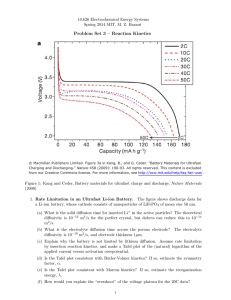
Butler-Volmer Equation in Electrochemistry: Origins & Applications
advertisement

Journal of Electroanalytical Chemistry 872 (2020) 114145 Contents lists available at ScienceDirect Journal of Electroanalytical Chemistry journal homepage: www.elsevier.com/locate/jelechem The Butler-Volmer equation in electrochemical theory: Origins, value, and practical application ⁎ Edmund J.F. Dickinson , Andrew J. Wain National Physical Laboratory, Hampton Road, Teddington TW11 0LW, United Kingdom A R T I C L E I N F O Article history: Received 18 December 2019 Received in revised form 6 April 2020 Accepted 8 April 2020 Available online 17 April 2020 Keywords: Butler-Volmer equation Electrode kinetics Overpotential Overvoltage Exchange current density Transfer coefficient Heterogeneous rate constant Electron transfer Cyclic voltammetry Lithium-ion battery Proton exchange membrane fuel cell Polymer electrolyte membrane fuel cell Proton exchange membrane water electrolyser Polymer electrolyte membrane water electrolyser PEMFC PEMWE Solid oxide fuel cell Solid oxide water electrolyser SOFC Redox flow battery A B S T R A C T We review the use of the Butler-Volmer equation to describe electrode kinetics. Across all fields of electrochemistry, this equation is deployed in diverse mathematical forms whose mutual interrelation is infrequently expressed and often unclear. The plurality of forms of the Butler-Volmer equation sometimes engenders misunderstanding in the literature, such as misuse of terminology or inappropriate mathematical expression of the kinetic equations. We express the basis of the Butler-Volmer equation as an empirical expression for a two-way activated redox process, and illustrate the constraints that limit interpretation of this empirical expression in a more physically specific manner, especially excluding many electrocatalytic processes not equating to a simple one-electron transfer. Preferences towards different parametric expressions of the Butler-Volmer equation between the fields of electroanalytical chemistry and electrochemical engineering are correlated to both the different types of electrochemical system commonly encountered in these fields, and their different scientific motivations. We further discuss briefly the scope of recent experimental efforts towards affirming the Butler-Volmer equation, considering the sensitivity (or relative lack thereof) of common experimental techniques to its component parameters. © 2020 Elsevier B.V. All rights reserved. 1. Introduction “I suppose you know you're doing that all wrong.” – man leaning nonchalantly over a wall, in a posthumously published sketch by “Pont” (Graham Laidler), Punch, 4th December 1940 The Butler-Volmer equation is widely used in electrochemical theory to describe the relation between electrode potential (vs. a suitable reference electrode) and current density. In spite of its ubiquity, however, the Butler-Volmer equation is quoted by different authors in a wide variety of ⁎ Corresponding author. E-mail address: edmund.dickinson@npl.co.uk. (E.J.F. Dickinson). http://dx.doi.org/10.1016/j.jelechem.2020.114145 1572-6657/© 2020 Elsevier B.V. All rights reserved. formulations, without uniformity in the precise meaning of variables and coefficients such as overpotential, exchange current density, transfer coefficient, etc. There is particular disparity between the use of the Butler-Volmer equation in electroanalytical chemistry (voltammetry and sensing) and its use in electrochemical energy conversion and storage science (batteries, fuel cells and electrolysers), reflecting not only the differing ideality of electrochemical systems encountered experimentally in the two disciplines, but also perhaps a lack of communication of standards or recognition of a unified kinetic theory. Confusion may arise because the Butler-Volmer equation is essentially an empirical relation, albeit one inspired by fundamental arguments from the transition state theory of electron transfer. Its constituent coefficients E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 can only be determined through electrochemical experiment rather than being available from independent predictions, and as recently noted by Batchelor-McAuley et al., “the Butler–Volmer equation is periodically damned due to being ‘phenomenological’ in its description of electron transfer, that is, it has no direct physical meaning” [1]. Rigorously derived formulations given in standard electrochemical textbooks [2–4] often appear to be misapplied in the literature – perhaps innocently. This is especially the case with respect to multistep electrochemical processes, such as the electrocatalytic reactions of hydrogen or oxygen. We also note that justifications for the Butler-Volmer equation from quantum theory can sometimes be pursued beyond the validity of their founding assumptions; meanwhile, support for the Butler-Volmer equation as an empirical expression is overlooked as a valid justification for its use in prediction and parameterisation of electrochemical systems. In this article, we aim to provide a consistent “user's guide” to the Butler-Volmer equation by linking its theoretical and practical basis to relevant instructions for its application. In support of this, we take a crosssectional sample of the application of the Butler-Volmer equation in different fields, considering electrochemical contexts where the Butler-Volmer equation provides a clear and measurable expression of electrochemical kinetics, or where, by contrast, it is inappropriate. drawn when a working electrode is at an equilibrium potential with respect to the solution chemistry at the electrode surface, such that the anodic and cathodic current densities are equal and opposite, and the total current is zero. Deviation from this equilibrium potential due to electrode polarisation is described by the overpotential η (V), which is the difference between the instantaneous electrode-electrolyte potential difference and its equilibrium value as given by the Nernst equation. The exact potential dependence is controlled by unitless transfer coefficients for both the anodic and cathodic current, which we denote here as αa and αc without at this stage constraining the values that these coefficients may take. The engineer's Butler-Volmer equation, as written, is compatible with multi-electron processes. We stress that according to contemporary recommendations [8,9], and in contrast to historic practice found in older textbooks [2,10,11], the values of the transfer coefficient should not be scaled by the value of “n” (number of electrons transferred). 1.1. Conventions of nomenclature The “practical” Butler-Volmer equation (Eq. (1.4)) is reported in a number of common reference works [3,12]. Its derivation from the engineer's Butler-Volmer equation was presented clearly by White et al. [13] It defines both exchange current density and overpotential as measured at chosen reference conditions, which need not equate to the local and instantaneous chemical environment of the electrode. Consequently, both exchange current density and overpotential with subscript “ref” are well-defined concentration-independent quantities, rather than possibly unknown functions of local chemistry. Here, ga and gc are functions of the electrode surface concentrations ci of all chemical species, measured with respect to their concentrations ci,ref under the given reference conditions, at which we define ga = gc = 1. This form retains aspects of the engineer's form in giving priority to measurable, quasi-empirical quantities such as exchange current density and overpotential, while making the concentration dependence conceptually obvious in the written form of the equation, and clearly stating a reference condition in terms of a relevant and experimentally accessible composition of the reference solution. Importantly, the reference condition may be far from standard conditions (1 bar, 273.15 K). As for the engineer's form, the practical equation is valid as written for multi-electron processes, provided transfer coefficients are defined correctly, as stated above [8,9]. 1.1.2. “Practical” Butler-Volmer equation i ¼ i0;ref ð expðαa f ηref Þ ga − expð−αc f ηref Þ gc Þ To initiate the discussion and provide some nomenclature for classifying different approaches, we begin by presenting three “textbook” formulations of the Butler-Volmer equation. We will later discuss the derivation of and relationship between the different forms, and hence illustrate the meaning and dependencies of the different possible coefficients with which the Butler-Volmer equation can be parameterised. Notation is summarised in the Appendix. Throughout, we consider a general redox process at a single electrode: e− ⇄ X vi i ð1:1Þ i so that species i with vi < 0 are species on the oxidised side of the half-cell reaction, acting as reactants with respect to reduction. It is widely accepted that, with rare exceptions, the Butler-Volmer equation is inadmissible for processes combining more than one electron transfer [2,4–6]. The only physically relevant exception would a concerted two-electron transfer; the plausibility of this situation for various redox systems has been discussed elsewhere [6,7]. We shall make a few remarks in this section to address the issue of multi-electron processes, and revisit this point again after further analysis under the section “Multistep processes: a comment”. For certain cases where we explicitly restrict our attention to unimolecular one-electron processes, we denote the oxidised and reduced forms of the redox couple as O and R respectively, so: − O þ e ⇄R 1.1.3. “Electroanalytical” Butler-Volmer equation i ¼ k 0 F exp αa f E−E0f cR − exp −αc f E−E0f cO ð1:2Þ ð1:5Þ The “electroanalytical” Butler-Volmer equation (Eq. (1.5)) is prominent in texts focused on analytical methods, especially techniques such as voltammetry [3,7]. This formulation expresses the current density as proportional to a heterogeneous rate constant k0 (m s−1) for the rate of electron transfer. The current density also depends explicitly upon the electrode polarisation E (V) against a fixed reference electrode, as well as the concentrations of a strictly unimolecular redox couple O and R (Eq. (1.2)), which is understood to have a formal potential E0f . If this equation is applied for the case of a unimolecular process with concerted multi-electron transfer, the total current density should be scaled by n, whereas the transfer coefficients as defined should not be altered [8,9]. and vO = −1, vR = +1. We now coin names for the three common forms of the Butler-Volmer equation. The notation f ≡ F/RT will be used throughout for brevity. 1.1.1. “Engineer's” Butler-Volmer equation i ¼ expðαa fηÞ− expð−αc fηÞ i0 ð1:4Þ ð1:3Þ The “engineer's” Butler-Volmer equation (Eq. (1.3)) emphasises the measurable, empirical quality of the equation, and is found in textbooks focusing on applied electrochemical science and engineering, such as Newman [4]. This equation defines the ratio between the instantaneous current density i and the exchange current density i0 (A m−2), where the latter is the magnitude of both the anodic and cathodic current densities 1.1.4. Comment on equivalence of equations All the forms of the Butler-Volmer equation share in common the expression of the current density as a sum of anodic and cathodic current density terms, each of which has an exponential dependence upon a measure of electrode polarisation, weighted by unitless transfer coefficients. All of the 2 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 solvent molecules). Gurney's “transfer coefficient” is then seen as a relative scaling between the vibrational strength of the two oxidation states of the redox-active species. These results accounted for the empirical Tafel slope from quantum mechanical first principles, including observed temperature dependence. Butler applied this work to the hydrogen oxidation reaction [16], considering adsorption of H(ads) from H2 to be at pre-equilibrium (this would today be considered a special case of the Tafel-Volmer mechanism! [21]).2 In the resulting kinetic expression, Butler considered both anodic and cathodic processes to be proceeding simultaneously, as at equilibrium. Hence we see the sum of two exponential terms, one for each direction of current density, in which the empirical transfer coefficient is present. Butler immediately recognised the possibility to linearise the equation about the equilibrium conditions, and linked this to experimental observation. above forms of the Butler-Volmer equation can be made equivalent to each other. Enforcing equivalence of the equations implies some relations between the parameters employed in each form, as will be discussed further in “Forms of the Butler-Volmer equation” below. 1.2. Historical context Consulting the primary literature may cause the interested electrochemist to question why the names Butler and Volmer are associated with the above equation(s), to the exclusion of some others. Citations given in modern textbooks typically isolate three works by Butler (two from 1924, one from 1932) [14–16] and one by Erdey-Grúz and Volmer from 1930 [17]; history has tended to excise the name Erdey-Grúz from the equation as the junior partner on the paper.1 A deeper historical discussion has been provided by Inzelt [18]. Butler's 1924 works [14,15] did not attempt to construct a theory of the rates of heterogeneous electron transfer processes, and as such did not directly engender his namesake equation. These works instead presented a “kinetic” (dynamical) theory of how the equilibrium potential of the Nernst equation can come into existence, through a very small degree of reduction or oxidation altering the Fermi level of the electrode. This explanation is commonly encountered by undergraduate chemists, especially through its appearance in the opening pages of the introductory primer by Compton and Sanders [19]; a reading of Butler's 1924 papers reveals that the Nernst equation was far from acceptance as immutable thermodynamical law at the time, and Butler's work (like that of Peters, a generation earlier [20]) did much to provide it with needed physical interpretation. Butler's first 1924 paper addressed specifically the dissolution and plating of metals [14], while the second considered heterogeneous electron transfer between a metal electrode and a solution-phase redox couple, in which specific and competitive adsorption of redox-active species is assumed [15]. These papers both invoked the concept of a “balance point” (i.e., transition state) of higher energy between the oxidised and reduced forms of the redox couple, in which an electron has been partially transferred. Butler only considered the equilibrium of the two states, however, and did not at this time consider how the energy of this transition state depends on the polarisation of the electrode. The paper by Erdey-Grúz and Volmer is specific to the discussion of the overpotential of the hydrogen evolution reaction [17]. They discussed a variety of possible mechanisms that could explain observed polarisation data for hydrogen evolution, with a focus on the case where the ratedetermining step is the reduction of protons to hydrogen atoms (hence, the “Volmer step” in the Tafel-Heyrovský-Volmer mechanism [21]). Their innovation was to consider that, while this step was considered ratedetermining, both the anodic and cathodic directions could contribute, and that at the equilibrium potential, their rates must be equal. Since the rate-determining step in question is a unimolecular, one-electron step, no complications due to the multistep nature of the overall process arose in the Erdey-Grúz-Volmer analysis. They did not, however, consider any broader implications of the form of their equation for other electrochemical reactions. Butler was apparently inspired to return to his 1924 equations by a 1931 work by R.W. Gurney, in which the rate of heterogeneous electron transfer was first considered using quantum mechanical arguments [22]. Gurney considered an irreversible electron transfer for the hydrogen evolution reaction in water, by means of integrating transition probabilities between two states; in discussing the Franck-Condon principle, he considered the case of an electron transfer from an excited vibrational state of a reactant such as H3O+ (there is certainly a foreshadowing of Marcus theory here, although Gurney does not consider the role of multiple 2. Derivation of the Butler-Volmer equation “It is a matter of taste [in constructing a derivation] whether to choose as a basis several empirical principles which make reference neither to atomic theory nor to quantum theory, or to choose a single principle superposed on atomic theory and quantum theory” – E.A. Guggenheim, Thermodynamics: An Advanced Treatment for Chemists and Physicists, Fifth Edition, 1967 [23] In standard textbooks on electroanalytical chemistry, it is common practice to introduce transition state theory in order to derive the Butler-Volmer equation from purely fundamental considerations, and then to illustrate Nernstian and Tafel behaviour as limiting cases [3,5,7,11,24–26]. Given the extremely wide range of electrochemical interfaces and reactions to which the general form the Butler-Volmer equation is habitually applied, we prefer to begin from an empirical standpoint, without confining ourselves to specific mechanistic or fundamental considerations at this stage. 2.1. Empirical derivation of the Butler-Volmer equation The below derivation is adapted from a similar derivation published previously by one of the authors in the specific context of polymer electrolyte membrane fuel cells (PEMFCs) [27]. Notation is summarised in the Appendix. In comparing or equating different forms of the Butler-Volmer equation, it is necessary to make some statement on what the Butler-Volmer equation is, for a redox system at a single electrode. We consider an appropriate definition as being in terms of the following series of phenomenological statements about the form of the equation: 1. The current density at an individual electrode surface due to a quasireversible electrochemical process equals the sum of independent anodic and cathodic current densities. 2. The potential and concentration dependences of each of the anodic and cathodic current density terms are separable. 3. The potential dependence of both the anodic and cathodic current densities is a Tafel equation, i.e. the electrode polarisation is proportional to the logarithm of current density, scaled by an empirical Tafel slope. Each assumption here is strictly empirical, with a validity only so good as the utility and accuracy of the result it yields; however, transition state theory as described below (under “Quantum mechanics-based derivation of the Butler-Volmer equation”, below) does support the given exponential dependence of the rate constants, under certain circumstances. 2 In [18], it seems that the author misinterpreted Butler's reference (in [16]) to “the exponential term in … (I)” as referring to reference 1 (the 1931 paper by Gurney, [22]). This seems to be a mistake; Butler's notation “(I)” applies instead to Eq. (I) within the paper [16], which is in turn taken from his own 1924 work [15]. As a result, the otherwise balanced historical discussion in [18] makes Butler sound perhaps more critical of his peers (and less so of himself) than is fair. 1 Erdey-Grúz Tibor (Hungarian style) went on to act as Minister of Education of Hungary (principally under Imre Nagy) until the 1956 Revolution. He published Kinetics of Electrode Processes in 1972 and served as President of the Hungarian Academy of Sciences during the last years of his life. Further information is available in Hungarian at http://mek.niif.hu/00300/ 00355/html/index.html. 3 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 we have We will also use the thermodynamic constraint that at zero net current density, a condition of dynamic equilibrium between anodic and cathodic processes exists, such that the polarisation under these conditions equals the equilibrium potential due to the Nernst equation. For constant concentrations of all species, the Tafel equation for anodic current density is: E−E ¼ Aa log10 ia ia i ¼ i0;ref ðexpðαa f ηref Þga expðαc f ηref Þgc Þ This is the practical Butler-Volmer equation! The various coefficients are all empirical. Eq. (2.8) must reproduce the Nernst equation when i = 0 for any solution composition at the electrode surface. Setting i = 0 in (2.8) gives a certain equal and opposite current for both anodic and cathodic processes, so that: ð2:1Þ and the Tafel equation for cathodic current density is: E−E ¼ −Ac log10 −ic ic i0;ref exp αa f ηeq;ref ga ¼ i0;ref exp −αc f ηeq;ref gc ð2:2Þ ¼ 1 ∂ lnim f ∂E where ηeq;ref ≡ Eeq Eeq;ref ð2:10Þ The value of both the left- and right-hand sides of (2.9) has been defined as the exchange current density i0. Substituting back into (2.8): i ¼ i0 exp αa f ηref −ηeq;ref − exp −αc f ηref −ηeq;ref ð2:11Þ From definition (Eqs. (2.7) and (2.10)): ηref ηeq;ref ¼ E Eeq ð2:3Þ ln10 Am f ð2:12Þ ≡η where η is the overpotential. Hence: To bring in the concentration dependence, we define a function gm for each current density, expressing the concentration dependence of the reaction as a function of the concentrations ci and the set of reference concentrations ci,ref. The quantities gm are defined to equal 1 when all ci = ci,ref. Since the functions gm can be any function, the use of concentration here does not imply any assumption of thermodynamic ideality. The empirical assumptions can then be combined mathematically in the following expression for current density: i ¼ ia þ ic ¼ ia expðαa f ðE−E ÞÞga −ic expð−αc f ðE−E ÞÞgc i ¼ i0 ð expðαa fηÞ− expð−αc fηÞÞ ia exp αa f Eeq;ref −E ¼ ic exp αc f Eeq;ref −E ð2:4Þ 2.2. Thermodynamic constraints on the Butler-Volmer equation Now we return to the situation at equilibrium. The thermodynamic Nernst equation can be written as follows: 1 Eeq ¼ Eeq;ref ln geq f ð2:5Þ ð2:14Þ where geq is a general function of the concentrations ci and reference concentrations ci,ref. This could be a nonlinear function over a broad range of ci, to account for non-ideality.3 Like ga and gc above, geq = 1 when all ci = ci,ref. From (2.10) and (2.14) we note that: ≡ i0;ref We have defined the equal quantities on the left- and right-hand sides of Eq. (2.5) as i0,ref, the exchange current density under the reference conditions. Now, by substituting into Eq. (2.4): i ¼ i0;ref exp −αa f E eq;ref −E expðαa f ðE−E ÞÞga −i0;ref exp αc f Eeq;ref −E expð−αc f ðE−E ÞÞgc ¼ i0;ref exp αa f E−Eeq;ref ga − exp −αc f E−Eeq;ref gc ð2:13Þ This is the engineer's Butler-Volmer equation. It is clearly just a rearrangement of the practical Butler-Volmer equation, since it can be reached with no further approximations. The empirical assumptions used to reach this point imply the independence of the exchange current density from overpotential; we note that this has been argued to be an approximation valid in practice only in the absence of double layer phenomena [25]. First, let us consider the condition of equilibrium at the reference conditions. When all ci = ci,ref, and the net current density is zero, the electrode potential E equals its equilibrium value Eeq,ref. Hence, setting i = 0, ga = gc = 1, and E = Eeq,ref in (2.4), and rearranging: ηeq;ref ¼ Eeq −Eeq;ref ð2:6Þ ð2:15Þ 1 ¼ − lngeq f 3 In separating the electrolyte potential (contained in E) from the chemical activity, we imply an activity defined under conditions of zero electric field; a discussion of the meaning of “electrolyte potential” exceeds the scope of our work here, but the reader is referred to Newman [4] for a deeper consideration of this important question. Defining now the polarisation with respect to reference conditions as: ηref ≡ E−Eeq;ref ð2:9Þ ≡ i0 where E∗ is any arbitrarily chosen reference value of electrode potential for measurement, defined with respect to a reference electrode, but not necessarily equalling the standard (or formal) reduction potential for the redox couple under consideration. The quantities im are reference current density magnitudes, which depend on the reference concentrations of the reactants. Although these reference current densities are sometimes called “exchange current densities”, we discourage this nomenclature due to ready confusion with the distinct quantity applied to a reversible reaction at equilibrium (as in Eq. (1.3)). The coefficients Aa and Ac are called Tafel slopes (V) and are strictly positive. The empirical Tafel slopes can be rewritten in terms of transfer coefficients, according to the IUPAC-recommended definition [8,9]: αm ≡ ð2:8Þ ð2:7Þ 4 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 The values of ga and gc from (2.19) give the following expression: Now, the equality (2.9) from the empirical derivations can be rearranged to: Eeq 1 1 g ¼ Eeq;ref − ln a ðαa þ αc Þ f gc i ¼ i0;ref ð2:16Þ ð2:21Þ In the special case where cR,ref = cO,ref = cref, we can now write directly in terms of concentration: For our empirically derived relation (2.16) (a corollary of the original assumptions) to be compatible with the thermodynamic relation (2.14), we require that: ga ¼ gðeqαa þαc Þ gc c R c O exp αa f E−E0f − exp −αc f E−E0f cR;ref cO;ref i¼ i0;ref exp αa f E−E0f cR − exp −αc f E−E0f cO cref ð2:22Þ Writing: i0;ref ¼ k 0 Fcref ð2:17Þ ð2:23Þ then For given values of αa and αc, this implies a constraint on the relative values of ga, gc, and geq. It is a consequence of the requirement of microscopic reversibility of the reaction. We emphasise that to reach this point we have required only the imposition of a Tafel-like potential dependence, as for an irreversible, globally well-mixed system, for linearly separable anodic and cathodic contributions to the current density of a reversible system with variable concentration at the electroactive surface. This assumption may be subject to criticism from a fundamental point-of-view and will not apply to all circumstances (see under “Multistep processes: a comment”, below). Nonetheless, the relatively modest requirements of deriving the Butler-Volmer equation from the above method may offer comfort to researchers considering materials or systems where specific arguments from transition state theory are less trustworthy than the empirical observation of Tafel behaviour under well-controlled conditions. Bockris and co-authors have proposed the following generic expressions for the concentration dependences [28]: ga ¼ Y i gc ¼ ci ci;ref i ¼ k 0 F exp αa f E−E0f cR − exp −αc f E−E0f cO This is the electroanalytical Butler-Volmer equation. Note that it is equivalent to the empirically derived Butler-Volmer equation only under the specific conditions where: (a) the law of mass action holds; and (b) there exists a common reference concentration cref where, when all redox-active species are present at this concentration, the equilibrium potential is a formal potential with respect to the range of concentrations of interest. Hamann has defined the standard exchange current density as the exchange current density in the case when all ci,ref = cref, and cref = c0 [29]. The exchange current density for the case of the electroanalytical Butler-Volmer equation is given from (2.9) and (2.23): i0 ¼ k 0 Fcref exp −αc f ηeq;ref gc cO i0 ¼ k0 Fcref exp αc lngeq cref ð2:18Þ ¼ k0 FcO gαeqc which is the expected expression [4]. If we are untroubled by non-integer exponentiation of quantities with units (but beware!), then following the convention that α = αc we can extend to the more commonly given form [3,4]: 2.3. Empirical requirements of the electroanalytical Butler-Volmer equation 1−α i0 ¼ k 0 FcαR cO To proceed from the above to the electroanalytical Butler-Volmer equation, further approximations are required. For the case of a unimolecular reaction (Eq. (1.2)), the law of mass action suggests: gc ¼ cR ð2:19Þ cO ¼ i0;ref cO;ref For constant transfer coefficients and assuming αa + αc = 1 (suggested by “Quantum mechanics-based derivation of the Butler-Volmer equation”, below), (2.19) is only compatible with the constraint (2.17) for the case where Eeq,ref is a formal potential; that is, where the Nernst equation can be written to a sufficient approximation, within the concentration ranges of interest, as: Eeq ¼ E0f − 1 cR ln f cO ð2:27Þ but to keep the unit syntax tidy, we prefer either (2.26) or: i0 ¼ k0 Fcref cR;ref ð2:26Þ αc cR ¼ k 0 FcO cO For αa + αc = n, and considering activity coefficients to be constant and equal to their values at ci = ci,ref, these formulas satisfy the constraint (2.17) by definition. They are also (thermodynamically) compatible with multistep, multi-electron processes. ga ¼ ð2:25Þ Using (2.15) and inserting the assumed form of the function gc from (2.19) gives: γi þαa vi Y ci γi −αc vi ci;ref i ð2:24Þ cR cref cR cref α α cO cref cO cref 1−α ð2:28Þ 1−α 2.4. Transition state theory-based derivation of the Butler-Volmer equation In the above, we have begun from an empirical standpoint, and progressed to introduce additional approximations only when needed. As noted above, most standard treatments instead begin from a simple quantum mechanical description and proceed to the Butler-Volmer equation from there, using a transition state theory. We will use this section to examine the implied approximations of this approach, by comparison to the strictly empirical method. ð2:20Þ 5 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 Hence by the same methods: The premise is that an electrode-electrolyte potential difference can be constructed theoretically, which for a correctly configured three-electrode cell [5,7] is correlated linearly with electrode polarisation. The common form of the argument follows the work of Horiuti and Polányi on transition state theory [30,31] with the thermodynamic notions of electrochemical potential introduced to the discussion by Parsons [32]. Within a transition state theory [30–34], the Eyring equation suggests that rate constants are proportional to a difference in chemical potential between reactants and transition state, as given by a Boltzmann factor in the form: ‡ −Δ μm k m ∝ exp RT a‡ ∝ am i ¼ F k a;ref expðð1−αc Þf ðE−E ÞÞga −kc;ref expð−αc f ðE−E ÞÞgc Can the Butler-Volmer equation be applied to processes whose electrochemical mechanism involves more than one elementary step? The quantum mechanics-based derivation and routes to the electroanalytical Butler-Volmer equation posit explicitly the case of a one-electron, unimolecular process, and so by definition exclude multistep processes. But there is nothing in our empirical assumptions for deriving the general engineering and practical forms of the Butler-Volmer equation that makes multistep processes formally inadmissible. Key, however, is our assumption that both the anodic and cathodic current density are simultaneously separable between polarisation-dependent and concentration-dependent terms. This would be very unlikely for a multistep process, such as hydrogen evolution or oxygen reduction. We can justify this as follows: in the case of a successive electron transfer and chemical step (in either order), the rate of the electron transfer is influenced by electrode polarisation, while that of the chemical step is not. We can therefore tune the rate of the electron transfer to be faster or slower than the chemical step; at some polarisation, a change of rate-determining step would be expected. Likewise, depending on the relative molecularity of the elementary steps, the local concentrations may affect the chemical step with different proportion to the effect on the electron transfer step, so a concentrationdependent change of rate-determining step is also possible. If the change of rate-determining step occurs within the accessible experimental potential window, it could be observed as a discontinuity in the value of the observed Tafel slope. On this basis, it is much more reasonable to suppose for multistep processes that the polarisation-dependence of current density is not indefinitely exponential, and that the polarisation-dependence is not separable from the concentration dependence for each of the anodic and cathodic current densities independently. In the further discussions below, we will focus on one-electron transfer processes, except in application-specific contexts. Several authors – ð2:31Þ ð2:32Þ ð2:33Þ For the corresponding partial reduction to the transition state, we have: ð2:34Þ 1 a‡ ln E ¼ E0‡;c − αc F aO ð2:35Þ ð2:39Þ 2.5. Multistep processes: a comment ð2:30Þ O þ αc e− ⇄f‡g ð2:38Þ which is equivalent to the empirical statement (2.4) but with the additional constraint (2.38) on the transfer coefficients, arising due to the physical consideration of the energetics of the transition state. That is, it is the identification of the transfer coefficient, previously defined empirically, with the number of electrons transferred at the transition state that leads to the constraint (2.38); the thermodynamic consistency requirements of the Butler-Volmer equation, as considered empirically, lead only to the more general constraint (2.17). From the quantum mechanics-based formula (2.39), the derivation of any of the particular forms of the Butler-Volmer equation would then proceed as under “Empirical derivation of the Butler-Volmer equation” above, with (2.39) in place of (2.4). Any expression in which a particular function of concentration (such as (2.18) or (2.19)) substitutes the empirical concentration dependences ga and gc will require an additional assumption, and may also require the assumption of constant activity coefficients over a concentration range of interest. and so according to transition state theory (Eq. (2.29)) k a ¼ ka;ref exp ðαa f ðE E ÞÞ ð2:37Þ Hence, introducing empirical concentration-dependences as before, and denoting α = α c: ð2:29Þ Hence: a‡ ¼ exp αa f E E0‡;a am k c ¼ k c;ref expð−αc f ðE−E ÞÞ αa þ αc ¼ 1 To determine the Boltzmann factor for occupation of the transition state, we use the Nernst equation to relate the activities to the electrode polarisation: 1 aR ln E ¼ E0‡;a − αa F a‡ ð2:36Þ It follows that the component processes (Eqs. (2.30) and (2.34)) must sum to the overall one-electron conversion (Eq. (1.2)). Therefore: where a‡ is the activity of the transition state and am is the activity of the reactant for process m (m = a,c). As noted by Newman, common derivations of the Butler-Volmer equation based on quantum mechanics frequently separate the electrochemical potential of the ionic species into “chemical” and “electrical” terms, in which the electrolyte potential representing the latter is “usually not a well-defined potential” [4]. There is a historical legacy here. Although the work of Guggenheim on electrochemical thermodynamics [35] predates the first development of a quantum mechanics-based kinetic theory of electrode reactions, the meaning of “electrolyte potential” has remained murky, and its relation to any potential expressed in a quantum mechanical theory is rarely expressed. The resolution of these disagreements lies well beyond the scope of this paper; instead, we rely on the ansatz of the Eyring transition state theory being that the transition state can be treated as having a thermodynamic activity that is meaningful according to (2.29), and that thermodynamic quantities in electrochemistry can be approached through the Nernst equation. Defining a half-cell electrode potential with respect to an ideal reference electrode avoids any need to ‘expose’ an electrolyte potential.4 By this method, we begin by considering the partial oxidation to the transition state in which αa electrons are transferred. This is the oxidative direction of the following redox couple: f‡g þ αa e− ⇄R a‡ ¼ exp αc f E E0‡;c aO 4 On this matter, the authors are happy to seek correction from experts in non-equilibrium statistical mechanics, so long as such experts concede to the requirements of electrochemical thermodynamics, from Guggenheim onwards! 6 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 especially Albery – have given clear discussions on the electrode kinetics of multistep electrochemical processes [2,5,7,29,36]. context-specific assessment of their validity in reproducing or predicting experimental results. We provide here a table of transformations to aid the conversions in each case Table 1. An especially common error noted in reviewing the literature [27] is the expression of overpotential as concentrationindependent (i.e., as η ref ) while neglecting to include the concentration-dependent terms ga and g c of the practical ButlerVolmer equation; normally a (meaningless) concentration dependence is still present because the concentration-dependent exchange current density is retained as the pre-factor. Why might one form or another be more suitable? In his work on Electrode Kinetics, John Albery attributed the conversion from k0 to i0 as an activity exclusive to “black-belt electrochemists” [5]! We suggest here that the choice whether or not to translate between two forms of the ButlerVolmer equation relates naturally to both the available input data and the expected outputs from the equation form. The engineer's Butler-Volmer equation is a particularly natural form if concentration dependence is to be neglected, since the implied concentration-dependence of the exchange current density and overpotential are complicating factors to this otherwise simple expression. By the same token, the practical Butler-Volmer equation can be seen as the most elegant expression of concentration dependence within the framework of the engineer's Butler-Volmer equation, such that the explicit concentration-dependent forms of the exchange current density and overpotential do not need to be defined. Instead, the concentration dependence is confined to empirical reaction orders contained in the functions ga and gc, and the thermodynamic behaviour in terms of the variable geq. This form of the equation does not constrain the choice of reference conditions; experimentally accessible and relevant reference conditions can be selected, in order to make a direct measurement of i0,ref and Eeq,ref. The relation of the quantities η and ηref to the cell overvoltage and electrode overpotential should be discussed. For an energy conversion system, it is common to consider the maximum electrochemical energy extractable as work, per unit charge passed. This quantity is the cell equilibrium potential, expressed in terms of the equilibrium potential of each electrode at relevant, uniform conditions of the reactants, such as the inflow concentrations to a fuel cell or flow battery: 3. Forms of the Butler-Volmer equation “Any empirical relation can be formally decomposed in a number of equally valid ways. Such a decomposition, however, is useful only if it does something more than restate the initial information.” – Walter Scheider, Journal of Physical Chemistry, 1975 [37] 3.1. Relating the three principal forms of the Butler-Volmer equation From the derivations above, we have demonstrated that the three formulations under “Conventions of nomenclature” are equivalent, provided that the values and, crucially, concentration-dependences of the coefficients are consistent between each case. A direct mathematical conversion is available between the engineer's Butler-Volmer equation and the practical ButlerVolmer equation (demonstrated under “Empirical derivation of the ButlerVolmer equation”, above): the choice of one over the other may depend on the availability of measured data but neither invokes any specific assumptions about the mechanistic character or physicochemical behaviour of the electron transfer. The electroanalytical Butler-Volmer equation, in which the concentrations appear directly, arises most naturally from the quantum mechanical conception of the process, but depends on the correctness of the law of mass action and agrees with the sum of two Tafel equations (with general concentration dependence) only over a concentration range sufficiently narrow that activity coefficients are approximately constant. We emphasise that if the transformation of the kinetic coefficients is not accomplished appropriately when moving from one formulation to another, the result will be a distinct equation which is not a Butler-Volmer equation, and does not enjoy the virtues of the latter in terms of empirical relation to Tafel-slope behaviour, arguable physical justification from transition state theory, or conceptual simplicity and consistency with standard thermodynamic and kinetic limiting cases. Such distinct kinetic expressions are numerous in the literature, as reviewed further under “Applications of the Butler-Volmer equation” below, but of doubtful utility and require a Eeq;cell ¼ Eeq;ref;cat −Eeq;ref;ano ð3:1Þ Table 1 Conversions between different forms of the Butler-Volmer equation (Eqs. (1.3)–(1.5)). From ↓ To → Engineering Practical Engineering i0;ref i0 i0 ¼ ¼ ga expðαa f ηeq;ref Þ gc expð−αc f ηeq;ref Þ Electroanalytical cO;ref ¼ cR;ref ¼ cref E0f ¼ Eeq ðcref Þ αa ¼ 1−α αc ¼ α ηref = E − Eeq(ci,ref) k0 ¼ Must satisfy (2.17). Practical i0 cR FcO cO cO;ref ¼ cR;ref ¼ cref i0 ¼ i0;ref ga expðαa f ηeq;ref Þ ¼ i0;ref gc expð−αc f ηeq;ref Þ η = E − Eeq(ci) E0f ¼ Eeq ðcref Þ Must satisfy (2.17). αa ¼ 1−α αc ¼ α k0 ¼ Electroanalytical i0 ¼ k 0 FcO cR cO !α i0,ref = k0Fcref ηref = E − Eeq(ci,ref) η = E − Eeq(ci) Must satisfy (2.17). Must satisfy (2.17). 7 !−α i0;ref Fcref E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 where the subscripts “ano” and “cat” refer to the distinct electrodes (anode and cathode) of the cell, as distinct from the subscripts “a” and “c” referring to anodic and cathodic current density contributions at a common electrode, as in the derivations above. Now, the real cell voltage is given: Ecell ¼ Ecat −Eano −ΔEIR (batteries, fuel cells, etc.). In combination with an interpretation through transition state theory, it has been called the “more insightful and easierto-grasp formulation of how the electrode potential influences chargetransfer rates” [38]. To yield this form from a purely empirical basis requires all reference concentrations to be equal to each other, and that the equilibrium potential for all species existing at this reference concentration be a formal potential over the concentration range of interest, to a sufficient approximation. Hence, the electroanalytical form is only naturally suitable to cases where the reactants are dilute, especially with respect to inert electrolyte concentration. For instance, to consider the activity coefficients of 1 mM analyte A in 1 M NaCl solution as equivalent to their infinitely dilute values (also in 1 M NaCl) seems to be a reasonable approximation; the validity of such an approximation can, of course, be tested experimentally. Conversely, for applications such as batteries (Li-ion or redox flow) where, typically, the only electrolyte is that containing the electroactive species (for instance, LiPF6 as electrolyte, containing Li+), activity effects are likely to become marked over any reasonable range of reactant concentration variation at the electroactive surface. If, on the other hand, the current densities remain sufficiently low in concentrated electrolyte that the reactant concentration is never significantly depleted at the electroactive surface, we have no need for a concentration-dependent equation! In summary, the electroanalytical Butler-Volmer equation offers pedagogical simplicity, but moreover emphasises the law of mass action, and suggests a primary concern with measuring the current density as a quantity that is sensitive to concentration. This aligns with the motivation and working practice of electroanalytical chemistry – hence our nomenclature. Similarly, the definition of a heterogeneous rate constant k0 rather than an exchange current density i0 is apparently more fundamental: k0 is intended to be concentration-independent, whereas i0 depends on solution composition. In any electrochemical experiment, though, it will be a current density or voltage that is the direct measurand, and not a concentration or rate constant. So, while i0 is less general, it is requires no auxiliary assumptions to be accessible experimentally, but instead can be assessed empirically from an equation such as (3.5). Since both k0 and i0 are expressed with reference to current density, they also become problematic when considering the behaviour of a composite, porous electrode, as encountered in batteries or fuel cell catalyst layers, since the meaning of “electroactive surface area” in a nanoscopically heterogeneous electrode is not meaningful [39,40]. In such cases, the specific exchange current density (A g−1) may be a preferred practical measure. ð3:2Þ where ΔEIR represents resistive losses in the cell. The total loss of energy extractable as work between the equilibrium condition and the operating condition is the difference between the equilibrium cell voltage and the operating voltage: ΔEtot ¼ Eeq;cell −Ecell ¼ Eano −Eeq;ref;ano − Ecat −E eq;ref;cat þ ΔEIR ð3:3Þ ¼ ηref;ano −ηref;cat þ ΔEIR Therefore, the quantities ηref represent contributions to the total cell overvoltage (noting that ηref,cat is typically a negative quantity). Since η is the overpotential considering the real concentrations present at any instant at the electrode surface, while ηref is measured with respect to fixed reference concentrations, η is commonly termed the activation overpotential, while the quantity (ηref − η) = ηeq,ref (see (2.12)) is the quantity classified as mass transport overpotential in the analysis of losses of galvanic cells. Since such losses are naturally relevant to energy conversion applications (batteries, redox flow batteries, etc.), it is unsurprising that the practical ButlerVolmer equation is often encountered in such settings (see also under “Applications of the Butler-Volmer equation” below). With relation to electrode kinetics rather than the analysis of the whole electrochemical cell, the engineer's Butler-Volmer equation has an important property: it can be linearised directly. While in the other forms, the exponential activation terms appear as products with concentrationdependent terms, the engineer's Butler-Volmer equation uniquely contains exponential terms of a single variable – the overpotential – which by definition equals zero at equilibrium. The behaviour for small perturbations from equilibrium is then directly accessed by expanding the exponentials as Taylor series about η = 0, and retaining terms only up to first order: i ≈ ð1 þ αa fηÞ−ð1−αc fηÞ i0 ð3:4Þ ¼ ðαa þ αc Þfη 3.2. Should the Butler-Volmer equation be expressed in terms of activity or concentration? Butler reported an analogous equation and remarked that it was obeyed in the limit of small current for hydrogen evolution on Pt, as well as for the “reduction of ferric salts, of methylene blue, and of quinone” [16] – all systems with notably fast electrode kinetics. The linear relation (3.4) between current density and overpotential under conditions close to open circuit is suggestive of Ohm's law, and hence led Vetter to define the charge transfer resistance [2]: Rct ≡ lim ∂η i→0 ∂i ¼ A commonly encountered misconception is that substituting activities for concentrations in the electroanalytical Butler-Volmer equation can be done freely, and that as in the Nernst equation doing so will make the results somehow ‘more accurate’ or ‘more correct’. As a thermodynamic quantity, the activity has no immediate bearing on rate or transport properties. Accordingly, while the activity is expected to appear in a rigorous definition of overpotential, there is no particular reason to expect the law of mass action to obey a simple equation in activity. As an example, a recent major review of theoretical methods for fuel cells [41] gives the Butler-Volmer equation in its practical form, but specifies concentration dependences according to the law of mass action with activities in place of concentration: ð3:5Þ 1 ðαa þ αc Þi0 f This charge transfer resistance is linked directly to the empirical quantity i0 and so has direct meaning with respect to the efficiency of electrolysis – it is also accessible through electrochemical impedance spectroscopy (EIS), which addresses the small current regime [6]. The linearisation of the kinetic equation is thus a concept with essential insight, and the engineer's Butler-Volmer equation is the form in which this linearisation is most succinctly accomplished. The electroanalytical Butler-Volmer equation stands out as the form most often encountered in elementary discussions of electrode kinetics, but also as a form almost never encountered in models of applied devices ga ¼ Y a j v j a j;ref j gc ¼ Y ak vk ak;ref k ð3:6Þ Like the Bockris-Reddy-Gamboa-Aldeco concentration dependences (Eq. (2.18)), if αa + αc = n, then the above expressions do satisfy the required thermodynamical constraint (2.17). There is no reason to consider them to do 8 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 parameters. The quality of the fit might offer some indication of the validity of the model; however, in the absence of a tangible relationship between these parameters and quantifiable measurands, it can be challenging to assess just how meaningful they are. Numerical methods are readily available for convenient simulation of quiescent voltammetry based on ButlerVolmer kinetics [46–49], but care should be exercised as overparameterisation of the model can lead to multiple non-unique fits and misleading results. In practice, it is critical to simplify and achieve ideal control over the mass transport characteristics of the experimental configuration in order to explore electron transfer kinetics with sufficient rigour. Hence, the uniform, planar electrode geometries commonly associated with electroanalytical chemistry have provided a strong foundation for fundamental kinetic studies. It is also important to recognise that the “kinetic window” available to the experimentalist is intrinsically limited by the rate of mass transport, which itself is intimately tied to experimental time and length scales. In the electrochemically reversible limit in which the rate of electron transfer greatly exceeds the rate of mass transport, the current-voltage profile encodes no information about the heterogeneous kinetics, and the response reflects purely a thermodynamic (i.e. Nernstian) response. No amount of “mass transport correction” of the data can change this. Acknowledging this intrinsic limitation is key to designing the appropriate experiment for characterising electron transfer behaviour. Furthermore, in the instance that the electron transfer kinetics are sufficiently fast as to never be ratelimiting, one might also question the utility of quantifying them; there may still, of course, be some value in establishing the minimum heterogeneous rate constant for a given electrode reaction. Some of the simplest experiments for studying electrode kinetics involve conventional (transient) cyclic voltammetry at planar macroelectrodes in the range of voltammetric scan rate (v) where v < 1 V s−1. Based on the classical work by Matsuda and Ayabe [50], the limit of reversibility for a simple one-electron process under such experimental conditions is k0 ≈ 0.3 cm s−1 (assuming D = 10−5 cm2 s−1 and v = 1 V s−1). As this limit is approached, however, the uncertainty associated with the deduced kinetic parameters becomes considerable, so in practice only electrode reactions with k0 in the order of 10−2 cm s−1 or slower tend to be accessible using this approach. Fig. 1 depicts this through simulations of cyclic voltammetry for a typical macroelectrode experiment (calculated by solving Fick's laws in 1D [47,48]) using a range of heterogeneous rate constants. In Fig. 1, the curves for k0 > 10−2 cm s−1 are largely indistinguishable. For smaller values of k0, when the rate of electron transfer is sufficiently slow compared to mass transport, the current becomes sensitive to heterogeneous kinetics and so uniquely, however. The implicit assumption that reaction order is identical to stoichiometric coefficient excludes any mechanistic interpretation of the rate law, and is quite unlikely to be true for the multi-electron electrocatalytic processes: the resulting equation implies implausible reaction orders for multi-step processes, such as a fourth-order dependence of oxygen reduction reaction rate on the activity of protons (for which vH = −4 when n = 4) in the proton-conducting phase in a polymer electrolyte membrane fuel cell. The concept of applying the law of mass action to activities seems to arise in the concept of treating chemical reactions as having rates proportional to a linear driving force corresponding to the chemical affinity, within the framework of non-equilibrium thermodynamics [42]. Since the linear assumption supposes that local departures from equilibrium conditions are small, this can only be the case for fast reactions. Extrapolation from the linearised Butler-Volmer regime, represented simply in terms of the overpotential, to the nonlinear regime has been suggested by some researchers [43,44]. This tends to amount, however, to the assumption that non-equilibrium thermodynamics can be used to infer the reaction orders of the kinetic rate law; since reaction orders relate to the mechanism of the reaction, they are not thermodynamic quantities, and so any “thermodynamic derivation” of the full, nonlinear Butler-Volmer equation is inappropriate. If a process is reversible, it is a necessary condition for a suitable choice of electrode kinetic equation that it satisfies the Nernst equation at i = 0; however, thermodynamics cannot yield any information on rate – including its order with respect to concentrations, which is fundamentally a function of mechanism. It is an obvious statement, but nonetheless one worth re-iterating, that a chemical process considered irreversible cannot achieve an equilibrium; hence, equations such as the Tafel equation that describe irreversible process are in no sense constrained to relate to thermodynamic quantities. 3.3. Need for notational alertness Throughout the electrochemical literature, it is possible to encounter forms of the Butler-Volmer equation that are superficially incorrect, but can be understood as actually representing one of the principal formulations faithfully, once the notation is understood. Especially common is the definition of overpotential with respect to equilibrium potential at given reference conditions, i.e. the quantity ηref in the practical Butler-Volmer equation, while retaining the nomenclature and conventional notation (η) of the true (local) overpotential. For example, in the derivation of the Butler-Volmer equation in the undergraduate textbook by Fisher, the meaning of the author's notation η is implicitly that of ηref throughout the derivation, even though this is never explicitly stated [45]. Accordingly, the central statements of the Butler-Volmer equation in [45] do not appear, at first glance, to match ours; in fact, the only difference is notational! 4. Verification of the Butler-Volmer equation “…the difficulty was encountered that the exponential term required by the experimental data was half that which appears in [my 1924 equation].” – J.A.V. Butler, Transactions of the Faraday Society, 1932 [16] 4.1. Experimental affirmation of the Butler-Volmer equation The use of electrochemical techniques to study electrode kinetic models is a vast topic, so a comprehensive coverage would be far beyond the scope of this review. It is instructive, however, to discuss experimental approaches that have been employed to validate the predictions of the Butler-Volmer equation, particularly in light of recent developments in this area. Due to the empirical nature of the Butler-Volmer formalism, its experimental verification has typically involved comparing the current-voltage behaviour of a model system (e.g. as measured via linear sweep voltammetry or cyclic voltammetry) to the response predicted by numerical simulation, in which the appropriate mass transport equations are solved using the Butler-Volmer equation as the boundary condition at the electrode surface [46–48]. Iterative fitting of theory to experiment then allows extraction of the kinetic Fig. 1. Simulated cyclic voltammetry for a one-electron transfer process at a macroelectrode, using the electroanalytical Butler-Volmer equation (c* = 1 mM, v = 0.1 V s−1, D = 10−9 m2 s−1, α = 0.5, T = 25 °C) for a range of k0 values. 9 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 voltammetric waves are shifted in the anodic direction with respect to the symmetric voltammogram with α = 0.5. The upper kinetic limit accessible via electroanalytical techniques can be extended by employing microelectrodes which exhibit enhanced rates of mass transport by virtue of radial diffusion, thus allowing steady-state measurements at conventional scan rates. The small capacitive currents and ohmic drop associated with microelectrodes further allow them to be combined with fast scan voltammetry (up to MV s−1) to gain access to even faster kinetics [51–54]. Hydrodynamic strategies, such as flow cells, impinging jets and rotating disc electrodes (RDEs), can similarly be introduced to enhance and control mass transport. Combining these two concepts to yield hydrodynamic microelectrode voltammetry has been shown to be particularly powerful approach to heterogeneous kinetic measurements, as exemplified by the microjet electrode [55–57] and the high speed channel microband electrode [58–60]. In addition to linear sweep and cyclic voltammetry, a variety of alternative electrochemical techniques have been applied for the study of Butler-Volmer kinetics. Potential step chronoamperometry can be performed and the current-time profile modelled, again using numerical methods, and once again provided the potential perturbation is small enough to avoid mass transport limitations. Building on the potential step concept, pulsed techniques such as square wave and differential pulse voltammetry can offer advantages compared to conventional voltammetry in the study of heterogenous kinetics [61–66]. These benefits derive from the differential or subtractive character of such methods, which allows elimination of non-faradaic contributions to the measured current and can provide heightened sensitivity to kinetics due to the forward and reverse electron transfer reactions being probed in rapid succession (pulse timescales in the range 10-1000 Hz are typically employed). More complex pulse methods, including differential double- and multi-pulse voltammetry, reverse pulse voltammetry and additive differential pulse voltammetry, have also been exploited for kinetic studies, offering further discrimination over non-faradaic contributions [1,67–71]. A theoretical comparison of various pulse techniques used in the context of heterogeneous kinetics has been published by Laborda and co-workers [72]. Sinusoidal waveforms, in which a driving potential or current may be alternated between anodic and cathodic directions at high frequency, are common in electrode kinetic analysis. Electrochemical impedance spectroscopy (EIS) is used in almost every field of electrochemistry [3,73]. As a small-amplitude technique, it is only capable of accessing linearised features of the electrode kinetics, but by oscillating about open circuit conditions it can in principle give direct experimental access to the exchange current density (as interpreted through Eq. (3.5)). Alternating current (AC) voltammetry develops the EIS principle by superimposing one or more sinusoidal potential waveforms onto a conventional voltage ramp, thereby combining the virtues of EIS and cyclic voltammetry and allowing both linear and nonlinear properties associated with differing timescales to be easily extracted. Over the last two decades, developments in instrumentation and theory have inspired considerable advances in this area, particularly in the use of large amplitude current perturbations in combination with Fourier transform filtering [74], as well as innovative approaches to data analysis [75–77]. Selected higher-order harmonic components of the measured currents have been shown to be particularly sensitive to electrode kinetics, offering enhanced mechanistic discrimination, whilst being insensitive to non-faradaic currents [78,79]. Scanning electrochemical microscopy (SECM) has also been used to monitor electrode kinetics; by using a micro- or nano-electrode to probe inside the diffusion layer of an electroactive interface, one can effectively measure its transient electrochemical properties using a steady-state method, with minimal contribution from non-faradaic and ohmic drop effects [80–83]. In this case, kinetic information is extracted by fitting of the probe current-distance approach curves and comparing with theory. Most notably, this approach has been employed to measure heterogeneous Fig. 2. Simulated cyclic voltammetry for a one-electron transfer process at a macroelectrode, using the electroanalytical Butler-Volmer equation (c* = 1 mM, v = 0.1 V s−1, D = 10−9 m2 s−1, T = 25 °C) for a range of α values with k0 = 10−2 cm s−1. one observes the characteristic shifting of voltammetric waves due to quasireversible electron transfer. The degree of sensitivity of the shape of the voltammetry to the value of α is of course a function of k0, and this can be seen clearly in Figs. 2 and 3. For values of k0 approaching the above-mentioned reversibility limit, the voltammetry is relatively unchanged by α (Fig. 2), with only a weakly reduced forward peak current and weakly elevated backward peak current with increasing α, for the voltammetric scan in the oxidative direction. The influence of the transfer coefficient naturally becomes more significant for slower kinetics, as shown in Fig. 3, where for k0 = 10−4 cm s−1 the role of α is clear. For the scan in the oxidative direction, increasing the value of α (a transition state implicitly more like the product) has the effect of making the forward peak appear ‘more irreversible’ – that is, lowering the peak current and displacing the voltammetric wave further from the formal potential – while, conversely, increasing α makes the backward peak appear ‘more reversible’ – the peak current is higher and the voltammetric wave shifts closer to the formal potential. Hence, at higher α, both Fig. 3. Simulated cyclic voltammetry for a one-electron transfer process at a macroelectrode, using the electroanalytical Butler-Volmer equation (c* = 1 mM, v = 0.1 V s−1, D = 10−9 m2 s−1, T = 25 °C) for a range of α values with k0 = 10−4 cm s−1. 10 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 the Butler-Volmer formalism; and (ii) it can account for potentialdependent transfer coefficients and curved Tafel slopes that are reported experimentally [103]. Thus, whilst being more challenging to handle theoretically or implement in numerical models, Marcus-Hush theory offers the potential advantage of allowing kinetics to be predicted based on the known physical properties of the system. The Compton research group has published a series of papers in which fitting of experimental data is compared between the Butler-Volmer equation and Marcus-Hush theory, using experiments drawn from a number of the methods described above [60,100,104–112]. Electrochemical systems studied include the reductions of europium(III), oxygen, cyclooctatetraene, tetraphenylethylene and various organic nitro compounds, and the oxidation of diphenylanthracene, in a range of solvents. In the majority of cases the experimental data demonstrated good agreement with the ButlerVolmer equation; in comparison, the Marcus-Hush theory demonstrated shortcomings when used in its symmetric form, for these redox systems, while an asymmetric Marcus-Hush theory gave broadly comparable capability for fitting the experimental data as the Butler-Volmer equation. In contrast, the Butler-Volmer equation did not give accurate fits to experimental data measured using a high speed channel electrode for the reduction of 2-nitropropane in acetonitrile [60]. In this case the deviation was attributed to a large potential difference between oxidation and reduction processes (> 1.5 V), such that relaxation of the requirement that αa + αc = 1 was necessary to allow acceptable fitting, since this relationship strictly only holds for measurements performed at the same potential [5]. As noted above, a Butler-Volmer equation viewed as the empirical overlay of two Tafel equations does not require a constraint on the transfer coefficients – this requirement arises due to a specific, fundamental interpretation of the electron transfer. Departure from classical Butler-Volmer behaviour has also been reported experimentally for electrodes coated with a thin insulating molecular monolayer, for which conditions of a slow heterogeneous rate constant combined with a small reorganisation energy can be realised [113]. For example, Li et al. recently characterised the oxidation of decamethylferrocene on octanethiol-modified Au electrodes using large amplitude Fouriertransformed AC voltammetry [114], and demonstrated that Marcus-Hush theory more accurately describes the behaviour than the Butler-Volmer model under these conditions. In discussing comparisons between the Butler-Volmer equation and Marcus-Hush theory, it is important to note that, given the intrinsically different origins of the two kinetic equations – one being phenomenological and the other derived from physical theory – their direct comparison should be treated with care and interpreted only in the context of the specific electrochemical system and conditions under investigation, rather than more generally. The greater physical descriptiveness of the Marcus-Hush theory, especially in its asymmetric form, also increases the degree of systemspecific parameterisation required for accurate description of voltammetric response for any particular redox system. electron transfer at the interface between two immiscible liquids, an experimental configuration that offers the further benefit of circumventing limitations associated with the electrochemical window by removing the need to externally bias the interface. 4.2. Experimental deviation from the Butler-Volmer equation The experimental techniques discussed above have demonstrated that the Butler-Volmer equation applies for a remarkably wide range of elementary one-electron processes, but deviations have been observed in a few cases, which typically can be accounted for empirically as potentialdependent transfer coefficients at high overpotentials. This embodies an inherent shortcoming of the Butler-Volmer model, which is equally present in the underlying Tafel equation in either direction of electron transfer: these equations predict the exponential relation between electron transfer rate and electrode overpotential to continue without limit – irrespective of how far the electrochemical interface is brought from equilibrium – which is not consistent with our modern understanding of electron transfer. As noted by Fletcher and Varley [84], however, definitive examples of significant deviation are actually rather rare. The most commonly cited examples are those reported by Savéant and Tessier [85,86], who examined the one-electron reduction of a range of organic nitro compounds in organic electrolytes using a mercury electrode. The fact that so few deviations of this kind have been reported experimentally may simply be because only a relatively narrow potential range exists over which measurements can be performed without mass transport or solvent window limitations becoming prohibitive, so nonlinearity of kinetic origin in the Tafel plots (as opposed to deviation from Tafel behaviour due to mass transport effects) is not discernible. This is not a criticism of the Butler-Volmer model, but more a testament to its wide applicability over a range of experimentally realisable conditions. One set of experimental conditions where deviation from Butler-Volmer kinetics might be expected is the case of nanoelectrodes [87–90]. When the diffusion layer thickness associated with a voltammetric experiment becomes comparable to the thickness of the electrical double layer, the charge on the electroactive species begins to interfere appreciably with the observed rate of electron transfer. Such double layer phenomena (“Frumkin effects” [91], amongst others) have been demonstrated experimentally to result in enhanced or attenuated currents: they are most pronounced in weakly-supported electrolyte solutions for electrodes as large as a few 10s of nm [92,93], but they have also been observed in fully supported media for electrodes with a critical dimension of a few nm [94,95]. However, it is unclear if these effects indicate a failure of the classical theory, or if they simply represent a modified (or Frumkin-corrected) heterogeneous rate constant within the Butler-Volmer model. Indeed, the altered mass transport regime associated with nanoelectrodes compared to microelectrodes may lead to a change in mechanism or rate-determining step, as reported for the electrochemical reduction of aqueous hydrogen peroxide [96], the reduction of 4-nitrophenol [97] and the hydrogen evolution reaction [98] at silver nanoparticle arrays. It is also worth noting that the enhanced mass transport at nanoelectrodes compared with microelectrodes extends the potential range under which the current is kinetically limited by electron transfer, so deviations associated with higher overpotentials are likely more easily observed. 5. Applications of the Butler-Volmer equation “…any study of chemical kinetics which ignores the mechanism is profitless.” – E.A. Guggenheim, Thermodynamics: An Advanced Treatment for Chemists and Physicists, Preface to the 3rd Edition, 1956 [23] 4.3. Fundamental alternatives to the Butler-Volmer equation: Marcus-Hush theory Experimental deviations from Butler-Volmer behaviour have inspired research into alternative theories of electron transfer. Most recently, Marcus-Hush theory has received considerable attention [99]. A detailed consideration of this theory falls beyond the remit of this review and can be found elsewhere [1,100–102], but two key distinctions of MarcusHush theory are that: (i) it is parameterised by the molecular reorganisation energy (λ) associated with electron transfer, a physical quantity carrying much more fundamental insight than the empirical transfer coefficient of 5.1. Voltammetric systems As discussed in the previous section, the Butler-Volmer equation is applied widely to describe ideal systems in voltammetry and chronoamperometry: especially in the case of relatively rapid (quasi-reversible) redox systems such as ferrocene, hexacyanoferrate (ferro/ferricyanide), etc. In this application, the electroanalytical Butler-Volmer equation has served as the central electrode kinetic rate law for the wider 11 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 Bernardi-Verbrugge formulation by Dickinson and Hinds [27], can be traced historically to an incorrect application [131] of the ‘general’ Butler-Volmer equation given in the Newman Electrochemical Systems textbook to multi-electron reactions (n > 1), which are in fact carefully excluded by Newman [4], combined with the use of a concentrationindependent expression for overpotential. Regrettably, this small error, which may violate the Nernst equation and alter the implied reaction order of the anodic and cathodic processes, has been repeated unchecked by over 100 subsequent publications. Similar applications of the Butler-Volmer equation to evaluate activation losses of the electrocatalytic reactions of hydrogen and oxygen may be found in the polymer electrolyte membrane water electrolyser (PEMWE) [132,133] and solid-oxide fuel cell / water electrolyser (SOFC / SOWE) fields [134–138]. The careful discussion in the early work of Austin on alkaline fuel cells [36] means that the concentration dependence implicit in the engineer's Butler-Volmer equation is often described consistently [139], even if the Butler-Volmer equation may not be always suitable to the multistep processes, or necessary for reactions at higher overpotential. Nonetheless, the apparent failure to recognise that overpotential depends on reactant concentrations at the electrode surface – and so may have a nonlinear, current density-dependent relation to polarisation – has led to ad hoc empirical modifications of the ButlerVolmer equation [132,140] that, while empirically successful in describing measured cell data, depart increasingly from the underlying electrochemical concept of simultaneous, activated anodic and cathodic processes. One recent, detailed PEMWE model seems to treat the exchange current density as a parameterisable constant, rather than as a quantity with a concentration dependence; possibly, the implied limitation to conditions varying negligibly from ideal mass transport is intentional [133]. As of November 2019, the commercial software COMSOL Multiphysics, which is widely used in modelling for electrochemical energy conversion devices, implements the Bockris-Reddy-Gamboa-Aldeco expressions (Eq. (2.18)) as a supplementary alternative to the law of mass action for Butler-Volmer equation implementation, guaranteeing thermodynamic consistency in the resulting expression [28,49]. science of voltammetric measurements: as, for example, in the semianalytical results of Nicholson and Shain [115]. A Butler-Volmer equation is behind the scenes in almost every voltammogram presented in the comprehensive exploration of electrochemical mechanism and electrode size effects in Understanding Voltammetry by Compton and Banks [7], as well as publications from the Compton research group on the study of electrochemically heterogeneous electrodes [116] and systems with scarce supporting electrolyte [117], amongst other topics. Further, the Butler-Volmer equation gives the rate of electron transfer in foundational works on mechanistic analysis of complex reaction systems by means of voltammetry, such as the “scheme of squares” for combined proton and electron transfers [118,119], and the method of protein film voltammetry for combined catalytic and electron transfer processes [120]. A relevant cautionary note relates to the use of voltammetry to characterise electrode kinetics at planar electrodes coated with films of micro- and nanoparticles. This is particularly common practice in electrocatalyst screening, in which a dispersion of supported nanoparticles is typically cast onto a RDE to create a porous layer and hydrodynamic voltammetry performed to quantify catalytic phenomena. However, it has been demonstrated recently both experimentally [40] and theoretically [121,122] that the complex mass transport within the porous layer can lead to voltammetric shifts uncorrelated to electrode kinetics, potentially undermining the validity of this method to rank electrocatalytic performance. This issue extends in principle to any electrode that exhibits heterogeneity or non-uniform accessibility of the electroactive surface – including the familiar microdisc electrode [123] – and underlines the necessity for a well-characterised mass transport regime at the electrode surface when undertaking kinetic studies, whether based on the Butler-Volmer model or otherwise [39]. 5.2. H2 and O2 electrocatalytic processes A recent review by Dickinson and Hinds on electrode kinetic equations used in polymer electrolyte membrane fuel cell (PEMFC) models [27] revealed that the majority of all models published historically for these systems used either the Butler-Volmer equation or a variant thereof, even though both the H2 and O2 electrocatalytic processes are multistep processes to which the Butler-Volmer equation need not necessarily apply. The Butler-Volmer equation for the PEMFC application had been reviewed previously by Mann [124]. Dickinson and Hinds argued that the Butler-Volmer equation introduces superfluous detail and its ‘generality’ in incorporating both anodic and cathodic current density is artificial [27] – while the hydrogen oxidation reaction is typically sufficiently fast to be treated using a linear approximation (Eq. (3.4)) under normal operating conditions for PEMFCs, the oxygen reduction reaction is sufficiently slow that the anodic term is negligible. Hence, the linear and Tafel simplifications respectively may apply; these may hold empirically for multistep processes even when the full ButlerVolmer equation is not expected to hold good. Broadly, the detailed kinetic study of multistep processes such as the hydrogen oxidation and oxygen reduction reactions require a specific mechanism to be postulated, including adsorbed intermediates. Deviations from the Butler-Volmer equation due to the multistep characteristics of the hydrogen oxidation and hydrogen evolution reactions have been discussed in detail by Kucernak and Zalitis [21], and the inapplicability of the Butler-Volmer equation to the general case of potential-dependent adsorbate coverage has long been understood [26]. Various researchers have recently explored more mechanistically detailed alternatives, for both fuel cell and electrolyser applications, in combination with a variety of experimental validation techniques [21,125–130]. One concerning trend noted in PEMFC models since 1992 is the common misuse of the engineer's Butler-Volmer equation with overpotential treated as a function only of a certain electrode polarisation, without reference to the concentration or activity dependence of the equilibrium potential of the half-cell. Many works also ignore the concentration-dependence of exchange current density in a model that is defined under conditions where concentration may vary. These approaches, collectively termed the 5.3. Energy storage device reactions In the “Newman model” for mass and charge transport in the concentrated electrolyte of a lithium-ion battery [141,142], the specific choice of the lithium-ion electrochemical potential as defining the electrolyte potential [4,141] yields some discrepancies of formulation for the Butler-Volmer equation, without altering any inherent assumptions. The practical consequence is that the overpotential loses its dependence on the lithium electrolyte concentration, so that: i ¼ expðð1−αÞ f ηins Þ− expð−αf ηins Þ i0 ð5:1Þ where i0 ¼ k 0 Fcs;sat ð1 θÞ1α θα ηins ¼ E−U ins cl cl;ref 1α ð5:2Þ ð5:3Þ in which cl is the electrolyte concentration, cs,sat is the concentration of inserted Li in the insertion compound at saturation, and θ is the ratio of inserted Li concentration to its saturated value. Uins is the equilibrium potential (open-circuit potential) of Li+ insertion to the active material, as a function of the degree of lithiation of the material and measured with respect to the Li/Li+ electrode. The specific form of the exchange current density in (5.2) arises due to the treatment of vacant sites for Li+ insertion as a ‘reactant’ in the insertion process [140]. Latz et al. have remarked that the singularities at θ = 0 and 1, at which ηins ➔ ± ∞ while i0 ➔ 0, create problems for numerical 12 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 CRediT authorship contribution statement implementation [143]. To circumvent this, they proposed a new ButlerVolmer equation form usable in the context of insertion reactions, derived from an empirical law of mass action for activities (but see the criticism on this topic under “Should the Butler-Volmer equation be expressed in terms of activity or concentration?” above) in combination with an appropriate thermodynamic treatment. Their result gives a qualitatively different concentration dependence for the exchange current density, due to the basis in a non-ideal law of mass action. While their proposed ButlerVolmer equation is thermodynamically consistent at equilibrium (as is Newman's Eq. (5.1)), thermodynamic arguments cannot indicate which form of the concentration dependence of exchange current density is more appropriate; this should be a matter for experimental investigation. The Butler-Volmer equation is also widely applied to electrode kinetics in vanadium redox-flow battery (VRFB) applications, since the redox species involved undergo one-electron electrode reactions. The typical form is either the engineer's Butler-Volmer equation [144], or later the practical Butler-Volmer equation [145,146]. When adopting the practical ButlerVolmer equation, it is common to use the (steadily varying) bulk concentrations of redox species in each tank solution as the reference state (see the discussion under “Relating the three principal forms of the Butler-Volmer equation” above). Confusingly, the notation η is used in this case as well as for the original engineer's Butler-Volmer equation – readers should be alert to the change in meaning! Electrode kinetics for various other redox flow battery (RFB) chemistries have also been considered using the Butler-Volmer equation, including zinc‑bromine [147], hydrogen‑bromine [148,149], and vanadium‑cerium [150]. Amongst these, we note that Kok et al. erroneously combine a concentration-independent ηref with the engineer's Butler-Volmer equation [149]. We note that the necessary involvement of protons in the interconversion of [VO]2+ and [VO2]+ ions in the VRFB (oxidation states +4 and +5 respectively) implies that the overall electrochemical reaction is not single-step, even if it is one-electron – as with the hydrogen oxidation reaction in the PEMFC context, the Butler-Volmer equation is neither necessarily correct [21] nor necessarily required [27] to describe the electrode kinetics of such a reaction. Edmund J.F. Dickinson: Conceptualization, Investigation, Methodology, Writing - original draft, Writing - review & editing. Andrew J. Wain: Conceptualization, Investigation, Methodology, Writing - original draft, Writing - review & editing Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. Acknowledgements E.J.F.D. and A.J.W. express their gratitude to their mentor and friend Richard Compton for his constant guidance, tuition, support and inspiration. They have him to thank for their careers in electrochemistry. This work was funded by the National Measurement System of the UK Department of Business, Energy and Industrial Strategy. Alan Turnbull (NPL), Gareth Hinds (NPL), and the anonymous reviewers provided helpful comments on the text. Appendix A Table of Abbreviations Abbreviation Meaning AC ads EIS PEMFC PEMWE RDE RFB SOFC SOWE VRFB 6. Conclusion alternating current adsorbed electrochemical impedance spectroscopy polymer electrolyte membrane fuel cell polymer electrolyte membrane water electrolyser rotating disc electrode redox flow battery solid oxide fuel cell solid oxide water electrolyser vanadium redox flow battery Table of Subscripts The Butler-Volmer equation is encountered in the analysis of electrochemical cells in a wide variety of disciplines. Almost equal to the number of diverse applications of the equation is the number of mathematical forms it can take. We have attempted to provide a clear summary of the various required concepts to interpret electrode kinetic equations, including exchange current density and overpotential. In particular, we have stressed the need for notational and derivational care in moving between a ButlerVolmer equation expressed in terms of the local overpotential (η), which depends on concentrations at the electrode surface, and an equation expressed in terms of overpotential with respect to reference conditions (ηref), in which case the exchange current density and open circuit potential are constants (defined at the reference conditions), but concentration-dependent factors are then required alongside. Additionally, we have demonstrated that the Butler-Volmer equation in its common form can be defined in terms of a limited set of empirical assumptions, without requiring recourse to quantum mechanics; fundamental interpretation of the transfer coefficients lends greater insight, of course, but will also constrain the implied range of physical applicability of the equation. As with any theoretical equation, when using the Butler-Volmer equation and considering its ‘validity’, it is worthwhile to consider the purpose of the kinetic analysis, as well as the evidence for suitability of the kinetic treatment. Careful experimental affirmation through the techniques discussed above underpins the selection of a suitable kinetic equation. Simpler equations may well be adequate in many applications; more complex, mechanistic equations may be required in others. Subscript Meaning a ano c cat cell ct eq i ins IR m O R ref ‡ in anodic direction at anode of electrochemical cell in cathodic direction at cathode of electrochemical cell of two-electrode cell charge transfer at equilibrium of species i for Li+ insertion (Newman model) with relation to resistive losses with respect to current density direction m oxidised species in redox couple reduced species in redox couple at defined reference conditions of transition state Table of Symbols Symbol Unit Meaning Am ai ai,ref c0 ci ci,ref cl cref V 1 1 mol m−3 mol m−3 mol m−3 mol m−3 mol m−3 Tafel slope in current density direction m Activity of species i Reference activity of species i Standard concentration (= 1 M) Concentration of species i (at electrode surface) Reference concentration of species i Lithium electrolyte concentration (Newman model) Reference concentration, electroanalytical Butler-Volmer equation (continued on next page) 13 E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 [15] [16] [17] [18] [19] (continued) Symbol Unit Meaning cs,sat D E E0f 0 E‡, m mol m−3 m2 s−1 V V V E Ecell Eeq,cell Eeq,ref ΔEIR F f gm geq i im i0 i0,ref i∗m V V V V V C mol−1 V−1 1 1 A m−2 A m−2 A m−2 A m−2 A m−2 km k0 km,ref m s−1 m s−1 m s−1 n R Rct T Uins 1 J K−1 mol−1 Ω m2 K V v vi V s−1 1 α 1 αm γi 1 1 η ηref V V ηeq,ref V ηins θ V 1 λ Δ‡μm J mol−1 J mol−1 Inserted Li concentration at saturation (Newman model) Diffusion coefficient Electrode polarisation Formal potential of redox couple Standard reduction potential between transition state and reactant in current density direction m Reference value of electrode polarisation for Tafel equation Cell voltage Equilibrium cell voltage Equilibrium potential of reference conditions Cell resistive losses Faraday constant Inverse thermal voltage, ≡ F/RT Concentration dependence in current density direction m Concentration-dependent function in Nernst equation Current density Component of current density in direction m Exchange current density Exchange current density at reference conditions Reference current density for Tafel equation, in current density direction m Heterogeneous rate constant in current density direction m Heterogeneous rate constant Heterogeneous rate constant in current density direction m at reference value of electrode polarisation Number of electrons transferred Gas constant Charge transfer resistance Temperature Equilibrium potential (open-circuit potential) for Li+ insertion, vs. Li/Li+ (Newman model) Voltammetric scan rate Stoichiometric coefficient of species i in redox process, in direction of reduction Transfer coefficient in reductive direction, electroanalytical Butler-Volmer equation and Newman model Transfer coefficient in current density direction m Empirical reaction order coefficient for species i, Bockris-Reddy-Gamboa-Aldeco equations Overpotential, = E − Eeq Overpotential with respect to reference conditions, = E − Eeq,ref Difference between equilibrium potential and its value at reference conditions, also identified as mass transfer overpotential, = Eeq − Eeq,ref Overpotential for Li+ insertion in Newman model Ratio of inserted Li concentration to saturation, Newman model Reorganisation energy in Marcus-Hush theory Electrochemical potential difference [20] [21] [22] [23] [24] [25] [26] [27] [28] [29] [30] [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] [46] [47] [48] [49] [50] [51] [52] [53] Note: all electrode potentials E defined for a single electrode are measured with respect to a choice of reference electrode. [54] [55] [56] References [57] [1] C. Batchelor-McAuley, E. Kätelhön, E.O. Barnes, R.G. Compton, E. Laborda, A. Molina, ChemistryOPEN 4 (2015) 224. [2] K.J. Vetter, Electrochemical Kinetics: Theoretical and Experimental Aspects, Academic Press Inc, New York / London, 1967. [3] A.J. Bard, L.R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Second ed. John Wiley & Sons, Inc, Hoboken, NJ, 2001. [4] J. Newman, K.E. Thomas-Alyea, Electrochemical Systems, Third ed. John Wiley & Sons, Inc, Hoboken, NJ, 2004. [5] J. Albery, Electrode Kinetics, Clarendon Press, Oxford, 1975. [6] D.H. Evans, Chem. Rev. 108 (2008) 2113. [7] R.G. Compton, C.E. Banks, Understanding Voltammetry, 2nd ed. Imperial College Press, London, 2011. [8] R. Guidelli, R.G. Compton, J.M. Feliu, E. Gileadi, J. Lipkowski, W. Schmickler, S. Trasatti, Pure Appl. Chem. 86 (2014) 245. [9] R. Guidelli, R.G. Compton, J.M. Feliu, E. Gileadi, J. Lipkowski, W. Schmickler, S. Trasatti, Pure Appl. Chem. 86 (2014) 259. [10] N. Tanaka, R. Tamamushi, Electrochim. Acta 9 (1964) 963. [11] E.J. Calvo, Fundamentals. The basics of electrode reactions, in: C.H. Bamford, R.G. Compton (Eds.), Electrode Kinetics: Principles and Methodology, Elsevier, 1986. [12] S. Srinivasan, Fuel Cells: From Fundamentals to Applications, Springer Science+Business Media, LLC, New York, 2006. [13] R.E. White, S.E. Lorimer, R. Darby, J. Electrochem. Soc. 130 (1983) 1123. [14] J.A.V. Butler, Trans. Faraday Soc. 19 (1924) 729. [58] [59] [60] [61] [62] [63] [64] [65] [66] [67] [68] [69] [70] 14 J.A.V. Butler, Trans. Faraday Soc. 19 (1924) 734. J.A.V. Butler, Trans. Faraday Soc. 28 (1932) 379. T. Erdey-Grúz, M. Volmer, Z. Physik. Chem. 150A (1930) 203. G. Inzelt, J. Solid State Electrochem. 15 (2011) 1373. R.G. Compton, G.H.W. Sanders, Electrode Potentials, Oxford University Press, Oxford, 1996. R. Peters, Z. Physik. Chem. 26 (1898) 193. A.R. Kucernak, C. Zalitis, J. Phys. Chem. C 120 (2016), 10721. R.W. Gurney, Proc. Roy. Soc. A 134 (1931) 137. E.A. Guggenheim, Thermodynamics: An Advanced Treatment for Chemists and Physicists, Fifth, Revised Ed, North-Holland Publishing Company, Amsterdam, 1967. J.O.M. Bockris, Electrode kinetics, in: J.O.M. Bockris, B.E. Conway (Eds.), Modern Aspects of Electrochemistry, Ch. 4, Butterworths Scientific Publications, London 1954, p. 180. P. Delahay, Double Layer and Electrode Kinetics, Interscience Publishers, New York, 1965. T. Erdey-Grúz, Kinetics of Electrode Processes, English language ed. Adam Hilger Ltd, London, 1972. E.J.F. Dickinson, G. Hinds, J. Electrochem. Soc. 166 (2019) F221. J.O.M. Bockris, A.K.N. Reddy, M.E. Gamboa-Aldeco, Modern Electrochemistry 2A: Fundamentals of Electrodics, Springer US, New York, 2000. C.H. Hamann, A. Hamnett, W. Vielstich, Electrochemistry, Second, Completely Revised and Updated Ed, Wiley-VCH, Weinheim, Germany, 2007. J. Horiuti, M. Polányi, Acta Physicochim. URSS 2 (1935) 505. J. Horiuti, M. Polanyi, J. Molec. Catal. A - Chemical 199 (2003) 185. R. Parsons, Trans. Faraday Soc. 47 (1951) 1332. H. Eyring, J. Chem. Phys. 3 (1935) 107. K.J. Laidler, M.C. King, J. Phys. Chem. 87 (1983) 2657. E.A. Guggenheim, J. Phys. Chem. 33 (1929) 842. L.G. Austin, The electrochemical theory of fuel cells, in: C. Berger (Ed.), Handbook of Fuel Cell Technology, Prentice-Hall, Inc, Englewood Cliffs, NJ 1968, p. 3. W. Scheider, J. Phys. Chem. 79 (1975) 127. P.M. Biesheuvel, M. van Soestbergen, M.Z. Bazant, Electrochim. Acta 54 (2009) 4857. J. Masa, C. Batchelor-McAuley, W. Schuhmann, R.G. Compton, Nano Res. 7 (2014) 71. L. Chen, E. Kätelhön, R.G. Compton, Appl. Mater. Today 16 (2019) 141. S. Balasubramanian, A.Z. Weber, Continuum, macroscopic modeling of polymerelectrolyte fuel cells, in: A.A. Franco, M.L. Doublet, W.G. Bessler (Eds.), Physical Multiscale Modeling and Numerical Simulation of Electrochemical Devices for Energy Conversion and Storage: From Theory to Engineering to Practice, Springer-Verlag, London 2016, p. 91 , Ch. 4. S.R. de Groot, P. Mazur, in: Dover (Ed.), Non-Equilibrium Thermodynamics, Dover Publications, Inc, New York, 1984. M. Pavelka, V. Klika, P. Vágner, F. Maršík, Appl. Energy 137 (2015) 158. W. Dreyer, C. Guhlke, R. Muller, Phys. Chem. Chem. Phys. 18 (2016), 24966. . A.C. Fisher, Electrode Dynamics, Oxford University Press, Oxford, 1996. M. Rudolph, D.P. Reddy, S.W. Feldberg, Anal. Chem. 66 (1994) 589A. R.G. Compton, E. Laborda, K.R. Ward, Understanding Voltammetry: Simulation of Electrode Processes, Imperial College Press, London, 2014. D. Britz, J. Strutwolf, Digital Simulation in Electrochemistry, Fourth ed. Springer International Publishing, New York, 2016. COMSOL AB, www.comsol.com 2019. H. Matsuda, Y. Ayabe, Z. Elektrochem. 59 (1955) 494. J.O. Howell, R.M. Wightman, Anal. Chem. 56 (1984) 524. C. Amatore, B. Fosset, J. Bartelt, M.R. Deakin, R.M. Wightman, J. Electroanal. Chem. Interf. Electrochem. 256 (1988) 255. C.P. Andrieux, D. Garreau, P. Hapiot, J. Pinson, J.M. Savéant, J. Electroanal. Chem. Interf. Electrochem. 243 (1988) 321. D.O. Wipf, E.W. Kristensen, M.R. Deakin, R.M. Wightman, Anal. Chem. 60 (1988) 306. J.V. Macpherson, S. Marcar, P.R. Unwin, Anal. Chem. 66 (1994) 2175. J.V. Macpherson, M.A. Beeston, P.R. Unwin, J. Chem. Soc. Faraday Trans. 91 (1995) 899. N.V. Rees, O.V. Klymenko, B.A. Coles, R.G. Compton, J. Phys. Chem. B 107 (2003), 13649. . N.V. Rees, R.A.W. Dryfe, J.A. Cooper, B.A. Coles, R.G. Compton, S.G. Davies, T.D. McCarthy, J. Phys. Chem. 99 (1995) 7096. A.D. Clegg, N.V. Rees, O.V. Klymenko, B.A. Coles, R.G. Compton, J. Am. Chem. Soc. 126 (2004) 6185. D. Suwatchara, M.C. Henstridge, N.V. Rees, R.G. Compton, J. Phys. Chem. C 115 (2011), 14876. R.L. Birke, M.-H. Kim, M. Strassfeld, Anal. Chem. 53 (1981) 852. W.S. Go, J.J. O'Dea, J. Osteryoung, J. Electroanal. Chem. Interf. Electrochem. 255 (1988) 21. Á. Molina, E. Laborda, E.I. Rogers, F. Martínez-Ortiz, C. Serna, J.G. Limon-Petersen, N.V. Rees, R.G. Compton, J. Electroanal. Chem. 634 (2009) 73. Á. Molina, F. Martínez-Ortiz, E. Laborda, R.G. Compton, Electrochim. Acta 55 (2010) 5163. M.A. Mann, J.C. Helfrick, L.A. Bottomley, Anal. Chem. 86 (2014) 8183. P. Dauphin-Ducharme, N. Arroyo-Currás, M. Kurnik, G. Ortega, H. Li, K.W. Plaxco, Langmuir 33 (2017) 4407. L. Camacho, J.J. Ruiz, A. Molina, C. Serna, J. Electroanal. Chem. 365 (1994) 97. A. Molina, M.M. Moreno, C. Serna, L. Camacho, Electrochem. Commun. 3 (2001) 324. E. Laborda, E.I. Rogers, F. Martínez-Ortiz, Á. Molina, R.G. Compton, Electroanalysis 22 (2010) 2784. E. Laborda, E.I. Rogers, F. Martínez-Ortiz, Á. Molina, R.G. Compton, Electrochim. Acta 55 (2010) 6577. E.J.F. Dickinson, A.J. Wain / Journal of Electroanalytical Chemistry 872 (2020) 114145 [110] E.E.L. Tanner, E.O. Barnes, P. Goodrich, C. Hardacre, R.G. Compton, J. Phys. Chem. C 119 (2015) 3634. [111] E.E.L. Tanner, E.O. Barnes, C.B. Tickell, P. Goodrich, C. Hardacre, R.G. Compton, J. Phys. Chem. C 119 (2015) 7360. [112] Y. Wang, E. Laborda, M.C. Henstridge, F. Martinez-Ortiz, A. Molina, R.G. Compton, J. Electroanal. Chem. 668 (2012) 7. [113] A.M. Becka, C.J. Miller, J. Phys. Chem. 96 (1992) 2657. [114] J. Li, G.F. Kennedy, A.M. Bond, J. Zhang, J. Phys. Chem. C 122 (2018) 9009. [115] R.S. Nicholson, I. Shain, Anal. Chem. 36 (1964) 706. [116] T.J. Davies, R.R. Moore, C.E. Banks, R.G. Compton, J. Electroanal. Chem. 574 (2004) 123. [117] E.J.F. Dickinson, J.G. Limon-Petersen, N.V. Rees, R.G. Compton, J. Phys. Chem. C 113 (2009) 11157. [118] J. Jacq, J. Electroanal. Chem. Interf. Electrochem. 29 (1971) 149. [119] D. Menshykau, C. Batchelor-McAuley, R.G. Compton, J. Electroanal. Chem. 651 (2011) 118. [120] J. Hirst, Biochim. Biophys. Acta Bioenerg. 1757 (2006) 225. [121] E. Kätelhön, L. Chen, R.G. Compton, Appl. Mater. Today 15 (2019) 139. [122] N. Blanc, K. Tschulik, J. Electroanal. Chem., this issue (2020). [123] S.R. Belding, E.I. Rogers, R.G. Compton, J. Phys. Chem. C 113 (2009) 4202. [124] R.F. Mann, J.C. Amphlett, B.A. Peppley, C.P. Thurgood, J. Power Sources 161 (2006) 775. [125] J.X. Wang, T.E. Springer, R.R. Adzic, J. Electrochem. Soc. 153 (2006) A1732. [126] J.X. Wang, J.L. Zhang, R.R. Adzic, J. Phys. Chem. A 111 (2007) 12702. [127] A. Holewinski, S. Linic, J. Electrochem. Soc. 159 (2012) H864. [128] M. Markiewicz, C. Zalitis, A. Kucernak, Electrochim. Acta 179 (2015) 126. [129] B. Jayasankar, K. Karan, Electrochim. Acta 273 (2018) 367. [130] J. Huang, J.B. Zhang, M. Eikerling, Phys. Chem. Chem. Phys. 20 (2018) 11776. [131] D.M. Bernardi, M.W. Verbrugge, J. Electrochem. Soc. 139 (1992) 2477. [132] K.S. Agbli, M.C. Péra, D. Hissel, O. Rallières, C. Turpin, I. Doumbia, Int. J. Hydrog. Energy 36 (2011) 1382. [133] F. Aubras, J. Deseure, J.-J.A. Kadjo, I. Dedigama, J. Majasan, B. Grondin-Perez, J.P. Chabriat, D.J.L. Brett, Int. J. Hydrog. Energy 42 (2017) 26203. [134] S.H. Chan, K.A. Khor, Z.T. Xia, J. Power Sources 93 (2001) 130. [135] S. Kakaç, A. Pramuanjaroenkij, X.Y. Zhou, Int. J. Hydrog. Energy 32 (2007) 761. [136] M. Ni, M.K.H. Leung, D.Y.C. Leung, Electrochim. Acta 52 (2007) 6707. [137] S.A. Hajimolana, M.A. Hussain, W.M.A.W. Daud, M. Soroush, A. Shamiri, Renew. Sust. Energ. Rev. 15 (2011) 1893. [138] K.N. Grew, W.K.S. Chiu, J. Power Sources 199 (2012) 1. [139] M. García-Camprubí, S. Izquierdo, N. Fueyo, Renew. Sust. Energ. Rev. 33 (2014) 701. [140] A. Doddathimmaiah, J. Andrews, Int. J. Hydrog. Energy 34 (2009) 8157. [141] M. Doyle, T.F. Fuller, J. Newman, J. Electrochem. Soc. 140 (1993) 1526. [142] K.E. Thomas, J. Newman, R.M. Darling, Mathematical modeling of lithium batteries, in: W.A. van Schalkwijk, B. Scrosati (Eds.), Advances in Lithium-Ion Batteries, Ch. 12, Kluwer Academic/Plenum Publishers, New York 2002, p. 345. [143] A. Latz, J. Zausch, Electrochim. Acta 110 (2013) 358. [144] A.A. Shah, M.J. Watt-Smith, F.C. Walsh, Electrochim. Acta 53 (2008) 8087. [145] D.J. You, H.M. Zhang, J. Chen, Electrochim. Acta 54 (2009) 6827. [146] K.W. Knehr, E. Agar, C.R. Dennison, A.R. Kalidindi, E.C. Kumbur, J. Electrochem. Soc. 159 (2012) A1446. [147] M.J. Mader, R.E. White, J. Electrochem. Soc. 133 (1986) 1297. [148] R. Zhang, J.W. Weidner, J. Appl. Electrochem. 41 (2011) 1245. [149] M.D.R. Kok, A. Khalifa, J.T. Gostick, J. Electrochem. Soc. 163 (2016) A1408. [150] S. Smith, I. Firdous, Q.H. Wang, S. Esmalla, W.A. Daoud, Electrochim. Acta (2019) 328. [71] D. Jadreško, M. Zelić, J. Electroanal. Chem. 707 (2013) 20. [72] E. Laborda, M.C. Henstridge, A. Molina, F. Martinez-Ortiz, R.G. Compton, J. Electroanal. Chem. 660 (2011) 169. [73] M.E. Orazem, B. Tribollet, Electrochemical Impedance Spectroscopy, John Wiley & Sons, Inc, 2008. [74] A.M. Bond, N.W. Duffy, S.-X. Guo, J. Zhang, D. Elton, Anal. Chem. 77 (186 A) (2005). [75] E. Mashkina, A.M. Bond, Anal. Chem. 83 (2011) 1791. [76] G.P. Morris, A.N. Simonov, E.A. Mashkina, R. Bordas, K. Gillow, R.E. Baker, D.J. Gavaghan, A.M. Bond, Anal. Chem. 85 (2013), 11780. [77] J. Li, G.F. Kennedy, L. Gundry, A.M. Bond, J. Zhang, Anal. Chem. 91 (2019) 5303. [78] G.P. Stevenson, R.E. Baker, G.F. Kennedy, A.M. Bond, D.J. Gavaghan, K. Gillow, Phys. Chem. Chem. Phys. 15 (2013) 2210. [79] J. Li, C.L. Bentley, A.M. Bond, J. Zhang, Anal. Chem. 88 (2016) 2367. [80] M. Tsionsky, A.J. Bard, M.V. Mirkin, J. Am. Chem. Soc. 119 (1997), 10785. . [81] A.L. Barker, P.R. Unwin, S. Amemiya, J. Zhou, A.J. Bard, J. Phys. Chem. B 103 (1999) 7260. [82] S. Amemiya, Z. Ding, J. Zhou, A.J. Bard, J. Electroanal. Chem. 483 (2000) 7. [83] P. Sun, F. Li, Y. Chen, M. Zhang, Z. Zhang, Z. Gao, Y. Shao, J. Am. Chem. Soc. 125 (2003) 9600. [84] S. Fletcher, T.S. Varley, Phys. Chem. Chem. Phys. 13 (2011) 5359. [85] J.M. Savéant, D. Tessier, J. Electroanal. Chem. Interf. Electrochem. 65 (1975) 57. [86] J.-M. Savéant, D. Tessier, Faraday Discuss. 74 (1982) 57. [87] P. Sun, M.V. Mirkin, Anal. Chem. 78 (2006) 6526. [88] S.M. Oja, M. Wood, B. Zhang, Anal. Chem. 85 (2013) 473. [89] S.L. Chen, Y.W. Liu, Phys. Chem. Chem. Phys. 16 (2014) 635. [90] P.H. Robbs, N.V. Rees, Phys. Chem. Chem. Phys. 18 (2016), 24812. . [91] A.N. Frumkin, G.M. Florianovich, Dokl. Akad. Nauk SSSR 80 (1951) 907. [92] S. Chen, A. Kucernak, J. Phys. Chem. B 106 (2002) 9396. [93] J.J. Watkins, H.S. White, Langmuir 20 (2004) 5474. [94] Y. Sun, Y. Liu, Z. Liang, L. Xiong, A. Wang, S. Chen, J. Phys. Chem. C 113 (2009) 9878. [95] Y. Liu, S. Chen, J. Phys. Chem. C 116 (2012) 13594. [96] F.W. Campbell, S.R. Belding, R. Baron, L. Xiao, R.G. Compton, J. Phys. Chem. C 113 (2009) 9053. [97] F.W. Campbell, S.R. Belding, R.G. Compton, ChemPhysChem 11 (2010) 2820. [98] F.W. Campbell, S.R. Belding, R. Baron, L. Xiao, R.G. Compton, J. Phys. Chem. C 113 (2009) 14852. [99] C.E.D. Chidsey, Science 251 (1991) 919. [100] M.C. Henstridge, Y.J. Wang, J.G. Limon-Petersen, E. Laborda, R.G. Compton, Chem. Phys. Lett. 517 (2011) 29. [101] M.C. Henstridge, E. Laborda, Y.J. Wang, D. Suwatchara, N. Rees, A. Molina, F. Martinez-Ortiz, R.G. Compton, J. Electroanal. Chem. 672 (2012) 45. [102] E. Laborda, M.C. Henstridge, C. Batchelor-McAuley, R.G. Compton, Chem. Soc. Rev. 42 (2013) 4894. [103] S.W. Feldberg, Anal. Chem. 82 (2010) 5176. [104] M.C. Henstridge, N.V. Rees, R.G. Compton, J. Electroanal. Chem. 687 (2012) 79. [105] E. Laborda, D. Suwatchara, N.V. Rees, M.C. Henstridge, A. Molina, R.G. Compton, Electrochim. Acta 110 (2013) 772. [106] E. Laborda, Y.J. Wang, M.C. Henstridge, F. Martinez-Ortiz, A. Molina, R.G. Compton, Chem. Phys. Lett. 512 (2011) 133. [107] D. Suwatchara, M.C. Henstridge, N.V. Rees, E. Laborda, R.G. Compton, J. Electroanal. Chem. 677 (2012) 120. [108] D. Suwatchara, N.V. Rees, M.C. Henstridge, E. Laborda, R.G. Compton, J. Electroanal. Chem. 665 (2012) 38. [109] D. Suwatchara, N.V. Rees, M.C. Henstridge, E. Laborda, R.G. Compton, J. Electroanal. Chem. 685 (2012) 53. 15