syndrome with short‐rib thoracic dysplasia, CSPP1‐related Atrial defects‐polydactyly‐multiple congenital malformation syndrome, PRKACA‐related Atrial defects‐polydactyly‐multiple congenital malformation syndrome, PRKACB‐related Mainzer‐Saldino syndrome, IFT140‐related Mainzer‐Saldino syndrome, IFT172‐related Mainzer‐Saldino syndrome, WDR19‐related Meckel syndrome, MKS1‐related Meckel syndrome, TMEM216‐related Meckel syndrome, TMEM67‐related Meckel syndrome, CEP290‐related Meckel syndrome, RPGRIP1L‐related Meckel syndrome, CC2D2A‐related Thoracolaryngopelvic dysplasia (Barnes) NOS 10‐0450 NOS 10‐0460 NOS 10‐0470 NOS 10‐0480 NOS 10‐0490 NOS 10‐0500 NOS 10‐0510 NOS 10‐0520 NOS 10‐0530 NOS 10‐0540 NOS 10‐0550 NOS 10‐0560 NOS 10‐0570 NOS 10‐0580 NOS 10‐0590 NOS 10‐0600 NOS 10‐0610 Inheritance SP AR AR AR AR AR AR AR AR AR AD, MOS AD AR AR AR AR AR AR Gene or locus CC2D2A RPGRIP1L CEP290 TMEM67 TMEM216 MKS1 WDR19 IFT172 IFT140 PRKACB PRKACA CSPP1 IFT43 IFT40 WDR19 WDR35 IFT122 MIM No. 187760 612284 611561 611134 607361 603194 249000 See 614376 266920 619143 619142 615636 614009 see 614620 614378 613610 218330 Dominant transmission reported, but diagnostic criteria not stringent. The existence pf this entity is disputed. WDR19 is also associated with MIM 614091, 614376, 614378, 615633 as well as with nephronopthisis (MIM 614377), and Senior‐Loken syndrome (MIM 616307) IFT172 also associated with Bardet‐Biedl syndrome (MIM 619471) and isolated retinitis pigmentosa (616394) IFT140 also associated with isolated retinitis pigmentosa (MIM 617781) OMIM created the name of “Cardioacrofacial syndrome 2”; one published patient later reclassified as “Ellis‐van Creveld syndrome”. OMIM created the name of “Cardioacrofacial syndrome 1”. In OMIM as “Joubert syndrome type 21”; not all cases have thoracic dysplasia see short rib thoracic dysplasia, IFT140‐associated, above WDR19 is associated with MIM 614091, 614376, 614378, 615633 as well as with nephronopthisis (MIM 614377), Senior‐Loken syndrome (MIM 616307) and Mainzer‐ Saldino syndrome (see below) Notes UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Given the common genetic basis of several entries in this group and the absence (so far) of clear genotype‐phenotype correlations, the distinction between chondroectodermal dysplasia, asphyxiating thoracic dystrophy (see below for name change), short rib‐polydactyly syndromes and related conditions is historical and restricted to the clinical phenotypes. We have followed MIM and used the term “short‐rib thoracic dysplasia” instead of “asphyxiating thoracic dysplasia”, to avoid the negative connotation and inaccuracy of “asphyxiating”. See also paternal UPD14 and Cerebro‐costo‐mandibular syndrome (rib gap syndrome), SNRPB‐related, both in Group 36. The Bardet‐Biedl syndrome with its large phenotypic spectrum has not been included in spite of minor skeletal involvement, as the predominant clinical features are non‐skeletal. Cranioectodermal dysplasia (Levin‐Sensenbrenner), IFT122‐related Name of group/name of disorder NOS 10‐0440 Group number/number of disorder TABLE 1 10 Metaphyseal dysplasia Schmid (MCS), COL10A1‐related Cartilage‐hair hypoplasia (CHH; metaphyseal dysplasia, McKusick type), RMRP‐related Metaphyseal dysplasia with short stature (CHH‐like), POP1‐related Metaphyseal dysplasia with short stature (CHH‐like), NEPRO‐related Metaphyseal dysplasia with pancreatic insufficiency and cyclic neutropenia (Shwachman‐Bodian‐Diamond syndrome), SBDS‐related Metaphyseal dysplasia with pancreatic insufficiency and cyclic neutropenia (SBDS type 2), EFL1‐related Metaphyseal dysplasia with pancreatic insufficiency and cyclic neutropenia, DNAJC21‐related Shwachman‐Diamond like syndrome, SRP54‐related Metaphyseal dysplasia Spahr, MMP13‐related Metaphyseal anadysplasia, MMP13‐related Metaphyseal anadysplasia, MMP9‐related Metaphyseal dysplasia with maxillary hypoplasia, RUNX2‐ related NOS 11‐0010 NOS 11‐0020 NOS 11‐0030 NOS 11‐0040 NOS 11‐0050 NOS 11‐0060 NOS 11‐0070 NOS 11‐0080 NOS 11‐0090 NOS 11‐0100 NOS 11‐0110 NOS 11‐0120 AD AR AD AR AD AR RUNX2 MMP9 MMP13 MMP13 SRP54 DNAJC21 EFL1 SBDS AR AR NEPRO POP1 RMRP COL10A1 Gene or locus AR AR AR AD Inheritance 156510 613073 602111 250400 618752 617052 617941 260400 618853 617396 250250 156500 MIM No. AD Odontochondrodysplasia (ODCD), TRIP11‐related Spondylometaphyseal dysplasia Sutcliffe (or “corner fractures” type), FN1‐related Spondylometaphyseal dysplasia with cone‐rod dystrophy, PCYT1A‐related Spondylometaphyseal dysplasia with corneal dystrophy, PLCB3‐related NOS 12‐0020 NOS 12‐0030 NOS 12‐0040 NOS 12‐0050 PLCB3 PCYT1A FN1 TRIP11 ACP5 618961 608940 184255 184260 607944 (Continues) Some cases are linked to COL2A1 but not the original family See also Achondrogenesis, TRIP11‐related (formerly type 1A) Frequently associated with intragenic duplication of exons 3 to 5 or 3 to 6. See also Cleidocranial dysplasia, RUNX2‐related (below; MIM 119600), as well as non‐ syndromic midline craniosynostosis, RUNX2‐related, below Dominant, monoallelic type; includes SEMD Missouri type Recessive, biallelic variants Known in OMIM as Neutropenia, severe congenital, 8 Known in OMIM as Bone Marrow Failure Syndrome 3; BMFS3 See also severe spondylodysplastic dysplasia, Sedaghatian‐ like Facial features and hypotrichosis reminiscent of Cartilage‐ Hair Hypoplasia The clinical spectrum is variable. The denomination of “anauxetic dysplasia 2” in MIM is confusing as anauxetic dysplasia is a variant of Cartilage‐Hair Hypoplasia The phenotype of CHH is variable and includes MIM 607095‐anauxetic dysplasia as well as MIM 250460‐ metaphyseal dysplasia without hypotrichosis Pathogenic variants are typically located in the C‐terminal domain of the protein. Notes 11 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License AR AR AR Spondyloenchondrodysplasia with immune dysregulation (SPENCD), ACP5‐related NOS 12‐0010 AR Spondylometaphyseal dysplasias (SMD) Group 12 See Rhizomelic spondylo‐metaphyseal dysplasia with remission, LBR‐related (Group 13), for another anadysplasia‐like disorder Name of group/name of disorder Metaphyseal dysplasias Group 11 (Continued) Group number/number of disorder TABLE 1 UNGER ET AL. (Continued) Inheritance AR Gene or locus HHAT MIM No. 600092 Notes Also known as Nivelon‐Nivelon‐Mabille syndrome (sic) Spondyloepi(meta)physeal dysplasias (SE(M)D) SED tarda, X‐linked (SED‐XL), TRAPPC2‐related SED with diabetes mellitus (Wolcott‐Rallison syndrome), EIF2AK3‐related Dyggve‐Melchior‐Clausen dysplasia, DYM‐related Smith‐McCort dysplasia, DYM‐related Smith‐McCort dysplasia, RAB33B‐related SEMD, BNIP1‐related SEMD, MATN3‐related SEMD, DDRGK1‐related (Shohat type) SEMD with leucodystrophy, AIFM1‐related SEMD, RSPRY1‐related SEMD, TMEM165‐related SEMD with microcephaly, retinal dystrophy and hearing loss, PISD‐related (Liberfarb syndrome) SEMD, UFSP2‐related SEMD, short limb–abnormal calcification type, DDR2‐ related Immuno‐osseous dysplasia, SMARCAL1‐related (Schimke type) SEMD with immune deficiency and intellectual disability, EXTL3‐related SEMD with immune deficiency, PGM3‐related SEMD with intellectual disability, NANS‐related SEMD with severe short stature, RPL13‐related SEMD with elevated lysosomal enzymes, MBTPS1‐related Short stature, skeletal dysplasia, liver failure, optic nerve atrophy and Pelger‐Huet anomaly, NBAS‐related Short stature, skeletal dysplasia and liver failure, RINT1‐ related NOS 13‐0010 NOS 13‐0020 NOS 13‐0030 NOS 13‐0040 NOS 13‐0050 NOS 13‐0060 NOS 13‐0070 NOS 13‐0080 NOS 13‐0090 NOS 13‐0100 NOS 13‐0110 NOS 13‐0120 NOS 13‐0130 NOS 13‐0140 NOS 13‐0150 NOS 13‐0160 NOS 13‐0170 NOS 13‐0180 NOS 13‐0190 NOS 13‐0200 NOS 13‐0210 NOS 13‐0220 AR AR AR AD AR AR AR AR AR AD AR AR AR XL AR AR AR AR AR AR AR XL RINT1 NBAS MBTPS1 RPL13 NANS PGM3 EXTL3 SMARCAL1 DDR2 UFSP2 PISD TMEM165 RSPRY1 AIFM1 DDRGK1 MATN3 BNIP1 RAB33B DYM DYM EIF2AK3 TRAPPC2 618641 616483 618392 618728 610442 615816 617425 242900 271665 142669, 617974 618889 614727 616723 300232 602557 608728 see 603291 615222 607326 223800 226980 313400 Combination of clinical features is variable; also known as infantile liver failure syndrome type 3 Combination of clinical features is variable; also known as infantile liver failure syndrome type 2 only two unrelated individuals known so far; in OMIM as “Kondo‐Fu type”; possible role of lysosomal dysfunction in pathogenesis is unclear Known in OMIM as “immunodeficiency 23” Also known as “Immunoskeletal dysplasia with developmental abnormalities”; includes Omenn syndrome with chondrodysplasia; see also SMARCAL1, above Nephrotic syndrome is an important manifestation; see also EXTL3 deficiency, below See also other dysplasias with stippling Includes Familial hip dysplasia (Beukes) Phenotypically variable; in some affected individuals hearing and vision may be unaffected Congenital disorder of glycosylation type IIk See also MATN3‐related MED in Group 9 See also SMD Kozlowski, TRPV4‐related, Severe spondylometaphyseal dysplasia (Sedaghatian type), GPX4‐related, as well as Axial spondylometaphyseal dysplasia, CFAP410‐related and Axial spondylometaphyseal dysplasia, NEK1‐related. In addition, there are many reports of sporadic patients with unclassified SMD variants. Chondrodysplasia‐pseudohermaphroditism syndrome, HHAT‐related Name of group/name of disorder UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 13 NOS 12‐0060 Group number/number of disorder TABLE 1 12 (Continued) Spondylar and nasal alterations with striated metaphyses (SPONASTRIME dysplasia), TONSL‐related Spondyloepiphyseal dysplasia, sensorineural hearing loss, impaired intellectual development, and Leber congenital amaurosis (SHILCA) syndrome, NMNAT1‐related Platyspondyly (brachyolmia) with amelogenesis imperfecta, LTBP3‐related Cerebral, ocular, dental, auricular, and skeletal anomalies (CODAS syndrome), LONP1‐related Epiphyseal and vertebral dysplasia, microtia, flat nose plus associated malformation (EVEN‐PLUS syndrome), HSPA9‐related Cataracts, growth hormone deficiency, sensory neuropathy, sensorineural hearing loss, and skeletal dysplasia (CAGSSS syndrome), IARS2‐related Steel syndrome, COL27A1‐related Rhizomelic spondylo‐metaphyseal dysplasia with remission, LBR‐related Rhizomelic spondylo‐epi‐metaphyseal dysplasia, GNPNAT1‐related NOS 13‐0240 NOS 13‐0250 NOS 13‐0260 NOS 13‐0270 NOS 13‐0280 NOS 13‐0290 NOS 13‐0300 NOS 13‐0310 NOS 13‐0320 Inheritance Gene or locus IARS2 AR AR AR GNPNAT1 LBR COL27A1 HSPA9 AR AR LONP1 AR LTBP3 NMNAT1 AR AR TONSL SLC39A13 AR AR MIM No. 619598 618019 615155 616007 616854 600373 601216 619260 271510 612350 Notes Mitochondrial chaperonopathy Mitochondrial chaperonopathy Nonsyndromic Leber congenital amaurosis (LCA9; MIM 608553) is also caused by biallelic NMNAT1 variants Possibly genetically heterogeneous SLC39A13/ZIP13 zinc transporter Achondrogenesis, TRIP11‐related (formerly type 1A) Schneckenbecken dysplasia, SLC35D1‐related Severe spondylometaphyseal dysplasia (Sedaghatian type), GPX4‐related Severe spondylometaphyseal dysplasia (SMD Sedaghatian‐like), SBDS‐related Opsismodysplasia, INPPL1‐related Spondylometaphyseal dysplasia, PAM16‐ related Carbohydrate deficient glycoprotein syndrome, ALG9‐ related (ALG9‐CDG; Gillessen‐Kaesbach‐Nishimura syndrome) NOS 14‐0010 NOS 14‐0020 NOS 14‐0030 NOS 14‐0040 NOS 14‐0050 NOS 14‐0060 NOS 14‐0070 AR PAM16 ALG9 AR INPPL1 SBDS GPX4 SLC35D1 TRIP11 AR AR AR AR AR 263210, 608776 613320 258480 260400 250220 269250 200600 Includes lethal and milder cases 13 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License (Continues) See also Thanatophoric dysplasia, FGFR3‐related; achondrogenesis and Torrance dysplasia, COL2A1‐related; Fibrochondrogenesis, COL11A1‐related; Achondrogenesis, SLC26A2‐related; and Metatropic Dysplasia, TRPV4‐related Severe spondylodysplastic dysplasias Group 14 See also: Opsismodysplasia, INPPL1‐related; Mucopolysaccharidosis type 4, GALNS‐related (type 4A; Morquio disease), as well as Progressive pseudorheumatoid dysplasia (PPRD), WISP3‐ related. See also the non‐genetic SEMD phenocopy, “Chondrodysplasia and growth failure following early hematopoietic stem cell transplantation”, ***https://doi.org/10.1002/ajmg.a. 62021, PMID: 33398909. Spondylodysplastic Ehlers‐Danlos syndrome (SDEDSS type 3), SLC39A13‐related Name of group/name of disorder NOS 13‐0230 Group number/number of disorder TABLE 1 UNGER ET AL. AD AR Mesomelic dysplasia (Langer type), SHOX‐related Omodysplasia, recessive type, GPC6‐related Omodysplasia, dominant type, FZD2‐related Robinow syndrome, WNT5A‐related Robinow syndrome, DVL1‐related Robinow syndrome, DVL3‐related Robinow syndrome, FZD2‐related Robinow syndrome, recessive type, ROR2‐related Robinow syndrome, recessive type, NXN‐related Mesomelic dysplasia, HOXD‐related (Kim or Korean type, Kantaputra type, Fryns type) Mesomelic dysplasia, Nievergelt type Mesomelic dysplasia, Kozlowski‐Reardon type Mesomelic dysplasia with acral synostoses (Verloes‐ David‐Pfeiffer type) Mesomelic dysplasia (Savarirayan type), ID4‐related Mesomelic dysplasia with digital anomalies and intellectual disability (KINSSHIP syndrome), AFF3‐related Oculo‐skeletal syndrome with rhizomelic shortening, MAB21L2‐related NOS 15‐0020 NOS 15‐0030 NOS 15‐0040 NOS 15‐0050 NOS 15‐0060 NOS 15‐0070 NOS 15‐0080 NOS 15‐0090 NOS 15‐0100 NOS 15‐0110 NOS 15‐0120 NOS 15‐0130 NOS 15‐0140 NOS 15‐0150 NOS 15‐0160 NOS 15‐0170 MAB21L2 AFF3 ID4 SULF1, SLCO5A1 HOXD NXN ROR2 FZD2 DVL3 DVL1 WNT5A FZD2 GPC6 SHOX SHOX Gene or locus 615877 619297 605274 600383 249710 163400 156232 268310 616894 616331 180700 164745 258315 249700 127300 MIM No. In OMIM as “Microphthalmia/Coloboma and skeletal dysplasia syndrome”. Skeletal involvement not in all individuals. Two brothers with biallelic variants (AR?) had ocular but no skeletal involvement. In spite of the acronym, this condition is quite different from both Nievergelt and Savarirayan mesomelic dysplasias Microdeletions on 6p22.3 Microdeletion syndrome involving two adjacent genes Duplications at HOXD gene cluster locus; phenotypes is variable also within families Includes previous COVESDEM (costo‐vertebral segmentation defect with mesomelia); see also brachydactyly type B Includes Reinhardt‐Pfeiffer dysplasia, MIM 191400. Clinical continuum with Idiopathic short stature (MIM 300582) Notes Acromesomelic dysplasias Acromesomelic dysplasia (type Maroteaux), NPR2‐related Acromesomelic dysplasia, PRKG2‐related NOS 16‐0010 NOS 16‐0020 AR AR PRKG2 NPR2 619636, 619638 602875 Condition associated with biallelic loss of function variants. Three brothers form one family were found to have a spondylo‐metaphyseal dysplasia phenotype (in OMIM as “619638 ‐ Spondylometaphyseal dysplasia, Pagnamenta type”. Needs to be confirmed) UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 16 See also Tibial hemimelia‐polysyndactyly‐triphalangeal thumb, ZRS‐related, also consider: mesomelic dysplasia, Camera type (MIM#611886), the status of which remains unconfirmed AD AD AD AD AD AR AR AD AD AD AD AD AR Pseudo‐AR Pseudo‐AD Dyschondrosteosis (Leri‐Weill), SHOX‐related NOS 15‐0010 Inheritance Name of group/name of disorder Mesomelic and rhizo‐mesomelic dysplasias Group 15 (Continued) Group number/number of disorder TABLE 1 14 Grebe dysplasia, GDF5‐related Grebe dysplasia, BMPR1B‐related Fibular hypoplasia and complex brachydactyly (Du Pan), GDF5‐related Fibular hypoplasia and complex brachydactyly (Du Pan), BMPR1B‐related Acromesomelic dysplasia, Osebold‐Remondini type Acromelic dysplasias Acrocapitofemoral dysplasia, IHH‐related Geleophysic dysplasia, ADAMTSL2‐related Geleophysic dysplasia, FBN1‐related Geleophysic dysplasia, LTBP3‐related Acromicric dysplasia, FBN1‐related Acromicric dysplasia, LTBP3‐related Weill‐Marchesani syndrome, dominant, FBN1‐related Weill‐Marchesani syndrome, ADAMTS10‐related Weill‐Marchesani syndrome, ADAMTS17‐related Weill‐Marchesani syndrome, LTBP2‐related Myhre dysplasia, SMAD4‐related Acrodysostosis, PDE4D‐related Acrodysostosis, PRKAR1A‐related Angel‐shaped phalango‐epiphyseal dysplasia (ASPED) Albright hereditary osteodystrophy, GNAS‐related Leri Pleonosteosis, linked to 8q22.1 SED with brachydactyly, MIR140‐related NOS 16‐0040 NOS 16‐0050 NOS 16‐0060 NOS 16‐0070 Group 17 NOS 17‐0010 NOS 17‐0020 NOS 17‐0030 NOS 17‐0040 NOS 17‐0050 NOS 17‐0060 NOS 17‐0070 NOS 17‐0080 NOS 17‐0090 NOS 17‐0100 NOS 17‐0110 NOS 17‐0120 NOS 17‐0130 NOS 17‐0140 NOS 17‐0150 NOS 17‐0160 NOS 17‐0170 Inheritance AD AD AD AD AD AD AD AR AR AR AD AD AD AD AD AR AR AD AR AR AR AR Gene or locus MIR140 8q22.1 GNAS PRKAR1A PDE4D SMAD4 LTBP2 ADAMTS17 ADAMTS10 FBN1 LTBP3 FBN1 LTBP3 FBN1 ADAMTSL2 IHH BMPR1B GDF5 BMPR1B GDF5 MIM No. 618618 151200 103580 105835 101800 614613 139210 614819 613195 277600 608328 See 617809 102370 617809 614185 231050 607778 112910 See 603248 228900 609441 200700 Brachydactylies (isolated) Brachydactyly type A1, IHH‐related Brachydactyly type A2, BMPR1B‐related Brachydactyly type A2, BMP2‐related NOS 18‐0010 NOS 18‐0020 NOS 18‐0030 AD BMP2 IHH BMPR1B AD AD 112500 112600 112600 Notes Duplication of BMP2 enhancer Brachydactyly with cone‐shaped epiphyses (Continues) Duplication at 8q22.1 encompassing GDF6 and SDC2 Overlaps with progressive osseous heteroplasia Possibly related or allelic to brachydactyly type C Includes acroscyphodysplasia (see PMID 30006632) Includes acrolaryngeal dysplasia, previously known as Fantasy Island dysplasia or Tattoo dysplasia, and Moore‐Federman syndrome See other conditions associated with the IHH gene in this table See also other GDF5‐related disorders Includes acromesomelic dysplasia Hunter‐Thompson type and acromesomelic dysplasia with genital anomalies; see also see other GDF5‐related disorders 15 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 18 See also Cartilage‐Hair Hypoplasia, RMRP‐related, and the brachdydactyly groups, below (Groups 18 and 19) Name of group/name of disorder NOS 16‐0030 (Continued) Group number/number of disorder TABLE 1 UNGER ET AL. Brachydactyly type A2, GDF5‐related Brachydactyly type B1, ROR2‐related Brachydactyly type B2, NOG‐related Brachydactyly type C, GDF5‐related Brachydactyly type D, HOXD13‐related Brachydactyly type E, HOXD13‐related Brachydactyly with anonychia (Cooks syndrome), KCNJ2‐ related Preaxial brachydactyly, PAX3 type, linked to 2q35‐36 NOS 18‐0050 NOS 18‐0060 NOS 18‐0070 NOS 18‐0080 NOS 18‐0090 NOS 18‐0100 NOS 18‐0110 Brachydactylies as part of syndromes Trichorhinophalangeal dysplasia types 1/3 Langer‐Giedion syndrome (Trichorhinophalangeal dysplasia type 2) Catel‐Manzke syndrome, TGDS‐related Deafness, onychodystrophy, osteodystrophy, retardation and seizures (DOORS) syndrome Brachydactyly—intellectual disability syndrome, HDAC4‐ related Hyperphosphatasia with intellectual disability, brachytelephalangy, and distinct face, PIGV‐related Brachydactyly‐short stature‐hypertension syndrome, PDE3A‐related (Bilginturan syndrome) Brachydactyly, obesity and intellectual disability syndrome, PRMT7‐related NOS 19‐0010 NOS 19‐0020 NOS 19‐0030 NOS 19‐0040 NOS 19‐0050 NOS 19‐0060 NOS 19‐0070 NOS 19‐0080 Inheritance AR AD AR AD AR AR AD AD AD AD AD AD AD AD AD AD Gene or locus PRMT7 PDE3A PIGV HDAC4 TBC1D24 TGDS TRPS1, EXT1 TRPS1 2q35‐36 KCNJ2 HOXD13 HOXD13 GDF5 NOG ROR2 GDF5 MIM No. 617157 112410 239300 600430 220500 616145 150230 190350, 190351 106995 113300 113200 113100 611377 113000 112600 Notes Phenotype reminiscent of Albright Hereditary Ostedystrophy (AHO), GNAS‐related (see above) but recessive. In OMIM as “617157—Short stature, brachydactyly, intellectual developmental disability, and seizures” Several other related defects of GPI synthesis known, most cases not known for skeletal changes; see for example, MIM 610293 for a summary The existence of this entity is questionable. HDAC4 variants alone may not be sufficient to produce either brachydactyly or intellectual disability. Some patients have microdeletions involving contiguous genes (2q37 deletion syndrome). HDAC4 variants have been associated with a developmental disorder (see MIM 619797) “Osteodystrophy” and “retardation” are misnomers Microdeletion syndrome; see also multiple cartilaginous exostoses TRPS1 and 3 are a phenotypic spectrum Deletions leading to disruption of TADs and abnormal expression of PAX3 Duplications of SOX9/KCNJ2 regulatory region Brachydactyly type D is often a component of Brachydactyly type E See other GDF5‐related disorders See also Robinow syndrome/COVESDEM See also Grebe dysplasia, GDF5‐related; Fibular hypoplasia and complex brachydactyly (Du Pan), GDF5‐ related; Brachydactyly type C, GDF5‐related; and Multiple synostoses syndrome, GDF5‐related UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 19 See also brachdactyly, PTHLH‐related (below) Name of group/name of disorder NOS 18‐0040 (Continued) Group number/number of disorder TABLE 1 16 (Continued) Hand‐foot‐genital syndrome, HOXA13‐related Rubinstein‐Taybi syndrome, CREBBP‐related Rubinstein‐Taybi syndrome, EP300‐related Brachydactyly, Temtamy type, CHSY1‐related Hyperphalangism, characteristic facies, hallux valgus and bronchomalacia (Chitayat syndrome), ERF‐related Hypoacusis with facial an digital anomalies (Keipert syndrome), GPC4‐related Christian type brachydactyly Coffin‐Siris syndrome, ARID1A‐related Coffin‐Siris syndrome, ARIB1B‐related Coffin‐Siris syndrome, SMARCB1‐related Coffin‐Siris syndrome, SMARCA4‐related Coffin‐Siris syndrome, SMARCE1‐related Cardiomyopathy and brachydactyly, LMNA‐related (Heart‐ hand syndrome type IV) NOS 19‐0100 NOS 19‐0110 NOS 19‐0120 NOS 19‐0130 NOS 19‐0140 NOS 19‐0150 NOS 19‐0160 NOS 19‐0170 NOS 19‐0180 NOS 19‐0190 NOS 19‐0200 NOS 19‐0210 NOS 19‐0220 Inheritance AD AD AD AD AD AD AD XL AD AR AD AD AD AD Gene or locus LMNA SMARCE1 SMARCA4 SMARCB1 ARID1B ARID1A GPC4 ERF CHSY1 EP300 CREBBP HOXA13 MYCN Campomelic dysplasia (CD), SOX9‐related Stüve‐Wiedemann syndrome, LIFR‐related Stüve‐Wiedemann syndrome, IL6ST‐related Kyphomelic dysplasia with facial dysmorphism, KIF5B‐ related Bent bone dysplasia, FGFR2‐related Bent bone dysplasia, LAMA5‐related NOS 20‐0010 NOS 20‐0020 NOS 20‐0030 NOS 20‐0040 NOS 20‐0050 NOS 20‐0060 AR AD AD AR AR AD LAMA5 FGFR2 KIF5B IL6ST LIFR SOX9 MIM No. 614592 211350 619751 601559 114290 610140 616938 614609 614608 135900 614607 112450 301026 617180 605282 613684 180849 140000 164280 Biallelic LAMA5 variants are associated with congenital or infantile nephrotic syndrome (MIM) The name “kyphomelic dysplasia” has been applied to heterogeneous conditions Includes former neonatal Schwartz‐Jampel syndrome or SJS type 2 Includes acampomelic campomelic dysplasia (ACD), mild campomelic dysplasia (MIM 602196); so‐called Ischio‐ pubic‐patellar dysplasia, as well as some cases of isolated Pierre‐Robin sequence in OMIM as “Heart‐Hand syndrome, Slovenian type” Variants in various components of the SWI/SNF complex have been reported in patients with a diagnosis of Coffin‐Siris syndrome Brachytelephalangy is the most consistent skeletal signs Typically a monoallelic Y89C substitution Includes Guttmacher syndrome Notes 17 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License (Continues) Bent bones is an unspecific finding, particularly in a prenatal setting, that can be observed in numerous other conditions, such as those with bone fragility; thus see the OI‐bone fragility group (group 26) as well as Hypophosphatasia, ALPL‐related. Bent bones dysplasia group Group 20 See also CDP, X‐linked recessive, ARSE‐related (brachytelephalangic type; CDPX1) Microcephaly‐oculo‐digito‐esophageal‐duodenal syndrome, MYCN‐related (Feingold syndrome) Name of group/name of disorder NOS 19‐0090 Group number/number of disorder TABLE 1 UNGER ET AL. Name of group/name of disorder Primordial dwarfism and slender bones group 3‐M syndrome, CUL7‐related 3‐M syndrome, OBSL1‐related 3‐M syndrome, CCDC8‐related Sanjad‐Sakati syndrome, recessive, TBCE‐related Kenny‐Caffey syndrome, dominant, FAM111A‐related Osteocraniostenosis, FAM111A‐related Hallermann‐Streiff syndrome Microcephalic osteodysplastic primordial dwarfism, RNU4ATAC‐related Roifman syndrome, RNU4ATAC‐related Microcephalic osteodysplastic primordial dwarfism, PCNT‐ related Microcephalic osteodysplastic primordial dwarfism, ATR‐ related Microcephalic osteodysplastic primordial dwarfism, RBBP8‐related Microcephalic osteodysplastic primordial dwarfism, CEP152‐related Microcephalic osteodysplastic primordial dwarfism, DNA2‐related Microcephalic osteodysplastic primordial dwarfism, TRAIP‐related Microcephalic osteodysplastic primordial dwarfism, NSMCE2‐related Microcephalic osteodysplastic primordial dwarfism, CENPE‐related Microcephalic osteodysplastic primordial dwarfism, CRIPT‐related Microcephalic osteodysplastic primordial dwarfism, XRCC4‐related Group 21 NOS 21‐0010 NOS 21‐0020 NOS 21‐0030 NOS 21‐0040 NOS 21‐0050 NOS 21‐0060 NOS 21‐0070 NOS 21‐0080 NOS 21‐0090 NOS 21‐0100 NOS 21‐0110 NOS 21‐0120 NOS 21‐0130 NOS 21‐0140 NOS 21‐0150 NOS 21‐0160 NOS 21‐0170 NOS 21‐0180 NOS 21‐0190 (Continued) AR AR AR AR AR AR AR AR AR AR AR AR AD AD AR AR AR AR Inheritance XRCC4 CRIPT CENPE NSMCE2 TRAIP DNA2 CEP152 RBBP8 ATR PCNT RNU4ATAC RNU4ATAC FAM111A FAM111A TBCE CCDC8 OBSL1 CUL7 Gene or locus 616541 615789 see 616051 617253 616777 615807 613823 606744 210600 210720 616651 210710 234100 602361 127000 241410 614205 612921 273750 MIM No. In MIM as short stature, microcephaly and endocrine dysfunction In MIM as short stature with microcephaly and distinctive facies In MIM as autosomal recessive primary microcephaly In MIM as Seckel syndrome 10 In MIM as Seckel syndrome 9 In MIM as Seckel syndrome 8. The DNA2 gene is also associated with autosomal dominant progressive external ophthalmoplegia with mitochondrial DNA deletions (MIM 615156) In MIM as Seckel syndrome 5. The CEP152 gene also causes primary microcephaly (MIM 614852) In MIM as Seckel syndrome 2. The RBBP8 gene is also associated with Jawad syndrome (microcephaly with intellectual disability and digital anomalies; MIM 251255) In MIM as Seckel syndrome 1 Was MOPD2, Majewski type See other RNU4ATAC‐related condition in this table Was MOPD 1/3; usually homozygous variants; includes Taybi‐Linder cephaloskeletal dysplasia Usually sporadic; some cases have phenotypic overlap with osteocraniostenosis, FAM111A‐related In OMIM as “Kenny‐Caffey type 1” but does not correspond to the disorder described by Kenny and Caffey which is the dominant form Includes dolichospondylic dysplasia and Yakut short stature syndrome Notes UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group number/number of disorder TABLE 1 18 (Continued) Microcephalic osteodysplastic primordial dwarfism, or microcephaly‐short stature‐micromelia‐limb abnormalities, DONSON‐related IMAGE syndrome (intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia, and genital anomalies) IMAGE syndrome/FILS syndrome, POLE‐related Saul‐Wilson syndrome, COG4‐related Short stature, facial dysmorphism, skeletal and dental anomalies syndrome, SCUBE3‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin), ORC4‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin), ORC4‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin), ORC6‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin), CDT1‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin), CDC6‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin), CDC45‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin), MCM3‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin), MCM5‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin), MCM7‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin), GMNN‐related Ear‐patella‐primordial short stature syndrome (Meier‐ Gorlin) with craniosynostosis, GINS2‐related Lysosomal storage diseases with skeletal involvement Mucopolysaccharidosis type 1, IDUA‐related Mucopolysaccharidosis type 2, IDS‐related Mucopolysaccharidosis type 3, SGSH‐related (type 3A) NOS 21‐0210 NOS 21‐0220 NOS 21‐0230 NOS 21‐0240 NOS 21‐0250 NOS 21‐0260 NOS 21‐0270 NOS 21‐0280 NOS 21‐0290 NOS 21‐0300 NOS 21‐0310 NOS 21‐0320 NOS 21‐0330 NOS 21‐0340 NOS 21‐0350 Group 22 NOS 22‐0010 NOS 22‐0020 NOS 22‐0030 Name of group/name of disorder AD AR XL AR AD AD AR AR AR AR AR AR AR AR AR AR SGSH IDS IDUA GINS2 GMNN MCM7 MCM5 MCM3 CDC45 CDC6 CDT1 ORC6 ORC4 ORC1 SCUBE3 COG4 POLE CDKN1C AD AR Gene or locus DONSON Inheritance AR MIM No. 252900 309900 607014, 607015, 607016 See 610609 613804 See 600592 602696 See 602693 603465 613805 605525 613803 613800 224690 619184 618150 618336, 615139 614732 251230, 617604 Notes Known as Sanfilippo A syndrome Known as Hunter syndrome (Continues) Was type 1H‐Hurler syndrome, type 1S‐Scheie syndrome A single case reported so far A single case reported so far in OMIM as “short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 2” The phenotype is variable and may include immune deficiency (OMIM 615139) Gene also known to cause Beckwith‐Wiedemann syndrome (MIM 130650). IMAGE‐associated variants are clustered in the PCNA‐binding region and are maternally transmitted (gene is imprinted with preferential maternal expression) Milder affected patients may fall into the Meier‐Gorlin syndrome spectrum 19 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License NOS 21‐0200 Group number/number of disorder TABLE 1 UNGER ET AL. Mucolipidosis III (Pseudo‐Hurler polydystrophy), GNPTAB‐related Mucolipidosis III (Pseudo‐Hurler polydystrophy), GNPTG‐ related Mucolipidosis, GCAF‐related Gaucher disease, GBA‐related NOS 22‐0230 NOS 22‐0240 NOS 22‐0250 NOS 22‐0260 Inheritance AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR AR Gene or locus GBA CGAF GNPTG GNPTAB GNPTAB SUMF1 SLC17A5 PPGB NEU1 GLB1 AGA MANBA MAN2B1 FUCA VPS33A ARSK GUSB ARSB GLB1 GALNS GNS HSGNAT NAGLU MIM No. 230800 619345 252605 252600 252500 272200 269920 256540 256550 230500 208400 248510 248500 230000 617303 610011 253220 253200 253010 253000 252940 252930 252920 Notes Long‐standing Gaucher disease can have bone changes that are different from the “dysostosis multiplex” pattern seen in other lysosomal diseases in this group The gene was previously known as TMEM251 and encodes for a “GNTAP cleavage and activity factor” (see GNTAP, above) In general somewhat milder phenotype than the GNPTAB‐ related form The old entity of Pacman dysplasia is the prenatal manifestation of mucolipidosis II with hyperparathyroidism Several forms of different severity Several forms of different severity Several forms, see also mucopolysaccharidosis type 4B (Morquio B) above Known as Sly syndrome Known as Maroteaux‐Lamy syndrome Known as Morquio B syndrome Known as Morquio A syndrome Known as Sanfilippo D syndrome Known as Sanfilippo C syndrome Known as Sanfilippo B syndrome Chondrodysplasia punctata (CDP) group CDP, X‐linked recessive, ARSE‐related (brachytelephalangic type; CDPX1) NOS 23‐0010 XL ARSE 302950 UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 23 See also familial arthritis with hyaluronidase deficiency (“mucopolysaccharidosis type 9”), HYAL1‐related; SEMD with elevated lysosomal enzymes, MBTPS1‐related, above; as well as Farber disease, ASAH1‐related, below. Mucolipidosis II (I‐cell disease), GNPTAB‐related NOS 22‐0220 beta‐Mannosidosis, MANBA‐related NOS 22‐0150 Multiple sulfatase deficiency, SUMF‐related alpha‐Mannosidosis, MAN2B1‐related NOS 22‐0140 Sialic acid storage disease (SIASD), SLC17A5‐related Fucosidosis, FUCA‐related NOS 22‐0130 NOS 22‐0210 Mucopolysaccharidosis‐plus syndrome, VPS33A‐related NOS 22‐0120 NOS 22‐0200 Mucopolysaccharidosis type 10, ARSK‐related NOS 22‐0110 Galactosialidosis, PPGB‐related Mucopolysaccharidosis type 7, GUSB‐related NOS 22‐0100 Sialidosis, NEU1‐related Mucopolysaccharidosis type 6, ARSB‐related NOS 22‐0090 NOS 22‐0190 Mucopolysaccharidosis type 4, GLB1‐related (type 4B) NOS 22‐0080 NOS 22‐0180 Mucopolysaccharidosis type 4, GALNS‐related (type 4A) NOS 22‐0070 Aspartylglucosaminuria, AGA‐related Mucopolysaccharidosis type 3, GNS‐related (type 3D) NOS 22‐0060 Gangliosidosis GM1, GLB1‐related Mucopolysaccharidosis type 3, HSGNAT‐related (type 3C) NOS 22‐0050 NOS 22‐0160 Mucopolysaccharidosis type 3, NAGLU‐related (type 3B) NOS 22‐0170 Name of group/name of disorder NOS 22‐0040 (Continued) Group number/number of disorder TABLE 1 20 (Continued) Congenital hemidysplasia, ichthyosis, limb defects (CHILD) syndrome, NSDHL‐related Keutel syndrome, MGP‐related Greenberg dysplasia, LBR‐related Rhizomelic CDP, PEX7‐related Rhizomelic CDP, DHPAT‐related Rhizomelic CDP, AGP5‐related Rhizomelic CDP, FAR1‐related Rhizomelic CDP, PEX5‐related CDP tibial‐metacarpal type NOS 23‐0030 NOS 23‐0040 NOS 23‐0050 NOS 23‐0060 NOS 23‐0070 NOS 23‐0080 NOS 23‐0090 NOS 23‐0100 NOS 23‐0110 Inheritance AR AR AR AR AR AR AR XL XL Gene or locus PEX5 FAR1 AGPS DHPAT PEX7 LBR MGP NSDHL EBP MIM No. 118651 616716 616154 600121 222765 215100 215140 245150 308050 302960 Some cases possibly caused by maternal auto‐immune disease MIM calls this entity “peroxisomal fatty acyl‐CoA reductase 1 disorder”; skeletal phenotype milder than other rCDP forms. The FAR1 gene is also associated with cataracts, spastic paraparesis, and speech delay (MIM 619338, AD) Includes hydrops‐ectopic calcification‐moth‐eaten appearance dysplasia (HEM) and dappled diaphyseal dysplasia; possibly includes also the ultrarare entity designed as Astley‐Kendall dysplasia. See also the non‐ lethal condition associated with LBR, above Notes Osteopetrosis and related osteoclast disorders Osteopetrosis, neonatal or infantile form, TCIRG1‐related Osteopetrosis, neonatal or infantile form, CLCN7‐related Osteopetrosis, neonatal or infantile form, SNX10‐related Osteopetrosis, infantile form, with nervous system involvement, OSTM1‐related Osteopetrosis, infantile form, osteoclast‐poor with immunoglobulin deficiency, TNFRSF11A‐related Osteopetrosis, intermediate form, TCIRG1‐related Osteopetrosis, intermediate form, TNFSF11‐related Osteopetrosis, intermediate form, PLEKHM‐related Osteopetrosis, intermediate form, CLCN7‐related Osteopetrosis, late‐onset, dominant form, CLCN7‐related NOS 24‐0010 NOS 24‐0020 NOS 24‐0030 NOS 24‐0040 NOS 24‐0050 NOS 24‐0060 NOS 24‐0070 NOS 24‐0080 NOS 24‐0090 NOS 24‐0100 AD AR AR AR AR AR AR AR AR AR CLCN7 CLCN7 PLEKHM1 166600 259710 611497 259700 259710 TCIRG1 612301 259720 615085 611490 259700 TNFSF11 TNFRSF11A OSTM1 SNX10 CLCN7 TCIRG1 (Continues) See also below in this group, dysosteosclerosis, TNFRSF11A‐related, as well as MIM 602080‐familial expansile osteolysis Includes former osteopetrosis with infantile neuraxonal dysplasia (MIM 600329) ‐ 21 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 24 Note: stippling can occur in several syndromes such as Zellweger cerebro‐hepato‐renal syndrome (see OMIM for the many genetic types), Smith‐Lemli‐Opitz (MIM 270400), in Mucolipidosis II (I‐cell disease), GNPTAB‐related, mild forms of Raine dysplasia, FAM20C‐related, and others. See also SEMD short limb‐abnormal calcification type, DDR2‐related. Stippling in the fetus is also observed as a consequence of maternal auto‐immune disease, sometimes presenting as “CDP tibial‐metacarpal type”. CDP, X‐linked dominant, EBP‐related (Conradi‐ Hünermann type; CDPX2) Name of group/name of disorder NOS 23‐0020 Group number/number of disorder TABLE 1 UNGER ET AL. Osteopetrosis with renal tubular acidosis, CA2‐related Osteopetrosis with ectodermal dysplasia and immune defect (OLEDAID), IKBKG‐related Osteopetrosis, moderate form, SLC4A2‐related Osteopetrosis, moderate form with defective leucocyte adhesion, FERMT3‐related Osteopetrosis, moderate form with defective leucocyte adhesion, RASGRP2‐related Osteosclerotic metaphyseal dysplasia, LRKK1‐related Pyknodysostosis, CTSK‐related Dysosteosclerosis, SLC29A3‐related Dysosteosclerosis, TNFRSF11A‐related Dysosteosclerosis with degenerative encephalopathy and brain malformation, CSF1R‐related NOS 24‐0120 NOS 24‐0130 NOS 24‐0140 NOS 24‐0150 NOS 24‐0160 NOS 24‐0170 NOS 24‐0180 NOS 24‐0190 NOS 24‐0200 Inheritance AR AR AR AR AR AR AR AR XL AR Gene or locus CSF1R TNFRSF11A SLC29A3 CTSK LRRK1 RASGRP2 FERMT3 SLC4A2 IKBKG CA2 MIM No. 618476 224300 224300 265800 615198 615888 612840 See 109280 300301 259730 In OMIM as “Brain abnormalities, neurodegeneration and dysosteosclerosis (BANDDOS)”; gene also associated with MIM 221820—leukoencephalopathy with spheroids. This entity probably forms a spectrum with Osteopetrosis, intermediate form, TNFSF11‐related (above) In some individuals, the features of pyknodyostosis are absent and the disorder mimics osteopetrosis The name may be misleading as the condition is best described as a form of osteopetrosis OMIM only includes bleeding disorder, platelet type, 18 (MIM 615888) for this gene In OMIM as Leucocyte Adhesion Deficiency 3 (LAD3)— MIM 612840 A single adult patient reported, phenotype may evolve Notes Osteosclerotic disorders Desmosterolosis, DHCR4‐related Raine dysplasia, FAM20C‐related Caffey disease, COL1A1‐related Caffey dysplasia (severe variants with prenatal onset) Dysplastic cortical hyperostosis, Kozlowski‐Tsuruta type Dysplastic cortical hyperostosis, Al‐Gazali type Osteopoikilosis, LEMD3‐related Melorheostosis with osteopoikilosis, LEMD3‐related Melorheostosis, MAP2K1‐related NOS 25‐0010 NOS 25‐0020 NOS 25‐0030 NOS 25‐0040 NOS 25‐0050 NOS 25‐0060 NOS 25‐0070 NOS 25‐0080 NOS 25‐0090 SP AD AD AR? AD AR AR MAP2K1 LEMD3 LEMD3 COL1A1 FAM20C DHCR24 155950 166700 166700 601356 114000 114000 259775 602398 Possibly locus heterogeneity Includes mixed sclerosing bone dysplasia Includes Buschke‐Ollendorff syndrome (same OMIM entry) Only a few cases known. Im OMIM as “Lethal short‐limb skeletal dysplasia, Al Gazali type” . Not to be confused with “Al‐Gazali syndrome”, a rare variant of B3GALT6 disorders (see above) A few sporadic cases known, phenotype consistent, molecular basis unknown Rare specific variants in COL1A1. See also osteogenesis imperfecta related to collagen 1 genes. Variable severity, many cases are perinatal severe, some cases show survival to adulthood; then often combined with FGF23 elevation and hypophosphatemic rickets See also other sterol‐metabolism related conditions UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 25 Note: osteomesopyknosis (MIM 166450) may represent a form of osteopetrosis. In a pattern similar to the ciliary disorders, the phenotypes from individual loci are variable and may overlap with those of other loci. Name of group/name of disorder NOS 24‐0110 (Continued) Group number/number of disorder TABLE 1 22 (Continued) Osteopathia striata with cranial sclerosis (OSCS), AMER1‐ related Pyle disease, SFRP4‐related Craniometaphyseal dysplasia, ANKH‐related Craniometaphyseal dysplasia, GJA1‐related Diaphyseal dysplasia Camurati‐Engelmann, TGFB1‐related Hyperostosis‐Hyperphosphatemia syndrome, GALNT3‐ related Hyperostosis‐Hyperphosphatemia syndrome, FGF23‐ related Hyperostosis‐Hyperphosphatemia syndrome, KL‐relathed Cerebellar hypoplasia‐endosteal sclerosis, POLR3B‐related Hematodiaphyseal dysplasia Ghosal, TBXAS1‐related Hypertrophic osteoarthropathy, HPGD‐related Hypertrophic osteoarthropathy, SLCO2A1‐related Oculodentoosseous dysplasia (ODOD), GJA1‐related, dominant, mild type Oculodentoosseous dysplasia (ODOD) GJA1‐related, recessive, severe type Osteoectasia with hyperphosphatasia (juvenile Paget disease), OPG‐related Osteosclerosis, LRP5‐related Sclerosteosis, SOST‐related Sclerosteosis, LRP4‐related Endosteal hyperostosis, van Buchem type, SOST‐related Endosteal hyperostosis, Worth type Craniodiaphyseal dysplasia, SOST‐related Craniodiaphyseal dysplasia, SP7‐related Trichodentoosseous dysplasia, DLX3‐related Diaphyseal medullary stenosis with malignant fibrous histiocytoma, MTAP‐related Craniotubular dysplasia, TMEM53‐related Craniometadiaphyseal dysplasia, Wormian bone type NOS 25‐0110 NOS 25‐0120 NOS 25‐0130 NOS 25‐0140 NOS 25‐0150 NOS 25‐0160 NOS 25‐0170 NOS 25‐0180 NOS 25‐0190 NOS 25‐0200 NOS 25‐0210 NOS 25‐0220 NOS 25‐0230 NOS 25‐0240 NOS 25‐0250 NOS 25‐0260 NOS 25‐0270 NOS 25‐0280 NOS 25‐0290 NOS 25‐0300 NOS 25‐0310 NOS 25‐0320 NOS 25‐0330 NOS 25‐0340 NOS 25‐0350 Name of group/name of disorder Inheritance Gene or locus AR TMEM53 MTAP AD AR DLX3 SP7 SOST LRP5 SOST LRP4 SOST LRP5 OPG GJA1 GJA1 SLCO2A1 HPGD TBXAS1 POLR3B KL AD AR AD AD AR AR AR AD AR AR AD AD, AR AR AR AR AR FGF23 GALNT3 AR TGFB1 AD GJA1 ANKH SFRP4 AMER1 AR AR AD AR XL MIM No. 269300 619727 112250 190320 See 606633 122860 144750 239100 614305 269500 144750, 607634 239000 257850 164200 614441 259100 231095 213002 617994 617993 211900 131300 218400 123000 265900 300373 Also known as Hardcastle disease (Continues) One family reported; SP7 variants also associated with Osteogenesis imperfecta (MIM 613849), see below Presumed dominant negative variant Specific 52 kb deletion downstream of SOST See also sclerosteosis, SOST‐related, below Includes previous AD osteopetrosis type 1 (OPTA1) Possibly homozygous form of mild ODOD Includes cranio‐osteoarthropathy, some cases of recessive pachydermoperiostosis, as well as recessively inherited isolated digital clubbing (MIM 119900) Formerly hyperphosphatemic tumoral calcinosis type 3 Formerly hyperphosphatemic tumoral calcinosis type 2 Formerly hyperphosphatemic tumoral calcinosis type 1 Gain‐of‐function variants Recessive type Dominant type The name “metaphyseal dysplasia, Pyle type” is misleading (no growth plate dysplasia) and should be avoided Notes 23 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License NOS 25‐0100 Group number/number of disorder TABLE 1 UNGER ET AL. Lenz‐Majewski hyperostotic dysplasia, PTDSS1‐related Osteochondrodysplasia with hypertrichosis (Cantu syndrome), ABCC9‐related Familial Paget disease of bone, SQSTM1‐related Inclusion body myopathy, Paget disease of bone and frontotemporal dementia Endosteal hyperostosis, oligodontia, short stature, facial dysmorphism and intellectual disability, POLR3GL‐ related Metaphyseal dysplasia, Braun‐Tinschert type Trichothiodystrophy with axial osteosclerosis NOS 25‐0370 NOS 25‐0380 NOS 25‐0390 NOS 25‐0400 NOS 25‐0410 NOS 25‐0420 Inheritance Gene or locus POLR3GL AR AR AD VCP SQSTM1 ABCC9 PTDSS1 AD AD AD AD MIM No. 605946 619234 167320 167250 239850 151050 A subset of patients with trichothiodystrophy have marked osteosclerosis but have not been molecularly characterized so far Phenotypic elements will need to be evaluated more precisely; one patient reported as Wiedemann‐ Rautenstrauch‐syndrome‐like Monoallelic variants in the VCP gene are also associated with MIM 616687‐Charcot‐Marie‐Tooth disease 2Y, and with MIM 613954‐Frontotemporal dementia and/ or amyotrophic lateral sclerosis 6. Notes Osteogenesis imperfecta and bone fragility group Osteogenesis imperfecta, non‐deforming (Sillence type 1), COL1A1‐related Osteogenesis imperfecta, non‐deforming (Sillence type 1), COL1A2‐related Osteogenesis imperfecta, severe perinatal form (Sillence type 2) COL1A1‐related Osteogenesis imperfecta, severe perinatal form (Sillence type 2), COL1A2‐related Osteogenesis imperfecta, severe perinatal form (Sillence type 2), CRTAP‐related Osteogenesis imperfecta, severe perinatal form (Sillence type 2), P3H1‐related Osteogenesis imperfecta, severe perinatal form (Sillence type 2), PPIB‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), COL1A1‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), COL1A2‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), IFITM5‐related NOS 26‐0010 NOS 26‐0020 NOS 26‐0030 NOS 26‐0040 NOS 26‐0050 NOS 26‐0060 NOS 26‐0070 NOS 26‐0080 NOS 26‐0090 NOS 26‐0100 AD AD AD AR AR AR AD AD AD AD IFITM5 COL1A2 COL1A1 PPIB P3H1 CRTAP COL1A2 COL1A1 COL1A2 COL1A1 610967 259420 259420 259440 610915 610682 166210 166210 166200 166200 In OMIM OI type III; phenotype is distinct but in some instances can minimize OI type III In OMIM as OI type III In OMIM as OI type III Formerly “perinatal lethal”; in OMIM as OI type IX Formerly “perinatal lethal”; in OMIM as OI type VIII Formerly “perinatal lethal”; in OMIM as OI type VII Formerly “perinatal lethal”; in OMIM as OI type II Formerly “perinatal lethal”; in OMIM as OI type II Usually with persistently blue sclerae, can have signs of connective tissue weakness (in MIM as OI type I) Usually with persistently blue sclerae, can have signs of connective tissue weakness (in MIM as OI type I) UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 26 See also the chondrodysplasia punctata group (group 23); as well as familial expansile osteolysis, TNFRSF11A‐related (below); and trichothiodystrophy with central osteosclerosis (PMID 15148554) Name of group/name of disorder NOS 25‐0360 (Continued) Group number/number of disorder TABLE 1 24 (Continued) Osteogenesis imperfecta, progressively deforming (Sillence type 3), SERPINF1‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), CRTAP‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), P3H1‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), PPIB‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), SERPINH1‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), FKBP10‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), TMEM38B‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), BMP1‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), WNT1‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), CREB3L1‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), SPARC‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), TENT5A‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), MBTPS2‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), MESD‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3) with neurodevelopmental features, KDELR2‐related Osteogenesis imperfecta, progressively deforming (Sillence type 3), CCD134‐related Osteogenesis imperfecta, moderate form (Sillence type 4), COL1A1‐related Osteogenesis imperfecta, moderate form (Sillence type 4), COL1A2‐related Osteogenesis imperfecta, moderate form (Sillence type 4), WNT1‐related NOS 26‐0120 NOS 26‐0130 NOS 26‐0140 NOS 26‐0150 NOS 26‐0160 NOS 26‐0170 NOS 26‐0180 NOS 26‐0190 NOS 26‐0200 NOS 26‐0210 NOS 26‐0220 NOS 26‐0230 NOS 26‐0240 NOS 26‐0250 NOS 26‐0260 NOS 26‐0270 NOS 26‐0280 NOS 26‐0290 Name of group/name of disorder Inheritance Gene or locus AR AD AD WNT1 COL1A2 COL1A1 CCD134 KDELR2 AR AR MESD MBTPS2 TENT5A SPARC CREB3L1 WNT1 BMP1 TMEM38B FKBP10 SERPINH1 PPIB P3H1 CRTAP SERPINF1 AR XLR AR AR AR AR AR AR AR AR AR AR AR AR MIM No. see 166220 166220 166220 619795 619131 618644 301014 617952 616507 616229 615220 614856 615066 610968 613848 see 259440 610915 610682 613982 Notes In OMIM as OI type XV In OMIM as OI type IV In OMIM as OI type IV In OMIM as OI typeXXII In OMIM as OI type XXI. Frequency of neurodevelopmental delay not clear yet. In OMIM as OI type XX In OMIM as OI type XIX In OMIM as OI type XVIII In OMIM as OI type XVII (Continues) In OMIM as OI type XVI. Has severe joint laxity and scoliosis, Ehlers‐Danlos‐like In OMIM as OI type XV. Biallelic variants; monoallelic variants may result in AD osteoporosis. In OMIM OI type XIII In OMIM OI type XIV In OMIM OI type XI In OMIM OI type X In OMIM OI type IX In OMIM OI type VIII In OMIM OI type VII In OMIM as OI type VI 25 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License NOS 26‐0110 Group number/number of disorder TABLE 1 UNGER ET AL. (Continued) AR XL Osteogenesis imperfecta, moderate form (Sillence type 4), PPIB‐related Osteogenesis imperfecta, moderate form (Sillence type 4), FKBP10‐related Osteogenesis imperfecta, moderate form (Sillence type 4), SP7‐related Osteogenesis imperfecta with calcification of interosseous membranes and/or hypertrophic callus (OI type 5), IFITM5‐related Osteogenesis imperfecta with craniosynostosis (Cole‐ Carpenter syndrome), P4HB‐related Osteogenesis imperfecta with craniosynostosis (Cole‐ Carpenter syndrome), SEC24D‐related Osteoporosis – X‐linked form, PLS3‐related Osteoporosis—X‐linked form, MBPTS2‐related NOS 26‐0320 NOS 26‐0330 NOS 26‐0340 NOS 26‐0350 NOS 26‐0360 NOS 26‐0370 NOS 26‐0380 NOS 26‐0390 Gene or locus AD AR AD Osteoporosis—AD form, ARHGAP25‐related Bruck syndrome type 1 (BS1), FKBP10‐related Bruck syndrome type 2 (BS2), PLOD2‐related Osteoporosis‐pseudoglioma syndrome, LRP5‐related Bone fragility with calvarial “doughnut” lesions, SGSM2‐ related Spondylo‐ocular dysplasia, XYLT2‐related NOS 26‐0420 NOS 26‐0430 NOS 26‐0440 NOS 26‐0450 NOS 26‐0460 NOS 26‐0470 PLS3 XYLT2 SGMS2 LRP5 PLOD2 FKBP10 ARHGAP25 LRP5 WNT1 MBTPS2 MIM No. 605822 126550 259770 609220 259450 see 610587 166710, 601884 615220 301014 300910 616294 112240 610967 613849 see 610968 see 259440 see 610682 166220 Notes Overlap with a spondylo‐metaphyseal dysplasia phenotype When eye involvement is absent, may mimic progressively deforming or moderate OI (Sillence types 3 and 4) See autosomal recessive OI, above; intrafamilial variability between OI type 3, arthrogryposis and Bruck syndrome 1 is documented Monoallelic variants; biallelic variants result in MIM 259770 osteoporosis‐pseudoglioma (see below); this gene is also associated with hyperostotic forms (see below) as well as with MIM 601813—exudative vitreoretinopathy, as well as MIM 617875—polycystic liver disease OMIM OI type XV In OMIM as OI type XIX; gene also associated with MIM 300918, MIM308205, MIM 308800 Was Cole‐Carpenter syndrome 2. Possibly misnomer, as most patients do not have craniosynostosis but rather large fontanels. Craniosynostosis is not well documented in this condition in spite of the name. When calcification of intraosseous membranes or hypertrophic callus are not observed, may mimic progressively deforming or moderate OI (Sillence types 3 and 4) In OMIM as OI type XII In OMIM as OI type XI In OMIM as OI type IX In OMIM as OI type VII in OMIM OI type IV UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License AR AR AR AD AD Osteoporosis—dominant form, WNT1‐related Osteoporosis – AD form, LRP5‐related NOS 26‐0400 NOS 26‐0410 XL SEC24D P4HB IFITM5 AD AD SP7 FKBP10 PPIB CRTAP IFITM5 AR AR AD AR Osteogenesis imperfecta, moderate form (Sillence type 4), CRTAP‐related NOS 26‐0310 Inheritance AD Osteogenesis imperfecta, moderate form (Sillence type 4), IFITM5‐related Name of group/name of disorder NOS 26‐0300 Group number/number of disorder TABLE 1 26 Gnathodiaphyseal dysplasia, ANO5‐related Osteoporosis with developmental delay and microcephaly, COPB2‐related Geroderma osteodysplasticum, GORAB‐related Cutis laxa, PYCR1‐related Cutis laxa, ATP6V0A2‐related Wiedemann‐Rautenstrauch syndrome, POLR3A‐related Singleton‐Merten dysplasia, IFIH‐related Singleton‐Merten dysplasia, DDX58‐related NOS 26‐0490 NOS 26‐0500 NOS 26‐0510 NOS 26‐0520 NOS 26‐0530 NOS 26‐0540 NOS 26‐0550 Inheritance AD AD AR AR AR AR AD AD Gene or locus DDX58 IFIH1 POLR3A ATP6V0A2 PYCR1 GORAB COPB2 ANO5 MIM No. 616298 182250 264090 278250, 219200 612940 231070 619884 166260 Notes Gene also associated with MIM 615846‐Aicardi‐Goutieres syndrome 7, and OMIM 619773‐Immunodeficiency 95 Gene also associated with MIM 607694‐ Leukodystrophy, hypomyelinating, with or without oligodontia and/or hypogonadotropic hypogonadism Autosomal recessive form, type 2A (ARCL2A); wrinkly skin syndrome. Skeletal features overlapping with progeroid EDS and geroderma osteodysplasticum Autosomal recessive form, type 2B (ARCL2B). Skeletal features overlapping with progeroid EDS and geroderma osteodysplasticum Clinically variable, microcephaly in some cases only Gene also associated with OMIM 613319‐Miyoshi muscular dystrophy 3, and OMIM 611307‐recessive limb‐girdle muscular dystrophy 12 Disorders of bone mineralisation Hypophosphatasia, ALPL‐related, recessive (biallelic) forms Hypophosphatasia, ALPL‐related, dominant (monoallelic) forms Hypophosphatemic rickets, PHEX‐related Hypophosphatemic rickets, FGF23‐related Hypophosphatemic rickets, DMP1‐related Hypophosphatemic rickets, ENPP1‐related Hypophosphatemic rickets, SGK3‐related Hypophosphatemic rickets with hypercalciuria, CLCN5‐ related Hypophosphatemic rickets with hypercalciuria, SLC34A3‐ related Vitamin D‐dependent rickets, CYP27B1‐related Vitamin D‐dependent rickets, CYP2R1‐related NOS 27‐0010 NOS 27‐0020 NOS 27‐0030 NOS 27‐0040 NOS 27‐0050 NOS 27‐0060 NOS 27‐0070 NOS 27‐0080 NOS 27‐0090 NOS 27‐0100 NOS 27‐0110 AR AR AR AR XL AD AR AR AD XL AD CYP2R1 CYP27B1 SLC34A3 CLCN5 SGK3 ENPP1 DMP1 FGF23 PHEX ALPL ALPL 600081 264700 241530 300554 see 607591 613312 241520 193100 307800 146300 241500 Formerly type 1B Formerly type 1A Autosomal recessive (HHRH) (Continues) X‐linked; part of Dent's disease complex (progressive proximal renal tubulopathy with hypercalciuria, low molecular weight proteinuria, and nephrocalcinosis; MIM 300009) Autosomal dominant Autosomal recessive (ARHR2) Autosomal recessive (ARHR1) Autosomal dominant X‐linked, most common genetic form of hypophosphatemic rickets Includes juvenile and adult forms as well as odontohypophosphatasia Includes perinatal, infantile and juvenile forms 27 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 27 Note: some of the recently discovered OI variants are still limited to very small numbers of patients; thus, the association with OI “Sillence type 3” is tentative and may be too restrictive as other phenotypes might emerge in the future. See also: Short stature, skeletal dysplasia, liver failure, optic nerve atrophy and Pelger‐Huet anomaly, NBAS‐related, above (Group 13); as well as all the Loeys‐Dietz syndrome variants and the Snyder‐Robinson syndrome, SMS‐related (Group 31). Name of group/name of disorder NOS 26‐0480 (Continued) Group number/number of disorder TABLE 1 UNGER ET AL. Vitamin D‐dependent rickets, VDR‐related Vitamin D‐dependent rickets, CYP3A4‐related Vitamin D‐dependent rickets, HNRNPC‐related Familial hyperparathyroidism, CDC73‐related Familial hyperparathyroidism linked to chromosome 2 Familial hyperparathyroidism, GCM2‐related Neonatal hyperparathyroidism, CASR‐related Neonatal hyperparathyroidism, TRPV6‐related Familial hypocalciuric hypercalcemia with transient neonatal hyperparathyroidism, CASR‐related Calcium pyrophosphate deposition disease (familial chondrocalcinosis), ANKH‐related Calcium pyrophosphate deposition disease (familial chondrocalcinosis), TNFRSF11B‐related Cutaneous skeletal hypophosphatemia syndrome Cutaneous skeletal hypophosphatemia syndrome NOS 27‐0130 NOS 27‐0140 NOS 27‐0150 NOS 27‐0160 NOS 27‐0170 NOS 27‐0180 NOS 27‐0190 NOS 27‐0200 NOS 27‐0210 NOS 27‐0220 NOS 27‐0230 NOS 27‐0240 Inheritance MOS MOS AD AD AD AR AR, AD AD AD AD AD? AD AR Gene or locus NRAS HRAS TNFRSF11B ANKH CASR TRPV6 CASR GCM2 2p14‐p13.3 CDC73 HNRNPC CYP3A4 VDR MIM No. see 602643 118600 145980 618188 239200 617343 610071 145000, 145001 see 164020 619073 277440 Notes Somatic mosaicism for activating variants in NRAS with elevated FGF23 levels Somatic mosaicism for activating variants in HRAS with elevated FGF23 levels Apparently monoallelic gain‐of‐function variants Loss‐of‐function variants (see also craniometaphyseal dysplasia, dominant type) Other forms of familial hypocalciuric hypercalcemia do not show significant skeletal phenotypes Transient form “Severe” form (but see below, transient form also CASR‐ related). Variants in the CASR gene can also result in autosomal dominant hypocalcemia (MIM 601198) Variants in this gene also cause familial isolated hypoparathyroidism (MIM 618883) Linkage studies; no gene identified With or without jaw tumors Formerly type 2B; molecular basis (supposed HNRNPC dominant negative) from a single patient Formerly type 3; specific monoallelic variants that increase enzyme activity leading to rapid degradation of active vitamin D Formerly type 2A Metaphyseal dysplasia, Jansen type, PTHR1‐related Metaphyseal dysplasia, Csukasi‐Krakow type, SIK3‐related Blomstrand dysplasia, PTHR1‐related Eiken dysplasia, PTHR1‐related Brachydactyly, PTHLH‐related (brachdactyly type E2) Osteolysis, PTHLH‐related NOS 28‐0010 NOS 28‐0020 NOS 28‐0030 NOS 28‐0040 NOS 28‐0050 NOS 28‐0060 PTHLH PTHLH PTHR1 PTHR1 SIK3 PTHR1 613382 600002 215045 618162 156400 Duplications of PTHLH causing acro‐osteolysis; see also Groups 30 and 18 Haploinsufficiency; with or without short stature Caused by recessive (biallelic) hypomorphic variants Caused by recessive (biallelic) loss‐of‐function variants disruption of mTOR signaling downstream of the PTH receptor Caused by activating variants UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Note: see also Acrodysostosis, PDE4D‐related and PRKAR1A‐related, above; and Albright hereditary osteodystrophy, GNAS‐related. Monoallelic loss‐of‐function variants in PTHR1 lead to primary failure of tooth eruption (MIM 125350) AD AD AR AR AR AD Skeletal disorders of the parathyroid hormone signaling cascade Group 28 Note: Hyperparathyroidism due to parathyroid adenoma occurs in a number of genetic disorders, for example, in Multiple Endocrine Neoplasias (see MIM for variants). See also Group 28, below, as well as Raine dysplasia, FAM20C‐related Name of group/name of disorder NOS 27‐0120 (Continued) Group number/number of disorder TABLE 1 28 Familial expansile osteolysis, TNFRSF11A‐related Mandibuloacral dysplasia, LMNA‐related Mandibuloacral dysplasia, ZMPSTE24‐related Mandiculoacral dysplasia, MTX2‐related Progeria, Hutchinson‐Gilford type, LMNA‐related Multicentric osteolysis, nodulosis and arthropathy (MONA), MMP2‐related Multicentric osteolysis, nodulosis and arthropathy (MONA), MMP14‐related Hajdu‐Cheney syndrome, NOTCH2‐related Multicentric carpal‐tarsal osteolysis with and without nephropathy, MAFB‐related Penttinen syndrome, PDGFRB‐related Nestor Guillermo progeria syndrome, BANF1‐related Farber disease, ASAH1‐related NOS 29‐0010 NOS 29‐0020 NOS 29‐0030 NOS 29‐0040 NOS 29‐0050 NOS 29‐0060 NOS 29‐0070 NOS 29‐0080 NOS 29‐0090 NOS 29‐0100 NOS 29‐0110 NOS 29‐0120 AR AR AD AD AD AR AR AD AR AR AR AD Inheritance ASAH1 BANF1 PDGFRB MAFB NOTCH2 MMP14 MMP2 LMNA MTX2 ZMPSTE24 LMNA TNFRSF11A Gene or locus 603811 601812 166300 102500 277950 259600 176670 619127 608612 248370 174810, 602080 MIM No. The chronic, adult form of Farber disease can present as osteolysis See also MIM 601812‐Kosaki overgrowth syndrome Includes the Serpentine fibula‐polycystic kidney syndrome (SFPKS) A biallelic homozygous MMP14 variant seen in the original Winchester syndrome family Includes Winchester‐Torg syndrome and nodulosis‐ arthropathy‐osteolysis syndrome See also Progeria (in this group), LMNA‐related cardiomyopathy and brachydactyly (Group 19) as well as many other conditions in MIM related to LMNA Includes early‐onset familial Paget disease of bone. See other TNFRSF11A‐related phenoytpes in Group 24 Notes Disorganized development of skeletal components group Multiple cartilaginous exostoses, EXT1‐related (MCE; or multiple osteochondromas, MO) Multiple cartilaginous exostoses, EXT2‐related (MCE; or multiple osteochondromas, MO) Cherubism, SH3BP2‐related Fibrous dysplasia, polyostotic form (McCune‐Albright syndrome), GNAS‐related Progressive osseous heteroplasia (POH), GNAS‐related Metachondromatosis, PTPN11‐related Osteoglophonic dysplasia, FGFR1‐related Fibrodysplasia ossificans progressiva (FOP), ACVR1‐ related Neurofibromatosis type 1, NF1‐related NOS 30‐0010 NOS 30‐0020 NOS 30‐0030 NOS 30‐0040 NOS 30‐0050 NOS 30‐0060 NOS 30‐0070 NOS 30‐0080 NOS 30‐0090 AD AD AD AD AD MOS AD AD AD NF1 ACVR1 FGFR1 PTPN11 GNAS GNAS SH3BP2 EXT2 EXT1 162200 135100 166250 156250 166350 174800 118400 133701 133700 Most cases sporadic but dominant transmission documented (Continues) Craniosynostosis is also an important feature (Group 34) Loss‐of‐function variants (in contrast to Noonan syndrome) with loss of heterozygosity in lesional tissue Germline loss‐of‐function of paternal allele Somatic mosaicism for gain‐of‐function variants; includes Mazabraud syndrome with intramuscular myxomas 29 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 30 Note: several neurologic conditions may cause acroosteolysis. See also Osteolysis, PTHLH‐related (above), Pyknodysostosis, CTSK‐related; cleidocranial dysplasia, RUNX2‐related; Keutel syndrome, MGP‐related; Singleton‐Merten dysplasia, IFIH‐related; and Singleton‐Merten dysplasia, DDX58‐related Name of group/name of disorder Osteolysis group Group 29 (Continued) Group number/number of disorder TABLE 1 UNGER ET AL. Cherubism with gingival fibromatosis (Ramon syndrome) Dysplasia epiphysealis hemimelica (Trevor) Lipomembraneous osteodystrophy with leukoencephalopathy, TREM2‐related (Nasu‐Hakola) Lipomembraneous osteodystrophy with leukoencephalopathy, TYROBP‐related (Nasu‐Hakola) Enchondromatosis, IDH1‐related (Ollier disease) Enchondromatosis, IDH2‐related (Ollier disease) Enchondromatosis with hemangiomas, IDH1‐related (Maffucci disease) Enchondromatosis with hemangiomas, IDH2‐related (Maffucci disease) Metaphyseal chondromatosis with D‐2‐hydroxyglutaric aciduria, IDH1‐related Primary intraosseous vascular malformation, ELMO2‐ related Osteofibrous dysplasia, MET‐related Genochondromatosis Gorham‐Stout disease and familial diffuse angiomatosis of bone NOS 30‐0110 NOS 30‐0120 NOS 30‐0130 NOS 30‐0140 NOS 30‐0150 NOS 30‐0160 NOS 30‐0170 NOS 30‐0180 NOS 30‐0190 NOS 30‐0200 NOS 30‐0210 NOS 30‐0220 SP (MOS?), AD AD AD, SP AR MOS MOS MOS MOS MOS AR AR SP AR Inheritance MET ELMO2 IDH1 IDH2 IDH1 IDH2 IDH1 TYROBP TREM2 Gene or locus see 123880 137360 607278 606893 614875 614569 614569 166000 166000 221770 618193 127800 266270 MIM No. Notes Severe Gorham‐Stout disease is mostly sporadic. Somatic KRAS variants have been found in rare cases. In contrast, milder cases may be familial (“familial diffuse cystic angiomatosis of bone”; see OMIM 123880). “Geno” from Greek “knee”, but upper limbs also affected. Probably includes the condition known as Vaandrager‐ Peña syndrome Possibly corresponding to the former “Campanacci dysplasia” Includes so‐called cheirospondyloenchondromatosis. Somatic mosaicism for IDH1 variants. Possibly also IDH2 variants but not yet well documented Somatic mosaicism for IDH2 variants; significantly rarer than IDH1 variants. See also MIM 147650 and 613657, D‐2‐hydroxyglutaric aciduria 2 Somatic mosaicism for specific IDH1 variants. See also MIM 147700 and 137800 Somatic mosaicism for IDH2 variants; significantly rarer than IDH1 variants. See also MIM 147650 and 613657, D‐2‐hydroxyglutaric aciduria 2 Somatic mosaicism for specific IDH1 variants. See also MIM 147700 and 137800 Also known as presenile dementia with bone cysts Also known as presenile dementia with bone cysts Some familial cases reported (“familial Trevor disease”) but probably represent a different condition Some similarities to primary intraosseous vascular malformation, ELMO2‐related (see below) Marfan syndrome, FBN1‐related Congenital contractural arachnodactyly (Beals‐Hecht syndrome), FBN2‐related NOS 31‐0010 NOS 31‐0020 FBN2 FBN1 121050 154700 See also as differential diagnosis: homocystinuria and marfanoid habitus with ID (Lujan Fryns syndrome): MED12; ZDHHC9; UPF3B UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License AD AD Overgrowth (tall stature) syndromes and segmental overgrowth Group 31 Note: PTEN‐related disorders are not included because the overgrowth is restricted to macrocephaly.‐ See also: Proteus syndrome, AKT1‐related; Spondyloenchondrodysplasia with immune dysregulation (SPENCD), ACP5‐related; Spondyloepimetaphyseal dysplasia, COL2A1‐related (“SED with marked metaphyseal changes”, including dysspondyloenchondromatosis); Cutaneous skeletal hypophosphatemia syndrome, HRAS‐ related and NRAS‐related. Some patients with SOX6 variants have osteochondromas. Name of group/name of disorder NOS 30‐0100 (Continued) Group number/number of disorder TABLE 1 30 AD AR Loeys‐Dietz syndrome, TGFBR2‐related Loeys‐Dietz syndrome, TGFB2‐related Loeys‐Dietz syndrome, TGFB3‐related Loeys‐Dietz syndrome, SMAD2‐related Loeys‐Dietz syndrome, SMAD3‐related Weaver syndrome, EZH2‐related Cohen‐Gibson (Weaver‐like) syndrome, EED‐related Imagawa‐Matsumoto (Weaver like) syndrome, SUZ12‐related Sotos syndrome, NSD1‐related Sotos syndrome, APC2‐related Malan (Sotos‐like) syndrome, NFIX‐related Luscan‐Lumish syndrome, SETD2‐related Tatton‐Brown‐Rahman syndrome, DNMT3A‐related Marshall‐Smith syndrome, NFIX‐related Beckwith‐Wiedemann syndrome Simpson‐Golabi‐Behmel syndrome, GPC3‐related Proteus syndrome, AKT1‐related Hypoinsulinemic hypoglycemia with hemihypertrophy (HIHGHH), AKT2‐related Congenital ipomatous overgrowth, vascular Malformations, epidermal Nevi, spinal/skeletal anomalies/scoliosis (CLOVES) syndrome, PIK3CA‐related Fibroadipose hyperplasia, PIK3CA‐related Snyder‐Robinson syndrome (intellectual disability, tall stature, osteoporosis and fractures), SMS‐related Overgrowth syndrome with 2q37 translocations Tall stature with long halluces, NPR2‐related Tall stature with long halluces, NPR3‐related Moreno‐Nishimura‐Schmidt syndrome NOS 31‐0040 NOS 31‐0050 NOS 31‐0060 NOS 31‐0070 NOS 31‐0080 NOS 31‐0090 NOS 31‐0100 NOS 31‐0110 NOS 31‐0120 NOS 31‐0130 NOS 31‐0140 NOS 31‐0150 NOS 31‐0160 NOS 31‐0170 NOS 31‐0180 NOS 31‐0190 NOS 31‐0200 NOS 31‐0210 NOS 31‐0220 NOS 31‐0230 NOS 31‐0240 NOS 31‐0250 NOS 31‐0260 NOS 31‐0270 NOS 31‐0280 Inheritance Loeys‐Dietz syndrome, TGFBR1‐related Gene or locus NPR3 NPR2 NPPC SMS MIM No. 608811 619543 615923 see 600296 309583 See 171834 612918 176920 312870 130650 602535 615879 616831 614753 617169 117550 606245 617561 277590 613795 619656 615582 614816 610168 609192 Notes (Continues) Biallelic loss‐of‐function variants in NPR3; in OMIM as Boudin‐Mortier syndrome Monoallelic gain‐of‐function variants in NPR2; in OMIM as epiphyseal chondrodysplasia, Miura type Overgrowth probably caused by overexpression of NPPC See MIM 171834 for the many conditions associated with somatic PIK3CA variants Also named PIK3CA‐related overgrowth syndrome (PROS); somatic variants; see MIM 171834 Gene also associated with OMIM 125853 diabetes mellitus type II Variant or deletion of imprinted genes within the chromosome 11p15.5 region See also Malan syndrome. The localization of the monoallelic variants determine the Malan vs. Marshall‐ Smith phenotype Some cases reported with NSD1, EED, and SUZ12 variants Osteopenia with propensity to fractures may be observed in all variants of the Loeys‐Dietz syndrome 31 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License SP AR AD SP XLR PIK3CA PIK3CA MOS MOS AKT2 AKT1 GPC3 11p15.5 region NFIX DNMT3A SETD2 NFIX APC2 NSD1 SUZ12 EED EZH2 SMAD3 SMAD2 TGFB3 TGFB2 TGFBR2 TGFBR1 AD MOS XL AD AD AD AD AD AD AD AD AD AD AD AD AD AD Name of group/name of disorder NOS 31‐0030 (Continued) Group number/number of disorder TABLE 1 UNGER ET AL. (Continued) Kosaki overgrowth syndrome, PDGFRB‐related Segmental odontomaxillary dysplasia, ACTB‐related NOS 31‐0300 NOS 31‐0310 Inheritance MOS AR AD, AR Gene or locus ACTB PDGFRB FGFR3 MIM No. see 102630 616592 610474 Notes See PMID 32585735; see also MIM 243310‐Baraitser‐ Winter syndrome See also MIM 601812‐Pentinnen syndrome Original family with monoallelic (dominant negative?) variant; a second family with biallelic variants (see Group 1) Sterile multifocal osteomyelitis, periostitis, and pustulosis (CINCA/NOMID‐like), IL1RN‐related Chronic recurrent multifocal osteomyelitis with congenital dyserythropoietic anemia (CRMO with CDA; Majeed syndrome), LPIN2‐related Familial juvenile arthritis with hyaluronidase deficiency, HYAL1‐related Hyaline Fibromatosis Syndrome, ANTXR2‐related NOS 32‐0030 NOS 32‐0040 NOS 32‐0050 NOS 32‐0060 ANTXR2 236490, 228600 601492 609628 See 147679 607115 208230 Previously known as infantile systemic hyalinosis, juvenile hyaline fibromatosis, and puretic syndrome Also known as mucopolysaccharidosis type 9, although clinically no storage Also known as SED with progressive arthropathy Cleidocranial dysplasia and related disorders Cleidocranial dysplasia, RUNX2‐related Cleidocranial‐like dysplasia, CBFB‐related CDAGS syndrome (craniosynostosis, delayed fontanel closure, parietal foramina, imperforate anus, genital anomalies, skin eruption), RNU12‐related Yunis‐Varon dysplasia, FIG4‐related Yunis‐Varon dysplasia, VAC14‐related Parietal foramina, MSX2‐related NOS 33‐0010 NOS 33‐0020 NOS 33‐0030 NOS 33‐0040 NOS 33‐0050 NOS 33‐0060 AD AR MSX2 VAC14 FIG4 RNU12 AR AR CBFB RUNX2 AD AD 168500 216340 603116 See 121360 119600 Only one case of VAC14‐related Yunis‐Varon reported so far, so association needs to be confirmed. Gene also associated with OMIM 617054 Striatonigral degeneration, childhood‐onset (several patients reported) Gene also causes OMIM 612577 amyotrophic lateral sclerosis 11, and OMIM 611228 CMT disease 4J See also MIM 601626, familial leukemia See also MIM 156510‐metaphyseal dysplasia with maxillary hypoplasia, as well as non‐syndromic midline craniosynostosis, RUNX2‐related, below UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 33 Farber disease, ASAH1‐related (osteolysis group 29, and MIM 228000) shows phenotypic overlap with the conditions in this group. AR HYAL1 LPIN2 AR AR IL1RN AR CIAS1 AD Chronic infantile neurologic cutaneous articular syndrome (CINCA), CIAS1‐related (neonatal onset multisystem inflammatory disease (NOMID)) NOS 32‐0020 WISP3 Progressive pseudorheumatoid dysplasia (PPRD), WISP3‐ related NOS 32‐0010 AR Genetic inflammatory or rheumatoid‐like osteoarthropathies Group 32 See also: Shprintzen‐Goldberg syndrome, SKI‐related, in the craniosynostosis group. Note: this group does not include disorders that cause overgrowth secondary to vascular malformations, such as Klippel‐Trenaunay syndrome, at least until a genetic (somatic) origin will have been demonstrated. Camptodactyly, tall stature and hearing loss syndrome (CATSHL), FGFR3‐related Name of group/name of disorder NOS 31‐0290 Group number/number of disorder TABLE 1 32 Parietal foramina, ALX4‐related Parietal foramina with cleidocranial dysplasia, MSX2‐ related NOS 33‐0080 Inheritance AD AD Gene or locus MSX2 ALX4 MIM No. 168550 609597 Notes MSX2 variants also cause craniosynostosis Boston type See also frontonasal dysplasia type 1 Syndromes featuring craniosynostosis Pfeiffer syndrome, FGFR1‐related Pfeiffer syndrome, FGFR2‐related Apert syndrome, FGFR2‐related Craniosynostosis with cutis gyrata (Beare‐Stevenson), FGFR2‐related Crouzon syndrome, FGFR2‐related Crouzon‐like craniosynostosis with acanthosis nigricans, FGFR3‐related Craniosynostosis, Muenke type, FGFR3‐related Antley‐Bixler syndrome, POR‐related Craniosynostosis Boston type, MSX2‐related Saethre‐Chotzen syndrome, TWIST1‐related Shprintzen‐Goldberg syndrome, SKI‐related Baller‐Gerold syndrome, RECQL4‐related Carpenter syndrome, RAB23‐related Carpenter syndrome, MEGF8‐related Craniosynostosis, TCF12‐related Craniosynostosis, SIX1‐related Complex craniosynostosis, ERF‐related Non‐syndromic midline (metopic / sagittal) craniosynostosis, SMAD6‐related Non‐syndromic midline craniosynostosis, RUNX2‐related NOS 34‐0010 NOS 34‐0020 NOS 34‐0030 NOS 34‐0040 NOS 34‐0050 NOS 34‐0060 NOS 34‐0070 NOS 34‐0080 NOS 34‐0090 NOS 34‐0100 NOS 34‐0110 NOS 34‐0120 NOS 34‐0130 NOS 34‐0140 NOS 34‐0150 NOS 34‐0160 NOS 34‐0170 NOS 34‐0180 NOS 34‐0190 AD AD? AD AD AD AR AR AR AD AD AD AR AD AD AD AD AD AD AD RUNX2 SMAD6 ERF SIX1 TCF12 MEGF8 RAB23 RECQL4 SKI TWIST1 MSX2 POR FGFR3 FGFR3 FGFR2 FGFR2 FGFR2 FGFR2 FGFR1 617439 600775 see 601205 615314 614976 201000 218600 182212 101400 604757 201750 602849 612247 123500 123790 101200 101600 101600 (Continues) Gain‐of‐function variants, duplications, triplications Rare SMAD6 variants and a common BMP2 polymorphism may interact to produce craniosynostosis; subject disputed Variants in ERF also cause Chitayat hyperphalangism syndrome (Group 19) Frequently sagittal and lambdoid synostosis. See also MIM 608389—Branchiootic syndrome 3, and 605192— deafness, autosomal dominant 23, for other SIX1‐ related phenotypes Frequently coronal craniosynostosis See other phenotypes associated with RECQL4 variants, above Variants in FGFR3, FGFR2, and TCF12 have been reported to cause phenotypes resembling Saethre‐Chotzen syndrome Heterozygous p.P148H variant in a two families Defined by specific FGFR3 p.P250R variant Defined by specific FGFR3 p.A391E variant; also known as Crouzonodermoskeletal syndrome Notably p.S372Y or p.Y375C variants Most have FGFR1 p.P252R variant; Includes Jackson‐ Weiss syndrome (MIM 123150) 33 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 34 See also: pyknodysostosis, CTSK‐related; cutis laxa, ATP6V0A2‐related; mandibuloacral dysplasia, LMNA‐related; progeria, Hutchinson‐Gilford type, LMNA‐related; and Hajdu‐Cheney syndrome, NOTCH2‐related, for similar clavicular defects or osteolysis. See also Crane‐Heise syndrome (MIM 218090), the nosologic status of which remains unclear. Name of group/name of disorder NOS 33‐0070 (Continued) Group number/number of disorder TABLE 1 UNGER ET AL. (Continued) Craniosynostosis and dental anomalies (CRSDA), IL11RA‐ related Craniosynostosis, retained deciduous teeth and intellectual disability, IL6ST‐related Cutis laxa with craniosynostosis, short stature, brachydactyly, and syndactyly, LTBP1‐related Bohring‐Opitz syndrome, ASXL1‐related Craniosynostosis, radiohumeral fusion and other skeletal defects, CYP26B1‐related Cardiac, facial and digital anomalies with developmental delay (CAFDADD), TRAF7‐related Craniosynostosis, hypertrichosis, progeroid appearance, bone dysplasia, characteristic face (Fontaine progeroid syndrome, Gorlin‐Chaudhry‐Moss syndrome), SLC25A24‐related Curry‐Jones syndrome, SMO‐related 3MC syndrome, MASP1‐related 3MC syndrome, COLEC11‐related 3MC syndrome, COLEC10‐related Weiss‐Kruszka syndrome, ZNF462‐related Au‐Kline syndrome, HNRNPK‐related Char syndrome, TFAP2B‐related Syndrome with developmental and speech delay, dysmorphic facies, craniosynostosis and T‐cell abnormalities NOS 34‐0210 NOS 34‐0220 NOS 34‐0230 NOS 34‐0240 NOS 34‐0250 NOS 34‐0260 NOS 34‐0270 NOS 34‐0280 NOS 34‐0290 NOS 34‐0300 NOS 34‐0310 NOS 34‐0320 NOS 34‐0330 NOS 34‐0340 NOS 34‐0350 Inheritance Gene or locus TFAP2B BCL11B AD HNRNPK ZNF462 COLEC10 COLEC11 MASP1 AD AD AD AR AR AR SMO SLC25A24 AD MOS TRAF7 CYP26B1 ASXL1 LTBP1 IL6ST IL11RA ZIC1 AD AR AD AR AR AR AD MIM No. 618092 169100 616580 618619 248340 265050 257920 601707 612289 618164 614416 605039 619451 see 600694 614188 618736 Craniosynostosis in some affected individuals Mainly with loss‐of‐function variants Craniosynostosis and vertebral anomalies in a significant proportion of cases Metopic ridging or CSO (metopic, lambdoid, 9/24) Craniosynostosis in 20%–30% Craniosynostosis in 20%–30% Craniosynostosis in 20%–30% Activating variant c.1234C>T (p.L412F) Multistructure craniosynostosis is one of the features CYP26B1 is a retinoid acid‐degrading enzyme, pathogenesis involves retinoic acid‐associated morphogenesis Single case reported, with preserved LIF signaling. See Stüve‐Wiedemann syndrome, IL6ST‐related (above, group 24) as well as IL6ST‐MIM 600694 for other phenotypes associated with IL6ST Notes Craniofacial dysostoses Mandibulofacial dysostosis, TCOF1‐related (Treacher‐ Collins, Franceschetti‐Klein) NOS 35‐0010 AD TCOF1 154500 UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 35 Craniosynostosis is not rare and may have a non‐genetic pathogenesis in many cases. It can also occur secondarily in any form of rickets. Conditions in which craniosynostosis is an occasional feature have not been included. See also: cranioectodermal dysplasia (several types in the ciliopathy group); SEMD, RSPRY1‐related; osteocraniostenosis, FAM111A‐related; Osteogenesis imperfecta with craniosynostosis (Cole‐Carpenter syndrome), P4HB‐related; CDAGS syndrome, RNU12‐related; syndactyly (Lueken type, with or without craniosynostosis), IHH‐related; and Multiple synostoses syndrome, FGF9‐related. Craniosynostosis can also be present in Loeys‐Dietz syndromes, Meier‐Gorlin syndrome, CDC45‐ related and GINS2‐related; Hypophosphatasia, ALPL‐related; Hypophosphatemic rickets, PHEX‐related; Greig cephalopolysyndactyly syndrome, GLI3‐related; and others. Structural brain anomalies with impaired ID and craniosynostosis / craniosynostosis type 6 Name of group/name of disorder NOS 34‐0200 Group number/number of disorder TABLE 1 34 (Continued) Frontonasal dysplasia, ALX3‐related Frontonasal dysplasia, ALX4‐related Frontonasal dysplasia, ALX1‐related Frontonasal dysplasia, SIX2‐related Frontonasal dysplasia with additional malformations (Sweeney‐Cox syndrome), TWIST1‐related Craniofrontonasal syndrome, EFN1B‐related Acromelic frontonasal dysostosis, ZSWIM6‐related Richieri‐Costa‐Pereira syndrome, EIF4A3‐related Auriculocondylar syndrome, GNAI3‐related (type 1) Auriculocondylar syndrome, PLCB4‐related (type 2) Auriculocondylar syndrome, EDN1‐related (type 3) NOS 35‐0120 NOS 35‐0130 NOS 35‐0140 NOS 35‐0150 NOS 35‐0160 NOS 35‐0170 NOS 35‐0180 NOS 35‐0190 NOS 35‐0200 NOS 35‐0210 NOS 35‐0220 Craniolenticulosutural dysplasia, SEC23A‐related Agnathia‐Otocephaly complex, PRRX1‐related NOS 35‐0110 Teebi hypertelorism syndrome, SPECCIL‐related Acrofacial dysostosis, SF3B4‐related (Nager syndrome) NOS 35‐0100 NOS 35‐0270 Postaxial acrofacial dysostosis, DHODH‐related (Miller syndrome) NOS 35‐0090 NOS 35‐0260 Burns‐McKeown syndrome, TXNL4A‐related NOS 35‐0080 Weyers acrofacial (acrodental) dysostosis, EVC2‐related Mandibulofacial dysostosis with alopecia, EDNRA‐related NOS 35‐0070 NOS 35‐0250 Mandibulofacial dysostosis with microcephaly, EFTUD2‐ related (Guion‐Almeida type) NOS 35‐0060 Orofaciodigital syndrome type I, OFD1‐related Mandibulofacial dysostosis with limb deficiencies, POLR1A‐related (Cincinnati type) NOS 35‐0050 Weyers acrofacial (acrodental) dysostosis, EVC1‐related Mandibulofacial dysostosis, POLR1D‐related (Treacher‐ Collins, Franceschetti‐Klein) NOS 35‐0040 NOS 35‐0230 Mandibulofacial dysostosis, POLR1C‐related (Treacher‐ Collins, Franceschetti‐Klein) NOS 35‐0030 Inheritance AR, AD AD AD AD XL AR AR, AD AD AR AD XL AD AD AR AR AR AD, AR AD, AR AR AR AD AD AD AD, AR AR AD Gene or locus SEC23A SPECC1L EVC2 EVC1 OFD1 EDN1 PLCB4 GNAI3 EIF4A3 ZSWIM6 EFNB1 TWIST1 SIX2 ALX1 ALX4 ALX3 PRRX1 SF3B4 DHODH TXNL4A EDNRA EFTUD2 POLR1A POLR1D POLR1C POLR1B MIM No. 607812 145420 193530 193530 311200 615706 614669 602483 268305 603671 304110 617746 See 604994 613456 613451 136760 202650 154400, 201170 263750 608572 616367 610536 616462 613717 248390 618939 Monoallelic and biallelic inheritance observed See also Group 10 See also Group 10 (Continues) Results from specific amino acid substitutions in TWIST1 Both monoallelic and biallelic variants are at the basis of a spectrum that also includes the former “Rodriguez type” of acrofacial dysostosis Some pathogenic variants are in the promoter region; severity is variable The original description was “acrofacial dysostosis: a mandibulofacial dysostosis with limb anomalies”. The limb anomalies are variable Notes 35 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License NOS 35‐0240 Mandibulofacial dysostosis, POLR1B‐related (Treacher‐ Collins, Franceschetti‐Klein) Name of group/name of disorder NOS 35‐0020 Group number/number of disorder TABLE 1 UNGER ET AL. (Continued) Baraitser‐Winter syndrome, ACTB‐related Baraitser‐Winter syndrome, ACTG1‐related Cerebrofaciothoracic dysplasia, TMCO1‐related Opitz GBBB syndrome, MID1‐related Arhinia microphthalmia syndrome, SMCHD1‐related (Bosma) Acrofrontofacionasal dysostosis Hemifacial microsomia NOS 35‐0290 NOS 35‐0300 NOS 35‐0310 NOS 35‐0320 NOS 35‐0330 NOS 35‐0340 NOS 35‐0350 Inheritance SP, AD AR AD XL AR AD AD XL Gene or locus SMCHD1 MID1 TMCO1 ACTG1 ACTB FGD1 MIM No. 164210 201180 603457 300000 213980 614583 243310 305400 Includes Goldenhar syndrome and Oculo‐Auriculo‐ Vertebral spectrum; genetically heterogeneous; SF3B2 haploinsufficiency identified in ~3% of sporadic and ~25% of familial cases; in some cases a microduplication on 14q23.1 Notes Vertebral and costal dysostoses Currarino syndrome, MNX1‐related Spondylocostal dysostosis, DLL3‐related Spondylocostal dysostosis, MESP2‐related Spondylocostal dysostosis, LFNG‐related Spondylocostal dysostosis, HES7‐related Spondylocostal dysostosis, TBX6‐related Spondylocostal dysostosis, RIPPLY2‐related Vertebral segmentation defect (congenital scoliosis) with variable penetrance, MESP2‐related Vertebral segmentation defect (congenital scoliosis) with variable penetrance, HES7‐related Short stature, cervical segmentation defects, and developmental delay, CDK10‐related Klippel‐Feil syndrome, GDF6‐related Klippel‐Feil syndrome, MEOX1‐related Klippel‐Feil syndrome, GDF3‐related Klippel‐Feil syndrome, MYO18B‐related NOS 36‐0010 NOS 36‐0020 NOS 36‐0030 NOS 36‐0040 NOS 36‐0050 NOS 36‐0060 NOS 36‐0070 NOS 36‐0080 NOS 36‐0090 NOS 36‐0100 NOS 36‐0110 NOS 36‐0120 NOS 36‐0130 NOS 36‐0140 AR AD AR AD AR AD AD AR AR, AD AR AR AR AR AD MYO18B GDF3 MEOX1 GDF6 CDK10 HES7 MESP2 RIPPLY2 TBX6 HES7 LFNG MESP2 DLL3 MNX1 616549 613702 214300 118100 617694 613686 608681 616566 122600 613686 609813 608681 277300 176450 Role of GDF6 variants in Klippel‐Feil syndrome as well as in AD spondylothoracic dysostosis remains unclear Possible role of CNVs in TBX6 Possible role of CNVs in TBX6 in modulating the phenotype? Possible clinical overlap with caudal regression syndrome (see MIM 600145; the role of heterozygous variants in VANGL1 remains to be confirmed) UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 36 See also orofaciodigital syndrome type 4 (Mohr‐Majewski), TCTN3‐related; Endocrine‐cerebro‐osteo dysplasia (ECO), CILK1‐related; the Cerebro‐Costo‐Mandibular syndrome, SNRPB‐ related (group 36, below); and Robinow syndrome (see variants in Group 15) Faciogenital dysplasia, FGD1‐related (Aarskog‐Scott syndrome) Name of group/name of disorder NOS 35‐0280 Group number/number of disorder TABLE 1 36 Cervico‐oculo‐acoustic (Wildervanck) syndrome Cerebro‐costo‐mandibular syndrome (rib gap syndrome), SNRPB‐related Cerebro‐costo‐mandibular‐like syndrome, COG1‐related Diaphanospondylodysostosis, BMPER‐related Spondylo‐megaepiphyseal‐metaphyseal dysplasia (SMMD), NKX3‐2‐related NAD deficiency syndrome, HAAO‐related NAD deficiency syndrome, KYNU‐related NAD deficiency syndrome, NADSYN1‐related VATER/VACTERL association VACTERL association with hydrocephalus (VACTERL‐H), FANCB‐related VACTERL association with hydrocephalus (VACTERL‐H), ZIC3‐related Uniparental disomy, paternal, for chromosome 14 (UPD14; Kagami‐Ogata syndrome) NOS 36‐0160 NOS 36‐0170 NOS 36‐0180 NOS 36‐0190 NOS 36‐0200 NOS 36‐0210 NOS 36‐0220 NOS 36‐0230 NOS 36‐0240 NOS 36‐0250 NOS 36‐0260 SP XL XL SP AR AR AR AR AR AR AD SP Inheritance 14q32? ZIC3 FANCB NADSYN1 KYNU HAAO NKX3‐2 BMPER COG1 SNRPB Gene or locus MIM No. 608149 314390 300514 192350 618845 617661 617660 613330 608022 611209 117650 314600 Notes Imprinted genes at 14q32 may have a role in this complex phenotype with skeletal malformations such as “coat‐ hanger ribs” FANCB‐related Fanconi anemia may present in hemizygous males with the VACTERL‐hydrocephalus phenotype In some cases VACTERL‐like In some cases VACTERL‐like With associated cardiac, limb and renal defects; VACTERL‐like Includes ischiospinal dysostosis, a term that has been used for milder cases Also known as CDG IIg Congenital perceptive deafness, Klippel‐Feil anomaly (see 118100), and abducens palsy with retractio bulbi Ischiopatellar dysplasia (small patella syndrome), TBX4‐ related Nail‐patella syndrome, LMX1B‐related Genitopatellar syndrome, KAT6B‐related NOS 37‐0010 NOS 37‐0020 NOS 37‐0030 AD AD AD KAT6B LMX1B TBX4 606170 161200 147891 See MIM 601360—posterior amelia for the biallelic phenotype Limb hypoplasia—reduction defects group Ulnar‐mammary syndrome, TBX3‐related Holt‐Oram syndrome, TBX5‐related Holt‐Oram/Ulnar Mammary blended phenotype NOS 38‐0010 NOS 38‐0020 NOS 38‐0030 AD AD AD TBX3, TBX5 TBX5 TBX3 142900 181450 (Continues) CNVs involving both TBX3 and TBX5 may result in combined phenotype 37 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 38 See also Meier‐Gorlin syndromes in the primordial dwarfism group (Group 21), and the pseudoachondroplasia/MED group (Group 9) for conditions with patellar changes; see also ischio‐ pubic‐patellar dysplasia as mild expression of campomelic dysplasia, SOX9‐related; RAPADILINO syndrome, RECQL4‐related. Patellar hypoplasia is variably present in Clubfoot with or without deficiency of long bones and/or mirror‐image polydactyly, PITX1‐related. Patellar dysostoses Group 37 VACTERL is nowadays defined as a “Recurrent Constellation of Embryonic Malformations” (RCEM; see Adam et al, AJMG 2020) without a single genetic basis. It may be mimicked by NAD deficiency syndrome, Fanconi anemia and others. The diagnosis is supported by negative genetic analysis. See also spondylocarpotarsal synostosis syndrome, FLNB‐related and RFLNA‐related, Robinow syndrome (variants in Group 15), and cerebrofaciothoracic dysplasia, TMCO1‐related (Group 35) Name of group/name of disorder NOS 36‐0150 (Continued) Group number/number of disorder TABLE 1 UNGER ET AL. Name of group/name of disorder Posterior Amelia, TBX4‐related Cornelia de Lange syndrome, NIPBL‐related Cornelia de Lange syndrome, SMC1A‐related Cornelia de Lange syndrome, SMC3‐related Cornelia de Lange syndrome, RAD21‐related Cornelia de Lange syndrome, HDAC8‐related Thrombocytopenia‐absent radius (TAR) syndrome, RBM8A‐related Thrombocythemia with distal limb defects, THPO‐related Okihiro syndrome (Duane syndrome with radial ray anomaly), SALL4‐related Cousin syndrome, TBX15‐related Roberts syndrome, ESCO2‐related Tibial hemimelia‐polysyndactyly‐triphalangeal thumb (Werner syndrome), ZRS‐related Clubfoot with or without deficiency of long bones and/or mirror‐image polydactyly, PITX1‐related Acheiropodia, LMBR1‐related Engrailed‐1 related dorsoventral syndrome (ENDOVES), limb‐brain type Engrailed‐1 related dorsoventral syndrome (ENDOVES), limb‐only type Tetra‐amelia, WNT3‐related Tetra‐amelia, RSPO2‐related Limb reduction syndrome, WNT7A‐related RAPADILINO syndrome, RECQL4‐related Rothmund‐Thompson syndrome, RECQL4‐related Rothmund‐Thompson syndrome, ANAPC1‐related Rothmund‐Thompson syndrome, DNA2‐related NOS 38‐0040 NOS 38‐0050 NOS 38‐0060 NOS 38‐0070 NOS 38‐0080 NOS 38‐0090 NOS 38‐0100 NOS 38‐0110 NOS 38‐0120 NOS 38‐0130 NOS 38‐0140 NOS 38‐0150 NOS 38‐0160 NOS 38‐0170 NOS 38‐0180 NOS 38‐0190 NOS 38‐0200 NOS 38‐0210 NOS 38‐0220 NOS 38‐0230 NOS 38‐0240 NOS 38‐0250 NOS 38‐0260 (Continued) Inheritance AR AR AR AR AR AR AR AR AR AR AD AD AR AR AD AD AR XL AD AD XL AD AR Gene or locus DNA2 ANAPC1 RECQL4 RECQL4 WNT7A RSPO2 WNT3 MAENLI EN1 LMBR1 PITX1 ZRS ESCO2 TBX15 SALL4 THPO RBM8A HDAC8 RAD21 SMC3 SMC1A NIPBL TBX4 MIM No. 618625 268400 266280 276820, 228930 618021 273395 619217 619218 200500 119800 188740 268300 260660 607323 187950 274000 300882 614701 610759 300590 122470 601360 Notes See also Baller‐Gerold syndrome, RECQL4‐related. See MIM 266280 for explanation of the RAPADILINO acronym Includes former Al‐Awadi Raas‐Rothschild limb‐pelvis hypoplasia‐aplasia as well as Fuhrmann syndrome MAENLI is a lncRNA regulating EN1 expression One single patient with a biallelic frameshift variant described The Brazilian founder allele is deletion spanning exon 4 of LMBR1 that probably affects the activity of ZRS, the limb‐specific enhancer of SHH. In a further patient, biallelic deletion of exons 1 to 16 of LMBR1, including the ZRS In some patients bilateral patellar hypoplasia (see Group 37) Monoallelic variants in ZRS, a limb‐specific enhancer of SHH that is located within intron 5 of the LMBR1 gene Includes IVIC syndrome Distal limb defects postulated as consequence of vascular occlusions Deletion and common SNP on other allele that has regulatory function See also ischiopatellar syndrome for the monoallelic TBX4‐related phenotype UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group number/number of disorder TABLE 1 38 (Continued) Hypoglossia‐hypodactylia (Hanhart syndrome) Poland syndrome Femoral facial syndrome (FFS) Femur‐fibula‐ulna syndrome (FFU) Fibular aplasia, tibial campomelia, and oligosyndactyly syndrome (FATCO) NOS 38‐0350 NOS 38‐0360 NOS 38‐0370 NOS 38‐0380 NOS 38‐0390 AR SP SP SP SP SP (several) TOP2B AD AD NOTCH1 EOGT DLL4 RBPJ DOCK6 AD AD AR AD Gene or locus ARHGAP31 MIM No. 227650 275220 246570 228200 134780 173800 103300 169550 609296 616028 615297 616589 614814 614219 100300 The complex genetic basis of Fanconi anemia and its complementation groups and loci is acknowledged but not further listed in this Nosology; please refer to MIM or to specialized reviews Rare cases reported as associated with monoallelic CDX2 variants with variable expressivity Possibly non‐genetic etiology Some phenotypic overlap with FFU syndrome (below) Most commonly sporadic and probably non‐genetic; some familial cases reported but no specific gene identified so far Also known as Hoffmann syndrome (see MIM 609296) Notes Split hand/foot with and without other manifestations Ankyloblepharon‐ectodermal dysplasia‐cleft palate (AEC) Ectrodactyly‐ectodermal dysplasia cleft‐palate syndrome Type 3 (EEC3) NOS 39‐0010 NOS 39‐0020 AD AD TP63 TP63 604292 106260 (Continues) See other TP63‐related disorders in this group (below) 39 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Group 39 There is overlap between this group and the split hand‐foot malformation group. See also Baller‐Gerold syndrome, RECQL4‐related; congenital hemidysplasia, ichthyosis, limb defects (CHILD) syndrome, NSDHL‐related; as well as the mesomelic and acromesomelic dysplasias groups (above). Some entities in this group (e.g. the Femoral‐facial syndrome and the Femur‐ fibula‐ulna [FFU] syndromes) might be considered (RCEM; see the note to VACTERL in Group 36) Fanconi anemia Scapulo‐iliac dysplasia (Kosenow syndrome) NOS 38‐0340 NOS 38‐0420 B‐cell immunodeficiency‐limb anomaly‐urogenital malformation syndrome (BILU syndrome), TOP2B‐ related NOS 38‐0330 Tibial hemimelia (isolated) Adams‐Oliver syndrome (aplasia cutis congenita and transverse limb defects), NOTCH1‐related NOS 38‐0320 Sirenomelia Adams‐Oliver syndrome (aplasia cutis congenita and transverse limb defects), EOGT‐related NOS 38‐0310 NOS 38‐0400 SP Adams‐Oliver syndrome (aplasia cutis congenita and transverse limb defects), DLL4‐related NOS 38‐0300 NOS 38‐0410 SP, AD Adams‐Oliver syndrome (aplasia cutis congenita and transverse limb defects), RBPJ‐related NOS 38‐0290 AR Adams‐Oliver syndrome (aplasia cutis congenita and transverse limb defects), DOCK6‐related NOS 38‐0280 Inheritance AD Adams‐Oliver syndrome (aplasia cutis congenita and transverse limb defects), ARHGAP31‐related Name of group/name of disorder NOS 38‐0270 Group number/number of disorder TABLE 1 UNGER ET AL. (Continued) Ectrodactyly‐ectodermal dysplasia‐macular dystrophy syndrome (EEM) Limb‐mammary syndrome (including ADULT syndrome) Split hand‐foot malformation, isolated form, type 4 (SHFM4) Split hand‐foot malformation, isolated form, type 1 (SHFM1) Split hand‐foot malformation, isolated form, type 1 (SHFM1) Split hand‐foot malformation, isolated form, type 3 (SHFM3) Split hand‐foot malformation, isolated form, type 6 (SHFM6) Split‐foot malformation with mesoaxial polydactyly (SFMMP) Split‐hand‐foot malformation with or without long bone deficiency (SHFLD), BHLHA9‐related Hartsfield syndrome, FGFR1‐related Split hand‐foot malformation, EPS15L‐related Aplasia cutis congenita with ectrodactyly, UBA2‐related Focal dermal hypoplasia (Goltz Syndrome), PORCN‐related Polydactyly‐Syndactyly‐Triphalangism group Preaxial polydactyly, SHH‐related Preaxial polydactyly, GLI1‐related Preaxial polydactyly, GLI3‐related Preaxial polydactyly type 3 (PPD3) Mirror‐image polydactyly of hands and feet (Laurin‐ Sandrow syndrome), SHH‐related Postaxial polydactyly, GLI1‐related NOS 39‐0040 NOS 39‐0050 NOS 39‐0060 NOS 39‐0070 NOS 39‐0080 NOS 39‐0090 NOS 39‐0100 NOS 39‐0110 NOS 39‐0120 NOS 39‐0130 NOS 39‐0140 NOS 39‐0150 Group 40 NOS 40‐0010 NOS 40‐0020 NOS 40‐0030 NOS 40‐0040 NOS 40‐0050 NOS 40‐0060 Name of group/name of disorder Inheritance AR AD AD AD AR AD XLD AD AD AD AD AR AR AD AD AD AD AD AR Gene or locus GLI1 SHH GLI3 GLI1 SHH PORCN UBA2 EPS15L1 FGFR1 BHLHA9 ZAK WNT10B 10q24 DLX6 DLX5 TP63 TP63 CDH3 MIM No. 618123 135750 174600 174700 174400 174400 305600 615465 612576 616890 225300 246560 183600 220600 605289 603543 225280 Duplication of ZRS (limb enhancer of SHH) Formerly preaxial polydactyly types 1 and 2 (with triphalangeal thumb); regulatory domain variant or duplication of ZRS (limb enhancer of SHH) 19q13.11 deletions may also cause this phenotype. In OMIM as “aplasia cutis congenita with ectrodactyly skeletal syndrome” (a redundant name) Structural variants (deletions) at this locus; one consanguineous family with homozygous point variant in EPS15L1 but inheritance still unclear Duplications at 17p13.3 that include BHLHA9. Phenotypic penetrance is less than 50% and shows markedly variable expressivity; includes the so‐called Gollop‐ Wolfgang complex Duplications at 10q24 encompassing LBX1, BTRC, POLL, DPCD and FBXW4 Structural variations at locus; also regulatory variants affecting exons of DYNC1I1 that regulate DLX5; association with deafness in a single family may be coincidental; a recessive DLX5 syndrome may exist Notes UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License NOS 39‐0030 Group number/number of disorder TABLE 1 40 Acrocallosal syndrome, KIF7‐related Acro‐pectoral‐vertebral dysplasia (F‐syndrome), WNT6‐ related Cenani‐Lenz syndactyly, LRP4‐related Cenani‐Lenz‐like syndactyly, GREM/FMN1‐related Oligosyndactyly, radio‐ulnar synostosis, hearing loss and renal defects syndrome, FMN1‐related Mesoaxial synostotic syndactyly with phalangeal reduction (Malik‐Percin), BHLHA9‐related STAR syndrome (syndactyly of toes, telecanthus, anal and renal malformations), FAM58A‐related NOS 40‐0180 NOS 40‐0190 NOS 40‐0200 NOS 40‐0210 NOS 40‐0220 NOS 40‐0230 NOS 40‐0240 Metacarpal 4‐5 fusion, FGF16‐related Lacrimo‐auriculo‐dento‐digital syndrome (LADD), FGF10‐ related NOS 40‐0170 NOS 40‐0300 Lacrimo‐auriculo‐dento‐digital syndrome (LADD), FGFR3‐ related NOS 40‐0160 Syndactyly (Lueken type, with or without craniosynostosis), IHH‐related Lacrimo‐auriculo‐dento‐digital syndrome (LADD), FGFR2‐ related NOS 40‐0150 NOS 40‐0290 Townes‐Brocks syndrome, SALL1‐related NOS 40‐0140 Syndactyly type 5 (Brachydactyly‐Syndactyly syndrome; syndactyly with metacarpal and metarsal fusion), HOXD13‐related Postaxial polydactyly, isolated (type A10), KIAA0825‐ related NOS 40‐0130 Syndactyly type 4 (I‐V) Haas type, SHH‐related Synpolydactyly, HOXD13‐related NOS 40‐0120 NOS 40‐0280 Synpolydactyly, FBLN1‐related NOS 40‐0110 NOS 40‐0270 Culler‐Jones syndrome, GLI2‐related NOS 40‐0100 Syndactyly type 1 (III‐IV) Hypothalamic hamartomas and polydactyly (Pallister‐Hall‐ like) syndrome, SMO‐related NOS 40‐0090 Syndactyly type 3 (IV‐V), GJA1‐related Pallister‐Hall syndrome, GLI3‐related NOS 40‐0080 NOS 40‐0260 Greig cephalopolysyndactyly syndrome, GLI3‐related Inheritance Gene or locus XLR FGF16 IHH HOXD13 AD SHH AD GJA1 FAM58A BHLHA9 FMN1 GREM1, FMN1 LRP4 WNT6 KIF7 FGF10 FGFR3 FGFR2 SALL1 KIAA0825 HOXD13 FBLN1 AD AD AD XLD AD AR AD AR AD AR AD AD AD AD AR AD AD GLI2 SMO AD GLI3 AR GLI3 AD AD MIM No. 309630 185900 186300, 610713 186200 186100 185900 300707 609432 see 212780 212780 102510 200990 149730 149730 149730 107480 618498 186000 608180 615849 241800 146510 175700 (Continues) Duplication of IHH and regulatory region on 2q35; includes syndactyly with craniosynostosis (Philadelphia type) Duplication of ZRS (limb enhancer of SHH) X‐linked dominant (only affected females known, possibly lethal in males) Biallelic deletion of the FMN1 gene (one individual) Monoallelic duplication of both GREM1 and FMN1 loci (one individual) Structural variations of locus resulting in ectopic activation of WNT6 Hypopituitarism Notes 41 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License NOS 40‐0250 Name of group/name of disorder NOS 40‐0070 (Continued) Group number/number of disorder TABLE 1 UNGER ET AL. (Continued) Synpolydactyly plus syndrome, MAPKAPK5‐related NOS 40‐0320 Inheritance AR AR Gene or locus MAPKAPK5 CKAP2L MIM No. 619869 272440 In OMIM as neurocardiofaciodigital syndrome Notes Multiple synostoses syndrome, NOG‐related Multiple synostoses syndrome, GDF5‐related Multiple synostoses syndrome, FGF9‐related Multiple synostoses syndrome, GDF6‐related Liebenberg syndrome, PITX1‐related Short stature, auditory atresia, mandibular hypoplasia, skeletal abnormalities (SAMS) syndrome, GSC‐related Radio‐ulnar synostosis with amegakaryocytic thrombocytopenia, HOXA11‐related Radio‐ulnar synostosis with amegakaryocytic thrombocytopenia, MECOM‐related Radio‐ulnar synostosis with microcephaly (Giuffré‐ Tsukahara syndrome) NOS 41‐0010 NOS 41‐0020 NOS 41‐0030 NOS 41‐0040 NOS 41‐0050 NOS 41‐0060 NOS 41‐0070 NOS 41‐0080 NOS 41‐0090 AD AD AR AD AD AD AD AD MECOM HOXA11 GSC PITX1 GDF6 FGF9 GDF5 NOG 603438 616738 605432 602471 186550 617898 612961 610017 186500, 186570 X‐linked recessive inheritance suggested Structural variants encompassing the H2AFY gene resulting in ectopic activation of PITX1 in upper limb See other GDF5‐related disorders Includes: Stapes ankylosis with broad thumbs and toes, Tarsal‐Carpal coalition syndrome, proximal Symphalangism 1A; see also Brachydactyly type B2, NOG‐related, in the brachydactyly group UNGER ET AL. 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License Note: The numbering system (first column) includes “NOS” for “Nosology, skeletal”, followed by the group number and the number of the disorder. The abbreviations are as follows: in the disorder names, SED in spondylo‐ ephyseal dysplasia; SEMD is spondylo‐epi‐metaphyseal dysplasia; MED is multiple epiphyseal dysplasia; CDP is chondrodysplasia punctata. In the “Inheritance” column: AD, autosomal recessive; AR, autosomal recessive; XL, X‐linked; MOS, somatic mosaicism; SP, sporadic and inheritance unknown. Pseudo‐AD and Pseudo‐AR refers to genes in the pseudoautosomal regions of chromosome X and Y. The “MIM No.” column shows the MIM number of the disorder; when the number is preceded by “see”, the MIM number is that of the underlying gene. See also Spondylocarpotarsal synostosis syndrome, FLNB‐related and RFLNA‐related; Cardiospondylocarpofacial syndrome, MAP3K7‐related; mesomelic dysplasia with acral synostoses (Verloes‐David‐Pfeiffer type); Baller‐Gerold syndrome, RECQL4‐related; and Antley‐Bixler syndrome, POR‐related Defects in joint formation and synostoses Group 41 Note: the Smith‐Lemli‐Opitz syndrome can present with polydactyly and/or syndactyly. The different variants of Meckel syndrome can have polydactyly and are included under the ciliopathies (see there). The Bardet‐Biedl syndromes may have polydactyly as a secondary feature and have not been included in this neither in this group nor in the ciliopathies. See also Clubfoot with or without deficiency of long bones and/or mirror‐image polydactyly, PITX1‐related. The entity called “Crossed polysyndactyly” not included as unclear whether or not it is a distinct entity. Syndactyly with microcephaly and mental retardation (Filippi syndrome), CKAP2L‐related Name of group/name of disorder NOS 40‐0310 Group number/number of disorder TABLE 1 42 1 | N A M I N G P RO B L E M S A N D TH E D Y A D I C A P P R O A C H A S A WA Y F O R W A R D Smith-McCort dysplasia); while for others the eponymic descriptors are too similar, leading to diagnostic confusion (e.g., Shprintzen-Goldberg and Goldberg-Shprintzen syndromes represent distinct disorders). MIM's Between 2010 and 2020, the technology of massively parallel choice of these eponyms may not reflect the most significant contribu- sequencing has taken center stage in medical genetics research and tion to the delineation of a phenotypic entity. By adopting the dyadic diagnostics. Among the many lessons learned from next-generation system, we have elected to describe each disorder with the name of the sequencing (NGS) results are (1) the large number of previously unrec- responsible gene rather than with an eponym. While MIM remains the ognized rare and ultrarare disorders in each domain of genetic medi- central reference database, the dyadic system allows the Nosology to cine, (2) the phenotypic heterogeneity arising from a single locus is group, lump, or dump disorders based on their molecular basis, especially much greater than previously suspected, and (3) for many dyadic enti- in the light of the lessons of NGS (see above), and is less bound by his- ties, individuals who present all the phenotypic criteria as described in torical constraints. Nevertheless, the Nosology curators have strived to the textbooks are the exception rather than the rule. maintain strong bridges to MIM: MIM numbers are included for all disor- On this background, it has been proposed that the phenotypic ders when available, and when not available, the MIM number for the descriptor of a genetic disorder (the “name” of a condition) is no longer responsible gene is included. Moreover, references are made to the sufficient to distinguish the disorder unequivocally. Furthermore, the MIM denomination of individual disorders as well as to other MIM disor- sequential numbering of conditions with the same name (as applied ders arising from pathogenic variants in the same gene. e.g., for osteogenesis imperfecta or ataxias in Mendelian Inheritance in Man [MIM]) may be unsatisfactory, as a number has no inherent information, making it necessary to consult the numbering reference. As a consequence, it has been suggested that instead of being attributed by 3 | T H E OR P H A N E T N O M E N C L A T U RE A N D T H E NO SO L O G Y a number (or an eponym, see below), the main phenotypic descriptor of a genetic disorder should best be coupled to the name of the underly- In the context of a joint collaboration between the International Skel- ing gene, allowing for more direct and pertinent information, less prone etal Dysplasia Society (ISDS), the European Reference Network on to ambiguities and errors; the so-called dyadic approach (Biesecker Rare Bone Disorders (ERN-BOND) and Orphanet, co-coordinated by et al., 2021). Of note, the dyadic concept has been pioneered by the Houda Ali (curator at Orphanet) and Geert Mortier (main curator of editors of the well-known resource GeneReviews starting in 2020 the 2019 Nosology), a detailed analysis of the Orphanet database in (Dr. M. Adam, personal communication; and Biesecker et al., 2021). comparison with the 2019 Nosology, resulted in a list of approxi- The Nosology has not been immune to the proliferation of num- mately 248 phenotypic entities that were present in the Orphanet bered lists, such as, for example, in osteogenesis imperfecta nomenclature of rare diseases but absent in the 2019 Nosology. To (Sillence & Rimoin, 1978; Sillence et al., 1979; Van Dijk & be included in the 2023 Nosology, disorders had to have a recogniz- Sillence, 2014). In this 2023 revision, the curators have decided to able phenotype and a clear inheritance pattern or molecular definition. adopt the dyadic naming approach as it allows for more precision both Approximately 30 of these disorders met inclusion criteria and have in the clinic and in the laboratory. However, compromises have been thus been included in the Nosology. Other disorders in this list had made in some instances to account for the historic evolution of the either been described in a single paper without molecular confirma- Nosology, as well as to maintain congruence and interoperability with tion, or represent historical descriptions with limited available infor- what is considered the most important reference database for genetic mation, reflecting the policy followed by Orphanet to represent all disorders, MIM (MIM and its online version, OMIM). disorders fitting the definition of a rare disease to the advantage of individuals affected by ultrarare presentations, as long as they constitute phenotypically distinct diagnoses (Ref. https://www.orpha.net/ 2 | T H E M E N D E LI A N I N H E R I T A N C E I N M A N CA T A L O G A N D TH E N O S O L O G Y orphacom/cahiers/docs/GB/eproc_disease_inventory_R1_Nom_Dis_ EP_04.pdf). On the other hand, it became evident that many entries in the list do not seem to represent distinct phenotypic entities in view The late Victor McKusick's opus, MIM, remains the single most impor- of the current knowledge and of the Nosology criteria. This has tant general reference database for genetic disorders. The way in which prompted an ongoing revision process by the Orphanet team to MIM was created and is still curated allows for a detailed documentation review and identify entries that need to be deactivated and subse- of the history of each and every disorder. For the same reason, it is less quently removed from the Orphanet nomenclature of rare diseases. well-suited to document the changes that occur in the nosography, for instance when one disorder is subsumed under another; several disorders listed in MIM have been subsumed under other conditions in the Nosology, or are altogether not recognized as distinct phenotypic enti- 4 | T H E NO SO L O G Y A N D T H E CL I N G E N CURATION INITIATIVE ties in the Nosology (e.g., mesomelic dysplasia, Camera type; MIM 611886). Also, MIM makes extensive use of eponyms to distinguish The ClinGen initiative (https://clinicalgenome.org/affiliation/40065/) related but distinct disorders (e.g., Dyggve-Melchior-Clausen disease and is currently working on a set of genes associated with skeletal 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License 43 UNGER ET AL. UNGER ET AL. disorders in order to provide strength of evidence for gene-disease disorders in the number or function of osteoclasts. The “Osteogenesis associations, and using strictly verified criteria to assess pathogenicity Imperfecta and decreased bone density group” was renamed “Osteo- of variants in genes found to have a definitive association with dis- genesis Imperfecta and bone fragility group” to reflect the fact that ease. It has already done so for other groups of genetic disorders, skeletal fragility is a hallmark of these disorders irrespective of the such as the cardiomyopathies and others. Several of the present bone mineral density (as in fact a small subset of osteogenesis imper- Nosology curators participate in this effort. However, the sheer num- fecta patients can have high bone mass). The name of the group of ber of genes involved in constitutional skeletal conditions is such that “Overgrowth (tall stature) syndromes with skeletal involvement” was the ClinGen section for skeletal dysplasia genes will take time to be changed to the more encompassing “Overgrowth (tall stature) syn- completed. In this context, the Nosology, with its list of curated genes dromes and segmental overgrowth”. The group of “Craniosynostosis and disorders (albeit not to the extent set out in the ClinGen initia- syndromes” was renamed “Syndromes featuring craniosynostosis”, as tive), will remain the best available resource for the foreseeable although disorders in this group frequently feature craniosynostosis, future. Of note, the ClinGen approach is much more “lumping” than this finding does not always represent the most salient feature. Other the Nosology, to the extent that phenotypes due to pathogenic vari- changes in group nomenclature included “Brachydactylies (without ants at a single locus must have significant qualitative differences to extraskeletal manifestations)” to “Isolated brachydactylies”; “Brachy- justify separate nosologic entries (phenotypic entities); a simple quan- dactylies (with extraskeletal manifestations)” to “Brachydactylies as titative difference (typically, more or less severe) is not enough to jus- part of syndromes”; “Ciliopathies with major skeletal involvement” to tify separation. This may lead us to reflect on what constitutes a “Skeletal disorders caused by abnormalities of cilia or ciliary signaling”; “dysplasia” or a “syndrome”. Some of us have also contributed to the “Abnormal mineralization group” to “Disorders of bone mineraliza- development of the “Nosology of inborn errors of metabolism” and tion”; and “Ectrodactyly with and without other manifestations” to the subsequent “International Classification of Inherited Metabolic “Split hand/foot with and without other manifestations”. Disorders” (ICIMD) (Ferreira et al., 2019, 2021). That nosology applies Some disorders were reassigned. As an example, trichorhinopha- the “one gene – one condition” principle, unless there are qualitative langeal dysplasia types 1/3 was moved from the “Acromelic dyspla- differences. However, it is more difficult to apply this principle to the sias” group to the group of “Brachydactylies as part of syndromes”. skeletal conditions. For example, children affected by methylmalonic The total number of disorders increased from 461 to 771, and the acidemia may have different urinary concentrations of methylmalonic number of genes from 437 to 552. Although we are aware of the acid but will be considered to have the same disorder (MIM 251000). problems inherent to numeric lists (as discussed above), we have ten- It would be more difficult to claim that a fetus with achondrogenesis tatively included a numbering system including the abbreviation type 1B (MIM 600972) and a child with recessive multiple epiphyseal “NOS” (for “Nosology, skeletal”), the group number, and a sequential dysplasia (MIM 226900) have the same disorder, even if the responsi- number withing the group, taking care to leave gaps that may allow ble gene is the same and the phenotypes represent opposite ends of for the inclusion of disorders in the future. Such a numbering system severity within the same spectrum, as the morphologic features and might prove helpful in cross-referencing with MIM, Orphanet and clinical prognosis are so radically distinct. other databases. 5 | C HA N G E S CO M P A R E D T O P R E V I O U S REVISIONS 6 | WH A T I S T H E U T I L I TY O F TH E NO SOL OG Y? Some changes in the structure of the Nosology deserve to be men- Since its early revisions, the Nosology has been helpful for pediatri- tioned. The total number of groups decreased from 42 to 41. This cians, geneticists, radiologists, and others as a reminder of the differ- decrease has to do with restructuring of a few groups. Specifically, ential diagnosis. Its original structure in groups of disorders with the previous “Perlecan group” and “Aggrecan group” were incorpo- similar radiographic features reflected the diagnostic approach of the rated into the new group of “Proteoglycan core protein disorders”, clinical geneticist and, even more, of the radiologist, to the osteochon- and the former groups of “Neonatal osteosclerotic dysplasias” and of drodysplasias. Over the years, the inclusion of brachydactylies, cranio- “Other sclerosing bone disorders” were fused into the group of (non- synostoses, craniofacial dysostoses, syndactylies, limb reductions, and osteopetrotic) “Osteosclerotic disorders”. A new group of “Skeletal other dysostoses, as well as primordial short stature and overgrowth disorders of parathyroid hormone signaling cascade” was added to syndromes has broadened its utility for the differential diagnosis the current Nosology. The two brachydactylies groups (isolated or as within these groups of disorders. The 2010 revision stated that “The part of syndromes; now groups 18 and 19) were found to be more aim is to provide the Genetics, Pediatrics and Radiology community with organically placed right after the acromesomelic and acromelic groups a list of recognized genetic skeletal disorders that can be of help in the (groups 16 and 17). diagnosis of individual cases, in the delineation of novel disorders, and in Several groups were renamed. The group of “Osteopetrosis and building bridges between clinicians and scientists interested in skeletal related disorders” is now named “Osteopetrosis and related osteo- biology. (…) The Nosology should be useful for the diagnosis of patients clast disorders”, to highlight the fact that the osteopetroses represent with genetic skeletal diseases, particularly in view of the information flood 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License 44 expected with the novel sequencing technologies; in the delineation of DBT/Wellcome Trust India Alliance Grant “Center for Rare Disease clinical entities and novel disorders, by providing an overview of estab- Diagnosis, Research and Training number: IA/CRC/20/1/600002” lished nosologic entities; and for scientists looking for the clinical corre- (to Katta M. Girisha), the Sigrid Jusélius Foundation (grant to Outi lates of genes, proteins and pathways involved in skeletal biology.” Makitie), the National Health and Medical Research Council of Thirteen years later, the Nosology may have an additional role in molec- Australia Leadership Fellow Grant #GNT2018081 (to Ravi Savarira- ular genetic diagnostic testing. In a pre-test setting, the Nosology may yan), and Curekids New Zealand (support to Stephen P. Robertson). inform the decision on which genes to include in a diagnostic panel tai- Open access funding provided by Universite de Lausanne. lored to a specific clinical situation. Current diagnostic workflows often involve plausibility verification of variants observed in panel or exome DATA AVAILABILITY STAT EMEN T sequencing tests (reverse phenotyping). Also, in the post-test setting, Data sharing is not applicable to this article as no new data were cre- the Nosology may be helpful for rapid reference and orientation. ated or analyzed in this study. The Nosology is also an illustration of the complexity of the human genome as demonstrated by the sheer number of genes and OR CID gene products required for normal skeletal development and growth. Débora R. Bertola The included table with its over 750 entries and its rows and columns Daniel H. Cohn is like the musical score for the orchestra of skeletal development and Katta M. Girisha https://orcid.org/0000-0002-0139-8239 growth that may be an inspiration to geneticists and basic scientists. Deborah Krakow https://orcid.org/0000-0001-9906-4968 Perhaps the hybrid nature of the Nosology, combining clinical, radio- Outi Makitie graphic, and molecular criteria, is a strength and not a weakness, Stephen P. Robertson https://orcid.org/0000-0002-5181-7809 allowing colleagues with varied backgrounds an approach to the data. Andrea Superti-Furga https://orcid.org/0000-0002-3543-7531 https://orcid.org/0000-0002-4701-6777 https://orcid.org/0000-0003-0388-4457 https://orcid.org/0000-0002-4547-001X It is clear that no nosology in medicine is perfect nor complete. They are always dynamic and evolving. However, the frequent cita- RE FE RE NCE S tions of previous versions of the Nosology suggest that, despite the A nomenclature for constitutional (intrinsic) diseases of bones. (1971). A nomenclature for constitutional (intrinsic) diseases of bones. The Journal of Pediatrics, 78(1), 177–179. https://doi.org/10.1016/s00223476(71)80286-x Beighton, P., Giedion, Z. A., Gorlin, R., Hall, J., Horton, B., Kozlowski, K., Lachman, R., Langer, L. O., Maroteaux, P., & Poznanski, A. (1992). International classification of osteochondrodysplasias. International working group on constitutional diseases of bone. American Journal of Medical Genetics, 44(2), 223–229. https://doi.org/10.1002/ajmg. 1320440220 Biesecker, L. G., Adam, M. P., Alkuraya, F. S., Amemiya, A. R., Bamshad, M. J., Beck, A. E., Bennett, J. T., Bird, L. M., Carey, J. C., Chung, B., Clark, R. D., Cox, T. C., Curry, C., Dinulos, M. B. P., Dobyns, W. B., Giampietro, P. F., Girisha, K. M., Glass, I. A., Graham, J. M., … Zarate, Y. A. (2021). A dyadic approach to the delineation of diagnostic entities in clinical genomics. American Journal of Human Genetics, 108(1), 8–15. https://doi.org/10.1016/j.ajhg.2020. 11.013 Bonafe, L., Cormier-Daire, V., Hall, C., Lachman, R., Mortier, G., Mundlos, S., Nishimura, G., Sangiorgi, L., Savarirayan, R., Sillence, D., Spranger, J., Superti-Furga, A., Warman, M., & Unger, S. (2015). Nosology and classification of genetic skeletal disorders: 2015 revision. American Journal of Medical Genetics. Part A, 167A(12), 2869–2892. https://doi.org/10.1002/ajmg.a.37365 Ferreira, C. R., Rahman, S., Keller, M., Zschocke, J., & ICIMD Advisory Group. (2021). An international classification of inherited metabolic disorders (ICIMD). Journal of Inherited Metabolic Disease, 44(1), 164– 177. https://doi.org/10.1002/jimd.12348 Ferreira, C. R., van Karnebeek, C. D. M., Vockley, J., & Blau, N. (2019). A proposed nosology of inborn errors of metabolism. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 21(1), 102–106. https://doi.org/10.1038/s41436-018-0022-8 Hall, C. M. (2002). International nosology and classification of constitutional disorders of bone (2001). American Journal of Medical Genetics, 113(1), 65–77. https://doi.org/10.1002/ajmg.10828 International nomenclature and classification of the osteochondrodysplasias. (1997). International working group on constitutional diseases of bone (1998). American Journal of Medical Genetics, 79(5), 376–382. many compromises necessary in its preparation the Nosology has been useful to, and widely adopted by, the medical and scientific community. Thanks to the continuous progress in delineating genetic conditions, the Nosology starts its obsolescence at the moment it is published. Nevertheless, may this new version encounter the same benevolent reception, and also be replaced in time by novel and more complete versions. AUTHORS' CONTRIBUTI ON S All authors conceptualized the revision work, contributed to the revision of several groups of disorders, and participated in the discussion and decision making. Sheila Unger, Carlos R. Ferreira, and Andrea Superti-Furga collated the contributions, compiled the final table and drafted the manuscript. The table and the manuscript were then revised by all authors and subsequently finalized by Sheila Unger, Carlos R. Ferreira, and Andrea Superti-Furga. ACKNOWLEDGMENTS We wish to acknowledge constructive exchanges with Dr. Ada Hamosh at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, USA, concerning the integration between the Nosology and the MIM catalog, as well as with Dr. Leslie G. Biesecker, Center for Precision Health Research, NHGRI, Bethesda, USA, concerning the adoption of the dyadic nomenclature system. The skillful assistance and graciousness of Mrs. Nathalie Zumstein in organizing the Nosology sessions in Lausanne (March 2022) is also gratefully acknowledged. This work was supported by the International Skeletal Dysplasia Society (www.isds.ch) with secretarial logistics, the University of Lausanne (support to Sheila Unger and Andrea Superti-Furga), the NIH/NHGRI (to Carlos R. Ferreira), the 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License 45 UNGER ET AL. https://doi.org/10.1002/(sici)1096-8628(19981012)79:5<376::aidajmg9>3.0.co;2-h [International nomenclature of constitutional bone diseases. Constitutional bone diseases without known pathogenesis]. (1971). International nomenclature of constitutional bone diseases. Constitutional bone diseases without known pathogenesis. Archives Francaises De Pediatrie, 28(5), 553–557. International nomenclature of constitutional diseases of bone. Revision, May 1983. (1983). International nomenclature of constitutional diseases of bone. Revision, May 1983. Annales de Radiologie, 26(6), 457–462. International nomenclature of constitutional diseases of bone: Revision-May 1977. (1979). International nomenclature of constitutional diseases of bone: Revision--May 1977. American Journal of Medical Genetics, 3(1), 21–26. https://doi.org/10.1002/ajmg.1320030107 [International nomenclature of constitutional diseases of bones]. (1970). International nomenclature of constitutional diseases of bones. Annales de Radiologie, 13(7), 455–464. Lachman, R. S. (1998). International nomenclature and classification of the osteochondrodysplasias (1997). Pediatric Radiology, 28(10), 737–744. https://doi.org/10.1007/s002470050458 McKusick, V. A., & Scott, C. I. (1971). A nomenclature for constitutional disorders of bone. The Journal of Bone and Joint Surgery. American, 53(5), 978–986. Mortier, G. R., Cohn, D. H., Cormier-Daire, V., Hall, C., Krakow, D., Mundlos, S., Nishimura, G., Robertson, S., Sangiorgi, L., Savarirayan, R., Sillence, D., Superti-Furga, A., Unger, S., & Warman, M. L. (2019). Nosology and classification of genetic skeletal disorders: 2019 revision. American Journal of Medical Genetics. Part A, 179(12), 2393– 2419. https://doi.org/10.1002/ajmg.a.61366 Sillence, D. O., & Rimoin, D. L. (1978). Classification of osteogenesis imperfecta. Lancet (London, England), 1(8072), 1041–1042. https://doi.org/ 10.1016/s0140-6736(78)90763-8 UNGER ET AL. Sillence, D. O., Rimoin, D. L., & Danks, D. M. (1979). Clinical variability in osteogenesis imperfecta-variable expressivity or genetic heterogeneity. Birth Defects Original Article Series, 15(5B), 113–129. Spranger, J. (1985). Pattern recognition in bone dysplasias. Progress in Clinical and Biological Research, 200, 315–342. Superti-Furga, A., & Unger, S. (2007). Nosology and classification of genetic skeletal disorders: 2006 revision. American Journal of Medical Genetics. Part A, 143A(1), 1–18. https://doi.org/10.1002/ajmg.a. 31483 Van Dijk, F. S., & Sillence, D. O. (2014). Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. American Journal of Medical Genetics. Part A, 164A(6), 1470–1481. https://doi.org/10. 1002/ajmg.a.36545 Warman, M. L., Cormier-Daire, V., Hall, C., Krakow, D., Lachman, R., LeMerrer, M., Mortier, G., Mundlos, S., Nishimura, G., Rimoin, D. L., Robertson, S., Savarirayan, R., Sillence, D., Spranger, J., Unger, S., Zabel, B., & Superti-Furga, A. (2011). Nosology and classification of genetic skeletal disorders: 2010 revision. American Journal of Medical Genetics. Part A, 155A(5), 943–968. https://doi.org/10.1002/ajmg.a.33909 How to cite this article: Unger, S., Ferreira, C. R., Mortier, G. R., Ali, H., Bertola, D. R., Calder, A., Cohn, D. H., Cormier-Daire, V., Girisha, K. M., Hall, C., Krakow, D., Makitie, O., Mundlos, S., Nishimura, G., Robertson, S. P., Savarirayan, R., Sillence, D., Simon, M., Sutton, V. R., … Superti-Furga, A. (2023). Nosology of genetic skeletal disorders: 2023 revision. American Journal of Medical Genetics Part A, 1–46. https://doi. org/10.1002/ajmg.a.63132 15524833, 0, Downloaded from https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63132 by Test, Wiley Online Library on [20/02/2023]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License 46