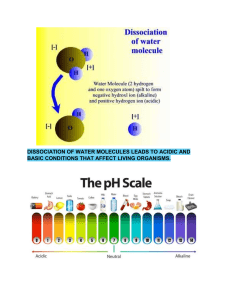
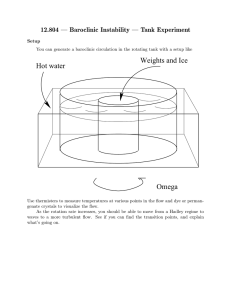
ARTICLE IN PRESS Chemical Engineering Science 65 (2010) 3709–3717 Contents lists available at ScienceDirect Chemical Engineering Science journal homepage: www.elsevier.com/locate/ces Manganese oxide dissociation kinetics for the Mn2O3 thermochemical water-splitting cycle. Part 1: Experimental Todd M. Francis, Paul R. Lichty, Alan W. Weimer Department of Chemical and Biological Engineering, University of Colorado, 1111 Engineering Dr., Campus Box 424, Boulder, CO 80309, USA a r t i c l e in fo abstract Article history: Received 3 July 2009 Received in revised form 20 February 2010 Accepted 2 March 2010 Available online 6 March 2010 It is shown that the dissociation of Mn2O3 to MnO in a short residence time aerosol flow reactor can achieve high conversions approaching 75% when the concentration of oxygen is kept below 0.25%. Significant recombination reaction occurs when the oxygen content exceeds 0.25% by volume. A dual reaction mechanism for Mn2O3 dissociation was found: Keywords: Chemical reactors Reaction engineering Kinetics Energy Manganese oxide Thermochemical water splitting RAvramiErofeev ¼ A1 eEa,1 =RT nð1XÞ½lnð1XÞðn1Þ=n ROrder_of _reaction ¼ A2 eEa,2 =RT ð1XÞn with the transition from one mechanism to the other occurring at an extent of reaction of approximately 0.6. Rate constants for the two mechanisms were calculated to be 1.8 107 7 1.3 107 and 5.6 103 7 4.1 103 s 1, respectively, for oxygen concentration o 0.25%. High levels of dissociation are achievable when the reaction is carried out in an inert gas environment using a reactor configuration that limits the reverse reaction. & 2010 Elsevier Ltd. All rights reserved. 1. Introduction Renewable energy is a reliable method to produce energy that is sustainable and environmentally friendly. In the past, energy from renewable sources has not been in the forefront of discussions. However, with recent increases in the price of fossil fuels, renewable energy is becoming an increasingly popular topic. Although the technology is still developing and not yet economically practical, many believe that the future of renewable energy is hydrogen. A three-step thermochemical water-splitting cycle to generate renewable hydrogen has been proposed, which uses manganese oxide and solar-based thermal energy to produce hydrogen (Nuesch, 1998; Sturzenegger et al., 1999; Sturzenegger and Nuesch, 1999): Mn2O3 solar energy ! 2MnO+ 1/2O2 (1) 2MnO+ 2NaOH-H2 + 2NaMnO2 (2) 2MnO2 + H2O-Mn2O3 + 2NaOH (3) H2O-H2 + 1/2O2 (4) Corresponding author. E-mail address: Alan.Weimer@Colorado.edu (A.W. Weimer). 0009-2509/$ - see front matter & 2010 Elsevier Ltd. All rights reserved. doi:10.1016/j.ces.2010.03.002 An efficiency study of this cycle was performed, where efficiency was defined as the amount of energy needed to split water into its constituents over the amount of energy input to the entire cycle (Sturzenegger and Nuesch, 1999). The maximum efficiency, which was the efficiency of an idealized system, was calculated to be 74%. When all unit operations and heat recoveries were included, the efficiency was then between 16 and 22%. This study lends optimism that this cycle has the potential to supply sustainable hydrogen efficiently. Most work on the sodium manganese oxide cycle has focused on the hydrogen generating step (Eq. (2)) and the product recovery step (Eq. (3)) with little work done on the reduction step (Eq. (1)) (Nuesch, 1998; Sturzenegger et al., 1999). It is essential to understand the reduction step because the entire feasibility of this thermochemical cycle is based on this reaction having a high overall conversion (Sturzenegger and Nuesch, 1999). The viability of preventing MnO and O2 recombination must be investigated in order to determine the degree to which recombination impacts the overall process (Ganz et al., 1998; Sturzenegger et al., 1999; Perkins and Weimer, 2004). A high-temperature aerosol flow reactor (AFR) was used to study the thermal dissociation of manganese oxide. This type of dilute phase particle phase reactor has shown to yield high dissociation reaction rates using other metal oxide systems. With the inherent importance of the thermal dissociation step (Eq. (1)), a rapid dissociation and maximum conversion are essential for the viability of a high-throughput thermochemical cycle for efficient ARTICLE IN PRESS 3710 T.M. Francis et al. / Chemical Engineering Science 65 (2010) 3709–3717 hydrogen production. Additionally, an AFR can easily be outfitted with a low-temperature quench zone, which can be used to rapidly cool the particles upon leaving the high-temperature zone. This quench zone reduces the potential for recombination and helps the reaction conditions to be tightly controlled. The dissociation of manganese oxide is reversible, as are most high temperature reduction steps from metal oxide thermochemical cycles (Otto, 1964; Matsushi and Thoburn, 1965; Ganz et al., 1998). In a proof of concept study, it was found that when Mn3O4 is reduced, MnO is only formed if the reaction was quickly quenched (Ganz et al., 1998; Sturzenegger et al., 1999). Conversions were between 5 and 15% MnO when the reaction was quickly quenched and 0% MnO when the reaction was not. Sturzenegger et al. were unsure if the reaction conditions or the reactor design led to low conversions (Sturzenegger et al., 1999), however a fast quench in a thermal gravimetric analyzer (TGA) was shown to increase the conversion of Mn2O3 to Mn3O4 from 34% to 95% (Otto, 1964). This result lends optimism that if a reactor can be designed to give a fast enough quench, high conversions may be retained. It was for this reason that an AFR was hypothesized to be a reactor capable of achieving high conversions. There are only a few examples of high-temperature AFRs for the processing of solids found in the literature, but those examples do provide insight into why an AFR would work well for the dissociation of manganese oxide. An AFR has been used to rapidly produce submicron sized boron carbide particles directly at a high operating temperature (Weimer et al., 1991). Particles that were uniform in size were produced at 2200 K via a rapid quench that limited grain growth. Residence times were between fractions of a second and only a few seconds. An expanded cooling zone, or quench zone, was used to effectively stop the reaction. Rapid cooling is a method that may help reduce the potential for recombination during the dissociation of manganese oxide. An AFR has also been used for reacting aluminum particles with nitrogen gas at 1873 K to form aluminum nitride (Weimer et al., 1994). The reaction was carried out to high conversions with little deposition of particles on the walls. This indicated that heating rates and residence times attainable in an AFR were sufficient for gas–solids reactions to be driven to high conversions. Carney et al. used an AFR for the decomposition of nickel oxalate (Carney et al. 2006a, b). Nickel oxalate was fed into the reactor and reduced in the presence of hydrogen to produce nickel powder. High conversions were achieved within short residence times, and were largely attributed to the fast heating rate in the reactor. Additionally, the product particles consisted of nanosized primary particles, which are desirable in thermochemical water-splitting cycles. This shows an additional advantage of studying the dissociation in an AFR. A solid particle receiver has been demonstrated to efficiently heat solids using concentrated solar energy (Hruby et al., 1984). A model of a flowing cloud of particles showed that temperatures of up to 1118 K could be achieved for micron size particles (Evans et al., 1987). Further, Dahl et al. showed an ‘‘on-sun’’ solar reactor achieving temperatures greater than 2000 K (Dahl et al., 2004a, b). Methane gas was dissociated to form carbon black and hydrogen and conversions up to 90% were achieved for a residence time of 0.1 s (Dahl et al., 2004a, b). Finally, an AFR has been used to study the reduction of metal oxides as well. ZnO was split into its constituent parts in an AFR at high temperatures (1873–2073 K) and over short residence times (1–2 s) (Perkins et al., 2008). High conversion to Zn metal was achieved, and the resulting primary particles were precipitated out of the gas phase (Zn was a vapor) and were nano-sized. These studies showed that an AFR has many advantages for dissociation reactions including rapid heating rates and the ability to control short residence times followed by rapid cooling to prevent particle growth. Hence, the AFR configuration provides for an opportunity to dissociate manganese oxide and to potentially utilize rapid cooling to limit the reverse reaction from occurring. Further, these types of reactors have been shown to be scalable for industrial production. 2. Materials and methods 2.1. Equipment Kinetic studies were completed using a Thermal Technology Inc. AFR, Fig. 1. The hot zone was maintained using a high-density graphite resistance heating element combined with high-purity graphite felt insulation that reduced heat losses. The reactor tube placed in the center of the furnace was a 99.8% slip cast alumina protection tube (ID¼0.09 m, L¼1.17 m) from McDanel Advanced Ceramic Technologies LLC. The alumina tube AFR has the capability to achieve reaction temperatures as high as 2173 K, limited by the upper temperature for using alumina. A Eurotherm 914 controller was utilized for temperature control within the reactor. Temperatures were measured on the wall of the hot zone using a Type-C thermocouple up to Sweep Nitrogen In Fluid Bed Feeder Feed Tube Hot Zone Graphite Element Quench Tube Alumina Tube Outer Cooling Zone HEPA Filter To Gas Analyzers and Vent Gravity Collection Vessel Fig. 1. Schematic of the aerosol flow reactor. ARTICLE IN PRESS T.M. Francis et al. / Chemical Engineering Science 65 (2010) 3709–3717 1073 K. From 1073 to 1373 K, the temperature measurement from the thermocouple was blended with that from an infrared pyrometer (Ircon Inc.) for seamless temperature control over the intermediate range. Above 1373 K, the temperature was measured only with the infrared pyrometer. All powder that was fed to the reactor and processed was then quickly quenched in a quench tube assembly, shown in Fig. 2. This quench effectively ‘‘terminated’’ the reaction for a more accurate calculation of the time the particles resided in the hot zone. The quench process also effectively inhibited the recombination reaction. The quench tube consisted of concentrically-mounted inner and outer copper tubes. The reason for this was to ensure uniform cooling along the copper tube. Because the quench tube was partially inserted into the hot zone, a siphon/pump system was employed to ensure that cooling water was always flowing. The copper tube was wrapped with zirconia insulation for thermal protection; a zirconia cap was mounted on the top of the quench assembly for further insulation. After the powder passed through the quench tube, it was collected in a gravity collection vessel. Any powder that remained entrained and left the collection vessel was trapped by a high efficiency particulate air (HEPA) filter. The filter was manufactured by General Electric H2O Process Technologies, had a 0.22 mm pore size, and was 0.14 m in diameter. After passing through the filter, gas was analyzed in an oxygen analyzer (Advanced Micro Instruments Model 201) and was subsequently vented. The Zirconia Cap Brass Weir Tube Inner Copper Tube Outer Copper Tube Zirconia Cloth Insulation Outer Cooling Zone 3711 oxygen analyzer had the ability to measure oxygen concentration over seven ranges, from 1000 ppm to 100%. A fluidized bed feeder was used to feed dispersed particles into the reactor. 2.2. Experimental plan to investigate reaction without oxygen The first experimental plan to study the kinetics of Mn2O3 dissociation was performed in an inert environment. This was done to determine if high conversion to MnO was feasible in the limited residence times in the reactor. Additionally, there are available rate laws for the dissociation without oxygen (Francis, 2008). The dissociation to MnO occurs in two reactions where Mn3O4 is an intermediary (Eqs. (5) and (6)). 3Mn2O3-2Mn3O4 + 1/2O2 (5) 2Mn3O4-6MnO+ O2 (6) Mn2O3 powder (Sigma-Aldrich, 99% purity), having a primary particle size of less than 44 mm was used to investigate the dissociation. Temperature and bulk gas flow rate were used as factors to study the kinetics of the dissociation. A central composite design (CCD) with the two factors (temperature and bulk gas flow rate) was performed where temperature was varied between 1673 and 1873 K and the bulk gas flow rate between 2.5 and 7.0 slpm. The lower temperature was selected because FACT thermodynamics software (Facility for the Analysis of Chemical Thermodynamics; McGill University) predicted significant conversion; the higher temperature was selected because it was approximately at the lower limit of the dissociation of popular two-step thermochemical cycles. If the reaction temperature for the dissociation of Mn2O3 needed to be much higher, then an alternative two-step water splitting cycle would likely become more desirable, due to inherent simplicity and potentially better process efficiency. The gas flow rate range was selected because initial calculations indicated that it would provide a desirable range of residence times ( 1–3 s). At the completion of an experimental run, the product powder was analyzed for oxygen content with a LECO Corporation TC600 oxygen determinator. X-ray diffraction (XRD) (Cu-k(a)) was used to identify product crystallinity, which helped to identify which forms of manganese oxide were in the product. By combining the results from the TC600 oxygen determinator and XRD, product conversion was calculated. See Appendix A for the calculation steps. In addition, a titration technique was used to determine the Mn + 2/Mn + 3 ratio. The titration technique supported conversions calculated from oxygen percent. 2.3. Experimental plan to study oxygen recombination effect An inherent challenge with the dissociation of metal oxides is the potential for recombination to occur in the quench step. In order to better understand the recombination effect in an AFR, two different experimental plans were executed. The first was a study of the recombination of MnO with O2. Eqs. (7) and (8) show the expected recombination reactions. Hot Water Out Cold Water In To Collection Vessel Fig. 2. Schematic of the quench tube assembly. 6MnO+ O2-2Mn3O4 (7) 2Mn3O4 + 1/2O2-3Mn2O3 (8) A 200 mesh, 99% MnO powder from Alfa Aesar was used to study the recombination. The same factors from the experimental plans in Section 2.2 (temperature, gas flow rate) along with the addition of oxygen, were used. A three-factor CCD was employed over ARTICLE IN PRESS T.M. Francis et al. / Chemical Engineering Science 65 (2010) 3709–3717 temperatures of 973–1573 K, bulk gas flow rates between 2.5 and 7.0 slpm and oxygen concentration between 0.25 and 1.0%. The temperature range was selected along with the oxygen concentration because these range combinations were predicted by FACT to give significant MnO oxidation at safe conditions in the reactor. Again, oxygen percents from the LECO TC600 oxygen determinator were used to calculate conversions. To add to the understanding of the recombination of MnO, an experimental plan of the dissociation of Mn2O3 was performed, with the addition of oxygen to the bulk gas flow. A 23 full-factorial design was executed with temperature, gas flow rate, and oxygen content as the factors. The ranges of temperature and gas flow rates were the same as those used in the experimental plan described in Section 2.2. The range for oxygen percentages (0.25–1.0%) was identical to that used during recombination studies. 3. Results and discussion 3.1. Mn2O3 to MnO forward reaction kinetics A typical oxygen generation curve of the dissociation reaction is presented in Fig. 3. The reactor temperature was 1873 K and had a bulk gas flow rate of 7.0 slpm. Several peaks may be seen in the center of the graph. These resulted from a small amount of vibration being applied to the feeder, which periodically increased the amount of powder fed to the reactor. Additionally, there were two discontinuities in the graph. The discontinuities occurred when the change in oxygen was so rapid that the O2 sensor range was not changed quickly enough. It is evident from Fig. 3 that the amount of oxygen in the system rapidly increased when manganese oxide was fed. All of the Mn2O3 powder from the experimental runs reacted completely through the first dissociation (Eq. (5)) and partially through the second dissociation (Eq. (6)). XRD data shown in Fig. 4 verified this, as the product spectrum showed the presence of both Mn3O4 and MnO spectra, with no evidence of peaks from the Mn2O3 spectrum. From the derivation in Appendix A, the conversion to MnO as a function of O% present in the sample is given as XMnO ¼ 2MWMnO A surface response analysis was done using the conversion as the response for the experimental plan. The p-values from this analysis, where a p-valueo0.05 corresponds to a significant factor, are displayed in Table 1. It is clear that the temperature and gas flow rate had a significant effect on the conversion over the response surface. A contour plot demonstrating the effect of gas flow rate and temperature on conversion is shown in Fig. 5. As expected, the conversion increased with an increase in temperature. Somewhat unexpectedly however, was that the conversion also increased with increased gas flow rate. An increase in gas flow rate inherently causes the residence time to AFR Product Relative Intensity 3712 MnO Standard Mn3O4 Standard Mn2O3 Standard 20 30 40 50 2θ 60 70 80 Fig. 4. XRD product spectra from the AFR. Table 1 Dissociation reaction response surface p-values. Parameter Constant Temperature Gas flow rate X 0.00 0.05 0.02 MWMn2 O3 ð0:23O%Þ 1 MWO2 1 MWO2 1þ þ 1þ ðO%0:27Þ 4 MWMn3 O4 4 MWMnO 60 70 7 ð9Þ Calculated conversions to MnO ranged between 50 and 74%, with the remainder of the product consisting of Mn3O4. Gas Flow (slpm) 0.35 0.3 0.25 Oxygen % 6 5 65 4 0.2 0.15 3 0.1 0.05 2 55 55 0 0 2 4 6 8 Time (min) 10 Fig. 3. O2 generation for Mn2O3 dissociation. 12 14 1650 1700 1750 1800 Temperature (K) 1850 Fig. 5. Contour plot of conversion to MnO. 1900 ARTICLE IN PRESS T.M. Francis et al. / Chemical Engineering Science 65 (2010) 3709–3717 decrease, which would typically lead to a decrease in conversion. However, a high gas flow rate could be limiting the recombination/reverse reaction, where an increased bulk gas flow in the system decreases the partial pressure of oxygen, which reduces the potential for any released oxygen to find a surface site to recombine with MnO to form Mn3O4 before leaving the system. In addition, the speed at which the powder flows through the quench tube is faster at higher flow rates, which in turn diminishes the possibility of a recombination occurring in the quench tube as well. In essence, there appeared to be a recombination reaction occurring and the effect of the recombination is presented further in the next section. 3713 Recombination conversion to Mn3O4 as a function of O% was derived in Appendix A and is XMn3 O4 ¼ 3 O%0:23 MWMnO MWMn3 O4 0:04 ð10Þ Calculations indicated the conversion of MnO to Mn3O4 ranged between 30 and 90%. Table 2 presents the p-value results of the surface response analysis. Unlike the dissociation, bulk gas flow rate did not have a significant effect on conversion. The factors that did have an effect on conversion were the concentration of oxygen and temperature. Fig. 8 is a contour plot of oxygen concentration versus T = 1270 O2 = 1.00% The manganese oxide dissociation results discussed in Section 3.1 appeared to show that the recombination reaction was present to an oftentimes significant degree. To investigate this, an experimental plan using MnO as the reactant was employed. The oxygen trace from a typical recombination reaction experimental run is shown in Fig. 6. If the additional oxygen was not interacting with the feed powder, a straight line would be expected. From Fig. 6, as soon as the feed powder was introduced into the reactor, the concentration of oxygen rapidly fell. After the onset of the reaction, the oxygen concentration began to rise again before two more distinct oxygen decreases. At the point of these decreases light vibration had been applied to the particle feeder to aid the flow of the powder. When the feed was shut-off, a rapid increase in the amount of oxygen was observed, until it leveled off at approximately the initial value. The observed decrease in oxygen passing through the reactor was certainly due to the recombination reaction occurring, as changes in the oxygen concentration profile coincided with feed powder entering the reactor. To confirm that the oxygen decrease seen in Fig. 6 was actually from a recombination reaction, XRD was performed on the samples that were collected from the gravity collection vessel. Product XRD spectra for different temperature oxygen concentration combinations are shown in Fig. 7. Comparing the product XRD spectra to the standardized manganese oxide spectra indicated that the product was a combination of Mn3O4 and MnO, without any Mn2O3. In addition, a trend of increasing temperature and oxygen concentration increased the amount of Mn3O4 in the sample. A surface response analysis was performed on the calculated conversions to investigate this trend statistically. T = 1270 O2 = 0.25% Relative Intensity 3.2. MnO to Mn3O4 recombination kinetics with oxygen T = 700 O2 = 0.25% MnO Standard Mn3O4 Standard Mn2O3 Standard 20 30 40 50 2Θ 60 70 80 Fig. 7. XRD product spectra from the recombination reaction. Table 2 Recombination reaction response surface p-values. Parameter Constant Temperature Gas flow rate Oxygen % X 0.00 0.01 0.73 0.03 80 1.2 80 60 1.0 0.8 0.3 O% 0.25 Oxygen % T = 700 O2 = 1.00% 0.6 0.2 0.4 0.15 40 0.1 70 0.2 0.05 0 2 4 6 8 Time (min) 10 Fig. 6. AFR O2 curve for recombination reaction. 12 14 50 30 0.0 0 800 900 50 1000 1100 1200 1300 1400 1500 1600 1700 Temperature (K) Fig. 8. Contour plot of recombination conversion to Mn3O4. ARTICLE IN PRESS 3714 T.M. Francis et al. / Chemical Engineering Science 65 (2010) 3709–3717 temperature with conversion as the response. The trend indicated that as oxygen concentration and temperature were increased the conversion of MnO to Mn3O4 increased. This result confirmed what was observed from the XRD product spectra in Fig. 7. It was particularly interesting that gas flow rate did not have an effect on the conversion. What this result indicated was that once the MnO was fed into the reactor, the recombination was occurring almost instantaneously, and was limited based on temperature and oxygen concentration. Additionally, the results of the recombination experimental plan show that during the quench of the full dissociation reaction, it is essential to maintain a low oxygen concentration while cooling the dissociation product as rapidly as possible to maximize the final Mn2O3 to MnO conversion. 3.3. Mn2O3 to MnO forward reaction kinetics with oxygen An experimental plan was executed to further understand the effect of oxygen and temperature on the dissociation of Mn2O3. Fig. 9 shows the oxygen generation curve normalized by the amount of mass fed for the dissociation of manganese oxide with and without oxygen added to the bulk gas (T¼ 1873 K, gas flow rate¼7.0 slpm). It was fairly obvious by looking at the comparison of the two curves, that the area under the curve without oxygen 0.2 With 1% Oxygen Oxygen % / Mass Fed 0.18 0.16 0.14 0.12 0.1 0.08 Without Oxygen 0.06 was larger than with oxygen. What this shows is that a higher conversion was attainable when oxygen was not present in the bulk gas flow. The reaction products are further compared in Fig. 10 using XRD spectra from each experimental condition. The XRD spectra shown in Fig. 10 confirmed what was observed from the oxygen generation curves. The main peak from the MnO spectrum is more intense in the product spectrum without oxygen in the bulk stream, suggesting a higher conversion. Conversions to MnO, calculated using Eq. (9), were between 9 and 48%. Table 3 presents the analyzed results of the factorial design. The significant factors from the factorial design for Mn2O3 dissociation with O2 in the bulk gas flow were the same as those from the recombination reaction. Both temperature and oxygen percent had a significant effect on conversion, while bulk gas flow rate did not. Although the statistical results indicated that gas flow rate did not have a significant effect on conversion, it showed an interesting trend. When the bulk oxygen percent was at 0.25%, the increase in gas flow rate caused an increase in conversion. However, when the bulk oxygen percent was increased to 1.00%, an increase in gas flow rate caused a decrease in conversion. This result is evident when looking at XRD spectra of the reaction products at 1673 K, as shown in Fig. 11. When comparing the XRD spectra at an oxygen concentration of 0.25%, it was evident that the main MnO peak (2y E411) at a gas flow (GF) of 2.5 slpm was not visible. This peak became visible when the bulk gas flow was increased to 7.0 slpm, which meant that the conversion increased with increasing gas flow rate. Conversely, when comparing the gas flow rates at a bulk O2 concentration of 1.00%, the Mn3O4 peak (2y E321) increased in intensity when the bulk flow rate was increased from 2.5 to 7.0 slpm, equating to a conversion decrease with a gas flow increase. Conversion calculations from experiments performed in the AFR agreed with these observations and are plotted on the design points in Fig. 12. 0.04 0.02 0 0 2 4 6 8 Time (min) 10 12 14 Fig. 9. O2 generation for Mn2O3 dissociation with and without O2 in the bulk gas flow. Table 3 Factorial design p-values for Mn2O3 dissociation with oxygen. Parameter Constant Temperature Gas flow rate Oxygen % X 0.00 0.00 0.56 0.05 GF = 7.0 O2 = 1.00% Product With O2 GF = 7.0 O2 = 0.00% Relative Intensity Relative Intensity GF = 7.0 O2 = 0.25% Product Without O2 MnO Standard GF = 2.5 O2 = 1.00% GF = 2.5 O2 = 0.25% GF = 2.5 O2 = 0.00% MnO Standard Mn3O4 Standard Mn3O4 Standard 20 20 30 40 50 2θ 60 70 Fig. 10. XRD product spectra of Mn2O3 dissociation with and without O2. 80 30 40 50 2Θ 60 70 80 Fig. 11. XRD product spectra of Mn2O3 dissociation at different bulk O2 percentages at 1673 K (GF represents total gas flow rate). ARTICLE IN PRESS T.M. Francis et al. / Chemical Engineering Science 65 (2010) 3709–3717 Fig. 12 confirms what was observed from XRD data. At low oxygen concentrations, increasing gas flow rate increased conversion. At higher oxygen concentrations, increasing gas flow rate decreased conversion. An increased conversion with increased gas flow rate is what was observed during experiments performed in an inert environment (Section 3.1). The results of the experiments predict that as long as the oxygen concentration is below 0.25%, the gas flow rate should have a significant effect on conversion. Additionally, it shows that conversion trends from two independent experimental plans were consistent. The other trend that may be taken from Fig. 12 is that when the reactor temperature was increased, the conversion increased; when the oxygen concentration increased, the conversion decreased. The trends are plotted in a contour plot in Fig. 13. An interesting trend was observed when comparing the contour plot in Fig. 13 with Fig. 8. In both contour plots, as the oxygen concentration approaches 1% and temperature approaches 30.9 9.73 47.9 21.3 26.0 Gas Flow (2.5-7 slpm) 3715 1700 K, both contours predicted a MnO fraction of approximately 20% and Mn3O4 fraction of approximately 80%. When comparing the results of the experimental plans of the recombination reaction with those from the forward reaction with a set O2 percent in the bulk gas flow, it was apparent that a limitation was being reached as the amount of oxygen was increased to 1%. From the statistical model, if a significant amount of oxygen is present in the system, the temperature must be increased significantly in order to garner high enough conversions for the Mn2O3 cycle to be feasible. Thus, it was concluded that for the cycle to be feasible in an AFR, Mn2O3 must be dissociated in an inert atmosphere. 3.4. Kinetic rate constant estimation from AFR data There are many factors in the rapid dissociation of manganese oxide that create difficulty in predicting the reaction kinetics of Mn2O3 dissociation from the AFR data. The first difficulty is the two step dissociation, with the second dissociation having a dual mechanism. It was previously hypothesized that an Avrami– Erofeev (Eq. (11)) nucleation and growth mechanism was occurring from the beginning of the dissociation to the end of the acceleration region of the second dissociation reaction (Francis, 2008). Because this is a thermal reduction and the Biot numbers are small, the Avrami–Erofeev mechanism predicts that the reaction can take place anywhere in the particle if there is sufficient energy. 39.9 11.9 RAvramiErofeev ¼ A1 eEa,1 =RT nð1XÞ½lnð1XÞðn1Þ=n 44.9 12.7 Oxygen (0.25-1%) Temperature (1673-1873 K) Fig. 12. Conversions from Mn2O3 dissociation with oxygen. Because the same reaction mechanism was occurring throughout the entirety of the Mn2O3 to Mn3O4 reaction, and the first portion of the Mn3O4 to MnO reaction, both of these mechanisms were grouped into a single mechanism when estimating a rate constant. The higher activation energy from the second dissociation Avrami–Erofeev mechanism was used in the calculation, to ensure a conservative estimate of the reaction rate constant. Therefore the pre-exponential rate constant was calculated (for conversions that were equal or less than 60%) using Eq. (12). 1.0 A¼ 20 ð11Þ ðlnð1XÞÞ1=n eEa=RT t ð12Þ The second issue that arose was that when the conversion exceeded 60%, the total degrees of freedom did not equal zero, as the mechanism shifted to an order of reaction mechanism: 0.9 0.8 RMn2 O3 ,2 ¼ A2 eEa,2 =RT ð1XÞn Oxygen % 0.7 0.6 40 0.5 To overcome this issue, the rate constant was calculated for all runs that had conversions equal to or less than 60%, which was approximately half of the experimental runs. For the runs that exceeded 60%, the time for the reaction to attain a conversion of 60% was calculated by rearranging Eq. (12). This time was then subtracted from the total residence time, to calculate the time in which the order of reaction mechanism controlled the reaction: t2 ¼ t 0.4 1700 1750 1800 Temperature (K) ðlnð0:4ÞÞ1=n eEa =RT A ð14Þ To then calculate the rate constant, the order of reaction mechanism was integrated from 0.6 to X, and subsequently divided by the residence time calculated from Eq. (14): 30 0.3 ð13Þ 1850 Fig. 13. Contour plot of dissociation conversion to MnO with oxygen. ð1XÞ1n ð0:4Þ1n Ea =RT e n1 A2 ¼ t2 ð15Þ ARTICLE IN PRESS 3716 T.M. Francis et al. / Chemical Engineering Science 65 (2010) 3709–3717 aspects of the industrial scale-up of the high temperature reduction step of metal oxide water-splitting cycles. Table 4 Predicted rate constants for Mn2O3 dissociation. TGA AFR A1 (s 1) A2 (s 1) 5.8 104 7 1.5 103 1.8 107 7 1.3 107 5.8 102 7 8.4 102 5.6 103 7 4.1 103 The kinetic rate parameters that were calculated in this section were based on the experimental plan that was performed in an inert environment as the oxygen was dilute enough to not significantly affect the reaction. Additionally, the reaction mechanisms were determined in an inert atmosphere. With that, approximately half of the runs yielded conversions of less than 0.6. The calculated rate constants are presented in Table 4. Values calculated from a TGA are included for comparison. The confidence intervals reported in Table 4 for the reaction kinetics parameters are rather large, but are typical for AFR analyses (Himmelblau, 1970; Galwey and Brown, 1999; Carney et al., 2006a, b; Perkins et al., 2008). Qualitatively, the reaction rates from the AFR were three orders of magnitude faster than from a TGA. This increase in reaction rate was believed to result from rapid oxygen diffusion away from dispersed particles in the AFR. The results indicate that an AFR is an excellent choice of equipment for the dissociation of manganese oxide. The increased reaction rate and high overall conversion are especially important for the feasibility of the sodium manganese thermochemical water-splitting cycle. 4. Conclusions Manganese oxide dissociation was investigated in an AFR, because of its importance as part of a thermochemical watersplitting cycle for the production of hydrogen using renewable means. The investigation showed that high Mn2O3 conversions are achievable with the use of an AFR. This is significant for implementation and scale up of a Mn2O3-based thermochemical cycle. Additionally, the results showed that by using an AFR, the reaction is rapid, with kinetic rate constants that are orders of magnitude greater than what was observed using a TGA. Overall conversions to MnO ranged between 50 and 74%, depending on the temperature and gas flow rate. The highest conversion was achieved with a combination of a high temperature and high bulk gas flow rate. It was clear that an increased temperature favored the forward reaction. Conversion remained high when high gas flow rates were used. This gave validity to the hypothesis that the propensity for the recombination reaction to occur could be effectively suppressed using high gas flow rates. The effect of oxygen on the dissociation was also investigated. When the oxygen concentration exceeded a critical point (0.25%), the recombination reaction apparently dominated the reaction and decreased the net conversion to MnO. The only way to obtain high conversions at oxygen contents exceeding 0.25% was to increase the temperature. Below the critical oxygen concentration, however, gas flow rate and temperature controlled the dissociation. What was especially interesting was that increasing gas flow rate, essentially decreasing residence time, increased conversion. This was again hypothesized to occur because of the faster quench step the reaction was undergoing, limiting the potential of the recombination reaction from occurring. Together, the results of these studies lead to the conclusion that high conversion was possible provided that the dissociation can be performed in an inert gas environment with a rapid quench. This work provides insight into the dissociation kinetics of Mn2O3, and Notation MWMn2 O3 MWMn3 O4 MWMnO MWO2 MWMn Msample NFe NFe,ex NKMnO4 NMn NMn3 þ NMn2 þ NMn2 O3 NMn3 O4 NMnO O% XMn3 O4 XMnO YMn3 O4 YO2 ,Mn3 O4 YMnO YO2 ,MnO molar mass of Mn2O3, g/mole molar mass of Mn3O4, g/mole molar mass of MnO, g/mole molar mass of O2, g/mole molar mass of O2, g/mole sample mass, g number of Fe2 + moles, moles number of excess Fe2 + moles, moles number of KMnO4 moles, moles moles of Mn, moles moles of Mn3 + , moles moles of Mn2 + , moles moles of Mn2O3, moles moles of Mn3O4, moles moles of MnO, moles oxygen percent from LECO TC-600, unitless conversion to Mn3O4, unitless conversion to MnO, unitless mass fraction of Mn3O4, unitless mass fraction of O2 from Mn3O4 transition, unitless mass fraction of MnO, unitless mass fraction of O2 from MnO transition, unitless Acknowledgments The authors wish to thank the United States Department of Energy (DOE) Grant number DE-F636-036013062 and the UNLV foundation for funding this project. Additionally, the authors would like to thank N. Brandon Hughes for the titration work. One of the authors wishes to acknowledge the GEM Consortium Fellowship program, National Science Foundation AGEP Fellowship program, and the United States Department of Education GAANN program for providing partial research scholarships. Appendix A A.1. Determining forward conversion with oxygen analysis In calculating the conversion of Mn2O3 to MnO, it was assumed that Mn2O3 did not exist in the sample. XRD patterns supported this assumption as there were only peaks attributed to the presence of Mn3O4 and MnO. A mass balance shows that when starting with 100% Mn2O3 there can only be Mn3O4, MnO, and oxygen released from the two reactions. Dividing by the initial mass of Mn2O3 the mass balance is shown in Eq. (A.1): 1 ¼ YMn3 O4 þ YO2 ,Mn3 O4 þ YMnO þYO2 ,MnO ðA:1Þ Oxygen percent, found using a LECO corp. TC-600 Oxygen Determinator, was put in terms of mass fraction of Mn3O4 and MnO (Eq. (A.2)). O% ¼ 0:27YMn3 O4 þ 0:23YMnO YMn3 O4 þ YMnO ðA:2Þ Oxygen fractions of Mn3O4 and MnO were 0.27 and 0.23, respectively. To find the mass fraction of O2 released during the transition to Mn3O4, an overall mole balance of the dissociation ARTICLE IN PRESS T.M. Francis et al. / Chemical Engineering Science 65 (2010) 3709–3717 Rearranging Eq. (A.12) leads to the mass fraction of Mn3O4 in the product: was performed: 6Mn2O3-4Mn3O4 + O2-12MnO+ 2O2 (A.3) YMn3 O4 ¼ From Eq. (A.3), for every four moles of Mn3O4 there will be one mole of O2 contributing to this fraction; for every twelve moles of MnO there will be on further mole of O2. Putting the mole balance in terms of the contributions from Mn3O4 and MnO, the mass fractions yields YO2 ,Mn3 O4 YMn3 O4 MWO2 YMnO MWO2 ¼ þ 4 MWMn3 O4 12 MWMnO ðA:4Þ A similar balance for the mass fraction of O2 released from the transition to MnO yields YO2 ,MnO ¼ YMnO MWO2 6 MWMnO ðA:5Þ Placing Eqs. (A.4) and (A.5) into Eq. (A.1) and simplifying, yields Eq. (A.6): 1 MWO2 1 MWO2 þ YMnO 1 þ ðA:6Þ 1 ¼ YMn3 O4 1 þ 4 MWMn3 O4 4 MWMnO Eqs. (A.2) and (A.6) provided two equations with two unknowns resulting in zero degrees of freedom. Solving Eq. (A.2) for YMn3 O4 and substituting into Eq. (A.6) yields YMnO ð0:23O%Þ 1 MWO2 1 MWO2 þ YMnO 1 þ 1þ 1¼ ðO%0:27Þ 4 MWMn3 O4 4 MWMnO ðA:7Þ Solving for YMnO gave Eq. (A.8): YMnO ¼ 1 ð0:23O%Þ 1 MWO2 1 MWO2 þ 1þ 1þ ðO%0:27Þ 4 MWMn3 O4 4 MWMnO ðA:8Þ Assuming 1 mole of Mn2O3 was reacted, conversion to MnO may be defined as XMnO ¼ YMnO MWMn2 O3 2 MWMnO ðA:9Þ Substituting Eq. (A.8) into Eq. (A.9) gave conversion to MnO as a function of O%: XMnO ¼ MWMn2 O3 ð0:23O%Þ 1 MWO2 1 MWO2 1þ 2MWMnO þ 1þ ðO%0:27Þ 4 MWMn3 O4 4 MWMnO ðA:10Þ A.2. Determining recombination conversion with oxygen analysis To calculate the conversion for the recombination of MnO to Mn3O4, only MnO and Mn3O4 need to be included, as verified by XRD. Eq. (A.2) was used again to relate the mass fractions of MnO and Mn3O4 to the amount of oxygen in the sample. The mass balance this time consists of MnO and Mn3O4: 1 ¼ YMn3 O4 þYMnO ðA:11Þ Eq. (A.11) was rearranged and substituted into Eq. (A.2): O% ¼ 0:27YMn3 O4 0:23YMn3 O4 0:23 3717 ðA:12Þ O%0:23 0:04 ðA:13Þ Assuming 1 mole of MnO was reacted conversion to Mn3O4 may be defined as XMn3 O4 ¼ 3YMn3 O4 MWMnO MWMn3 O4 ðA:14Þ Substituting Eq. (A.13) into Eq. (A.14) gave conversion for the recombination from MnO to Mn3O4 as a function of O%. XMn3 O4 ¼ 3 O%0:23 MWMnO MWMn3 O4 0:04 ðA:15Þ References Carney, C.S., Gump, C.J., et al., 2006a. Rapid nickel oxalate thermal decomposition for producing fine porous nickel metal powders part 2: global kinetics. Materials Science and Engineering a-Structural Materials Properties Microstructure and Processing 431 (1–2), 13–25. Carney, C.S., Gump, C.J., et al., 2006b. Rapid nickel oxalate thermal decomposition for producing fine porous nickel metal powders part 1: synthesis. Materials Science and Engineering a-Structural Materials Properties Microstructure and Processing 431 (1–2), 1–12. Dahl, J.K., Buechler, K.J., et al., 2004a. Rapid solar-thermal dissociation of natural gas in an aerosol flow reactor. Energy 29 (5–6), 715–725. Dahl, J.K., Buechler, K.J., et al., 2004b. Solar-thermal dissociation of methane in a fluid-wall aerosol flow reactor. International Journal of Hydrogen Energy 29 (7), 725–736. Evans, G., Houf, W., et al., 1987. Gas-particle flow within a high-temperature solar cavity receiver including radiation heat-transfer. Journal of Solar Energy Engineering-Transactions of the ASME 109 (2), 134–142. Francis, T.M., 2008. Dissociation of Manganese(III) Oxide as part of a thermochemical water splitting cycle. Chemical and Biological Engineering. Boulder, University of Colorado, 250. Galwey, A.K., Brown, M.E., 1999. Thermal Decomposition of Ionic Solids. Elsevier, Amsterdam, New York. Ganz, J., Steiner, E., et al. 1998. Powder cloud reactors. An attractive concept to run solar high-temperature reactions. In: 9th SolarPaces International Symposium on Solar Thermal Concentrating Technologies, EDP Sciences, Font-Romeu, France. Himmelblau, D.M., 1970. Process Analysis by Statistical Methods. Wiley, New York. Hruby, J.M., Steele, B.R., et al. 1984. Solid particle receiver experiments: radiant heat test. S.N. Laboratories. Matsushi, T., Thoburn, W.J., 1965. Application of differential thermal analysis to dissociation of MnO2 and Mn2O3. Canadian Journal of Chemistry 43 (6), 1723–1728. Nuesch, P., 1998. Wasserstoff asu Soonenenergie-Ein neuer Thermochmisher Zyklus auf der Basis von Manganoxiden, Universitat Zurich. Otto, E.M., 1964. Equilibrium pressures of oxygen over Mn2O3–Mn3O4 at various temperatures. Journal of the Electrochemical Society 111 (1), 88–92. Perkins, C., Lichty, P.R., et al., 2008. Thermal ZnO dissociation in a rapid aerosol reactor as part of a solar hydrogen production cycle. International Journal of Hydrogen Energy 33 (2), 499–510. Perkins, C., Weimer, A.W., 2004. Likely near-term solar-thermal water splitting technologies. International Journal of Hydrogen Energy 29 (15), 1587–1599. Sturzenegger, M., Ganz, J., et al., 1999. Solar hydrogen from a manganese oxide based thermochemical cycle. Journal De Physique Iv 9 (P3), 331–335. Sturzenegger, M., Nuesch, P., 1999. Efficiency analysis for a manganese-oxidebased thermochemical cycle. Energy 24 (11), 959–970. Weimer, A.W., Cochran, G.A., et al., 1994. Rapid process for manufacturing aluminum nitride powder. Journal of the American Ceramic Society 77 (1), 3–18. Weimer, A.W., Roach, R.P., et al., 1991. Rapid carbothermal reduction of boronoxide in a graphite transport reactor. AICHE Journal 37 (5), 759–768.