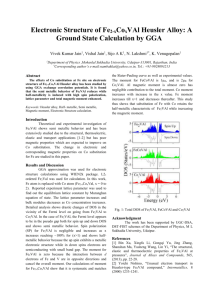
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/350567489 Spintronic properties of the Ti 2 CoB Heusler compound by density functional theory Article in Solid State Communications · January 2011 DOI: 10.1016/j.ssc.2011.05.00 CITATION READS 1 60 3 authors, including: Selçuk Kervan Nazmiye Kervan Gazi University, Polatlı Science and Arts Faculty Eskişehir Technical University 116 PUBLICATIONS 904 CITATIONS 40 PUBLICATIONS 15 CITATIONS SEE PROFILE All content following this page was uploaded by Nazmiye Kervan on 01 April 2021. The user has requested enhancement of the downloaded file. SEE PROFILE Solid State Communications 151 (2011) 1162–1164 Contents lists available at ScienceDirect Solid State Communications journal homepage: www.elsevier.com/locate/ssc Spintronic properties of the Ti2 CoB Heusler compound by density functional theory Selçuk Kervan ∗ , Nazmiye Kervan Nevşehir University, Faculty of Arts and Sciences, Physics Department, 50300, Nevşehir, Turkey article info Article history: Received 4 April 2011 Received in revised form 29 April 2011 Accepted 10 May 2011 by F. Peeters Available online 17 May 2011 Keywords: A. Magnetically ordered materials D. Electronic band structure abstract The electronic structure and magnetic properties of the Ti2 CoB Heusler compound with a high-ordered CuHg2 Ti structure were investigated using the self-consistent full potential linearized augmented plane wave (FPLAPW) method within the density functional theory (DFT). Spin-polarized calculations show that the Ti2 CoB compound is half-metallic ferromagnetic with a magnetic moment of 2 µB at the equilibrium lattice constant, a = 5.74 Å. The Ti2 CoB Heusler compound is ferromagnetic below the equilibrium lattice constant and ferrimagnetic above the equilibrium lattice constant. A large peak in majority-spin DOS and an energy gap in minority-spin DOS are observed at the Fermi level, yielding a spin polarization of 100%. A spin polarization higher than 90% is achieved for a wide range of lattice constants between 5.6 and 6.0 Å. © 2011 Elsevier Ltd. All rights reserved. 1. Introduction 2. Computational method Half-metallic (HM) materials, which have a complete spin polarization at the Fermi level, attract increasing interest for their potential spintronic device applications such as nonvolatile magnetic random access memories (MRAM) and magnetic sensors [1–4]. Half-metallic properties have been observed in many materials, for example Heusler compounds [5–10], ferromagnetic metallic oxides [11–13], dilute magnetic semiconductors [14,15] and zincblende transition-metal pnictides and chalcogenides [16–20]. The Heusler compounds are ternary X2 YZ intermetallics, where X and Y are transition metals and Z is a main group element [21]. The X2 YZ Heusler compounds crystallize in the cubic AlCu2 Mn-type structure with the space group Fm3m. In this structure, X, Y and Z atoms are placed on the Wyckoff positions 8c (1/4, 1/4, 1/4), 4a (0, 0, 0) and 4b (1/2, 1/2, 1/2), respectively. If the number of 3d electrons of Y atom is more than that of X atom, CuHg2 Ti-type structure with the space group F 43m is observed. In this structure, X atoms occupy the nonequivalent 4a (0, 0, 0) and 4c (1/4, 1/4, 1/4) positions, while Y and Z atoms are located on 4b (1/2, 1/2, 1/2) and 4d (3/4, 3/4, 3/4) positions, respectively [7]. Although many Heusler compounds have been theoretically predicted to be half-metallic [5–10,22–26], the electronic structure calculations of the Ti2 -based Heusler compounds have not been widely studied up to now. In the present paper, the electronic structure and magnetism of the Ti2 CoB Heusler compound with CuHg2 Ti-type structure are studied by means of the self-consistent full potential linearized augmented plane wave (FPLAPW) method. Electronic structure calculations were performed using the self-consistent full potential linearized augmented plane wave (FPLAPW) method [27] implemented in WIEN2K code [28] within the density functional theory (DFT). The Perdew–Burke–Ernzerhof generalized gradient approximation (GGA) [29,30] was used for the exchange correlation correction. In this method, the space is divided into non-overlapping muffin-tin (MT) spheres separated by an interstitial region. The basis functions are expanded into spherical harmonic functions inside the muffin-tin sphere and Fourier series in the interstitial region. The muffin-tin sphere radii were 2.15 a.u. for Ti and Co, 2.0 a.u. for B. The convergence of the basis set was controlled by a cut off parameter Rmt Kmax = 7, where Rmt is the smallest of the MT sphere radii and Kmax is the largest reciprocal lattice vector used in the plane wave expansion. The magnitude of the largest vector in charge density Fourier expansion (Gmax ) was 12. The cutoff energy, which defines the separation of valence and core states, was chosen as −6 Ry. We select the charge convergence as 0.0001e during self-consistency cycles. In these calculations, we neglected the effect of spin–orbit coupling. For the Brillouin zone (BZ) integration, the tetrahedron method [28] with 72 special k points in the irreducible wedge (2000 k-points in the full BZ) was used to construct the charge density in each self-consistency step. 3. Results and discussion ∗ Corresponding author. Tel.: +90 384 215 3900; fax: +90 384 215 3948. E-mail address: selcuk.kervan@nevsehir.edu.tr (S. Kervan). 0038-1098/$ – see front matter © 2011 Elsevier Ltd. All rights reserved. doi:10.1016/j.ssc.2011.05.008 The calculated total energy as a function of lattice constant for both ferromagnetic and non-magnetic configurations is plotted S. Kervan, N. Kervan / Solid State Communications 151 (2011) 1162–1164 Fig. 1. The calculated total energy as a function of lattice constant for the Ti2 CoB Heusler compound. Fig. 2. The spin-polarized total densities of states (DOS) and atom-projected DOS. Fig. 3. The band structures of the Ti2 CoB compound for the spin-up and spin-down electrons. 1163 Fig. 4. The dependence of the half-metallic state on the lattice constant. in Fig. 1 for the CuHg2 Ti-type structure. It can be seen that the ferromagnetic state is more stable. The equilibrium lattice constant is 5.74 Å and there is no experimental or theoretical lattice constant value to compare with our value. The energy difference between the ferromagnetic and non-magnetic states increases slightly with increasing lattice constant. Fig. 2 presents the spin-polarized total densities of states (DOS) and atom-projected DOS of Ti2 CoB at its equilibrium lattice constant. The calculated total magnetic moment of Ti2 CoB compound is 2 µB , while the atomic magnetic moments are 0.981 µB for Ti (1), 0.575 µB for Ti (2), 0.004 µB for Co and −0.023 µB for B. It is clear that the majority-spin band is metallic, while the minority-spin band shows a semiconducting gap around the Fermi level. In minority-spin band, the valance band maximum is located at −0.59 eV and the conduction band minimum is at 0.05 eV. The energy gap for spin-down electrons at around the Fermi level is 0.64 eV. This energy gap in the minority-spin band gap leads to 100% spin polarization at the Fermi level, resulting in the halfmetallic behavior at equilibrium state. In the spin-down band, the total density of states around the Fermi level are predominantly due to Co-d, Ti(1)-d and Ti(2)-d electrons. The projected density of states of Co atom lies mainly below the Fermi level and has the main contribution to the total DOS. The spin-down densities of states of Ti(1) and Ti(2) atoms lie mainly above the Fermi level. The energy region between −9 and −7 eV consists mainly of s electrons of B atoms. The band structures of the Ti2 CoB compound for the spin-up and spin-down electrons is plotted in Fig. 3. It is obviously seen that the spin-up band structure has metallic intersections at the Fermi level which is a sign of metallic nature. However, the spindown band structure has energy gap near the Fermi level. The width of the energy gap can be calculated using the energies of the highest occupied band at the Γ point and the lowest unoccupied band at the L point. The Fermi level lies 0.59 eV above the highest spin-down valance band. Therefore, spin-flip gap, which is the minimum energy required to flip a minority-spin electron from the valance band maximum to the majority-spin Fermi level, is of 0.59 eV. The non-zero spin-flip gap indicates that the Ti2 CoB compound is a true half-metallic ferromagnet (HMF). This spinflip gap is close to the spin-flip gap of the Co2 MnP compound [25]. In order to investigate the dependence of the half-metallic state on the lattice constant, the electronic structure calculations were performed for the lattice parameters between 5.5 and 6.0 Å. It is clearly seen in Fig. 4 that the Ti2 CoB Heusler compound has half-metallic nature above the lattice constant value of 5.65 Å. Therefore, the lattice expansion does not change half-metallic behavior of the Ti2 CoB compound. 1164 S. Kervan, N. Kervan / Solid State Communications 151 (2011) 1162–1164 4. Conclusions Using the first-principles full potential linearized augmented plane waves (FPLAPW) method, the spintronic and magnetic properties have been calculated for the Ti2 CoB Heusler compound. The spin-polarized calculations show that the Ti2 CoB Heusler compound is a half-metallic ferromagnet with a magnetic moment of 2 µB at the equilibrium lattice constant. The Ti2 CoB Heusler compound has half-metallic character above the lattice constant value of 5.65 Å. References Fig. 5. The calculated total magnetic moment, the magnetic moments of the Ti(1), Ti(2) and Co atoms and the spin polarization as a function of lattice constant. The calculated total magnetic moment, the magnetic moments of the Ti(1), Ti(2) and Co atoms and the spin polarization as a function of lattice constant is given in Fig. 5. The calculated total magnetic moment is 2 µB above the lattice constant value of 5.55 Å. The calculated magnetic moments of the Ti(1) and Ti(2) atoms increase with increasing lattice constant, while the magnetic moment of the Co atom decreases. The Ti2 CoB Heusler compound is ferromagnetic up to the lattice constant value of 5.75 Å before becoming ferrimagnetic. The Ti2 CoB Heusler compound maintains the half-metallic character having 100% polarization above the lattice constant value of 5.65 Å. [1] R.A. de Groot, F.M. Mueller, P.G. van Engen, K.H.J. Buschow, Phys. Rev. Lett. 20 (1983) 2024. [2] R.A. de Groot, K.H.J. Buschow, J. Magn. Magn. Mater. 54-57 (1986) 1377. [3] S.A. Wolf, D.D. Awschalom, R.A. Buhrman, J.M. Daughton, S. von Molnar, M.L. Roukes, A.Y. Chtchelkanova, D.M. Treger, Science 294 (2001) 1488. [4] I. Zutic, J. Fabian, S. Das Sarma, Rev. Modern Phys. 76 (2004) 323. [5] X.-Q. Chen, R. Podloucky, P. Rogl, J. Appl. Phys. 100 (2006) 113901. [6] K. Özdoğan, I. Galanakis, E. Şasıoğlu, B. Aktaş, Solid State Commun. 142 (2007) 492. [7] H.C. Kandpal, G.H. Fecher, C. Felser, J. Phys. D: Appl. Phys. 40 (2007) 1507. [8] G.D. Liu, X.F. Dai, H.Y. Lui, J.L. Chen, Y.X. Li, G. Xiao, G.H. Wu, Phys. Rev. B 77 (2008) 14424. [9] K. Özdoğan, I. Galanakis, J. Magn. Magn. Mater. 321 (2009) L34. [10] V. Sharma, A.K. Solanki, A. Kashyap, J. Magn. Magn. Mater. 322 (2010) 2922. [11] Z. Szotek, W.M. Temmerman, A. Svane, L. Petit, G.M. Stocks, H. Winter, J. Magn. Magn. Mater. 272–276 (2004) 1816. [12] W. Song, J. Wang, Z. Wu, Chem. Phys. Lett. 482 (2009) 246. [13] S. Lv, H. Li, D. Han, Z. Wu, X. Liu, J. Meng, J. Magn. Magn. Mater. 323 (2011) 416. [14] Y. Zhang, W. Liu, H. Niu, Solid State Commun. 145 (2008) 590. [15] Y. Saeed, S. Nazir, A. Shaukat, A.H. Reshak, J. Magn. Magn. Mater. 322 (2011) 3214. [16] I. Galanakis, P. Mavropoulos, Phys. Rev. B 67 (2003) 104417. [17] Y.-Q. Xu, B.-G. Liu, D.G. Pettifor, Physica B 329–333 (2003) 1117. [18] K.L. Yao, G.Y. Gao, Z.L. Liu, L. Zhu, Solid State Commun. 133 (2005) 301. [19] K.L. Yao, G.Y. Gao, Z.L. Liu, L. Zhu, Y.L. Li, Physica B 366 (2005) 62. [20] X.-F. Ge, Y.-M. Zhang, J. Magn. Magn. Mater. 321 (2009) 198. [21] Fr. Heusler, Verh. Deutsch. Phys. Ges. 5 (1903) 219. [22] N. Xing, H. Li, J. Dong, R. Long, C. Zhang, Comput. Mater. Sci. 42 (2008) 600. [23] H. Luo, Z. Zhu, G. Liu, S. Xu, G. Wu, H. Liu, J. Qu, Y. Li, J. Magn. Magn. Mater. 320 (2008) 421. [24] N. Xing, Y. Gong, W. Zhang, J. Dong, H. Li, Comput. Mater. Sci. 45 (2009) 489. [25] Z. Yao, S. Gong, J. Fu, Y.-S. Zhang, K.-L. Yao, Solid State Commun. 150 (2010) 2239. [26] U. Kanbur, G. Gökoğlu, J. Magn. Magn. Mater. 323 (2011) 1156. [27] D. Singh, Planes Waves, Pseudo-Potentials and the LAPW Method, Kluwer Academic Publishers, Boston, Dortrecht, London, 1994. [28] P. Blaha, K. Schwarz, G.K.H. Madsen, D. Hvasnicka, J. Luitz, WIEN2k, An augmented plane wave + local orbitals program for calculating crystal properties, Karlheinz Schwarz, Techn. Universit Wien, Austria, 2001, ISBN: 3-9501031-1-2. [29] J.P. Perdew, K. Burke, Y. Wang, Phys. Rev. B 54 (1996) 16533. [30] J.P. Perdew, S. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865.