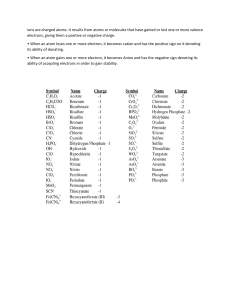
Gaffney-Jeffrey-S. -Marley-Nancy-A-General-chemistry-for-engineers-Elsevier-2018
advertisement

GENERAL CHEMISTRY FOR ENGINEERS GENERAL CHEMISTRY FOR ENGINEERS JEFFREY S. GAFFNEY NANCY A. MARLEY Elsevier Radarweg 29, PO Box 211, 1000 AE Amsterdam, Netherlands The Boulevard, Langford Lane, Kidlington, Oxford OX5 1GB, United Kingdom 50 Hampshire Street, 5th Floor, Cambridge, MA 02139, United States # 2018 Elsevier Inc. All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, or any information storage and retrieval system, without permission in writing from the publisher. Details on how to seek permission, further information about the Publisher’s permissions policies and our arrangements with organizations such as the Copyright Clearance Center and the Copyright Licensing Agency, can be found at our website: www.elsevier.com/permissions. This book and the individual contributions contained in it are protected under copyright by the Publisher (other than as may be noted herein). Notices Knowledge and best practice in this field are constantly changing. As new research and experience broaden our understanding, changes in research methods, professional practices, or medical treatment may become necessary. Practitioners and researchers must always rely on their own experience and knowledge in evaluating and using any information, methods, compounds, or experiments described herein. In using such information or methods they should be mindful of their own safety and the safety of others, including parties for whom they have a professional responsibility. To the fullest extent of the law, neither the Publisher nor the authors, contributors, or editors, assume any liability for any injury and/or damage to persons or property as a matter of products liability, negligence or otherwise, or from any use or operation of any methods, products, instructions, or ideas contained in the material herein. Library of Congress Cataloging-in-Publication Data A catalog record for this book is available from the Library of Congress British Library Cataloguing-in-Publication Data A catalogue record for this book is available from the British Library ISBN: 978-0-12-810425-5 For information on all Elsevier publications visit our website at https://www.elsevier.com/books-and-journals Publisher: John Fedor Acquisition Editor: Kostas Marinakis Editorial Project Manager: Amy M. Clark Production Project Manager: Prem Kumar Kaliamoorthi Cover Designer: Victoria Pearson Typeset by SPi Global, India Preface (11) use the techniques, skills, and modern engineering tools necessary for engineering practice. In 2000, the Accreditation Board for Engineering and Technology, Inc (ABET) released its “Engineering Criteria 2000,” which became the standard for accreditation of all ABET accredited Engineering Programs primarily in the United States, but also in 30 other countries. The focus of the new criteria was to be on outcomes, or what the student has learned. This was a departure from the previous focus on inputs, or what was being taught. The important learning outcomes are listed as a set of 11 skills or abilities that an engineering student must possess after graduation in order to assure that they are adequately prepared to enter the profession and perform well under the challenges of the modern world. These are: The curriculum requirements listed in the Engineering Criteria 2000 were given as general subject areas only. Details of specific required courses were not given. The curriculum requirements included: (1) one year of a combination of college level mathematics and basic sciences, with some experimental experience (2) one and one-half years of engineering topics, consisting of engineering sciences and engineering design (3) a general education component that complements the technical content of the curriculum (1) apply the knowledge of mathematics, science, and engineering (2) design and conduct experiments, as well as to analyze and interpret the data obtained from experiment (3) design a system, component, or process to meet desired needs (4) function on multidisciplinary teams (5) identify, formulate, and solve engineering problems (6) demonstrate an understanding of professional and ethical responsibility (7) communicate effectively (8) obtain the broad education necessary to understand the impact of engineering solutions in a global and societal context (9) recognize the need for and an ability to engage in life-long learning (10) acquire a knowledge of contemporary issues The “Criteria for Accrediting Engineering Programs” released by ABET for 2012–13 is a bit more specific as to the required curricula for each of its 28 Engineering Areas, listing a broad or thorough knowledge of chemistry as a requirement for 11 of the Programs. An additional 10 programs are listed as requiring a solid knowledge of materials properties and/or environmental interactions with materials as well as environmental impacts and sustainability. With the new ABET focus on ethics, communication skills, team building, and global awareness, many programs are turning to additional courses and/or including study abroad programs to address the newly added learning outcomes. This additional curriculum requirement has led many schools to reduce the two semesters of general chemistry ix x PREFACE requirement to only one semester. This has led to a problem. The first semester of any two semester general chemistry course designed for chemistry students will not fulfill the needs of an engineering student. This is because many topics important to engineering studies such as kinetics, thermodynamics, etc. are taught during the second semester and not covered in the first semester. To remedy this problem, some schools have adopted a separate one semester accelerated course for all areas of engineering requiring a knowledge of chemistry with the exception being chemical engineering programs which require more semesters of chemistry education. Since this trend is likely to continue, a one semester course in chemistry should be specially designed to adequately address the needs of engineering students, that is, with a major focus on materials, sustainable energy, safety, and the environment. To meet these goals, General Chemistry for Engineers takes an approach that first establishes a solid foundation in chemical principles for the engineering student and then focuses on how these principles can be applied to lead to the understanding of the chemistry and properties of materials by using examples that reinforce the learning process throughout the book. By understanding materials on a molecular level, a more thorough understanding of their formation and bulk properties can be achieved and predicted. The study of the reactions of materials with each other and the environment leads to the understanding of their durability and mechanisms of possible failure as well as the production of environmental hazards. Additionally, an understanding of the formation of materials that includes the energy required to produce them is key to developing sustainable energy and safe methods for their production and usage. Special attention is also given to the learning outcomes added by ABET in 2000 and specifically identified in 2012–13. Professional and ethical responsibility in the global community is addressed by the introduction of the concepts of green engineering and sustainability. Care has been given to stress the advantages of interdisciplinary teams working together and is demonstrated by presenting chemists (as well as other scientists) and engineers as partners in solving the problems of society. The engineers of the future working with chemists and other scientific disciplines will be instrumental in the development of solutions to problems facing the world today and tomorrow. These include new, safe, and more efficient energy sources, as well as new designs and processes to adapt to the consequences of climate change. The answers to these problems will only be achieved through interdisciplinary teams working together and chemistry and engineering will be at the forefront of this effort. Recognizing the need for a focused and logically organized textbook for the chemical education of our future engineers, we have attempted to combine the ABET goals and learning outcomes with the fundamental chemical understanding of thermodynamics, kinetics, materials, and their applications with specific examples. General Chemistry for Engineers is organized to start the engineering student out with the fundamentals of chemistry and each chapter builds upon the previous chapter in order for the development of a thorough understanding of the basics. Each chapter contains case studies that show how chemistry is applied to a process or measurement and how it is used to solve problems related to materials and engineering. Example problems show the student how to perform critical thinking skills in setting up chemistry problems and PREFACE determining the correct answer stressing the correct use of units. After each chapter, a list of important terms, study questions, and problems are given to assist the student in mastering the material. The book begins with covering why chemistry is important for engineers and going over basic units of measurements and the physical states of matter. This textbook takes an atom’s first approach by presenting the simple shell model of the atom, covering the atomic electronic configurations and showing how these relate to the periodic table of the elements along with periodic trends of the elements in Chapter 2. Once the student has an understanding of atom basics, Chapter 3 addresses the types of chemical bonding of atoms to form molecules after first discussing the difference between atoms and ions. Ionic, covalent, and mixed covalent/ionic bonding are covered along with molecular orbitals, geometry, polarity, and describe intermolecular forces that result from the molecular structures. The first three chapters give the students a fundamental basis of the reasons for chemical reactions that are then introduced in Chapter 4. This chapter covers the basic concepts of the mole, stoichiometry, and balancing chemical equations. The concepts of limiting reagents and percent yield are addressed, along with aqueous solubility of ionic compounds, precipitation reactions, and concentrations in aqueous solutions. Chapter 5 defines acids and bases in aqueous solutions and covers their reactions along with developing the concept of pH and other p functions used in chemistry. Buffers are explained and the method of chemical titration is introduced to the student. A historical perspective of the development of the gas laws is given in Chapter 6, leading to the Ideal Gas Law. Nonideal gas behavior, the determination and use of partial gas pressures, and gas phase chemical xi reactions are also covered in this chapter. Reversible reactions resulting in an equilibrium process and how to determine the reversible reaction equilibrium constants are detailed in Chapter 7. Le Chatelier’s principle and the use of a reaction quotient to determine concentrations after an equilibrium is disturbed is examined as well. Given the foundation set in the first seven chapters, the textbook then covers the basics in thermodynamics in Chapter 8 and chemical kinetics in Chapter 9. Chapter 10 introduces the students to oxidation-reduction chemical reactions and balancing them. The use of standard cell potentials and the Nernst equation are presented and their use in chemical electrolysis and the development and use of batteries and fuel cells that are important engineering are stressed. The properties and types of solids are reviewed in Chapter 11 keeping with the materials focus. Solution chemistry and the effects of temperature and pressure on solutions, the process of dissolution, ionic solids solubility, complexing agents, and surfactants are reviewed in Chapter 12. The chemistry of carbon is briefly covered in Chapter 13. This material is usually not covered in this detail in a normal general chemistry course, but is needed for the engineering student to have a basic understanding of how organic reactions can lead to the formation of organic polymers so often used as materials in engineering today. This chapter stresses the importance of orbital hybridization in the chemistry of carbon, and goes over the bonding and geometry of sp, sp2, and sp3 carbon atoms interactions with the common reactants, i.e., H, N, O, and halogens. This allows the student to then understand free radical and condensation reaction mechanisms involved in basic organic polymer formations. Also, it allows for the students to understand their chemical reactivity based on xii PREFACE their chemical structure and their physical properties. Nuclear and radiochemistry including radioactive decay processes, nuclear fission, and nuclear fusion are addressed in Chapter 14. This chapter gives the students an important knowledge base concerning the use of nuclear energy, along with radiation safety and the application of radioisotopes. Basic types of separation and instrumentation that are commonly used in the laboratory and in undergraduate teaching laboratories are described in Chapter 15. Considering that a typical semester is 15 weeks in length and involves 3 h of lecture accompanied by 3 h of laboratory, this book was written and organized in this manner to allow the instructor some flexibility in the material covered during the course. For example, it would be reasonable for the instructor to make use of Chapter 15 as background material for many of the engineering chemistry laboratory assignments. To that end, we have also added three appendices. The first is working with units, the second is Using Excel, and the third is a list of Standard Half-cell Potentials. All of which should be useful in connection with the student working with problems in the classroom as well as the laboratory. Having worked together with numerous engineers during our professional careers, we have recognized and agreed with ABET that it is extremely important for all engineering disciplines to have a basic understanding of chemistry and the common methods used in chemical analysis. This basic chemical knowledge is needed for engineers to be able to communicate and work effectively with chemists in solving current and future problems involving materials and sustainable energy. Working together, chemists and engineers must also attempt to make sure that those solutions are environmentally safe and sound. We hope that General Chemistry for Engineers will serve as the foundation for a one semester chemistry course for engineering students in the United States and other countries. We believe that this chemistry textbook will not only assist in the training of future engineers as per ABET accreditation, but will also be a very useful reference for trained engineers throughout their careers when a chemistry basics refresher is needed. Jeffrey S. Gaffney Nancy A. Marley Acknowledgments General Chemistry for Engineers evolved from a one semester course developed and taught by Dr. Gaffney while he was the Chair and Professor of Chemistry at the University of Arkansas at Little Rock. During the development of this course, it became clear that there was not a suitable textbook available for the engineering students in chemical basics that they needed as most of the students had minimal exposure to chemistry before taking the course. During the course of writing this book, many questions arose concerning the best ways to present the chemistry and how it would best connect with engineering. We wish to acknowledge all of the faculty members of Department of Chemistry in the College of Arts, Letters, and Science at the University of Arkansas at Little Rock for numerous helpful conversations and their constant encouragement in completing this book. We also want to acknowledge the assistance of College of Engineering at the University of Arkansas at Little Rock faculty for their encouragement. Finally, we want to thank the students in CHEM 1406 who used the first three chapters of this book in their class and gave us very useful and positive feedback that helped us to know we were on the right track in writing and publishing this book for the chemical training of future engineers. xiii C H A P T E R 1 Introduction O U T L I N E 1.1 The Role of Chemistry in Engineering 1 1.6 Separation of Mixtures 26 1.2 Green Engineering 4 Important Terms 34 1.3 Measurement and Calculations 5 Study Questions 35 1.4 The Physical States of Matter 14 Problems 37 1.5 Classification of Matter 21 1.1 THE ROLE OF CHEMISTRY IN ENGINEERING Engineering has been called an applied science. The various disciplines of engineering focus on the design and construction of structures, machines, apparatus, or processes to solve problems. This requires an in-depth knowledge of the properties of materials and a broad knowledge of science and mathematics. Although engineers use scientific principles in their designs, they must also consider economics and safety issues as well as efficiency, reliability, and ease of construction. In many cases, the best choice of materials for a design may not be economically feasible and compromises must be made. The Accreditation Board for Engineering and Technology (ABET) lists 28 different areas of engineering in their 2012–13 “Criteria for Accrediting Engineering Programs.” These range from Chemical, Biochemical, Biomolecular Engineering, which use chemical principles on a daily basis, to Systems Engineering, which is typically not directly involved with chemical principles. In between these two extremes are disciplines that require a basic knowledge of chemistry, materials science, and/or environmental science to fully understand the problems they are to solve. Even those areas of engineering that do not use chemical principles directly are involved with them indirectly because chemical principles dictate the properties and behavior of materials, electrical systems, and electronic devices, as well as energy production and environmental impacts. General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00001-1 1 # 2018 Elsevier Inc. All rights reserved. 2 1. INTRODUCTION An engineer uses the macroscopic properties of materials such as the hardness, strength, malleability, or conductivity to determine the appropriate choice of materials for a specific project. Many times, these macroscopic properties are experimentally measured. Chemistry is the study of the properties and behavior of matter on the microscopic or the atomic/molecular scale. This description of matter on the microscopic scale lays the foundation for the underlying reasons for the properties and behavior of materials observed on the macroscopic scale. By understanding the atomic and molecular forces that lead to the basic properties of materials, their macroscopic properties and their reactions to external stresses can be better understood and predicted. CASE STUDY: SCRATCH HARDNESS The scratch hardness of a material is the measure of its resistance to being scratched or abraded by a sharp object. This is a prime consideration in choosing materials for construction of mechanical tools or machines where friction is an issue. One of the oldest measures of hardness is the Mohs scale, created in 1812 by the German geologist Friedrick Mohs. This scale was originally designed to describe the hardness of minerals with diamond as the hardest known mineral at a value of 10 and talc as the softest known mineral with a value of 1. Other minerals were given intermediate values based on the ability of one mineral to scratch another mineral. Table 1.1 lists the scratch hardness of some common materials. The minerals specified by Mohs are listed in the Table along with some other materials which were determined later to fall between the levels designated by Mohs. As we shall see in later Chapters, this trend in scratch hardness can be explained by an understanding of the types of bonding between atoms in the materials on the microscopic scale. Although diamonds and graphite are both composed of carbon atoms, the hardness of the two materials differs by almost a factor of 10 on the Mohs scale. However, the Mohs scale is a relative designation and is not a linear scale. The difference between the hardness of graphite and diamond is much larger on the linear scale of Absolute Hardness. Absolute hardness is a modern scale of scratch hardness measured by a sclerometer, an instrument that determines the width of a scratch produced by a moving diamond stylus under a constant force. The scratch hardness number on the Absolute Hardness scale is the applied force divided by the scratch width multiplied by a geometrical constant determined for the instrument. The key to understanding this large difference in the scratch hardness of different forms of carbon lies in the structure and bonding between the carbon atoms at the microscopic level, which can only be explained using chemical principles. These differences in microscopic structure also give the different forms of carbon large differences in other useful properties as will be explained in Chapter 11. Because of their extreme hardness, diamonds are used in cutting tools for the manufacture of high precision auto parts, stone building materials, and for cutting cores in well drilling applications. Graphite is used in pencils and drawing materials due to its softness and ease of removal from the parent crystal. The same microscopic properties that produce its softness also make it a good lubricant. Another form of carbon, carbon nanotubes have become more and more important in engineering due to their excellent mechanical properties, chemical stability, and electrical and thermal conductivities. The scratch hardness of carbon nanotubes cannot be measured on the macroscopic level with a sclerometer due to their small size. However, the scratch hardness can be estimated on the relative scale by understanding the nature of the carbon bonds as well as the crystal structure of the nanotubes. This has led to the estimation of the scratch hardness of nanocrystalline diamond as greater than 10 on the Mohs scale. 3 1.1 THE ROLE OF CHEMISTRY IN ENGINEERING TABLE 1.1 The Mohs Scale of Hardness Compared to Absolute Hardness as Measured by a Sclerometer. The original minerals used by Mohs to define the scale are shown in bold Material Mohs Hardness Absolute Hardness Cesium, rubidium 0.2–0.3 Lithium, sodium, potassium 0.5–0.6 Talc 1 1 Graphite, tin, lead 1.5 2 Gypsum, calcium, cadmium 2 3 Calcite, copper, arsenic 3 9 Fluorite, iron, nickel 4 21 Steel, platinum 4.5 25 Apatite, cobalt, palladium 5 48 Beryllium, molybdenum 5.5 50 Feldspar, manganese, uranium 6 72 Glass, silicon, opal 6.5 85 Quartz, porcelain, osmium 7 100 Emerald, hardened steel, tungsten 7.3 150 Topaz, cubic zirconia 8 200 Chromium, silicon nitride 8.5 300 Corundum, silicon carbide 9 400 Boron, boron nitride 9.5 1 000 Diamond 10 1 600 Nanocrystalline diamond >10 Not available An important property of a material, which may limit its usefulness, is its ability to react with other materials or the environment. A metal that is easily corroded in air may not be the best choice for an outdoor structure. The ability of metals to react with oxygen in the air can also be determined by their molecular or atomic properties. In Chapter 10, corrosion is explained as a transfer of electrons from the metal to oxygen in the air, resulting in degradation of the metal and formation of the metal oxide. The same chemical principles are also responsible for the sometimes unexpected corrosion of metals in contact with each other as with electroplated materials. This reaction can be explained by understanding the atomic structure of the metals and the chemical principles that drive the reaction. Armed with this knowledge, the probability of corrosion occurring under different conditions can be determined and used in selecting materials for different applications in different environments. 4 1. INTRODUCTION 1.2 GREEN ENGINEERING There is an increasing awareness among scientists and engineers that the Earth’s resources are limited and the ever-growing world population and rapid technological development places increasing stress on the available global resources. The rapid advancement of technology without regard to the negative impacts on human health and the environment will eventually have disastrous consequences. This realization has led scientists and engineers to promote the “Green Chemistry” and “Green Engineering” initiatives. Green Chemistry focuses primarily on the elimination or reduction of the use and generation of substances that are hazardous to human health and the environment. Green Engineering has a somewhat broader view. It focuses on the design, development and use of products, processes, and systems that are economically feasible, while minimizing the risks to human health and the environment. Basically, green engineering seeks to conduct its professional activities in a manner that lessens the negative impacts on the planet and its ability to sustain life and focuses on how to achieve economic, social, and environmental sustainability through the application of green science and technology. These goals go beyond the impacts of the products or processes themselves and must include the entire life cycle of the materials and forms of energy used in the design. Life cycle design begins with the impacts of the acquisition of the necessary raw materials. These could include the environmental effects of the mining, drilling, or harvesting techniques used to obtain the raw materials. The life cycle assessment continues through the manufacturing, distribution, use, and eventual disposal or recycle of a product. Many times, products are presented as green because the product itself has no negative impacts on the environment. However, a thorough life cycle analysis can sometimes reveal toxic effects in its manufacture or disposal. Green Engineering seeks to assure that all materials and energy sources involved in a process as well as final products are as nonhazardous as possible, while minimizing energy consumption and material use and preventing waste generation. Materials and energy sources should be renewable rather than depleting and final products, processes, and systems should be designed from production to disposal or recycle with this concept in mind. The principles of Green Engineering have been outlined by the American Chemical Society and the Environmental Protection Agency. These principles are intended to be used as guidelines to achieve the optimization of sustainability in the design of products, processes, and systems. The major points in both these lists of green principles are: • • • • • • • • The use of materials and energy sources that are inherently nonhazardous. To prevent waste rather than rely on after treatment. To minimize consumption of energy and materials. The use of renewable materials and energy. To minimize the number of material types used in a process. To make use of local sources of materials and energy. To design for recycle. To maximize efficiency. 1.3 MEASUREMENT AND CALCULATIONS 5 • To choose simplicity over complexity. • To maximize durability without environmental immortality. • To meet needs while minimizing excess. These principles of Green Engineering are built on the idea that it is easier, more efficient, and less costly to incorporate the prevention of negative environmental impacts at the beginning of a design rather than relying on clean up after they occur. The laws of thermodynamics, outlined in Chapter 8, tell us that there is no process totally without waste and no energy source that is totally without environmental impacts. Choices must be made to minimize these impacts as much as possible while still meeting the needs of society. Engineers make decisions daily that have potentially significant impacts on the environment. These decisions can either contribute to our environmental problems and the depletion of our natural resources or they can reduce the negative effects of technology on the environment and help us achieve environmental, social, and economic sustainability. The National Academy of Engineering lists mitigation and adaptation to climate change and the development of clean energy sources at the top of the “Grand Challenges for Engineering” in the next millennium. These global problems are listed as threatening the future itself. Slowing the effects of climate change will require new methods for capturing carbon produced during the combustion of fuels and sequestering it in an environmentally benign form. At the same time, the emissions of carbon and other greenhouse gases need to be reduced as much as possible. Adapting to the impacts of climate change will require designing infrastructure and agricultural processes that can withstand more severe storms, heat waves, floods, and drought. Although use of carbon-free energy sources such as solar or nuclear are attractive, their wide use will require the development of more efficient, economical, and less hazardous manufacturing and operating processes. These challenges bridge across the fields of engineering and chemistry and meeting them will require a broad knowledge of both disciplines. The development of sustainable solutions to meet these challenges will require engineers and chemists working together in interdisciplinary teams on a global scale. 1.3 MEASUREMENT AND CALCULATIONS The disciplines of chemistry and engineering are both quantitative in nature. That is, they deal primarily with numerical measurements of the amount of a material or its properties. Chemists then search for relationships between the physical and chemical properties of a material to determine the molecular basis for the observed results. Recording the measurements of any physical property requires both a number and a unit. This involves working with a variety of different units and a wide range of numerical values. In order to more easily perform calculations involving very large or very small numbers, scientists, mathematicians, and engineers use a compact standard format called scientific notation, which expresses the numbers as powers of 10. This format simplifies the calculations, makes it easier to estimate the magnitude of the result, and decreases the chance of errors in transcribing very large or very small decimal numbers. It is also more specific than other numerical formats as to the number of significant figures in a measurement value. Any number written in scientific 6 1. INTRODUCTION notation is expressed as a number from 1 to less than 10 multiplied by a power of 10. To write any number in scientific notation: 1. shift the decimal point so that there is only one nonzero digit to the left of the decimal point, 2. multiply the number by a power of 10 that is equal to the number of places the decimal point has been moved, 3. use a positive power of 10 if the decimal point is moved to the left and a negative power of ten if the decimal point is moved to the right. For example, the number 1246 is expressed as 1.246 103 and the number 0.00046 is expressed as 4.6 10–4 in scientific notation. Engineering notation is a variant of scientific notation where the powers of ten are restricted to be multiples of three. Therefore, in engineering notation, the restriction of having only one nonzero digit to the left of the decimal point is dropped in order to maintain the exponent as a multiple of three. In engineering notation, 1246 will still be expressed as 1.246 103 since it is a multiple of 3. However, 0.00046 would be expressed as 460 10–6. The advantage of engineering notation is that it is in line with the commonly used units of measure, which have different names for every 103 increase in value. So, in this format, numbers are always stated in terms of thousands, millions, billions, etc. The main disadvantage of using engineering notation is that the number of significant figures in a measurement is not as easily determined as it is with scientific notation. For this reason, all measurements and calculations in this text will use scientific notation. Measurements of physical and chemical properties are not exact. They are subject to uncertainties, which are caused by errors inherent to the methods of measurements used. These measurement uncertainties are described by the accuracy and precision of the measurement method. The accuracy of a measurement is the degree of closeness of the measurements to the actual or true value. Large deviations from the true value are described as a low degree of measurement accuracy. This is caused by systematic errors in the measurement method. Systematic errors are constant and always of the same sign. They result in a constant offset, either positive or negative, from the true value. Examples of a systematic error would be mass measurements made with a balance that does not read zero with nothing on it or time measurements made with a clock that has not been set properly. The measurement accuracy is determined by calibration of the measurement method through comparison of the measured result obtained on a known standard to its certified true value. Calibration of a balance would involve recording the measurement of a sample of known mass, such as a calibrated weight. Once this comparison is made, the systematic error can be corrected and the accuracy of the results improved by either electronically or manually subtracting or adding the offset value from the measurement. The precision of a measurement is the degree to which repeated measurements give the same value whether or not that value is true. Precision is often called reproducibility and is an indicator of the scatter in a series of measured values made on the same sample. The accuracy of a measurement can never be any greater than its precision. Large differences in the values of repeated measurements are described as a low degree of measurement precision. This is caused by random errors in the measurement method. A random error is one that produces both positive and negative variations from an average value. They are caused by unpredictable fluctuations in the measurement apparatus, in the environmental conditions, or in the operator’s interpretation of the instrumental reading. Examples of some causes of 1.3 MEASUREMENT AND CALCULATIONS 7 random errors are: the electronic noise in an electrical instrument, the effect of temperature changes, vibrations and air movement on balances, and using volumetric glassware with poor resolution to measure volumes. The measurement precision can be determined by repeating the measurement several times on the same sample and recording the variation in the values. Since random errors produce both positive and negative variations in the measurement, the measurement precision can be improved by reporting the average of a series of measurements made on the same sample. When the result of a measurement is recorded, the precision of the measurement is indicated in the number of significant figures used in the numerical value. The number of significant figures given in a reported measurement is simply the number of digits in the value that are known with some degree of reliability. The last significant digit reported in the measured value is the one that is uncertain. All other reported digits are known with a high level of certainty. For example, the length of a steel bar is reported to be 25.34 cm. This result has four significant figures, which implies a value of 25.3 to be known with certainty and the last digit 25.34 to be uncertain. EXAMPLE 1.1: DETERMINING SIGNIFICANT FIGURES Determine the number of significant figures in the following measurements of length: (a) 1000 m, (b) 124.50 km, (c) 200.4 m, (d) 3450 mm, (e) 0.5 m. 1. Convert all measurements to scientific notation: (a) 1000 m ¼ 1 103 m (b) 124.50 km ¼ 1.2450 102 km (c) 200.4 m ¼ 2.004 102 m (d) 3450 mm ¼ 3.45 103 mm (e) 0.5 m ¼ 5 10–1 m 2. Determine the number of significant figures by counting the number of digits used in the scientific notation. (a) 1 103 ¼ 1 significant figure (b) 1.2450 102 ¼ 5 significant figures (c) 2.004 102 ¼ 4 significant figures (d) 3.45 103 ¼ 3 significant figures (e) 5 10–1 ¼ 1 significant figure Remember that trailing zeros are only kept in scientific notation if a decimal point is present in decimal notation. If a decimal point is not used, the trailing zeros are assumed not to be significant. The margin of error in last significant digit is determined by repeating the measurement several times on the same sample and recording the variation in the measured values. The result is then reported as the average of the series of measurements with a plus or minus () value written after the measurement. The result of 25.34 0.02 cm for the steel bar means that all the values obtained were between 25.32 and 25.36 cm. If the measurement method has been adequately calibrated, it is also implied that the true value lies within this same range. The number of significant figures in a recorded measurement is equal to the number of digits used when the value is expressed in scientific notation format. 8 1. INTRODUCTION EXAMPLE 1.2: SIGNIFICANT FIGURES AND MEASUREMENT ERRORS Determine the number of significant figures in the following measurements of length: (a) 1000 10 m, (b) 124.50 0.05 km, (c) 200.4 0.02 m, (d) 3450 1 mm, (e) 0.5 0.1 m. 1. Convert all measurements to scientific notation based on the uncertainties given. (a) 1000 10 m ¼ 1.00 103 m (b) 124.50 0.05 km ¼ 1.2450 102 km (c) 200.4 0.02 m ¼ 2.004 102 m (d) 3450 1 mm ¼ 3.450 103 mm (e) 0.5 0.1 m ¼ 5 10–1 m 2. Determine the significant figures by counting the digits used in scientific notation. (a) 1.00 103 ¼ 3 significant figures (b) 1.2450 102 ¼ 5 significant figures (c) 2.004 102 ¼ 4 significant figures (d) 3.450 103 ¼ 4 significant figures (e) 5 10–1 ¼ 1 significant figure The addition of absolute measurement errors to the measurements removes the ambiguity associated with trailing zeros. The results of a calculation based on measurements are only as certain as the least certain measurement used in the calculation. When adding and subtracting measurements, the uncertainty of the result is determined by the uncertainty of the least precise measurement. The least precise measurement is the one with the least significant decimal place. For example, addition of the measurements: 0.132 + 1.25 + 1.0 gives a result of 2.4 (rounded up from 2.38). The least precise measurement is 1.0, with a least significant decimal place of tenths. So the result also contains one decimal place (rounded to the tenths place). Addition of the measurements: 240 + 100 + 1.10 gives a result of 400 (rounded up from 350.1). In this case, the least precise measurement is 100, with a least significant decimal place of hundreds. So the result is rounded to the hundreds place. If the measurements were given as: (249 1) + (100 10) + (1.10 0.02), the result would be 350 since the measurement error indicates that the uncertainty is in the tens place. EXAMPLE 1.3: DETERMINING SIGNIFICANT FIGURES IN ADDITION AND SUBTRACTION OF MEASUREMENTS Give the result of each of the following calculations to the appropriate number of significant figures: (a) 100 + 2.5 + 20.55, (b) 15 – 12.82, (c) 125.173 + 129.2 + 52.25, (d) 5.671 102 + 6.3 10–3, (e) 1.4 103–5.5 10–1. Continued 1.3 MEASUREMENT AND CALCULATIONS 9 1. Determine the result of each calculation. Scientific notation is not required for this determination. (a) 100 + 2.5 + 20.55 ¼ 123.05 (b) 15 – 12.82 ¼ 2.18 (c) 125.173 + 129.2 + 52.25 ¼ 133.45 (d) 5.671 102 + 6.3 10–3 ¼ 567.1063 (e) 1.4 103 – 5.5 10–1 ¼ 1399.45 2. Determine the least significant decimal place in each calculation. Least significant digit is highlighted in red: (a) 100 + 2.5 + 20.55 (b) 15 – 12.82 (c) 125.173 + 129.2 + 52.25 (d) 5.671 102 + 6.3 10–3 (e) 1.4 103 – 5.5 10–1 3. Determine the number of significant figures from the least significant decimal place. Least significant digit is highlighted in red: (a) 100 + 2.5 + 20.55 ¼ 1 significant figure (b) 15 – 12.82 ¼ 1 significant figure (c) 125.173 + 129.2 + 52.25 ¼ 4 significant figures (d) 5.671 102 + 6.3 10–3 ¼ 4 significant figures (e) 1.4 103 – 5.5 10–1 ¼ 2 significant figures 4. Round the result to the appropriate number of significant figures. (a) 100 (rounded from 123.05) (b) 2 (rounded from 2.18) (c) 133.5 (rounded from 133.45) (d) 567.1 (rounded from 567.1063) (e) 1400 (rounded from 1399.45) When multiplying and dividing measurements, the result must contain the same number of significant figures as the measurement with the least number of significant figures. Thus, (1.3 10–3) (1.25 102) ¼ 1.7 10–1 (rounded from 1.625 10–1). Since 1.3 10–3 has two significant figures and 1.25 102 has three significant figures, the result should have two significant figures. Since significant figures are used to express experimental uncertainty, only experimentally measured numbers or results of calculations that use experimentally measured numbers use significant figures. Numbers that are defined to be a specific value, such as conversion factors, or the result of a count are exact numbers. Since they do not have measurement uncertainties associated with them, they are considered to have an infinite number of significant figures. When used in calculations with numbers generated from measurements, they do not affect the number of significant figures in the result. 10 1. INTRODUCTION EXAMPLE 1.4: DETERMINING SIGNIFICANT FIGURES IN MULTIPLICATION AND DIVISION OF MEASUREMENTS Determine the number of significant figures in each of the following calculations: (a) 2000/115, (b) (136.50) (0.0014), (c) 200.6/0.25, (d) 3450 2.0, (e) 0.5/2. 1. Convert all values to scientific notation to determine the measurement with the smallest number of significant figures. (a) 2000/115 ¼ (2 103)/(1.15 102) (b) (136.50) (0.0014) ¼ (1.3650 102) (1.4 10–3) (c) 200.6/0.25 ¼ (2.006 102)/(2.5 10–1) (d) 3450 2.0 ¼ (3.45 103) 2.0 (e) 0.5/2 ¼ (5 10–1)/(2) 2. Determine the result of each calculation in scientific notation. (a) (2 103)/(1.15 102) ¼ 2 105 (b) (1.3650 102) (1.4 10–3) ¼ 1.9 10–2 (c) (2.006 102)/(2.5 10–1) ¼ 8.0 102 (d) (3.45 103) 2.0 ¼ 6.9 103 (e) (5 10–1)/(2) ¼ 3 10–1 3. Determine the number of significant figures from the scientific notation. (a) 2 105 ¼ 1 significant figure (b) 1.9 10–2 ¼ 2 significant figures (c) 8.0 102 ¼ 2 significant figures (d) 6.9 103 ¼ 2 significant figures (e) 3 101 ¼ 1 significant figure The global standard and most widely used system of measurement units in both science and engineering is the International System of Units, abbreviated SI from the French “Le Système International d’Unites.” These SI units are based on seven “base units,” which are defined in an absolute way without referring to any other units. All other SI units, called SI “derived units,” can be defined algebraically in terms of the fundamental base units, although their definitions based on other derived units are more commonly used. For example, the SI unit of pressure is the pascal (Pa). It is commonly defined to be a force per unit area equal to one newton per square meter (N/m2) using the SI derived unit for force (newton). However, the technical definition in terms of the SI base units is one kilogram per meter per second squared (kg • m1• s2). Currently, there are 22 accepted SI derived units for a total of 29 base and derived units. The base units and the most common derived SI units are listed in Table 1.2. Other SI derived units not included in Table 1.2 are simple mathematical combinations of the base units that do not have special names attached to them. For example, the SI unit of area is m2, arising directly from the algebraic formula for area; length (m) width (m) ¼ area (m m ¼ m2). In a similar manner, the unit for volume is m3, the unit for velocity is m/s, and the unit for density is kg/m3. 11 1.3 MEASUREMENT AND CALCULATIONS TABLE 1.2 The SI System of Units; Base Units Are Listed in Bold Unit Symbol Measured Property Base Equivalent kilogram kg Mass – mole mol Amount of substance – meter m Distance – radian rad Plane angle m/m ¼ dimensionless steradian sr Solid angle m2/m2 ¼ dimensionless second s, sec Time – kelvin K Temperature – degree Celsius o Temperature K – 273.15 ampere A, amp Electric current – coulomb C Electric charge A•s farad F Electric capacitance kg1 • m2 • s4 • A2 Ohm Ω seimens Volt C S V Electric resistance Electrical conductance Electric potential 3 2 kg • m • s 1 kg 2 2 3 •m •s •A 3 2 •A Derived Equivalent kg • m • s •A 1 C/V V/A 2 A/V W/A 2 newton N Force kg • m • s pascal Pa Pressure kg • m1 • s2 N/m2 joule J Energy, work kg • m2 • s2 N•m watt webber W Wb Power Magnetic flux 2 3 2 2 kg • m • s kg • m • s 2 J/s •A 1 1 V•s tesla T Magnetic field strength kg • s •A Wb/m2 henry H Inductance kg • m2 • s2 • A2 Wb/A candela cd Intensity of light – lumen lm Flux of light cd • sr Lux lx Flux per unit area cd • m2 hertz Hz Frequency s–1 becquerel Bq Radioactive decay rate s1 gray Gy Radiation dose m2 • s2 sievert katal Sv kat Equivalent dose Catalytic activity 2 2 m •s s 1 • mol lm/m2 J/kg J/kg 12 1. INTRODUCTION The SI units use a set of prefixes that precede the basic unit of measure which act as decimal-based multipliers of the unit. These prefixes are listed in Table 1.3. Each prefix name has an associated symbol which can be used in combination with the symbols for the units of measure. For example, the prefix “milli” is used to indicate that the unit of measurement is multiplied by a factor of 10–3. Thus, 1 mm is equal to 1 10–3 m ¼ 0.001 m. Since the symbol for meter is “m” and the symbol for the prefix “milli” is “m,” the symbol for millimeter is “mm” (0.001 m ¼ 1 mm). Only one prefix can be used at a time with a unit of measure. This also applies to units of mass. The SI base unit for mass is the kilogram (not the gram!) and this unit already uses a designated prefix. So, the prefixes are used with the derived unit of mass the “gram,” which is defined as the base unit kilogram divided by 1000 (1 g ¼ 1 kg/1000 ¼ 0.001 kg). So, 0.000001 kg (1 10–6 kg) is equal to 0.001 g, which is called the milligram (mg) not the microkilogram (μkg). TABLE 1.3 SI Unit Prefixes Used to Define Powers of Ten Prefix yotta zetta exa peta tera giga mega kilo hecto deka Symbol Y Z E P T G M k h da Multiplier Decimal Equivalent English 24 1,000,000,000,000,000,000,000,000 Septillion 21 1,000,000,000,000,000,000,000 Sextillion 18 1,000,000,000,000,000,000 Quintillion 15 1,000,000,000,000,000 Quadrillion 12 1,000,000,000,000 Trillion 9 1,000,000,000 Billion 6 1,000,000 Million 3 1000 Thousand 2 100 Hundred 1 10 Ten 0 1 One 0.1 Tenth 0.01 Hundredth 0.001 Thousandth 0.000001 Millionth 0.000000001 Billionth 0.000000000001 Trillionth 0.000000000000001 Quadrillionth 0.000000000000000001 Quintillionth 0.000000000000000000001 Sextillionth 0.000000000000000000000001 Septillionth 10 10 10 10 10 10 10 10 10 10 10 deci centi milli micro nano pico femto atto zepto yocto d c m μ n p f a z y –1 10 –2 10 –3 10 –6 10 –9 10 –12 10 –15 10 –18 10 –21 10 –24 10 1.3 MEASUREMENT AND CALCULATIONS 13 EXAMPLE 1.5: CONVERTING BETWEEN UNIT PREFIXES The distance between carbon atoms in a diamond is 0.154 nm. What is this distance in meters? In picometers? 1. Convert from nanometers to meters. The conversion factor from nanometers to meters is: 1 nm ¼ 1 10–9 m 0.154 nm (1 10–9 m/nm) ¼ 1.54 10–10 m 2. Convert from meters to picometers. The conversion from meters to picometers is: 1 m ¼ 1 1012 pm 1.54 10–10 m (11012 pm/m) ¼ 1.54 102 pm ¼ 154 pm Remember that conversion factors are exact numbers and do not affect the number of significant figures in the result. EXAMPLE 1.6: CONVERTING BETWEEN POWERS OF TEN The mass of the earth is 5.97219 1022 kg. What is this mass in teragrams? 1. Convert from kilograms to grams. 1 kg ¼ 1 103 g (5.97219 1022 kg) (1 103 g/kg) ¼ 5.97219 1025 g 2. Convert from grams to teragrams. 1 g ¼ 1 10–12 Tg 5.97219 1025 g(1 10–12 g/Tg) ¼ 5.97219 1013 Tg Remember that ratios of units are cancelled in the same manner as numbers. This same calculation can be done in one step. (5.97219 1022 kg) (1 103 g/kg) (1 10–12 Tg/g) ¼ 5.97219 1013 Tg Be sure that all units cancel except for Tg. For further help with working with units and powers of 10 see Appendix I. Although the SI system of units can be used to make any physical measurement, some non-SI units are still widely used in science and engineering. These units have been used for so long that they most likely will be continued to be used for the foreseeable future. A limited number of these other units have been defined in terms of the SI units in a similar manner as the SI derived units in order to assure that their continued use will be consistent across the globe. These commonly used non-SI units are listed in Table 1.4 with their SI definitions. 14 1. INTRODUCTION TABLE 1.4 Commonly Used Units Not Included in the SI System of Units Unit Symbol Measured Property Base Equivalent Derived Equivalent minute min Time 60 • s hour h Time 3600 • s 60 • min day d Time 86,400 • s 24 • h degrees Fahrenheit °F Temperature (1.8 • (K – 273.15)) + 32 (1.8•°C) + 32 degree ° Plane angle π/180 • rad minute ’ Plane angle π/10,800 • rad 1/60•° second 00 Plane angle π/64,800 • rad 1/60•’ 3 liter L Volume 0.001 • m metric ton t Mass 1000 • kg electronvolt eV Energy 1.602 10–19 • J atomic mass unit u Mass 1.660 10–27 • kg ångstr€ om Å Distance 1 10–10 • m bar bar Pressure 1 105 • Pa curie Ci Radioactive decay rate 3.7 1010 • Bq rad rad Radiation dose 1 10–2 • Gy rem rem Equivalent dose 1 10–2 • Sv 1.4 THE PHYSICAL STATES OF MATTER Matter is traditionally defined as anything that has mass (m) and volume (V). A material is defined as anything that is made of matter. Although the term “matter” is most often used by chemists and physicists in a general sense and the term “material” is most often used by engineers in a more specific sense, in many ways they are the same. The ratio of the mass to the volume (m/V) of a material is the density, which is most commonly symbolized with the Greek letter ρ. ρ ¼ m=V (1) The SI units for density are given in terms of mass/volume, such as kilograms/cubic meter (kg/m3) or grams/cubic centimeter (g/cm3). The commonly used non-SI unit of density is grams/milliliter (g/mL). Since a liter is defined as equal to 0.001 m3, 1 mL is equal to 1 cm3. The density of a material varies with temperature and pressure. Increasing the pressure on a compressible material decreases the volume, and since the mass remains constant, its density is increased according to Eq. (1). Increasing the temperature of a material generally increases its volume and therefore decreases its density. The density of a material may change abruptly as the temperature rises as shown in Fig. 1.1. Each of these rapid decreases 1.4 THE PHYSICAL STATES OF MATTER FIG. 1.1 The changes in the volume and density of a pure substance as the temperature increases. Liquid Volume Density Vapor 15 Crystalline Temperature in density with increasing temperature corresponds to a rapid increase in volume as the material undergoes a change in phase or state. CASE STUDY: NUCLEAR FUEL RODS A nuclear power plant is powered by a fissionable fuel material, usually uranium dioxide (UO2), contained in a nuclear reactor. The nuclear reaction produces heat which is then used to produce energy by more conventional means such as operation of multisteam turbines. Early commercial nuclear power plant designs incorporated Generation II reactors. Generation II reactors used uranium fuel that is formed into rods and canned inside of zirconium alloy tubes. These tubes are grouped together into clusters of 20–40 evenly spaced rods. As the nuclear reaction takes place, the high energies produced by the reaction cause the temperature of the fuel material to increase. A circulating water cooling system is used to prevent overheating and to maintain a stable operating temperature inside the fuel rods. In this design, if the cooling system should fail, the temperature inside the fuel rods would rapidly increase and cause the solid uranium fuel to expand. Due to the fixed volume of the fuel tubes, the rising temperatures would cause pressures inside the tubes to increase. This rising pressure would eventually lead to rupture of the containment vessel and rapid vaporization of the remaining water coolant. These early plant designs relied exclusively on active safety features involving electrical or mechanical operations, initiated automatically or by plant operators, to attempt to shut down the reactor and stop the nuclear reaction and the runaway temperature rise during coolant failures. In the case of complete electrical failure, such as during the Fukushima tsunami event of 2011, the rapidly rising temperatures within the fuel could eventually reach melting temperatures of the solid fuel. Today’s more advanced nuclear power plant designs include passive safety features which do not require actions of an operator or electrical mechanisms to shut down the reactor in case of coolant failure. Instead, they rely on the inherent expansion properties of the fuel and cladding under rising temperatures to act as an automatic feedback mechanism. If the coolant system should fail and the temperature of the fuel begins to increase, the fuel and cladding are designed to allow the solid fuel to expand as the temperature rises. As shown in Fig. 1.2, when the fuel expands, the volume of the solid fuel increases and the density of the fuel decreases. Under normal operating conditions, the density of the uranium fuel is sufficient to maintain the nuclear chain reaction. Any decrease in 16 10 Solid Density (g/cm3) FIG. 1.2 Density change in uranium dioxide nuclear fuel with increasing temperature. Data from W.D. Downing, CONF-810696-1. 1. INTRODUCTION 9.5 9 Liquid 8.5 8 2750 2800 2850 2900 2950 3000 3050 Temperature (°C) density of the fuel interferes with the fission process as will be discussed in Chapter 14. Therefore, the decrease in fuel density with the expansion of the fuel as temperature rises automatically slows down the nuclear reaction which allows the temperature of the fuel to decrease spontaneously. This shuts down the nuclear reaction automatically and keeps the fuel from reaching melting temperatures. This passive failsafe design was tested in 1986 by nuclear engineers at Argonne National Laboratory. A coolant failure was simulated by shutting off the coolant pumps with the reactor at full power. The reactor power dropped to near zero within about 300 s of coolant shut down and no damage to the fuel or the reactor was observed. This test demonstrated that even with a loss of all electrical power and no capability to shut down the reactor using the normal systems, this reactor design will shut down automatically without danger or damage to the system. There are some important exceptions to this trend of decreasing density with increasing temperature, the most notable being water. The density of water increases as the temperature is increased from below 0oC up to 100oC, implying a decrease in volume as the temperature rises. This behavior seems opposite to that which would be expected from the observation of the macroscopic properties of water alone and can only be explained with a thorough understanding of the forces and properties that control the behavior of water on a molecular level. This anomalous behavior of water arises from some very important properties that set it apart from other materials, which will be discussed in detail in Chapter 3. The most readily observable macroscopic property of matter is its physical state. The five physical states of matter are: superfluids, solids, liquids, gases, and plasmas. There are four states of matter observable in everyday life. These are, in the order of increasing temperature and decreasing densities: solid, liquid, gas, and plasma. A fifth state of matter has most recently been identified that is important to engineers because of its close relationship to superconductivity. This state, which exists only at very low temperatures, is called a superfluid. The three classical states of matter important to chemistry are solid, liquid, and gas. These classical states of matter were first defined by the differences in their observed macroscopic properties and the changes in these properties as the temperature was increased (Table 1.5). A solid has a fixed shape and volume, is noncompressible, and maintains a constant density 17 1.4 THE PHYSICAL STATES OF MATTER TABLE 1.5 The Macroscopic Physical Properties of the Five Physical States of Matter Superfluid Solid Liquid Gas Plasma Shape Variable Fixed Variable Variable Variable Volume Fixed Fixed Fixed Variable Variable Compressibility Slightly None Slightly High High Conductivity Thermal high Variable Variable None Electrical high Density Medium High Medium Low Low Temperature Super cooled Variable Variable Variable Super heated Movement Very low Low Medium Rapid Very rapid Other No viscosity Rigid Fluid Diffusible Ionized under pressure. It is rigid and can only change shape by an applied force such as when it is cut. A liquid has a fixed volume but a variable shape. It is able to flow and take the shape of the container that holds it with one free surface not determined by the container. It is only slightly compressible and shows only minor changes in density at very high pressures. A gas has no fixed shape or volume, but expands or diffuses to completely fill its container. It is readily compressible with a much lower density than solids or liquids. The density of a gas is strongly influenced by changes in pressure or temperature. As will be shown in Chapter 6, the density of a gas can be doubled by doubling the pressure. The more extreme states of matter are plasmas and superfluids. Plasmas exist at very high temperatures and are formed by super heating a gas. Similar to gases, plasmas also have no fixed shape or volume and will expand to completely fill the container. They are also readily compressible with a density similar to the gas from which they were formed. In contrast to gases, which have very low electrical conductivities and are often used as insulators, plasmas have very high electrical conductivities. They are important in many products today including plasma display screens, fluorescent and arc lamps, and arc welders. They occur naturally in lightning and auroras. In contrast, superfluids exist only at extremely low temperatures and can be formed by rapidly super cooling a gas, liquid, or solid. They have the properties of a liquid with the addition that they exhibit frictionless flow and have a very high thermal conductivity. The most common superfluid is liquid helium, which has been used in the study of superconductors and superconducting magnets. Liquids, gases, plasmas, and superfluids are all considered to be fluids since they all will continually flow under an applied shear stress. In an attempt to explain the properties and behavior of the three classical states of matter, the early Greek philosopher Democritus described matter as made up of tiny microscopic particles called atomos meaning “not to be cut.” He envisioned these tiny particles as the smallest building blocks of all matter and the properties and behavior of these tiny particles on the microscopic level gave rise to the observable macroscopic properties of materials. This particulate view of matter held five major points: 1. All matter is composed of tiny indivisible particles. 18 1. INTRODUCTION 2. Each pure substance has its own kind of particle, different from every other pure substance. 3. There are spaces between the particles that are very large compared to the size of the particles. 4. The particles are in constant motion. 5. Particles at higher temperatures move faster than particles at lower temperatures. This particulate theory of matter was successful in explaining the behavior and properties of the classical states of matter as the temperature was increased (Table 1.6). As shown in Fig. 1.3, the particles in solids are closely packed in fixed positions with the least amount of space between them. They have a minimum amount of movement around these fixed positions. This is the reason solids have fixed, rigid shapes. In liquids, the particles are more loosely packed with more space between them. They have a larger range of motion and are free to slide past each other. This larger range of motion allows them to flow to fit the shape of their container. In gases, the particles are widely spread with rapid movement and larger spaces between them. This rapid movement allows them to quickly expand to fill the shape and volume of the container. The larger free space between the particles also makes them more easily compressible with changes in volume or pressure. This particulate view of matter can also help describe the behavior of the two more recently identified states of matter. As the temperature of a gas is increased, the particles move faster and collide with each other at very high speeds. At very high temperatures, these energetic collisions cause the particles to become charged. The charged particles give rise to the high electrical conductivity of plasmas. At the opposite extreme, as temperature rapidly decreases, the particles begin to move very slowly and bunch very close together with little space between them. At extremely low temperatures, the individual movement of the particles virtually stops and the space between them approaches zero. In this state, all of the particles behave in the same manner as if they were a single particle. If one particle moves, they will all move. The viscosity of a superfluid, the degree to which the fluid resists movement, TABLE 1.6 Particulate Descriptions of the Five States of Matter Superfluid Solid Liquid Gas Plasma Packing Bunched Tight Close Well separated Widely separated Free space None Little Little Large Large Order High High None Random Random Movement None Fixed Slow Rapid Rapid + − + − + − − + + − + Superfluid Solid Liquid FIG. 1.3 Particulate representations of the five states of matter. Gas Plasma 19 1.4 THE PHYSICAL STATES OF MATTER approaches zero. Because of this property, superfluids appear to be able to self-propel and travel in a way that defies the forces of gravity and surface tension. Liquid helium in the superfluid state shown in Fig. 1.4 will creep up the inside wall of its container as a thin film. It then comes down the outside of the container forming a drop which will fall into the liquid below. This will continue until the container is empty. A substance can convert from one state to another with changes in temperature and pressure, as shown in Fig. 1.5. These state changes occur abruptly and are known as phase transitions. The transition from a solid to a liquid is known as melting and the temperature at which this occurs is the melting point of the solid. The melting point of a pure solid material is characteristic of that substance and can be used to identify it. The transition from a liquid to a solid is known as freezing and the temperature at which this occurs is the freezing point. The melting point and the freezing point of a pure substance is the same temperature. The transition from a liquid to a gas is called vaporization and the temperature at which this phase change occurs is the boiling point of the liquid. The reverse transition from a gas to FIG. 1.4 The superfluid liquid helium in a cup. Photograph by Alfred Leitner, Wikimedia Commons. Sublimation Melting Vaporization lonization Gas Solid Freezing Super fluid Plasma Liquid Condensation Deposition Coalescence Temperature FIG. 1.5 Phase transitions between the five states of matter. Recombination 20 1. INTRODUCTION a liquid is known as condensation and also occurs at the same temperature as the boiling point of the substance. The boiling point of a pure liquid is dependent on the pressure above the liquid. The boiling point of water decreases with decreasing pressure. Since atmospheric pressure decreases with increasing altitude, the boiling point of water also decreases with increasing altitude as shown in Fig. 1.6. While the boiling point of water at sea level is 100oC, it would be 96oC in Denver and only 73oC at the summit of Mt. Everest. Although cooking water would reach a boil faster in Denver due to the lower required temperature, the water may not ever be hot enough for proper cooking even with rigorous boiling. Conversely, at points below sea level, atmospheric pressures would be higher and so would the boiling point of water. On the shores of the Dead Sea, at an altitude of 423 m below sea level, water would need to reach a temperature of 101oC to reach the boiling point. Pressure cookers are used to speed up the cooking time of food because, at the high pressures inside the cooker, the temperature of water can reach 120oC before boiling. This higher water temperature cooks food faster than the water outside the cooker at 100oC. Substances can also transition from a solid directly to a gas without passing through the liquid state, called sublimation. The transition from a gas to a solid directly without passing through the liquid state is called deposition. The formation of a plasma by super heating a gas is known as ionization and the reverse transition from a plasma back to the classical gas state is known as recombination. Superfluids can be formed by super cooling any of the three classical states of matter (e.g., solids, liquids, or gases) known as coalescence. The phase changes of pure substances as a function of temperature and pressure are commonly presented as a phase diagram. A generic phase diagram is shown in Fig. 1.7. The curves in the diagram show the phase boundaries between the three classical states of matter: solid, liquid, and gas. The red curve in Fig. 1.7 represents the phase boundary between the solid and liquid phase and the temperature and pressure conditions where melting and freezing occur. Similarly, the blue curve represents the boundary between the liquid and gas 10000 Mt. Everest 200 Pressure (mm Hg) 400 6000 500 4000 600 Denver 2000 700 Sea level 800 0 Dead sea −2000 105 100 95 90 85 80 75 70 65 Boiling point (°C) FIG. 1.6 The boiling point of water at different altitudes. Data from EngineeringToolBox.com. Altitude (m) 8000 300 1.5 CLASSIFICATION OF MATTER 21 FIG. 1.7 A typical phase diagram of a pure substance. Pressure Solid Supercritical fluid Liquid Pcr cp The red curve is the phase boundary between the solid and liquid phase. The blue curve is the phase boundary between the liquid and gas phase. The green curve is the phase boundary between the solid and gas phase. tp, triple point; cp, critical point; Pcr, critical pressure; Tcr, critical temperature. Gas tp Tcr Temperature phases and the temperatures and pressures where vaporization and condensation occur. The green line is the phase boundary between the solid and gas phases and the temperature and pressure conditions where sublimation and deposition occur. The point where all three boundaries join is known as the triple point (tp) of the pure substance. The temperature and pressure of the triple point are the conditions where all three classical phases of matter can coexist simultaneously. The phase boundary between the liquid and gas phases ends at a point called the critical point (cp). The temperature at this point is known as the critical temperature (Tcr) and the pressure at this point is the critical pressure (Pcr). Above this point, the liquid and gas phases become indistinguishable and the substance becomes a supercritical fluid with properties of both gas and liquid phases. Supercritical fluids are compressible and diffuse rapidly like gases, but with densities similar to liquids. Near the critical point, a small change in pressure or temperature results in a large change in the density of the supercritical fluid. In addition, since there is no liquid-gas phase boundary above the critical point, there is no surface tension. The most common supercritical fluid is carbon dioxide, which is used for the decaffeination of coffee beans, the extraction of hops for the manufacture of beer, and as a replacement for organic solvents in “greener” dry cleaning procedures. 1.5 CLASSIFICATION OF MATTER Although the particulate theory of matter was first presented by Democritus in 400 BC, it was John Dalton who is credited with first developing the modern atomic theory of matter in the early 1800s. The theory presented by Democritus was predominately a philosophical statement and therefore not widely accepted. In contrast, Dalton’s theory was based on careful experimental observations and measurements and so became the first atomic theory of matter founded on scientific concepts and principles. The main concepts in Dalton’s atomic theory of matter are: 22 1. INTRODUCTION 1. 2. 3. 4. 5. All matter is composed of extremely small particles called atoms. Atoms cannot be subdivided, created, or destroyed. Atoms of the same element are identical in size, weight, and other properties. Atoms of different elements are different in size, weight, and other properties. Atoms of different elements combine in simple whole number ratios to form chemical compounds. 6. Atoms can be combined, separated, or rearranged in chemical reactions. Dalton’s atomic theory includes two important additions to that postulated by Democritus. The first is the concept that the atoms that make up a particular element have a weight specific to that element and different from all other elements. The other important addition is that atoms can combine in simple whole number ratios in a chemical reaction to form chemical compounds. This idea explained the law of definite proportions proposed earlier by Joseph Proust, which stated that a chemical compound always contains exactly the same proportion of elements by mass and the mass of the compound is equal to the sum of the masses of the component elements. The smallest particles that make up a chemical compound were called molecules, which were described as composed of the atoms of the combining elements bonded tightly together. According to the modern atomic theory, matter can be classified according to both its physical and chemical state as shown in Fig. 1.8. Each classification is defined both by the observable macroscopic properties and the atomic or molecular properties. The nature of the physical state involves only the orientation and movement of the atoms or molecules making up the substance with no effect on the composition of the substance. Matter Physical state Superfluid Solid Liquid Gas Plasma Chemical state Pure substance Mixture Homogeneous Heterogeneous Crystalline Amorphous Element Metal FIG. 1.8 Classification of matter. Compound Nonmetal Metalloid Organic Inorganic 1.5 CLASSIFICATION OF MATTER 23 In addition to the classification of matter by the major five physical phases, the solid phase can be further subdivided into crystalline solids and amorphous solids. These will be described in detail in Chapter 11. Briefly, crystalline solids have a long range highly ordered three dimensional arrangements of the atoms or molecules. The structure of the entire solid can be described as being made up of a regular repeating pattern. This long range molecular order results in characteristic macroscopic geometric shapes that are bounded by planes or faces as shown in Fig. 1.9 upper left. Amorphous solids lack this long range order. Although they may have a short range localized order of the atoms or molecules, they still appear as randomly oriented when viewed in three dimensions. Some examples of amorphous solids include glasses, thin films, and polymers. The chemical state of a material is divided into pure substances and mixtures of pure substances. Pure substances are either elements or compounds. The elements are divided into metals, metalloids, and nonmetals. Most elements are classified as metals. In the solid state, metals are shiny, malleable (able to be pressed permanently out of shape without breaking or cracking), ductile (able to be deformed under applied stress), and fusible (able to be fused together at fairly low temperatures). They also have high electrical and thermal conductivities. Some familiar examples of metals are aluminum, copper, gold, iron, lead, and mercury. Nonmetals are poor conductors of heat and electricity. They are dull and brittle and are generally soft and have lower densities, boiling points, and melting points than metals. There are 16 elements classified as nonmetals. Included in these are the elements that are most commonly found in the compounds that make up living organisms: carbon, hydrogen, oxygen, nitrogen, sulfur, and phosphorous. Metalloids have properties that are intermediate between those of metals and nonmetals. They usually have a metallic appearance, but they are brittle and only fair conductors of electricity. There are seven elements commonly classified as metalloids: boron, silicon, germanium, arsenic, antimony, tellurium, and astatine. The metalloids are most commonly used in glasses, metal alloys, and semiconductors, which will be discussed in detail in Chapter 11. Compounds are also pure substances because they are composed of one kind of molecule. They have a definite set of properties that are different from the properties of the elements FIG. 1.9 Crystalline iron pyrite (left) and amorphous black metallic glass (right) are shown in the macroscopic view with a schematic representation of their microscopic molecular order. Photographs by Vassil-left and George Stobbart-right; diagram adapted from Cristal, Wikimedia Commons. 24 1. INTRODUCTION from which they are formed. But, compounds can be separated into the simpler elements by chemical reactions. Compounds are classified as organic or inorganic. Organic compounds are those that contain one or more atoms of carbon bonded to atoms of other nonmetal elements, commonly hydrogen, oxygen, or nitrogen. Organic compounds, discussed in Chapter 13, include those that make up living organisms and those that make up most synthetic polymers. Inorganic compounds include all compounds except the organic compounds. A few simple carbon containing compounds are classified as inorganic. These include carbonates, carbides, cyanides, and simple oxides of carbon such as carbon monoxide and carbon dioxide. A chemical mixture contains two or more pure substances in an indefinite ratio. The pure substances are mixed together, but are not combined chemically. Mixtures can be either homogeneous or heterogeneous. A homogeneous mixture is one in which the components are thoroughly mixed so that the atoms or molecules that make up the mixture are uniformly distributed throughout the mixture. It is uniform in appearance, composed of only one physical phase, and the components cannot be separated by mechanical methods. Solutions are a special case of a homogeneous mixture, which will be discussed in detail in Chapter 12. Most solutions are composed of a solid substance, called a solute, dissolved in a liquid substance, called the solvent. One example of a solution is white crystalline table salt-dissolved water to make a clear salt solution as shown in Fig. 1.10. A heterogeneous mixture is one that is not thoroughly mixed and is not uniform in composition or appearance. Instead, it is made of different substances that remain physically separate in two or more distinct regions or phases. Because the components of a heterogeneous mixture are physically isolated, they can be separated by using simple mechanical methods. A mixture of milled flour with water will result in a heterogeneous mixture of the solid flour particles dispersed throughout the liquid water phase as shown in Fig. 1.10. Although it may appear to be uniform, on a microscopic level it is composed of separate solid particles suspended in the water. This heterogeneous mixture of solid particles and a liquid is known as a suspension. Suspensions are composed of suspended particles >0.1 μm. Heterogeneous mixtures with particles <0.1 μm are called colloids. A heterogeneous mixture of solid particles or liquid droplets in a gas is called an aerosol. FIG. 1.10 A solution of salt in water (left) and a suspension of flour in water (right). Photographs by Chris73, Wikimedia Commons. Solution Suspension 25 1.5 CLASSIFICATION OF MATTER CASE STUDY: SILICON MONOXIDE Silicon dioxide, also called silica, is a chemical compound that is a combination of silicon and oxygen chemically bonded together in the ratio of 1 silicon atom to 2 oxygen atoms. The element silicon is a metalloid with a high metallic luster and is very brittle. It is a crystalline solid with bluish reflective faces. The element oxygen is a nonmetal, a colorless gas at room temperature, and a pale blue liquid below 183oC. The compound silicon dioxide is a hard and strong solid. In the crystalline form, it is colorless and transparent. It has good abrasion resistance, but will fracture under an imposed stress. It is a nonconductor of electricity, with high electrical insulation and high thermal stability. Silicon dioxide is abundant in nature and commonly found as the quartz minerals or as sand (Fig. 1.11). Silicon monoxide is a chemical compound that is a combination of silicon and oxygen chemically bonded together in the ratio of 1 silicon atom to 1 oxygen atom. It has been detected by astronomers in young stars, but has not been found to exist naturally on earth. In 1905, Henry Noel Potter, a Westinghouse engineer, reported to have formed silicon monoxide from the decomposition of silicon dioxide in the presence of carbon at very high temperatures. When the heated reaction chamber was cooled rapidly, the evolved gas condensed to form a blackish brown amorphous solid. After combustion analysis, the unknown solid substance was identified as silicon monoxide. Today, silicon monoxide is produced in the vapor phase as a byproduct of the commercial production of purified silicon for the solar energy market and is sold commercially under the name “silicon monoxide.” The properties of the pure compound silicon dioxide are very different from the properties of either element from which it is made. However, the reported properties of silicon monoxide are not widely different from that of silicon dioxide. Also, the vapor phase silicon monoxide produced in this high temperature process has been shown to react quickly with oxygen in the air to reform silicon dioxide. Recent studies of the solid silicon monoxide using modern microscopic and spectroscopic techniques have shown that instead of a pure chemical compound, the amorphous silicon monoxide is actually a heterogeneous mixture of amorphous silicon dioxide and amorphous silicon in an approximate ratio of 1 to 1. This results in an overall ratio of silicon to oxygen in the mixture of 1 to 1, even though the individual atoms are not bonded in the ratio of 1 to 1. This example demonstrates the importance and sometimes the difficulties of correctly classifying pure substances and mixtures. It is not always enough to determine the macroscopic ratio of Silicon Liquid oxygen Silicon dioxide FIG. 1.11 The element silicon (left), liquid oxygen (center), and silicon dioxide, shown as the mineral quartz (right). Photographs by Enricoros–left, Senior Airman Matthew Lotz-center, and Dr. Warwick Hillier-right, Wikimedia Commons. 26 1. INTRODUCTION elements in a substance and assume that ratio holds on a molecular level. A thorough understanding of the molecular bonding in a pure compound and how that affects the macroscopic properties is necessary to correctly classify an unknown substance. When necessary, modern separation techniques used to purify mixtures, which will be discussed in Chapter 15, can also be used to determine the number of pure substances in an unknown material. 1.6 SEPARATION OF MIXTURES Almost every element or compound occurs naturally in a mixture that includes unwanted components. Compounds synthesized in a laboratory or industrial process almost always are associated with impurities or unwanted substances to some degree. Frequently, these mixtures must be separated into their pure components before they can be used. Separation methods are an essential part of industrial processes from energy production to the manufacture of building materials. Some processes require complete separation of the mixture into its pure components, while others may require only partial separation. Since the pure substances in a chemical mixture retain their individual identities and properties, the differences in these properties can be used to separate the pure substances from the mixture. The more similar the properties of the pure substances, the more difficult it is to achieve a separation. Separation methods used for a particular mixture should be based on the properties of the pure substances that have the largest differences. Also, some methods may not be suitable for some mixtures because of the harsh conditions of the separation, such as heating to the boiling or melting point, which may cause chemical alteration of the pure substances. The separation of mixtures into their pure components can be carried out based on the differences in either the physical or the chemical properties. Although very important in industrial processes, a detailed description of chemical separation methods requires an understanding of molecular structure and types of bonding and so will be covered later in Chapters 12 and 15. Some commonly used separation methods that are based on the differences in the physical properties of the pure substances are listed in Table 1.7. Because a heterogeneous mixture is made up of different substances that remain physically separate, a TABLE 1.7 Mixture Separation Methods Based on Differences in Physical Properties of the Pure Substances Method Mixture Type Property Sedimentation Solid-liquid Heterogeneous Density Filtration Solid-liquid; solid-gas Heterogeneous Size Distillation Solid-liquid; liquid-liquid Homogeneous Boiling point Fractional distillation Liquid-liquid; gas-gas Homogeneous Boiling point Sublimation Solid-solid Both Boiling point Effusion Gas-gas Homogeneous Mass 1.6 SEPARATION OF MIXTURES 27 physical separation can be carried out using a mechanical process to separate the different phases. These include: sedimentation and filtration. The physical separation of a homogeneous mixture can be carried out by inducing a phase change of one component in the mixture, resulting in its separation from the other components. These separation methods include: distillation, fractional distillation, sublimation, and effusion. Sedimentation is a mechanical method of separation based on the differences in densities between the particles and the fluid phase. If the solid particles are denser than the fluid in which they are suspended and of sufficient size, they will fall under the force of gravity and settle at the bottom of the fluid separating them from the mixture. After sedimentation has been completed, the top fluid layer can be carefully poured off leaving the solids behind, a process called decanting. But, a small amount of liquid is usually left in the container with the solids and care must be used to prevent solids from flowing out of the container with the last of the liquid. The rate of particle sedimentation in a fluid is described by Stokes Law of Sedimentation; (2) v ¼ g ρp ρ d2 =18μ where v ¼ sedimentation rate, g ¼ gravitational force, ρp ¼ density of the solid particle, ρ ¼ density of the fluid, d ¼ diameter of the particle, and μ ¼ viscosity of the fluid. From this relationship, it can be shown that the sedimentation rate of particles suspended in a liquid or gas: increases as the particle size (d) increases, increases as the difference in density between the particle and the fluid (ρp – ρ) increases, is zero when the particle density (ρp) is the same as the fluid density (ρ), decreases as the fluid viscosity (μ) increases, and increases as the gravitational force (g) increases. Also, if ρ, d, and μ are known, the measurement of the sedimentation velocity, or sedimentation rate, of the particle can provide a measure of the size of the particles. Measurement of sedimentation rate is commonly used to determine the size of large biochemical molecules and other colloids. Particles suspended in denser fluids (large ρ) must be larger (large d) to be removed by sedimentation, while smaller particles can be removed by sedimentation in less dense fluids. Thus, particles suspended in air, which has a density of 1.18 kg/m3 at 25oC, can be removed quickly by sedimentation if they are larger than about 3 μm. However, particles suspended in water, which has a density of 997 kg/m3 at 25oC, must be larger than about 100 μm to be separated by sedimentation in 1 h. Also, larger particles settle faster than smaller particles in the same fluid. The smaller the particle, the longer the time required to achieve total sedimentation. Sedimentation is an important first step in wastewater treatment processes. Solid particles that are kept suspended in the waste stream by the turbulence of moving water can be removed by sedimentation in the still water of settling ponds as shown in Fig. 1.12. Large open settling basins or tanks are often built with mechanical means for continuous removal of the solids that are deposited at the bottom of the tank, while the clarified water is removed from the top of the tank. The normal force of gravity is sufficient to separate many types of particles over time. However, the length of time required for this sedimentation to occur makes this method of separation impractical for mixtures of smaller particles. According to Stokes Law of Sedimentation, the rate of sedimentation can be increased by increasing the effective 28 1. INTRODUCTION FIG. 1.12 Settling tanks of the Newton Creek sewage treatment plant in Brooklyn, New York. Photograph by Hope Alexander, Wikimedia Commons. gravitational force (g). This is accomplished by rapidly spinning the suspension, a process known as centrifugation and the apparatus used for this process is called a centrifuge. When a suspension is rotated rapidly, the centrifugal force causes the particles to move away from the axis of rotation. The larger the centrifugal force, the faster the sedimentation can be accomplished and the smaller the particles that can be separated. The centrifugal force generated by a centrifuge is commonly expressed as a ratio relative to the earth’s gravitational force called the relative centrifugal force (RCF). The types of centrifuges commonly used in laboratory environments are listed in the upper portion of Fig. 1.13. Centrifuges are classified as low-speed centrifuges with a RCF up to 8000, high-speed centrifuges with a RCF up to 20,000, or ultracentrifuges with a RCF up to 2,000,000. While low-speed centrifuges are capable of separating particles as small as 3–4 μm suspended in water, ultracentrifuges are capable of separating colloids as small as viruses. Centrifuges used in industrial applications are common either in low-speed or high-speed varieties. Low-speed centrifuges are used in batch separators to speed up particle sedimentation with removal of the solids from the bottom of the separator as needed. One common application is the separation of soy bean curd (tofu) from coagulated soy milk. High-speed centrifuges are used for the removal of small amounts of solids to clarify liquids, such as in the purification of fuel oils. They are also used in processes designed for the recovery of finely divided metal particles in dilute suspensions as in the recycling of platinum from spent catalytic converters. Ultracentrifuges are almost exclusively used in microbiology and molecular biology including separations of proteins, DNA, or cell components. They are also used in nanotechnology to separate colloidal nanoparticles. Suspensions and aerosols can also be separated by filtration, a mechanical method of separation based on particle size. Macrofiltration methods are accomplished by passing the fluid 29 1.6 SEPARATION OF MIXTURES Sedimentation methods Low speed High speed Ultracentrifuge Macro particle filtration Microfiltration Filtration methods Ultrafiltration Nanofiltration Colloid Mixture type Suspension Silt Clay Viruses Bacteria Fine sand Course sand Protozoa Proteins Particle type Yeast Milled flour Colloidal silica Animal and plant cells Nanoparticles Fulvic acids Humic acids Asbestos Oil smoke 0.001 0.01 0.1 Coal dust Fly ash Pollen 1 10 Particle size (mm) 100 1000 10000 FIG. 1.13 Particle size ranges for sedimentation and filtration separation methods with some examples of the types of particles in each range. through a permeable filter medium with a pore size that captures the larger solid particulates while allowing the fluid to pass through the filter medium. However, the particles retained by the filter can be contaminated with a small amount of fluid. The fluid that has passed through the filter, known as the filtrate, may also contain fine particles that were not retained by the filter. The degree of contamination depends on the pore size of the filter and the size distribution of the particles in the mixture. With liquids, the simplest way to achieve filtration is by gravity flow. But, the filtration rate can be increased by increasing the pressure on the feed side or decreasing the pressure on the filtrate side of the filter medium. The filter medium can be either a surface filter or a depth filter. Depth filters can be granular, such as a bed of sand, or composed of randomly oriented sheets of polymeric, inorganic, or metallic fibrous materials. The multiple layers of randomly oriented grains or fibers in depth filters create inconsistent pore sizes, so their pore size rating is typically only approximate. They are relatively thick compared to their pore size and rely on a torturous path of flow channels through the filter medium to capture particles within the matrix of the filter. The particles will travel along the pore until they reach a point where the pore narrows down to a size too small for the particles to travel further and they become trapped. The particles collected with depth filters are captured within the flow channels of the fibrous material and so the particulates are not easily recovered. Depth filters are less prone to clogging in very high particulate levels and they are commonly used in applications where the solid particulates are the unwanted phase of the mixture. 30 1. INTRODUCTION The pore sizes of surface filters are better defined than that of depth filters. Macro-particle surface filters can be constructed of cellulose filter paper, which is used for the filtration of the larger particles up to 25 μm. For the smaller particles from 1.0 to 2.7 μm, the filters are commonly constructed of glass or quartz fibers. These combine fast flow rates, high loading capacities, and good retention of smaller particles. Common uses are the removal of pollen, dust, and fly ash from air, and separation and isolation of large suspended solids, single cells, and some bacteria from water. Microfiltration, ultrafiltration, and nanofiltration are all surface filtration methods, which make use of microporous membrane filters with specific pore ratings. These are typically constructed of synthetic polymers, such as polycarbonate, polysulfone, or cellulose acetate. Microfiltration is effective for removing larger colloids within the approximate range of 0.1–1.0 μm. It is used to remove smoke particles from air and small bacteria from water. Ultrafiltration removes colloids in the range of 0.002–0.1 μm in size, reaching into the range of viruses and large macromolecules. Nanofiltration is used to remove the very smallest colloids less than 0.002 μm. Since ultrafiltration and nanofiltration membranes with pore sizes below 0.01 μm are capable of removing colloids in the macromolecular size range, they are typically rated by molecular weight instead of by particle size, where 0.01 μm is equal to a molecular weight of approximately 10,000 Daltons. Ultrafiltration and nanofiltration require high feed pressures to force the fluid through the very small pores in the membrane. The smaller the pore size, the higher the feed pressure required for separation. Also, unlike low pressure filtration methods, they do not remove all the fluid from the mixture. The trapped colloids are recovered as a concentrate in a small amount of fluid. The colloids in the concentrate can be separated into smaller size ranges by using a series of membranes with decreasing pore sizes. This method is used in industry for purifying and concentrating colloidal and macromolecular mixtures such as proteins. Ultrafiltration and nanofiltration methods are being used more often for water treatment in place of chemical disinfection due to their ability to physically remove all microbiological species including viruses. Distillation is a physical separation method that involves the vaporization of a liquid component followed by recondensation to the liquid phase. The purified substance obtained by distillation is called the distillate and the distillation apparatus is called a still. Distillation is often used to separate solutes from solvents in liquid solutions such as in the desalination of seawater or the removal of impurities from freshwater. The industrial use of distillation methods requires large amounts of energy. Since the boiling point of a liquid decreases with decreasing pressure, distillation can be achieved at lower temperatures with less energy required if the pressure in the still is reduced. This process is called vacuum distillation. Distillation can also be used to separate homogeneous mixtures of liquids with different boiling points. If the boiling points of the liquids in the mixture differ by more than 25oC, separation can be achieved by raising the temperature of the mixture stepwise, beginning with the lowest boiling point, allowing the component with that boiling point to vaporize followed by condensation of the purified liquid. The temperature of the liquid is then raised to the next lowest boiling point and the process continued until the desired separation is complete. If the boiling points of the liquids in the mixture are closer together than 25oC, the gas phase will not be composed of the pure component only, but will also contain a proportion of the liquid with the next nearest boiling point. When this occurs, the resulting liquid must be redistilled to 1.6 SEPARATION OF MIXTURES 31 achieve separation of the two components. This process is called fractional distillation. The number of distillation times required for complete separation of a mixture depends on the number of components in the mixture and the differences in their boiling points. Fractional distillation is one of the most widely used separation methods in industry. It is used to separate crude oil into heavier and lighter fractions in oil refining, and in the distillation of fermented solutions to increase the alcohol content. Although commonly used for liquid mixtures, fractional distillation can also be used to separate mixtures of gases by first condensing the gas mixture into a liquid, followed by low temperature distillation. This process is called cryogenic distillation. The most common use of cryogenic distillation is the separation of air into its components; nitrogen, oxygen, and argon. CASE STUDY: OIL REFINING Most industrial processes that separate mixtures of liquids based on fractional distillation make use of distillation columns like those shown in Fig. 1.14. These columns are typically large, vertical cylindrical towers ranging from about 1 to 10 m in diameter and 10 to 60 m or more in height. The liquid mixture is heated in a boiler and added to the bottom of the column. Each level of the column is maintained at a constant temperature, which decreases with height. As the mixture boils, it FIG. 1.14 Industrial fractional distillation columns. Photograph by Luigi Chiesa, Wikimedia Commons. 32 1. INTRODUCTION vaporizes and rises up the column. When each component reaches the height of the column corresponding to the temperature of its boiling point, it condenses and the resulting liquid is captured in trays. These distillation columns are operated in a continuous steady state. That is, the mixture is always being added to the bottom of the distillation column and the separated components are always being removed from the trays at the same rate. This maintains a steady concentration of components in the mixture. One common application of fractional distillation is in oil refining, where crude oil is refined and processed into more useful products. Crude oil, or petroleum, is a mixture of hundreds of different components from gases to very heavy waxes. The various components of crude oil have wide ranges of different sizes, molecular weights, and boiling points. So, it is not very useful in the raw form. Due to the large complexity of the mixture, it is impractical to attempt to separate crude oil into all of the pure components. However, the first step in refining is to separate it into more useful fractions. Each fraction is a mixture of limited components with a restricted boiling point range and similar physical properties. The crude oil is heated in a boiler to a high temperature and the resulting hot liquid and vapor is then pumped into the distillation tower as shown in Fig. 1.15. The hot gases rise up the tower through trays with holes in them until they reach their condensation points. As the gases cool, the different components condense back into several distinct liquid fractions and are collected in the trays based on a range of similar boiling points. Each fraction is itself a mixture of pure compounds with similar physical properties. Lighter liquid fractions, like kerosene and naphtha, have lower boiling points and collect near the top of the tower. Heavier liquid fractions, like lubricants and waxes, have higher boiling points and collect in trays at the bottom of the tower. The desired liquid fractions are drawn off the distillation column at various heights. In some cases, distillation columns are operated at low pressures to lower the temperature at which the mixture boils. This vacuum distillation reduces the chance of thermal decomposition of the liquid fractions from overheating of the mixture. Most refinery products can be grouped into three classes: light distillates (liquefied petroleum gas, naphtha, and gasoline), middle distillates (kerosene and diesel), and heavy distillates 20 °C Crude oil Gas (LPG) Naptha Increasing temperature FIG. 1.15 Refining crude oil by using a fractional distillation column. Gasoline Kerosene Diesel oil Lubricating oil Fuel oil Residuals 400 °C Boiler Distillation column 1.6 SEPARATION OF MIXTURES 33 (lubricating oil, fuel oil, waxes, and tar). The gases, methane, ethane, propane, and butane have a boiling range of less than 40oC and are collected at the top of the column in the gas form. These are often liquefied under pressure to form liquefied petroleum gas (LPG), which is commonly used for residential heating and cooking. The residuals: coke, asphalt, tar, and waxes, have a boiling range of greater than 400oC and are left in the bottom of the column as a solid mixture. Their common uses include adhesives, roofing compounds, and asphalt manufacture. The remaining six fractions are all liquids with varying sizes, weights, and boiling points. Naphtha is the lightest liquid fraction and has the lowest range of boiling points of all the liquid fractions. It is most commonly used as an additive for high octane gasoline. The second lightest liquid fraction is gasoline, followed by the middle distillates kerosene and diesel oil. The heaviest and highest boiling liquid fractions are lubricating oil and fuel oil. Many of the fractions obtained from the distillation column are further chemically processed to make other more profitable products. For example, only 40% of the refined crude oil is recovered directly in the gasoline distillate fraction. Since gasoline is the major product of oil companies, in order to increase the gasoline yield other distillate fractions are further chemically processed into gasoline. This increases the yield of gasoline from each barrel of crude oil processed instead of continually distilling large quantities of raw crude oil, which requires large amounts of energy and depletes a limited resource. Sublimation is a physical separation method based on the volatilization of a solid at low pressures. The solid mixture is heated under a vacuum in a sublimation chamber to the temperature of the lowest boiling component. The low pressure causes the solid with the lower boiling point to vaporize without passing through the liquid phase leaving the higher boiling components behind. The gas condenses as the purified solid compound on a cooled surface, called a cold finger, located at the top of the sublimation chamber. The purified solid component is then collected from the cooled surface. Sublimation is typically used to separate low molecular compounds from higher molecular weight impurities. A type of sublimation method commonly used in industrial applications is called freeze drying. A mixture of solid substances in water is first frozen and then the surrounding pressure is reduced to allow the frozen water in the mixture to transition from a solid directly to a gas. The water vapor is then evacuated from the chamber. Freeze drying is essentially a dehydration process used to preserve perishable materials or to make them easier to transport. This produces a dry product which can be returned to its original form simply by adding water. The greatly reduced water content inhibits the action of microorganisms and enzymes that would normally cause spoilage. It also drastically reduces the weight of the material. Freeze drying is often used instead of other higher temperature dehydration methods to avoid thermal decomposition of the mixtures from overheating. In addition, flavors, smells, and nutritional content generally remain the same after rehydration, making the process popular for preserving food. But other volatile compounds such as vinegar and alcohols can also be lost through sublimation sometimes giving undesirable results. Freeze drying was originally developed during World War II to transport serum without the need for refrigeration. Since that time, freeze drying has been used as a preservation or processing technique for a wide variety of products. It is most often used in the food industry. The largest application is freeze dried coffee. The freeze drying process also has important 34 1. INTRODUCTION applications in the pharmaceutical industry. It is now a standard process used to stabilize, store, or increase the shelf life of drug products and other biological materials. Another important application is in freeze dried starter microbiological cultures used in fermentation reactions. Effusion can be used to separate mixtures of gases based on the differences in mass of the gas molecules. When the mixture of gases is allowed to pass through a porous membrane into an area of low pressure, the speed of the gas molecules through the membrane from the high pressure side to the low pressure side depends on their mass. Heavier molecules move slower than lighter molecules. Therefore, gases of lighter mass will diffuse through the membrane pores faster than those with heavier mass as long as the pore size is small compared to the distance between the molecules of the gas. The rate of gas flux through the pore is proportional to the velocity of the molecule and inversely proportional to the square root of the mass of the molecules. For a mixture of 2 gases; ½ (3) rate of gas 1 = rate of gas 2 ¼ mass of gas 2 = mass of gas 1 The separation efficiency through a single membrane is only moderate. However, gas mixtures can be partially separated by allowing them to effuse across multiple membrane barriers. During World War II, gaseous effusion was used to separate two isotopes of uranium having different masses for the production of the first atomic bomb. Effusion has also been used to separate hydrogen from oxygen for use in fuel cells. IMPORTANT TERMS Accuracy the degree of agreement between a measured value of a physical quantity and the actual or true value for that quantity. Aerosol the heterogeneous mixture of solid particles or liquid droplets in a gas that are sufficiently large for sedimentation. Amorphous solid a solid material whose atoms or molecules lack a long range ordered pattern. Centrifugation a method of increasing the rate of sedimentation by rapidly spinning the mixture increasing the effective gravitational force. Centrifuge an apparatus that puts an object in rotation around a fixed axis resulting in an increase in the effective gravitational force. Coalescence the formation of a superfluid by super cooling any of the three classical states of matter (solids, liquids, or gases). Colloid the heterogeneous mixture of very small solid particles in a liquid that are not readily settleable. Compound a pure chemical substance consisting of two or more different chemical elements in a simple whole number ratio that can be separated into simpler substances by chemical reactions. Condensation the transition from a gas phase to a liquid phase. Crystalline solid a solid material whose atoms or molecules are arranged in a highly ordered pattern extending in all three dimensions. Critical point (cp) the temperature and pressure above which the liquid and gas phases become indistinguishable and the substance becomes a supercritical fluid with properties of both gas and liquid phases. Cryogenic distillation the separation of a mixture of liquefied gases by distillation at very low temperatures. Decanting pouring off the top fluid layer leaving the solids behind. Density the ratio of the mass to the volume of a material. Deposition the transition from a gas phase to a solid phase directly without passing through the liquid state. STUDY QUESTIONS 35 Distillate the purified components of a liquid mixture that has been separated by distillation. Distillation a physical method of separating liquid mixtures based on vaporization of the liquid components depending on the differences in their boiling points, followed by recondensation. Ductile the ability to deform under applied stress. Effusion a physical method of the separation of a mixture of gases based on the rate of flux through a porous membrane, which is dependent on the differences in mass of the gas molecules. Filtration a mechanical method of separating solids from fluids by using a medium through which only the fluid can pass. Filtrate a liquid that has passed through a filter. Fractional distillation the separation of liquid mixtures by distillation in which the products are collected in a series of separate fractions, each with a higher boiling point than the previous fraction. Freezing the transition from a liquid phase to a solid phase. Green engineering the design, development and use of products, processes, and systems that are economically feasible while minimizing the risks to human health and the environment. Heterogeneous mixture one that is not thoroughly mixed and is not uniform in composition or appearance. Homogeneous mixture one in which the components are thoroughly mixed so that the atoms or molecules that make up the mixture are uniformly distributed and the macroscopic properties are the same throughout the mixture. Ionization the formation of a plasma by super heating a gas. Melting the transition from a solid phase to a liquid phase. Molecule an electrically neutral group of two or more atoms held together by chemical bonds. Phase diagram a graphical representation of the phase changes of pure substances as a function of temperature and pressure Plasma an ionized gas occurring typically at low pressures or at very high temperatures. Precision the degree to which repeated measurements of a physical quantity made by the same method give the same value. Quantitative dealing with numerical measurements of the amount of a material or its properties. Random error fluctuations in a measurement method that produces both positive and negative variations from an average value of the measured quantity. Recombination the transition from a plasma to a classical gas phase. Sedimentation the tendency for particles in a suspension to settle out of the fluid in which they are mixed, based on the differences in densities between the particles and the fluid phase. Solute a substance that is dissolved in a solvent, resulting in a solution. Solvent a substance that dissolves a solute, resulting in a solution. Solution a homogeneous mixture composed of only one phase formed by solvation of the solute by the solvent. Sublimation the transition from a solid phase directly to a gas phase without passing through the liquid state. Supercritical fluid a substance at a temperature and pressure above its critical point where distinct liquid and gas phases do not exist. Superfluid a state of matter which behaves like a fluid with zero viscosity. Suspension the heterogeneous mixture of solid particles in a liquid that are sufficiently large for sedimentation to occur. Systematic error bias in a measurement method that leads to the situation where the average of many separate measurements differs significantly from the actual value of the measured quantity. Triple point (tp) the temperature and pressure where all three classical phases, solid, liquid, and gas, can coexist simultaneously. Vacuum distillation separation of a liquid mixture by distillation under reduced pressure. Vaporization the transition from a liquid phase to a gas phase. STUDY QUESTIONS 1.1 1.2 1.3 What is the main focus of Green Chemistry? What is the main focus of Green Engineering? What is the difference between scientific notation and engineering notation? 36 1.4 1.5 1.6 1.7 1.8 1.9 1.10 1.11 1.12 1.13 1.14 1.15 1.16 1.17 1.18 1.19 1.20 1.21 1.22 1.23 1.24 1.25 1.26 1.27 1.28 1.29 1.30 1.31 1.32 1.33 1.34 1.35 1.36 1. INTRODUCTION What are the advantages of scientific notation? What are the advantages of engineering notation? What is the difference between accuracy and precision? (a) What types of measurement errors cause low accuracy? (b) What types of errors cause low precision? (a) How is measurement accuracy determined? (b) How is measurement precision determined? What is the difference between systematic errors and random errors? Which digit in a reported measurement is uncertain? (a) What determines the margin of error in the last significant digit of a measurement? (b) How is it expressed? What determines the number of significant figures in the result obtained from the sum of a series of measurements? What determines the number of significant figures in the result obtained from the multiplication of two measurements? How many significant figures are there in an exact number? What is the most widely used system of measurement units? In the SI system of measurements, what is the difference between the base units and the derived units? What are the seven SI base units? What properties do they measure? (a) What is the derived SI unit for temperature? (b) How is it derived from the base unit? What are the SI base units for: (a) pressure, (b) energy, (c) force, (d) radioactive decay? What are the numerical equivalents for the following SI unit prefixes: (a) deca, (b) milli, (c) nano, (d) mega, (e) pico, (f) tera? What are the abbreviations used for the following units: (a) microgram, (b) milliliter, (c) centimeter, (d) picovolt, (e) femtosecond, (f) gigawatt? What are the numerical equivalents for each of the units listed in Question 1.21? What are the SI prefixes for the following: (a) 1 102, (b) 1 10–1, (c) 1 1015, (d) 1 10–18, (e) 1 10–2, (f) 1 10–6? What are the SI unit names for the following: (a) 0.1 L, (b) 0.000000001 m, (c) 1000 g, (d) 1,000,000,000,000,000 W, (e) 0.000000000000001 curie, (f) 0.000001 L? What are the abbreviations for the SI unit names for the values listed in Question 1.24? What are the common units for: (a) pressure, (b) temperature, (c) volume, (d) distance? What are the SI base equivalents of the common units in Question 1.26? What is the unit for density in terms of the SI base units? (a) What is the common unit for density? (b) What are the base equivalents? How does increasing the pressure on a compressible material affect the density? How does increasing the temperature of a material affect the density? How does increasing the temperature of water from below 0oC to above 0oC affect the density? List the five states of matter in order of increasing particulate movement. What is the physical difference between a liquid and a gas? What are the terms for the following transitions: (a) gas to solid, (b) solid to gas, (c) solid to liquid, (d) liquid to gas, (e) gas to plasma, (f) gas to liquid. (a) What four states of matter are considered to be fluids? (b) Why? PROBLEMS 37 1.37 The boiling point of a pure liquid increases or decreases with: (a) increasing pressure, (b) increasing altitude? 1.38 At what temperature and pressure does a supercritical fluid exist? 1.39 What is the triple point of a substance? 1.40 What is the difference between a crystalline solid and an amorphous solid? 1.41 (a) The elements are divided into what three categories? (b) Most elements fall into which category? 1.42 What is the difference between an organic compound and an inorganic compound? 1.43 What is the difference between a homogeneous mixture and a heterogeneous mixture? 1.44 What is a solution? 1.45 (a) What is a heterogeneous mixture of solid particles and a liquid called? (b) What is a heterogeneous mixture of solid particles and a gas called? 1.46 Define the chemical state of the following substances according to Fig. 1.8: (a) green tea, (b) aluminum foil, (c) diamond, (d) table sugar, (e) milk, (f) soda pop, (g) clean air, (h) salad dressing, (i) brass. 1.47 How does particles size affect the sedimentation rate of particles suspended in a liquid or gas? 1.48 What process is used to increase sedimentation? 1.49 What are the two methods used for separation of heterogeneous mixtures of a solid in a liquid? 1.50 What is the method used for separation of homogeneous mixtures of a solid in a liquid? 1.51 What is the method used to separate homogeneous mixtures of gases? 1.52 What process is used to separate homogeneous mixtures of liquids? 1.53 What process is used to decrease the time of liquid separation? 1.54 What filtration methods can separate particulates as small as viruses from a liquid? 1.55 The separation of a mixture of gases by effusion is based on what physical property? 1.56 What is a molecule? 1.57 If the following parameters are increased and everything else remains constant, will the sedimentation rate of the particles in a suspension increase or decrease? (a) relative gravitational force, (b) particle size, (c) particle density, (d) fluid viscosity, (e) fluid density. PROBLEMS 1.58 Express 24,000,000 in: (a) scientific notation and (b) engineering notation. 1.59 Listed below are distances in kilometers. Each distance is the result of a calculation based on simple measurements. Convert each result to scientific notation. (a) 384,403 (Earth to Moon), (b) 149,600,000 (Earth to Sun), (c) 88,200,000 (Earth to Mars at perigee), (d) 501,000,000 (Earth to Jupiter at perigee), (e) 38,000,000 (Earth to Venus at perigee), (f) 2,570,000,000 (Earth to Uranus at perigee) 1.60 What is the number of significant figures in each of the measurements given in Problem 1.59? 38 1. INTRODUCTION 1.61 The speed of light is defined as 299,792.458 km/s. If this constant were used to calculate the time it would take for light to travel each of the distances listed in Problem 1.59, how many significant figures would each result have? 1.62 The following atomic radii have been measured by X-ray crystallography with an accuracy of 5 pm. Determine the number of significant figures in the measurements of each of the following atomic radii: (a) hydrogen ¼ 25 pm, (b) carbon ¼ 70 pm, (c) aluminum ¼ 125 pm, (d) sulfur ¼ 100 pm, (e) zirconium ¼ 155 pm. 1.63 Determine the results for the following calculations to the correct number of significant figures: (a) 248.65 g + 245 g, (b) 378 cm + 2.36 cm + 200 cm, (c) 469.01 kg – 33.2264 kg, (d) 271 m + 1 m + 16 m, (e) 500 m – 20 m, (f) 0.110 m – 0.04 m. 1.64 Determine the results for the following calculations to the correct number of significant figures, express the results in scientific notation, and give the units of the results: (a) 33.25 g/1.1 mL, (b) 84 m/s 25.2 s, (c) 450 m/242 s, (d) 3.0 105 km/s 9.3 107 km, (e) 1.22 103 J/32 s. 1.65 Express the results of the following conversions in scientific notation using the correct number of significant figures: (a) 239.0 cm to km, (b) 100.6 mg to g, (c) 0.004 g to μg, (d) 0.75 L to mL, (e) 10.5 mL to μL, (f) 65 s to ms. 1.66 Convert each of the measurements to kg expressing each in scientific notation: (a) 1.24 L to mL, (b) 2.63 pg to ng, (c) 3.6 103 km to m, (d) 460 Tg. 1.67 Express each of the distances listed in Problem 1.59 in meters with the appropriate SI prefix. 1.68 Listed below are masses in kilograms of some solar objects. Express each in scientific notation in yottagrams. (a) 5.9736 1024 (Earth), (b) 7.35 1022 (Moon), (c) 4.8685 1024 (Venus), (d) 1.8986 1027 (Jupiter), (e) 6.4185 1021 (Mars), (f) 1.9891 1030 (Sun). 1.69 Calculate the density of the solar objects in Problem 1.68 using the volumes listed below in km3. Express the result in petagrams/km3 with the correct number of significant figures. (a) 1.08321 1012 (Earth), (b) 2.1958 1010 (Moon), (c) 9.2843 1011 (Venus), (d) 1.43128 1015 (Jupiter), (e) 1.6318 1011 (Mars), (f) 1.412 1018 (Sun). 1.70 Convert the densities in Problem 1.69 to g/cm3 using the conversion factor; 1 km3 ¼ 1 1015 cm3. 1.71 The atmospheric pressures at the surface of some solar objects are listed below in SI derived units. What are these pressures in bar? Express the results in scientific notation with the correct number of significant figures. (a) 101 kPa (Earth), (b) 800 Pa (Mars), (c) 300 pPa (Moon light), (d) 800 pPa (Moon dark). 1.72 The mean surface temperatures of some solar objects are given below in the SI base unit. What are these temperatures in degrees Celsius? (a) 220 K (Moon), (b) 210 K (Mars), (c) 737 K (Venus). 1.73 What are the temperatures listed in Problem 1.72 in degrees Fahrenheit? 1.74 An unknown solid material in the shape of a rod is measured and found to be 22.5 mm in diameter and 7.4 cm long. The mass of the material is measured and found to be 79.4523 g. Calculate the density of the solid with the correct number of significant figures. 1.75 Calculate the density of an unknown liquid with a measured mass of 23.724 g and a measured volume of 30.0 mL. 1.76 A laboratory experiment requires 63.2 g of the liquid in Problem 1.75. How much volume of the liquid should be measured in order to obtain the required mass? PROBLEMS 39 1.77 The volume of the liquid in Problem 1.75 was measured at a temperature of 25°C. When the temperature of the liquid was increased to 50°C, the volume of the liquid increased to 31.0 mL. What is the decrease in density of the liquid with this 25°C increase in temperature? 1.78 A solution is made up of table salt in water by measuring three salt samples of 1.2, 10.0, and 4.36 g. All three samples are dissolved in 100 mL of water. What is the final concentration of the salt in the solution in g/mL? 1.79 What physical separation methods should be used to separate the following mixtures: (a) a homogeneous mixture of hydrogen and oxygen, (b) a heterogeneous mixture of clay and water, (c) a homogeneous mixture of ethanol in water, (d) a heterogeneous mixture of sand in water, (e) a heterogeneous mixture of proteins in ice? C H A P T E R 2 The Periodic Table of the Elements O U T L I N E 2.1 Atomic Structure 41 2.5 Periodic Trends 64 2.2 The Shell Model of the Atom 48 Important Terms 70 2.3 Electron Assignments 54 Study Questions 71 Problems 72 2.4 The Periodic Table of the Elements 58 2.1 ATOMIC STRUCTURE As we saw in Chapter 1, chemists have over the years identified the 98 naturally occurring elements that make up all of the molecules and materials that we find on our planet. Each of these elements is defined by its distinctive chemical and physical properties, which set it apart from all the other elements. Each element was named as it was discovered. Some of the names of the elements were derived from their observed properties or their method of discovery. For example, the element hydrogen was discovered by Henry Cavendish in 1766. He noted that it generated water when reacted with oxygen and named it from the Greek words for water (hydros) and generator (genes). Helium was named from the Greek word for sun (Helios) as it was originally discovered in 1895 through studies of the solar emission spectrum. Other elements were named for their discoverers or their place of discovery. Europium, discovered by a French chemist Eugène-Anatole Demarcay in 1896, was named for the European continent where it was discovered while americium was named for America, the continent where it was discovered. Similar histories can be found for all of the elements. Only one element has been named for a chemist while he was still alive. This element is Seaborgium, named for Dr. Glenn Seaborg who led the discovery of a number of short-lived elements that were artificially generated using nuclear reactions at the University of California, Berkeley. We will discuss these elements and their radioactivity in Chapter 14. In an attempt to determine the scientific basis for the observed differences in the properties of the elements, chemists looked to the internal structure of the atom that makes up each of the General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00002-3 41 # 2018 Elsevier Inc. All rights reserved. 42 2. THE PERIODIC TABLE OF THE ELEMENTS FIG. 2.1 Structure of the atom. Nucleus + Neutron − Proton Electron elements. It was determined that atoms are made up of three types of particles: electrons, protons, and neutrons. The atomic nucleus consists of positively charged protons and uncharged neutrons held tightly together in a small volume at the center of the atom as shown in Fig. 2.1. The negatively charged electrons “orbit” this nucleus forming the electron cloud surrounding the nucleus. Since the proton and neutron have the highest mass of the three particles (see Table 2.1), the nucleus makes up most of the mass of the atom while the electrons in their orbits make up most of the volume of the atom. The number of protons in the nucleus of the atom is the atomic number of the element and is commonly designated by the symbol Z. For a neutral atom, the number of electrons equals the number of protons. The atomic number is specific to each element as it also determines the number of electrons, which determines the properties of the element. The sum of the number of protons and neutrons in the nucleus is called the mass number of the element since these two heaviest particles determine the approximate mass of the atom. The mass number is commonly designated by the symbol A. • Z ¼ number of protons ¼ number of electrons • A ¼ number of protons + number of neutrons • A–Z ¼ number of neutrons TABLE 2.1 Properties of the Subatomic Particles Particle Proton Neutron Electron Symbol p n e Charge +1 0 1 Mass (g) Mass Relative to Neutron 24 1.6726 10 24 1.6749 10 28 9.1094 10 0.9986 1.0 0.0005439 43 2.1 ATOMIC STRUCTURE An atomic symbol is used to abbreviate the atomic structures of each element as; where X is the symbol of the element as listed in Table 2.2, Z is the atomic number of the element, and A is the mass number. The use of the atomic number (Z) in the atomic symbol is optional since the number of protons is specific to an element and can be obtained from the element symbol itself. In this case, the atomic symbol is written as; TABLE 2.2 Atomic Symbols of the Elements Name Symbol Name Symbol Name Symbol Actinium Ac Germanium Ge Potassium K Aluminum Al Gold Au Praseodymium Pr Americium Am Hafnium Hf Promethium Pm Antimony Sb Hassium Hs Protactinium Pa Argon Ar Helium He Radium Ra Arsenic As Holmium Ho Radon Rn Astatine At Hydrogen H Rhenium Re Barium Ba Indium In Rhodium Rh Berkelium Bk Iodine I Roentgenium Rg Beryllium Be Iridium Ir Rubidium Rb Bismuth Bi Iron Fe Ruthenium Ru Bohrium Bh Krypton Kr Rutherfordium Rf Boron B Lanthanum La Samarium Sm Bromine Br Lawrencium Lr Scandium Sc Cadmium Cd Lead Pb Seaborgium Sg Cesium (Caesium) Cs Lithium Li Selenium Se Calcium Ca Livermorium Lv Silicon Si Continued 44 2. THE PERIODIC TABLE OF THE ELEMENTS TABLE 2.2 Atomic Symbols of the Elements—cont’d Name Symbol Name Symbol Name Symbol Californium Cf Lutetium Lu Silver Ag Carbon C Magnesium Mg Sodium Na Cerium Ce Manganese Mn Strontium Sr Chlorine Cl Meitnerium Mt Sulfur S Chromium Cr Mendelevium Md Tantalum Ta Cobalt Co Mercury Hg Technetium Tc Copernicium Cn Molybdenum Mo Tellurium Te Copper Cu Neodymium Nd Terbium Tb Curium Cm Neon Ne Thallium Tl Darmstadtium Ds Neptunium Np Thorium Th Dubnium Db Nickel Ni Thulium Tm Dysprosium Dy Niobium Nb Tin Sn Einsteinium Es Nitrogen N Titanium Ti Erbium Er Nobelium No Tungsten W Europium Eu Osmium Os Uranium U Fermium Fm Oxygen O Vanadium V Flerovium Fl Palladium Pd Xenon Xe Fluorine F Phosphorous P Ytterbium Yb Francium Fr Platinum Pt Yttrium Y Gadolinium Gd Plutonium Pu Zinc Zn Gallium Ga Polonium Po Zirconium Zr Most of the symbols of the elements are derived directly from their common name using the first letter (capitalized) often coupled with a second letter. Twelve of the elements use only the first letter of the element name. These are Boron, Carbon, Fluorine, Hydrogen, Iodine, Nitrogen, Oxygen, Phosphorous, Sulfur, Uranium, Vanadium, and Yttrium. Forty four of the elements use the first and second letters of their name. Still others use the first and third letters, etc. The symbols for a few of the elements can be quite different from their current names. This generally occurred for elements discovered very early and originally given Latin names, which described the element’s properties. Later, these elements were given names equivalent to the English translation of the original Latin names. The seemingly strange atomic symbol was then derived from the original Latin name of the element. A list of the elements whose atomic symbols were derived from names different from their current names is given in Table 2.3. All the names in this list were Latin except for wolfram (tungsten), which is German. 45 2.1 ATOMIC STRUCTURE TABLE 2.3 Atomic Symbols of Elements Derived From Names Other Than Their Current Name Current Name Symbol Original Name Antimony Sb Stibium Copper Cu Cuprum Gold Au Aurum Iron Fe Ferrum Lead Pb Plumbus Mercury Hg Hydrargyrum Potassium K Kalium Silver Ag Argentium Sodium Na Natrium Tin Sn Stannum Tungsten W Wolfram Atoms with the same number of protons and electrons, but with different numbers of neutrons, are called isotopes. Since they have the same number of protons and electrons, they are the same element and have the same chemistry. But because they have different numbers of neutrons, they have different masses. For example, hydrogen has one proton and one electron with an atomic number of 1 and a mass number of 1. If we add a neutron to the proton in the hydrogen nucleus, the atomic number is still 1. However, the mass number will now be 2 and this isotope will have a mass about twice that of the hydrogen atom. Hydrogen is the only element whose isotopes have different names. The isotope of hydrogen with a mass number of 2 is called deuterium. Another isotope of hydrogen has a mass number of 3 and is called tritium. Both deuterium and tritium have the same chemistry as hydrogen, but deuterium weighs twice as much and tritium weighs three times as much as hydrogen. So, an isotope is an atomic form of an element that has a different mass, but retains the same basic chemistry. Note that although the chemistry is the same, the change in mass can affect the rates at which the element will react. This is known as a kinetic isotope effect, which we will discuss in detail in Chapter 9. Most elements occur naturally as a mixture of isotopes all with different masses. Normally, one isotope will be the most abundant and will make up most of the naturally observed form of the element. The mass number (A) in the atomic symbol is used to distinguish between the different isotopes of an element. Chemists usually will represent a specific isotope of an element by using only AX, as the difference between A and Z are the number of neutrons, which defines the isotope, and the element itself defines the atomic number (Z). For example, the stable isotopes of carbon are represented as 12C and 13C. Both of these isotopes have an atomic number of 6, as carbon has 6 protons in all of its isotopes. The atomic symbols for hydrogen, deuterium, and tritium are 1H, 2H, and 3H. The mass number is usually left off the atomic symbol for the most abundant isotope unless other isotopes need to be identified. For example, H is always assumed to be 1H. The atomic mass of an element, also called atomic weight, is different from the mass number and represents an average of all the naturally occurring isotopes. The atomic mass is 46 2. THE PERIODIC TABLE OF THE ELEMENTS determined as the sum of the masses of each isotope of the element multiplied by the fraction of its natural abundances. Since naturally occurring carbon consists of about 99% 12C (six neutrons, six protons, and six electrons) and about 1% 13C (seven neutrons, six protons, and six electrons), the atomic mass of carbon is determined from the following; Atomic mass C ¼ mass 12 C Þ ð0:99Þ + mass 13 C Þð0:01Þ (1) Even though we know the exact masses in grams of the three fundamental subatomic particles (Table 2.1), it is more convenient to use the atomic mass unit (amu), also called a Dalton, to define an element’s mass. The atomic mass unit is defined as one twelfth of the mass of a single carbon atom containing six neutrons, six protons, and six electrons (12C). Carbon-12 is assigned a mass of 12.000 amu and 1 amu ¼ 1.66054 1024 g. The atomic mass of the element carbon calculated from the average of its two isotopes is slightly higher than 12. The average of 12C with a natural abundance of 0.99 and 13C with a natural abundance of 0.01 gives the element carbon an atomic mass of 12.0107 amu. • The important fact to remember in chemistry is that atoms with the same number of protons will have the same chemistry because the number of protons determines the number of electrons and the electrons control the chemistry. Added neutrons only add mass, but do not change the chemical behavior of an element. CASE STUDY: DETERMINING THE ATOMIC MASS AND ISOTOPE ABUNDANCES OF XENON Xenon is a colorless, odorless gas that occurs in the atmosphere in trace amounts. It has an atomic number of 54 (54 protons and 54 electrons). In order to determine the atomic mass of xenon, we need to know its naturally occurring isotopes with each of their masses and their natural abundances. This is done experimentally with a mass spectrometer. The mass spectrometer separates the isotopes according to their mass through a series of steps shown in Fig. 2.2. These steps are: (1) ionization, (2) acceleration, (3) deflection, and (4) detection. Magnet Gas inlet Electron beam e− Detector Step 1 Step 2 Ionization Acceleration Step 3 Deflection Step 4 Detection FIG. 2.2 A schematic diagram of a mass spectrometer showing the four steps used in determining isotopic natural abundances. 47 2.1 ATOMIC STRUCTURE The xenon sample is introduced into the mass spectrometer as a gas stream where it passes through a high energy electron beam. The high energy electrons collide with the xenon atoms causing an electron to be stripped from its orbit around the xenon nucleus resulting in xenon atoms with 54 protons and 53 electrons, giving them a positive charge. An atom with an unequal number of protons and electrons is called an ion and the process of creating the ion from the uncharged atom is called ionization (step 1). The stream of positively charged ions is then passed through a series of negatively charged plates, causing the positively charged ions to become accelerated (step 2). The rapidly moving xenon ions then pass through a magnetic field that is perpendicular to the flow of the ions. The magnetic field causes the stream of xenon ions to curve and be deflected from their original path (step 3). The extent of this deflection depends on the mass of the ions. The lighter isotopes are deflected more severely than the heavier isotopes separating them according to their masses. The extent of deflection of the isotopes is also dependent on the strength of the magnetic field. As the strength of the magnetic field is slowly changed, the isotopic streams of differing masses are each focused in turn on the surface of a detector (step 4). Since the ions are positive, they pick up electrons from the detector and the amount of ions hitting the detector is measured as an electric current. The strength of the magnetic field at the time each isotope is detected determines its mass and the magnitude of the electric current determines its natural abundance. A plot of the relative abundances of the isotopes versus their mass is called a mass spectrum. The mass spectrum of xenon is shown in Fig. 2.3 and the results are summarized in Table 2.4. The mass spectrum shows that xenon has nine naturally occurring isotopes; 124Xe, 126Xe, 128Xe, 129 Xe, 130Xe, 131Xe, 132Xe, 134Xe, and 136Xe. Since all the xenon isotopes have 54 protons, the number of neutrons for each isotope is equal to: Mass number 54 ¼ 70, 72,74,75, 76,77, 78,80, and 82: 12 Except for C, which is defined as having a mass of 12.0 amu, isotopic masses are not integer values. But, they are always very close to the mass number. The actual mass of the nine xenon isotopes as measured by the mass spectrometer in Table 2.4 are from 0.09 to 0.1 amu less than their mass 25 20 15 10 Mass FIG. 2.3 The mass spectrum of xenon. 136 134 132 131 130 129 128 0 126 5 124 Relative abundance (%) 30 48 2. THE PERIODIC TABLE OF THE ELEMENTS TABLE 2.4 Relative Abundances of the Xenon Isotopes as Determined From the Mass Spectrum Mass Number Isotopic Mass (amu) Number of Neutrons Abundance (%) 124 123.91 70 0.1 126 125.90 72 0.1 128 127.90 74 1.9 129 128.91 75 26.4 130 129.90 76 4.1 131 130.91 77 21.2 132 131.90 78 26.9 134 133.91 80 10.4 136 135.91 82 8.9 numbers. This is due in part to the mass of the electrons, which is not included in the mass number. It is also due to the contribution of binding energy between the neutrons and protons, which will be discussed in Chapter 14. Since the atomic mass of xenon is equal to the sum of the masses of each isotope multiplied by the fraction of its natural abundances, the atomic mass for xenon is; ð123:91Þð0:001Þ + ð125:90Þð0:001Þ + ð127:90Þð0:019Þ + ð128:91Þð0:264Þ + ð129:90Þð0:041Þ + ð130:91Þð0:212Þ + ð131:90Þð0:269Þ + ð133:91Þð0:104Þ + ð135:91Þð0:089Þ ¼ 131:29amu (2) 2.2 THE SHELL MODEL OF THE ATOM In order to further explain the properties of the elements, scientists struggled to determine the exact structure of the electron cloud surrounding the nucleus of an atom. Why was it that the attractive forces between the negatively charged electrons and the positively charged protons did not result in the electrons falling into the nucleus? Niels Bohr addressed this question by proposing that the electrons orbit the nucleus in circular orbits of fixed energy. These orbits exist at stable distances from the nucleus and the electrons must remain in these orbits to prevent them from collapsing into the nucleus. The electrons cannot exist between orbits. There are a series of these stable orbits of different energies and at different distances from the nucleus. The orbit closest to the nucleus is of lowest energy, while the one farthest from the nucleus is of highest energy. This arrangement results in the energy of the electrons being restricted to certain allowed values. This restriction of electrons to a limited number of possible energy values is known as being quantized. The allowed orbits are each designated by a positive integer n called the principal quantum number. The value of n is equal to 1 for the orbit closest to the nucleus and increases with distance from the nucleus as shown in Fig. 2.4. 49 2.2 THE SHELL MODEL OF THE ATOM FIG. 2.4 Neils Bohr’s model of the atom with electrons confined to n=4 discrete stable orbitals. n=3 n=2 n=1 As n increases and the electrons occupy orbits further from the nucleus, the energy of the electron increases. So each orbit is also known as the energy level of the electron. CASE STUDY: THE EMISSION SPECTRUM OF HYDROGEN When hydrogen gas is exposed to an electric current or heated to high temperatures, it emits light that appears blue to the human eye. However, when this light is separated into its component wavelengths by passing it through a prism, it is found that it is actually composed of several discrete lines at different wavelengths with only darkness in between. This type of light spectrum is called a line emission spectrum. The line emission spectrum of hydrogen is shown in Fig. 2.5. Since the visible spectrum is from 390 to 700 nm, only four of these lines can be seen without using infrared and ultraviolet detectors. These visible lines appear at 656 (red), 486 (blue green), 434 (blue violet), and 410 (violet). Because these lines could be detected by the human eye, they were the first to be discovered. They were called the Balmer series after Johann Balmer who first predicted their existence. A second set of lines appears in the ultraviolet at 122, 103, 97, 95, and 94 nm. These lines are called the Lyman series after their discoverer Theodore Lyman. A third set of lines appears in the Lyman series Balmer series 0 500 Paschen series 1000 Wavelength (nm) FIG. 2.5 The line emission spectrum of hydrogen. 1500 2000 50 2. THE PERIODIC TABLE OF THE ELEMENTS FIG. 2.6 The transition of an electron from a lower orbit to a higher orbit due to the absorption of energy followed by the electron returning to the lower orbit with the release of the excess energy in the form of light of a specific wavelength (λ). n=3 Energy n=2 l1 l2 n=1 infrared at 1875, 1282, and 1094 nm. They are called the Paschen series also after their discoverer Friedrich Paschen. It should be noted that these three series of lines differ not only by the wavelength range in which they occur, but also by the number of lines in each series and the separation between them. The Lyman Series appears at shorter wavelengths and is composed of five lines. These five lines are very close together, being separated by an average distance of 7 nm. The Balmer Series, in the visible wavelength range, is composed of four lines with an average separation of 82 nm. The Paschen series appears at longer wavelengths and is composed of only three lines separated by much larger distances with an average distance of 390 nm. The atomic number of hydrogen is 1. It has one electron in the lowest energy level (n ¼ 1). Bohr attempted to explain the occurrence of the hydrogen line emission spectrum by suggesting that the energy added to the hydrogen atoms when exposed to the electric current caused the hydrogen electron to “jump” to higher orbits as shown in Fig. 2.6. It then would return to its original orbit by releasing the excess energy in the form of light. Since the allowed orbits available to the electron are quantized, the energy released is also quantized and appears as narrow lines at specific wavelengths in the line emission spectrum. The wavelength of each line is dependent on the difference in energy between the two electronic orbits. An electron falling from the n ¼ 2 orbit to the n ¼ 1 orbit would release light of wavelength λ1 in Fig. 2.6, while an electron falling from n ¼ 3 orbit to the n ¼ 1 orbit would release light of wavelength λ2. Since n ¼ 3 is of higher energy than n ¼ 2, the light emitted from the n ¼ 3 to n ¼ 1 transition would be of shorter wavelength than that from the n ¼ 2 to n ¼ 1 transition (λ1 > λ2 in Fig. 2.6). The energy released when the electron falls back to a lower orbit is related to the wavelength of light observed by; E ¼ hc=λ ¼ hν (3) where h is Plank’s constant (6.626 1034 J • s), c is the speed of light (2.998 108 m/s), λ is the light emission wavelength in nm, and ν is the emission frequency. (So, when someone asks you “What’s new” you can answer by saying “c over lambda.”) The emission line observed at the shortest wavelength (highest energy) in the hydrogen line emission spectrum arises from electrons falling from the highest orbit (n ¼ 6) back to the lowest orbit (n ¼ 1). This line of highest energy appears at 94 nm in the Lyman Series. The lines occurring at longer wavelengths (lower energy) occur from electrons falling between orbits closer together in energy. 51 2.2 THE SHELL MODEL OF THE ATOM FIG. 2.7 Electronic transitions responsible for the hydrogen line emission spectrum. Modified from Szdori, Wikimedia Commons. 12 2n m 10 3 nm Lyman series 97 95 nm nm m 656 n 486 nm 434 nm n=1 n=2 94 nm 410 nm Balmer series 187 5 nm 12 82 nm n=3 10 94 nm Paschen series n=4 n=5 n=6 The Lyman series appears in the ultraviolet (highest energy) because all five lines arise from electrons in the higher orbits (n ¼ 2, 3, 4, 5, and 6) falling back to their original n ¼ 1 orbit as shown in Fig. 2.7. The Balmer series has four lines (four electronic transitions) caused by electrons in orbits n ¼ 3, 4, 5, and 6 falling to the n ¼ 2 orbit. The three lines in the Paschen series are from electrons in orbits n ¼ 4, 5, and 6 falling to the n ¼ 3 orbit. Both the Balmer and Paschen series require a second transition for the hydrogen atoms to return to the lowest energy and most stable state. This lowest possible energy state is known as the ground state of the atom. Higher energy states arising from the absorption of energy and the transition of electrons to higher orbits are known as excited states. The energy difference between the two orbits in an electronic transition (n2 to n1) responsible for each emission line in the spectrum can be calculated from the wavelength of the emission line as; ΔE ¼ Ef Ei ¼ hc=λ (4) where Ei is the energy of the initial state and Ef is the energy of the final state. The wavelength of light emitted from an electronic transition (nf to ni) can be calculated from; 1=λ ¼ R 1=nf 2 1=ni 2 (5) Balmer developed this relationship to help explain the source of the visible lines in the Balmer series. The constant R is known as the Rydberg constant (1.0974 107 m1) and the equation is known as the Balmer equation. For the red line in the Balmer series, the wavelength of the transition ni ¼ 3 to nf ¼ 2 is; 1 1:0974 107 1=22 1=32 ¼ 6:56 107 m ¼ 656nm: (6) 52 2. THE PERIODIC TABLE OF THE ELEMENTS It was later determined by X-ray spectroscopy that each Bohr orbit can contain more than one electron. The negative charges of electrons occupying the same orbit would cause them to repel each other resulting in an energy difference between the electrons. In order to explain these differences in energy, the electrons were then described as being in close orbits within the same principal energy level, also called an electron shell. These electron shells were designated by the principal quantum number n as before. In addition, the electron shells were also given letter designations which came from X-ray notation. These new designations were an alphabetical series of letters beginning with the letter K (K, L, M, N, …..) where n ¼ 1 is the K shell, n ¼ 2 is the L shell, n ¼ 3 is the M shell, and so on. Each electron shell was divided into subshells denoted by the letters s, p, d, f with increasing energies (s < p < d < f). So the s subshell is the lowest in energy and the f subshell is the highest in energy within an electron shell. These letter designations come from the original description of a series of alkali metal spectroscopic lines as sharp, principle, diffuse, and fine. Each electron shell can contain the number of subshells equal to the value of its principal quantum number. The K shell with n ¼ 1 contains only one subshell (s). The L shell with n ¼ 2 contains two subshells (s and p). The M shell (n ¼ 3) contains three subshells (s, p, and d), and so on. The subshells are also designated by a quantum number called the azimuthal quantum number, l. The value of l for the different subshells is an increasing positive integer beginning with 0. So, the value of l for the s subshell is 0, for the p subshell is 1, for the d subshell is 2, and for the f subshell is 3. The values of the azimuthal quantum numbers are outlined in Table 2.5. Each subshell then contains orbitals to which the electrons are confined. The number of orbitals in each subshell is equal to 2l + 1. So, the s subshell has 2(0) + 1 ¼ 1 orbital, the p subshell has 2(1) + 1 ¼ 3 orbitals, and so on. These are also outlined in Table 2.5. The orbitals within a subshell have a characteristic shape specific to that subshell shown in Fig. 2.8. This shape is defined mathematically by the azimuthal quantum number and represents the probability of locating the electron within the area of each orbital. The orbitals in each of the s subshells are spherical in shape and increase in diameter as n increases. The p orbitals all have a “dumb bell” shape with a nodal plane (a region of zero probability) passing through the nucleus. The number of nodal planes in an orbital is equal to the value of l for the subshell. So, the orbital in an s subshell has no nodal planes (l ¼ 0), the three orbitals in the p subshell (l ¼ 1) have one nodal plane, all five orbitals in the d subshell (l ¼ 2) have two nodal planes, and all seven orbitals in the f subshell (l ¼ 3) have three nodal planes. TABLE 2.5 Properties of the Electron Subshells Subshell l Number Orbitals (2l + 1) Maximum Electrons 2(2l + 1) Shape Nodal Planes (l) s 0 1 2 Spherical 0 p 1 3 6 Dumb bell (2 lobes) 1 d 2 5 10 Double-dumb bell (4 lobes) 2 f 3 7 14 Complex (6–8 lobes) 3 53 2.2 THE SHELL MODEL OF THE ATOM z z y z y px x py s orbital pz p orbitals y z y y x x x FIG. 2.8 Shapes of the s, p, and d atomic orbitals. z y x z x z y x z dxy dxz z y y x x dyz d orbitals dx2−y2 dz2 Each orbital can contain a maximum of two electrons. Since negatively charged electrons will repel each other, in order for the orbital to accept two electrons they must be of opposite spin. The electron spin is also assigned a quantum number called the spin quantum number (s). The spin quantum number has the values of +½ or ½. The complete description of the energy level of any electron within an atom requires all four quantum numbers (n, l, m, and s) and no two electrons in the same atom can have the same four quantum numbers. This rule is called the Pauli Exclusion Principle. The properties of the four quantum numbers are summarized in Table 2.6. The first two quantum numbers (n and l) also determine the maximum number of electrons in each electron shell. The K shell (n ¼ 1) can have only one subshell (s with l ¼ 0) with one orbital (2l + 1) and a maximum of two electrons [2(2l + 1)]. So, the maximum number of electrons in the K shell is equal to two. The L shell (n ¼ 2) can have two subshells (s with l ¼ 0 and p with l ¼ 1) with a total of four orbitals (1 + 3) and eight electrons (2 + 6). TABLE 2.6 The Properties of the Four Quantum Numbers That Describe the Energy Level of an Electron Name Symbol Value Property Relationships Principal quantum number n 1, 2, 3,⋯ Energy level n ¼ number of subshells in shell Azimuthal quantum number l n 1 0, 1, 2, 3 Orbital shape l ¼ number of nodal planes, 2l + 1 ¼ number of orbitals in subshell 2(2l + 1) ¼ number of electrons in subshell Magnetic quantum number m +l…0….l Orbital spatial orientation Spin quantum number s +½, ½ Electron spin 54 2. THE PERIODIC TABLE OF THE ELEMENTS EXAMPLE 2.1: DETERMINING THE MAXIMUM NUMBER OF ELECTRONS IN AN ELECTRON SHELL What is the maximum number of electrons in the M and N electron shells? 1. Determine the number of subshells. M shell: n ¼ 3, subshells ¼ 3 (s, p, d) N shell: n ¼ 4, subshells ¼ 4 (s, p, d, f) 2. Determine the number of orbitals per subshell. Number of orbitals ¼ 2l + 1: s ¼ 2(0) + 1, p ¼ 2(1) + 1, d ¼ 2(2) + 1, f ¼ 2(3) + 1 M shell: 2l + 1 ¼ 1 + 3 + 5 ¼ 9 N shell: 2l + 1 ¼ 1 + 3 + 5 + 7 ¼ 16 3. Multiply the total number of orbitals by two to obtain the number of electrons. M shell: 2(9) ¼ 18 N shell: 2(16) ¼ 32 2.3 ELECTRON ASSIGNMENTS The electrons of any element are assigned to the shells, subshells, and orbitals in the order of increasing energy. Each electron will occupy the orbital of lowest energy available. They will enter an orbital of higher energy only when the orbitals of lower energy are all filled. This procedure for assigning electrons to shells, subshells, and orbitals in the order of increasing energy is known as the Aufbau principle (aufbau is the German word for “building up”). The Aufbau principle is guided by: 1. The Pauli Exclusion Principle: no more than two electrons can occupy any one orbital. 2. Hund’s Rule: within a subshell, electrons will occupy the orbitals individually (with parallel spins) before filling them in pairs (with opposite spins). The energy of any orbital is predicted by both the n and l quantum numbers, which determine the energies of the electronic shell and subshell. In general, the electrons fill all the subshells in a shell in the order of increasing energy s < p < d < f before beginning to fill the next higher shell. This method of assigning electrons to subshells according to the increasing principal quantum number holds strictly for elements of atomic number 1–18. The trend is more complex for elements with atomic numbers higher than 18. An element with an atomic number of 1 (hydrogen) has only one electron in the single orbital of the s subshell of the n ¼ 1 shell. The element with an atomic number of 2 (helium) has two electrons in the single orbital of the s subshell of the n ¼ 1 shell each with opposite spin. Since the n ¼ 1 shell has only one orbital and can only have two electrons, the element with atomic number 3 (lithium) has two electrons in the 1s subshell and one electron in the 2s subshell. This assignment of electrons to the electronic shells, subshells, and orbitals is called the electronic configuration of the atom and is most often represented in a symbolic notation known as subshell notation. This form lists the principal quantum number of the electron shell, followed by the subshell type, and the number of electrons in each subshell as shown in Fig. 2.9. Each subshell in the atom containing electrons is listed in this manner beginning with the 1s. The electronic configurations in subshell notation for the elements with atomic 2.3 ELECTRON ASSIGNMENTS Electron shell quantum number 55 Number of electrons Subshell type FIG. 2.9 Subshell notation used to describe the electronic configuration of an atom. TABLE 2.7 Electron Configuration in Subshell Notation for Elements With Atomic Numbers 1–18 Atomic Number Electron Configuration 1 1s1 2 1s2 3 1s22s1 4 1s22s2 5 1s22s22p1 6 1s22s22p2 7 1s22s22p3 8 1s22s22p4 9 1s22s22p5 10 1s22s22p6 11 1s22s22p63s1 12 1s22s22p63s2 13 1s22s22p63s23p1 14 1s22s22p63s23p2 15 1s22s22p63s23p3 16 1s22s22p63s23p4 17 1s22s22p63s23p5 18 1s22s22p63s23p6 numbers 1–18 are shown in Table 2.7. Elements with atomic numbers 1–10 complete the filling of electrons in both the K (n ¼ 1) and L (n ¼ 2) shells, while elements with atomic numbers 11–18 fill the s and p subshells of the M (n ¼ 3) shell. So, the order of energies for the subshells for elements with atomic numbers 1–18 follows the simple trend of 1s < 2s < 2p < 3s < 3p. At atomic number 19, the filling order of the subshells becomes more complex and the 4s subshell is filled before the 3d subshell. This may seem like a departure from the simple trend that holds for atomic numbers 1–18, but in fact it is not. This is because the order of the energy of the subshells, which determines the order of the electron filling order, is determined by the sum of the n + l quantum numbers (Madelung’s Rule). The value of n + l for both the 3p subshell and the 4s subshell is equal to 4. 56 2. THE PERIODIC TABLE OF THE ELEMENTS However, the value of n + l for the 3d subshell is equal to 5. So, the 4s subshell is actually of lower energy than the 3d subshell because it has a lower n + l value. This is why electrons are placed in the 4s subshell before the 3d subshell. The Aufbau principle is then modified to account for the electron assignments of the elements with atomic numbers above 18 as follows: 1. 2. 3. 4. Electron subshells are filled in the order of increasing n + l. Where two subshells have the same n + l, the subshell with the lowest n is filled first. No more than two electrons can occupy any one orbital. Electrons will fill all orbitals in a subshell individually (with parallel spins) before filling them in pairs (with opposite spins). So, the resulting order of energies for the electronic subshells according to the increasing n + l values is; 1s < 2s < 2p < 3s < 3p < 4s < 3d < 4p < 5s < 4d < 5p < 6s < 4f < 5d < 6p < 7s < 5f < 6d < 7p < 8s Table 2.8 details the calculation of n + l values for all available subshells and determines the electron filling order of each subshell. Fig. 2.10 summarizes the electron filling order in TABLE 2.8 Electron Filling Order According to the n + l Rule Subshell n l n+l Filing Order 1s 1 0 1 1 2s 2 0 2 2 2p 2 1 3 3 3s 3 0 3 4 3p 3 1 4 5 3d 3 2 5 7 4s 4 0 4 6 4p 4 1 5 8 4d 4 2 6 10 4f 4 3 7 13 5s 5 0 5 9 5p 5 1 6 11 5d 5 2 7 14 5f 5 3 8 17 6s 6 0 6 12 6p 6 1 7 15 6d 6 2 8 18 7s 7 0 7 16 7p 7 1 8 19 8s 8 0 8 20 57 2.3 ELECTRON ASSIGNMENTS n+l = Electron shell n 1 2 3 4 1 1s 2 2s 2p 3 3s 3p 3d 4 4s 4p 4d 4f 5 5s 5p 5d 5f 6 6s 6p 6d 7 7s 7p 8 8s l= 0 1 2 5 6 7 8 FIG. 2.10 Diagrammatic representation of the electron filling order according to the n + l rule. 3 diagram form. Subshells above the 6d, 7p, and 8s are not known to exist in the ground state of any known element. Although the subshell notation is a simple way to represent the electronic configuration of an atom, it holds no information regarding the electronic orbitals or electron spin. A more detailed way of describing the electronic configuration is by an orbital diagram where the orbitals are designated by boxes and the electrons are shown by arrows inside the boxes. Electron spin is represented by the direction of the arrow in each box. An upward pointing arrow represents a spin of +½ and a downward pointing arrow represents a spin of ½. Elements with atomic numbers 1–10 would be represented in orbital diagrams shown in Fig. 2.11. Atomic number Orbitals 1s 2s 2p 1 2 3 4 5 6 7 8 9 10 FIG. 2.11 Orbital diagrams for the elements with atomic numbers 1–10. 58 2. THE PERIODIC TABLE OF THE ELEMENTS EXAMPLE 2.2: DESCRIBING THE ELECTRONIC CONFIGURATION OF AN ELEMENT Show the electronic configuration of nickel (atomic number 28) in subshell notation and in an orbital diagram. 1. Subshell notation. The first 18 electrons fill the subshells in order: 1s22s22p63s23p6 Since the n + l value of the 4s subshell ¼ 4 and the value for the 3d subshell ¼ 5, the 4s fills first followed by the 3d. The electronic configuration of nickel is: 1s22s22p63s23p64s23d8 2. Orbital diagram. The orbital diagram has the addition of electron spin, which follows Hund’s Rule of electron pairing. The electron configuration is represented as: 1s 2s 2p 3s 3p 4s 3d 2.4 THE PERIODIC TABLE OF THE ELEMENTS As the Bohr concept of stable circular orbits eventually evolved to consider orbitals of other shapes, which basically explained the electronic structures of atoms and their chemical and physical properties, these theories also evolved further as chemists continued to discover new elements and detail their properties. By the early 19th century, chemists had acquired enough knowledge of the elements and their electronic structures to begin to recognize similarities among groups of elements and a regularity or periodicity of some properties that appeared to be tied to similarities in their electronic structures. In the 19th century, a Russian chemist, Dimitri Mendeleev, and a German chemist, Julius Meyer, both recognized this periodicity of chemical properties of the elements. Both chemists had determined correctly that the properties of the elements follow basic rules which are determined by their electronic structure and that this periodicity would be an extremely useful tool in explaining chemical reactions, molecular structures, and predicting elemental class behavior. Mendeleev and Meyer both developed a periodic law independently stating; “the properties of the elements are a periodic function of their atomic masses.” Both chemists attempted to express this periodic law in the form of a chart grouping together elements with similar properties. While Meyer chose to arrange the elements as a function of atomic volume (the atomic mass of an element divided by the density of its solid form), Mendeleev chose to arrange the elements according to atomic mass. Using this arrangement, Mendeleev published the first “periodic table” and predicted that some elements must be undiscovered at that time. He left unassigned places for these undiscovered elements in his table. For example, at that time there was no element known with an atomic 2.4 THE PERIODIC TABLE OF THE ELEMENTS 59 mass between 40 (calcium) and 48 (titanium) and Mendeleyev left a vacant space for it in his table. Later in 1879, the element scandium was discovered with an atomic mass of 45 and properties that were consistent with giving it the position in the previously unassigned spot in the table. The discovery of scandium was one of a series of elemental discoveries which validated the periodic law, thus leading to Mendeleev being given primary credit for the development of the Periodic Table of the Elements. However, some inconsistencies remained with Mendeleev’s version of the periodic table. In 1913, Henry Moseley, a British physicist, was able to determine the atomic numbers of all the known elements using X-ray emission spectroscopy. He then proceeded to rearrange the elements in Mendeleev’s periodic table according to increasing atomic numbers. This arrangement seemed to clear up the contradictions and inconsistencies in Mendeleev’s arrangement by atomic masses. Since atomic number defines the number of protons in the nucleus and therefore the number of electrons in the neutral atom, this arrangement was more directly related to electronic structure and more clearly predicted properties of the elements. Moseley restated the periodic law originally proposed by Mendeleev and Meyer as a function of the atomic number of the elements. Moseley’s periodic law as stated below is now considered the current Periodic Law. • The physical and chemical properties of the elements recur periodically in a systematic and predictable way when the elements are arranged in order of increasing atomic number. An understanding of this periodicity of the chemical properties of the elements allowed chemists to predict how elements would combine and how they would behave when reacting with other elements. The development of the Periodic Table of the Elements can thus be compared to the discovery of the “Rosetta Stone,” which provided the key to the translation of ancient languages into modern text and for the first time allowed scholars to be able to understand the meaning of ancient documents. Similarly, the periodic table effectively organizes the elements into classes according to their electronic configurations, providing the key to understanding and predicting the chemical behavior of the elements. In order to effectively use the periodic table to predict chemical properties and behavior of the elements, it is important to first know how its structure relates to electronic configurations. The general layout of the modern periodic table is shown in Fig. 1.12. It arranges the elements in vertical columns called groups. There are 18 groups in the periodic table numbered 1–18. Elements in the same group generally share the same chemical properties with clear trends associated with increasing atomic number going down a group. Some groups are known by family names to indicate the similar properties that they share. Group 1 is called the alkali metals (lithium, sodium, potassium, rubidium, cesium, and francium). Group 2 is known as the alkaline earth metals (beryllium, magnesium, calcium, strontium, barium, and radium). Group 17 is the halogens (fluorine, chlorine, bromine, iodine, and astatine) and group 18 is the noble gases (helium, neon, argon, krypton, xenon, and radon). Elements in groups 3–12 are known collectively as the transition metals. The elements are also arranged in seven horizontal rows called periods because the periodic properties of the elements increase systematically with increasing atomic number going across a row. There are seven periods in the periodic table numbered 1–7. The number of each period corresponds to the principal quantum number of the outermost electron shell containing electrons for all elements in the period. So, the elements in period 1 contain 60 2. THE PERIODIC TABLE OF THE ELEMENTS Group 1 1 Period 2 3 4 5 6 7 1 H 3 Li 11 Na 19 K 37 Rb 55 Cs 87 Fr 2 4 Be 12 Mg 20 Ca 38 Sr 56 Ba 88 Ra * ** 3 4 5 6 7 8 ** 22 Ti 40 Zr 72 Hf 104 Rf 23 24 25 Cr Mn V 41 42 43 Nb Mo Tc 73 74 75 Ta W Re 105 106 107 Db Sg Bh 57 La 89 Ac 58 Ce 90 Th 59 Pr 91 Pa 21 Sc 39 Y * 60 Nd 92 U 9 10 11 12 13 14 15 16 17 18 26 27 28 29 Fe Co Ni Cu 44 45 46 47 Ru Rh Rd Ag 76 77 78 79 Pt Au Os lr 108 109 110 111 Hs Mt Ds Rg 30 Zn 48 Cd 80 Hg 112 Cn 65 Tb 97 Bk 66 Dy 98 Cf 61 62 63 64 Pm Sm Eu Gd 93 94 95 96 Np Pu Am Cm Alkali metals Alkaline earth metals Transition metals 5 B 13 Al 31 Ca 49 ln 81 TI 113 Lanthanide metals Actinide metals Post-transition metals 6 C 14 Si 32 Ge 50 Sn 82 Pb 114 FI 7 N 15 P 33 As 51 Sb 83 Bi 115 8 9 O F 16 17 S CI 34 35 Se Br 52 53 Te I 84 85 Po At 116 117 Lv 2 He 10 Ne 18 Ar 36 Kr 54 Xe 86 Rn 118 67 68 69 70 71 Ho Er Tm Yb Lu 99 100 101 102 103 Es Fm Md No Lr Metalloids Non metals Noble gases FIG. 2.12 The Periodic Table of the Elements. The atomic number of each element is displayed above the element’s symbol. Modified from Sandbh, Wikimedia Commons. electrons only in the n ¼ 1 shell, while the elements in period 2 contain electrons in the n ¼ 1 and n ¼ 2 shells, and elements in period 3 contain electrons in the n ¼ 1, 2, and 3 shells, and so on. Since the periodic table is arranged according to atomic number, increasing the atomic number by one corresponds to adding one electron to the electronic configuration of the previous element in the period. Some areas of the periodic table are called blocks shown in Fig. 2.13. The blocks indicate the filling sequence of the electron shells and each block is named according to the subshell in which the outermost electrons reside for elements in that block. The s-block includes elements in group 1 (alkali metals) and group 2 (alkaline earth metals). It also includes helium (atomic number 2). All of these elements have outermost electrons in the s-subshell in the shell corresponding to the number of the period. The p-block includes groups 13–18 and contains the metalloids, post-transition metals, nonmetals, and the noble gases. The elements in this block have their outermost electrons in p orbitals. Elements in groups 3–12 make up the dblock which contains all of the transition metals. The f-block has no group numbers, but includes the lanthanide series excluding lanthanum and the actinide series excluding actinium. The lanthanide and actinide series are often shown offset below the rest of the periodic table for simplicity, as in Fig. 2.12. However, lanthanum and actinium are actually in group 3 and part of the d-block elements. The rest of the lanthanide and actinide series belong between groups 2 and 3 in periods 6 and 7. The blocks appear in the periodic table according to the electron filling sequence following Madelung’s rule described in Section 2.3. The d-block appears in periods 4–7 before the p-block since the d subshell fills before the p subshell after the n ¼ 3 shell. Similarly, the f-block 61 2.4 THE PERIODIC TABLE OF THE ELEMENTS Group 1 2 Period 1 3 4 5 6 7 8 9 10 11 12 131415161718 1s 1s 2 2s 2p 3 3s 3p 4 4s 3d 4p 5 5s 4d 5p 6 6s 4f 5d 6p 7 7s 5f 6d 7p s-block f-block d-block p-block FIG. 2.13 Subshell blocks indicating the electron filling order in the Periodic Table of the Elements. Modified from DePiep, Wikimedia Commons. appears before the d-block for periods 6 and 7 corresponding to the electron filling order. So, the elements in the periodic table appear in the same sequence as the electron filling order and the periodic table can be used as a guide to determine electronic configurations of any element. EXAMPLE 2.3: USING THE SUBSHELL BLOCKS OF THE PERIODIC TABLE TO FIND THE ELECTRONIC CONFIGURATION OF AN ELEMENT Give the electronic configuration of iodine in subshell notation. 1. Locate iodine in the periodic table. Iodine is atomic number 53. Period 5, group 17 of the periodic table. 2. Starting from period 1, group 1, and continuing through up to iodine determine the sequence of blocks. 1s-block, 2s-block, 2p-block, 3s-block, 3p6 4s-block, 3d-block, 4p-block, 5s-block, 4d-block, 5p-block. 3. Determine the electrons that each block will contribute to the electronic configuration of iodine. All the blocks will be filled except for the last block: the 5p-block. Since iodine is in the fifth space in the 5p-block, it will have 5p electrons. Stringing all these together gives the complete electronic configuration of iodine: 1s2 2s2 2p6 3s2 3p6 4s2 3d10 4p6 5s2 4d10 5p5 Generally, all the elements in the same group have the same electron configurations in their outermost occupied electron shell called the valence shell. For example, all the 62 2. THE PERIODIC TABLE OF THE ELEMENTS elements in group 1 have only one electron in the valence shell. All elements in group 2 have two electrons in the valence shell and elements in group 3 all have three electrons in the valence shell, and so on. The electrons in the valence shell are called the valence electrons and it is these electrons that take part in chemical bonding and will predict how elements will behave in chemical reactions. So, this similarity of valence electron configurations for all elements in a group gives them similar chemical properties. The electrons in shells between the nucleus and the valence electrons are called the core electrons. The core electrons are not involved in chemical bonding and so do not contribute to the chemical properties of an element. In general, the number of valence electrons for an element is the same as its group number except for elements in the p-block. For elements in groups 13–18, the number of valence electrons is the same as the group number minus 10. • The period number corresponds to the principal quantum number of the valence shell for all elements in the period. • The group number corresponds to the number of valence electrons for all elements in the group for groups 1–12. • The group number minus 10 corresponds to the number of valence electrons for all elements in the group for groups 13–18. Using these rules, it is easy to determine the electron configurations of the valence shells for any element except the lanthanides and actinides (excluding La and Ac), which do not have group numbers. The electron configurations of the valence shells for each group are listed in Table 2.9. Note that the valence electrons of the transition metals in groups 3–12 also include electrons in the (n 1)d subshell, even though they do not reside in the outermost n shell. This is because the (n 1)d subshell is very close in energy to the ns subshell and so the electrons in this subshell can also take part in chemical bonding until the subshell becomes completely filled at group 12. The (n 1)d electrons therefore behave as valence electrons in groups 3–12 and are important in determining the chemical properties of the transition metals. Once the d subshell is completely filled, its electrons no longer act as valence electrons and are considered as core electrons for elements in groups 13–18. A very simple shorthand method of writing electron configurations based on the element’s place in the periodic table is called the noble gas notation. This notation uses the symbol for the noble gas in the period preceding the element in brackets to represent the electron configuration for all periods before the element. This leaves only the electrons in the last period to be written in subshell notation. For example, the electron configuration for carbon (period 2, group 14) is 1s22s22p2, while the electron configuration for helium (the noble gas in period 1) is 1s2. So, the electron configuration for carbon written in noble gas notation would be; [He]2s22p2. Similarly, the electron configuration for zinc (period 4, group 12) is 1s22s22p63s23p63d104s2, while the electron configuration for argon (the noble gas in period 3) is 1s22s22p63s23p6. So, the electronic configuration for zinc in noble gas notation would be: [Ar]3d104s2. For elements in groups 1–11, the electronic configuration of the noble gas are the core electrons of the element and the electrons written in subshell notation are the valence electrons. This does not hold for elements in groups 12–17 since the 10 electrons in the filled d subshell are not valence electrons. 63 2.4 THE PERIODIC TABLE OF THE ELEMENTS TABLE 2.9 The Valence Electron Configurations for the Elements in Each Group of the Periodic Table; where n ¼ The Period Number Group Valence Electron Configurations Number of Valence Electrons ns 1 2 ns 2 3 ns (n 1)d 3 4 ns (n 1)d 4 5 ns (n 1)d 5 6 ns (n 1)d 6 7 ns (n 1)d 7 8 ns (n 1)d 8 9 ns (n 1)d 9 10 ns (n 1)d 10 11 ns (n 1)d 11 12 ns (n 1)d 12 1 1 2 2 1 2 2 2 3 2 4 2 5 2 6 2 7 2 8 2 9 2 10 13 2 ns np 1 3 14 2 ns np 2 4 15 2 ns np 3 5 16 2 ns np 4 6 17 2 ns np 5 7 18 ns2np6 8 EXAMPLE 2.4: DESCRIBING THE ELECTRONIC CONFIGURATION OF AN ELEMENT IN NOBLE GAS NOTATION Show the electronic configuration of nickel (atomic number 28) in noble gas notation. 1. Determine the noble gas before nickel. Nickel is in period 4. The noble gas in period 3 is Ar. 2. Determine the electronic configuration from the noble gas to nickel. The remaining electrons in period 4 up to and including nickel would be two electrons in the s-block and eight electrons in the d-block: 4s23d8 Remember that the 3d subshell fills before the 4p subshell as shown in Fig. 2.13. 3. Write the noble gas in brackets followed by the electronic configuration after the noble gas. Electronic configuration of nickel: [Ar]4s23d8 64 2. THE PERIODIC TABLE OF THE ELEMENTS TABLE 2.10 Electron Dot Structures for the Elements Group Number Electron Dot Structure 1 Li 2 Be 3 and 13 B_ 4 and 14 _ C _ 5 and 15 € N _ 6 and 16 € : O _ 7 and 17 € : F € 8 and 18 € : Ne: € The valence electrons can be represented symbolically by electron dot structures, also called Lewis dot structures after the chemist G. N. Lewis. In this notation, the valence electrons are represented by dots placed around the chemical symbol of the element arranged by the following rules. 1. Electrons are placed on each side of the elemental symbol for a maximum of eight (four on each side), which is the number of electrons in filled s and p subshells (s2 + p6). 2. A single electron is placed on each side before pairing them up according to Hund’s rule with the exception of helium. Helium’s two electrons are shown paired because its valence shell is completely filled. Since all the elements in a group have the same number of valence electrons, they all have the same electron dot structures. The electron dot structures for elements in the s- and p-blocks are shown in Table 2.10. Since electron dot structures of the elements are limited to a valence of eight electrons, they cannot be used for elements in groups 9–12. 2.5 PERIODIC TRENDS As described by the Periodic Law, there are trends in many properties of the elements in the periodic table which increase or decrease systematically as you move across a period or down a group. These periodic trends can be explained by the changes in electronic configuration of the elements within the periods or groups. Some important periodic trends are: atomic radius, ionization energy, electron affinity, and electronegativity. These observed trends give chemists a useful tool that allows them to predict an element’s properties, chemical reactivities, and types of compounds that it will form from their position in the periodic table. 65 2.5 PERIODIC TRENDS 400 Fr Atomic radius (pm) 350 Cs 300 Rb K 250 Na 200 Li 150 100 Ar 50 0 Kr He 0 Rn Xe Ne 10 20 30 40 50 60 Atomic number 70 80 90 100 FIG. 2.14 Atomic radius in picometers as a function of atomic number for elements 1–95. Perhaps the first periodic trend to be observed is that of changing atomic radius. The atomic radius is the distance from the nucleus to the outermost occupied electron orbital in an atom. As shown in Fig. 2.14, the atomic radii follow a fairly regular pattern of a minimum followed immediately by a maximum and then decreasing steadily to the next minimum as atomic number is increased. The maximum radii are found for the alkali metals in group 1; lithium (atomic number 3), sodium (atomic number 11), potassium (atomic number 19), rubidium (atomic number 37), cesium (atomic number 55), and francium (atomic number 87). These maxima are followed by a steady decrease in atomic radius with increasing atomic number until reaching a minimum. The minimum radii are for the noble gases in group 18; helium (atomic number 2), neon (atomic number 10), argon (atomic number 18), krypton (atomic number 36), xenon (atomic number 54), and radon (atomic number 86). So, the atomic radius decreases from left to right across a period. The reason for this decreasing trend in atomic radii occurs because as atomic number increases proceeding from left to right across a period, the electrons are added to the same electron shell. This has little effect on increasing the atomic radius. But, as the electrons are added to the shell, protons are also added to the nucleus. Increasing the number of positively charged protons in the nucleus causes the attraction between the valence electrons and the nucleus to become stronger, pulling the valence shell closer to the nucleus. So, the atomic radius decreases across a period as protons are added to the nucleus. In Fig. 2.14, both the maximum values (group 1) and the minimum values (group 18) for atomic radii increase as the atomic number increases. So, as you move from top to bottom in the same group in the periodic table, the atomic radius increases. This is because the electrons are added to a higher electron shell farther from the nucleus, thus increasing the atomic radius. So, the valence electrons for elements in a group occupy higher energy levels with increasing principal quantum number (n) going down the group. As a result, the valence electrons are further away from the nucleus as n increases. In general, the atomic radius decreases from left to right within a period and increases from top to bottom within a group. 2. THE PERIODIC TABLE OF THE ELEMENTS FIG. 2.15 Ionization Energy in kJ/mol as a function of atomic number for elements 1–98. 2500 Ionization energy (kJ/mol) 66 He Ne 2000 Ar 1500 Kr Xe Rn 1000 500 Li Na K Rb Cs Fr 0 0 20 40 60 Atomic number 80 100 A similar periodic trend is seen for the relationship between the first ionization energy for the elements versus atomic number shown in Fig. 2.15. The first ionization energy is the amount of energy required to remove the outermost electron from an atom (X) in the gas phase (g), resulting in a positively charged ion X+1. XðgÞ + energy ! X +1 ðgÞ + e The lower the ionization energy, the more readily the atom can lose an electron and become a positively charged ion. The higher the ionization energy, the more unlikely the atom will lose an electron and become a positively charged ion. The ionization energies for the elements also follow a fairly regular pattern. However, a comparison of Figs. 2.14 and 2.15 shows a pattern opposite to that seen for atomic radius. Ionization energy shows a pattern with a maximum followed immediately by a minimum and a steady increase back to a maximum. So, the periodic trend in ionization energies is opposite to that observed with the atomic radii. The highest ionization energies are seen for the noble gases in group 18 (atomic numbers 2, 10, 18, 36, 54, and 86), while the lowest ionization energies are seen for the alkali metals in group 1 (atomic numbers 3, 11, 19, 37, 55, and 87). So, the ionization energy increases from left to right across a period. Although opposite to the trend observed in atomic radius, the trend in ionization energy occurs because of the same electronic effects responsible for the trend in atomic radius. Ionization energy tends to increase as you move across a period from the alkali metals to the noble gases because the increasing number of protons in the nucleus results in a stronger attraction for the valence electrons. This stronger attraction increases the energy required to remove a valence electron from the valence shell. Also, it has been observed that an atom whose valence shell contains the maximum number of electrons allowed by the Pauli Exclusion Principle, called a closed shell, is especially stable. The alkali metals in group 1 have the lowest ionization energies because their single electron in the valence shell requires less energy to remove. The noble gases have very high ionization energies because their valence shells are completely full and the nuclear attraction for the valence electrons is at a maximum. Notice that helium (atomic number 2) has the highest ionization energy of all the elements. 2.5 PERIODIC TRENDS 67 Both the maxima (group 18) and minima (group 1) decrease as atomic number increases. So, the ionization energy decreases from top to bottom in a group. As atomic number increases in a group, the electrons are added to electron shells farther from the nucleus (increasing n). The valence electrons experience a weaker attraction to the nucleus’s positive charge and require less energy to remove them. The higher the principal quantum number of the outermost electrons, the lower the ionization energy required to remove them. In general, ionization energy increases from left to right within a period and decreases from top to bottom within a group. The properties and chemical reactivities of metals depend on their ability to lose a valence electron. This dependence will be discussed in detail in Chapter 12. Put simply, ionization potential is a measure of metallic character of an element and the periodic trends in metallic character follow those of ionization potential. Metallic character of the elements decreases as you proceed across a period from left to right. The most reactive metals are the alkali and alkaline earth metals in groups 1 and 2. These are followed by the transition metals (groups 3 through 12), the post-transition metals, the metalloids, and finally the nonmetals at the far right. The alkali metals, alkaline earth metals, and transition metals all fall in groups containing metals with similar properties. However, the post-transition metals, metalloids, and nonmetals do not all fall in groups of their own. For example, group 15 contains two nonmetals (N and P), two metalloids (As and Sb), and one post-transition metal (Bi) as atomic number increases. Group 16 contains three nonmetals (O, S, and Se), one metalloid (Te), and one post-transition metal (Po). The reason for this is that, like ionization energy, metallic character also increases as you proceed from top to bottom down a group. This is why at the far right of the periodic table (groups 13–16), metallic character changes within a group. Electron affinity can be considered to be the opposite of ionization energy. It is defined as the amount of energy released when an electron is added to a neutral atom in the gas phase to form a negative ion. XðgÞ + e ! X1 ðgÞ + energy In simple terms, it is the ability of an atom to gain an electron. Electron affinity is shown in Fig. 2.16 as a function of atomic number. The halogens in group 17; fluorine (atomic FIG. 2.16 Electron affinity in kJ/mol as a function of atomic number for elements 1–85. Nobel gases ( ), alkali metals ( ), alkaline earth metals ( ). 400 Electron affinity (kJ/mol) CI F Br I 300 At 200 100 0 0 20 40 60 Atomic number 80 100 68 2. THE PERIODIC TABLE OF THE ELEMENTS number 9), chlorine (atomic number 17), bromine (atomic number 35), iodine (atomic number 53), and astatine (atomic number 85) have the largest electron affinities. This is because the addition of one electron to their valence shell would close the shell and give them a stable electronic configuration. The noble gases in group 18 have electron affinities of zero, because all the orbitals in their valence shell are completely filled. It would be impossible to add an electron to the closed shell. The alkali metals in group 1 (atomic numbers 3, 11, 19, 37, 55, and 87) have a very low but measurable electron affinity. Although the addition of an electron to the alkali metals would not complete the electron shell, it would complete the s orbital adding extra stability to the electron configuration. The alkaline earth metals in group 2 (atomic number 4, 12, 20, 38, 56, and 88) have electron affinities at or very close to zero. The electron affinities for the halogens in Fig. 2.16 generally decrease as atomic number increases. As you proceed down the group from fluorine to astatine, the valence electrons are placed in electron shells that are farther away from the nucleus (increasing n). As the distance between the negatively charged electron and the positively charged nucleus becomes larger, the force of attraction between the nucleus and a valence electron becomes weaker and the electron affinity decreases. So, in general, electron affinity decreases going down a group. However, this trend holds strictly only for group 1 and 17. The trend is less clear for other groups. It can be seen from Fig. 2.16 that electron affinity varies more across the periodic table than some of the other elemental properties. However, some trends can be seen. Nonmetals have higher electron affinities than metals. In general, electron affinity increases moving from left to right across a period from the alkali metals to the halogens. The forces of attraction between the nucleus and valence electrons become stronger with added protons as atomic number increases resulting in an increase of electron affinity from left to right across a period. Electronegativity is a measure of the tendency of an atom to attract electrons from another atom in a chemical bond. The most commonly used scale for calculating electronegativity is that originally proposed by Linus Pauling. This is a dimensionless number on a relative scale from around 0.7–4.0 based on bond energies and is known as the Pauling scale. Electronegativity of the elements is shown in Fig. 2.17 as a function of atomic number. Fluorine, the element with the highest electronegativity, is assigned a value of 4.0, and values range down to cesium (or caesium) and francium which are the least electronegative at 0.7. The halogens (group 17) have the highest electronegativity and the alkali metals (group 1) have the lowest electronegativity. Many of the noble gases have not been measured. FIG. 2.17 Electronegativity on the Pauling Scale as a function of atomic number for elements 1–102. F Electronegativity 4 CI 3 Br I At 2 1 Li Na K Rb Cs Fr 40 60 Atomic number 80 0 0 20 100 69 2.5 PERIODIC TRENDS Electronegativity slowly increases from the group 1 elements to the halogens. So, electronegativity generally increases going from left to right across a period. This is because the greater nuclear charge has a greater attraction for the valence electrons and would result in a higher tendency to attract electrons from the open valence shell of another atom in a chemical bond. Electronegativity decreases for the halogens from fluorine to astatine. In general, electronegativity decreases going from top to bottom in a group. This is also due to the increasing atomic radii resulting in a weaker attraction for the valence electrons and a weaker attraction for electrons in a chemical bond. In summary, many of the chemical properties and reactivities of an element can be determined from its position in the periodic table. This position determines: 1. The number of protons in the nucleus (atomic number)—more protons result in greater attraction between the nucleus and the valence electrons. 2. The distance of the valence shell from the nucleus (principal quantum number)—the larger the distance (higher principal quantum number), the weaker the attraction between the nucleus and the valence electrons. 3. The number of electrons in the valence shell—how close is the valence configuration to that of a closed shell. This results in the following general trends in the elemental properties: • • • • atomic radius—decreases across a period, increases down a group ionization energy—increases across a period, decreases down a group electron affinity—increases across a period, decreases down a group electronegativity—increases across a period, decreases down a group These trends are shown graphically in Fig. 2.18. Atomic radius Ionization energy Electron affinity Electronegativity 1 H 2 He 6 C 3 Li 4 Be 5 B 7 N 8 O 9 F 10 Ne 11 Na 12 Mg 13 AI 14 Si 15 P 16 S 17 Cl 18 Ar 19 K 20 Ca 21 Sc 22 Ti 23 V 24 Cr 25 Mn 26 Fe 27 Co 28 Ni 37 Rb 38 Sr 39 Y 40 Zr 41 Nb 42 Mo 43 Tc 44 Ru 45 Rh 55 Cs 56 57– Ba 71 72 Hf 73 Ta 74 W 75 Re 76 Os 77 Ir 87 Fr 88 89– Ra 103 104 105 106 107 108 Rf Db Sg Bh Hs 30 Zn 48 Cd 31 Ga 32 Ge 33 As 34 Se 35 Br 36 Kr 46 Pd 29 Cu 47 Ag 49 In 50 Sn 51 Sb 52 Te 53 I 54 Xe 78 Pt 79 Au 80 Hg 81 Tl 82 Pb 83 Bi 84 Po 85 At 86 Rn 109 110 111 112 113 114 115 116 117 118 Mt Ds Rg Cn Fl Lv Atomic radius Ionization energy Electron affinity Electronegativity FIG. 2.18 Periodic trends for atomic radius, ionization energy, electron affinity, and electronegativity. The trends generally increase in the direction of the arrows. Modified from Sandbh, Wikimedia Commons. 70 2. THE PERIODIC TABLE OF THE ELEMENTS IMPORTANT TERMS Atomic mass the average mass of the naturally occurring isotopes of an element. Atomic mass unit (amu) the standard unit of mass on an atomic scale defined as one twelfth of the mass of a single carbon atom containing six neutrons, six protons, and six electrons. Atomic nucleus the very small region at the center of an atom consisting of protons and neutrons. Atomic number the number of protons in the nucleus of an atom. Atomic radius the distance from the nucleus to the outermost occupied electron orbital in an atom. Atomic symbol a one or two letter code for a chemical element, usually derived from the name of the element, with numerical superscripts and subscripts to indicate the atomic number and mass number of the element. Aufbau principle the procedure for assigning electrons to shells, subshells, and orbitals in the order of increasing energy. Azimuthal quantum number the number designation (l) of an electron subshell. Closed shell a valence shell in an atom that has the maximum number of electrons allowed by the Pauli Exclusion Principle. Core electrons the electrons in all the electron shells between the nucleus and the valence shell of an atom. Electron a negatively charged particle that orbits the nucleus in all atoms. Electron affinity the amount of energy released when an electron is added to a neutral atom in the gas phase. Electron dot structure a diagram of the valence electrons of an element where the electrons are represented by dots placed around the chemical symbol of the element. Electron shell a group of electron subshells all having the same value of the principal quantum number. Electron subshell a group of electron orbitals with the same value of the principal quantum number. Electronegativity a measure of the tendency of an atom to attract electrons from another atom in a chemical bond. Electronic configuration the assignment of the electrons in an atom to electronic shells, subshells, and orbitals. Excited state higher energy state of an atom arising from the absorption of energy and the transition of electrons to higher orbitals. Ground state the lowest possible energy state of an atom. Groups vertical columns in the Periodic Table of the Elements. Hund’s Rule electrons will occupy orbitals individually before filling them in pairs. Ion an atom with an unequal number of protons and electrons. Ionization the process of creating an ion from an uncharged atom. Ionization energy the amount of energy required to remove an electron from an atom in the gas phase. Isotopes atoms with the same number of protons, but different numbers of neutrons in the nucleus. Line emission spectrum the pattern of bright lines separated by darkness emitted by a substance in an excited state as the electrons return to the ground state. Madelung’s Rule the order of the energy of the electron subshells, which determines the electron filling order, is determined by the sum of the n + l quantum numbers. Magnetic quantum number a number (m) that mathematically defines the shape of an electron orbital in a subshell. Mass number the sum of the number of protons and neutrons in the nucleus of an atom. Neutron an uncharged particle present in all atomic nuclei. Noble gas notation a shorthand method for writing the electron configuration of an atom that uses the symbol for the preceding noble gas in brackets to represent the core electrons. Nodal plane a region in the shape of a plane where the probability of finding an electron is zero. Orbital diagram a method for describing the electronic configuration of an atom that uses boxes to represent the subshells and arrows inside the boxes to represent the electrons. The direction of the arrow represents the electron spin. Pauli Exclusion Principle no two electrons in the same atom can have the same four quantum numbers. Pauling Scale a dimensionless number on a relative scale used to measure electronegativity. Periodic Law the physical and chemical properties of the elements recur periodically in a systematic manner when arranged in the order of increasing atomic number. Periods horizontal rows in the Periodic Table of the Elements. Principal energy level the energy level or electron shell denoted by the principal quantum number. STUDY QUESTIONS 71 Principal quantum number the number designation (n) of an electron shell. Proton a positively charged particle present in all atomic nuclei. Quantized a quantity that is restricted to certain allowed values. Spin quantum number a value (s) of +½ or ½ that describes the spin of an electron. Subshell notation a method for writing the electron configuration of an atom which lists the principal quantum number of the electron shell, the subshell type, and the number of electrons in each subshell. Valence electrons the electrons in the outermost occupied electron shell of an atom which take part in chemical bonding. Valence shell the outermost occupied electron shell of an atom. STUDY QUESTIONS 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 2.9 2.10 2.11 2.12 2.13 2.14 2.15 2.16 2.17 2.18 2.19 2.20 2.21 2.22 2.23 2.24 2.25 2.26 2.27 2.28 2.29 2.30 How many naturally occurring elements are there? What three particles make up an atom and what are their charges? Where is the majority of the mass of an atom located? Where is the majority of the volume of an atom located? What is the atomic number of an atom equal to? What is the mass number of an atom equal to? What are the atomic symbols of the following elements: (a) boron, (b) antimony, (c) silver, (d) tungsten, (e) astatine, (f) iron? What are the names of the following elements: (a) Cl, (b) Cu, (c) Au, (d) Tl, (e) Na, (f) Rn? What are isotopes? How atomic mass different from mass number? How is the atomic mass unit defined? What determines the chemistry of an atom? What instrument is used to measure atomic mass? What is meant by electrons being quantized? What is the quantum number that designates an electron shell? What is its letter designation? What is the quantum number that designates an electron subshell? What is its letter designation? What is the quantum number that designates the spatial orientation of an orbital? What is its letter designation? What is the Pauli Exclusion Principle? List the four quantum numbers, their letter designations, and their allowed values. What is the ground state of an atom? What creates an atomic excited state? What is released when an electron falls from a higher orbital to a lower one? What is the Aufbau principle? What is Hund’s Rule? List the electron subshells in the order of increasing energy. What are the shapes of the orbitals in each subshell? How many orbitals are in each subshell? How many electrons are in each subshell? What is an electronic configuration? What is an orbital diagram? 72 2. THE PERIODIC TABLE OF THE ELEMENTS 2.31 2.32 2.33 2.34 2.35 2.36 2.37 2.38 2.39 2.40 2.41 2.42 2.43 2.44 2.45 2.46 2.47 What does the modern Periodic Law state? What are the vertical columns in the Periodic Table of the Elements called? What are the horizontal rows in the Periodic Table of the Elements called? What groups are in the s-block of the periodic table? In the p-block? What periods fill the d subshell before the p subshell? What periods fill the f subshell before the d subshell? What is the valence shell of an atom? What are valence electrons? What are the electrons between the nucleus and the valence electrons called? Which group in the Periodic Table of the Elements has a closed valence shell? What does the period number correspond to? What does the group number correspond to? What is an electron dot structure? What is the ionization energy of an atom? What is the difference between the electron affinity and the electronegativity of an atom? What is a closed shell? What elemental properties increase across a period? Which ones decrease across a period? 2.48 What elemental properties increase down a group? Which ones decrease down a group? PROBLEMS 2.49 Determine the number of protons, electrons, and neutrons for: (a) 3H, (b) 13C, (c) 30Si, (d) 65 Cu. 2.50 Determine the number of protons, electrons, and neutrons for: (a) 113Cd, (b) 210Pb, (c) 68Ga, (d) 238U. 2.51 Write the atomic symbol for an isotope having six electrons, six protons, and eight neutrons. 2.52 Write the atomic symbol for an isotope having 20 electrons, 20 protons, and 13 neutrons. 2.53 Boron has two isotopes: 10B with an isotopic mass of 10.013 amu and a natural abundance of 19.9% and 11B with an isotopic mass of 11.01 amu and a natural abundance of 80.1%. Calculate the atomic mass of boron. 2.54 Silicon has three isotopes: 28Si with an isotopic mass of 27.977 amu and a natural abundance of 92.23%, 29Si with an isotopic mass of 28.976 amu and a natural abundance of 4.67%, 30Si with an isotopic mass of 29.9738 amu and a natural abundance of 3.10%. Calculate the atomic mass of silicon. 2.55 What is the wavelength of light released when an electron falls from the n1 ¼ 4 to the n2 ¼ 1 energy levels in a hydrogen atom? 2.56 If a hydrogen electron falls from ni ¼ 7 to nf ¼ 5, what wavelength of light would be released? 2.57 The hydrogen first Paschen line occurs at 1094 nm. What is the energy difference between the two energy levels (ni and nf) in the electronic transition that releases light of this wavelength? 2.58 Determine the maximum number of orbitals in the (a) K, (b) L, (c) M electron shells. PROBLEMS 73 2.59 Determine the maximum number of electrons in the electron shells listed in Problem 2.58. 2.60 Write the electronic configurations for the following elements in subshell notation: (a) beryllium, (b) aluminum, (c) chlorine. 2.61 Write the electronic configuration of the elements in Problem 2.60 in noble gas notation. 2.62 Write the electronic configurations for the following elements in subshell notation: (a) calcium, (b) nickel, (c) strontium. 2.63 Write the electronic configuration of the elements in Problem 2.62 in noble gas notation. 2.64 Draw an orbital diagram for magnesium. 2.65 Draw an orbital diagram for vanadium. 2.66 How many valence electrons do the following elements have: (a) potassium, (b) fluorine, (c) argon, (d) gallium, (e) barium? 2.67 Give the valence electron configurations for the elements in Problem 2.66. 2.68 Give the core electron configurations for the elements in Problem 2.66. 2.69 What noble gases have the electron configurations in Problem 2.68? 2.70 Draw electron dot structures for the following elements: (a) potassium, (b) chlorine, (c) phosphorous, (d) silicon. 2.71 Arrange the following elements in the order of increasing atomic radius: potassium, zinc, cesium, krypton, francium. 2.72 Arrange the following elements in the order of increasing ionization energy: neon, sodium, phosphorous, rubidium, chlorine. 2.73 Arrange the following elements in the order of decreasing electron affinity: iodine, helium, astatine, bromine, antimony. 2.74 Arrange the following elements in the order of decreasing electronegativity: fluorine, bromine, lithium, francium, silicon. 2.75 Arrange the following elements in the order of increasing metallic character: potassium, oxygen, aluminum, barium, nickel. C H A P T E R 3 Chemical Bonding—The Formation of Materials O U T L I N E 3.1 Atoms and Ions 75 3.7 Molecular Polarity 105 3.2 Ionic Bonding 82 3.8 Intermolecular Forces 107 3.3 Covalent Bonding 86 Important Terms 113 3.4 Mixed Covalent/Ionic Bonding 92 Study Questions 114 3.5 Molecular Orbitals 95 Problems 115 3.6 Molecular Geometry 100 3.1 ATOMS AND IONS A neutral atom that loses one or more electrons becomes a positively charged ion. This positively charged ion is known as a cation (from the Greek word katá, meaning “down”). A neutral atom that gains one or more electrons has a negative charge and is known as an anion (from the Greek word ánō, meaning “up”). The number of electrons an element will gain or lose is also a periodic property and can generally be predicted from its position in the periodic table as shown in Fig. 3.1. Atoms will gain or lose electrons to form ions that have electronic configurations which are more stable than the electronic configurations of the parent atoms. For most elements, this means that they will either gain or lose the number of electrons needed to achieve a closed valence shell. Remember from Table 2.9 of Chapter 2 that the number of electrons in an atom’s valence shell can be determined from its group number. Since the noble gases in group 18 already have a closed valence shell with eight valence electrons (ns2np6), they do not form ions. Other elements (except for those in groups 8 through General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00003-5 75 # 2018 Elsevier Inc. All rights reserved. 76 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS FIG. 3.1 The Periodic Table of the Elements showing the common ions formed by the elements in each group. Elements indicated by a * have more than one ionic form. 12) tend to gain or lose electrons to achieve the ns2np6 closed valence shell of a noble gas. Most of the elements can easily achieve this by losing the extra electrons in their valence shell and become cations with the new valence shell of ns2np6. However, elements in groups 15 through 17 have valence shells that are nearly full and so can complete the ns2np6 closed valence shells by gaining the missing electrons and become anions. The alkali metals in group 1 have one electron in their valence shell (ns1). The loss of this electron forms a cation with a stable electronic configuration of the noble gas in the period previous to it. For example, sodium has the electronic configuration 1s22s22p63s1. By losing the electron from the 3s valence orbital, sodium acquires the stable electronic configuration of the noble gas neon (1s22s22p6) with a new closed valence shell of 2s22p6. So, the alkali metals will always lose the ns electron to form 1+ cations. Although hydrogen is normally placed in group 1, it is unusual in that it is the only element in the periodic table with only one electron. However, like the alkali metals, hydrogen will also lose this 1s electron to form a 1+ cation, even though it is now only a single proton with no electrons. The alkaline earth metals in group 2 have two electrons in their valence shell (ns2). They can achieve a closed valence shell with the electronic configuration of the noble gas in the period just above them by losing both valence electrons. The valence shell then becomes ns2np6.The alkaline earth metals always form 2+ cations. The transition metals in group 3 have three valence electrons [ns2(n 1)d1] and will lose these three electrons to achieve the closed valence shell of a noble gas and become 3+ cations. The transition metals in groups 4 through 7 can also lose their valence electrons as indicated by their group numbers, to get to the electronic configuration of the noble gas just before them in the periodic table. However, many transition metals can also form cations with other charges as shown in Table 3.1. For example, all the transition metals in group 4 form 77 3.1 ATOMS AND IONS TABLE 3.1 Elements With Multiple Ionic Forms Element Group Ionic Forms Titanium 4 4+, 3+, 2+ Vanadium 5 5+, 4+, 3+, 2+ Niobium 5 5+, 3+ Chromium 6 6,+ 3+, 2+ Manganese 7 7+, 4+, 3+, 2+ Technetium 7 7+, 6+, 4+ Rhenium 7 7+, 6+, 4+ Iron 8 3+, 2+ Osmium 8 3+, 4+ Cobalt 9 3+, 2+ Iridium 9 4+, 3+ Nickel 10 2+, 3+ Palladium 10 2+, 3+ Platinum 10 2+, 4+ Copper 11 1+, 2+ Gold 11 1+, 3+ Mercury 12 2+, 1+ Thallium 13 3+, 1+ Germanium 14 4+, 2+ Tin 14 4+, 2+ Lead 14 4+, 2+ Arsenic 15 5+, 3+, 3 Antimony 15 5+, 3+, 3 Bismuth 15 5+, 3+ Tellurium 16 6+, 4+, 2 Polonium 16 6+, 4+, 2+ 4+ cations, but titanium can also form a 3+ cation and a 2+ cation. Similarly, the transition metals in group 5 all form 5+ cations, but vanadium can also form a 4+ cation, a 3+ cation, and a 2+ cation. Niobium can also form a 3+ cation. This variability in the ionic forms of many of the transition metals is because the removal of all the valence electrons to get to the noble gas configuration would require 78 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS a great deal of energy for transition metals with many valence electrons. The additional ionic forms are achieved by losing a smaller number of electrons, which is more energy-efficient. Although this does not give the transition metal a closed valence shell, the resulting electronic configuration is still more stable than that of the parent atom. Elements in groups 8 through 12 are unable to lose all their valence electrons and are restricted to cations with lower charges. Looking at Table 3.1, the most common cations for the transition metals are 2+ and 3+. The valence electrons of the transition elements are in the ns and the (n 1)d subshells. The orbitals in these two subshells are very close in energies and the electrons in both subshells can be used in bond formation. The 2+ cation is common among the transition metals because the two electrons in ns subshell are lost first, before the (n 1)d electrons. The 3+ and 4+ cations are also common because there is a tendency for the transition metals to lose unpaired d electrons since unpaired electrons are less stable than paired electrons. These trends in the d-block elements lead to lower ionic charges than expected from their position in the periodic table. They also contribute to the multiple ionic forms of many of the transition metals. The elements in groups 13 and 14 in the p-block of the periodic table have three (ns2np1) and four (ns2np2) electrons in their valence shells. They will lose these valence electrons to achieve the valence shell of the noble gases and become 3+ and 4+ cations, respectively. The exceptions in these two groups are boron, carbon, and silicon, which do not form ions. These elements prefer to gain a closed shell by sharing electrons with another atom rather than losing them entirely. The sharing of electrons will be covered in Section 3.3. Thallium in group 13 can form a 1+ cation by losing the single unpaired p electron and leaving the filled ns2 subshell. Similarly, germanium, tin, and lead in group 14 can form 2+ cations by losing their two p electrons leaving the filled ns2 subshell. The elements in groups 15 (ns2np3), 16 (ns2np4), and 17 (ns2np5) have five, six, and seven valence electrons and so require three, two, and one electrons to achieve the closed shell of eight electrons. Because they can reach the stable configuration of eight electrons more easily by gaining electrons than by losing them, they have a high electron affinity (see Fig. 2.16). They will gain the required electrons to achieve the closed valence shell of eight. So, elements in group 15 will form 3 anions, those in group 16 will form 2 anions, and those in group 17 will form 1 anions. Notice from Fig. 2.16 that the exceptions to this pattern are the elements in the higher periods which have lower electron affinities. It is easier for these elements to lose electrons than to gain them because the outer shell is so far from the nucleus. So, arsenic and antimony in group 15 can lose all their valence electrons to form 5+ cations or they can lose only the three p electrons to form 3+ cations. Similarly, tellurium in group 16 can form 6+ and 4+ cations. Bismuth and polonium in period 6 have such low electron affinities that they cannot form anions at all, but are restricted to the cationic forms. The chemical symbol for an ion consists of the symbol for element it is derived from followed by a sign showing the number of electrons it has lost or gained. Cations are designated by a plus sign, while anions are designated by a minus sign. The charge is written in superscript immediately after the atomic symbol. For ions with charges >1, the magnitude of the charge is written before the sign. For singly charged ions, the magnitude of the charge is omitted. For example, the sodium cation is written as Na+ (not Na1+), while the calcium cation is Ca2+. 79 3.1 ATOMS AND IONS EXAMPLE 3.1: DETERMINING THE ELECTRONIC CONFIGURATION OF AN ION What is the electronic configuration of O2? 1. Determine the electronic configuration of oxygen. Oxygen is in group 16 of period 2. The noble gas in period 1 is helium, so the electronic configuration of oxygen in noble gas notation is: [He]2s22p4. 2. Determine the electronic configuration of O2. Since O2 has a 2 charge it has gained two electrons. So, the electronic configuration of O2 will be that for oxygen with two electrons added to the p subshell: [He]2s22p6. Notice that this is also the electronic configuration for the noble gas neon. Anions are named by adding the suffix “ide” to the root of the name of the parent element. So, Cl is the chloride ion, N3 is the nitride ion, O2 is the oxide ion, and so on. The names of all the anions are listed in Table 3.2. Cations of elements that have only one ionic form are named by simply adding the word “ion” after the element name. For example, Na+ is called the sodium ion and Zn2+ is called the zinc ion. Cations of elements that have multiple ionic forms start with the name of the element followed immediately by a Roman numeral in parentheses, called the Stock number, to indicate the charge of the ion. For example, Fe2+ is iron (II) and Fe3+ is iron(III). TABLE 3.2 The Names of the Anions Symbol 3 N P 3 O Arsenide ion Antimonide ion 2 S Selenide ion Telluride ion Br I Sulfide ion 2 Cl Oxide ion 2 Te F Phosphide ion 3 2 Se Nitride ion 3 As Sb Name Fluoride ion Chloride ion Bromide ion Iodide ion At Astatide ion 80 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS The use of Roman numerals to denote the charge of the cation is the method in use today. However, an older, and possibly more confusing, system of naming cations with multiple charges may be found that appends the suffixes “ous” and “ic” to the root name of the element to denote the charge. The ion with the lesser charge ends with “ous” and the one with greater charge ends with “ic.” For example, in this system Fe2+ is called the ferrous ion while Fe3+ is called the ferric ion, Cu+ is called the cuprous ion while Cu2+ is the cupric ion, and Sn2+ is called the stannous ion while Sn4+ is called the stannic ion. Although this naming system is outdated, it can still be found in older textbooks. The gain or loss of electrons by an atom to form an ion has a very large effect on the chemical and physical properties of the atom. Since the electronic configurations of the ions are more stable than that of the parent atom, ions are less reactive than neutral atoms. For example, sodium is a soft metal which reacts rapidly and explosively with water. In Fig. 3.2, the reaction of sodium metal with water is shown to be so violent that it breaks the glass container. In contrast, the sodium cation dissolves readily in water without any reaction. Chlorine is a greenish yellow gas that is so reactive it corrodes every metal and is toxic to all living things. It reacts with almost all elements including other chlorine atoms to form a diatomic chlorine molecule. On the other hand, chlorine anions are colorless and also dissolve readily in water without reaction. Both sodium and chlorine ions are so unreactive that they are found in most food products in the form of table salt and are an essential ingredient for life. As with the atomic radius, the trend in changing ionic radius (the distance from the nucleus to the outermost occupied electron orbital in an ion) is also a periodic property. A comparison between atomic radius and ionic radius for the most common ions of the elements is shown in Fig. 3.3. Since anions have more electrons than the parent atom and more electrons than protons in the nucleus, the attraction between the valence electrons and the nucleus is weaker resulting in a larger ionic radius than the parent atom. This can be seen in Fig. 3.3 for elements in groups 15 through 17 just before the noble gases. Cations have fewer electrons than protons in the nucleus causing the attraction between the valence electrons and the nucleus to become stronger, pulling the valence shell closer to the nucleus. So, cations have smaller radii than the parent atom. The cations also have smaller radii than most of the anions. Notice in Fig. 3.3 that the ionic radius of the transition metals is more variable than the atomic radius of the neutral atoms. This is because ionic charge also affects the ionic radius. The higher the ionic charge, the stronger the nuclear attraction and the smaller the ionic radius. Since the transition metals have multiple ionic states that vary within a group, the ionic radius FIG. 3.2 Right: Sodium metal reacting with water. Left: Sodium and chloride ions dissolving in water. Photographs by Naatriumi and Chris 73, Wikimedia Commons. 81 3.1 ATOMS AND IONS FIG. 3.3 Atomic radius (blue) and ionic radius (green) in picometers as a function of atomic number for elements 1 through 95. also varies, although it is consistently smaller than the parent atom and also smaller than the anions. Overall, the ionic radius follows the same general periodic trends as the atomic radius and for the same reasons. Both the atomic radius and the ionic radius increase going down a group as long as the charge on the ions remains the same. This is due to the addition of an extra principal electron shell with each increasing period. Also, like the atomic radius, the ionic radius generally decreases going across a period for ions of the same charge due to the stronger nuclear attraction with the increasing number of protons. This decreasing trend across a period is not as apparent for some transition metals due to the large variation in ionic charge in the d-block. In summary: • • • • • • Cations are smaller than their parent atoms. Anions are larger than their parent atoms. Anions are generally larger than cations. Increasing charge leads to decreasing ionic radius. Ionic radius increases going down a group. Ionic radius generally decreases going across a period. EXAMPLE 3.2: DETERMINING THE RELATIVE SIZE OF IONS List the following ions in order of increasing size: Cs+, K+, F, and Cl. 1. Determine the position of the ions in the periodic table. Cs+ and K+ are both in group 1; K+ is in period 4 and Cs+ is in period 6. F and Cl are both in group 17; F is in period 1 and Cl is in period 2. 2. Determine how the position of the ions is related to their size. Since ionic radius increases going down a group: Cs+ > K+ and Cl > F. Cs+ and K+ in periods 4 and 6 have more electronic shells than F and Cl in period 1 and 2. 3. Determine how the ionic charge is related to their size. Anions are smaller than cations: F < Cl < K+ < Cs+. 82 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS 3.2 IONIC BONDING When a chemical reaction occurs between two atoms, their valence electrons are rearranged to achieve a more stable electronic configuration for both of them. The simplest way to accomplish this is to completely transfer one or more electrons from one atom to another. This creates two oppositely charged ions, a cation that has lost the electrons and an anion that has accepted the electrons. The electrostatic attractions between the oppositely charged positive and negative ions create an ionic bond which holds the atoms together forming an ionic compound. The force (F) acting between two charged particles with charges q1 and q2 is described by Coulomb’s law as; F¼k q1 q2 d2 (1) where “d” is the distance between the two charged particles and “k” is a constant with the value of 9 109 N • m2 • C2. Chemists use a modified version of this law to describe the potential energy (E) between two ions, with charges q1 and q2. E¼k q1 q2 d (2) A negative potential energy (E ¼ ) means that the ions will attract each other, while a positive potential energy (E ¼ +) means that the ions will repel each other. If one ion is a cation (q1 ¼ +) and the other ion is an anion (q2 ¼ ), the energy of their interaction will be negative and the ions will attract each other. The potential energy (E) as a function of the distance (d) between two oppositely charged ions is shown in Fig. 3.4. When the ions are very far apart, there is a very small energy of attraction between the two ions. But as they get closer to each other (d decreases), the energy between them decreases. So, the two ions of opposite charge will have a lower energy and be more stable when they are close together than when they are far apart. An energy minimum is reached at the ionic bond FIG. 3.4 Potential energy (E) as a function of the distance (d) between two oppositely charged ions. The values of the ionic bond length and the ionic bond strength are shown by dotted lines. 3.2 IONIC BONDING 83 length of the resulting ionic compound. If the ions move closer than the bond length, they will repel each other and the energy increases rapidly. This happens because of the repulsion between the positively charged nuclei at very small distances. The bond length represents the distance where the attractive and repulsive forces between the two oppositely charged ions are balanced. The magnitude of the energy minimum in Fig. 3.4 gives a measure of the ionic bond strength between the two ions and the distance at which the energy minimum occurs is the bond length. So, the strength of an ionic bond can be predicted by using Coulomb’s law where the distance between the two ions in an ionic compound is equal to the sum of the two ionic radii (d ¼ r1 + r2). From Eq. (2), the magnitude of the bond strength depends on the magnitude of the product of the charges (q1 q2) and the bond length (d) where the energy is at a minimum. Larger charges will have stronger interactions and result in lower potential energies and a stronger bond. Also, smaller ions will be able to get closer together and have smaller bond lengths. They will also have lower potential energies and form stronger bonds. In summary: • The smaller the ionic radius, the shorter the bond length and the stronger the ionic bond. • The larger the ionic radius, the longer the bond length and the weaker the ionic bond. • The larger the ionic charge, the stronger the ionic bond. EXAMPLE 3.3: DETERMINING THE RELATIVE STRENGTH OF IONIC BONDS List the following ionic bonds in the order of increasing bond strength: NadF, NadCl, CadF, NadI, and ScdF. 1. Determine the product of the ionic charges for each ion pair. NadF ¼ +1 1 ¼ 1. NadCl ¼ + 1 1 ¼ 1. CadF ¼ +2 1 ¼ 2. NadI ¼ +1 1 ¼ 1. ScdF ¼ +3 1 ¼ 3. According to the charges of the ions: ScdF > CadF, > NadI, NadF, NadCl. 2. Determine the remaining order of increasing bond strength by comparing the size of the ions. NadF, NadCl, and NadI have the same ionic charge product and the same cation, The size of the anion is: I > Cl > F. 3. The order of increasing bond strength: NadI < NadCl < NadF < CadF < ScdF. Since there are few elements in the periodic table that form anions, ionic bonds are restricted to the anions of the nonmetals in groups 15, 16, and 17 binding with the cations of metals. Most ionic bonds occur between elements with very different electronegativities (Fig. 2.17). The further they are apart in the periodic table, the more different their electronegativities and the more likely they are to form ionic bonds. One simple ionic compound formed from the elements in groups 1 and 17 is sodium chloride. When sodium and chlorine react, the 3s valence electron of sodium is transferred to the 3p valence orbital of chlorine. Sodium has 84 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS the electronic configuration [Ne]3s1. By losing the electron from the 3s valence orbital, sodium acquires the stable electronic configuration of the noble gas neon (1s22s2sp6). By gaining the electron, chlorine ([Ne]3s23p5) will complete its valence shell and acquire the electronic configuration of the noble gas argon (1s22s2sp63s23p6). Na 1s2 2s2 sp6 3s1 1e ! Na + 1s2 2s2 sp6 Cl 1s2 2s2 sp6 3s2 3p5 1e ! Cl 1s2 2s2 sp6 3s2 3p6 This tendency for atoms in the s- and p-blocks to combine in such a way that each atom acquires eight electrons in its valence shell is known as the octet rule. Since the formation of chemical bonds involves the rearrangement of the valence electrons of the atoms, it can be represented by electron dot structures. The electron dot structures of the formation of an ionic bond between a metal and a nonmetal shows the transfer of electrons from the metal to the nonmetal to form a stable electronic configuration for both. The electron dot representation of the formation of the ionic compound sodium chloride from the elements sodium and chlorine would be; Na + Cl [Na] + [ Cl ] − The electron dot structure of the ionic compound is written with the dot structures of the ions in brackets. The charge of the ions is placed in the upper right hand corner outside the brackets. The anion is shown with the stable configuration of eight valence electrons. Although the cation now also has eight valence electrons in the new outermost electronic shell, it is shown with an empty valence shell to indicate the loss of its old valence electrons to the anion. EXAMPLE 3.4: DETERMINING THE ELECTRON DOT REPRESENTATION OF AND IONIC COMPOUND Write the ionic reaction of magnesium and oxygen to form magnesium oxide using electron dot representations. 1. Determine the electron dot representations for magnesium and oxygen. Magnesium is in group 2 of the periodic table with two valence electrons. Mg Oxygen is in group 16 of the periodic table with six valence electrons. O 2. Determine the number of electrons gained and lost in the reaction. In order for oxygen ([He]2s22p4) to achieve the noble gas valence shell of eight electrons, it must gain two electrons. Similarly, in order for magnesium ([Ne]3s2) to achieve a noble gas electronic configuration, it must lose two electrons. 3. The electron dot representation of this reaction would be: Mg + O [Mg]2+ [ O ]2− 3.2 IONIC BONDING 85 Electron dot representations can also be used to predict the chemical formula of an ionic compound. A chemical formula is a way of expressing the bonding between atoms and ions in a compound using a single line of the element symbols along with numeric subscripts to indicate the number of atoms of each element in the compound. For the reaction between calcium ([Ar]4s2) and chlorine ([Ne]3s23p5), the electron dot structures for the neutral atoms are; Ca + Cl Calcium needs to lose its two 4s valence electrons to achieve the stable noble gas electronic configuration, but chlorine only needs one electron to complete the octet. So the ionic compound that forms between calcium and chlorine will need two chlorine atoms for every one calcium atom in order for calcium to lose both valence electrons. The electron dot representation for the ionic compound is; [ Cl ]− [Ca]2+ [ Cl ]− When writing the chemical formula for the ionic compound, the atomic symbol of the cation is always written first followed by the atomic symbol for the anion. Numeric subscripts are used to indicate the number of atoms of each ion in the compound. The charges of the ions are not included in the chemical formulas of ionic compounds since the overall charge of the compound is zero. The chemical formula for the ionic compound formed between calcium and chlorine is CaCl2. EXAMPLE 3.5: DETERMINING THE CHEMICAL FORMULA OF AN IONIC COMPOUND USING ELECTRON DOT REPRESENTATIONS What is the chemical formula of the ionic compound formed from the reaction between sodium and sulfur? 1. Determine the electron configurations of the neutral atoms. sodium ¼ [Ne]3 s1. sulfur ¼ [Ne]3s23p4. 2. Determine the electron dot representations for the neutral atoms. S and Na 3. Determine the combination of atoms that will result in a closed valence shell. Sulfur needs two electrons to complete the valence octet. This requires two sodium atoms. 4. Write the chemical formula. Na2S. To determine the chemical formula for ionic compounds that have more complicated valence configurations, such as the transition metals, the ions must be combined so that the resulting compound is electrically neutral. That is, the total positive charge of the cations must equal the total negative charge of the anions. For the compound formed by a combination of Zn2+ and Cl, there must be two chloride ions for every zinc ion in order for the positive and negative charges to balance and to achieve a total charge of zero. So, the 86 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS chemical formula is ZnCl2. Similarly, for the compound formed from Al3+ and O2, there must be three oxygen anions for every two aluminum cations for the compound to be electrically neutral. The aluminum contributes a 6+ charge and the oxygen contributes a 6 charge resulting in a total charge of zero and the chemical formula for the ionic compound is Al2O3. Ionic compounds are named by simply combining the names of the ions without using the term “ion.” As with the chemical formula, the name of the cation is first followed by the name of the anion. For example, NaCl is sodium chloride, MgO is magnesium oxide, Na2S is sodium sulfide, and CaCl2 is calcium chloride. For compounds containing metals with multiple ionic states such as iron, the charge on the cation can be determined from the chemical formula. For example, the iron cation in FeCl2 must have a 2+ charge in order to balance out the two negative charges of the chlorine atoms. So, FeCl2 is iron(II) chloride. In the same way, FeCl3 is iron(III) chloride. 3.3 COVALENT BONDING Two atoms with very different electronegativities (those far apart on the periodic table) will form an ionic compound since the atom with a high electronegativity has the ability to capture electrons from the atom with low electronegativity. But, elements with similar electronegativities (those closer together in the periodic table) cannot completely transfer electrons from one atom to another. They can still achieve a closed valence shell by sharing their electrons. For example, two hydrogen atoms have the same electronegativity and cannot transfer electrons from one hydrogen atom to the other to form an ionic bond. But they can both achieve the closed valence shell of a helium atom (1s2) if both atoms share their single valence electron to form a shared electron pair. This can be represented by an electron dot structure as; H + H H H or H−H The pair of electrons that is shared between the two hydrogen atoms forms a covalent bond between them resulting in a diatomic molecule of hydrogen with the formula H2. Similarly, two fluorine atoms ([He]2s22p5) can both achieve the closed valence shell of neon ([He]2s22p6) by sharing one valence electron each. F + F F F or F F The shared electron pair, which forms the covalent bond between the two fluorine atoms, is known as bonding pair electrons. The three pairs of valence electrons around each fluorine atom that are not shared are known as lone pair electrons. Covalent bonds are formed when two or more valence electrons are attracted by the positively charged nuclei of both atoms and so are shared between the two atoms. This description of covalent bonds as involving shared pairs of valence electrons that are localized in a bond between two atoms is known as valence bond theory. Fig. 3.5 shows the potential energy between two atoms of hydrogen as they approach each other. In a similar manner 87 3.3 COVALENT BONDING FIG. 3.5 Potential energy change during HdH covalent bond formation from isolated hydrogen atoms. A representation of the two atoms with their 1s electron shell is shown above the graph. as in the formation of an ionic bond (Fig. 3.4), the potential energy decreases as the hydrogen atoms get closer together and the attraction between the valence electrons and the nuclei of each atom becomes stronger. The covalent bond is formed when the two hydrogen atoms reach the lowest potential energy, which is the covalent bond length. The bond length is determined by the sum of the atomic radii of the two atoms. For hydrogen, this occurs at a distance of 74 pm. At this point, the overlap of the 1s orbitals of the two hydrogen atoms concentrates the electron density between the two nuclei and the attractive and repulsive forces are balanced. If the nuclei get closer together than the bond length, the repulsive forces become dominant and the potential energy increases. If the formation of a single bonding electron pair between two atoms does not result in each atom achieving the stable valence octet, it is possible for two atoms to share more than one electron pair. For example, oxygen has six valence electrons and needs two electrons to achieve the stable valence octet of neon ([He]2s22p6). If two oxygen atoms ([He]2s22p4) shared one electron pair, they would each only gain one electron. But if they shared two electron pairs, each oxygen atom would then have eight valence electrons resulting in a diatomic oxygen molecule with the formula O2. + O O O O or O O Similarly, if two atoms share two electron pairs and still do not have eight valence electrons, they can share three electron pairs. An example of this is the formation of N2. Each atom of nitrogen has five valence electrons and needs three more electrons to get to the stable valence octet of neon ([He]2s22p6). This can be achieved if each nitrogen atom shares three electron pairs. N + N N N or N N The covalent bond that results when two atoms share one electron pair is called a single bond. The bond resulting from sharing two electron pairs is a double bond and that resulting 88 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS from sharing three electron pairs is a triple bond. Double bonds are shorter and stronger than single bonds and triple bonds are shorter and stronger than double bonds. The triple bond in N2 is very strong and requires a lot of energy to break it. In fact, diatomic nitrogen is so stable it is considered to be chemically inert. The only elements that normally exist in nature as stable diatomic molecules are the gases H2, N2, O2, F2, Cl2, Br2, and I2. Since a compound must contain at least two different elements and these molecules are only composed of a single element, these diatomic gases are not considered to be chemical compounds. Instead, they are known as diatomic elements. EXAMPLE 3.6: DETERMINING THE ELECTRON DOT STRUCTURES OF COVALENT COMPOUNDS What is the electron dot structure for CO2? 1. Determine the number of valence electrons for carbon and oxygen. Carbon is in period 2, group 14 with four valence electrons. Oxygen is in period 2, group 16 with six valence electrons. 2. Draw the electron dot structures for both atoms. C O 3. Determine the number of electrons each atom needs to achieve a stable valence configuration. Carbon needs four electrons and each oxygen atom needs two electrons. In order for carbon to obtain four electrons, it would have to share four electron pairs. Since there are two oxygen atoms in CO2, this could be achieved by sharing two electron pairs from each oxygen atom. 4. The electron dot structure would then be: O C O or O C O When two bonded atoms have the same electronegativities, they will share the electrons equally. The bonds between atoms where electrons are shared equally are known as nonpolar covalent bonds. In molecules where two bonded atoms have different electronegativities, the electrons are shared unequally. This difference in electronegativities between the two atoms results in the electron density being shifted towards the more electronegative atom giving it a partial negative charge, while the less electronegative atom acquires a partial positive charge. This type of bond where the electrons are unequally shared between two atoms is called a polar covalent bond. For example, in the molecule HF, hydrogen and fluorine share a pair of valence electrons. However, the fluorine atom has a much higher electronegativity (3.98 on the Pauling scale) than the hydrogen atom (2.20 on the Pauling scale). So the fluorine atom attracts the shared electrons more than the hydrogen atom. Since the hydrogen atom has its single valence electron pulled away from it, it has a partial positive charge while the fluorine atom has a partial negative charge. A bond (or molecule) that has a partial positive charge on one end and a partial negative charge on the other end is called a dipole. In molecular 89 3.3 COVALENT BONDING structures, the bond polarity is indicated by a “δ+” written by the less electronegative atom and a “δ” written by the more electronegative atom as; δ− δ+ F H The larger the difference in electronegativities of the two atoms, the more polar the bond will be. For example, as shown in Fig. 3.6, the halogens have electronegativities on the Pauling scale of: F ¼ 3.98, Cl ¼ 3.16, Br ¼ 2.96, I ¼ 2.66, At¼ 2.20. If each of the halides is bonded to hydrogen (2.20), the differences in electronegativities between the halogen and hydrogen, shown in Table 3.3, decrease from a value of 1.78 for HdF to 0.46 for HdI. Consequently, the hydrogendhalogen bond polarity decreases in the order of HdF > HdCl > HdBr > HdI. In addition, since hydrogen and astatine have the same electronegativity (2.20), the HdAt bond will be nonpolar. The measure of the bond polarity is the dipole moment of the bond. The bond dipole is modeled as two partial opposite charges δ+ and δ that are equal in magnitude and separated Period Group 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 1 H 2.20 2 Be Li 0.98 1.57 3 P S Cl Al Si Na Mg Ar 1.61 1.90 2.19 2.58 3.16 0.93 1.31 As Se Br Kr Co Ni Cu Zn Ga Ge Ti V Cr Mn Fe K Ca Sc 0.82 1.00 1.36 1.54 1.63 1.66 1.55 1.83 1.88 1.91 1.90 1.65 1.81 2.01 2.18 2.55 2.96 3.00 4 He N O F B C 2.04 2.55 3.04 3.44 3.98 Ne 5 Sb Te I Xe Tc Ru Rh Pd Ag Cd In Sn Sr Y Zr Nb Mo Rb 0.82 0.95 1.22 1.33 1.60 2.16 1.90 2.20 2.28 2.02 1.93 1.69 1.78 1.96 2.05 2.10 2.66 2.60 6 Bi Po At Rn Re Os Ir Pt Au Hg Tl Pb Ba La Hf Ta W Cs 0.79 0.89 1.10 1.30 1.50 2.36 1.90 2.20 2.20 2.28 2.54 2.00 1.62 2.33 2.02 2.00 2.20 2.20 7 Ra Ac Fr 0.70 0.90 1.10 <1 Rf Db Sg Bh Hs Mt Ds Rg Cn Fl 2.0−2.9 1−1.9 Lv 3.0−3.9 FIG. 3.6 Electronegativities of the elements on the Pauling scale. TABLE 3.3 Properties of the Hydrogen Halide Bonds Molecule Electronegativity Difference Dipole Moment (D) Bond Length (Å) HF 1.78 1.82 0.92 HCl 0.96 1.08 1.27 HBr 0.76 0.82 1.41 HI 0.46 0.44 1.61 HAt 0.0 0.0 1.72 90 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS δ+ d δ− The model for a covalent bond dipole where δ+ and δ are the partial charges on the bonded atoms and d is the bond length. FIG. 3.7 FIG. 3.8 Electron density surfaces for the covalent bonds between hydrogen and the halogens. The bar and circle inside each figure represents the molecule with the hydrogen atom shown in blue and the halogens in orange (F), green (Cl), red (Br), and purple (I). The outer shape describes the extent of the electron density in each molecule. The red color represents the partial negatively charged regions (δ) and the blue color represents the partial positively charged regions (δ+). Ben Mills, Wikimedia Commons. HF HCl HBr HI by a distance (d) as shown in Fig. 3.7. The dipole moment (μ) of the bond is given by the product of the magnitude of the charge on either atom (δ) and the bond length (d) as; μ¼δd (3) The SI unit for a dipole moment is the coulomb-meter (C • m). However, a more convenient unit for covalent bond polarities is the debye (D), which is defined as 1D ¼ 3.34 1030C • m. The dipole moments for the hydrogendhalide bonds in Table 3.3 decrease from 1.82D for HdF to 0.44D for HdI. At the same time, the bond length increases from 0.92 Å for HdF to 1.61 Å for HdI and 1.72 Å for the nonpolar HdAt. So, the bond length increases with decreasing bond polarity. The bond polarity trend for the polar hydrogendhalide bonds is demonstrated in Fig. 3.8. The bar and circle shape inside each figure represents the center of the HdF, HdCl, HdBr, and HdI molecules. The electron density around the atoms is shown as the outer shape. The red color represents the partial negatively charged regions (δ) caused by the electrons spending more time near the halogen atoms in each bond. The blue color represents the partial positively charged regions (δ+) surrounding the hydrogen atoms. The colors are strongest for HdF and progressively weaker for each halogen because the HdF bond is strongest and more polar, while the HdI bond is weakest and less polar. The bond length also increases as the bond polarity decreases. As shown in Table 3.3, it is the electronegativity differences between atoms that control the dipole moment and the polarity of the bond. For bonds where the electronegativity differences are nonzero but very small, they can be considered as virtually nonpolar for purposes of determining the chemical reactivity of the bond. One such bond that receives much attention in chemistry is the CdH bond in organic molecules. Carbon has an electronegativity of 2.55 on the Pauling scale and hydrogen has an electronegativity of 2.20. So, the electronegativity difference for a CdH bond is 0.35, which is very small. The bond would therefore have a small dipole moment (0.3D) and could be considered as virtually nonpolar for the purpose of determining its chemical reactivity. This will become very important in Chapter 13. 3.3 COVALENT BONDING 91 A “rule of thumb” for determining how a bond will behave in chemical reactions is: • If the electronegativity difference between the two bonded atoms is >0.4, the bond will behave as polar. • If the electronegativity difference is 0.4 or less, the bond will behave as nonpolar. • If the electronegativity difference is >2, the bond will behave as ionic. All the examples given above have used electron dot structures to determine the number of electrons shared in covalent bonds. Since electron dot formulas can only be used to show eight valence electrons per atom, all the examples have followed the octet rule of acquiring eight valence electrons to form a stable molecule (two for hydrogen). However, the octet rule only holds for elements in the s- and p-blocks of the periodic table with a few other exceptions. Elements in periods 3 and higher can have more than eight valence electrons when bonding to fluorine, chlorine, or oxygen. This is because the electron shells with n ¼ 3 and higher can also have empty d orbitals, which can accommodate additional valence electrons. Some examples are: SF6 where sulfur has 12 valence electrons, PF5 where phosphorous has 10 valence electrons, ClF3 where chlorine has 10 valence electrons including two lone pairs, and BrF5 where bromine has 10 valence electrons. Another exception to the octet rule is boron, which can form compounds with other nonmetals with three covalent bonds giving boron six valence electrons, two short of the octet. However, these compounds are very reactive and often react with other molecules or ions that can provide the missing electron pair. A more general notation for representing covalent bonds in a molecule is bonding notation where the lone pair electrons are omitted and the bonding electrons are represented by lines. In this notation, the molecular structure for F2, O2, and N2 then becomes FdF, O]O, and N^N. In addition to being simpler than electron dot structures, bonding notation can be used for all molecules including those with more than eight valence electrons. SF6 could be represented in bonding notation as; F F F S F F F The chemical formula for a covalent compound is called a molecular formula because, unlike ionic compounds, covalent compounds can exist as separate, distinct molecules. The molecular formula is written with the least electronegative element (the one further to the left on the periodic table) placed first followed by the more electronegative element. One important exception to this order is when the compound contains oxygen bonded to a halogen. For these compounds, the halogen is written first. In naming covalent compounds, the first element in the molecular formula is named first using the neutral element name. The last element is named as an anion with the suffix “ide.” A Greek prefix is used in front of each element name to indicate how many atoms of each element are in the compound. The most common prefixes are: “mono” (one), “di” (two), “tri” (three), “tetra” (four), “penta” (five), and “hexa” (six). For example, N2O3 is dinitrogen trioxide. Some exceptions are that the “mono” prefix is not used for the first element in the formula as in carbon dioxide (CO2) or silicon tetrafluoride (SiF4). Also, if the use of the prefix 92 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS places two vowels next to each other, the “a” or “o” of the prefix is dropped. For example, NO is nitrogen monoxide, N2O is dinitrogen monoxide, and N2O5 is dinitrogen pentoxide. But, the “i” of “di” and “tri” are never dropped. Some simple covalent compounds have common names rather than systematic names. The most often encountered are water (H2O), ammonia (NH3), and methane (CH4). Methane is an organic compound. The naming of organic compounds will be covered in Chapter 13. 3.4 MIXED COVALENT/IONIC BONDING Polyatomic ions, also called molecular ions, are ions that are made up of two or more different atoms covalently bonded tightly together so that they can behave as a single unit. Polyatomic ions have a charge because the group of atoms making up the molecule has either gained or lost electrons. So, the polyatomic ion is a covalently bonded molecule that behaves like an ion in forming ionic compounds since it has a charge. Some common polyatomic ions are listed in Table 3.4. TABLE 3.4 Common Polyatomic Ions With Their Chemical Formulas. The Ions are Listed in Groups or Families Depending on the Parent Element. Although Not Listed in the Table, Bromine and Iodine Form the Same Four Polyatomic Ions as Chlorine Parent Element Polyatomic Ion Formula Hydrogen Hydroxide OH Hydronium H3O+ Carbonate CO32 Hydrogen carbonate (bicarbonate) HCO3 Cyanate CNO Thiocyanate CNS Cyanide CN Acetate H3C2O2 Oxalate C2O4 Nitrate NO3 Nitrite NO2 Ammonium NH4+ Sulfate SO42 Hydrogen sulfate HSO4 Thiosulfate S2O32 Sulfite SO32 Hydrogen sulfite HSO3 Carbon Nitrogen Sulfur 93 3.4 MIXED COVALENT/IONIC BONDING TABLE 3.4 Common Polyatomic Ions With Their Chemical Formulas. The Ions are Listed in Groups or Families Depending on the Parent Element. Although Not Listed in the Table, Bromine and Iodine Form the Same Four Polyatomic Ions as Chlorine—cont’d Parent Element Phosphorous Chlorine Metals Polyatomic Ion Formula Thiosulfite S2O22 Phosphate PO43 Hydrogen phosphate HPO42 Dihydrogen phosphate H2PO4 Phosphite PO33 Hypophosphite PO23 Hydrogen phosphite HPO32 Dihydrogen phosphite H2PO3 Chlorate ClO3 Perchlorate ClO4 Chlorite ClO2 Hypochlorite ClO Aluminate AlO2 Arsenate AsO43 Arsenite AsO33 Chromate CrO42 Chromite CrO2 Dichromate Cr2O72 Permanganate MnO4 The names of the polyatomic ions follow some general, although sometimes confusing, conventions. Most of the common polyatomic ions are anions. There are only two cations listed in Table 3.4. These cations are H3O+ (hydronium ion) and NH4+ (ammonium ion), both of which end in the suffix “ium.” For the polyatomic anions, the names of the ions containing one or more oxygen atoms (oxyanions) are determined by the number of oxygens in the ion. The name ending in the suffix “ate” is considered to be the base name for the polyatomic ions containing oxygen. If there are only two possible polyatomic ions in a group that contain oxygen atoms, the ion containing the most oxygens ends with the suffix “ate,” such as nitrate (NO3 ). The ion that has one less oxygen atom than the “ate” form ends with the suffix “ite,” such as nitrite (NO2 ). For groups with more than two ions containing oxygen atoms, the ions with a prefix of “per” combined with a suffix of “ate” have one more oxygen atom than the “ate” form. Ions with a prefix of “hypo” and a suffix of “ite” have one less oxygen atom than the “ite” form. For example, the chlorine group of polyatomic ions has four forms containing oxygen: perchlorate (ClO4 ) with four oxygen atoms, chlorate (ClO3 ) with three oxygen 94 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS atoms, chlorite (ClO2 ) with two oxygen atoms, and hypochlorite (ClO) with one oxygen atom. The phosphorous group only has three forms containing oxygen. The form containing the most oxygen atoms (PO4 3 ) is named phosphate. Then the last two ions in the series are named phosphite (PO3 3 ) and hypophosphite (PO2 3 ). Notice that in all cases, the charge on the oxyanions remains the same. In addition to the naming conventions for the oxyanions, the prefix “thio” indicates that one oxygen atom has been replaced with a sulfur atom. For example, in thiosulfate (S2 O3 2 ) the prefix “thio” indicates that it is a sulfate ion (SO4 2 ) with one oxygen atom replaced by sulfur. Again, the charge on the ion is unchanged. Also, some polyatomic oxyanions can add hydrogen ions (H+). These are named simply by adding the name “hydrogen” to that of the oxyanion as in hydrogen carbonate (HCO3 ). An older convention indicates the addition of a hydrogen atom by using the prefix “bi” naming HCO3 bicarbonate instead of hydrogen carbonate. For the polyatomic ions containing hydrogen, each added hydrogen ion neutralizes one negative charge on the parent anion. So, carbonate has a 2 charge while hydrogen carbonate has a 1 charge. There are only a few exceptions to these naming conventions. For example, the names of the hydroxide (OH) and cyanide (CN) ions have the “ide” suffix because they were once thought to be monatomic ions. EXAMPLE 3.7: DETERMINING THE ELECTRON DOT STRUCTURES OF POLYATOMIC IONS What are the electron dot structure for NO3 ? 1. Determine the number of valence electrons in the group. Nitrogen has five valence electrons, each oxygen atom has six valence electrons. Since the charge on the group is 1, there is one extra electron. The number of valence electrons is: 5 + (3 6) + 1 ¼ 24. 2. Determine the central atom in the structure. Almost always the central atom will be the least electronegative atom. It is also the atom that will require the most bonds to achieve a closed valence shell. Nitrogen will need to share three electron pairs, while each oxygen will share only one electron pair. Nitrogen is also less electronegative than oxygen. Nitrogen will be the central atom. 3. Determine the bonding to the central atom. In order for nitrogen to achieve a valence shell of eight electrons, it must share four electron pairs with each of the three oxygen atoms. 4 bonds ¼ two single bonds and one double bond. 4. Determine the electron placement in the electron dot structure. 24 electrons 8 bonding electrons ¼ 16 electrons ¼ 8 lone pair electrons. The remaining 16 electrons are placed as lone pairs around the oxygen atoms. As with the monatomic ions, the electron dot structure is written in brackets with the charge of the polyatomic ion written outside the brackets in the upper left corner. The electron dot structure for NO3 is: − − O N O O or O N O O 95 3.5 MOLECULAR ORBITALS While the structure for the nitrate ion given in Example 3.7 is correct, it is not the only correct structure. Because of the symmetry of the NO3 ion, it does not matter which of the oxygen atoms receives the double bond. Since there are three different oxygen atoms that could form the double bond to nitrogen, there will be three different correct electron dot structures, called resonance structures. Each resonance structure shows a different oxygen atom with a double bond to the nitrogen atom. It is conventional to use double-headed arrows placed between the multiple resonance structures to indicate that the structures are equivalent. − O N O O − O N O − O O N O O Resonance structures are two or more equivalent structures which differ only in the position of their electrons (not the position of the atoms). This does not imply that the structure of the ion (or molecule) switches between these different forms. In reality, the ion exists as an average of these structures where the bonding electrons are delocalized between the oxygens. This electron delocalization makes the ion more stable than if it existed as any of the single resonance structures. Resonance can often exist when multiple atoms of the same type surround a central atom. Electron dot representations as well as the chemical formulas of ionic compounds containing polyatomic ions are much the same as for ionic compounds containing only monoatomic ions. In determining the chemical formula, the ions must be combined so that the resulting compound is electrically neutral. That is, the total positive charge of the cations must equal the total negative charge of the anions. For the compound formed by a combination of Na+ and SO4 2 , there must be two sodium ions for every sulfate ion in order for the positive and negative charges to balance and achieve a total charge of zero. So, the chemical formula is Na2SO4. Similarly, for the compound formed from Al3+ and SO4 2 , there must be three sulfate anions for every two aluminum cations in order for the compound to be electrically neutral. The aluminum contributes a 6+ charge and the sulfate contributes a 6 charge resulting in a total charge of zero for the ionic compound. Parentheses are used in the chemical formula to separate the numeric subscripts used to indicate the number of atoms in the ion from the subscript used to indicate the number of ions in the compound. So, the chemical formula for aluminum sulfate is Al2(SO4)3. 3.5 MOLECULAR ORBITALS When atoms share electrons in covalent bonds, the electrons no longer reside in the atomic orbitals. Instead, they occupy molecular orbitals that are formed when the atomic orbitals overlap. The total number of molecular orbitals is equal to the total number of atomic orbitals involved in bonding. So, when the 1s orbitals of two hydrogen atoms overlap to form the covalent bond in an H2 molecule, two molecular orbitals are formed. One of these molecular orbitals is lower in energy than the 1s atomic orbitals and is called a bonding molecular orbital. The other molecular orbital is higher in energy than the 1s atomic orbitals and is called an antibonding molecular orbital. Bonding molecular orbitals are formed when the atomic orbitals combine in phase. Filled bonding orbitals result in an increase in the electron density 96 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS between the atoms. The electrons in these bonding orbitals stabilize the bonded atoms since the orbitals are of less energy than the atomic orbitals. Antibonding molecular orbitals are formed when the atomic orbitals combine out of phase. Filled antibonding orbitals result in a decrease in the electron density between the atoms. The electrons in the antibonding orbitals destabilize the molecule since they are higher in energy than the electrons in the atomic orbitals. The valence electrons of the two atoms are assigned to molecular orbitals of increasing energy according to the Aufbau principle, the Pauli Exclusion Principle, and Hund’s Rule in the same way that electrons are assigned to atomic orbitals. As with atomic orbitals, each molecular orbital can have only two electrons, each with an opposite spin. Fig. 3.9 shows a schematic diagram of the molecular orbitals (molecular orbital diagram) for the diatomic H2. The diagram shows the two 1s atomic orbitals which combine to form one bonding orbital and one antibonding orbital. The two paired valence electrons are placed in the bonding molecular orbital, which is lower in energy than the atomic orbitals. This stabilizes the molecule. The higher energy antibonding molecular orbital remains empty. There are two types of molecular orbitals. These are sigma orbitals and pi orbitals as shown in Fig. 3.10. Molecular orbitals that are symmetrical about the axis of the bond are called sigma molecular orbitals, denoted by the Greek letter “σ.” They lie between the nuclei of two atoms where there is an increase in electron density along the bond axis. This increase in electron density causes the nuclei of the two atoms to be drawn closer together. In sigma antibonding orbitals (σ*), there is a low electron density between the nuclei of the two atoms, which destabilizes the bond. Sigma molecular orbitals can be formed from the combination of two s atomic orbitals, as in H2, or from the combination of two p atomic orbitals that lie along the axis of the bond (pz). Pi orbitals are formed from the combination of two atomic orbitals that lie outside of the bond axis (px, py). This results in a side to side overlap of the atomic orbitals. The pi bonding orbital, denoted by the Greek letter “π,” thus has no electron density along the bond axis. Instead, the electron density lies above and below the bond axis. The electron density in the pi antibonding orbital (π*) lies entirely outside the bond axis. FIG. 3.9 A molecular orbital diagram for the H2 molecule. The combination of two 1s atomic orbitals results in one bonding molecular orbital (σ) and one antibonding molecular orbital (σ*). 3.5 MOLECULAR ORBITALS Atomic orbitals Molecular orbitals Bonding Antibonding s s s∗ pz s s∗ py p 97 FIG. 3.10 The shapes of bonding and antibonding molecular orbitals formed from the combinaton of two of the atomic orbitals shown at left. The black dots indicate the location of the nuclei. p∗ Single covalent bonds consist of one sigma molecular orbital. Double bonds consist of one sigma molecular orbital and one pi molecular orbital. Triple bonds are made up of one sigma molecular orbital and two pi molecular orbitals. The number of bonds between two atoms is called the bond order. The bond order can be calculated from a molecular orbital diagram as; Bond order ¼ ½ ðb aÞ m (4) where “b” is the number of electrons in bonding molecular orbitals and “a” is the number of electrons in antibonding molecular orbitals. If the bond order is zero, no bonds are produced and the molecule is not stable. If the Bond Order is equal to 1, the molecule contains a single covalent bond. If the bond order is equal to 2, the molecule contains a double bond and if the bond order is equal to 3 the molecule contains a triple bond. So, the bond order also indicates the strength of the bond. The greater the bond order, the stronger the bond. A bond order that is not a whole number indicates that resonance is present and the bond order is an average of the possible resonance structures. The bond order of H2 calculated from Fig. 3.9 is ½ (2 0) ¼ 1, indicating a single bond between the two hydrogen atoms. Compare this to the molecular orbital diagram for the He2 molecule shown in Fig. 3.11. The molecular orbitals formed from the combination of the two 1s atomic orbitals are the same as for H2. However, each He atom has two valence electrons (1s2) giving the molecule four valence electrons. These four electrons fill both the bonding and the antibonding molecular orbitals, which destabilizes the bond. The bond order for He2 calculated from the molecular orbital diagram is ½ (2 2) ¼ 0. This indicates that no stable bonds are formed between the two helium atoms and He2 does not occur in nature. Fig. 3.12 shows the molecular orbital diagrams for the diatomic oxygen (O2) and nitrogen (N2) molecules. Both oxygen and nitrogen have valence electrons in the 2s, 2px, 2py, and 2pz atomic orbitals. The combination of these four atomic orbitals on one atom with the same set of four atomic orbitals on a second atom leads to the formation of a total of eight molecular orbitals: two σ orbitals, two σ* orbitals, two π orbitals, and two π* orbitals. The σ2s molecular orbitals are formed from the atomic 2s orbitals. The σ2p and π2p molecular orbitals are formed from the atomic 2p orbitals. Since the 2s atomic orbitals are lower energy than the 2p orbitals, the σ2s and σ*2s molecular orbitals both lie at lower energies than the molecular orbitals 98 FIG. 3.11 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS The molecular orbital diagram for the He2 molecule. FIG. 3.12 The molecular orbital diagram for diatomic oxygen and nitrogen. The subscripts on the molecular orbital designations indicate the atomic orbitals that formed each molecular bond. formed from the combination of the 2p atomic orbitals. Of the three 2p atomic orbitals, the 2pz orbitals lie in the bond axis, while the 2px and 2py orbitals lie outside the bond axis (see Fig. 3.10). So, the interaction between the two 2pz orbitals is stronger than the interaction between the 2px or 2py orbitals. This normally results in the molecular σ2p orbital, formed from the combination of the atomic 2pz orbitals, being at a lower energy than the π2p orbitals, formed from the 2px and 2py orbitals. Also, the σ2p* orbital is higher energy than the π2p* orbitals. This trend is shown in Fig. 3.12 for diatomic oxygen. Diatomic nitrogen has a slightly different molecular orbital sequence than diatomic oxygen. This is because the 2s and the 2p atomic orbitals are close in energy, making it possible for them to interact. This interaction results in s-p atomic orbital mixing when the molecular orbitals are formed. This orbital mixing is called hybridization and is very important to the understanding of the chemistry of carbon covered in Chapter 13. The result of this orbital hybridization is a slight change in the relative energies of the molecular orbitals and the 3.5 MOLECULAR ORBITALS 99 π2p orbitals become of lower energy than the σ2p orbitals. Notice from Fig. 3.12 that this orbital mixing only affects the order of the bonding orbitals and the order of the antibonding orbitals remains the same. Each nitrogen atom has five valence electrons (2s22p3) giving 10 valence electrons in the N2 molecule. When placed in the molecular orbitals, the σ2s, σ*2s, σ2p, and the two π2p orbitals are completely filled. There is no net bonding from the σ2s orbitals, because the number of bonding electrons equals the number of antibonding electrons. Since none of the σ*2p or π*2p antibonding orbitals are filled, this leaves six total bonding electrons. For N2, the bond order is ½ (8 2) ¼ 3. The bond order of 3 indicates a triple bond with one sigma bond (σ2p) and two pi bonds (π2p). Each oxygen atom has six valence electrons (2s22p4) with a total of 12 valence electrons in the O2 molecule. When placed in the molecular orbitals, the σ2s, σ*2s, σ2p, and the two π2p orbitals are completely filled and the two π*2p orbitals are half filled with one electron each. There is also no net bonding in the σ2s orbitals for O2 because the number of bonding electrons equals the number of antibonding electrons. Also, the two unpaired electrons in the π*2p orbital cancels one additional pair of bonding electrons in the π2p orbitals, leaving a total of four electrons in bonding orbitals and two bonds between the two oxygen atoms. The bond order is ½ (8 4) ¼ 2 indicating a double bond. The number of unpaired electrons in the molecular orbitals determines the magnetic properties of the molecule. Molecules which have all electrons paired, such as N2, are diamagnetic, meaning that they are weakly repelled by a magnetic field. Molecules that have one or more unpaired electrons, like O2, are paramagnetic. They are strongly attracted to a magnetic field. Liquid oxygen, with its two unpaired electrons in the π*2p orbitals, is attracted to a magnetic field as shown in Fig. 3.13. The valence electron configuration for molecules can be written in the same way that electron configurations are written for atoms, by listing the molecular orbitals and the number of electrons in them. The valence electron configuration for the H2 molecule would be simply FIG. 3.13 A stream of liquid O2 is deflected in a magnetic field. Photograph by Pieter Kuiper, Wikimedia Commons. 100 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS TABLE 3.5 Electron Configurations and Predicted and Observed Properties of Diatomic Molecules Formed From the Elements in the Second Period of the Periodic Table Electron Configuration Bond Order Bond Energy (kJ) Bond Length (pm) Unpaired Electrons Li2 (σ2s)2 1 105 267 0 d Be2 2 2 0 ― ― 0 ― 2 2 1 1 1 289 159 2 p 2 2 2 2 2 598 131 0 d 2 2 2 2 2 3 946 110 0 d 2 2 2 2 2 1 1 2 498 121 2 p 2 2 2 2 2 2 2 1 158 143 0 d 2 2 2 2 2 2 2 0 ― ― 0 ― B2 C2 N2 O2 F2 Ne2 (σ2s) (σ*2s) (σ2s) (σ*2s) (π2p) (π2p) (σ2s) (σ*2s) (π2p) (π2p) (σ2s) (σ*2s) (π2p) (π2p) (σ2p) (σ2s) (σ*2s) (π2p) (π2p) (σ2p) (π*2p) (π*2p) (σ2s) (σ*2s) (π2p) (π2p) (σ2p) (π*2p) (π*2p) (σ2s) (σ*2s) (π2p) (π2p) (σ2p) (π*2p) (π*2p) (σ*2p) 2 d ¼ diamagnetic while; p ¼ paramagnetic. (σ1s)2 with the 2 superscript indicating the number of electrons in the molecular orbital. Similarly, the valence electron configuration for N2 and O2 are: 2 2 2 N2 : ðσ2s Þ2 ðσ∗ 2s Þ2 π2p π2p σ2p 1 1 2 2 2 O2 : ðσ2s Þ2 ðσ∗ 2s Þ2 π2p π2p σ2p π∗ 2p π∗ 2p The order of molecular orbitals is important in molecular orbital diagrams because it denotes the order of increasing energies of the molecular orbitals. However, it is not important in electron configurations since the lower energy orbitals are completely filled. The electron configuration of molecules can be used to predict molecular properties. For example, the number of unpaired electrons determines magnetic properties and bond order determines bond strength, bond length, molecular stability, and in some cases the presence of resonance structures. Some examples from the possible diatomic elements in period 2 are shown in Table 3.5. Of the eight possible diatomic molecules listed in the table, two have a bond order of 0 indicating that no bonds are formed and so the molecules do not exist. Those with bond orders from 1 to 3 have increasing bond energies and decreasing bond length. Only two of the molecules listed have unpaired electrons, predicting that they will be paramagnetic. 3.6 MOLECULAR GEOMETRY Molecules are three-dimensional groups of atoms. The chemical formulas or electron dot structures of the molecules can tell us about the arrangement and bonding of the atoms, but they tell us nothing about their three-dimensional shapes. The shape of a molecule is important because it determines several molecular properties including: chemical reactivity, polarity, physical state, boiling point, melting point, and many more. The molecular shape 3.6 MOLECULAR GEOMETRY 101 depends on the bond lengths, the angles between the bonds, and the positioning of the electron pairs. The repulsions of electron pairs held in the molecular bonds or as lone pairs control the angles between bonds and the positions of the atoms in the molecule. Both the electrons held in covalent bonds, regardless of the bond order, and those as lone pairs are considered as electron groups. Since electrons repel each other, the electron groups will seek to be as far apart as possible. So, a molecule will have a geometry that minimizes the repulsion between its electron groups. The prediction of molecular shape from the repulsion between bonding and nonbonding electron groups is known as the valence shell electron pair repulsion (VSEPR) model of molecular geometry. There are five basic arrangements of electron groups around a central atom that is surrounded by two to six electron groups shown in Fig. 3.14. When a molecule has two electron groups around a central atom, they will occupy positions opposite each other in order to minimize the repulsion between the electron groups. The resulting geometry is linear with the bond angles of 180 degrees. When three electron groups surround a central atom, the most stable geometry will be in the shape of a triangle with the bond angles of 120 degrees. This geometry is called trigonal planar since all atoms lie in the same plane. With four electron groups around the central atom, the most stable geometry will be in the shape of a tetrahedron with the bond angles of 109.5 degrees. With five electron groups surrounding a central atom, the electron groups will be in the shape of two tetrahedra connected at the bases with the central atom positioned in the center of the base. This geometry is called trigonal bipyramidal. The atoms that lie along the vertical axis of the molecule are known as the axial positions and those that lie in the horizontal plane of the molecule, perpendicular to the vertical axis, are known as the equatorial positions. The bond angles between the axial atoms are 90 degrees, while the bond angles between the equatorial atoms are 120 degrees. With six electron groups around a central atom, the most stable geometry will be in the shape of two square base pyramids connected at the bases with the central atom at the center of the base. This geometry is called octahedral because the resulting figure has eight sides. The bond angles in an octahedral geometry are all 90 degrees. These electron group geometries cannot easily be explained by simple overlap of s and p atomic orbitals to form molecular orbitals. They can, however, be explained by including orbital hybridization. The hybrid orbitals, formed by mixing of s and p atomic orbitals, which Linear 109.5 degrees 120 degrees 180 degrees Trigonal planar Tetrahedral 90 degrees 90 degrees 120 degrees Trigonal bipyramidal Octahedral FIG. 3.14 The electron group geometries predicted by the VSEPR model for a central atom that is surrounded by two to six electron groups. 102 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS are close in energy, are directed towards the bond axis leading to better overlap and stronger bonds. The number of hybrid orbitals formed will be equal to the number of atomic orbitals that were combined. The types of hybridization for the first three geometries shown in Fig. 3.14 are; • sp. hybridization—The mixing of one s atomic orbital with one p atomic orbital on the central atom to give two hybrid orbitals separated by 180 degrees with a linear geometry. • sp2 hybridization—The mixing of one s atomic orbital with two p atomic orbitals on the central atom to give three sp2 hybrid orbitals separated by 120 degrees with a trigonal planar geometry. • sp3 hybridization—The mixing of one s atomic orbital with three p atomic orbitals on the central atom to give four sp3 hybrid orbitals separated by 109.5 degrees with a tetrahedral geometry. The geometries shown in Fig. 3.14 are for electron groups surrounding a central atom. Since molecular geometry describes the arrangement of atoms around a central atom, the molecular geometry will be the same as the electron group geometry only if all the electron groups are contained in bonds. Although the lone pairs on a central atom occupy space and are part of the electron group geometry, they are not part of the molecular geometry. So, molecules containing lone pairs of electrons on the central atom will not have the same molecular geometry as that of the electron groups. However, the molecular geometry can be derived from one of the five basic shapes. EXAMPLE 3.8: PREDICTING MOLECULAR GEOMETRIES OF MOLECULES WITH ONLY SINGLE BONDS SURROUNDING A CENTRAL ATOM What is the electron group geometry, molecular geometry, and bond angles of methane (CH4). 1. Draw the electron dot structure of methane. Carbon has four valence electrons and hydrogen has one valence electron. If carbon and hydrogen each share one valance electron, there will be four single bonds to carbon. The electron dot structure of methane is: H H C H H 2. Determine the electron group geometry. Methane has four electron groups surrounding a central carbon atom. The electron group geometry will be tetrahedral. 3. Determine the molecular geometry. Since all the electron pairs are contained in bonds, the molecular geometry will also be tetrahedral with bond angles of 109.5 degrees. 3.6 MOLECULAR GEOMETRY 103 In order to better represent the molecular geometry, a three-dimensional form of bonding notation is used in which the structures are drawn with solid lines representing bonds in the plane of the paper, dotted lines representing bonds extending behind the plane of the paper, and wedge-shaped lines representing bonds extending in front of the plane of the paper. The geometric structure of methane would then be drawn as; H C H H H For molecules that have one or more lone pair electrons on the central atom, the lone pairs are also considered as an electron group and occupy a position in the electron pair geometry. For example, the ammonia molecule (NH3) has four electron groups surrounding the central nitrogen atom. Three of these are bonding electrons and one is a lone pair. The electron dot structure for ammonia is; H N H H The predicted electron group geometry for four electron groups would be tetrahedral, similar to methane. However, since lone pairs are not part of the molecular geometry, the electron group geometry and the molecular geometry will not be the same. The atoms in the ammonia molecule form the shape of a pyramid with the hydrogens at the base and the nitrogen at the peak of the pyramid. This molecular geometry is called trigonal pyramidal (half of a trigonal bipyramidal geometry). N H H H Similarly, the water molecule with the electron dot structure of; H O H has two bonding electron groups and two nonbonding electron groups. With four electron groups surrounding the central oxygen atom, the electron group geometry is tetrahedral, but the molecular geometry is known as a bent shape represented as; O H H Since the electron group geometries of both ammonia and water are tetrahedral, it would be expected that the bond angles would still be 109.5 degrees. However, the bond angles in the ammonia molecule are actually 107.5 degrees and those in the water molecule are 104.5 degrees. This is because the lone pairs occupy more space than bonded pairs. This increases 104 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS TABLE 3.6 Predicted Electron Group Geometries and Molecular Geometries for Molecules with Two to Six Electron Groups and a Varying Number of Lone Pairs Surrounding a Central Atom Electron Groups Lone Pairs Bonds Electron Group Geometry Molecular Geometry Bond Angle (Degrees) 2 0 2 Linear Linear 180 3 0 3 Trigonal planar Trigonal planar 120 3 1 2 Trigonal planar Bent <120 4 0 4 Tetrahedral Tetrahedral 109.5 4 1 3 Tetrahedral Trigonal pyramidal <109.5 4 2 2 Tetrahedral Bent <109.5 5 0 5 Trigonal bipyramidal Trigonal bipyramidal 90, 120 5 1 4 Trigonal bipyramidal See-saw 90, 120 5 2 3 Trigonal bipyramidal T-shaped 90 5 3 2 Trigonal bipyramidal Linear 180 6 0 6 Octahedral Octahedral 90 6 1 5 Octahedral Square pyramidal 90 6 2 4 Octahedral Square planar 90 the repulsion between electron groups and forces the atoms closer together than predicted for molecules without lone pairs, resulting in smaller bond angles between the atoms. Table 3.6 describes the difference between the electronic group geometry and molecular geometry for molecules with two to six electron groups surrounding a central atom and a varying number of lone pairs. The shapes that occur in molecular geometries that do not occur in electron group geometries are: bent, trigonal pyramidal, see-saw, T-shaped, square pyramidal, and square planar. The bent and trigonal pyramidal geometries have been described above for water and ammonia; the remaining four are shown in Fig. 3.15. FIG. 3.15 The molecular geometries for a central atom surrounded by five electron groups with one lone pair (seesaw) and two lone pairs (T-shaped) and by six electron groups with one lone pair (square pyramidal) and two lone pairs (square planar). 90 degrees 90 degrees T-shaped Square planar 90 degrees 90 degrees 120 degrees Square pyramidal See-saw 3.7 MOLECULAR POLARITY 105 3.7 MOLECULAR POLARITY As explained in Section 3.3, polar covalent bonds occur when two bonded atoms have different electronegativities and share the electrons unequally, while nonpolar covalent bonds occur when two bonded atoms have the same electronegativities and share the electrons equally. Molecules can also be polar or nonpolar depending on the overall polarity of the bonds in the molecule. The polarity of the molecule is the sum of all of the individual bond polarities in the molecule. Since the dipole moment of each bond is a vector quantity with both magnitude and direction, the dipole moment of the molecule is determined as the vector sum of the individual bond dipole moments. Also, because the molecular polarity is a vector sum, the polarity of all bonds, however small, must be considered unless they are truly nonpolar with a dipole moment of 0.0D. A molecule that contains no polar covalent bonds (all bonds ¼ 0.0D) will have no charge difference between one part of the molecule and another part and the molecule will be nonpolar. A polar molecule occurs when electron density accumulates in one part of the molecule giving it a partial negative charge (δ). The other side of the molecule will then have an equally partial positive charge (δ+). Since diatomic molecules have only one bond, if that bond is polar, the molecule will be polar, as in HBr. Or, if the bond is nonpolar the molecule will be nonpolar, as in HAt. In polyatomic molecules, the molecular polarity is determined from the sum of all of the individual bond polarities and how they are oriented in space (molecular geometry). For example, consider CO2 with the electron dot structure; + O δ− δ+ C + O δ− With two electron groups around the central carbon atom, the electron group geometry will be linear and, since there are no lone pairs on the central atom, the molecular geometry will also be linear. Since carbon and oxygen have different electronegativities, both bonds in the CO2 molecule are polar. However, the molecule is symmetrical and the polarity of the two bonds is identical and oriented in exactly opposite directions. This is shown by arrows signifying the direction and strength of the bond polarities. The “+” sign at the beginning of the arrow indicates that the carbon end of the bond has the partial positive charge. The vector sum of the dipole moments of these two bonds is zero because they are exactly equal, but orientated in exactly opposite directions. So, even though the individual C]O bonds are polar, the CO2 molecule is nonpolar. In contrast, SO2 has three electron groups around the central sulfur atom with a trigonal planar electron group geometry. But, since one of the electron groups is a lone pair, the molecular geometry is bent. In this case, the SdO bond polarities contribute to the molecular polarity because the bent shape of the molecule is not symmetrical. The vector sum of the individual bond dipole moments gives a molecular polarity that is directed from the less electronegative sulfur atom towards the more electronegative oxygen atoms as indicated by the red arrow in the structure; 106 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS S O O In summary: • A polyatomic molecule is nonpolar if all of its terminal atoms have the same electronegativities and they are arranged symmetrically around the central atom. • A polyatomic molecule is polar if its terminal atoms have different electronegativities or they have the same electronegativities, but are not arranged symmetrically around the central atom. • The direction and strength of the polarity of a polyatomic molecule is determined by the vector sum of the dipole moments of all the bonds. EXAMPLE 3.9: PREDICTING WHETHER A MOLECULE IS POLAR OR NONPOLAR What is the electron group geometry, molecular geometry, and polarity of: H2S, COS, HCN, and CCl4. 1. First write the electron dot structures for each compound. Cl H S H O C S H C N Cl C Cl Cl 2. Determine the electron group geometry for each compound. H2S—with four electron groups is tetrahedral. COS—with two electron groups is linear. HCN—with two electron groups is linear. CCl4—with four electron groups is tetrahedral. 3. Determine the molecular geometry for each compound. H2S—with two lone pairs is bent. COS—with no lone pairs remains linear. HCN—with no lone pairs remains linear. CCl4—with no lone pairs remains tetrahedral. 4. Determine the molecular polarity of each compound. H2S: Sulfur (2.58) is more electronegative than hydrogen (2.20), so the SdH bonds are polar. The vector sum of the two HdS bond dipole moments results in a molecular dipole moment directed from the less electronegative hydrogens towards more electronegative sulfur. The H2S molecule will be polar with the partial negative charge on the sulfur. S H H COS: Sulfur (2.58) has very similar electronegativity as carbon (2.55), so the C]S bond has a dipole moment 0.0 and is nonpolar. Oxygen (3.44) is more electronegative than carbon, so the C]O bond is polar. The COS molecule will be polar with the partial negative charge on oxygen. 3.8 INTERMOLECULAR FORCES 107 EXAMPLE 3.9: PREDICTING WHETHER A MOLECULE IS POLAR OR NONPOLAR— CONT’D O C S HCN: Carbon (2.55) is more electronegative than hydrogen (2.20). The CdH bond is polar with the partial negative charge on carbon. Nitrogen (3.04) is also more electronegative than carbon. The vector sum of both bond dipole moments results in a molecular dipole moment that is directed from the less electronegative hydrogen to the most electronegative nitrogen. H C N CCl4: Chlorine (3.16) is more electronegative than carbon (2.55), so each CdCl bond will be polar. However, the tetrahedral geometry of the molecule is symmetrical. Since CCl4 has a symmetric charge distribution around the central carbon atom, the polar CdCl bonds all have dipole moments of equal magnitude with each pair directed opposite each other. The vector addition of all the CdCl dipole moments will be zero and the CCl4 molecule will be nonpolar. Cl C Cl Cl Cl 3.8 INTERMOLECULAR FORCES Molecular polarity is the source of interactions between molecules, which are important in the determination of many of the compound’s physical properties. The forces of attraction and repulsion between molecules are known as intermolecular forces. The attractive forces are known as van der Waals forces, named after the Dutch scientist Johannes Diderik van der Waals. These van der Waals forces are weak compared to the forces that hold the atoms of a molecule together, such as covalent or ionic bonding. However, they still can affect boiling point, freezing point, vapor pressure, evaporation rate, viscosity, surface tension, solubility, and other molecular properties. What determines the physical state of a substance, whether it exists as a solid, liquid, or gas, is based on both the strength of the van der Waals forces and the thermal energy of the system. At a given temperature, substances that contain strong van der Waals forces are more likely to be solids or liquids, while those with weak van der Waals forces will tend to be gases. There are two types of van der Waals forces: dipole-dipole forces and London dispersion forces. Dipole-dipole forces are the strongest type of van der Waals forces. A dipole-dipole force is an attractive force that occurs between molecules with permanent dipoles. Polar molecules, which have permanent dipole moments, will attract each other when the partially positive region of one molecule is near the partially negative region of another molecule. 108 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS FIG. 3.16 An example of a dipole-dipole interaction in liquid HCl where the partially positive hydrogen attracts the partially negative chlorine resulting in the alignment of the HCl molecules. An example of a dipole-dipole interaction is seen in HCl shown in Fig. 3.16. Since chlorine is more electronegative than hydrogen and the molecular geometry is linear, the HdCl bond is polar and so has a permanent dipole moment. The positive region of the molecule will attract the negative region of a neighboring molecule and affect its position. This attraction results in all the molecules aligning so that the attractions between molecules are maximized and the repulsions are minimized. A very special type of dipole-dipole interaction is hydrogen bonding. This force is referred to as bonding because it is the strongest type of van der Waals force. It occurs when the permanent dipole in a molecule results from a covalent bond between a hydrogen atom and one of the very small, highly electronegative atoms: fluorine, oxygen, or nitrogen. The very large difference in electronegativities between hydrogen (2.20) and fluorine (3.98), oxygen (3.44), and nitrogen (3.04) causes the bond between them to be extremely polar. The electronegative atom attracts the bonding electrons towards itself and away from the hydrogen so strongly that that the hydrogen atom essentially becomes an exposed proton. An attractive force then forms between the hydrogen atom of one molecule and a lone pair of electrons on the electronegative atom of another molecule. The hydrogen becomes the “hydrogen bond donor” and the lone pair becomes the “hydrogen bond acceptor” forming a bridge between the hydrogen and the highly electronegative element. This can occur between molecules of the same kind, between molecules of different kinds, or between different sites on the same molecule if the molecule is very large. This bridging between hydrogen and a lone pair on oxygen or nitrogen is responsible for the folding of protein molecules giving them their three-dimensional structures. CASE STUDY: WATER Water (H2O) is the most abundant molecular compound found on Earth. It covers 70% of the Earth’s surface and is essential for all life, making up 65% of the human body. Water is the only substance found naturally in all three of the common states of matter: solid, liquid, and gas. The gas form is found in the atmosphere as water vapor or on the Earth’s surface as steam from hydrothermal vents. Liquid water is in the atmosphere as clouds and also in precipitation as rain. It is found on the surface as both fresh and salt water and also in groundwater aquifers. Ice, the solid form, is seen as snow or hail in precipitation, as icebergs in the polar oceans, and as glaciers in higher elevations. As described in Section 3.6, the water molecule has two bonding electron groups and two nonbonding electron groups. With four electron groups surrounding the central oxygen atom, the electron group geometry is tetrahedral but the molecular geometry is bent. With a difference in electronegativity between oxygen (3.44) and hydrogen (2.20) and with two lone pairs on the oxygen atom, water is a very polar molecule with a dipole moment of 1.85D. O H H 109 3.8 INTERMOLECULAR FORCES Water has many unusual properties including its boiling point, freezing point, surface tension, viscosity, and cohesion. Table 3.7 lists the boiling and freezing points of the compounds formed from the elements in group 16 of the periodic table bonded to hydrogen. Going down the group from hydrogen sulfide (H2S) to hydrogen telluride (H2Te), the boiling points increase from 60°C to 2°C as the molecular size increases. But water, the smallest of the group, has a boiling point of 100°C, which is very high for its molecular size. The same trend is seen in the freezing points of this group. Going down the group from H2S to H2Te, the freezing points increase from 82°C to 49°C as the molecular size increases, but water has a very high freezing point of 0°C. These inconsistent boiling and freezing points of water are due to the fact that water forms very strong hydrogen bonds. In liquid water, a lone pair on the oxygen of one water molecule can form a hydrogen bond with a hydrogen atom on another water molecule. In liquid water, this hydrogen bonding is repeated until every water molecule in the liquid is bonded to four other water molecules, two through the two lone pairs on the oxygen atom and two through the two hydrogen atoms as shown in Fig. 3.17. Water is unique in that it can form such a large number of hydrogen bonds per molecule. This is because the number of lone pairs and hydrogen atoms in each molecule are equal. In contrast, hydrogen fluoride has three lone pairs on the fluorine atom, but only one hydrogen atom. So there are three times as many lone pairs as there are hydrogen atoms in liquid HF and the number of hydrogen bonds it can form is limited to the number of hydrogen atoms. Each HF molecule can only form two hydrogen bonds, one with the hydrogen atom and one with one of the lone pairs on the fluorine atom. The other two lone pairs remain unbonded due to the lack of hydrogen atoms. Similarly, ammonia has only one lone pair on nitrogen and three hydrogen atoms. This time the number of hydrogen bonds formed in liquid NH3 is limited by the number of lone pairs. So, each TABLE 3.7 Boiling and Freezing Points in °C of the Hydrides of Group 16 Elements Compound Molecular Formula Boiling Point Water H2O 100 0 Hydrogen sulfide H2S 60 82 Hydrogen selenide H2Se 41 66 Hydrogen telluride H2Te 2 49 FIG. 3.17 δ− δ+ δ+ δ− δ+ Hydrogen bonds H O δ− H δ+ δ− Freezing Point Hydrogen bonding in liquid water where each water molecule forms hydrogen bonds with four other water molecules, two through the two lone pairs on the oxygen atom and two through the two hydrogen atoms. Modified from Qwerter, Wikimedia Commons. 110 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS ammonia molecule can also only form two hydrogen bonds, one with the lone pair and one with one of the hydrogen atoms. The unusually high boiling point of water is due to the high number of hydrogen bonds each molecule can form relative to its small size. This extensive hydrogen bonding prevents water molecules from being easily released from the surface of the liquid, so a higher temperature is required to achieve vaporization. The large number of hydrogen bonds in water also leads to the unusually high freezing point. Although the liquid water molecules are mobile and the hydrogen bonds are in flux, the hydrogen-bonded structure approximates an extended tetrahedral arrangement as shown in Fig. 3.17. Since the molecules are already in an extended regular structure in liquid water, the transition from a liquid to a solid does not require a large drop in temperature as with similar molecules that are not so strongly hydrogen-bonded. The hydrogen-bonded structures of liquid water and ice are compared in Fig. 3.18. Liquid water has a partially ordered tetrahedral structure in which the hydrogen bonds are constantly being formed and broken. On the other hand, ice has a rigid structure where each water molecule remains hydrogen-bonded to the same four water molecules. So, when water becomes a solid at the freezing point, the water molecules transition into a crystalline structure which has an open cage-like form that contains a lot of empty space. Because of this, ice increases in volume by about 9% over liquid water. Water is the only known chemical compound that expands when it freezes and since water is ubiquitous in nature, the effects of this behavior can be powerful. The expansion of ice as water freezes is the basic cause of water pipes bursting from the pressure of the expanding ice inside the pipes. It is also the major cause of damage to building foundations and roadways. Liquid water can seep into cracks in building materials, expanding and widening the cracks when the temperature drops to the freezing point at night. The subsequent rise in temperatures during the day can result in an expansion-contraction cycle during winter months, widening cracks and increasing damage. This freeze-thaw cycle is also responsible for the natural weathering of rocks. Along with the increasing volume when water freezes, the density of ice also decreases over that of the liquid water phase. All substances become less dense when they are heated and denser when they are cooled. So, when liquid water is cooled its density increases. The density continues to increase until it reaches a maximum density at 4°C (1.00 g/cm3) as shown in Fig. 3.19. This is the point where molecular motion is significantly decreased and the extended tetrahedral structure of liquid water becomes most compact. As water is cooled below 4°C, it becomes less dense as it begins to freeze and transition into the open cage-like structure of the solid form. The density of ice is 0.917 g/cm3 at 0°C, while the density FIG. 3.18 The structures of liquid water and ice. P99am, Wikimedia Commons. Liquid water Ice 111 3.8 INTERMOLECULAR FORCES FIG. 3.19 The temperature dependence of the densities of ice and water. 1.01 1 Maximum density = 4 °C 0.99 Density (g/cm3) 0.98 0.97 0.96 0.95 0.94 0.93 0.92 0.91 –100 –50 0 50 100 150 Temperature (°C) of liquid water is 0.9998 g/cm3 at 0°C. So, the density of ice is approximately 8.3% less than the density of water at the same temperature. The density of ice increases slightly with decreasing temperature and has a value of 0.927 g/cm3 at 80°C. Since the water molecules are more tightly packed in the liquid state than in the solid state, this explains why ice floats on liquid water. The weakest type of van der Waals forces is the London dispersion force named after the German American physicist Fritz London. The London dispersion force is a weak van der Waals force arising from the formation of an induced polarization in molecules that do not have a permanent dipole moment. As two nonpolar molecules approach each other, the repulsion between their electrons leads to a distortion of the electron clouds. This distortion results in a temporary polarization of the molecules, creating a temporary dipole in each molecule. These temporary dipoles can cause a temporary dipole to form in nearby molecules due to the attraction between their partially positive regions and the electron clouds of other molecules. The temporary attractions between the partially positive region of one dipole with the partially negative region of another dipole causes the molecules to line up as shown in Fig. 3.20, stabilizing the system. This stabilization also affects chemical properties such as boiling points, freezing points, and solubilities. However, since London dispersion forces are much weaker than dipole-dipole interactions, the effects are not as strong. London dispersion forces are the only van der Waals forces that act on nonpolar molecules including the diatomic elements (N2, O2, and the halogens) as well as nonpolar hydrocarbons such as methane. London forces can cause nonpolar substances to condense into liquids and to freeze into solids when the temperature is lowered sufficiently. The strength of the London dispersion force depends on how easily the electrons in a molecule can be polarized, called the polarizability of a molecule. In general, London forces increase with an increasing 112 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS FIG. 3.20 The formation of an induced dipole in a nonpolar diatomic molecule. The temporary distortion of the electron cloud results in induced temporary dipoles in neighboring molecules, resulting in weak attractive forces called London dispersion forces. δ− δ+ δ− δ+ δ− δ+ TABLE 3.8 Boiling Points (°C) and Number of Electrons in the Diatomic Elements. The Boiling Point of At2 Has Not Been Measured and is Predicted From Chemical Properties Element Electrons Boiling Point (°C) N2 14 196 O2 16 183 F2 18 188 Cl2 34 34 Br2 70 59 I2 106 184 At2 170 337 number of electrons. The larger the atoms, the more electrons in the molecule, the more easily they can be polarized, and the stronger the London dispersion force. Also, in a larger atom, the electrons occupy higher electron shells that are farther away from the nucleus, so the electrons will be less tightly held by the nuclear attractive forces and are able to move more freely. This is why the London dispersion force increases with increasing atomic mass. London dispersion forces affect boiling points of nonpolar substances. Table 3.8 lists the boiling points of the diatomic elements. It is clear from the trend of the halogens that the boiling point increases as the number of electrons increases. For example, Cl2 with 34 electrons has a higher boiling point than F2 with 18 electrons due to the higher polarizability, and thus, the stronger London dispersion forces between the larger Cl2 molecules than for the smaller F2 molecules. This trend continues down the group from F2 to I2. Although the boiling point of the last member of the group has not been measured directly due to the very high radioactivity of At2, it has been predicted from other observed chemical properties to be very high (337°C). The first three diatomic elements shown in Table 3.8 (N2, O2, and F2) are all in period 2 of the periodic table. They have very similar boiling points ranging from d196°C for N2 to 183°C for O2, a difference of only 13°C compared to a difference of 247°C for F2 (188°C) and Br2 (59°C). Since N2, O2, and F2 are in adjacent groups (15 through 17) in the same period, they each have a very similar number of electrons (14 to 18) and also their electron configurations are similar with all the electrons occupying the same electron shells. So, it is expected that they would have very similar polarizabilities and very similar London dispersion forces leading to their similarity in boiling points. IMPORTANT TERMS 113 IMPORTANT TERMS Anion a negatively charged ion. Antibonding molecular orbital a molecular orbital resulting from the combination of atomic orbitals out of phase. Axial the atoms that lie along the vertical axis of a molecule. Bond order the number of bonds between two atoms. Bonding pair electrons the shared electron pair that forms a covalent bond between two atoms. Bonding molecular orbital a molecular orbital resulting from the combination of atomic orbitals in phase. Bonding notation a representation of the structure of a molecule using element symbols connected by lines to represent bonding electrons. Cation a positively charged ion. Chemical formula a way of expressing the bonding between atoms and ions in a compound using a single line of element symbols along with numeric subscripts to indicate the number of atoms of each element. Coulomb’s Law the force of the electrostatic interaction between two charged particles is proportional to the product of the charges divided by the square of the distance between them. Covalent bond sharing of a pair of electrons between two atoms which holds them together. Diatomic molecule a molecule made up of two atoms. Diamagnetic having the property of being weakly repelled by a magnetic field. Dipole a bond (or molecule) that has a partial positive charge on one end and partial negative charge on the other. Dipole-dipole forces an attractive force between molecules with permanent dipole moments. Dipole moment a measure of the polarity of a covalent bond. Double bond a covalent bond that results from the sharing of two electron pairs between two atoms. Equatorial the atoms that lie along the horizontal plane of a molecule. Hybridization a mixing of s and p atomic orbitals when molecular orbitals are formed. Hydrogen bonding a type of dipole-dipole force that occurs between molecules containing a covalent bond between a hydrogen atom and a very electronegative atom with at least one lone pair of electrons, usually fluorine, oxygen, or nitrogen. Intermolecular forces the forces of attraction and repulsion between molecules. Ionic bond electrostatic attractions between oppositely charged positive and negative ions which holds the atoms together to form an ionic compound. Ionic radius the distance from the nucleus to the outermost occupied electron orbital in an ion. London dispersion force a weak attractive force arising from the formation of an induced instantaneous polarization in molecules that do not have a permanent dipole moment. Lone pair electrons a pair of electrons surrounding an atom in a molecule that is not shared with another atom. Molecular formula the chemical formula for a covalent compound. Molecular orbitals orbitals formed when the atomic orbitals overlap during covalent bonding. Nonpolar covalent bond a covalent bond where electrons are shared equally between two atoms. Octet rule atoms in the s- and p-blocks of the periodic table tend to combine in such a way that each atom acquires eight electrons in its valence shell, giving it the same electronic configuration as a noble gas. Paramagnetic having the property of being strongly attracted by a magnetic field. Polar covalent bond a covalent bond where electrons are shared unequally between two atoms. Polarizability the ability for a molecule to be polarized. Resonance structures two or more equivalent chemical structures which differ only in the position of their electrons (not the position of the atoms). Single bond a covalent bond that results from the sharing of one electron pair between two atoms. Stock number a system of naming cations that uses the name of the element followed by a Roman numeral in parentheses to indicate the charge of the ion. Triple bond a covalent bond that results from the sharing of three electron pairs between two atoms. Valence bond theory the description of covalent bonds as involving shared pairs of electrons which are localized in a bond between two atoms. van der Waals forces the forces of attraction between molecules. 114 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS STUDY QUESTIONS 3.1 What is the difference between a cation and an anion? 3.2 What determines the number of electrons an atom will gain or lose? 3.3 Which group in the periodic table does not form ions? Give the group number and its name. 3.4 Which group in the periodic table forms 1+ ions? Give the group number and its name. 3.5 Which group in the periodic table forms 1 ions? Give the group number and its name. 3.6 Which group in the periodic table forms 2+ ions? Give the group number and its name. 3.7 What is the common name for the groups in the periodic table that contain elements which can have multiple ionic forms? 3.8 What are the most common cations for the transition metals? 3.9 What is an ionic radius? 3.10 Which ion has a larger radius than the parent atom; anion or cation? Why? 3.11 Which ion has a smaller radius than the parent atom? Why? 3.12 Which ion has the larger radius; anion or cation? 3.13 How does ionic charge affect ionic radius? 3.14 How does ionic radius vary within a group? 3.15 How does ionic radius vary within a period? 3.16 What creates an ionic bond? 3.17 What does Coulomb’s Law describe? 3.18 The potential energy between two charged particles is directly proportional to what? Inversely proportional to what? 3.19 How does ionic radius affect ionic bond strength? 3.20 How does ionic charge affect ionic bond strength? 3.21 Ionic bonds generally occur between which anions and cations? 3.22 What is the octet rule? 3.23 What is a chemical formula? 3.24 (a) What is a covalent bond? (b) How is it different from an ionic bond? 3.25 (a) What is a lone pair of electrons? (b) What is a bonding pair? 3.26 What is valence bond theory? 3.27 (a) What is a single bond? (b) Double bond? (c) Triple bond? 3.28 What makes a covalent bond polar? 3.29 What is a dipole? 3.30 What is the dipole moment of a bond or molecule? 3.31 What electronegativity difference between two bonded atoms will result in the bond behaving as: (a) polar, (b) nonpolar, and (c) ionic? 3.32 What is bonding notation? Why is it used? 3.33 Why is the chemical formula of a covalent compound called a molecular formula? 3.34 What is a polyatomic ion? 3.35 What is a resonance structure? 3.36 How are molecular orbitals formed? 3.37 How are bonding molecular orbitals formed? 3.38 How are antibonding molecular orbitals formed? 3.39 What are the two types of bonding molecular orbitals? How do they differ? PROBLEMS 3.40 3.41 3.42 3.43 3.44 3.45 3.46 3.47 3.48 3.49 3.50 3.51 3.52 3.53 3.54 3.55 3.56 3.57 3.58 3.59 115 What is the bond order of two bonded atoms? What determines the magnetic properties of a molecule? What causes molecules to be diamagnetic? paramagnetic? What is bond hybridization? What are the three types of bond hybridization? How is each formed? What are the five possible electron group geometries? What determines the direction and strength of the polarity of a polyatomic molecule? What are intermolecular forces? What are van der Waals forces? What are two types of van der Waals forces? What is a hydrogen bond? Which van der Waals force is the strongest? What property of water is responsible for its unusual boiling point? How many hydrogen bonds can a single water molecule form? Why does water expand when freezing? At what temperature is water at its maximum density? What is the weakest van der Waals force? What is the only van der Waals force that acts on nonpolar molecules? What are London dispersion forces? What is polarizability? PROBLEMS 3.60 What are the charges of the ions formed from the following elements: (a) Na, (b) F, (c) Ca, (d) Br, and (e) Cs? 3.61 What are the electronic configurations in noble gas notation for the following ions: (a) Mg2+, (b) N3, (c) Al3+, and (d) S2? 3.62 Name the following anions: (a) P, (b) O2, (c) Cl, (d) S2, and (e) I. 3.63 Name the following cations: (a) Al3+, (b) As3+, (c) Cd2+, (d) Cr6+, and (e) Cu1+. 3.64 List the following ions in the order of increasing radius: As3, N3, Sb3, N3, Sb3+. 3.65 List the following ions in the order of decreasing radius: Sc3+, V3+, K+, V5+, Cr6+. 3.66 List the following ions in the order of increasing size: S 2, Sr2+, Te2, Be2. 3.67 Name the following ionic compounds: (a) CrF2, (b) FeCl3, (c) Al2S3, (d) PbO, (e) Mg3P2, and (f) TiI4. 3.68 Give the chemical formulas for the following ionic compounds: (a) silver cyanide, (b) iron(II) oxide, (c) calcium oxide, (d) sodium bromide, (e) copper(I) arsenide, and (f) beryllium chloride. 3.69 List the following ionic bonds in the order of increasing bond strength: KdBr, KdI, MgdCl, ZrdCl, KdF. 3.70 List the ionic bonds in Problem 3.69 in the order of increasing bond length. 3.71 Write the following ionic reactions using electron dot representations: (a) lithium + chlorine, (b) calcium + sulfur, (c) magnesium + bromine, (d) barium + oxygen and (e) strontium + iodine. 116 3. CHEMICAL BONDING—THE FORMATION OF MATERIALS 3.72 Write the chemical formula of the ionic compounds formed from each reaction in Problem 3.71. 3.73 List the following ionic compounds in the order of increasing melting points: KBr, MgCl2, TiCl4, KF, KI. 3.74 Name the following covalent compounds: (a) SbBr3, (b) ClO2, (c) N2O3, (d) PI3, (e) CCl4, and (f) CH4. 3.75 Give the molecular formulas for the following covalent compounds: (a) nitrogen trifluoride, (b) dinitrogen pentoxide, (c) hydrogen sulfide, (d) sulfur hexafluoride, (e) nitrogen dioxide, and (f) carbon disulfide. 3.76 Draw the electron dot structures for the following molecules: (a) BF3, (b) CS2, (c) PCl3, (d) NO2, and (e) CHCl3. 3.77 Which molecules in Problem 3.76 contain double bonds? 3.78 What are the electron group geometries of the molecules listed in Problem 3.76? 3.79 What are the molecular geometries of the molecules listed in Problem 3.76? 3.80 Are the molecules listed in Problem 3.76 polar or nonpolar? 3.81 Write the molecular formulas in Problem 3.76 in three-dimensional bonding notation. 3.82 Name the following polyatomic ions: (a) OH, (b) CNS, (c) SO4, (d) ClO4, and (e) CrO4. 3.83 Write the chemical formulas for the following polyatomic ions: (a) hydronium, (b) carbonate, (c) nitrate, (d) phosphate, and (e) nitrite. 3.84 Write the electron dot structure for the following polyatomic ions: (a) CO32, (b) OH, (c) NO2, and (d) H3O+. 3.85 Which polyatomic ions in Problem 3.84 have resonance structures? Draw the resonance structures. 3.86 In the molecular orbital diagram for Li2: (a) How many electrons occupy the bonding molecular orbitals? (b) How many electrons occupy the antibonding molecular orbitals? (c) What is the bond order for Li2? and (d) Is Li2 a stable molecule? 3.87 In the molecular orbital diagram for Be2: (a) How many electrons occupy the bonding molecular orbitals? (b) How many electrons occupy the antibonding molecular orbitals? (c) What is the bond order for Be2? and (d) Is Be2 a stable molecule? 3.88 List the following molecules in order of increasing boiling points: CH4, HCl, N2, NH3, H2, H2O. C H A P T E R 4 Chemical Equations and Mass Balance O U T L I N E 4.1 The Mole 117 4.2 The Empirical Formula 119 4.3 Chemical Equations 124 4.4 Stoichiometry 127 4.5 Limiting Reactant and Percent Yield 128 4.6 Aqueous Solubility of Ionic Compounds 4.7 Precipitation Reactions in Aqueous Solution 135 4.8 Concentrations in Aqueous Solution 139 Study Questions 142 Problems 143 133 4.1 THE MOLE Since chemistry deals with the properties of individual atoms and molecules and how these atoms form chemical bonds to create compounds, it is important for chemists to determine the amount of a substance on the molecular scale. Although the common use of the term “amount of substance” used in Engineering may be interpreted as the weight (in grams) or the volume (in cm3) of a substance, in chemistry the amount of a substance is a measure of the number of fundamental particles, such as atoms, molecules, or ions that are present in a given mass of substance. The SI base unit for the amount of a substance is the mole. The exact definition of a mole is the mass of any substance, which contains the same number of fundamental units as there are atoms in exactly 12.000 g of 12C. Carbon-12 was chosen to serve as the reference standard of the mole unit for the International System of Units since it was also chosen to serve as the reference standard for atomic mass and the atomic mass unit, as described in Chapter 1. The fundamental units of a substance may be atoms, molecules, ions, or chemical formula units, depending on the type of substance. The number of atoms in 12.000 g of 12C has been determined to be equal to 6.022 1023. So, one mole of any element always contains 6.022 1023 atoms, one mole of any covalent General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00004-7 117 # 2018 Elsevier Inc. All rights reserved. 118 4. CHEMICAL EQUATIONS AND MASS BALANCE compound always contains 6.022 1023 molecules, and one mole of any ionic compound always contains 6.022 1023 chemical formula units. The number 6.022 1023 by itself is known as Avogadro’s number (NA), after the nineteenth century scientist Amedeo Avogadro. It is actually a conversion factor between the number of moles and the number of fundamental units in a substance. NA ¼ 6:022 1023 or 6:022 1023 mol1 mol (1) The importance of the mole concept to chemistry is that it can be extended to the molecular level. If one mole of oxygen reacts with two moles of hydrogen to form two moles of water, O2 + H2 ! 2H2 O then one atom of oxygen reacts with two atoms of hydrogen to form one molecule of water. O + 2H ! H2 O The mass of one mole of a substance is called the molar mass of that substance. Since the element 12C was used to define the mole as the number of atoms in 12.000 g, which is also the atomic mass of 12C, then the molar mass of any element is equal to the atomic mass of that element in units of grams/mole. Similarly, the molar mass of any covalent compound is equal to its molecular mass (also called molecular weight) in units of grams/mole and the molar mass of an ionic compound is equal to its formula mass (or formula weight) in units of grams/mole. The molecular mass is the mass of a molecule calculated as the sum of the atomic masses multiplied by the number of atoms of each element in the molecule. For example, the molecular mass of methane with the molecular formula of CH4 is calculated as; ð12:011 amu 1Þ + ð1:008 amu 4Þ ¼ 16:043 amu Since there is no molecular formula for an ionic compound, the molar mass of an ionic compound is calculated similarly from the chemical formula as the sum of the atomic masses multiplied by the number of atoms of each element in the chemical formula. The molar mass is used to convert the mass of a substance in grams to the number of moles or to convert the number of moles of a substance to its mass in grams. For example, if the mass of substance A is known, the number of moles of substance A can be calculated by; grams A ¼ moles A molar massA g=mol (2) Similarly, if the number of moles of substance B is known, the mass of substance B can be calculated by; moles B molar mass B g=mol ¼ grams B (3) 4.2 THE EMPIRICAL FORMULA 119 EXAMPLE 4.1: DETERMINING THE NUMBER OF MOLES OF A COMPOUND FROM ITS MASS How many moles are there in 15.0 g of sodium chloride? How many moles of sodium ions are there in the same sample? How many moles of chloride ions? 1. Determine the molar mass of NaCl. The atomic mass of Na ¼ 23.0 amu, the atomic mass of Cl ¼ 35.5 amu The molar mass of NaCl ¼ 23.0 + 35.5 ¼ 58.5 g/mol 2. Convert grams of NaCl to moles of NaCl. 15:0g ¼ 0:256 moles Nacl 58:5g=mol 3. Determine the number of moles of ions. NaCl has one sodium ion and one chloride ion. 15.0 g NaCl ¼ 0.256 moles of sodium ions and 0.256 moles of chloride ions. EXAMPLE 4.2: DETERMINING THE MASS OF A COMPOUND FROM THE NUMBER OF MOLES How many grams are in 0.700 moles of hydrogen peroxide (H2O2)? 1. Determine the molar mass of H2O2: The atomic mass of H ¼ 1.01 amu, the atomic mass of O ¼ 16.0 amu. The molar mass of H2O2 ¼ (1.01 2) + (16.0 2) ¼ 34.0 g/mol 2. Convert moles to grams: 0.700 mol 34.0 g/mol ¼ 23.8 grams of H2O2 in 0.700 moles. 4.2 THE EMPIRICAL FORMULA While the molecular formula of a compound shows the number of each type of atom in a molecule, the empirical formula shows the simplest positive integer ratio of atoms present in the compound. As such, the empirical formula does not necessarily represent the actual numbers of atoms present in a single molecule of the compound. For example, the molecular formula for sulfur monoxide is SO and the molecular formula for disulfur dioxide is S2O2. However, both compounds have the same empirical formula, which is SO. So, the molecular formula can either be the same as the empirical formula (as with sulfur monoxide) or it can be a multiple of the empirical formula (as with disulfur dioxide). Since there is no molecular formula for an ionic compound, the chemical formula and the empirical formula are always the same. An empirical formula is important because it is determined from elemental composition data obtained on a compound directly by experiment. This composition data is either given 120 4. CHEMICAL EQUATIONS AND MASS BALANCE as the weight percentage of each of the elements present in the compound or as the number of grams of each element present in the sample. In either case, in order to determine the empirical formula from this experimental data, the results must first be converted to moles by using the molar mass of each element. After converting the composition values to moles, the empirical formula is then derived by determining the smallest positive whole number ratio of the moles present in the compound. This is done by dividing the number of moles of each element by the smallest molar value. If this does not result in a whole number, fractional values that are close to a whole number can be rounded to the nearest whole number. For example, 2.03 can be rounded off to 2.00 and 6.98 can be rounded up to 7.00. However, a fractional value that is greater than 0.1 or less than 0.9 must be multiplied by a small integer to obtain a whole number as shown in Table 4.1. All values must be multiplied by the same integer to get the lowest whole number multiple. Be sure to use at least three significant figures when calculating empirical formulas to minimize these round-off problems. In summary, to determine the empirical formula from compositional data: 1. Determine the number of grams of each element in the compound. If the data are given in weight percentages, assume that the total mass of the sample is 100 g. 2. Convert the mass of each element to moles using its molar mass. 3. To obtain the smallest whole number ratio, divide the number of moles of each element by the smallest molar value. 4. Round fractional values to the nearest whole number. If the fractional value is greater than 0.1 or less than 0.9, multiply each value by a small integer to get the lowest whole number multiple. TABLE 4.1 Converting Fractional Values to Whole Numbers in Empirical Formula Calculations Fractional Value Multiplier Whole Number 1.20 5 6 1.25 4 5 1.33 3 4 1.35 6 8 1.40 5 7 1.50 2 3 1.67 3 5 1.75 4 7 1.80 5 9 4.2 THE EMPIRICAL FORMULA 121 EXAMPLE 4.3: DETERMINING THE EMPIRICAL FORMULA OF A COMPOUND FROM COMPOSITIONAL DATA What is the empirical formula of a compound that has been found by experiment to contain 32.8% chromium and 67.2% chlorine. 1. Determine the number of grams of each element in the sample: In 100 g of sample, there would be 32.8 g of chromium and 67.2 g of chlorine. 2. Convert the masses of chromium and chlorine to number of moles. (32.8 g)(51.0 g/mol) ¼ 0.643 mol chromium (67.2 g)(35.5 g/mol) ¼ 1.893 mol chlorine 3. Divide each result by the smallest molar value. In this case the smallest molar value is for chromium ¼ 0.643. For chromium: (0.643)/(0.643) ¼ 1 For chlorine: (1.893)/(0.643) ¼ 2.94 3 4. The empirical formula is: CrCl3. The molecular formula of a covalent compound can be determined from the empirical formula if the molecular mass is known. If the empirical formula and the molecular formula are not the same, the actual molecular formula is some multiple (f) of the empirical formula. In order to determine the multiplication factor f, the molecular mass is divided by the empirical mass, which is calculated as the sum of the atomic masses in the empirical formula multiplied by the number of atoms of each element. f¼ Molecular mass Empirical mass (4) EXAMPLE 4.4: DETERMINING THE MOLECULAR FORMULA OF A COMPOUND FROM COMPOSITIONAL DATA The compositional analysis of an unknown compound showed it to be 82.67% carbon and 17.33% hydrogen. The molar mass was determined to be 58.11 g/mol. What is the molecular formula of the compound? 1. Convert the compositional data from grams to moles. Number of moles of carbon ¼ (82.67 g)/(12.01 g/mol) ¼ 6.88 mol Number of moles of hydrogen ¼ (17.33 g)/(1.01 g/mol) ¼ 17.16 mol 2. Determine the empirical formula. The smallest whole number ratio is 6.88 mol For carbon: (6.88 mol)/(6.88 mol) ¼ 1 For hydrogen: (17.16 mol)/(6.88 mol) ¼ 2.49 multiply by 2 to get whole numbers ¼ 2 carbons to 5 hydrogens The empirical formula is: C2H5 Continued 122 4. CHEMICAL EQUATIONS AND MASS BALANCE EXAMPLE 4.4: DETERMINING THE MOLECULAR FORMULA OF A COMPOUND FROM COMPOSITIONAL DATA— CONT’D 3. Determine the empirical mass. empirical mass ¼ (12.01 2) + (1.01 5) ¼ 29.07 4. Determine the molecular formula. The multiplication factor (f) ¼ (58.11)/(29.07) ¼ 1.99 2 The molecular formula is equal to the empirical formula 2 ¼ C4H10 CASE STUDY: THE DETERMINATION OF THE EMPIRICAL FORMULA OF A COMPOUND BY GRAVIMETRIC ANALYSIS The empirical formula of a compound can be obtained by experimental techniques that determine the masses of the elements which make up a known amount of the compound. An experimental technique that involves the quantitative determination of a substance based on the measurements of mass is known as a gravimetric analysis. This type of analysis is routinely done whenever a new compound is made or when the purity of an existing compound is in question. One of the most widely used methods for determining the amount of each element in a compound involves the conversion of the compound to other compounds of known composition. A known mass of a compound whose formula is unknown is converted to a compound or compounds whose formulas are known by chemical reactions. The weight percentage of the elements and the empirical formula of the starting compound can be determined from the masses of both the starting compound and the compounds of known composition. This approach can be used for both ionic and molecular compounds. The analysis of binary ionic compounds containing a metal and a nonmetal is often accomplished by converting the nonmetal to an insoluble compound such as AgCl or BaSO4. The insoluble compound can then be separated and the mass determined. Since the formula and molar mass of the insoluble compound is known, the mass of the nonmetal in the starting compound can be determined. The mass of the metal in the starting compound is then determined by difference. For example, an unknown compound is determined to be composed of chlorine and iron. So, the compound is either FeCl3 or FeCl2. A 0.984 g sample is dissolved in water and silver nitrate is added to the solution to form insoluble silver chloride (Fig. 4.1). After precipitation is complete, the sample is filtered and the solid is dried, weighed, and the mass found to be equal to 0.2633 g. The mass and percent of both the chloride and iron can then be calculated as: 0:2633g AgCl ¼ 1:84 103 mol AgCl ¼ 1:84 103 mol Cl 143:323g=mol 1:84 103 mol Cl 35:5g=mol ¼ 0:0653g Cl 0:0984g sample 0:0653g Cl ¼ 0:0331g Fe 123 4.2 THE EMPIRICAL FORMULA FIG. 4.1 When a solution of silver nitrate is added to a solution containing chloride, a white silver chloride solid is formed. Photograph by Luisbrunda, Wikimedia Commons. 0:0653g Cl 100 ¼ 66% Cl and 33% Fe 0:0984g sample So the empirical formula of the compound is FeCl3 The empirical formula of organic hydrocarbons, compounds that are made up of only carbon and hydrogen, is often determined by combustion analysis. As described in Section 1.6, hundreds of hydrocarbons have been isolated from crude oil. But often a single hydrocarbon is desired in its pure form. Since obtaining the pure compounds from the hydrocarbon mixtures requires many distillation steps, combustion analysis is routinely performed at each step to determine the identity and the purity of the separated compounds. A typical experimental apparatus, known as a combustion train, is shown in Fig. 4.2. A measured amount of an unknown hydrocarbon is contained within a furnace and burned in the presence of oxygen. In the furnace, all of the carbon in the sample is converted to carbon dioxide gas and all of the hydrogen in the sample is converted to water vapor. These gas phase products flow from the furnace into a chamber where H2O is absorbed by a hydrophilic substance, typically magnesium perchlorate. The CO2 then flows through a second chamber where it is absorbed by sodium hydroxide. The change in weight of each chamber is determined to FIG. 4.2 Hydrocarbon O2 O2 Furnace H2O absorber CO2 absorber Schematic diagram of a combustion train used for the determination of the amounts of carbon and hydrogen in a hydrocarbon. 124 4. CHEMICAL EQUATIONS AND MASS BALANCE calculate the mass of H2O and CO2 absorbed. After the masses of H2O and CO2 have been determined, they can be used to calculate the weight percentage of the elements and the empirical formula of the hydrocarbon in the original sample. (See Fig. 4.1.) The combustion of a generic hydrocarbon with the formula CaHb would yield; Ca Hb + O2 ! aCO2 + b=2 H2 O The number of moles of CO2 and H2O produced in the reaction can be calculated from the masses of CO2 and H2O measured in the absorption chambers divided by their molar masses. Since one mole of CO2 is produced for every mole of carbon atoms in the original sample, the number of moles of carbon (a) in the original sample is equal to the number of moles of CO2 produced. Similarly, one mole of H2O is produced for every two moles of hydrogen atoms in the original sample. So, the number of moles of hydrogen (b) in the original sample is equal to the number of moles of H2O produced multiplied by 2. From this information, the empirical formula of the original hydrocarbon can be determined. For example, in a controlled fractional distillation for the purification of hexane from a petroleum distillate, the hydrocarbon produced from the distillation is analyzed by combustion to determine the purity of the product. The amount of hydrocarbon placed in the furnace is 0.5000 g and the amount of CO2 and H2O recovered on the absorbers is 1.5315 and 0.7316 g. 1:5315g CO2 ¼ 0:0348mol CO2 ¼ 0:0348mol C in the original sample 44:01g=mol 0:7316g H2 O ¼ 0:0404mol H2 O ¼ 0:0808mol H in the original sample 18:02g=mol Dividing each by the smallest molar value gives 1 CO2 to 2.32 H2O. According to Table 4.1, a multiplier of 3 is required to obtain whole numbers. So, a ¼ 3 and b ¼ 7 and the empirical formula for the hydrocarbon is C3H7. This corresponds exactly to the molecular formula of hexane (C6H14) and the purity of the product is confirmed. If the result had been far from C3H7, then the sample is determined to be impure and further separation would be required. 4.3 CHEMICAL EQUATIONS A chemical equation is the symbolic representation of a chemical reaction using symbols of the elements and chemical formulas. These equations are written according to the following format: 1. The reactants, the substances that are present before the chemical change takes place, are written on the left side of the equation and the products, the species formed from the chemical reaction, are written on the right side of the equation. The reactants and products are separated by an arrow which indicates the direction of the reaction. If there are multiple products and reactants, a plus sign (+) is placed between them. A+B!C+D 4.3 CHEMICAL EQUATIONS 125 2. Small whole numbers are placed in front of the symbols and formulas to show the number of units (atoms, molecules, ions, moles) that react or are produced. When no number is shown, one unit is implied. 2A + 3B ! 2C + D 3. The physical state of each substance can be indicated by the following symbols: s ¼ solid state , l ¼ liquid state , g ¼ gaseous state, aq ¼ aqueous solution. The symbols are written in parentheses following the symbol or formula of the substance. AðsÞ + BðlÞ ! C g + DðsÞ Chemical reactions involve change. Elements are combined into compounds, compounds are transformed into new compounds or are decomposed back into elements. During chemical reactions, bonds are broken and new bonds are formed. However, the number of atoms of each element must be present before and after any chemical reaction. The principle that atoms are neither created nor destroyed in a chemical reaction is known as the conservation of mass. The conservation of mass is represented in a chemical equation by a process called “balancing the equation.” This is done by adjusting the coefficients so that the number of atoms of each element on the right side of the equation equals the number of atoms of the same element on the left side of the equation. For example, consider the chemical equation for the combustion of methane (CH4). Writing the reaction as shown in the previous Case Study where a ¼ 1 and b ¼ 4 gives; CH4 + O2 ! CO2 + 2H2 O This equation is not balanced because there are two oxygen atoms on the right side of the equation and four oxygen atoms on the left side of the equation. So, there needs to be a coefficient of 2 in front of O2. The equation then becomes; CH4 + 2O2 ! CO2 + 2H2 O (5) Now there are four oxygen atoms on the left side of the equation and four oxygen atoms on the right side of the equation (two in CO2 and two in 2H2O). There are also four hydrogen atoms on the left side and four hydrogen atoms on the right side as well as one carbon atom on the left side and one carbon atom on the right side. So, none of the atoms are created or destroyed in the reaction and the equation is now balanced. EXAMPLE 4.5: BALANCING A CHEMICAL EQUATION Balance the chemical equation for the incomplete combustion of methane, which gives the products CO and H2O. The balanced chemical Eq. (5) is for the complete combustion of methane. This occurs only when there is sufficient oxygen to convert all the carbon atoms in the hydrocarbon to carbon dioxide. When there is not enough oxygen present, the combustion will be incomplete giving the products carbon monoxide and water. Continued 126 4. CHEMICAL EQUATIONS AND MASS BALANCE EXAMPLE 4.5: BALANCING A CHEMICAL EQUATION— CONT’D 1. Write the unbalanced equation using the correct chemical formulas for the reactants and product. CH4 + O2 ! CO + H2 O 2. Begin by inserting coefficients to balance an atom that appears in the fewest formulas. Hydrogen and carbon appear in the fewest formulas, but carbon is already balanced. So, insert coefficients that will balance the hydrogen atoms: CH4 + O2 ! CO + 2H2O Now hydrogen has four atoms on the right side and four atoms on the left side. 3. Insert coefficients to balance the remaining atoms. The remaining unbalanced atom is oxygen. Oxygen has two atoms on the left side and three atoms on the right side. Whenever there is a combination of two atoms on one side of the equation and three of the same atoms on the other side of the equation, use coefficients that will give six atoms on each side of the equation. Use coefficients of 3 for O2, 2 for CO, and 4 for H2O: CH4 + 3O2 ! 2CO + 4H2 O Now there are six oxygen atoms on the left side (3O2) and six oxygen atoms on the right side (2CO + 4H2O). 4. Check to see if any of the atoms has become unbalanced. Hydrogen now has four atoms on the left side and eight atoms on the right side. Also, carbon has one atom on the left side and two atoms on the right side. This unbalancing is resolved by using a coefficient of 2 for methane: 2CH4 + 3O2 ! 2CO + 4H2O Now the reactants ¼ 2C, 8H, 6O and the products ¼ 2C, 8H, 6O. The equation is balanced. Always use the smallest possible whole number coefficients when balancing chemical equations. Chemical equations are balanced by using only the coefficients in front of the chemical formulas of the compounds. Never try to balance an equation by changing the subscripts within a chemical formula. Also, when balancing an equation containing polyatomic ions, the polyatomic ions are treated as the same as individual atoms if the same polyatomic ions appear on both sides of the equation. Balance the equation by balancing the ions beginning with the nonpolyatomic ions followed by balancing the polyatomic ions. For example, the reaction of ammonium phosphate with barium hydroxide to give ammonium hydroxide and barium phosphate ðNH4 Þ3 PO4 + BaðOHÞ2 ! NH4 OH + Ba3 ðPO4 Þ2 has one Ba2+ on the right and three Ba2+ on the left, and so, begin to balance the equation by adding a coefficient of 3 in front of barium on the left. ðNH4 Þ3 PO4 + 3BaðOHÞ2 ! NH4 OH + Ba3 ðPO4 Þ2 4.4 STOICHIOMETRY 127 Balance the OH by placing a 6 in front of Ba(OH)2. ðNH4 Þ3 PO4 + 3BaðOHÞ2 ! 6NH4 OH + Ba3 ðPO4 Þ2 Next balance the PO43 by placing a 2 in front of (NH4)3PO4. 2ðNH4 Þ3 PO4 + 3BaðOHÞ2 ! 6NH4 OH + Ba3 ðPO4 Þ2 The equation is now balanced. 4.4 STOICHIOMETRY The relationship between the amounts of reactants and products in a chemical reaction is called stoichiometry and the coefficients in a balanced chemical equation are called stoichiometric coefficients. The stoichiometric coefficients can be interpreted as the number of atoms or molecules that are consumed and produced in the chemical reaction. They can also be interpreted as the number of moles of each reactant and product that are consumed and produced in the chemical reaction. For example, the balanced chemical equation for the complete combustion of methane in Eq. (5) can mean that one molecule of methane reacts with two molecules of oxygen to yield one molecule of carbon dioxide and two molecules of water. It also means that one mole of methane reacts with two moles of oxygen to yield one mole of carbon dioxide and two moles of water. For practical purposes, it is the masses of the reactants and products that are most useful because these are the quantities that can be measured directly. The stoichiometric coefficients can also be used to determine the masses of the substances consumed or produced in a chemical reaction. Multiplying the number of moles by the molar mass of each substance gives the mass of each substance consumed or produced. For the complete combustion of one mole of methane; 16:04g CH4 + 2 16:00g O2 ! 44:01g CO2 + 2 18:015g H2 O 16:04g CH4 + 32:00g O2 ! 44:01g CO2 + 36:03g H2 O So, the mole ratios in a balanced chemical equation obtained from the stoichiometric coefficients allow for the calculation of the amount or mass of one substance in a chemical reaction when given the mass or amount of another substance in the chemical reaction. EXAMPLE 4.6: DETERMINING THE AMOUNT AND MASS OF REACTANTS AND PRODUCTS IN A CHEMICAL REACTION If 0.400 mol of methane undergo complete combustion in the presence of oxygen, what is the mass of water formed? What is the mass of oxygen consumed? 1. Determine the balanced chemical equation: CH4 + O2 ! CO2 + 2H2 O Continued 128 4. CHEMICAL EQUATIONS AND MASS BALANCE EXAMPLE 4.6: DETERMINING THE AMOUNT AND MASS OF REACTANTS AND PRODUCTS IN A CHEMICAL REACTION— CONT’D 2. Calculate the number of moles of water formed using the mole ratios in the balanced equation. Two moles of water are formed from one mole of methane, so the mole ratio of water to methane is 2/1: 0:400mol CH4 2mol H2 O ¼ 0:800mol H2 O produced 1mol CH4 3. Calculate the mass of water formed. 0:800mol H2 O 18:015g=mol ¼ 14:4g H2 O produced 4. Calculate the number of moles of oxygen consumed using the mole ratios in the balanced equation. One mole of oxygen is consumed for every one mole of methane, so the mole ratio of oxygen to methane is 1/1: 0:400mol CH4 1mol O2 ¼ 0:400mol O2 consumed 1mol CH4 5. Calculate the mass of oxygen consumed. 0:400mol O2 16:0g=mol ¼ 6:40g O2 consumed 4.5 LIMITING REACTANT AND PERCENT YIELD Chemical reactions are usually carried out with one reactant “in excess” (more than the stoichiometric amount). This is done to assure that the critical reactant is consumed completely. For example, in the combustion analysis of hydrocarbons for the purpose of determining the empirical formula, it is critically important that the entire hydrocarbon sample be combusted. Otherwise the masses of carbon dioxide and water determined after combustion will yield incorrect results. In order for complete combustion to be guaranteed, the oxygen must be in an excess amount. Then the amount of products formed is not determined by the amount of oxygen; it is determined by the amount of hydrocarbon. The hydrocarbon is the limiting reactant because its amount determines (or limits) the amount of the products. Any increase in the amount of the limiting reactant leads to an increase in the amount of products, while an increase in the excess reactant will not affect the amount of product because there is already enough present to consume all of the limiting reactant. There are several practical reasons why most chemical reactions have a limiting reactant. In gravimetric analysis as well as other analytical techniques, the reactions are purposely set up with the analyte, the substance whose chemical constituents are being identified and 129 4.5 LIMITING REACTANT AND PERCENT YIELD measured, as the limiting reactant. This assures that it is completely consumed in the reaction in order to yield accurate masses of the products. In industrial processes, the limiting reactant is often the most expensive or least abundant substance. Other less expensive or more plentiful reactants are present in excess to assure that none of the expensive reactant is wasted. A limiting reactant can also occur if the reactants are mixed in arbitrary or nonstoichiometric amounts. In this case, the limiting reactant must be identified in order to determine the maximum possible amounts of the products formed. EXAMPLE 4.7: DETERMINING THE LIMITING REACTANT The first step of steel production is to heat solid iron(III) oxide with solid carbon to form elemental iron and carbon dioxide gas by the following balanced equation; 2Fe2 O3 + 3C ! 4Fe + 3CO2 What mass of iron is produced if 150 g of Fe2O3 is added to 30.0 g of carbon? What is the mass of the excess reactant after the reaction is completed? 1. Determine the number of moles of each reactant. 150g Fe2 O3 = 159:7g=mol ¼ 0:939mol Fe2 O3 30:0g C = 12:0g=mol ¼ 2:50mol C 2. Determine the limiting reactant. The stoichiometric mole ratio is: 2mol Fe2 O3 0:667mol Fe2 O3 or 3mol C 1mol C The current mole ratio is: 0:939molFe2 O3 0:376molFe2 O3 or 2:50molC 1molC Since the amount of Fe2O3 is much smaller than the stoichiometric amount. Fe2O3 is the limiting reactant. 3. Calculate the number of moles of iron formed based on the amount of the limiting reactant. 0:939molFe2 O3 4molFe ¼ 1:88molFe formed 2molFe2 O3 4. Calculate the mass of iron formed. 1:88mol Fe 55:9g=mol ¼ 105g Fe formed 5. Determine the mass of carbon left after the reaction is complete. First calculate the amount of carbon required to complete the reaction of Fe2O3 using the stoichiometric mole ratios: Continued 130 4. CHEMICAL EQUATIONS AND MASS BALANCE EXAMPLE 4.7: DETERMINING THE LIMITING REACTANT— CONT’D 0:939mol Fe2 O3 3mol C ¼ 1:41mol C required 2mol Fe2 O3 Amount of carbon left: 2.50 mol C 1.41 mol C ¼ 1.09 mol C left Mass of carbon left: 1.09 mol C 12.0 g/mol ¼ 13.1 g C remaining The maximum mass of products that can be produced from a chemical reaction is the theoretical yield for that reaction. The theoretical yield can only be achieved if the reaction goes completely according to the stoichiometry of the reaction. The actual yield, the mass of products actually obtained from a chemical reaction in a laboratory or industrial process, is almost always less than the theoretical yield. There are several reasons for this. The reaction may not go completely to the products, but may stop before completion leaving unconsumed reactants or the products may decompose back to reactants. Unexpected reactions may sometimes occur which give products other than those expected. Also, the processes of separation and purification of the products almost always leads to product loss. The difference between the theoretical yield and the actual yield can be expressed as a percent yield. The percent yield specifies how much of the theoretical yield was actually obtained. It is calculated as the ratio of the actual yield to the theoretical yield multiplied by 100. Percent yield ¼ Actual yield 100 Theoretical yield (6) In industrial processes, it is important to achieve the highest percent yield possible in order to maximize the desired product and minimize the amount of reactants consumed. This in turn reduces the cost of the process. It also reduces the amount of undesired, and sometimes environmentally hazardous, products. Many times, industrial processes require several successive reactions followed by several purification steps to achieve a desired product. Determining the percent yield at each step can help to identify inefficiencies and increase the overall product yield giving a higher profit margin. CASE STUDY: PERCENT YIELD AND ATOM ECONOMY IN THE STEEL INDUSTRY The industrial production of steel begins with the production of iron from iron ore. The most common iron ore used is hematite (iron(III) oxide). The ore is converted to the pure metal in a reaction with carbon at high temperatures. The reaction is typically conducted in a blast furnace shown in Fig. 4.3 at temperatures from about 1500 to 2000°C. Carbon is added to the furnace in the form of coke, which is derived from decomposition of low ash, low sulfur coal. The process also requires a flux such as limestone, which is used to remove silicate minerals in the ore. The iron ore, coke, and limestone are fed into the top of the furnace, while a blast of superheated air is forced into the bottom of the furnace. The downward moving column of the reactants must be porous enough for the heated gas to pass through. This requires the solid particles to be large enough to be gas-permeable. 4.5 LIMITING REACTANT AND PERCENT YIELD Iron ore, coke, limestone 131 FIG. 4.3 Schematic diagram of a blast furnace. Adapted from Robert Blazek, Wikimedia Commons. Gas collection Exhaust gases Preheating zone Column of ore, coke Reaction of Fe2O3 Reaction of FeO Fe + slag Melting zone Hot air blast Hot air blast Slag removal Fe collection The oxygen in the heated blast air reacts with the carbon in the coke to produce carbon monoxide. 2CðsÞ + O2 g ! 2CO g The hot carbon monoxide then reacts with the iron ore (Fe2O3). Since the temperature varies in the different parts of the furnace, with the hottest at the bottom and the coolest at the top, the Fe2O3 reacts in several steps as it falls through the column. At the top of the furnace, where the temperature is usually in the 200–700°C range, the Fe2O3 reacts partially with the carbon monoxide to form Fe3O4 and carbon dioxide; 3Fe2 O3 ðsÞ + CO g ! 2Fe3 O4 ðsÞ + CO2 g Further down the furnace, at temperatures around 850°C, the Fe3O4 reacts further with carbon monoxide to give iron(II) oxide (FeO) and carbon dioxide; Fe3 O4 ðsÞ + CO g ! 3FeOðsÞ + CO2 g As the FeO moves down the furnace to the area where temperatures are highest, it reacts further with carbon monoxide to form molten iron metal. FeOðsÞ + CO g ! FeðsÞ + CO2 g At these highest temperatures, the limestone flux is decomposed to calcium oxide and carbon dioxide. CaCO3 ðsÞ ! CaOðsÞ + CO2 g The calcium oxide formed from this decomposition reacts with silicate impurities in the iron ore to form a calcium silicate slag (Fig. 4.4). 132 4. CHEMICAL EQUATIONS AND MASS BALANCE FIG. 4.4 Slag runoff from a blast furnace (Bundesarchiv, Wikimedia Commons). SiO2 + CaO ! CaSiO3 The gaseous products of these reactions, hot carbon dioxide, unreacted carbon monoxide, and nitrogen from the air pass up through the furnace and are collected at the top. It is easy to see how the determination of percent yield for this process can be difficult. The carbon monoxide produced from the coke is in excess, so the Fe2O3 is the limiting reactant. Since the overall reaction; Fe2 O3 ðsÞ + 3CO g ! 2FeðlÞ + 3CO2 g actually occurs in three steps; the different reactions that occur at the varying temperatures in the furnace will have different actual yields. In addition, some of the iron that is produced as well as unconsumed reactants are carried out with the slag and lost. The iron produced is called pig iron because it contains 4%–5% of unreacted carbon. This carbon content is too high for use in steel production. So, further processing must be done to reduce the carbon levels in the pig iron. This would further reduce the actual product yield. While the concept of percent yield is useful in determining the amount of product that can actually be obtained from the given reactants, it does not give the whole picture of reaction efficiency. It ignores the production of waste products that can be costly or environmentally hazardous. A reaction can have a high percent yield, but result in a lot of waste products. A major concept of both Green Chemistry and Green Engineering encourages process designs that assure the maximum amount of all reactants ending up in the desired product while a minimum amount of waste is produced. With these goals of increasing product yield while at the same time reducing waste and increasing sustainability, industrial processes have begun to include the concept known as atom economy along with that of percent yield. Atom economy is the conversion efficiency of a chemical reaction in terms of all the atoms involved. It is calculated as the ratio of the formula weight of the desired product(s) to the total formula weight of all the reactants multiplied by 100. 4.6 AQUEOUS SOLUBILITY OF IONIC COMPOUNDS Atom economy ð%Þ ¼ Formula weight of desired productðsÞ 100 Formula weight of all reactants 133 (7) The atom economy of the overall reaction for the conversion of iron ore to pig iron in a blast furnace is 45.8%. Any reaction that has an atom economy less than 50% is considered to be poor. In addition, this calculation does not take into account the excess reagents used to drive the reactions to completion or the production of waste slag from the limestone flux. Including these waste products would greatly reduce the atom economy of the blast furnace process. One of the biggest problems with the use of blast furnaces is the high levels of carbon dioxide produced. Three moles of CO2 are produced for every two moles of iron. In addition, CO2 is also formed from the decomposition of the limestone flux. Currently, there are no known reactions that can convert iron ore to pig iron with a better atom economy on as high a production scale as the blast furnace. However, some suggested design modifications may result in reductions in energy costs as well as environmental impacts. For example, it might be possible to further intensify the smelting operation through the use of oxygen instead of air, increasing the pressure in the top of the furnace and recycling waste gases back into the furnace. The carbon dioxide formed in this process could be reconverted to carbon monoxide by the coke under high pressure and temperature, thus reducing the amount of coke and CO required. 4.6 AQUEOUS SOLUBILITY OF IONIC COMPOUNDS An aqueous solution is a solution where water is the solvent. It is indicated by the abbreviation (aq) following the chemical formula of the solute in the chemical equation. Ionic compounds behave differently in aqueous solution from molecular compounds containing only covalent bonds. Covalent compounds exist as individual molecules both in the pure state (solid or liquid) and in aqueous solution. However, as described in Chapter 3, solid ionic compounds are made up of cations and anions held tightly together in a three-dimensional structure. Dissolving an ionic compound in water involves breaking the ionic bonds between each ion and the oppositely charged ions surrounding it. Water is particularly good at accomplishing this because of its polarity and its ability to hydrogen bond. Since water is a polar molecule, it has a permanent dipole, as described in Section 3.8. The oxygen atom has a partial negative charge (δ ) and the hydrogen atoms have a partial positive charge (δ+). When an ionic compound is dissolved in water, the water dipoles attract the positive and negative ions releasing them from the solid structure, a process called solvation. The anions become surrounded by the Hδ+ of the water molecules, while the cations become surrounded by the Oδ of the water molecules. The attraction between the ions and the partial charges on the water dipoles take the place of the oppositely charged ions in the solid. This solvation shell of water molecules surrounding the ions in solution reduces the attractions between the ions and they are said to be “solvated.” The ions and their solvation shell of water molecules are held together by ion-dipole forces. This allows the ions to move freely in the aqueous environment (Fig. 4.5). 4. CHEMICAL EQUATIONS AND MASS BALANCE FIG. 4.5 A schematic diagram of an ionic compound, such as sodium chloride, dissolved in water. When an ionic compound is dissolved in water, each ion is surrounded by water molecules with the oxygen (δ) atoms closest to the cations and the hydrogen (δ+) atoms closest to the anions. This allows the ions to move about freely in the solution. H H H H H O H H O H + O H O H H H O H H H H H O H H O H – H O O H – H H O H O + H O H H O H O H H O O H H O H H O H H O H – H O + H O H H H H 134 H O H O Many ionic compounds dissolve completely in water; some dissolve only to a small extent, and others are essentially insoluble. In general, the solubility of an ionic compound in water is determined by the strength of the bonds between the ions in the solid. The stronger the bonds are between the oppositely charged ions, the lower the solubility of the compound. As shown in Chapter 3, this ionic bond strength is directly proportional to the magnitudes of the charges on the ions. The greater the ionic charges on the ions, the stronger the ionic bonds will be. So, the greater the ionic charges, the lower the solubility of the ionic compound. Using this generality, the following set of rules can be used as guidelines to predict whether an ionic compound will be soluble or insoluble in water: 1. Compounds containing an ion with a 1+ or 1 charge are soluble. Exceptions: Halides of Ag+, Hg22+, Pb2+ are insoluble. Fluorides of Mg2+, Ca2+, Sr2+, Ba2+, Pb2+ are insoluble. 2. Compounds containing an ion with a 3+, 3 , charge or higher are insoluble. Exceptions: Cations combined with SO42, or Cr2O72 are soluble. 3. Compounds containing ions with a 2 charge are normally insoluble. Exceptions: Cr2O72, all sulfates except Sr2+, Ba2+, Pb2+, Ca2+, Hg22+ are soluble. The solubility rules above are listed in an order of decreasing importance. For example, if rule #1 applies, it has priority over rules 2 and 3. Similarly, if rule # 2 applies, it has priority over rule #3. Rule #3 applies only if both rules #1 and #2 do not apply. These rules should be considered only as guidelines, as they list only the more common ionic compounds. There are other exceptions to the solubility rules not listed above that apply to less common ionic compounds. 4.7 PRECIPITATION REACTIONS IN AQUEOUS SOLUTION 135 EXAMPLE 4.8: DETERMINING THE AQUEOUS SOLUBILITY OF IONIC COMPOUNDS Determine the solubility of the following compounds: (a) Fe2(SO4)3, (b) MgSO4, (c) Ba(OH)2, (d) AgCl, (e) Ba(NO3)2, and (f) BaSO4. 1. Determine the charges on the ions: (a) Fe2(SO4)3 ¼ Fe3+ and SO42, (b) MgSO4 ¼ Mg2+ and SO42 (c) Ba(OH)2 ¼ Ba2+ and OH, 2+ (d) AgCl ¼ Ag+ and Cl, (e) Ba(NO3)2 ¼ Ba2+ and NO and SO42. 3 , and (f) BaSO4 ¼ Ba 2. Determine which rule and/or exception applies. (a) Fe2(SO4)3; Rule 2, exception; soluble, (b) MgSO4; Rule 3, exception; soluble, (c) Ba(OH)2; Rule 1; soluble, (d) AgCl; Rule 1, exception insoluble, (e) Ba(NO3)2; Rule 1; soluble, and (f) BaSO4; Rule 3; insoluble. 4.7 PRECIPITATION REACTIONS IN AQUEOUS SOLUTION Chemical reactions that occur in aqueous solution are some of the most important reactions in chemistry. There are two types of reactions that occur in aqueous solutions: ion exchange reactions and electron transfer reactions. In electron transfer reactions, electrons are transferred from one species to another to form new species. Electron transfer reactions, also called oxidation-reduction reactions, are covered in detail in Chapter 10. In ion exchange reactions, the ions are exchanged from one ionic compound to another to form new compounds. Ion exchange reactions include precipitation reactions and acid–base reactions. Acid–base reactions are covered in detail in Chapter 5. Here we will focus on precipitation reactions. Ion exchange reactions involve an exchange of cations and anions of the ionic compounds in aqueous solution. So, in effect, the ions of the original compounds “change partners” such as; A + B ðaqÞ + C + D ðaqÞ ! A + D ðaqÞ + C + B ðsÞ (8) A precipitation reaction occurs when two different ionic salts are mixed together in aqueous solution and a cation from one compound combines with an anion from the other compound to form an insoluble ionic salt. The insoluble product is called the precipitate and the liquid phase is called the supernate. The precipitate can be separated from the supernate by sedimentation or centrifugation followed by decanting or it can be separated by filtration as described in Section 1.6. The balanced chemical equation shown in Eq. (8), which shows all the ionic compounds participating in the reaction, is known as the formula equation. The formula equation does not, however, show the ions present in the solution. In order to more completely describe the aqueous reaction, an equation can be written that fully shows all the dissolved ions in solution, such as; A + ðaqÞ + B ðaqÞ + C + ðaqÞ + D ðaqÞ ! A + ðaqÞ + D ðaqÞ + C + B ðsÞ (9) 136 4. CHEMICAL EQUATIONS AND MASS BALANCE In this equation, C+ B is determined to be the insoluble compound by the solubility rules listed in Section 4.5. This equation, which shows all the dissolved ionic species present during the reaction plus any insoluble ionic compound present as a solid, is called the complete ionic equation. In the complete ionic equation, there are several ions that appear in the same form on both sides of the equation. These ions are unchanged during the reaction and are called spectator ions (i.e., they don’t participate, they only sit and watch). Little chemical information is lost by omitting them from the equation altogether. So, the ionic equation can be simplified by not including the spectator ions. In the above complete ionic equation, A+(aq) and D(aq) occur on both sides of the equation and so are spectator ions and can be omitted from the chemical equation. The ionic equation then becomes; B ðaqÞ + C + ðaqÞ ! + C + B ðsÞ (10) The ionic equation that omits the spectator ions and only shows those chemical species participating in the chemical reaction is called the net ionic equation. An example of a precipitation reaction is the reaction of silver nitrate with sodium chloride to yield soluble sodium nitrate and insoluble silver chloride. In this reaction, the NO 3 anion trades places with the Cl anion to form an insoluble compound with silver. The formula equation for this reaction is; AgNO3 ðaqÞ + NaClðaqÞ ! AgClðsÞ + NaNO3 ðaqÞ The complete ionic equation for this reaction is; Ag + ðaqÞ + NO3 ðaqÞ + Na + ðaqÞ + Cl ðaqÞ ! AgClðsÞ + Na + ðaqÞ + NO3 ðaqÞ In the complete ionic equation, the sodium cations and the nitrate anions appear on both sides of the equation unchanged. So, they are spectator ions and can be omitted from the net ionic equation, which is; Ag + ðaqÞ + Cl ðaqÞ ! AgClðsÞ If all of the products on the right side of the equation are soluble and remain in the same ionic form as the reactants, no chemical reaction has occurred and all the ions are considered to be spectator ions. For example, when calcium chloride and magnesium nitrate are dissolved in water, the formula equation is; CaCl2 ðaqÞ + MgðNO3 Þ2 ðaqÞ ! CaðNO3 Þ2 ðaqÞ + MgCl2 ðaqÞ The reactants and products are all soluble ionic compounds, so the complete ionic equation is; Ca2 + ðaqÞ + 2Cl ðaqÞ + Mg2 + ðaqÞ + 2NO3 ðaqÞ ! Ca2 + ðaqÞ + 2NO3 ðaqÞ + Mg2 + ðaqÞ + 2Cl ðaqÞ All ions appear in the same form on both sides of the equation, so they are all spectator ions and are removed to yield no net ionic equation. This means that no chemical reaction takes place between CaCl2 and Mg(NO3)2. This is often indicated by the designation; “NR.” 4.7 PRECIPITATION REACTIONS IN AQUEOUS SOLUTION 137 EXAMPLE 4.9: DETERMINING THE NET IONIC EQUATION FOR PRECIPITATION REACTIONS What is the net ionic equation for the reaction of sodium phosphate and calcium chloride? 1. Determine the balanced formula equation. (Hint: Balance the PO43 ion first) 2Na3(PO4) + 3CaCl2 ! 6NaCl + Ca3(PO4)2 2. Determine the insoluble species. Ca3(PO4)2 is insoluble according to Rule 1, exception. 3. Determine how many ions are present in solution from each compound: 2Na3(PO4) ¼ 6Na+ and 2PO43, 3CaCl2 ¼ 3Ca2+ and 6Cl, 6NaCl ¼ 6Na+ and 6Cl Ca3(PO4)2 ¼ no ions in solution. 4. Write the complete ionic equation including all ions and their states. 6Na + ðaqÞ + 2PO43 ðaqÞ + 3Ca2 + ðaqÞ + 6Cl ðaqÞ ! 6Na + ðaqÞ + 6Cl ðaqÞ + Ca3 ðPO4 Þ2 ðsÞ 5. Identify the spectator ions. 6Na+ and 6Cl 6. Write the net ionic equation omitting the spectator ions. PO43(aq) + 3Ca2+(aq) + ! Ca3(PO4)2(s) CASE STUDY: PRECIPITATION REACTIONS IN WATER TREATMENT Hard water is water that has a high concentration of cations with a 2 + charge, primarily calcium and magnesium. The Ca2+ and Mg2+ ions enter the water supply by leaching from subsurface minerals in the aquifer such as calcite (CaCO3) and gypsum (CaSO4) in the case of Ca2+ or dolomite [CaMg(CO3)2] in the case of Mg2+. Although these compounds are listed as insoluble in water (Rule 3, Section 4.5), their dissolution can be promoted by reaction with dissolved CO2 in the water. In the case of CaCO3, the reaction is; CaCO3 ðsÞ + CO2 ðaqÞ + H2 OðlÞ ! Ca2 + ðaqÞ + 2HCO3 ðaqÞ These ions do not pose a health threat, but they are the main reactant in aqueous chemical reactions that leave behind off white, chalky, insoluble mineral deposits called “limescale.” Limescale is made up of calcium and magnesium carbonate, which are formed by a series of reactions involving dissolved carbon dioxide, bicarbonate, and carbonate. The buildup of limescale can restrict the flow of water in pipes (Fig. 4.6) and is responsible for the calcium deposits often seen on water faucets. 138 4. CHEMICAL EQUATIONS AND MASS BALANCE FIG. 4.6 Limescale buildup inside a heat exchanger pipe reduces liquid flow through the pipe reducing the heating efficiency. Photograph by Александр Юрьевич Лебедев, Wikimedia Commons. In boilers, limescale deposits act as an insulator, impairing the flow of heat into water and causing the metal boiler components to overheat. In a pressurized system, this overheating can lead to failure of the boiler. So, in industrial settings water hardness is closely monitored to avoid costly breakdowns in boilers, cooling towers, and other equipment that handle large amounts of water. The 2 + cations in hard water can also react with other substances, such as soap. Soap solutions in hard water form a white precipitate known as “soap scum” instead of producing suds. This occurs because the 2 + cations undergo an ion exchange reaction with the ionized fatty acids in the soap (C17H35COO Na+). This destroys the sudsing properties of the soap by forming the insoluble precipitate [(C17H35COO)2Ca]; 2C17 H35 COO Na + ðaqÞ + Ca2 + ðaqÞ ! ðC17 H35 COOÞ2 CaðsÞ + 2Na + ðaqÞ In fact, water hardness can be measured as the soap consuming capacity of a water sample, or the capacity to form the insoluble precipitate in a soap solution. Because of these problems involved with the use of hard water, a variety of methods have been developed to remove the calcium and magnesium ions in order to “soften the water.” One of the methods used in large scale municipal and industrial operations is known as the “lime soda process.” This process, shown schematically in Fig. 4.7, involves ion exchange reactions, which result in the formation of insoluble precipitates. The hard water is first treated with Ca(OH)2 (slaked lime ), which removes the dissolved CO2 from the water in order to prevent any redissolution of the Ca2+ in step 2. CaðOHÞ2 ðaqÞ + CO2 ðaqÞ ! CaCO3 ðsÞ + H2 OðlÞ 2+ At the same time, the Mg is removed by ion exchange with the Ca(OH)2, forming the insoluble precipitate Mg(OH)2. The formula equation for this precipitation reaction is; CaðOHÞ2 ðaqÞ + MgCl2 ðaqÞ ! MgðOHÞ2 ðsÞ + CaCl2 ðaqÞ 139 4.8 CONCENTRATIONS IN AQUEOUS SOLUTION Ca(OH)2(aq) NaCO3(aq) Sedimentation Hard water Sedimentation Filtration Rapid mix Rapid mix Mg(OH)2(s) Softened water CaCO3(s) FIG. 4.7 Schematic diagram of the lime soda process for removing calcium and magnesium ions from hard water. The complete ionic equation is; Ca2 + ðaqÞ + 2OH ðaqÞ + Mg2 + ðaqÞ + 2Cl ðaqÞ ! MgðOHÞ2 ðsÞ + Ca2 + ðaqÞ + 2Cl ðaqÞ Omitting the spectator ions, the net ionic equation is; 2OH ðaqÞ + Mg2 + ðaqÞ ! MgðOHÞ2 ðsÞ All the Ca2+ ions are then removed by precipitation using Na2CO3 (soda ash); Na2 CO3 ðaqÞ + CaCl2 ðaqÞ ! CaCO3 ðsÞ + 2NaClðaqÞ The complete ionic equation for this reaction is; 2Na + ðaqÞ + CO3 2 ðaqÞ + Ca2 + ðaqÞ + 2Cl ðaqÞ ! CaCO3 ðsÞ + 2Na + ðaqÞ + 2Cl ðaqÞ The net ionic equation is; CO3 2 ðaqÞ + Ca2 + ðaqÞ ! CaCO3 ðsÞ The lime soda process is also effective at removing bacteria and particulate organic matter from the water by flocculation, a process in which the fine particles of the precipitate aggregate to form larger particles. As the particles of the precipitate come together to form larger particles, other particles in the water, such as bacteria and particulate organic matter, become entrained in the mass of the precipitate and are carried out with the solid. 4.8 CONCENTRATIONS IN AQUEOUS SOLUTION Chemical reactions in living systems, as well as chemical reactions in the laboratory and industrial processes, generally take place in aqueous solution. An aqueous solution consists of at least two components, the solvent (water) and the solute. The chemical properties and behavior of solutions are determined by their composition. There are several methods used to 140 4. CHEMICAL EQUATIONS AND MASS BALANCE specify the composition of a solution. These will all be discussed in detail in Chapter 12, but the most useful method in chemistry is to define the concentration of the solute in the solution as the amount of solute per unit volume of the total solution. The SI units of concentration would be; moles per cubic meter (mol/m3). However, since 1 m3 ¼ 0.001 L, this unit is inconvenient for practical use in laboratory chemical reactions. Instead, the concentration unit of molarity (M) is used. Molarity is defined as the number of moles of solute per liter of solution. Molarity ðMÞ ¼ moles of solute ðmolÞ liters of solution ðLÞ (11) It is important to note that the molarity of a solution is defined as the number of moles of solute per liter of solution, not as the number of moles of solute per liter of solvent. This is because the solute adds volume to the solution when it is added to a measured volume of water. So, when you add a chemical substance to water, the volume of the resulting solution will be different than the original volume of the water. To avoid this problem, chemists make up solutions in volumetric flasks, which are very accurately calibrated to contain a precise volume at a particular temperature. The solute is added to the flask first and then water is added until the solution reaches the calibrated etched mark on the neck of the flask (Fig. 4.8). FIG. 4.8 A 500 mL volumetric flask. To make 500 mL of an aqueous solution, a measured amount of solute is added to the flask. Then enough water is added to fill the flask just to the blue mark on the neck of the flask. Photograph by Hannes Grobe, Wikimedia Commons. Fill line IMPORTANT TERMS 141 EXAMPLE 4.10: DETERMINING THE CONCENTRATION OF A SOLUTION What is the concentration of 100 mL of solution containing 25.0 g of silver nitrate? 1. Determine the amount of silver nitrate. 25:0g ¼ 0:147mol AgNO3 170g=mol 2. Convert volume of solution to liters. 0:147mol ¼ 1:47M 0:100L IMPORTANT TERMS Actual yield the mass of products actually obtained from a chemical reaction. Amount of substance a measure of the number of fundamental particles, such as atoms, molecules, or ions that are present in a given mass of substance. Analyte a substance whose chemical constituents are being identified and measured. Aqueous solution a solution where water is the solvent. Atom economy the ratio of the formula weight of the desired product(s) to the total formula weight of all the reactants multiplied by 100. Avogadro’s number (6.022 1023 mol1) a conversion factor between the number of moles and the number of fundamental particles in a substance. Chemical equation the symbolic representation of a chemical reaction using symbols of the elements and chemical formulas. Complete ionic equation a chemical equation for a reaction in aqueous solution that shows all the dissolved ionic species present during the reaction. Conservation of mass the principle that atoms are neither created nor destroyed in a chemical reaction. Empirical mass the sum of the atomic masses in the empirical formula multiplied by the number of atoms of each element. Empirical formula the simplest positive integer ratio of atoms present in the compound. Flocculation a process in which the fine particles of a precipitate aggregate to form larger particles. Formula equation a chemical equation which shows all the ionic compounds and their states during an aqueous reaction, but does not specifically show their ionic forms in solution. Gravimetric analysis an experimental technique that involves the quantitative determination of a substance based on the measurements of mass. Hydrocarbons compounds made up of only carbon and hydrogen. Limiting reactant the reactant whose amount determines, or limits, the amount of the products formed. Molar mass the mass of one mole of a substance. Mole (mol) the mass of any substance which contains the same number of fundamental particles as there are atoms in exactly 12.000 g of 12C. Molarity the number of moles of solute per liter of solution. Molecular mass the mass of a molecule calculated as the sum of the atomic masses multiplied by the number of atoms of each element in the molecule. Net ionic equation a chemical equation for a reaction in aqueous solution that omits the spectator ions and only shows those chemical species participating in the chemical reaction. 142 4. CHEMICAL EQUATIONS AND MASS BALANCE Percent yield the ratio of the actual yield of a chemical reaction to the theoretical yield multiplied by 100. Precipitate the insoluble product of a chemical reaction. Products the species formed from a chemical reaction. Reactants the substances that are present before the chemical reaction takes place. Solvation the process of attraction and association of the molecules of a solvent with the molecules of a solute. Solvation shell water molecules surrounding ions in solution. Spectator ions ions that are present but unchanged during a chemical reaction. Stoichiometric coefficients the coefficients in a balanced chemical equation. Stoichiometry the relationship between the amounts of reactants and products in a chemical reaction. Supernate the liquid phase above a precipitate in a chemical reaction. Theoretical yield the maximum mass of products that can be produced from a chemical reaction. STUDY QUESTIONS 4.1 4.2 4.3 4.4 4.5 4.6 4.7 4.8 4.9 4.10 4.11 4.12 4.13 4.14 4.15 4.16 4.17 4.18 4.19 4.20 4.21 4.22 4.23 4.24 4.25 4.26 4.27 What is the SI base unit for the amount of a substance? How many atoms are there in one mole of any element? What is the molar mass of 12C? What is the difference between a molecular formula and an empirical formula? Can a molecular formula ever be the same as an empirical formula? Why? An experimental technique that is based on the measurement of mass is called what? The complete combustion of a hydrocarbon produces what two products? Incomplete combustion of a hydrocarbon produces what two products? A symbolic representation of a chemical reaction using symbols of the elements and chemical formulas is called what? The species present before a chemical reaction takes place are called what? The species formed from a chemical reaction are called what? What symbols identify the following physical states of the species in a chemical equation: (a) solid, (b) liquid, (c) gas, and (d) aqueous solution? What is meant by the conservation of mass? How do you balance a chemical equation? What is stoichiometry? What are the stoichiometric coefficients? What do the stoichiometric coefficients represent? What is a limiting reactant? In gravimetric analysis, which reactant is set up as the limiting reactant? What is the difference between a theoretical yield and an actual yield? What is the difference between percent yield and atom economy? What is an aqueous solution? The water molecules surrounding an ion in solution are called what? Compounds containing an ion with a 1+ or 1 charge are soluble except what compounds? Compounds containing an ion with a 3+ or 3 charge are insoluble except what compounds? Compounds containing an ion with a 2 charge are generally insoluble except for what compounds? An insoluble product of a chemical reaction is called what? PROBLEMS 143 4.28 4.29 4.30 4.31 4.32 4.33 The liquid phase above a solid product is called what? What is the difference between a formula equation and a complete ionic equation? What is the difference between a complete ionic equation and a net ionic equation? What is a spectator ion? What is flocculation? In the water softening process called the soda lime process what two ions are removed by precipitation? 4.34 What chemical reactants are used to remove the ions in Question 4.33? 4.35 What is the most commonly used unit of concentration in chemistry? 4.36 How is the unit of concentration in Question 4.35 converted to SI units? PROBLEMS 4.37 4.38 4.39 4.40 4.41 4.42 4.43 4.44 4.45 4.46 4.47 4.48 4.49 4.50 4.51 4.52 4.53 4.54 4.55 4.56 How many atoms are there in 0.50 mol of gold? How many molecules are there in 2.0 mol of water? How many ions are there in a solution containing 1.5 mol of sodium chloride? What is the molar mass of the following compounds: (a) NaCl, (b) Ca(OH)2, (c) C2H4O2, and (d) NaHCO3? How many moles of calcium hydroxide are there in 15 g of calcium hydroxide? How many moles of calcium ions? How many moles of hydroxide ions? How many moles of sodium ions are there in 2.5 g of sodium chloride? How many moles of hydrogen peroxide are there in 17.0 g H2O2? How many moles of ammonium sulfate are there in 99.1 g of ammonium sulfate? What is the mass of 0.200 mol of hydrogen sulfide? What is the mass of 3.40 105 moles of sodium carbonate? What is the mass of 0.05 moles of silver nitrate? What is the mass of 1.2 moles of iron(III) hydroxide? How many molecules in 2.05 g of water? Which contains more molecules, 10.0 g of O2 or 10.0 g of N2? What are the empirical formulas of the following compounds: (a) H2O2 (hydrogen peroxide), (b) C6H6 (benzene), (c) CH2O (formaldehyde), (d) C6H14 (hexane), and (e) C6H12O6 (glucose). What is the empirical formula of a compound which contains 13.5 g Ca, 10.8 g O, and 0.68 g H. What is the empirical formula of a compound that contains 50.1% sulfur and 50.0% oxygen. What is the empirical formula of a compound that contains 12 g of carbon, 2 g of hydrogen, and 16 g of oxygen. If the molar mass of the compound is 60 g/mol, what is the molecular formula? What is the molecular formula of a compound that has an empirical formula of CH and a molar mass of 78.11 g/mol? What is the molecular formula of a compound that has an empirical formula of C3H7 and a molar mass of 86.2 g/mol? 144 4. CHEMICAL EQUATIONS AND MASS BALANCE 4.57 The compound ethylene glycol is often used as antifreeze. It contains 38.7% carbon, 9.75% hydrogen, and the rest oxygen. The molecular weight of ethylene glycol is 62.07 g. What is the molecular formula of ethylene glycol? 4.58 The combustion of a hydrocarbon produced 38.8 mg of CO2 and 31.7 mg of water. What is the empirical formula of the hydrocarbon? 4.59 What is the balanced chemical equation for the reaction of nitrogen gas with hydrogen gas to produce ammonia? 4.60 Write the balanced chemical equation for the complete combustion of propane (C3H8). 4.61 Balance the following chemical equations: (a) Fe + Cl2 ! FeCl3 (b) Li + H3PO4 ! H2 + Li3PO4 (c) Na3PO4 + MgCl2 ! NaCl + Mg3(PO4)2 (d) ZnS + O2 ! ZnO + SO2 (e) NH3 + N2O ! N2 + H2O (f) KMNO4 + HCl ! KCl + MnCl2 + H2O + Cl2 4.62 Potassium chlorate decomposes to give potassium chloride and oxygen gas. What is the mass of oxygen produced from the decomposition of 138.6 g of potassium chlorate? 4.63 Iron reacts with superheated steam to form hydrogen gas and the oxide Fe3O4. (a) What is the balanced chemical equation for this reaction? (b) How many moles of hydrogen gas will be produced from the reaction of 10.0 g of iron? 4.64 What is the mass of CO that can react with 0.150 kg of Fe2O3 in a blast furnace? What mass of iron is produced? 4.65 In the combustion reaction of ammonia with oxygen, water vapor and nitrogen monoxide are produced. (a) What is the balanced chemical equation for this reaction? (b) If a 2.00 g sample of ammonia is mixed with 4.00 g of oxygen, what is the limiting reactant? (c) What is the mass of excess reactant remaining after the reaction is completed? 4.66 Aluminum reacts with chlorine gas to form aluminum chloride. (a) What is the balanced chemical equation for this reaction? (b) If 34.0 g of aluminum is mixed with 39.0 g of chlorine gas, what is the limiting reactant? (c) What is the mass of aluminum chloride produced? 4.67 Iron(IV) sulfide reacts with oxygen to form ferric oxide and sulfur dioxide according to the reaction; 4FeS2 ðsÞ + 11O2 g ! 2Fe2 O3 ðsÞ + 8SO2 g (a) If a 22 g of iron(IV) sulfide is reacted with 1.1 moles of oxygen, what is the mass of each product produced? (b) What is the limiting reactant? (c) How many moles of the excess reactant are left after the reaction is complete? PROBLEMS 145 4.68 15.00 g aluminum sulfide reacts with 10.00 g of water to form hydrogen sulfide and aluminum hydroxide. The reaction proceeds until the limiting reactant is used up. (a) Write the balanced equation for the reaction. (b) Which is the limiting reactant? (c) What is the maximum mass of hydrogen sulfide that can be formed from these reactants? (d) How much excess reactant remains after the reaction is complete? 4.69 Copper(II) sulfate reacts with zinc metal to yield zinc sulfate and copper metal. (a) What is the theoretical yield of Cu if 79.8 grams of copper(II) sulfate reacts with 32.7 grams of zinc metal? (b) If 28.5 grams of copper are actually obtained from this reaction, what is the percent yield? 4.70 (a) How much iron can theoretically be obtained in a blast furnace process from 100 metric tons of pure hematite ore? (b) If only 66.2 metric tons of iron is produced, what is the percent yield of the blast furnace process? 4.71 Calcium carbonate decomposes on heating to calcium oxide and carbon dioxide. If the reaction of 20.7 grams of CaCO3 produces 6.81 grams of CaO, what is the percent yield? 4.72 Hydrogen can be produced from the reaction of carbon in the form of coal with steam according to the reaction; CðsÞ + 2H2 O g ! CO2 g + 2H2 g : What is the percent atom economy of this reaction? 4.73 Hydrogen can also be produced from the reaction of methane and steam according to the reaction; CH4 + 2H2 O ! CO2 + 4H2 What is the atom economy of this reaction? 4.74 Comparing the results of Problem 4.72 and Problem 4.73, which is the greener process for the production of hydrogen? 4.75 Which of the following compounds are soluble in water: NaCl, AgIO3, (NH4)2CO3, NaClO3, ZnS? 4.76 Which of the following compounds are insoluble in water: FeAsO4, NaOH, Ag2S, NH4IO3, PbCl2? 4.77 What is the insoluble product formed from the aqueous reaction of iron(III) nitrate and sodium hydroxide? 4.78 Give a) the balanced chemical equation, b) the complete ionic equation, and c) the net ionic equation for the reaction in Problem 4.77. 4.79 Give a) the complete ionic equation, and b) the net ionic equation for the reaction; 2KIðaqÞ + PbðNO3 Þ2 ðaqÞ ! 2KNO3 ðaqÞ + PbIðsÞ 4.80 What is the insoluble product formed from the aqueous reaction of barium nitrate and sodium carbonate? 4.81 What are the spectator ions in the aqueous reaction of barium nitrate and sodium carbonate? 146 4. CHEMICAL EQUATIONS AND MASS BALANCE 4.82 (a) Identify the spectator ions and (b) write the net ionic equation for the reaction; Bottom of Form HClðaqÞ + NaHCO3 ðaqÞ ! NaClðaqÞ + H2 OðlÞ + CO2 g 4.83 Give: (a) the complete ionic equation, and (b) the net ionic equation for the reaction; 2NaClðaqÞ + CuSO4 ðaqÞ ! Na2 SO4 ðaqÞ + CuCl2 ðsÞ 4.84 Write the (a) balanced molecular, (b) complete ionic, and (c) net ionic equations for the aqueous reaction of aluminum nitrate with potassium chloride to produce aluminum chloride and potassium nitrate. 4.85 What is the molarity of a liter of solution containing 100 g of copper (II) chloride? 4.86 What mass of NaCl is required to prepare 250 mL of a 0.50 M solution? 4.87 What is the molarity of 5.30 g of Na2CO3 dissolved in 400.0 mL of solution? 4.88 What volume of 18.0 M H2SO4 is needed to contain 2.45 g H2SO4? 4.89 How many grams of Ca(OH)2 are needed to make 100.0 mL of 0.250 M solution? 4.90 Determine the number of moles of solute to prepare the solutions: (a) 500 mL of a 2.00 M Cu(NO3)2 solution, (b) 250 mL of a 0.500 M MgCO3 solution, (c) 1.00 L of a 2.50 M Na2O solution, (d) 25 mL of a 0.0150 M solution of Pb(NO3)2. 4.91 Determine the mass of solute required to make each of the solutions in Problem 4.90. C H A P T E R 5 Acids and Bases O U T L I N E 5.1 Defining Acids and Bases 147 5.2 Acids and Bases in Aqueous Solution 156 5.3 The pH Scale 160 5.4 Other “p” Functions 165 5.5 Buffer Solutions 168 5.6 The Titration 172 Important Terms 176 Study Questions 177 Problems 178 5.1 DEFINING ACIDS AND BASES The first modern attempt at defining acids and bases was by a Swedish chemist named Svante Arrhenius in 1887. Arrhenius defined an acid as a material that releases hydrogen ions (H+) when dissolved in water. Similarly, he defined a base as a material that releases hydroxide ions (OH) when dissolved in water. This definition only held for ionic compounds containing hydrogen or hydroxide ions and did not apply to many acids and bases that we deal with today. Since this early definition of acids and bases was so limited, two more sophisticated and general definitions of acids and bases have since been developed, which are in wide use today. These are known as the Brønsted-Lowry definition and the Lewis definition. In 1923, both J.N. Brønsted of Denmark and Thomas Lowry of England, working independently, defined an acid as a species that can donate a hydrogen ion to a base. A base was defined as a species that can accept a hydrogen ion from an acid. So, a Brønsted-Lowry acid is a proton (H+) donor and a Brønsted-Lowry base is a proton (H+) acceptor. Both acids and bases are divided into two categories, strong and weak. The Brønsted-Lowry measure of the strength of an acid is determined by the ability of the acid to give up a proton. The measure of the strength of a base is the ability of the base to attract a proton. The relative strength of an General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00005-9 147 # 2018 Elsevier Inc. All rights reserved. 148 5. ACIDS AND BASES acid is often described quantitatively in terms of an acid ionization constant (Ka), which will be covered in detail in Section 5.3. An acid-base reaction, then, involves the transfer of a proton from an acid to a base to form a new acid and a new base. When an acid loses its proton, it becomes a conjugate base and when a base gains a proton it becomes a conjugate acid. The term “conjugate” comes from Latin and means “joined together.” It particularly refers to things that are joined together in pairs. The Brønsted-Lowry acid-base reaction can be summarized by the generalized chemical equation; HA + B ! Acid Base A Conjugate base + HB Conjugate acid (1) So, a conjugate acid is a species formed by the addition of a proton (H+) to a base. A conjugate base is a species formed by the removal of a proton from an acid. The acid and its conjugate base as well as the base and its conjugate acid are known as conjugate pairs, whose chemical formulas are related by the gain or loss of a hydrogen ion. Some examples of acids and their conjugate bases are given in Table 5.1. Notice how the conjugate pairs in Table 5.1 differ by just one proton. Acid strength decreases and conjugate base strength increases going down the table. Since a strong acid is a molecule that has a strong ability to give up a proton, its conjugate base will have a weak tendency to attract a proton. Similarly, since a strong base has a strong ability to attract a proton, its conjugate acid will have a weak tendency to donate a proton. So, there is an inverse relationship between the strength of an acid and the strength of its TABLE 5.1 Some Examples of Acids and Their Conjugate Bases Strong acids Weak acids Acid Name Acid Formula Conjugate Base Formula Hydrochloric acid HCl Cl Weak bases Sulfuric acid H2SO4 HSO4 Nitric acid HNO3 NO3 Hydronium ion H3O+ H2O SO4 2 Hydrogen sulfate ion HSO4 Phosphoric acid H3PO4 H2 PO4 Acetic acid CH3CO2H CH3 CO2 Carbonic acid H2CO3 HCO3 Dihydrogen phosphate ion H2 PO4 HPO4 2 Ammonium ion NH4 + NH3 CO3 2 Hydrogen carbonate ion HCO3 Hydrogen phosphate ion HPO4 2 PO4 3 Water H2O OH Strong bases 5.1 DEFINING ACIDS AND BASES 149 conjugate base, and likewise, there is an inverse relationship between the strength of a base and the strength of its conjugate acid. Strong acids produce weak conjugate bases and strong bases produce weak conjugate acids (see Table 5.1). Also, weak acids produce strong conjugate bases and weak bases produce strong conjugate acids. • • • • Strong acid ! Weak conjugate base Strong base ! Weak conjugate acid Weak acid ! Strong conjugate base Weak base ! Strong conjugate acid Since a weak acid has a weaker tendency to give up its proton and the conjugate base it produces has a stronger tendency to accept its proton, a competition results between the acid and the conjugate base for the proton. So, only a fraction of the weak acid molecules will donate a proton to the base, giving rise to a significant concentration of both the acid and conjugate base forms in solution. The reaction is said to be in chemical equilibrium, the state in which both reactants and products are present in concentrations which have no further tendency to change with time. This equilibrium is represented in the chemical equation by the presence of a double arrow between products and reactants, indicating that the reaction can go in both directions and both products and reactants are present; HA + B A + HB The concentrations of all the species depend on the strengths of the acids and bases. The Brønsted-Lowry definition of acids and bases also describes more clearly what actually happens when an acid dissolves in water. From this point of view, the dissolution of acids, such as HCl, in aqueous solution is seen to involve an acid-base reaction between the acid and water. Although HCl is a covalent compound, it can donate a proton to a water molecule to form the cation H3O+; HClðaqÞ + H2 OðlÞ ! H3 O + ðaqÞ + Cl ðaqÞ The common name for this cation is the hydronium ion. It belongs to a family of ions called oxonium ions, which is any oxygen cation containing three-bonded species. Other oxonium ions have three organic groups bonded to the oxygen and will be discussed in Chapter 13. Recall from Chapter 3 that the water molecule has two nonbonding electron groups on the central oxygen atom. It also has a difference in electronegativity between oxygen (3.44) and hydrogen (2.20) of 1.24. This electronegativity difference and the two lone pairs on the oxygen atom make water a very polar molecule with a dipole moment of 1.85 D. Hydrochloric acid, on the other hand, has a difference in electronegativity between hydrogen (2.20) and chlorine (3.16) of only 0.96 and a dipole moment of 1.08 D. The lone pair electrons on the oxygen atom are then capable of pulling away the hydrogen from the HCl molecule leaving behind its electrons, a process called ionization. The hydrogen ion forms a covalent bond with one of the lone pair electrons on the oxygen atom resulting in the H3O+ cation (Fig. 5.1). So, the dissolution of a covalently bonded acid in water is an acid-base reaction where H2O is the base and the hydronium ion is the conjugate acid. 150 5. ACIDS AND BASES + O O H+ H H H H Conjugate acid H Base FIG. 5.1 The aqueous cation H3O+, called the hydronium ion, has a trigonal pyramid geometry with the oxygen atom at its apex. The HdOdH bond angle is approximately 113 ° and the center of mass is very close to the oxygen atom. From Bensaccount, Wikimedia Commons. Some compounds can act as both a Brønsted-Lowry acid and a Brønsted-Lowry base. These are known as amphoteric compounds. For example, in Table 5.1; HSO4 , H2 PO4 , HCO3 , HPO4 2 , and H2O appear in both the acid and the conjugate base columns. Water is the most common example of an amphoteric compound because it can either accept a proton, becoming H3O+ or it can donate a proton, becoming OH. This property allows it to react with itself. The Brønsted-Lowry definition of acids and bases explains this autoionization of water to form very low concentrations of both hydronium and hydroxide ions in aqueous solution. H2 O + H2 O > Acid Base H3 O + Conjugate acid + OH Conjugate base (2) In the autoionization reaction, one molecule of water acts as the acid, donating a hydrogen ion to a second molecule of water and forming the conjugate base OH. The second molecule of water acts as a base, accepting the hydrogen ion from the first water molecule forming the conjugate acid H3O+. However, the conjugate acid H3O+ is a very strong acid, and the conjugate base OH is a very strong base. So, they rapidly react with each other reforming H2O (Fig. 5.2). The result of this autoionization is that a chemical equilibrium is established where a very small number of H3O+ and OH ions are present at any given time. The exact concentration of each ion in pure water can be calculated to be 1.00 107 mol dm3 at room temperature. This will be covered in more detail in Section 5.3. Since acids such as HCl or HNO3 can donate only one proton to a base, they are known as monoprotic acids. Several common acids, such as H2SO4, H3PO4, or H2CO3, can donate two or more protons and so are called polyprotic acids. They can also be called diprotic acids (donating two protons), triprotic acids (donating three hydrogens), etc. When dissolved in FIG. 5.2 The autoionization of water to give hydronium ions and hydroxide ions. From Cdang, Wikimedia Commons. 5.1 DEFINING ACIDS AND BASES 151 aqueous solution, the polyprotic acids ionize in several steps donating one proton at each step. Each successive proton ionizes less readily than the one before. For example, sulfuric acid is a strong acid because the first proton is completely ionized in aqueous solution as; H2 SO4 ðaqÞ + H2 OðlÞ ! H3 O + ðaqÞ + HSO4 ðaqÞ The conjugate base HSO4 can also act as an acid donating its proton to a water molecule, but it is a weaker acid than the parent H2SO4. HSO4 ðaqÞ + H2 OðlÞ > H3 O + ðaqÞ + SO4 2 ðaqÞ Since the acid HSO4 is a weak acid, the conjugate base (SO4 2 ) of this second ionization step is a strong base with a stronger tendency to attract a proton. So, the acid HSO4 is not completely ionized because it is competing with the stronger base SO4 2 for the available proton. As with the autoionization of water, significant concentrations of both the acid (HSO4 ) and conjugate base (SO4 2 ) forms remain in solution. Not all acid-base reactions take place in aqueous solution. The Brønsted-Lowry definition also applies to proton transfer in nonaqueous solvents. For example, the reaction of HCL with excess liquid ammonia is; HClðin NH3 Þ + NH3 ðlÞ ! NH4 + ðin NH3 Þ + Cl ðin NH3 Þ In this reaction, the NH3 acts as both base and solvent in a similar manner as H2O acts as both base and solvent. The HCl acid donates a proton to the NH3 base to form the conjugate acid NH4 + and the conjugate base Cl. In 1923, G.N. Lewis developed a more general definition of acids and bases which includes the Brønsted-Lowry definition as a special case. While the acid-base reaction of HCl in water is described by Brønsted-Lowry as the donation of a proton to the water molecule, the Lewis definition describes the same reaction as the donation of a pair of electrons on the oxygen atom to the H+ to form a covalent bond. The Lewis acid-base definition is therefore based on the sharing of an electron pair between an acid and a base instead of on the donation of a proton (Fig. 5.3). FIG. 5.3 Comparison of the Brønsted-Lowry and Lewis definitions of acids and bases as they apply to the reaction of HCl with water. 152 5. ACIDS AND BASES A Lewis acid is a substance that can accept a lone pair of electrons to form a new covalent bond. So, a Lewis acid is an electron pair acceptor, sometimes called an electrophile (meaning “electron loving”). A Lewis base is a substance that can donate a lone pair of electrons to form a new covalent bond. So, a Lewis base is an electron pair donor, sometimes called a nucleophile (meaning “nucleus loving”). The new covalent bond formed between the Lewis acid and the Lewis base is called a coordinate covalent bond, a bond where both electrons are provided by a lone pair on the Lewis base. The product of the Lewis acid-base reaction is called an acid-base adduct. A + B : ! B! A Acid Base Adduct (3) Often, the coordinate covalent bond in the acid-base adduct is shown in the chemical formula as an arrow instead of a line (B–A) or dots (B:A) commonly used in an electron dot structure. The arrow points from the donor atom to the acceptor atom. At first glance, the Lewis definition of acids and bases appears to be a minor twist on the Brønsted-Lowry definition. However, since the Lewis definition focuses on the sharing of electron pairs instead of on proton transfer, it expands the number of acids to include species other than proton donors and so expands the number of acid-base reactions. In the Lewis theory, an acid is any ion or molecule that can accept a pair of nonbonding valence electrons. While this includes all Brønsted-Lowry acids, it also includes other electron-deficient species not covered by the Brønsted-Lowry concept. A large number of species can act as Lewis acids. These include the following electron-deficient species: • • • • All cations. An atom, ion, or molecule with an incomplete octet of valence electrons. Molecules where the central atom can have more than eight valence shell electrons. Molecules that have double or triple bonds between two atoms of different electronegativities. All cations can act as Lewis acids since they are electron-deficient and are able to accept electrons. Examples include the metal cations such as Cu+2, Co+3, Fe+2, and Fe+3, the hydrogen ion (H+), protonated compounds such as NH4 + , H3O+, and also CH3+, a species very important to organic chemistry and will be discussed in Chapter 13. Metal cations have two properties that allow them to act as Lewis acids: (1) their positive charge attracts electrons and (2) they have at least one empty orbital that can accept an electron pair. When a metal cation (Lewis acid) reacts with a species containing a lone pair of electrons (Lewis base), a coordination complex is formed. A coordination complex is a product of a Lewis acid-base reaction in which neutral molecules or anions bond to a central metal cation by coordinate covalent bonds. The molecules or anions that bond to the central metal cation are called ligands, from the Latin meaning “to tie or bind.” When a metal salt is dissolved in water, it ionizes to release the metal cations. Because the metal cations are electron-deficient, they react with water molecules (an electron pair donor) to form coordinate covalent bonds. These metal-water coordination complexes contain the central metal ion with water as the ligands. They are the predominant species in aqueous solutions of many metal salts, such as metal nitrates, sulfates, and perchlorates. They have the general stoichiometry of [M(H2O)n]z+, where M is the metal cation with a charge of z +. Since 5.1 DEFINING ACIDS AND BASES 153 FIG. 5.4 The structure of an octahedral metal-water coordination complex. the coordination complex carries a net charge, it is called a complex ion. The properties of these metal-water coordination complexes are important in many aspects of environmental, biological, and industrial chemistry. The most common of the metal-water coordination complexes are those where the metal cation is bonded to six water molecules with the formulas [M(H2O)6]2+ and [M(H2O)6]3+ (Fig. 5.4). This makes the molecular geometry of the complex ion octahedral, as described in Section 3.7. An example of a common metal-water coordination complex is one that is formed when Co(NO3)3 is dissolved in water. The Co3+ cation reacts with six water molecules to form CoðH2 OÞ6 3 + as; Co3 + + 6H2 O ! CoðH2 OÞ6 3 + In this reaction, Co3+ is the Lewis acid because it has empty valence shell orbitals that can accept pairs of electrons. The valence shell of Co3+, [Ar] 4s03d6, can accept an additional 12 electrons, two electrons in the 4s orbital, four electrons in the 3d subshell, and an additional six electrons in the 4p subshell. So, Co3+ accepts the lone pairs of electrons from six water molecules, which act as the Lewis base, to form the coordination complex CoðH2 OÞ6 3 + . The Lewis acid-base concept can also explain why aqueous solutions of many metal ions show acidic properties. When the water molecule forms a coordination complex with the positively charged metal ion, electron density is drawn away from the oxygen atom of the water molecules. This causes the OdH bond to become more polarized and the water molecules bound to the metal ion become more acidic than those in the bulk solvent. This enhanced polarization allows the metal cation-water complex to act as a Brønsted-Lowry acid, giving up a proton as; 2 + 3 + CoðH2 OÞ6 ðaqÞ + H2 OðlÞ ! CoðH2 OÞ5 ðOHÞ ðaqÞ + H3 O + ðaqÞ The amount of the acidity of a metal-water coordination complex is dependent on the strength of the metal-water bond. The stronger the metal-water bond, the more polar the OdH bond and the greater the acidity of the coordination complex. The larger the ionic charge and the smaller the ionic radius of the metal ion, the stronger the metal-water bond, the more polar the OdH bond, and the greater the acidity of the coordination complex. Conversely, metal ions with a small charge and/or a relatively large ionic radius have complex ions with little or no acidity in water. Molecular compounds of some elements from groups 2 and 13, such as Be, Al, and B, can form compounds with incomplete valence octets (less than eight electrons in the valence 154 5. ACIDS AND BASES shell). Structures with incomplete valence octets are electron-deficient and can act as Lewis acids, reacting with molecules that have lone pairs of electrons. For example, boron trifluoride (BF3) has the structure; F B F F with only six electrons in the valence shell of boron. So, boron trifluoride is electron-deficient and can react with a Lewis base to gain the extra two electrons needed to complete the valence octet of boron. In the Lewis acid-base reaction of boron trifluoride with ammonia, the lone pair on nitrogen is used to form a coordinate covalent bond between boron and nitrogen to complete the valence octet of boron; BF3 g + : NH3 g ! NH3 BF3 ðsÞ with the electronic structure; H F N— B H F F H Molecules with a central atom that are capable of having more than eight electrons in the valence shell (called hypervalency) can also act as Lewis acids by accepting additional lone pair electrons. As shown in Section 3.4, elements in periods 3 and higher can have more than eight valence electrons when bonding to fluorine, chlorine, or oxygen. The tetrahalides of silicon, germanium, and tin and the pentahalides of phosphorous, arsenic, and antimony behave in this same manner. Although the tetrahalides have a complete valence octet, they can add Lewis bases to give the adducts: MX4L and MX4L2, where M is Si, Ge, or Sn, X is the halogen, and L is a Lewis base. These adducts have valence electrons of 10 and 12, respectively. This occurs because of the empty d orbitals on Si, Ge, and Sn which can also accept electrons. For example, the electron configuration of silicon is [Ne]3s23p2. So, silicon needs four electrons to complete the octet in the s and p subshells. It achieves this by sharing four electrons with four fluorine atoms. F F Si F F But the M electron shell (n ¼ 3) of silicon also has five empty d orbitals. These empty d orbitals can accommodate up to two extra ligands, contributing two to four electrons for a total of 10 and 12 valence electrons on silicon; SiF4 + L ! SiF4 L SiF4 + 2L ! SiF4 L2 5.1 DEFINING ACIDS AND BASES 155 Although this configuration does not completely fill all the empty d orbitals, the formation of additional coordinate covalent bonds is prevented by repulsion of the existing atoms surrounding the central silicon atom. L F F Si F F L Molecules that have multiple bonds between two atoms of different electronegativities, such as CO2 and SO2, can also act as Lewis acids. Since oxygen (3.44) is more electronegative than carbon (2.55) with an electronegativity difference of 0.89 on the Pauling scale, both bonds in the linear CO2 molecule are polar The electrons are drawn toward the oxygen atoms leaving the carbon atom with a partial positive charge, as described in Section 3.8. + O δ− + δ+ C O δ− In the reaction with a Lewis base, such as OH, the carbon atom of CO2 accepts the lone pair of electrons from the oxygen in OH to form a new coordinate covalent bond resulting in the bicarbonate ion. In a similar manner, SO2 reacts with OH to form HSO3 . There are significantly fewer Lewis bases than there are Lewis acids. The most common Lewis bases are: • • • • Simple anions, such as F. Complex anions, such as OH, SO4 2 , and NO3 2 . Molecular species containing atoms with lone pairs, such as H2O and NH3. Electron rich π-systems, such as ethylene and benzene (covered in Chapter 13). The hydroxide ion (OH) is an excellent Lewis base since it has three lone pairs of electrons on the oxygen atom. So, it can bind to metal cations (Lewis acids) to yield metal hydroxides. An important property of metal hydroxides is that they are amphoteric. For example, solid aluminum hydroxide behaves as a Lewis acid, dissolving in basic solution to form a complex ion with one additional hydroxide as; AlðOHÞ3 ðsÞ + OH ðaqÞ ! AlðOHÞ4 ðaqÞ Aluminum hydroxide can also act as a Brønsted-Lowry base, reacting with a BrønstedLowry acid, such as H3O+. The product of this reaction is the hexaaqua aluminum(III) complex ion; AlðOHÞ3 ðsÞ + 3H3 O + ðaqÞ ! AlðH2 OÞ6 3 + ðaqÞ 156 5. ACIDS AND BASES EXAMPLE 5.1: DETERMINING LEWIS ACIDS AND LEWIS BASES Which of the following compounds can act as Lewis acids and which can act as Lewis bases: (a) PH3, (b) H2S, (c) Ni2+, (d) BBr3, (e) NO3 ? 1. Determine which species are electron-deficient. Hint: To decide if the molecular species are electron-deficient, look at the electron dot structures. Ni2+ is electron-deficient. BBr3 is electron-deficient because it has only six valence electrons on boron. B Br Br Br 2. Determine which species are electron-rich. NO3 is electron-rich. PH3 is electron-rich because it has a lone pair on phosphorous. H H P H H2S is electron-rich because it has two lone pairs on sulfur. S H H 3. Determine the Lewis acids and Lewis bases. Lewis acids: Ni+, BBr3. Lewis bases: NO3 , PH3, H2S. 5.2 ACIDS AND BASES IN AQUEOUS SOLUTION Acids and bases are normally used as aqueous solutions and so acid-base reactions generally occur in aqueous solution. It is important to consider the role of water in acid-base reactions. As described in Section 5.1, water is not only the solvent, but it can also act as a reactant. The strength of an acid or base in aqueous solution is determined by the degree that it ionizes. A strong acid or base is completely (100%) ionized. According to the BrønstedLowry definition, a strong acid (HAs) in aqueous solution has a very strong ability to donate its proton to water converting entirely to the conjugate acid (H3O+) and the conjugate base (A); Hn As ðaqÞ + nH2 OðlÞ ! nH3 O + ðaqÞ + A ðaqÞ (4) There are seven strong Brønsted-Lowry acids, which are all covalent compounds. These include: nitric acid (HNO3), sulfuric acid (H2SO4), perchloric acid (HClO4), chloric acid (HClO3), and the hydrogen halides: hydrochloric acid (HCl), hydrobromic acid (HBr), and hydroiodic acid. 5.2 ACIDS AND BASES IN AQUEOUS SOLUTION 157 A strong base (Bs) will also completely ionize in aqueous solution. All of the hydroxides of the alkali metals in group 1 are considered to be strong bases. The hydroxides of the alkaline earth metals in group 2 that are soluble in water are also considered to be strong bases. These are: calcium hydroxide, strontium hydroxide, and barium hydroxide. Since all these strong bases are ionic, they will completely ionize in aqueous solution to give the metal cation and the hydroxide ion; MOHn ðaqÞ ! M +z ðaqÞ + nOH ðaqÞ Some nonhydroxide bases include: sodium hydride (NaH), sodium amide (NaNH2), and sodium ethoxide (NaC2H5O). These ionic salts completely dissociate in water to give a very strongly basic anion, which reacts violently with water to produce hydroxide ion. NaBðaqÞ ! Na + ðaqÞ + B ðaqÞ B ðaqÞ + H2 O ! BH + OH Since strong acids and strong bases are completely ionized in aqueous solution, the concentration of the ionization products (the conjugate acid and the conjugate base) is equal to the initial concentration of the strong acid or base before ionization; ½HAs i ¼ ½H3 O + f ¼ ½A f ½BOHi ¼ ½B + f ¼ ½OH f Each chemical formula is placed in square brackets to indicate the molar concentration of the species inside the bracket. The subscript “i” stands for the initial concentration of the strong acid or strong base, and the subscript “f” stands for the final concentration of each ionization product in aqueous solution. For example, a 0.02 M aqueous solution of hydrochloric acid will completely ionize to give 0.02 M Cl, 0.02 M H3O+, and 0.00 M HCl. A weak acid only partially ionizes in water, as described by the equilibrium equation; HAw ðaqÞ + H2 OðlÞ > H3 O + ðaqÞ + A ðaqÞ (5) Since different weak acids ionize in water to different degrees, there are different levels of acid weakness. The strength of a weak acid in an aqueous solution is defined by the extent that it ionizes in water. The extent that a weak acid ionizes in water is expressed quantitatively by an acid ionization constant (Ka), which is a form of equilibrium constant described in detail in Chapter 7. The acid ionization constant is equal to the ratio of the molar concentrations of the ionized products (conjugate acid and conjugate base) to the molar concentration of the unionized acid at equilibrium; Ka ¼ ½H3 O + ½A ½HAw (6) Although water is a reactant in the ionization of a weak acid, the concentration of water is not used in the expression of the acid ionization constant. This is because, as the solvent, the concentration of water is in great excess compared to the concentrations of the acid and the ionization products. So, the concentration of water remains constant during the reaction. 158 5. ACIDS AND BASES Only the concentrations of the species changed by the reaction are included in the expression for the acid equilibrium constant. The stronger the acid and the more it is ionized, the higher the concentrations of the conjugate acid and conjugate base (numerator of Ka) and the smaller the concentration of the unionized acid (denominator of Ka), resulting in a larger Ka. The weaker the acid and the less it is ionized, the lower the concentrations of the conjugate acid and conjugate base and the higher the concentration of the unionized acid, resulting in a smaller Ka. This means that the larger the Ka the stronger the acid and the smaller the Ka the weaker the acid. A weak base (Bw), such as ammonia, partially ionizes in aqueous solution by reaction with water resulting in the protonated base (conjugate acid) and the hydroxide ion (conjugate base). Bw ðaqÞ + H2 OðlÞ > BH + ðaqÞ + OH ðaqÞ (7) The strength of the weak base and the extent that it ionizes in water is expressed quantitatively by a base ionization constant (Kb), which is the ratio of the concentrations of the ionized products (conjugate acid and conjugate base) to the concentration of the unionized base in the aqueous solution at equilibrium; Kb ¼ ½BH + ½OH ½ Bw (8) As with the weak acid, the stronger the weak base the more it is ionized in aqueous solution; the higher the concentrations of the conjugate acid and base, the larger the Kb. The weaker the base, the less it is ionized in aqueous solution; the lower the concentrations of the conjugate acid and base, the smaller the Kb. So, the larger the Kb, the stronger the base and the smaller the Kb the weaker the base. The ionization constants for some common weak acids and their conjugate bases are listed in Table 5.2. The strength of the acids decrease going down the table and the strength of the conjugate bases increase going down the table. Comparing the values of Ka and Kb in Table 5.2, you can see that the weaker the acid (the smaller the Ka), the stronger its conjugate base (the larger the Kb). For example, nitrous acid (HNO2) is above carbonic acid (H2CO3) in Table 5.2 with a larger Ka value. The Ka for nitrous acid is 4.5 104, while the Ka for carbonic acid is 4.2 107. This tells us that carbonic acid is 103 times weaker than nitrous acid. Also, the Kb for the conjugate base of carbonic acid (HCO3 ) is 2.4 108 and the Kb for the conjugate base of nitrous acid (NO2) is 2.2 1011. So, the conjugate base of carbonic acid is about 103 times stronger than the conjugate base of nitrous acid. The autoionization of water shown in Eq. (2) can also be expressed quantitatively by an autoionization constant, which is given the symbol Kw. The autoionization constant of water is equal only to the product of the concentration of the hydronium ion and the hydroxide ion. As with Ka and Kb, the concentration of water is not used in determining the autoionization constant because its concentration is very much larger than that of the ionized products and is therefore a constant. So, Kw has no denominator and the units of Kw are mol2/L2. Kw ¼ ½H3 O + ½OH (9) In pure water, the autoionization of water is the only source of hydronium and hydroxide ions. So, [H3O+] ¼ [OH]. Electrical conductivity measurements have determined that 159 5.2 ACIDS AND BASES IN AQUEOUS SOLUTION TABLE 5.2 Ionization Constants for Some Weak Acids and Their Conjugate Bases in Water at 25°C Acid Formula + Ka (mol/L) Conjugate Base Kb (mol/L) 1.0 H2O 1.0 1014 Hydronium ion H3O Sulfurous acid H2SO3 1.2 102 HSO3 8.3 1013 Hydrogen sulfate ion HSO4 1.2 102 SO4 2 8.3 1013 Phosphoric acid H3PO4 7.5 103 H2 PO4 1.3 1012 Hydrofluoric acid HF 7.2 104 F 1.4 1011 Nitrous acid HNO2 4.5 104 NO2 2.2 1011 Formic acid HCO2H 1.8 104 HCO2 5.6 1011 Benzoic acid C6H5CO2H 6.3 105 C6 H5 CO2 1.6 1010 Acetic acid CH3CO2H 1.8 105 CH3 CO2 5.6 1010 Carbonic acid H2CO3 4.2 107 HCO3 2.4 108 Hydrogen sulfide H2S 1.0 107 HS 1.0 107 Dihydrogen phosphate ion H2 PO4 6.2 108 HPO4 2 1.6 107 Hydrogen sulfite ion HSO3 6.2 108 SO3 2 1.6 107 Hypochlorous acid HClO 3.5 108 ClO 2.9 107 Ammonium ion NH4 + 5.6 1010 NH3 1.8 105 Hydrocyanic acid HCN 4.0 1010 CN 2.5 105 Hydrogen carbonate ion HCO3 4.8 1011 CO3 2 2.1 104 Hydrogen phosphate ion HPO4 2 3.6 1013 PO4 3 2.8 102 Water H2O 1.0 1014 OH 1.0 19 Hydrogen sulfide ion HS 1.0 10 2 S 1.0 105 the concentrations of [H3O+] and [OH] are; 1.0 107 M at 25°C. This gives a value for Kw of; (10) Kw ¼ ½H3 O + ½OH ¼ 1:0 107 M 1:0 107 M ¼ 1:0 1014 mol2 L2 The equation for Kw applies not only to pure water, but it also applies to all aqueous solutions. Although the value of Kw is temperature-dependent, as shown in Fig. 5.5, at any given temperature the product of [H3O+][OH] in any aqueous solution is always the same, and at 25°C, it is always equal to 1.0 1014. If an acid or base is added to pure water, the concentration of [H3O+] or [OH] will be increased. However, the product of [H3O+] [OH] must always be equal to 1.0 1014 at 25°C. So, for aqueous solutions at 25°C: • In a neutral solution, [H3O+] ¼ [OH] ¼ 1.0 107 M • In an acidic solution, [H3O+] > [OH], [H3O+] > 1.0 107 M, and [OH] < 1.0 107 M. 160 5. ACIDS AND BASES FIG. 5.5 Temperature dependence of the autoionization constant for water. • In a basic solution, [H3O+] < [OH], [H3O+] < 1.0 107 M, and [OH] > 1.0 107 M. • In any aqueous solution, if either [H3O+] or [OH] is known, the other can be calculated from the equation: Kw ¼ [H3O+][OH] ¼ 1.0 1014. EXAMPLE 5.2: DETERMINING HYDRONIUM AND HYDROXIDE ION CONCENTRATIONS FOR AN AQUEOUS SOLUTION OF A STRONG ACID OR A STRONG BASE What are the hydronium and hydroxide ion concentrations in a 0.015 M solution of hydrochloric acid at 25°C? 1. Write the chemical equation for the ionization of the acid. Since HCl is a strong acid, it is totally ionized in aqueous solution: HCl(aq) + H2O(l) ! H3O+(aq) + Cl(aq) 2. Determine the hydroxide ion concentrations from the autoionization constant for water. Since HCl is completely ionized; [H3O+] ¼ 0.015 M Kw ¼ 1.0 1014 mol2 L–2 ¼ [H3O+][OH] ¼ (0.015 M)[OH] [OH] ¼ 1:0 1014 M2 ¼ 6.7 1013 M 1:5 102 M 5.3 THE pH SCALE The concentrations of hydronium ions in aqueous solutions can vary from less than 1015 M in concentrated strong bases to greater than 10 M in concentrated strong acids. So, the concentrations of hydronium ions in everyday aqueous solutions can span over 16 orders of magnitude. In order to more easily deal with these very large variations in concentrations, scientists use a logarithmic scale, called the pH scale. The pH scale is defined as the negative of the base –10 logarithm (log) of the hydronium ion concentration in an aqueous solution. 5.3 THE pH SCALE 161 pH ¼ log ½H3 O + (11) The negative logarithm is used because it gives a result in positive pH values for the very small concentrations with negative exponents. Unfortunately, this results in a scale that may seem to be reversed, a low pH value indicating a high acid concentration and a high pH indicating a low acid concentration. It is important to remember that since pH is on a logarithmic scale, each one unit change in pH corresponds to a tenfold change in hydronium ion concentration, as shown in Table 5.3. So, a change of 2 in pH represents a 100-fold change in [H3O+]. Although theoretically the pH scale is open-ended, most pH values fall in the range from 0 to 14. The pH of pure water at 25°C can be calculated from the concentration of [H3O+] in pure water, which is equal to 1.0 107 M. pH ¼ log 1 107 ¼ log ð1:0Þ + log 107 ¼ ½0 + ð7Þ ¼ 7:00 Acidic solutions will have a higher [H3O+] than pure water (values >1 107). So, the pH values of acidic solutions will be less than 7.0. Since basic solutions will have a lower concentration of [H3O+] than pure water (values <1 107), the pH values of basic solutions will be greater than 7.0. The lower the pH value, the higher the hydronium ion concentration and the stronger the acid solution. Similarly, the higher the pH value, the lower the hydronium ion concentration and the stronger the basic solution (Fig. 5.6). TABLE 5.3 Molar Hydronium and Hydroxide Ion Concentrations for pH Values From 0 to 14 pH [H+] [OH2] 0 1.0 0.00000000000001 1 0.1 0.0000000000001 2 0.01 0.000000000001 3 0.001 0.00000000001 4 0.0001 0.0000000001 5 0.00001 0.000000001 6 0.000001 0.00000001 7 0.0000001 0.0000001 8 0.00000001 0.000001 9 0.000000001 0.00001 10 0.0000000001 0.0001 11 0.00000000001 0.001 12 0.000000000001 0.01 13 0.0000000000001 0.1 14 0.00000000000001 1.0 162 Battery acid Tomato juice, beer Coffee Stomach acid 8 7 6 Lemon juice, vinegar Basic 14 13 12 11 10 9 Pure water, blood Milk, urine Sea water, baking soda Neutral Detergent Ammonia Soapy water Bleach, oven cleaner Drain cleaner substances. Toothpaste, hand soap FIG. 5.6 pH values for some common Grapefruit and orange juice 5. ACIDS AND BASES 2 1 0 Acidic 5 4 3 EXAMPLE 5.3: DETERMINING HYDRONIUM ION AND HYDROXIDE ION CONCENTRATION FROM PH What are the hydronium ion and hydroxide ion concentrations of an aqueous solution that has a pH of 6.10 at 25°C? 1. Determine hydronium ion concentration as a power of 10. Since pH ¼ log[H3O+]; log[H3O+] ¼ pH [H3O+] ¼ 10pH ¼ 106.10 2. Determine the value of 10–pH. On a calculator; calculate 106.10, or “inverse” log (6.10). [H3O+] ¼ 106.10 ¼ 7.9 107 M 3. Determine the hydroxide ion concentration. Since [H3O+][OH] must always be equal to 1.0 1014 at 25°C: ½OH ¼ 1:0 1014 M2 ¼ 1:3 108 M 7:9 107 M CASE STUDY: OCEAN ACIDIFICATION The level of CO2 in the atmosphere is rapidly increasing, primarily due to increased combustion of fossil fuels such as coal and petroleum. As of 2013, the atmospheric concentration of CO2 was 43% above preindustrial levels. While the impacts of increasing CO2 on global temperatures and climate change have received much attention, its effects on surface waters have received less. The increased levels of CO2 in the atmosphere also increase the amount of CO2 that is taken up by surface waters. Fig. 5.7 shows the increase in atmospheric CO2 from the years 1958 to 2015. Over this time period, the atmospheric CO2 levels have increased from about 315 ppm (parts per million) to about 395 ppm. 163 5.3 THE pH SCALE 8.33 400 8.28 375 8.23 350 8.18 325 8.13 300 8.08 FIG. 5.7 Time series of increasing CO2 in the atmosphere (red), measured at Mauna Loa, Hawaii, and in seawater (green), measured at nearby Aloha Station, accompanied by a decrease in seawater pH (blue). From R.A. Feely, NOAA’s Pacific Marine Environmental Laboratory carbon program. pH CO2 425 275 1955 1965 1975 1985 Year 1995 2005 8.03 2015 This increase is mirrored by an increase in the amount of CO2 dissolved in sea water as shown by open ocean measurements made from 1989 to 2013. After CO2 is dissolved in seawater, a Lewis acid-base reaction takes place where the water molecule acts as the Lewis base and the CO2 acts as the Lewis acid. When the carbon atom accepts a pair of electrons from the water molecule, it no longer needs to form double bonds with both of the oxygen atoms to complete its valence octet. An intermediate adduct is formed where carbon has one double-bonded oxygen atom and two single-bonded oxygen atoms. One of the single-bonded oxygen atoms carries a positive charge and the other carries a negative charge. A proton is then transferred from the positively charged oxygen atom to the negatively charged oxygen atom, resulting in an electrically neutral compound. The final product of this Lewis acid-base reaction between CO2 and water is the Brønsted-Lowry acid H2CO3 (carbonic acid). Carbonic acid is a diprotic acid, which rapidly ionizes in two steps losing up to two protons through Brønsted-Lowry acid-base reactions; H2 CO3 ðaqÞ + H2 OðlÞ > H3 O + ðaqÞ + HCO3 ðaqÞ HCO3 ðaqÞ + H2 OðlÞ > H3 O + ðaqÞ + CO3 2 ðaqÞ The lost protons are released into the seawater increasing the acidity and lowering the pH. So, dissolving CO2 in seawater increases the hydronium ion concentration, a process known as “ocean 164 5. ACIDS AND BASES acidification.” A decreasing trend in seawater pH, which accompanies the increase in CO2 uptake over the years 1989 to 2013, is also shown in Fig. 5.7. Before the industrial revolution began, at the end of the 18th century, the pH of seawater was about 8.2. In the 1990s, the pH of seawater had fallen to a value of 8.1. Remember that since pH is a logarithmic scale, this decrease of 0.1 pH unit represents an increase of H3O+ concentration of 18.9% over preindustrial times. At present, the pH of seawater is about 8.07 for an H3O+ increase of 28.8%. By the end of the century, the ocean pH is expected to drop by a further 0.4 pH units representing an increase in H3O+ concentration of 126.5% over preindustrial levels. The acid ionization constants for the three reactions that follow the dissolution of CO2 in seawater are: Ka1 ¼ ½H2 CO3 ¼ 1:2 105 ½CO2 ðaqÞ Ka2 ¼ ½H3 O + ½HCO3 ¼ 9:3 107 ½H2 CO3 ½H3 O + CO3 2 Ka3 ¼ ¼ 6:5 1010 ½HCO3 Collectively, CO2(aq), H2CO3, H2 CO3 , and CO3 2 are known as dissolved inorganic carbon (DIC). As can be seen in the acid ionization constants, the relative concentrations of the DIC species depend on the [H3O+] concentrations and therefore the pH of the seawater. This dependence of the DIC species on pH is shown in Fig. 5.8. The impacts of this lowering of the ocean’s pH reach beyond increasing the H3O+ content. As shown in Fig. 5.8, when the pH of seawater is lowered, the concentration of CO3 2 is reduced. This is because, since the ionization of carbonic acid produces two H3O+ ions for every one CO3 2 , FIG. 5.8 The distribution of dissolved inorganic carbon species (DIC) in seawater as a function of pH. The predicted change in pH between preindustrial times (1700s) and the turn of the century (2100) is shown by the vertical blue lines. The predicted change in free carbonate concentration is shown by horizontal dotted lines. 5.4 OTHER “p” FUNCTIONS 165 the percentage increase in H3O+ is larger than the percentage increase in carbonate, creating an imbalance in the reaction; HCO3 > CO3 2 + H + In order to maintain chemical equilibrium, some of the carbonate ions in the ocean combine with some of the hydronium ions to make further bicarbonate, lowering the free CO3 2 content of the oceans. The chemical principles behind this effect will be further discussed in Chapter 7. As shown in Fig. 5.8, the fraction of free carbonate content of the oceans in preindustrial times was about 0.14% or 14% of the total DIC. The fraction of free carbonate at the turn of the next century is predicted to decrease to about 0.05% or 5% of the total DIC, a reduction of 50%. This reduction in the free carbonate in the oceans will make it difficult for marine calcifying organisms to form biogenic calcium carbonate structures and existing structures will be more easily dissolved. Some of the vulnerable calcifying organisms are corals, coccolithophore algae, coralline algae, foraminifera, pteropods, shellfish, echinoderms (starfish and sea urchins), and crustaceans (crabs, lobsters, shrimp, etc.). Needless to say that declines in shellfish and crustacean populations would dramatically impact commercial fisheries. In addition, since many of the calcifying planktonic organisms, including foraminifera and pteropods, are at the base of the ocean’s food chains, their decline could potentially lead to negative feedbacks that will impact higher members of the food chain. Ocean acidification is a rising global problem as over a billion people around the world rely on some type of seafood as their primary source of protein. In addition, many jobs and world economies depend on the harvesting of fish and shellfish from our oceans. Some scientists consider ocean acidification a more important threat than global climate change. In order to prevent such a possible disaster, our emissions of carbon dioxide to the atmosphere must be substantially reduced. This will include the development of new sustainable energy sources as well as changing wasteful practices. This is one example of why the creative use of green engineering principles by future engineers will play a pivotal role in protecting our threatened resources while maintaining a thriving economy as well as a comfortable way of living. 5.4 OTHER “p” FUNCTIONS As with pH, a “p” function of any variable X is written as pX and is defined as the negative of the base 10 logarithm (log) of X. pX ¼ log ðXÞ (12) So, the hydroxide ion concentration, which also spans many orders of magnitude, can be represented by a logarithmic scale similar to that used for [H3O+]. This scale is called pOH and it is defined as the negative of the base 10 logarithm (log) of the hydroxide ion concentration in an aqueous solution as; pOH ¼ log ½OH Since, in pure water at 25°C, [H3O+] ¼ [OH] ¼ 1.0 107 M; (13) 166 5. ACIDS AND BASES pH ¼ pOH ¼ log 1:0 107 ¼ 7:00 In addition, we can also define a logarithmic (“p”) function for Kw as; pKw ¼ log ðKw Þ (14) + and since the values of [H3O ] and [OH ] are related by Kw, log Kw ¼ log ½H3 O + + log ½OH ¼ log 1:0 1014 or pKw ¼ pH + pOH ¼ 14:0 (15) This relationship between pH and pOH can be used to determine one of the values when the other is known. For example, a 0.001 M solution of the strong acid HCl has a [H3O+] of 0.001 M and the pH is given by; pH ¼ log(0.001) ¼ 3.00. So, the pOH is given by: pOH ¼ pKw pH ¼ 14:00 3:00 ¼ 11:00 There is an inverse relationship between the pH and pOH scales. The pOH scale actually runs opposite to the pH scale as shown in Fig. 5.9. The pH is equal to the pOH only for pure water at 25°C where pH ¼ pOH ¼ 7.0. EXAMPLE 5.4: DETERMINING THE PH AND POH FOR AN AQUEOUS SOLUTION OF A STRONG ACID OR A STRONG BASE What is the pH and pOH of an aqueous solution of 0.025 M sodium hydroxide at 25°C? 1. Determine the hydroxide ion concentration. Since NaOH is a strong base, it is totally ionized in aqueous solution. [NaOH] ¼ [OH] ¼ 0.025 M 2. Determine the pOH. pOH ¼ log(0.025) ¼ 1.60 3. Determine the pH. pKw ¼ pH + pOH ¼ 14 pH ¼ 14 1.60 ¼ 12.4 FIG. 5.9 The relationship between the pH and pOH scales. Red indicates acidic solutions and blue indicates basic solutions. From Patricia R, Wikimedia Commons. 167 5.4 OTHER “p” FUNCTIONS The acid ionization constants also span many orders of magnitude. Those listed in Table 5.2 vary from 1.0 to 1010. Because of this, it is also convenient to express the Ka values as “p” functions when comparing the relative strengths of acids. pKa ¼ log ðKa Þ ¼ log ½H3 O + ½A ½HAw (16) Table 5.4 lists the pKa values for the same acids listed in Table 5.2. You can see that it is much easier to evaluate the strength of the acids by looking at the pKa values than the Ka values. The larger the value of the pKa, the smaller the value of the Ka and the smaller the extent of dissociation of the acid in aqueous solution. That is, the larger the pKa the weaker the acid. As the pKa of weak acids become smaller, the acid strength increases. Conversely, as the pKa becomes larger, the strength of the acid decreases. TABLE 5.4 Ionization Constants (Ka) and pKa Values for Some Weak Acids in Water at 25°C Acid Hydronium ion Sulfurous acid Hydrogen sulfate ion Phosphoric acid Hydrofluoric acid Nitrous acid Formic acid Formula + H3O 1.0 H2SO3 HSO4 pKa Ka H3PO4 HF HNO2 HCO2H 0.0 2 1.2 10 2 1.2 10 3 7.5 10 4 7.2 10 4 4.5 10 4 1.8 10 5 1.92 1.92 2.12 3.14 3.35 3.74 Benzoic acid C6H5CO2H 6.3 10 4.20 Acetic acid CH3CO2H 1.8 105 4.74 Carbonic acid H2CO3 4.2 107 6.38 Hydrogen sulfide H2S 1.0 107 7.0 Dihydrogen phosphate ion H2 PO4 6.2 108 7.21 Hydrogen sulfite ion HSO3 6.2 108 7.21 Hypochlorous acid HClO 3.5 108 7.46 Ammonium ion NH4 + 5.6 1010 9.25 Hydrocyanic acid HCN 4.0 1010 9.40 Hydrogen carbonate ion HCO3 4.8 1011 10.32 Hydrogen phosphate ion HPO4 2 3.6 1013 12.44 Water H2O 1.0 1014 14.0 19 Hydrogen sulfide ion HS 1.0 10 19.0 168 5. ACIDS AND BASES A weak acid has a pKa value in the approximate range of 0–12 in water. Acids with a pKa value of less than 0 are considered to be strong acids since they are completely dissociated in water and the concentration of the undissociated acid is too small to be measured. EXAMPLE 5.5: DETERMINING THE RELATIVE STRENGTHS OF ACIDS List the following acids in the order of increasing acid strength: HNO2, H2CO3, HCN, HF, H2SO3, and HBr. 1. Determine the pKa values. HBr is a strong acid pKa < 1.0. pKa: HNO2 ¼ 3.35, H2CO3 ¼ 6.38, HCN ¼ 9.40, HF ¼ 3.14, H2SO3 ¼ 1.92 2. List the acids in order of decreasing pKa. HCN > H2CO3 > HNO2 > HF > H2SO4 > Br The strength of the acid increases with decreasing pKa. 3. List the acids in the order of increasing acid strength. HCN < H2CO3 < HNO2 < HF < H2SO4 < HBr 5.5 BUFFER SOLUTIONS A buffer solution (or simply a buffer) is a solution which resists changes in pH when small quantities of a strong acid or a strong base are added to it. So, buffer solutions are used as a means of keeping the pH at a nearly constant value. They are used widely in both biochemical and industrial applications. In biochemistry, buffers are necessary for assays used to study the activity of enzymes. Since enzyme activity varies with pH, the pH must remain constant during the assay to get accurate results. Buffer solutions are also used in medicines that require a constant pH to maintain their activity. The textile industry uses buffer solutions to keep the pH within narrow limits during fabric dyeing and processing. Many laundry detergents also use buffers to prevent their natural ingredients from breaking down. Buffer solutions usually consist of a weak acid and its conjugate base in relatively equal concentrations. Practically, this is achieved by mixing a soluble compound that contains the conjugate base with an aqueous solution of the acid. For example, an acetate buffer is made by the addition of sodium acetate to an aqueous solution of acetic acid. Buffer solutions achieve their resistance to pH change because of the excess amount of the conjugate base and the equilibrium between the weak acid (HA) and its conjugate base (A). When a small amount of a strong acid is added to a buffer solution, the excess conjugate base present in the buffer consumes the added hydronium ion from the strong acid converting it into water and the weak acid of the conjugate base. 5.5 BUFFER SOLUTIONS 169 A ðaqÞ + H3 O + ðaqÞ ! H2 OðlÞ + HAðaqÞ This results in a decrease in the amount of the excess conjugate base and an increase in the amount of the unionized weak acid in the solution. So, the pH of the buffer solution remains relatively stable and may decrease by only a very small amount when a small amount of the strong acid is added to it. Similarly, when a small amount of a strong base is added to a buffer solution, the added hydroxide ions are consumed by the weak acid forming water and the conjugate base of the acid. OH ðaqÞ + HAðaqÞ ! H2 OðlÞ + A ðaqÞ The result is that the amount of the weak acid decreases and the amount of the conjugate base increases. This consumption of hydroxide prevents the pH of the solution from rising significantly, which would occur if the buffer system was not present. The pH of a buffer solution can be calculated from the concentrations of the various components and the acid ionization constant. For example, in an aqueous solution containing acetic acid, the equilibrium between the unionized acid and its conjugate base is; CH3 CO2 HðaqÞ + H2 OðlÞ $ H3 O + ðaqÞ + CH3 CO2 ðaqÞ with an ionization constant of; Ka ¼ ½CH3 CO2 ½H3 O + ¼ 1:8 105 ½CH3 CO2 H In an aqueous solution containing only acetic acid, the concentrations of the hydronium ions and the acetate ions are equal because the ionization of one acetic acid molecule results in one hydronium ion and one acetate ion. This is no longer true for an acetate buffer solution because of the added sodium acetate. In the buffer solution, the concentration of acetate ions coming from the ionization of the weak acetic acid is negligible compared to the concentration of acetate ions from the added sodium acetate. So, the ½CH3 CO2 is assumed to be the same as the sodium acetate concentration. Similarly, the [CH3CO2H] is assumed to be the same as the concentration of the original acetic acid solution because very little of the acid is ionized due to the presence of the excess acetate. The [H3O+] concentration can then be calculated from the ionization constant and the concentrations of the acetic acid solution and the sodium acetate. Ka ¼ ½NaCH3 CO2 ½H3 O + ¼ 1:8 105 ½CH3 CO2 H 1:8 105 ½CH3 CO2 H ½H3 O ¼ ½NaCH3 CO2 + The pH of the buffer solution is determined from the [H3O+]. 170 5. ACIDS AND BASES EXAMPLE 5.6: DETERMINING THE PH OF A BUFFER SOLUTION What is the pH of a buffer solution made with 10.00 g of sodium acetate in 200.00 mL of 1.00 M acetic acid? 1. Calculate the concentration of sodium acetate in solution. (10.00g)/(82.03 g/mol) ¼ 0.12 mol (0.12 mol)/(0.20 L) ¼ 0.61 M 2. Determine the hydronium ion concentration. ½CH3 CO2 ¼ 1:8 105 ð0:61M=1:00MÞ ½H3 O + ¼ Ka ½CH3 CO2 H ¼ 1:09 105 3. Calculate the pH. pH ¼ log(1.10 105) ¼ 4.96 EXAMPLE 5.7: DETERMINING THE PH OF A BUFFER SOLUTION AFTER THE ADDITION OF A SMALL AMOUNT OF A STRONG ACID OR BASE What is the pH of the buffer solution in Example 5.6 after the addition of 2.00 mL of 6.00 M HCl? 1. Determine the initial amount of acid and conjugate base. 0.12 mol sodium acetate (10.00 g)/(60.05 g/mol) ¼ 0.17 mol acetic acid 2. Determine the moles of H3O+ added. (6.00 mol/L)(0.002 L) ¼ 0.012 mol H3O+ 3. Determine the new concentrations. Since all the H3O+ reacts with the conjugate base to give acetic acid; 0.17 + 0.012 ¼ 0.182 mol acetic acid 0.12 – 0.012 ¼ 0.108 mol acetate 4. Determine new molarity. (Remember that the new volume is 202 mL.) (0.182 mol)/(0.202 L) ¼ 0.90 M acetic acid (0.108 mol)/(0.202 L) ¼ 0.53 M acetate 5. Determine the new pH. ð0:90MÞ ½CH3 CO2 ¼ 1:8 105 ½CH3 CO2 H ð0:53MÞ [H3O+] ¼ 3.06 10–5 pH ¼ log(3.06 105) ¼ 4.51 ½H3 O + ¼ Ka It is the actual concentrations of the weak acid and its conjugate base (HA and A–) that determine the effectiveness of a buffer. The higher the concentrations of A– and HA, the less the effect of the addition of a strong acid or strong base on the pH of the solution. As long as there is excess conjugate base in the solution, the addition of a strong acid will not significantly affect the pH. But, if enough strong acid is added to the solution, the conjugate base 5.5 BUFFER SOLUTIONS 171 will eventually be depleted by reaction of the hydronium ion with the conjugate base. At this point, the buffer will no longer function and the pH will rapidly decrease. Similarly, a buffer solution will cease to function if enough strong base is added to the solution. All the weak acid will be consumed by the reaction of hydroxide ion with the weak acid. Then the buffer will also no longer function and the pH will rapidly increase. The amount of strong acid or strong base that a buffer solution can absorb before it ceases to function is called the buffer capacity. The useful pH range of any buffer is determined by the Ka of the weak acid and the ratio of the concentrations of the weak acid and the conjugate base. The buffer will be most effective for stabilizing the pH of the solution when the ratio [A]/[HA] is equal to one. This is because if either concentration is in excess over the other, the chemical equilibrium will cause the concentration of the lower species to further decrease, reducing the effectiveness of the buffer. So, the maximum buffer capacity for any buffer solution exists when [HA] ¼ [A]. According to the acid ionization constant (Ka ¼ [H3O+][A–]/[HA]), when these concentrations are equal; Ka ¼ ½H3 O + and pKa ¼ pH (17) This relationship is useful in choosing a buffer solution for a particular application. For example, if you need a buffer to cover a pH range of 4.5–5.0, the best choice would be an acetic acid/sodium acetate buffer with [CH3CO2H] ¼ ½CH3 CO2 because the pKa of 4.74 for acetic acid is in the desired pH range. Eq. (17) is a special case of the Henderson-Hasselbalch equation, an equation that describes the relationship between the pH of a solution with the pKa and the ratio of the concentrations of the acid and conjugate base; pH ¼ pKa + log ½A ½HA (18) This equation is useful in calculations of buffers that do not have the acid and conjugate base in equal concentrations. In these cases, the pH can be calculated directly by the HendersonHasselbalch equation without the need of first determining [H3O+] from the Ka. EXAMPLE 5.8: DETERMINING THE PH OF A BUFFER SOLUTION USING THE HENDERSON-HASSELBALCH EQUATION Determine the pH of a buffer solution containing 0.10 M carbonic acid and 0.5 M sodium hydrogen carbonate? 1. Write the chemical equation for the buffer equilibrium. The acid is H2CO3 and the conjugate base is HCO3 . H2 CO3 ðaqÞ + H2 OðlÞ > H3 O + ðaqÞ + HCO3 ðaqÞ 2. Determine the pH from the Henderson-Hasselbalch equation. pKa of H2CO3 ¼ 6.38 ½A pH ¼ pKa + log ½HA pH ¼ 6.38 + log(0.5/0.10) ¼ 6.38 + log(5.0) ¼ 6.38 + 0.70 ¼ 7.08 172 5. ACIDS AND BASES The Henderson-Hasselbalch equation can also be used to design a buffer with a specific pH. For example, if a buffer is required to maintain a pH value of 4.60, the most appropriate acid system would be acetic acid with a pKa of 4.74 (from Table 5.4). The required ratio of the concentrations of acid [CH3CO2H] and conjugate base [NaCH3CO2] needed to give a pH value of 4.60 are given by; pH ¼ pKa + log pH pKa ¼ log ½NaCH3 CO2 ½CH3 CO2 H ½NaCH3 CO2 ¼ 4:60 4:74 ¼ 0:14 ½CH3 CO2 H ½NaCH3 CO2 ¼ 100:14 ¼ 0:72 ½CH3 CO2 H The ratio of 0.72 can be obtained in several ways. If we choose a concentration of acetic acid to be 0.1 M, then the required concentration of sodium acetate would be; ½NaCH3 CO2 ¼ ð0:72Þ ð0:10 MÞ ¼ 0:072 M 5.6 THE TITRATION A titration is a common laboratory method used to determine the unknown concentration of an analyte. A solution of known concentration, called the titrant, is added slowly to a known volume of the analyte until the reaction is complete. The added volume of titrant is accurately measured and the concentration of the analyte is determined from the concentration and volume of titrant added and the volume of the analyte. Since the measurement of volumes plays a key role, this type of analysis is known as a volumetric analysis. Titrations are most commonly associated with acid-base reactions, but they can also be used to determine the concentrations of reactants in other types of chemical reactions. An acid-base titration involves the determination of the concentration of an acid or base by exactly neutralizing the acid or base with an acid or base of known concentration. Neutralization is a type of chemical reaction in which an acid and a strong base react completely with each other resulting in a solution that is neither acidic nor basic. The net ionic equation for the neutralization reaction between an acid and a strong base is just the reaction of hydronium ion with hydroxide ion to give water, which is the reverse of the autoionization of water. H3 O + + OH ! 2H2 O The complete reaction of the acid and a base allows for determination of the concentration of the unknown acid or basic solution when the concentration of the other is known. A typical acid-base titration begins by dispensing an accurately known volume of the acid or base whose concentration is unknown into a flask with a small amount of indicator. The indicator is designed to change color at the exact point that the reaction is complete. The titrant is contained in a calibrated burette shown in Fig. 5.10, a device used for dispensing accurately measured amounts of a solution. The burette is suspended above the flask containing the analyte and small volumes of the titrant are added to the analyte and indicator until the indicator changes color, signaling that the reaction is complete. This point in the titration is called the end 5.6 THE TITRATION 173 FIG. 5.10 A typical acid-base titration. In this experiment, the titrant in the burette is a base and the analyte in the flask is an acid. The indicator is phenolphthalein, which turns pink at the end point of the acid-base titration. From Theresa Knott, Wikimedia Commons. point or equivalence point. When the endpoint of the reaction is reached, the volume of reactant consumed is measured and used to calculate the concentration of analyte as; Ca Va ¼ Ct Vt (19) where Ca is the molar concentration of the analyte, Va is the volume of the analyte in liters, Ct is the molar concentration of the titrant, and Vt is the volume of the titrant in liters. EXAMPLE 5.9: DETERMINING THE CONCENTRATION OF AN ACID BY TITRATION The titration of 50.0 mL of HCl with 0.15 M NaOH reaches an endpoint at 26.50 mL. What is the concentration of the HCl? CHCl VHCl ¼ CNaOH VNaOH CHCl ¼ CNaOH VNaOH ð0:15MÞð0:0265LÞ ¼ ¼ 0:075 M HCl VHCl ð0:050LÞ 174 5. ACIDS AND BASES CASE STUDY: BIODIESEL MANUFACTURING Biodiesel is a renewable replacement for petroleum diesel that has the potential to reduce US dependence on foreign oil, create jobs, and improve the environment. It is produced at plants in nearly every state in the country. Biodiesel can be manufactured from recycled cooking oil, soybean oil, and animal fats. It can be used in existing diesel engines without modification while meeting strict technical fuel quality and engine performance specifications (Fig. 5.11). The use of biodiesel is covered by the warranties of all major engine manufacturers, although most often for blends of up to 5%–20% biodiesel in petroleum diesel. The manufacture of biodiesel involves a reaction of vegetable oil or animal fats with methanol. This reaction requires an alkaline reagent, usually sodium hydroxide, for the reaction to proceed. (The complete reaction of methanol with oils to form biodiesel will be covered in more detail in Chapter 13.) The waste oils used to manufacture biodiesel also contain free fatty acids (FFA), which will interfere with the biodiesel reaction as they will react with the sodium hydroxide, reducing its concentration before all the oils are converted to the biodiesel product. The reaction of vegetable oils with methanol to produce biodiesel requires 3.5 g sodium hydroxide per liter of oil. So, in order for all of the vegetable oil to react to form biodiesel, more than the required 3.5 g/L sodium hydroxide must be added to react with the FFA. A titration is performed on the oil in order to determine how much of the FFA is present and how much extra sodium hydroxide is required to drive the biodiesel reaction to completion. The titration is performed by dripping 0.025 M NaOH into a mixture of 1 mL of the waste vegetable oil in an isopropyl alcohol solvent. Phenolphthalein can be used as an end point indicator, as it changes from colorless to bright pink when the end point is reached. However, a more accurate way of determining the end point of an acid-base titration is to measure the change in pH of the solution after every addition of the titrant. The results are then plotted as pH vs volume of titrant added. The resulting graphical plot is called a titration curve. An example of a titration curve for the titration of waste vegetable oil with 0.025 M NaOH is shown in Fig. 5.12. You can see in the titration curve that the pH rises very slowly until the reaction is near the equivalence point. As the reaction approaches the equivalence point, there is a steep slope in the curve. Once the equivalence point is passed, the curve returns to a relatively constant pH with the addition of the titrant. The equivalence point is FIG. 5.11 Bus running on soybean biodiesel. From U.S. Department of Energy; Energy Efficiency and Renewable Energy, via Wikimedia Commons. 5.6 THE TITRATION 175 FIG. 5.12 A titration curve for the titration of waste vegetable oil with 0.025 M NaOH equivalence point is at the halfway point in the steep portion of the curve. The volume of titrant at the equivalence point is the amount required for the reaction FFA + NaOH to go to completion. The green lines are used to determine the exact pH of the midpoint of the titration, which is halfway between the beginning of the titration and the equivalence point. determined as the halfway point between the two flat portions of the curve. The blue lines in Fig. 5.12 are used to determine the exact position of the equivalence point from the titration curve. The volume of NaOH solution used to reach the equivalence point in the titration represents the amount of extra NaOH needed to be added to the biodiesel reaction in order to completely neutralize the FFA. This amount will be added to the reaction in addition to the 3.5 g/L required for the reaction of the oil with methanol. For example, according to Fig. 5.12, it takes 3.5 mL of 0.025 M NaOH solution to reach the equivalence point of the titration. ð0:0035 LÞð0:025 mol=LÞ ¼ 8:75 105 mol NaOH needed to react with FFA 8:75 105 mol 40g=mol ¼ 0:0035g NaOH for 1mL of oil ¼ 3:5g NaOH for 1L of oil So, 3.5 g of NaOH per liter of oil will be required to completely neutralize the FFA. This amount of NaOH is in addition to the 3.5 g/L required for the methanol-oil reaction, for a total of 6.5 g NaOH per liter of waste oil required for the biodiesel reaction to go to completion. The use of a titration curve instead of an equivalence point indicator gives much more information than just the concentration of the acid or the amount of titrant needed. As shown in Section 5.5, when the concentrations of a weak acid and the conjugate base are equal ([A] ¼ [HA]), the ratio [A]/[HA] ¼ 1. So, from the Henderson-Hasselbalch equation, pH ¼ pKa From the titration curve in Fig. 5.12, the concentrations of the FFA and their conjugate base are equal at the midway point between the beginning of the titration (0 mL) and the equivalence point (3.5 mL). This occurs at the addition of 1.75 mL of titrant, which is at a pH ¼ 4.0. So, the pKa of the FFA in the vegetable oil is 4.0. 176 5. ACIDS AND BASES IMPORTANT TERMS Acid-base adduct the product of a Lewis acid-base reaction. Acid ionization constant (Ka) the ratio of the concentrations of the ionized products (conjugate acid and conjugate base) to the concentration of the unionized acid in the aqueous solution at equilibrium. Amphoteric a compound that can act as both a Brønsted-Lowry acid and as a Brønsted-Lowry base. Autoionization an amphoteric compound which can react with itself. Autoionization constant of water (Kw) the product of the concentrations of the ionized products (conjugate acid and conjugate base). Base ionization constant (Kb) the ratio of the concentrations of the ionized products (conjugate acid and conjugate base) to the concentration of the unionized base in the aqueous solution at equilibrium. Brønsted-Lowry acid a proton donor. Brønsted-Lowry base a proton acceptor. Buffer capacity the amount of strong acid or strong base that a buffer solution can absorb before it ceases to function Buffer solution a solution which resists changes in pH when small quantities of a strong acid or a strong base are added to it. Burette a device used for dispensing variable, accurately measured amounts of a solution. Chemical equilibrium the state in which both reactants and products are present in concentrations which have no further tendency to change with time. Complex ion a coordination complex with a metal ion at its center and a number of other neutral molecules attached to it by coordinate covalent bonds. Conjugate acid an acid that has lost a proton. Conjugate base a base that has gained a proton. Conjugate pairs an acid and its conjugate base or a base and its conjugate acid. Coordination complex the product of a Lewis acid-base reaction in which neutral molecules or anions bond to a central metal atom or ion by coordinate covalent bonds. Coordinate covalent bond a bond where both electrons are provided lone pair on a Lewis base. Diprotic acid a Brønsted-Lowry acid that can donate two protons per molecule to an aqueous solution. Electrophile a chemical species that is attracted to electrons. End point the point in a titration when the chemical reaction is exactly complete. Equivalence point the point in a titration when the equal quantities of reactants have been combined. Henderson-Hasselbalch equation an equation that describes the relationship between the pH of a solution with the pKa and the ratio of the concentrations of the acid and conjugate base. Hydronium ion the common name for the aqueous cation H3O+ produced by protonation of water. Hypervalency a molecule that contains one or more main group elements formally bearing more than eight electrons in their valence shells. Indicator a chemical compound that changes color at the end point of a titration. Ionization the process by which an atom or a molecule acquires a negative or positive charge by gaining or losing electrons to form ions. Lewis acid a chemical species that can accept a lone pair of electrons to form a new covalent bond. Lewis base a chemical species that can donate a lone pair of electrons to form a new covalent bond. Ligands molecules or anions that bond to a central metal atom by coordinate covalent bonds. Monoprotic acid a Brønsted-Lowry acid that can donate only one proton per molecule to an aqueous solution. Neutralization is a type of chemical reaction in which an acid and base react completely with each other. Nucleophile a chemical species that donates an electron pair to an electrophile to form a chemical bond. pH scale the negative of the base-10 logarithm (log) of the hydronium ion concentration in an aqueous solution. Polyprotic acid a Brønsted-Lowry acid that can donate more than one proton per molecule to an aqueous solution. Strong acid a Brønsted-Lowry acid with very strong ability to donate its proton to water converting entirely to the conjugate acid and the conjugate base. Strong base a Brønsted-Lowry base that will completely ionize in aqueous solution. Titrant a solution of known concentration used in a titration. STUDY QUESTIONS 177 Titration a common laboratory method used to determine the unknown concentration of an analyte by measuring the volume of a solution of known concentration required to completely react with the analyte. Titration curve a graphical plot of pH of solution vs volume of titrant added. Triprotic acid a Brønsted-Lowry acid that can donate three protons per molecule to an aqueous solution. Volumetric analysis an experimental technique that involves the quantitative determination of a substance based on the measurements of volumes. STUDY QUESTIONS 5.1 5.2 5.3 5.4 5.5 5.6 5.7 5.8 5.9 5.10 5.11 5.12 5.13 5.14 5.15 5.16 5.17 5.18 5.19 5.20 5.21 5.22 5.23 5.24 5.25 5.26 5.27 5.28 5.29 5.30 5.31 5.32 What is the definition of a Brønsted-Lowry acid? What is the definition of a Brønsted-Lowry base? What is a conjugate acid? What is a conjugate base? An acid and its conjugate base are known as what? When is a chemical reaction in equilibrium? As the strength of an acid increases, does the strength of its conjugate base increase or decrease? What is the chemical formula for a proton in aqueous solution? What is the molecular geometry for the species in Question 5.8? (Refer to Table 3.7) What is the name of the species in Question 5.8? What is an amphoteric compound? What is meant by autoionization? Which are the polyprotic acids in the following: H3PO4, HCl, NH4 + , H2PO4, HNO3, H3O+, H2O, HCO3 ? Which are the monoprotic acids in Question 5.11? What is the definition of a Lewis acid? What is the definition of a Lewis base? What is the definition of an electrophile? A chemical species that reacts with an electrophile is called what? Is an electrophile a Lewis acid or a Lewis base? Is a nucleophile a Lewis acid or a Lewis base? How is a coordinate covalent bond different from other covalent bonds? What is the product of a Lewis acid-base reaction called? What four types of chemical species can act as Lewis acids? How is a coordination complex different from other acid-base adducts? What is a Lewis base that is bonded to a central metal cation by a coordinate covalent bond called? What is a complex ion? What is the molecular geometry of a complex ion formed by a metal cation bonded to six water molecules? Why do aqueous solutions of some metal ions have acidic properties? What is hypervalency? What four types of chemical species can act as Lewis bases? What determines the strength of a Brønsted-Lowry acid or base? Name the seven strong Brønsted-Lowry acids. 178 5.33 5.34 5.35 5.36 5.37 5.38 5.39 5.40 5.41 5.42 5.43 5.44 5.45 5.46 5.47 5.48 5.49 5.50 5.51 5.52 5.53 5.54 5.55 5.56 5.57 5. ACIDS AND BASES What are the strong Brønsted-Lowry bases? What is the difference between a strong acid and a weak acid? What is an acid ionization constant (Ka) equal to? What is a base ionization constant (Kb) equal to? What is the autoionization constant of water (Kw) equal to? What is the value of Kw at 25°C? What is the hydronium ion concentration in a neutral aqueous solution at 25°C? What is the hydroxide ion concentration in a neutral aqueous solution at 25°C? What is pH? What is the pH of a neutral aqueous solution at 25°C? Will the value of the pH of acidic aqueous solutions at 25°C be higher or lower than that of a neutral aqueous solution? Will the pH of basic aqueous solutions at 25°C be higher or lower than that of a neutral aqueous solution? What is pOH? What is the value of the pOH of a neutral aqueous solution at 25°C? What is the relationship between pH, pOH, and pKw? What is the value of pKw at 25°C? What is pKa? What is a buffer solution? What is buffer capacity? The maximum buffer capacity for any buffer is at what concentration of acid and conjugate base? If the buffer is at maximum capacity (concentrations in Question 5.52), what is the pH of the buffer solution? What is a titration? What is the titrant? What is a titration indicator? What is the end point of a titration? PROBLEMS 5.58 List the conjugate bases for the following acids: (a) H3PO4, (b) HCl, (c) NH4 + , (d) H2SO4, (e) H3O+. 5.59 List the conjugate acids for the following bases: (a) OH, (b) PO4 3 , (c) HCO3 , (d) SO4 2 , (e) H2O. 5.60 What are the products of each of the following reactions? (a) HSO4 + H2 O ! (b) H2CO3 + OH ! (c) HNO3 + H2O ! (d) H3O+ + F ! (e) NH4 + + H2 O ! (f) HCO3 + OH ! PROBLEMS 179 5.61 List the acid and conjugate base pairs for each of the reactions in Problem 5.60. 5.62 List the base and conjugate acid pairs for each of the reactions in Problem 5.60. 5.63 Which of the following compounds are amphoteric: HSO4 , OH, H2O, HCl, H2 PO4 , NH4 + ? 5.64 Write the balanced chemical equation for the autoionization of water. 5.65 Which is the weaker acid: (a) HSO4 or H2SO4, (b) H3O+ or H2O, (c) H3PO4 or H2 PO4 , (d) H2CO3 or HCO3 ? 5.66 Which of the following compounds can act as Lewis acids and which can act as Lewis bases: (a) BCl3, (b) Be2+, (c) Cl, (d) CO3 2 , (e) CO2, (f) H2O? 5.67 Which of the following acid-base reactions can only be described by the Lewis definition: (a) (b) (c) (d) HCl(aq) + NH3(aq) ! NH4Cl(aq) HNO3(aq) + NaOH(aq) ! NaCl + H2O(l) Zn(OH)2(s) + 2OH(aq) ! [Zn(OH)4]2(aq) H2 SO4 ðaqÞ + H2 OðlÞ ! H3 O + + SO4 2 5.68 Write the chemical equation for the aqueous ionization and the expression of the acid ionization constant for the following weak acids: (a) HF, (b) HSO4 , (c) H2CO3, (d) H2 PO4 . 5.69 Write the chemical equation for the aqueous ionization and the expression of the base ionization constant for the reaction of each of the following weak bases: (a) F, (b) HPO4 2 , (c) H2 PO4 , (d) CO3 2 . 5.70 A 2.0 M solution of HCl contains what concentration of chloride ions? 5.71 How many Na+ ions are there in 100 mL of an aqueous solution of 0.05 M sodium hydroxide? 5.72 Which is the stronger acid: H2SO3 with a Ka of 1.2 102 or HF with a Ka of 7.2 104? 5.73 Which is the stronger base: NO2 with a Kb of 2.2 1011 or NH3 with a Kb of 1.8 105? 5.74 What are the hydroxide and hydronium ion concentrations in a 0.0015 M aqueous solution of NaOH at 25°C? 5.75 What are the hydroxide and hydronium ion concentrations in a 0.013 M aqueous solution of HNO3 at 25°C? 5.76 What are the hydroxide and hydronium ion concentrations in a 5.0 103 M aqueous solution of H2SO4 at 25°C? 5.77 What is the pH of 250 mL of an aqueous solution containing 0.465 g of HNO3 at 25°C? 5.78 What is the pH of a 0.0012 M aqueous solution of NaOH at 25°C? 5.79 What is the hydronium ion and hydroxide ion concentrations in an aqueous solution with a pH of 4.32 at 25°C? 5.80 A solution of the strong base Sr(OH)2 has a pH of 10.46 at 25°C. What is the initial concentration of Sr(OH)2? 5.81 What is the pH and pOH of a 0.0023 M solution of HNO3 at 25°C? 5.82 List the following acids in order of increasing strength: NH4 + (pKa ¼ 9.25), HNO2 (pKa ¼ 3.35), H2S (pKa ¼ 7.0), H2SO3 (pKa ¼ 1.29), HNO3 (pKa ¼ –1.3), and H2O (pKa ¼ 14.0). 5.83 Which is the strongest acid in aqueous solution: HF, H2CO3, H3O+, or H3PO4? 180 5. ACIDS AND BASES 5.84 What is the pH of a buffer solution which is 2.00 M in CH3CO2H and 2.00 M in NaCH3CO2? 5.85 What is the [H3O+] and the pH of a buffer solution made from 500 mL of 0.30 M hydrofluoric acid and 500 mL of 0.70 M sodium fluoride? 5.86 What is the [H3O+] and pH of the buffer in Problem 5.86 after the addition of 0.08 mol of NaOH? 5.87 What is the [H3O+] and pH of the buffer in Problem 5.86 after the addition of 0.04 mol of HCl? 5.88 How many grams of sodium benzoate (NaC6H5CO2 ) must be added to 750 mL of 0.200 M benzoic acid (C6H5CO2H) to make a buffer solution with a pH of 4.00? 5.89 What buffer systems (acid and conjugate base) would be best to obtain the following pHs: (a) 6.45, (b) 3.45, (c) 4.65, (d) 9.00? 5.90 What ratio of conjugate base to acid (A–/AH) is required to give exactly the pHs listed in Problem 5.89? 5.91 If it takes 18.3 mL of a 0.115 M sodium hydroxide solution to completely titrate 25.0 mL of a nitric acid solution, what is the molar concentration of the nitric acid? 5.92 If 11.6 mL of a 3.0 M sulfuric acid are required to neutralize 25.00 mL of a NaOH solution, what is the molarity of the NaOH solution? C H A P T E R 6 Properties of Gases O U T L I N E 6.1 A Historical Perspective 181 6.7 Partial Pressures 204 6.2 Boyle’s Law 185 6.8 Chemical Reactions With Gases 207 6.3 Charles’ Law 188 Important Terms 208 6.4 Gay-Lussac’s Law 195 Study Questions 209 6.5 The Ideal Gas Law 197 Problems 210 6.6 Nonideal Gas Behavior 201 6.1 A HISTORICAL PERSPECTIVE Many early chemists in the 18th century concentrated their efforts on the study of different gases, which they called “airs,” and how they were produced in chemical reactions. This widespread study of gases was prompted by the invention of the pneumatic trough by Stephen Hales (Fig. 6.1). The pneumatic trough was an apparatus used to collect the gas given off by a chemical reaction for further study. It consisted of a large glass dish containing water and a gas bottle suspended above the dish to hold the collected gas. The bottle was filled with water, inverted, and placed into the water-filled dish. The outlet tube from the chemical reaction was inserted into the opening of the bottle so that the gas products from the reaction could bubble up into the bottle displacing the water and trapping the gas in the bottle. Because of the extensive use of this apparatus in the 1700s, the group of chemists involved in the study of gases was known as pneumatic chemists. Some of the pneumatic chemists who were credited with discovering some important gas species using the pneumatic trough include: Joseph Black, Daniel Rutherford, Joseph Priestly, and Henry Cavendish. The gases that each chemist discovered are listed in Table 6.1 along with the original names given to them. Joseph Black discovered carbon dioxide in 1757 by exposing limestone (CaCO3) to acids. This gas, which he called “fixed air,” General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00006-0 181 # 2018 Elsevier Inc. All rights reserved. 182 6. PROPERTIES OF GASES FIG. 6.1 The pneumatic trough invented by Stephen Hales for the collection of gases from chemical reactions. From “Vegetable Statics” by Stephen Hales, via Wikimedia Commons. TABLE 6.1 The Gases Discovered by the Pneumatic Chemists From 1757 to 1776 Along With the Original Names Given to Them Gas Year Discoverer Original Name CO2 1757 Joseph Black Fixed air N2 1772 Daniel Rutherford Noxious air NO 1772 Joseph Priestly Nitrous air NO2 1772 Joseph Priestly Red nitrous vapor N2O 1772 Joseph Priestly Diminished nitrous air HCl 1772 Joseph Priestly Acid air CO 1772 Joseph Priestly Inflammable air NH3 1773 Joseph Priestly Alkaline air O2 1774 Joseph Priestly Dephlogisticated air SO2 1774 Joseph Priestly Vitriolic air SiF4 1775 Joseph Priestly Fluor acid air H2 1776 Henry Cavendish Inflammable air 6.1 A HISTORICAL PERSPECTIVE 183 was observed to be denser than atmospheric air and was seen to extinguish a flame and not support animal life. Daniel Rutherford, a student of Black’s, isolated nitrogen gas in 1772, which he called “noxious air.” The isolation procedure consisted of systematically exposing atmospheric air to a series of experiments that were known to destroy other gases, such as a burning candle (for oxygen) or bubbling through a lime solution (for carbon dioxide). Joseph Priestly, perhaps the most prolific of the pneumatic chemists, is credited with discovering a total of nine gases from 1772 to 1775. These are: nitrogen monoxide (or nitric oxide), nitrogen dioxide, dinitrogen monoxide (or nitrous oxide), hydrochloric acid, carbon monoxide, ammonia, oxygen, sulfur dioxide, and silicon tetrafluoride. His success was primarily due to the use of mercury instead of water in the pneumatic trough. This simple adaptation allowed him to isolate and examine gases that were soluble in water. He also invented a procedure for making soda water by dissolving Black’s “fixed air” into an agitated bowl of water. Priestley published the invention in the paper “Impregnating Water with Fixed Air” and it has been said that he referred to it as his “happiest discovery.” Henry Cavendish is credited with the discovery of hydrogen gas in 1776 while studying the properties of the gas produced when metals were dissolved in acids. He unfortunately called this gas “inflammable air,” which was the name Priestly had given to carbon monoxide. The pneumatic chemists who were more interested in studying the properties of gases, including the effects of pressure and temperature, were aided by the invention of the barometer by Evangelista Torricelli in 1643. Torricelli was the first to suggest that atmospheric air had weight and that the force of the atmosphere pushing down on the surface of a liquid could drive it into an evacuated tube up to a certain height. He constructed an apparatus made of a tube approximately 1 m long, sealed at the top, and filled with mercury (Fig. 6.2). He inverted the tube into a basin of mercury. After inverting the tube, the mercury in the tube dropped to a height of about 76 cm, leaving an empty space at the top of the tube above the column of mercury. Torricelli explained that the weight of air pushing down on the dish of mercury prevented the mercury in the tube from draining out completely and the height (h) of the column of mercury could be used as a measure of the pressure of the atmospheric air. He noticed that the level of the fluid in the tube changed slightly each day and concluded that this was due to the changing pressure of the atmosphere. This device was the first barometer, and adaptations of this approach allowed the pneumatic chemists to measure the pressure of gases in their experiments. In 1660, Robert Boyle published the paper “New Experiments Physico–Mechanical, Touching the Spring of Air and its Effects,” which detailed the relationship between the volume of a gas and its pressure. This relationship between the volume and pressure of a gas, later called Boyle’s law, is described in detail in Section 6.2. In 1801, John Dalton presented data describing the relationship between the volume of a gas and its temperature, which is described in detail in Section 6.3. This relationship was later known as Charles’ law after chemist Jacques Charles, who first outlined the concept in unpublished work in the 1780s. Finally, Joseph Louis Gay-Lussac first outlined the relationship between the temperature of a gas and its pressure in 1802. This relationship, later known as Gay-Lussac’s law, is detailed in Section 6.4. These three studies on the relationships between the volume, temperature, and pressure of a gas are now collectively known as 184 6. PROPERTIES OF GASES FIG. 6.2 The design of Torricelli’s barometer. “the gas laws” and have since been combined into “the combined gas law,” yielding the relationship between the pressure, volume, and temperature for a fixed mass of a sample of gas. In addition to these three important laws, Amedeo Avogadro hypothesized in 1811 that if the volume, temperature, and pressure of two gas samples are the same, they also contain the same number of molecules. This is now known as Avogadro’s law and, when combined with the three gas laws, the result is the general “ideal gas law” described in Section 6.5. Since these chemists were so interested in gases, they were also interested in identifying the composition and properties of Earth’s atmosphere. In 1804, Gay-Lussac, accompanied by his friend Jean-Baptiste Biot, ascended to a height of 4000 m (about 13,000 ft) in a hot air balloon shown in Fig. 6.3, in order to record differences in temperature, pressure, and moisture content in the atmosphere as a function of height. Gay-Lussac later made a second ascent by himself in order to lessen the weight of the balloon and reach a greater height. He reached a calculated height of 7016 m above sea level, a record not equaled for half a century. He collected air samples at different heights in evacuated flasks and the analysis of these samples showed that the proportion of oxygen in the air was identical at all heights with that at ground level. The discovery that the composition of the atmosphere does not change with increasing altitude was important to show that the atmosphere at these heights is well mixed. 185 6.2 BOYLE’S LAW FIG. 6.3 Gay-Lussac and Jean-Baptiste Biot ascend to 4000 m in a hot air balloon in 1804 to study the composition of the atmosphere. Illustration from the late 19th century via Wikimedia Commons. 6.2 BOYLE’S LAW The behavior of a gas of fixed mass can be described entirely by three variables, the volume (V), the pressure (P), and the temperature (T). Boyle’s law deals with the relationship between the pressure and volume of a gas when the temperature and mass remain constant. The gas volume is simply the amount of space that the gas occupies measured in liters (L). The gas pressure is the force (F), measured in newtons, exerted by the gas per unit area (A) on the surfaces surrounding it, measured in square meters (N/m2 ¼ Pa). F (1) A Gas molecules inside a container are constantly moving and so they frequently collide with each other and with the walls of the container as shown in Fig. 6.4. This large number of collisions between gas molecules and the container walls exerts a force on the surface. The larger the number of collisions per area of the container surface, the larger the force and the larger the gas pressure. P¼ 186 6. PROPERTIES OF GASES FIG. 6.4 The pressure exerted on the walls of a container by gas molecules colliding on the surface of the walls. The collisions are shown in red. Modified from Becarlson, Wikimedia Commons. Hg 140 120 Pressure 100 h2 Air 80 60 40 h3 20 h1 0 0 0.02 0.04 0.06 1/Volume 0.08 FIG. 6.5 Robert Boyle’s experimental apparatus for measuring the volume and pressure of a gas (left). When the mercury is at h1, the air sample is at atmospheric pressure. As mercury is poured into the open end of the tube, the resulting pressure of the gas is obtained by h2 and the volume of the air sample is obtained by h3. The results of his study are shown on the right. Robert Boyle’s experiment consisted of a sample of air sealed in a J-shaped tube closed at one end as shown in Fig. 6.5. At the beginning of the experiment, the trapped air was at atmospheric pressure (h1). Mercury was poured into the open side of the tube forcing the volume of the air on the closed side to contract under the weight of the added mercury. The added pressure of the air sample was measured as the height of the mercury on the open end of the tube (h2), in a similar manner to Torricelli’s barometer. The volume of the air on the closed side of the tube was measured as the height of the sealed air chamber (h3). By 187 6.2 BOYLE’S LAW repeating the experiment using different amounts of mercury, he found that the pressure of the gas was inversely proportional to its volume. The exact statement of Boyle’s law is; • The absolute pressure and the volume of a gas are inversely proportional at constant temperature and fixed mass. P¼k 1 V (2) This means that since the product of pressure and volume for any gas at constant temperature is a constant (PV ¼ k), if the pressure or temperature of the gas is changed, the effect on the other variable can be calculated by: P1 V1 ¼ P2 V2 (3) The SI unit for pressure is the pascal (Pa), which is equal to 1 N/m2 from Eq. (1). Some other common units for pressure and their conversion factors are listed in Table 6.2. In the field of science, the unit of one standard atmosphere (atm) is an established constant defined as 1.013 105 Pa. It is approximately equal to air pressure at earth mean sea level. Most scientists use the atmosphere (atm) as the standard unit of pressure because the SI unit of pascals results in very large numbers under normal conditions. The bar and the millibar were introduced by the British meteorologist William Napier Shaw in 1909 and are still widely used by meteorologists. The bar is equal to 1.0 105 Pa, slightly less than a standard atmosphere. It was originally defined as simply the pressure exerted by the atmosphere at the Earth’s surface and is considered to be approximately equal to the standard atmosphere. Pressure can also be expressed as millimeters of mercury (mmHg), since it is commonly measured using a mercury barometer or its cousin the manometer. The unit mmHg is also called the torr, after Evangelista Torricelli who invented the barometer. One torr is equal to 1 mmHg and is 1/760 of a standard atmosphere. So, 1 atm is equal to 760 mmHg, or 760 Torr. It is interesting to note the unit itself (torr) is written in lower case, while the unit’s symbol (Torr) is always written with an upper case “T.” To convert from units listed on the left to units listed at the top, the value is multiplied by the conversion factors listed in Table 6.2. The unit of mmHg is not included in the table because the conversion factors are the same as for the torr. TABLE 6.2 Conversion Factors Between Commonly Used Units of Pressure Pascal (Pa) Bar (bar) Standard Atmosphere (atm) Torr (Torr) Pounds Per Square Inch (psi) Pa 1.0 105 9.869 106 7.501 103 1.450 104 bar 105 0.9869 750.1 14.50 atm 1.013 10 760 14.70 1.0 5 1.00 10 5 1.0 3 3 Torr 133.3 1.333 10 1.316 10 1.0 1.934 102 psi 6.895 103 6.895 102 6.805 102 51.71 1.0 188 6. PROPERTIES OF GASES EXAMPLE 6.1: DETERMINING THE VOLUME OF A GAS FROM A CHANGE IN PRESSURE AT CONSTANT TEMPERATURE AND MASS If a 1.5 L gas sample at a pressure of 3.0 atm is reduced to a pressure of 0.5 atm, what is the final volume of the gas sample if the temperature remains the same? According to Boyle’s law: P1 ¼ 3:0atm, P2 ¼ 0:5atm, V1 ¼ 1:5L So P1 V1 ¼ P2 V2 V2 ¼ ðP1 V1 Þ=P2 ¼ ð3:0atmÞð1:5LÞ ¼ 9:0 L 0:5atm EXAMPLE 6.2: DETERMINING THE PRESSURE OF A GAS FROM A CHANGE IN VOLUME AT CONSTANT TEMPERATURE AND MASS A gas sample occupies 10.5 L at 0.8 atm. What is the pressure of the gas sample if the volume is increased to 5.0 L at the same temperature? According to Boyle’s law: P1 ¼ 0:8atm, V1 ¼ 10:5L, V2 ¼ 5:0L So P1 V1 ¼ P2 V2 P2 ¼ ðP1 V1 Þ=V2 ¼ ð0:8atmÞð10:5LÞ ¼ 1:7L 5:0atm 6.3 CHARLES’ LAW Charles’ law deals with the relationship between the temperature and volume of a gas when the pressure and mass remain constant. Temperature is viewed as an objective measure of how hot or cold something is. However, like pressure, it can be related to the motion of the molecules in the gas sample. The faster the molecules move, the higher the temperature. The measure of the relative speed of the molecular movement in the gas sample is known as the average kinetic energy of the gas, which depends only on the temperature of the gas. So, the average kinetic energy of the gas molecules increases as the temperature increases and the gas molecules move faster. If the gas pressure remains constant, this increased motion causes an increase in the volume, or the amount of space occupied by the gas. This view of temperature and pressure as related to the motion of the gas molecules is known as the kinetic-molecular theory of gases. 6.3 CHARLES’ LAW 189 The study of the effect of temperature upon the properties of gases took much longer to accomplish than the study of the effect of pressure. This was primarily because there was no quantitative temperature scale available until 1724–42, when the Fahrenheit and Celsius scales were developed. Both of these temperature scales were empirically based. That is, they were based on the direct measurement of the physical properties of materials. The Fahrenheit scale was originally based on the measurement of three fixed temperatures using the mercury thermometer invented by Daniel Fahrenheit shown in Fig. 6.6. Interestingly enough, this thermometer operates on the principle that the volume of a liquid, specifically mercury, varies with temperature. It consists of a bulb containing mercury attached to a narrow glass tube. The volume of mercury changes slightly with temperature and since the volume of mercury FIG. 6.6 Daniel Fahrenheit’s mercury thermometer displayed on his memorial plaque at his burial site in The Hague, Netherlands. Modified from Donarreiskoffer, Wikimedia Commons. 190 6. PROPERTIES OF GASES in the tube is much less than the volume of mercury in the bulb, a small increase in volume with increasing temperature drives the mercury a relatively long way up the tube. The lowest point on Fahrenheit’s scale was the temperature of a solution of equal parts of ice and ammonium chloride, which was set to be 0°F. The second point was the melting point of ice, set at 32°F, and the third point was the average temperature of the human body, set at 96°F. The scale was later simplified to be defined by only two fixed points, the freezing and boiling points of water at 32°F and 212°F. This new definition was more easily divided into equal degrees than the original scale using three points and resulted in 180°F separation between the two fixed points. The Celsius scale was also based on the boiling and freezing points of water measured by a mercury thermometer. It was originally set so that 0°C was defined as the boiling point of water and 100°C was defined as the freezing point of water, which was easily divided into 100°C. The scale was later inverted so that 0°C was set at the freezing point of water and 100°C was set at the boiling point. Since Anders Celsius was aware that the boiling point of water varied with changing pressure, he required that the temperature of boiling water be measured at atmospheric pressure at sea level (one standard atmosphere). This temperature scale was originally called Centigrade from the Latin “centum,” which means 100, and “gradus,” which means steps. The name was changed to Celsius in 1948 after its inventor, Anders Celsius. The Fahrenheit and Celsius temperature scales are interconverted by the following relationships: ° F ¼ 1:8 ° C + 32 (4) ° C ¼ ° F 32 =1:8 (5) The factor 1.8 is used to correct for the differences in the number of divisions in the scales, 180 in the Fahrenheit scale and 100 in the Celsius scale. A direct comparison between the Fahrenheit and Celsius temperature scales is shown in Fig. 6.7. Although the Fahrenheit scale was the primary temperature standard for meteorological, industrial, and medical purposes, the Celsius scale was more convenient for scientific studies. This was due to its easy division into 100 instead of 180. It remains a derived SI unit of temperature today. Around 1787, Jacques Charles designed an experiment to measure the change in volume of a gas sample with changes in temperature, using the Celsius temperature scale. The equipment used by Charles was similar to that used by Boyle shown in Fig. 6.5. A sample of gas was trapped in a J-shaped tube sealed at one end and filled with mercury. The tube was immersed in a water bath, which was used to change the temperature of the gas. The volume of the gas was measured by the height of the trapped sample. The pressure of the gas sample was held FIG. 6.7 A comparison between the Fahrenheit (°F) and Celsius (°C) temperature scales. Modified from TheVovaNik, Wikimedia Commons. 191 6.3 CHARLES’ LAW constant by adjusting the height of the mercury column so that the two columns of mercury were at the same height (h1), causing the pressure of the gas sample to be held equal to atmospheric pressure. The results of these experiments showed that at a fixed pressure, the volume of a gas is directly proportional to the temperature of the gas. However, when the volume is plotted versus the temperature in °C shown in Fig. 6.8A, the intercept was always equal to approximately 273°C. Any temperature below this would result in negative volumes, which were impossible. So, 273°C became known as absolute zero because it is the lowest possible temperature a gas can achieve. Later, it was discovered that if any gas is cooled by 1°C, the volume of the gas decreases by about 1/273, and for every additional degree of cooling, the volume would be reduced by an additional 1/273. Also, the equation of the line in Fig. 6.8A; t ° C ¼ ðm V Þ 273:15 gives the direct proportionality between temperature and volume, which is complicated by the very large intercept on the Celsius scale. These observations led Lord Kelvin to propose his absolute temperature scale where 0 K was set to absolute zero (273.15°C) and (6) T ðKÞ ¼ t ° C + 273:15 300 500 200 400 Temperature (K) Temperature (°C) The size of one unit on the Kelvin scale is the same as the size of one degree on the Celsius scale. Water freezes at 273.15 K (0°C) and boils at 373.15 K (100°C). So, the Kelvin temperature scale is simply the Celsius scale shifted by 273.15 degrees. The Kelvin temperature scale is an absolute scale instead of an empirical one. That is, the Kelvin scale is based on thermodynamic principles discussed in Chapter 8, since it uses absolute zero as its zero point. In the thermodynamics, absolute zero is the temperature at which all molecular motion stops and so there can be no temperature below absolute zero. Since Kelvin is an absolute temperature scale and the SI base unit for temperature, it is now used exclusively for scientific temperature calculations. It should be noted that there is a second absolute temperature scale, which is used by some engineering fields. This temperature scale is called the Rankine scale, after its inventor 100 0 −100 −200 −300 (A) 300 200 100 0 0 10 20 30 Volume (mL) 40 50 0 (B) 10 20 30 Volume (mL) 40 FIG. 6.8 The volume of a gas sample as a function of temperature in degrees Celsius (A) and Kelvin (B). 50 192 6. PROPERTIES OF GASES William Rankine. It also uses absolute zero as its zero point, but the Rankine degree is defined as equal to 1°F instead of the 1°C used by the Kelvin scale. The conversion factors of the Rankine temperature scale to the other three scales are: ° R ¼ ° F + 459:67 ° R ¼ ° C + 273:15 9=5 ° R ¼ K 9=5 A comparison of some standard fixed temperatures for each of the four temperature scales is listed in Table 6.3. A temperature of 459.67°R is exactly equal to 0°F. The freezing point of water is 32°F ¼ 0°C ¼ 273.15 K ¼ 491.67°R and the boiling point of water is 211.97°F ¼ 100°C ¼ 373.13 K ¼ 671.64°R. The use of the Kelvin scale in studies of the effects of temperature on the volume of a gas, shown in Fig. 6.8B, changes the linear relationship to; T ðKÞ ¼ m V which assures that temperature and volume are simply and directly proportional and the volume intercept is always zero at 0 K. The exact statement of Charles’ law is; • At constant pressure, the volume of a fixed mass of any gas is directly proportional to the absolute temperature in degrees Kelvin. V ¼kT (7) This means that since the ratio of volume to temperature for any gas at constant pressure is (V/T ¼ k), if the volume or temperature of the gas is changed, the effect on the other variable can be calculated by; ðV1 =T1 Þ ¼ ðV2 =T2 Þ (8) TABLE 6.3 Fixed Temperatures of Thermal Properties of Common Materials as Measured by the Two Empirical (Fahrenheit and Celsius) and Two Absolute (Kelvin and Rankine) Temperature Scales Fahrenheit Celsius Kelvin Rankine Absolute zero 459.67°F 273.15°C 0K 0°R Freezing point of NH4Cl 0°F 17.78°C 255.37 K 459.67°R Freezing point of water 32°F 0°C 273.15 K 491.67°R Triple point of water 32.02°F 0.01°C 273.16 K 491.69°R Boiling point of water 211.97°F 100°C 373.13 K 671.64°R 6.3 CHARLES’ LAW 193 EXAMPLE 6.3: DETERMINING THE VOLUME OF A GAS AFTER A CHANGE IN TEMPERATURE AT CONSTANT PRESSURE AND FIXED MASS A sample of carbon dioxide has a volume of 25.0 mL at a temperature of 20.0°C. What is the volume of the same sample at the same pressure and a temperature of 37.5°C? According to Charles’ Law: V1 ¼ 25:0mL, T1 ¼ 20:0° C ¼ 20:0 + 273:15 ¼ 293:2K T2 ¼ 37:5° C ¼ 37:5 + 273:15 ¼ 310:7K So ðV1 =T1 Þ ¼ ðV2 =T2 Þ V2 ¼ ðV1 T2 =T1 Þ ¼ ð25:0mLÞð310:7KÞ ¼ 26:5mL 293:2K CASE STUDY: HOT AIR BALLOONS Charles was an avid balloonist, having invented the hydrogen-filled balloon. His interest in the effects of temperature on the volume of gases was partially fueled by the desire to understand the scientific principles behind the hot air balloon. Charles’ law describes how a gas expands as the temperature increases or contracts as the temperature decreases. This is also the principle behind the hot air balloon. Since a gas expands when heated, hot air occupies a larger volume than the same mass of colder air. So hot air is less dense than cold air. When the air in a balloon is hot enough, the weight of the balloon plus the hot air is less than the weight of the same volume of colder air surrounding it, and the balloon starts to rise. When the gas in the balloon is allowed to cool and contract to a smaller volume, its density increases and it descends to the ground. A hot air balloon consists of a bag, called the envelope, capable of containing heated air. Modern envelopes are made of nylon and come in various sizes. The larger the envelope, the greater the volume of hot air it can contain and the larger the mass it can lift. The smallest, one person, basketless balloons have envelopes as small as 600 m3. Balloons used by commercial tourism companies may be able to carry well over two dozen people with envelopes up to 17,000 m3. The most often used size is about 2800 m3 carrying three to five people. Suspended beneath the envelope is a gondola or wicker basket, which carries the passengers, and a source of heat, usually an open flame. The mass of a common hot air balloon system including passengers is given in Table 6.4. The amount of lift provided by a hot air balloon depends primarily upon the difference between the temperature of the air inside the envelope and the temperature of the air outside the envelope. The amount of lift generated by 2800 m3 of dry air heated to various temperatures can be calculated from the density of the air at each temperature. At a typical atmospheric temperature of 20°C, the density of air is 1.024 kg/m3. The mass of 2800 m3 of this air is; 1:024kg=m3 2800m3 ¼ 3371kg 194 6. PROPERTIES OF GASES TABLE 6.4 The Mass of Various Components of a Hot Air Balloon Component Kilograms 3 113 2800 m 5 passenger basket 63 Double burner 23 3 fuel tanks full of propane (75.7 L) 184 5 passengers 340 Total 723 Similarly, the density of air heated to 100°C is 0.946 kg/m3 and a mass of 2800 m3 of this air is 2649 kg. The lift generated from the difference in masses of the air outside the envelope at 20°C and the air inside the envelope at 100°C is; 3371kg 2649kg ¼ 722kg This is barely enough to generate neutral buoyancy for the total mass of the system in Table 6.4. For most envelopes, the maximum internal temperature is limited to approximately 120°C in order to prevent deterioration of the nylon. The total lift for a 2800 m3 envelope heated to 120°C (air density of 0.898) would be 857 kg. On a warm day, a balloon cannot lift as much as on a cool day. If the temperature of the surrounding air is closer to 25°C, the lift at 120°C would only be 56 kg and the temperature required for launch would exceed the maximum sustainable for the nylon envelope. This is why balloon launches generally happen at sunrise when the air temperature is cooler. Also, the calculations of lift were based on the assumption that the temperature of the air inside the balloon is the same throughout the 2800 m2 volume. In reality, the air contained in the envelope is not all the same temperature, as shown in Fig. 6.9. The temperature of the envelope in flight is commonly measured by a single temperature gauge placed near the top of the envelope where the air is hottest. Since the air nearer the bottom of the envelope is cooler, this will reduce the expected lift. The amount of lift also depends on the altitude above sea level and the humidity of the air surrounding the balloon. The density of air decreases with altitude due to decreasing atmospheric pressure, so the differences in the density of the outside air and the air inside the envelope decrease as the balloon ascends. In the lower atmosphere, the lift provided by a hot air balloon at a constant temperature decreases about 3% for each 1000 m of altitude gained. Also, the molecular mass of water vapor (18.02 amu) is considerably less than the average molecular mass of air (28.57 amu). So, the average mass of humid air is less than the average mass of the same volume of dry air at the same temperature. This also decreases the difference in density of the outside air and the hot air inside the envelope decreasing the expected lift. 6.4 GAY-LUSSAC’S LAW 195 FIG. 6.9 Thermal image of a hot air balloon in flight showing the temperature variation in the envelope. From Luftfahrer, Wikimedia Commons. 6.4 GAY-LUSSAC’S LAW Gay-Lussac’s law deals with the relationship between the temperature and pressure of a gas when the mass and volume remain constant. It is sometimes called Amonton’s law after Guillaume Amonton, who first observed the relationship while building an air thermometer in 1702. According to the kinetic-molecular theory of gases, when the temperature of a gas is increased, the average kinetic energy of the gas increases, the gas molecules move faster, and so they will impact the walls of the container more often. Since pressure is a measure of the number of collisions per area of the container surface, as the number of collisions increase the pressure increases. Gay-Lussac’s experiments showed that the pressure of a gas held in a rigid container was directly proportional to the temperature. However, when the variation of pressure with increasing temperature was measured using the Celsius temperature scale shown in Fig. 6.10A, a graphical plot was obtained similar to that obtained for the temperature-volume relationship. The temperature at zero pressure was also found to be approximately 273°C. When the temperature is converted to the absolute Kelvin temperature scale as in Fig. 6.10B, the pressure and temperature become directly proportional and pressure becomes equal to zero at 0 K. 196 6. PROPERTIES OF GASES 400 100 Temperature (K) Temperature (°C) 200 0 −100 −200 −300 200 100 0 0 0.5 1 Pressure (atm) (A) FIG. 6.10 300 0 1.5 (B) 0.5 1 Pressure (atm) 1.5 The pressure of a gas sample as a function of temperature in degrees Celsius (A) and Kelvin (B). The modern statement of Gay-Lussac’s law is; • At constant volume, the pressure of a fixed mass of any gas is directly proportional to the absolute temperature in degrees Kelvin. P¼kT (9) This means that since the ratio of pressure to temperature for any gas at constant pressure is (P/T ¼ k), if the pressure or temperature of the gas is changed, the effect on the other variable can be calculated by; ðP1 =T1 Þ ¼ ðP2 =T2 Þ (10) EXAMPLE 6.4: DETERMINING THE PRESSURE OF A GAS AFTER A CHANGE IN TEMPERATURE AT CONSTANT VOLUME AND MASS If a gas contained in a steel tank at 21.4°C has a pressure of 5.17 atm. What will the pressure be if it is heated to a temperature of 89.6°C? According to Gay-Lussac’s Law: P1 ¼ 5:17atm, T1 ¼ 21:4° C ¼ 21:4 + 273:15 ¼ 294:6K T2 ¼ 37:5° C ¼ 37:5 + 273:15 ¼ 310:7K So ðP1 =T1 Þ ¼ ðP2 =T2 Þ P2 ¼ ðP1 T2 =T1 Þ ¼ ð5:17atmÞð310:7KÞ ¼ 5:45 atm 294:6K 6.5 THE IDEAL GAS LAW 197 6.5 THE IDEAL GAS LAW Boyle’s law, Charles’ law, and Gay-Lussac’s law each describe relationships between pairs of the three important variables that determine the behavior of a gas (temperature, pressure, and volume). In order to determine the values of all three variables when more than one is changing, the three gas laws can be combined into a single law. This gives a relationship between pressure, volume, and temperature for a fixed amount of any gas expressed as a single equation called the combined gas law; The exact statement of the combined gas law is; • The ratio between the pressure-volume product and the absolute temperature of a fixed mass of gas measured in Kelvin remains constant; PV ¼k T (11) This means that since the ratio of the pressure-volume product to the temperature for any gas with a fixed mass is constant pressure (PV/T ¼ k), if any of the three variables is changed, the effect on the other two can be calculated by; ðP1 V1 =T1 Þ ¼ ðP2 V2 =T2 Þ (12) EXAMPLE 6.5: DETERMINING THE PRESSURE, TEMPERATURE, AND VOLUME OF A GAS AFTER A CHANGE IN TWO OF THESE VARIABLES AT CONSTANT MASS A gas sample is initially at a pressure of 12.5 atm, a volume of 23.0 L, and a temperature of 200 K. If the pressure is raised to 14.0 atm and the temperature is raised to 300 K, what is the new volume of the gas? According to the combined gas law: P1 ¼ 12:5atm,P2 ¼ 14:0atm,V1 ¼ 23:0L,T1 ¼ 200K,T2 ¼ 300K So ðP1 V1 =T1 Þ ¼ ðP2 V2 =T2 Þ V2 ¼ ðP1 V1 T2 =P2 T1 Þ ¼ ð12:5atmÞð23:0LÞð300KÞ ¼ 30:8L ð14:0atmÞð200KÞ However, the combined gas law applies only to situations where the amount of the gas does not change. In order to understand how a change in amount affects the other three variables, a fourth variable needed to be included in the combined gas law. The relationship between volume and the amount of gas in a sample was first observed by Amedeo Avogadro in 1811. He stated that; “…equal volumes of gases at the same temperature and pressure contain the same number of molecules regardless of their chemical nature and physical properties.” 198 6. PROPERTIES OF GASES V ¼ kNA (13) This number of molecules (NA), known as Avogadro’s number, is 6.022 10 . Avogadro’s number has been defined as the number of atoms in 12.000 g of carbon-12. It is also the number of molecules in 1 mol of any substance, as described in Section 4.1. Since 1 mol of any gas contains 6.022 1023 molecules, 1 mol of any gas has the same volume at the same temperature and pressure. The direct consequence of this observation is that the volume of a gas at a given temperature and pressure is directly proportional to the amount of gas in moles (n); 23 V ¼kn (14) So, since the ratio of the volume to the number of moles of any gas at constant temperature and pressure is constant (V/n ¼ k), if the volume or amount of the gas is changed, the effect on the other variable can be calculated by; ðV1 =n1 Þ ¼ ðV2 =n2 Þ (15) This relationship is now known as Avogadro’s law because it comes as a direct result of Avogadro’s proposal that equal volumes of gases contain the same number of molecules. Including Avogadro’s hypothesis in the combined gas law results in the following relationship between the pressure, temperature, volume, and number of moles of a gas; PV ¼ kn T (16) By combining the three gas laws with Avogadro’s hypothesis, all four variables controlling the physical behavior of a gas are defined. The most important consequence of this relationship is that the constant (k) has the same value for all gases. So, this constant is a universal constant, called the ideal gas constant, which can be used to determine the properties of any gas. Because it is based on the amount of the gas instead of the mass, it has the same value for all gases independent of the size or mass of the gas molecules. The ideal gas constant is given the designation “R” and the equation; PV ¼ nRT (17) is called the ideal gas law. The exact statement of the ideal gas law is; • The product of the pressure and the volume of an ideal gas is equal to the product of the absolute temperature of the gas, the amount of the gas, and the universal gas constant. The ideal gas law describes the behavior of an ideal gas, which is a theoretical gas composed of many randomly moving point size particles that do not interact with each other except when they collide elastically. Although, in reality, there is no such thing as an ideal gas, most gases behave close enough to ideal that the ideal gas law can effectively describe their behavior. Generally, a gas behaves more like an ideal gas at higher temperatures and lower pressures. The major exceptions to the assumption of ideality are discussed in Section 6.6. The value of the ideal gas constant has been determined experimentally by precisely measuring the pressure, volume, temperature, and amount of a gas sample. This value will 6.5 THE IDEAL GAS LAW 199 depend on the units used for temperature, pressure, volume, and amount. For the standard SI units of Kelvin for temperature, liters for volume, and moles for amount, as well as the conventional scientific unit for pressure of atmospheres; R ¼ 0:08206 L atm K mol (18) When performing calculations using the ideal gas law, other values for R are available for cases where the values of the four variables are given in units different from the standard units. For example, if pressure is given in torr instead of atmospheres, the value of R would be 62.37 (L•torr)/(K•mol). But, it is usually easier to first convert the values to the standard units and use the value of R as 0.08206 (L•atm)/(K•mol) than to use alternative values of R. EXAMPLE 6.6: DETERMINING THE AMOUNT OF A GAS IF THE TEMPERATURE, PRESSURE, AND VOLUME ARE KNOWN What is the amount of a gas sample with a volume of 575 mL at a pressure of 750 Torr and a temperature of 22.0°C? 1. Convert all values to standard units. 750 Torr P¼ ¼ 0:987atm 760Torr=atm T ¼ 22. 0°C + 273.15 ¼ 295.15 K 575mL ¼ 0:575L V¼ 1000mL=L 2. Determine the amount of sample using the ideal gas law and the ideal gas constant in SI units. PV ¼ nRT ð0:987atmÞð0:575LÞ ¼ 0:0240mol n ¼ ðPV=RTÞ ¼ 0:08206L atm mol1 K1 ð295:15KÞ The ideal gas equation is also useful for determining the density of a gas when the temperature and pressure of the gas is known. Since the amount of a gas is equal to the mass divided by the molar mass (n ¼ m/M), the ideal gas equation can be written as; PV ¼ m RT M Since density (ρ) is mass/volume in g/L; ρ¼ m PM ¼ V RT (19) So, the density of a gas is directly proportional to the pressure and molar mass and inversely proportional to the temperature of the gas. 200 6. PROPERTIES OF GASES CASE STUDY: COMPRESSED GAS CYLINDER SAFETY Compressed gases are commonly used in a wide range of industries, including petrochemicals, mining, steelmaking, power, pharmaceuticals, electronics, metallurgy, oil and gas, and many more. Some common industrial gases are: ammonia, carbon dioxide, carbon monoxide, hydrogen chloride, dinitrogen monoxide, nitrogen trifluoride, sulfur dioxide, sulfur hexafluoride, and methane. Industrial gases are transported and stored in compressed gas cylinders, which are pressure vessels used to store gases at very high pressures well above 1 atm. This allows for easy storage and transportation of large amounts of gases in a small volume. For a large compressed gas cylinder measuring 58.5 in. tall and 9 in. in diameter, the internal volume would be 50.0 L. If the internal pressure of the gas is 2900 psi (197.3 atm) at 25°C, the amount of carbon dioxide in the cylinder would be; n¼ PV ð197:3atmÞð50:0LÞ 9865 ¼ ¼ ¼ 403mol RT ½0:08206ðL atmÞ=ðK molÞ½298:15K 24:47 While the very high pressures inside the gas cylinders create a convenience of storage, the large pressure differences inside and outside the cylinder can cause significant safety hazards if not operated, stored, or maintained properly. There are special safety regulations for handling compressed gas cylinders to prevent catastrophic release of the contents. These include securing bottles to a wall or other fixed object with a strap or chain to prevent falling and proper ventilation in storage areas to prevent exposure should leaks occur. If a compressed gas cylinder should fall over causing the valve block to be sheared off, the rapid release of the high pressure gas inside the cylinder could cause it to be powerfully accelerated becoming like a missile projectile. Run away cylinders have been known to penetrate concrete block walls. Also, a sudden release of even nonhazardous gases such as carbon dioxide or nitrogen can cause asphyxiation of anyone in a nonventilated area. Storage as in Fig. 6.11 is not considered proper because, when stored in a bunch, one cylinder cannot be removed without releasing the whole group and possibly causing a cascade fall. Also, the gas cylinders should not be stored in an area where they could be exposed to high ambient temperatures since an increase in temperature at constant volume would cause an increase in the already high pressure inside the cylinder. If the 50 L compressed gas cylinder of carbon dioxide at 197.3 atm of pressure were to be exposed to conditions which caused a release of the gas into a closed room at 1 atm and 25°C, the volume of the released gas would be; V2 ¼ P1 V1 ð197:3atmÞð50LÞ ¼ ¼ 9865L or 9:9m3 P2 1atm An average room size is approximately 4 m 5 m 2.5 m, or a volume of 50 m3. Assuming that the room contains no furniture or other storage, the room would contain 50 m3 of air before the CO2 release. Of this, only 21%, or 10.5 m3, is oxygen. So, a sudden release of 9.9 m3 of CO2 near ground level could easily displace breathable oxygen causing asphyxiation of anyone in the immediate area. The available volume of air would be drastically reduced if the room was partially filled with furniture or other laboratory equipment. Carbon dioxide or nitrogen are not considered to be harmful industrial gases compared to other common industrial gases, such as ammonia, hydrogen chloride, or carbon monoxide, which are toxic and/or corrosive. However, asphyxiation by nitrogen and carbon dioxide are the most common means of death due to sudden release of the compressed gases. 6.6 NONIDEAL GAS BEHAVIOR 201 FIG. 6.11 Improper storage of compressed gas cylinders. Cylinders should be chained to the wall individually so that one can be selected without releasing the others. Cylinders should also never be stored in direct sunlight or places where high temperatures may cause an increase in pressure and possible release of the compressed gas. Photograph by Ildar Sagdejev, Wikimedia Commons. 6.6 NONIDEAL GAS BEHAVIOR The behavior of real gases can usually be predicted by the ideal gas law to within about 5% at normal temperatures and pressures. But, at low temperatures or high pressures, real gases deviate significantly from ideal gas behavior. The kinetic-molecular theory of gases, which is based on the behavior of an ideal gas, makes some assumptions that do not hold at low temperatures and high pressures. These are: • Collisions between gas molecules and the walls of a container are perfectly elastic. That is, there is no loss of kinetic energy in molecular collisions. • Gas molecules do not have any volume. • There are no repulsive or attractive forces between molecules. These assumptions define ideal gases. So, deviations from ideal behavior become important when kinetic energy is lost in collisions, when molecular volume is significant compared to the volume of the container, and when intermolecular forces become important. Gases do not behave ideally at very low temperatures because the molecules move relatively slowly allowing for the effects of intermolecular forces to increase. Gases also do not 202 6. PROPERTIES OF GASES behave ideally at high pressures because the gas molecules are closer together at high pressures. This makes the volume occupied by the gas molecules significant compared to the total volume of the container. Also, the intermolecular forces become more important and collisions between the molecules and the walls of the container are more energetic and become less elastic. Dutch physicist Johannes van der Waals developed an equation to correct for the deviations from ideal behavior at low temperatures and high pressures. This allowed him to explain the behavior of real gases over a much wider range of pressures and temperatures than could be achieved using the ideal gas law alone. This equation, known as the van der Waals equation, is; P + a n2 =V 2 ½ðV nbÞ ¼ nRT (20) This equation is of a similar form as the ideal gas law and approaches the ideal gas law as the values of a and b approach zero. The constants a and b, known as the van der Waals constants, have positive values and are characteristic of each individual gas. They are determined experimentally by observing the behavior of a gas sample as a function of pressure, volume, and temperature. The constant a, in units of atm•L2•mol2, provides a correction for the attractive forces between the molecules and becomes important for polar molecules that strongly interact with each other. The constant b, in units of L/mol, is a correction for the finite molecular size and becomes more important for larger molecules. The constant a is multiplied by the factor “n2/V2” to account for the fact that the increase in attractive forces between the molecules causes a reduction in the number of collisions with the container walls. This reduction in wall collisions is observed as a decrease in pressure of the gas and is proportional to the volume of the container (V) and the amount of gas (n) in the container. So, the measured pressure of a real gas is lower than the measured pressure of an ideal gas by the factor, a(n2/V2), and this value must be added to the measured pressure to correct for the nonideal behavior of the gas. The constant b is the volume of the gas molecules per mole of gas. Since real gas molecules have finite volumes, the measured volume includes the volume of the molecules as well as the volume of the container. The value nb must be subtracted from the measured volume to correct for the volume taken up by the gas molecules. This correction results in a more accurate measure of the empty space available for the gas molecules. Unlike the ideal gas constant, which is the same for all gases, the van der Waals constants a and b are different for each gas. This is because the attractive forces and molecular sizes are different for each gas. Table 6.5 lists the van der Waals constants for some common gases. In summary, the ideal gas equation adequately explains the behavior of a gas when the attractions between gas molecules are not significant and the gas molecules themselves do not occupy a major part of the total volume. This is usually true when the pressure is around one atmosphere and the temperature is between 0°C and 100°C. Under conditions of high pressures and/or low temperatures, the ideal gas law may give results that are significantly different from what is observed experimentally. In these cases, the van der Waals equation is used to correct for the nonideal behavior. A comparison of the results obtained from the ideal 6.6 NONIDEAL GAS BEHAVIOR TABLE 6.5 203 Van der Waals’ Constants for Some Common Gases Gas a [(L2•atm)/mol2] b (L/mol) Helium 0.035 0.02370 Neon 0.214 0.01709 Hydrogen 0.248 0.02661 Nitrogen monoxide 1.358 0.02789 Argon 1.363 0.03219 Oxygen 1.378 0.03183 Nitrogen 1.408 0.03913 Carbon monoxide 1.505 0.03985 Methane 2.283 0.04278 Krypton 2.349 0.03978 Carbon dioxide 3.640 0.04267 Hydrogen chloride 3.716 0.04081 Dinitrogen monoxide 3.832 0.04415 Ammonia 4.225 0.03707 Xenon 4.250 0.05105 Hydrogen sulfide 4.490 0.04287 Hydrogen bromide 4.510 0.04431 Nitrogen dioxide 5.354 0.04424 Water 5.536 0.03049 Hydrogen iodide 6.309 0.05303 Chlorine 6.579 0.05622 Sulfur dioxide 6.803 0.05636 Hydrogen fluoride 9.433 0.07390 gas equation and the van der Waals equation for the same gas under the same conditions will give a measure of the magnitude of the deviation from ideal behavior. The low temperatures required for significant deviations from ideal behavior are not usually encountered in engineering applications. But, the high pressures responsible for nonideal behavior of real gases are often encountered in applications using compressed gases. The corrections to the ideal gas law as given by the van der Waals equation become especially important for these cases. 204 6. PROPERTIES OF GASES EXAMPLE 6.7: DETERMINING THE DEVIATION FROM IDEAL BEHAVIOR OF A GAS AT HIGH PRESSURE What is the deviation from ideal gas behavior for 300.0 mol of helium in a 50.00 L container at 25.00°C? 1. Calculate the pressure of the gas using the ideal gas law. T ¼ 25.00 + 273.2 ¼ 298.2 K, V ¼ 50.00 L, n ¼ 300.0 mol, R ¼ 0.0821 atm•mol1•K1 PV ¼ nRT nRT ð300:0molÞ 0:0821atm mol1 K1 ð298:2KÞ ¼ 146:9atm ¼ 50:00L V 2. Calculate the pressure of the gas using the van der Waals equation. From Table 6.5: a ¼ 1.358 and b ¼ 0.02789 [P + a(n2/V2)][(V nb)] ¼ nRT nRT an2 ð300:0molÞ 0:0821atm mol1 K1 ð298:2KÞ ð1:358Þð300:0molÞ2 P¼ 2¼ V nb V 50:00L ð300:0molÞð0:02789Þ ð50:00LÞ2 P¼ P ¼ 127.5 atm 3. Calculate the deviation from ideal behavior. The deviation from ideal behavior under these conditions is; 146:9atm 127:5atm 100% ¼ 13% deviation 146:9atm 6.7 PARTIAL PRESSURES Under conditions where a gas behaves ideally, the ideal gas law applies to mixtures of gases in the same manner as for pure gases because it is dependent only on the number of particles and not the identity of the gas. So, in a mixture of ideal gases, each gas has a partial pressure, which is the pressure the gas would exert if it alone occupied the same volume as the mixture at the same temperature. The total pressure of a mixture of ideal gases is the sum of the partial pressures of each individual gas in the mixture. For the case of a mixture of three gases; P T ¼ PA + PB + PC where PT is the total pressure and PA, PB, and PC, are the partial pressures of the gases A, B, and C, respectively, in the mixture. This relationship was first observed by John Dalton and is now known as Dalton’s law of partial pressures. The exact statement of Dalton’s law of partial pressures is; • The total pressure of a mixture of gases equals the sum of the pressures that each would exert if it were present alone. Since, in a mixture of ideal gases, each gas acts independently of the others, the partial pressure of each gas in the mixture can be calculated from the ideal gas law as; 6.7 PARTIAL PRESSURES 205 PT ¼ PA + PB + PC … ¼ ðnA RT=V Þ + ðnB RT=V Þ + ðnC RT=V Þ + … PT ¼ ðnA + nB + nC …ÞðRT=V Þ (21) PT ¼ nT ðRT=V Þ So, at constant temperature and volume, the total pressure of a gas sample is determined by the total number of moles of gas in the sample, whether the sample is composed of a pure gas or a mixture of gases. EXAMPLE 6.8: DETERMINING THE PARTIAL PRESSURES OF GASES IN A MIXTURE A mixture of 10.0 g of oxygen and 5.50 g of methane is placed in a 10.0 L container at 25°C. What is the partial pressure of each gas, and what is the total pressure in the container? 1. Determine the partial pressure of oxygen. V ¼ 10.0 L, T ¼ 25 + 273 ¼ 298 K nO2 ¼ 10:0g ¼ 0:313mol 32:0g=mol nRT ð0:313molÞ 0:0821atm mol1 K1 ð298KÞ ¼ 0:766atm PO2 ¼ ¼ 10:0L V 2. Determine the partial pressure of methane. V ¼ 10.0 L, T ¼ 25 + 273 ¼ 298 K nCH4 ¼ 5:50g ¼ 0:344mol 16:0g=mol nRT ð0:344molÞ 0:0821atm mol1 K1 ð298KÞ ¼ 0:842atm ¼ 10:0L V 3. Determine the total pressure. PCH4 ¼ PT ¼ PO2 + PCH4 ¼ 0.766 atm + 0.842 atm ¼ 1.61 atm The ratio of the partial pressure of one gas in a mixture to the total pressure gives; PA nA ðRT=V Þ nA ¼ ¼ PT nT ðRT=V Þ nT So, the ratio of the partial pressure of gas A in a mixture of gases to the total pressure of the mixture is equal to the ratio of the amount of gas A to the total amount of gas in the mixture. This ratio of amounts (nA/nT) is called the mole fraction of gas A in the mixture of gases and is designated by the symbol χ A. This means that the partial pressure of a component gas (PA) in a mixture of gases is equal to its mole fraction (χ A) times the total pressure of the mixture (PT). PA ¼ nA PT ¼ χ A PT nT (22) 206 6. PROPERTIES OF GASES CASE STUDY: COLLECTING A GAS SAMPLE OVER WATER A common way to determine the amount of a gas produced by a chemical reaction is to collect the gas over water, as long as the gas is not soluble in the water. The modern apparatus for the collection of a gas over water, shown in Fig. 6.12, is called a eudiometer and is very similar to Stephen Hales’ pneumatic trough. A graduated gas collection tube, sealed at the top, is filled with water and inverted in a reservoir of water. The gas evolved from the chemical reaction is collected by attaching one end of a hose to the reactioncontainerandinsertingtheotherendupintotheinvertedgascollectiontube.Asthegasisreleased,it displaces the water from the tube. The volume of gas produced by the reaction is then determined by measuring the volume of water that was displaced by the gas by using the graduated markings on the tube. During the gas collection, the water level in the collection tube adjusts so that the pressure inside and outside the tube are both equal to atmospheric pressure. However, the total pressure inside the collection tube is partially due to the dry gas produced from the chemical reaction and partially due to water vapor that has escaped from the surface of the water in the collection tube. From Dalton’s law of partial pressures, the total pressure of the gas in the collection tube is equal to the sum of the partial pressure of the dry gas produced from the chemical reaction and the partial pressure of the water vapor, called the vapor pressure of water. PT ¼ Pgas + PH2 O The vapor pressure, the partial pressure of a gas in contact with its liquid form, is dependent on the temperature. An example of the temperature dependence of the vapor pressure of water is shown in Fig. 6.13 and the exact value of the vapor pressure of water for each experiment can be determined from vapor pressure charts or tables. 50 100 150 200 250 FIG. 6.12 A eudiometer used to collect and measure the volume of gas produced from a chemical reaction. The gas is collected over water and the volume of gas is determined by the volume of water displaced. From Danielchemik, Wikimedia Commons. 6.8 CHEMICAL REACTIONS WITH GASES 207 FIG. 6.13 The temperature dependence (°C) of the vapor pressure of water (torr). When a sample of potassium chlorate (KClO3) is heated, it will decompose to potassium chloride (KCl) and oxygen gas. If the volume of gas collected in a eudiometer is 250 mL at 25°C (PH2O ¼ 23.8 Torr) and a pressure of 765 Torr, the partial pressure of the dry oxygen gas is; PO2 ¼ PT PH2 O ¼ 765Torr 23:8Torr ¼ 741:2Torr=760Torr=atm ¼ 0:975atm The amount of oxygen gas collected is; n¼ PV ð0:975atmÞð0:250LÞ ¼ 9:97 103 molO2 ¼ RT 0:0821atm mol1 K1 ð298KÞ 6.8 CHEMICAL REACTIONS WITH GASES The use of stoichiometric coefficients in chemical equations to determine the amounts of reactants and products in a chemical reaction was described in Chapter 4. In chemical reactions involving gaseous reactants and products, the amounts are often given as a volume at a specified temperature and pressure. Conversions between volume and amount of gases can easily be determined by using the molar volume of a gas. The molar volume is the volume occupied by 1 mol of an ideal gas at standard temperature and pressure. Standard temperature and pressure (abbreviated STP) is defined as 1 atm of pressure and a temperature of 273 K. The molar volume at STP has been experimentally determined to be 22.41 L. This can be used as a conversion factor to relate the number of moles to a volume of gas. For most cases, gases are measured at conditions other than STP, so the volumes must first be converted to STP in order to use the molar volume in stoichiometric calculations. 208 6. PROPERTIES OF GASES EXAMPLE 6.9: DETERMINING THE AMOUNT OF PRODUCTS IN A GAS PHASE CHEMICAL REACTION FROM THE VOLUMES What is the amount of water vapor formed when 1.25 L of hydrogen gas reacts with oxygen gas at STP? 1. Write the balanced chemical equation. 2H2(g) + O2(g) ! 2H2O(g) 2. Determine the amount of hydrogen gas. 1:25L ¼ 0:0558molH2 22:4L=mol 3. Determine the amount of water produced. According to the chemical equation, 2 mol of H2O is produced from 2 mol of H2. So, 0.0558 mol of H2 will form 0.558 mol of H2O. In a balanced chemical equation, the stoichiometric coefficients for gas species represent both the number of moles and the number of molar volumes that take place in the reaction. So, at a given temperature and pressure, the volumes of the gas reactant species are proportional to the number of moles. This is known as the law of combining volumes. For chemical reactions involving gases where the temperature and pressure do not change, volume relationships are the same as molar relationships. For cases where the temperature and pressure change during the reaction, the ideal gas law must be used to determine the amounts of products and reactants in moles. EXAMPLE 6.10: DETERMINING THE VOLUME OF PRODUCTS IN A GAS PHASE CHEMICAL REACTION How many liters of nitrogen monoxide are required to produce 3.0 L of nitrogen dioxide from the reaction with oxygen? 1. Write the balanced chemical equation. 2NO(g) + O2(g) ! NO2(g) 2. Determine the volume of product formed. According to the chemical equation, 2 L of NO will produce 1 L of NO2. So, it will require 2 3.0 L ¼ 6.0 L of NO to produce 3.0 L of NO2. IMPORTANT TERMS Absolute zero the lowest temperature that is theoretically possible, at which all molecular motion stops. Avogadro’s law equal volumes of gases at the same temperature and pressure contain the same number of molecules regardless of their chemical nature and physical properties. Barometer an instrument measuring atmospheric pressure. Boyle’s law the absolute pressure and the volume of a gas are inversely proportional at constant temperature and fixed mass. STUDY QUESTIONS 209 Charles’ law at constant pressure, the volume of a fixed mass of any gas is directly proportional to the absolute temperature in degrees Kelvin. Combined gas law the ratio between the pressure-volume product and the absolute temperature of a fixed mass of gas measured in Kelvin remains constant. Dalton’s law of partial pressures the total pressure of a mixture of gases equals the sum of the pressures that each would exert if it were present alone. Gay-Lussac’s law at constant volume, the pressure of a fixed mass of any gas is directly proportional to the absolute temperature in degrees Kelvin. Ideal gas a theoretical gas composed of many randomly moving point particles that do not interact except when they collide elastically. Ideal gas constant (R) a universal physical constant used in the equation for the ideal gas law. Ideal gas law the product of the pressure and the volume of an ideal gas is equal to the product of the absolute temperature of the gas, the amount of the gas, and the universal gas constant. Kinetic energy energy that a body possesses by virtue of being in motion. Kinetic-molecular theory of gases the view that the temperature and pressure of a gas is related to the motion of the gas molecules. Law of combining volumes at a given temperature and pressure, the volumes of the gaseous species reacting are proportional to the number of moles. Molar volume the volume occupied by 1 mol of an ideal gas at standard temperature and pressure. It is equal to 22.41 L. Mole fraction (χ A) the ratio of the amount of gas A to the total amount of gas in the mixture. Partial pressure the pressure the gas would exert if it alone occupied the same volume as the mixture at the same temperature. Standard temperature and pressure (STP) 1 atm of pressure and a temperature of 273 K. van der Waals constants experimentally derived constants characteristic of each individual gas, which correct the pressure and volume of a gas for nonideal behavior. van der Waals equation an equation that corrects for the deviations of a gas from ideal behavior at low temperatures and high pressures. Vapor pressure the partial pressure of a gas in contact with its liquid form. STUDY QUESTIONS 6.1 6.2 6.3 6.4 6.5 6.6 6.7 6.8 6.9 6.10 6.11 6.12 6.13 6.14 What is a pneumatic trough and why was its invention important? Who invented the barometer? What unit bears his name? What determines the pressure of a gas? What is the general statement of Boyle’s law? For any gas at constant temperature what is the equation that describes Boyle’s law? What is the SI unit for pressure? What is the standard scientific unit for pressure? How are they interconverted? How is the pressure unit torr related to the pressure unit atmospheres? What is the kinetic-molecular theory of gases? What are the freezing and boiling points of water in degrees Celsius? Degrees Fahrenheit? What is the conversion factor between degrees Fahrenheit to degrees Celsius? What is the conversion factor between degrees Celsius and degrees Fahrenheit? Why are the Celsius and Fahrenheit temperature scales said to be empirical? What is the temperature of absolute zero in degrees Celsius? What is thought to happen at absolute zero? 210 6. PROPERTIES OF GASES 6.15 Why is the Kelvin temperature scale an absolute scale? 6.16 What is the conversion factor between degrees Celsius and degrees Kelvin? 6.17 What is the absolute temperature scale used in some engineering fields? How does it differ from the Kelvin scale? 6.18 What is the general statement of Charles’ law? 6.19 What is the equation that describes Charles’ law? 6.20 What is the general statement of Gay-Lussac’s law? 6.21 What is the equation that describes Gay-Lussac’s law? 6.22 What is the equation of the combined gas law? 6.23 What is the general statement of Avagadro’s law? 6.24 What is the equation that describes Avagadro’s law? 6.25 What is the equation of the ideal gas law? 6.26 What is an ideal gas? 6.27 What is the value of the gas constant in L•atm/K•mol? 6.28 Under what conditions do gases not behave ideally? 6.29 What are the three assumptions of the kinetic-molecular theory of gases that do not hold at low temperatures and high pressures? 6.30 What is the van der Waals equation? 6.31 What do the van der Waals constants correct for? 6.32 Which van der Waals constant is most important for polar molecules? Which constant is more important for larger molecules? 6.33 What is the statement of Dalton’s law of partial pressures? 6.34 What is a mole fraction? 6.35 What is STP? 6.36 What is the volume of one mole of a gas at STP? 6.37 What is the law of combining volumes? PROBLEMS 6.38 Convert the following pressures to atmospheres, (a) 1.4 105 Pa, (b) 2.53 bar, (c) 1520 Torr, (d) 19.1 psi, (e) 836 mmHg, (f) 507 millibar. 6.39 If the volume of gas in a piston is 5.63 L at a pressure of 1.5 atm and the gas is compressed to a volume is 4.8 L, what will be the new pressure inside the piston if the temperature remains constant? 6.40 A gas occupies 12.3 L at a pressure of 40.0 mmHg. What is the volume when the pressure is increased to 60.0 mmHg at constant temperature? 6.41 A 1.5 L container is filled with nitrogen at a pressure of 10 atm. What size container would be required to hold this gas at a pressure of 2.0 atm at the same temperature? 6.42 A gas occupies 1.56 L at 1.00 atm. What will be the volume if the temperature remains the same and the pressure is increased to 3.00 atm? 6.43 If the pressure on a gas is decreased by one half, how will the volume change if the temperature does not? PROBLEMS 211 6.44 A weather balloon contains 15 L of air at sea level. What will be the volume of the balloon when it ascends to an altitude where the air pressure is 0.85 atm at the same temperature? 6.45 A gas occupies 77.0 L at 18.0 Torr at standard temperature. What will its volume be at STP? 6.46 Convert the following temperatures to Fahrenheit: (a) 15.5°C, (b) 25°C, (c) 52.3°C, (d) 75°C, (e) 83°C. 6.47 Convert the following to Celsius: (a) 167 °F, (b) 131°F, (c) 86°F, (d) 298 K, (e) 324 K. 6.48 Convert the following to Kelvin: (a) 25.5°C, (b) 50.3°C, (c) 98.0°C, (d) 256°R, (e) 536°R. 6.49 What is the decrease in temperature when 2.00 L of a gas at 25.0°C is compressed to 1.00 L if the pressure remains the same? 6.50 What is the volume of a gas at 100°C and standard pressure if it occupies 1.00 L at STP? 6.51 A 5.00 L sample of gas is collected at 100 K and then allowed to expand to 20.0 L. What is the new temperature of the gas if the pressure does not change? 6.52 What is the volume of a gas sample at 150°C if it has a volume of 6.00 L at 27.0°C at the same pressure? 6.53 A balloon has a volume of 2500 mL during the day at a temperature of 28.8°C. If the temperature at night falls to 10.0°C, what will the volume of the balloon be, in liters, if the pressure remains constant? 6.54 The volume of a gas in a sealed cylinder is 30 cm3 at a temperature of 30°C. As the gas is heated to 60°C, the piston moves to maintain the same pressure. What is the new volume of the gas in liters? 6.55 If 50.0 L of oxygen at 20.0°C is compressed to 5.00 L, what must the new temperature be to maintain constant pressure? 6.56 What is the final pressure of a gas sample in a rigid container at 1.00 atm when it is heated from 25.0°C to 40.0°C. 6.57 What is the final pressure, in standard atmospheres, of a gas sample in the steel tank at 3000 Torr when the gas is cooled from 25.0°C to 0.00°C. 6.58 A gas has a pressure of 640 Torr at 40.0°C. What is the temperature at standard pressure? 6.59 A gas is collected in a rigid container at 22.0°C and 745 Torr. If the temperature decreases to 0.0°C, what is the pressure in the container in atmospheres? 6.60 A gas sample is collected in a rigid cylinder under 1.25 atm pressure. At 36.5°C, that same gas sample has a pressure of 2.50 atm. What was the initial temperature of the gas in the cylinder? 6.61 A gas sample has a volume of 17 L, a pressure of 2.3 atm, and a temperature of 299 K. If the temperature of the gas is raised to 350 K and the pressure is lowered to 1.5 atm, what is the new volume of the gas sample? 6.62 A gas sample has a volume of 17 L, a temperature of 67°C, and a pressure of 88.89 atm, what will be the pressure of the gas sample if the temperature is raised to 94°C and the volume is decreased to 12 L? 6.63 A 2.00 L gas sample is collected at 25.0°C and 745.0 Torr. What is the volume of the sample at STP? 6.64 The volume of a gas sample at STP is 500 mL. What volume would the same gas occupy when at a pressure of 3.00 atm and temperature of 100°C? 212 6. PROPERTIES OF GASES 6.65 If the absolute temperature of a gas sample is doubled and the pressure tripled, how does the volume change? 6.66 If 4 mol of a gas is contained in a 12 L container at a pressure of 5.6 atm, what is the temperature of the gas? 6.67 What is the volume of 20.3 g of carbon dioxide gas at STP? 6.68 How many moles are there in a gas sample at STP and a volume of 56.2 L? 6.69 What is the molecular weight of a 64.1 g sample of gas that occupies 22.4 L at STP? 6.70 What is the molecular weight of a gas that occupies 19.2 L at STP with a mass of 12.0 g? 6.71 What is the pressure of 2.4 mol of a gas sample in a 45 L container at a temperature of 97°C? 6.72 What is the density of ammonia gas at 1.00 atm and 298 K? 6.73 What is the molar mass of a gas that has a density of 1.853 g/L at 745.5 Torr and 23.8°C? 6.74 Which of the following gases has the smallest van der Waals “a” constant: NH3, N2, H2O, CCl4? 6.75 Which of the gases in Problem 6.74 has the largest van der Waals “b” constant? 6.76 Compare the calculated pressure of 1.00 mol of carbon dioxide gas in a 536 mL container at 373 K using: (a) the ideal gas equation and (b) the van der Waals equation (a ¼ 3.64 L2•atm•mol2; b ¼ 0.0427 L/mol). 6.77 What is the deviation from ideal behavior for carbon dioxide under the conditions of Problem 6.76? 6.78 What is the total pressure of a mixture of 2.00 g of hydrogen, 8.00 g of nitrogen, and 12.0 g of argon in a 10.0 L container at 273 K? 6.79 A mixture of 1.01 g of hydrogen and 17.7 g of chlorine in a container at 300 K has a total gas pressure of 0.975 atm. What is the partial pressure of hydrogen in the mixture? 6.80 A 1.00 L flask contains 1.10 g of carbon dioxide and an unknown amount of oxygen at 373 K and 608 Torr. What is the mass of oxygen in the flask? 6.81 A tank contains 480 g of oxygen and 80 g of helium at a total pressure of 7.0 atm. (a) What is the mole fraction of oxygen in the tank? (b) What is the partial pressure of oxygen? 6.82 What is the amount of oxygen produced from the decomposition of 4.00 g hydrogen peroxide (H2O2 ! H2O + O2)? 6.83 If 2.0 mol of methane (CH4) are completely combusted at STP, how many liters of CO2 are produced (see Section 4.4)? 6.84 Ozone decomposes in water to yield hydrogen and oxygen gas. What volume of ozone is required to produce 5 L of oxygen gas at STP? 6.85 Ammonia gas reacts with oxygen to yield NO2 and H2O. (a) What is the balanced equation for this reaction? (b) What volume of NO2 is produced from 100 g of NH3(g) at STP? C H A P T E R 7 Chemical Equilibrium O U T L I N E 7.1 Reversible Reactions 213 7.5 The Reaction Quotient 232 7.2 The Equilibrium Constant 215 Important Terms 235 7.3 Relationships Between Equilibrium Constants 220 Study Questions 236 Problems 237 7.4 Le Chatelier’s Principle: Disturbing a Chemical Equilibrium 222 7.1 REVERSIBLE REACTIONS The idea that a chemical reaction can be reversible was introduced by Claude Louis Berthollet in 1803 when he observed the formation of sodium carbonate crystals at the edge of a limestone salt lake in Egypt (Fig. 7.1). Since the salt lake was a landlocked body of water with a very high concentration of dissolved sodium chloride (>3 g/L) and other minerals, he knew that the formation of these crystals must be a result of the following chemical reaction; 2NaCl + CaCO3 ! Na2 CO3 + CaCl2 (1) Berthollet also knew that laboratory experiments conducted with sodium carbonate and calcium chloride as reactants always gave the products sodium chloride and calcium carbonate. Na2 CO3 + CaCl2 ! 2NaCl + CaCO3 (2) From these observations, he concluded that the chemical reaction forming the sodium carbonate crystals in the lake was the reverse of the reaction observed in the laboratory experiments and was caused by the excess sodium chloride concentration present in the lake. Before this observation, chemical reactions were thought to always proceed in one direction, from reactants to products. General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00007-2 213 # 2018 Elsevier Inc. All rights reserved. 214 7. CHEMICAL EQUILIBRIUM FIG. 7.1 Sodium carbonate crystals at the € edge of a salt lake in Chad. From Stefan Thungen, Wikimedia Commons. A reversible reaction is a chemical reaction where the reactants first form products, which then react together to give back the reactants. Most of all chemical reactions are reversible to some extent even if the back reaction is very slow and the concentrations of the reformed reactants are very small. The reversible chemical reaction is represented in a chemical equation by the presence of a double arrow (>) between the reactants and products. The double arrow indicates that the reaction can go in both directions and both products and reactants are present in the reaction system at the same time. In the generic reversible reaction; aA + bB > cC + dD (3) the reactants A and B can react to form the products C and D and the products C and D can also react to reform the original reactants A and B. The reaction that proceeds from left to right, forming products C and D, is known as the forward reaction, while the reaction that proceeds from right to left reforming the original reactants A and B is known as the reverse reaction. Even though reversible chemical reactions occur in both directions, the chemical species designated as products and reactants are determined by the forward reaction. So, the species on the left side of the chemical equation are the reactants and the species on the right side of the chemical equation are the products of the reaction. As previously described in Section 5.1, weak acids and bases undergo reversible reactions. A weak Brønsted-Lowry acid (HAw) partially ionizes in water to form the hydronium ion and the conjugate base (A), which can then react to reform the weak acid and water. HAw ðaqÞ + H2 OðlÞ >H3 O + ðaqÞ + A ðaqÞ The hydronium ion, the conjugate base, and the molecular form of the weak acid all exist in solution at the same time. So, both the forward reaction, ionization of the weak acid, and the reverse reaction, reformation of the weak acid, occur in the solution simultaneously. As shown in Fig. 7.2, when the reactants of a reversible reaction are first combined, the forward reaction occurs rapidly increasing the concentrations of the products and decreasing the concentrations of the reactants. As the reaction proceeds and the concentration of products increases, the forward reaction slows and the reverse reaction begins to occur. After some time, an equilibrium is reached where the concentrations of reactants and products do not 215 7.2 THE EQUILIBRIUM CONSTANT FIG. 7.2 A reversible chemical reaction reaches dynamic equilibrium when the rate of the forward and reverse reactions are equal (left) and the concentrations of reactants and products do not change with time (right). change with time and the forward and reverse reaction rates are equal. This is known as a dynamic equilibrium because, although forward and reverse reactions still occur, the rate of the forward reaction equals the rate of the reverse reaction. So, the ratio of concentrations of the products to the concentrations of the reactants remains constant, although the reaction is constantly proceeding in both directions. 7.2 THE EQUILIBRIUM CONSTANT Since the concentrations of reactants and products of a reversible reaction at equilibrium are constant, an expression can be written for an equilibrium constant (Keq) that describes the concentrations of the products and reactants at equilibrium. For the generic reversible reaction (3) at equilibrium, the equilibrium constant expression is; Keq ¼ ½Cc ½Dd ½Aa ½Bb (4) As with the acid ionization constants described in Section 5.2, the equilibrium constant for a reversible reaction is expressed as the ratio of the equilibrium concentrations of the products in the numerator to the equilibrium concentrations of the reactants in the denominator. The superscript letters are the stoichiometric coefficients of the reactants and products in the balanced chemical equation. The exact form of an equilibrium constant expression for any reversible reaction is determined according to the following rules: 1. 2. 3. 4. All concentrations (indicated by square brackets) must be equilibrium values. The concentrations of the products are written in the numerator. The concentrations of the reactants are written in the denominator. For homogeneous reactions, the numerator contains a term for every reactant and the denominator contains a term for every product. 5. For heterogeneous reactions involving a solid as reactant or product, the concentration of the solid is not included in the equilibrium constant expression. 6. For heterogeneous reactions involving a liquid that acts both as a solvent and as a reactant or product, the concentration of the liquid is not included in the equilibrium constant expression. 216 7. CHEMICAL EQUILIBRIUM 7. The concentrations of the products and the reactants are raised to a power equal to the stoichiometric coefficient in the balanced chemical equation for the reversible chemical reaction. Rule 4 includes the restriction that the reaction be homogeneous. Homogeneous reactions are chemical reactions that involve reactants and products all present in the same phase. This usually occurs where reactants and products are all in the gas phase or are all in the solution phase. A heterogeneous reaction is one that has reactants and/or products that are present in more than one phase, such as reactions involving liquids and gases, or liquids and solids. The exceptions that apply to equilibrium constant expressions for heterogeneous reactions are outlined in rules 5 and 6. According to rule 5, reversible reactions involving a solid as a reactant or product do not include the concentration of the solid in the equilibrium constant expression. For example, the reaction of hot steam with carbon in a closed container yields hydrogen and carbon monoxide gas; CðsÞ + H2 O g > H2 g + CO g Since carbon is a solid, the concentration is fixed by its density and so is constant. This means that the concentration of carbon is not changed by the reaction and the amount of carbon present does not affect the reaction. So, carbon is omitted from the equilibrium constant expression. The equilibrium constant expression for the reaction of solid carbon with hot steam becomes; Keq ¼ ½H2 ½CO ½H2 O Also, according to rule 6, the concentration of a solvent such as water that also acts as a reactant or product is not included in the equilibrium constant expression. When a liquid acts as both a solvent and as a reactant or product, the concentration of the solvent is very much larger than the small amount of it that is taken up or produced by the reaction. So, the concentration of the solvent is not changed by the reaction and remains essentially constant. For this reason, it also is not included in the equilibrium constant expression for the reaction. For example, in the reaction of the weak base ammonia with water; NH3 ðaqÞ + H2 OðlÞ >NH4 + ðaqÞ + OH ðaqÞ water is a reactant, but it also acts as a solvent for both reactants and products, as indicated by the notation “(aq)” in the chemical equation. The density of water is approximately 1000 g/L and its molar mass is 18.02 g/mol. So, the molar concentration of water is: 1000 g=L ½H2 O ¼ ¼ 55:5mol=L: 18:02 g=mol The concentration of water, as the solvent, remains at 55.5 mol/L during the chemical reaction with ammonia and so it is not changed by the reaction. It is not included in the equilibrium constant expression for the same reasons that a solid is not included. The equilibrium constant expression for the reaction of ammonia with water becomes; Keq ¼ ½NH4 + ½OH ½NH3 7.2 THE EQUILIBRIUM CONSTANT 217 The value of the equilibrium constant for any reversible chemical reaction is always the same at a given temperature, regardless of the initial amounts of the reactants. The magnitude of the value of the equilibrium constant is a measure of the extent that the forward and reverse reactions take place, as shown in Fig. 7.3. For Keq > 1, the concentrations of the products are greater than the concentrations of the reactants at equilibrium. For Keq < 1, the concentrations of reactants are greater than the concentrations of products at equilibrium. A very large equilibrium constant (Keq ≫ 1) indicates that the forward reaction goes essentially to completion and the equilibrium lies in favor of the production of the products. A very small equilibrium constant (Keq ≪ 1) indicates that there is little tendency for the forward reaction to take place and the reverse reaction goes nearly to completion. The equilibrium lies in favor of the reactants. In summary: • Keq > 1; [products] > [reactants] at equilibrium. • Keq < 1; [reactants] > [products] at equilibrium. • Keq ≫ 1; The forward reaction dominates and essentially all reactants are converted to products at equilibrium. • Keq ≪ 1; The reverse reaction dominates and essentially only reactants are present at equilibrium. The units for Keq depend on the units used for the concentrations of the reactants and products. For the equilibrium constants of reactions in aqueous solution, the concentration units are molar and the unit for the equilibrium constant in Eq. (4) is; Keq ðsolutionÞ ¼ ½Mc ½Md ½Ma ½Mb ¼ Mðc + dÞða + bÞ (5) The exponent, (c + d) (a + b), represents the change in the number of moles when going from reactants to products. For cases where the number of moles of products equal the number of moles of reactants, (a + b) ¼ (c + d), the exponent is zero and Keq is unitless. FIG. 7.3 The magnitude of Keq describes the concentrations of the products (blue) and reactants (red) at equilibrium. 218 7. CHEMICAL EQUILIBRIUM EXAMPLE 7.1: DETERMINING THE EQUILIBRIUM CONSTANT FROM KNOWN EQUILIBRIUM CONCENTRATION VALUES Calculate the value of the equilibrium constant, Keq, for the reaction of 0.4 mol of sulfur dioxide with 1.0 mol of oxygen to form 1.4 mol of sulfur trioxide in a 2 L vessel at equilibrium. 1. Write the balanced chemical equation. 2SO2 g + O2 g > 2SO3 2. Write the equilibrium constant expression for the reaction. Keq ¼ ½SO3 2 ½SO2 2 ½O2 3. Convert all concentrations to molar. ½SO2 ¼ ½O2 ¼ 0:4 mol ¼ 0:2M 2L 1:0 mol ¼ 0:5M 2L ½SO3 ¼ 1:4 mol ¼ 0:7M 2L 4. Solve for Keq. Keq ¼ ½SO3 2 2 ½SO2 ½O2 ¼ ð0:7 MÞ2 ð0:2 MÞ2 ð0:5 MÞ ¼ 24:5M1 EXAMPLE 7.2: DETERMINING THE EQUILIBRIUM CONCENTRATIONS FROM INITIAL CONCENTRATIONS OF REACTANTS AND THE EQUILIBRIUM CONSTANT What are the equilibrium concentrations for the decomposition of 0.100 M phosphorous pentachloride to phosphorous trichloride and chlorine gas (Keq ¼ 0.030 M at 250°C)? 1. Write the balanced chemical equation. PCl5 g > PCl3 g + Cl2 g 2. Write the equilibrium constant expression for the reaction. Keq ¼ ½PCl3 ½Cl2 ¼ 0:030M ½PCl5 7.2 THE EQUILIBRIUM CONSTANT 219 EXAMPLE 7.2: DETERMINING THE EQUILIBRIUM CONCENTRATIONS FROM INITIAL CONCENTRATIONS OF REACTANTS AND THE EQUILIBRIUM CONSTANT—CONT’D 3. Derive expressions for the equilibrium concentrations. At equilibrium, an unknown amount (x) of PCl5 will be decomposed to PCl3 and Cl2 ½PCl5 ¼ 0:100 xM ½PCl3 ¼ xM ½Cl2 ¼ xM 4. Insert concentration values into the equilibrium constant expression and solve for x. Keq ¼ ½PCl3 ½Cl2 x2 ¼ 0:030 M ¼ ½PCl5 ð0:10 xÞ The Keq expression takes the form of a quadratic equation: x2 + 0.030 x 0.003 ¼ 0 Solving the quadratic equation: ½ b b2 4ac x¼ 2a x¼ 0:030 ð0:114Þ ¼ 0:042 M 2 5. Calculate the equilibrium concentrations. ½PCl5 ¼ 0:100 x M ¼ 0:10 0:042 ¼ 0:058 M ½PCl3 ¼ x M ¼ 0:042 M ½Cl2 ¼ x M ¼ 0:042 M For equilibrium constants of reactions in the gas phase, the units can also be expressed as partial pressures in atmospheres or Torr. The unit for the equilibrium constant expressed as partial pressures is; Keq ðatmÞ ¼ ½Pc ½Pd ½Pa ½Pb ¼ Pðc + dÞða + bÞ (6) The Keq expressed in partial pressures can be converted to Keq expressed in molarity by using the ideal gas equation. Rearranging the ideal gas equation; P ¼ (n/V)(RT) and since n/V is the concentration in moles/L, P ¼ MRT. Keq ðatmÞ ¼ Pðc + dÞða + bÞ ¼ ðMÞðc + dÞða + bÞ ðRT Þðc + dÞða + bÞ Keq ðatmÞ ¼ Keq ðMÞ ðRT Þðc + dÞða + bÞ (7) 220 7. CHEMICAL EQUILIBRIUM EXAMPLE 7.3: DETERMINING THE EQUILIBRIUM CONSTANT AND EQUILIBRIUM CONCENTRATIONS OF A GAS REACTION IN PARTIAL PRESSURES FROM THE EQUILIBRIUM CONSTANT AND CONCENTRATIONS IN MOLAR UNITS Determine the value of the equilibrium constant and partial pressures of the reactants and products in Example 7.2. 1. Determine Keq(atm). Keq ðatmÞ ¼ Keq ðMÞ ðRTÞðc + dÞða + bÞ ¼ 0:030mol=L 0:08206L atm=K mol 523K ¼ 1:29atm 2. Determine the concentration of the reactant in atmospheres. P¼ nRT ¼ MRT V P ¼ ð0:100 mol=LÞð0:08206L atm=K molÞð523 KÞ ¼ 4:29atm 3. Derive expressions for the equilibrium concentrations in atmospheres. ½PCl5 ¼ 4:29 x atm ½PCl3 ¼ x atm ½Cl2 ¼ x atm 4. Insert concentration values into the equilibrium constant expression and solve for x. Keq ¼ ½PCl3 ½Cl2 x2 ¼ 1:29atm ¼ ½PCl5 ð4:29 xÞ The Keq expression takes the form: x2 + 1.29x 5.53 ¼ 0 Solving the quadratic equation gives: x ¼ 1.79 atm 5. Solve for the equilibrium concentrations. ½PCl5 ¼ 4:29 x atm ¼ 4:29 1:79 ¼ 2:50atm ½PCl3 ¼ x atm ¼ 1:79atm ½Cl2 ¼ xatm ¼ 1:79atm 7.3 RELATIONSHIPS BETWEEN EQUILIBRIUM CONSTANTS The equilibrium constant expressions for the forward and reverse reactions in a reversible chemical reaction are related to each other. Consider Berthollet’s chemical equations for the formation of sodium carbonate crystals (1) and the laboratory reaction for the formation of calcium carbonate (2) in Section 7.1. The equilibrium constant expression for the formation of sodium carbonate crystals would be; 7.3 RELATIONSHIPS BETWEEN EQUILIBRIUM CONSTANTS Keq ð1Þ ¼ 221 ½Na2 CO3 ½CaCl2 ½NaCl2 ½CaCO3 In this chemical equation, the formed sodium carbonate and calcium chloride are the products and the sodium chloride and calcium carbonate are the reactants of the chemical reaction. The equilibrium constant expression for the chemical reaction that forms calcium carbonate would be; Keq ð2Þ ¼ ½NaCl2 ½CaCO3 ½Na2 CO3 ½CaCl2 In this reaction, sodium chloride and calcium carbonate are the products and sodium carbonate and calcium chloride are the reactants. The equilibrium constant expression for the chemical reaction (1) is the inverse of the equilibrium constant expression for the chemical reaction (2). Since Eq. (1) is considered to be the forward reaction and Eq. (2) is considered to be the reverse reaction of the same chemical equilibrium; • The value of the equilibrium constant for a forward chemical reaction Keq(f) and the value of the equilibrium constant for the reverse chemical reaction Keq(r) are the reciprocals of each other. Keq ð f Þ ¼ 1=Keq ðrÞ (8) This means that if a forward chemical reaction has a very large equilibrium constant, the reverse chemical reaction will have a very small equilibrium constant. Or, if a forward chemical reaction has a very small equilibrium constant, the reverse reaction will have a very large one. For cases where an overall chemical reaction can be considered stepwise, as the sum of two or more separate reactions, the equilibrium constant for the overall reaction can be determined from the equilibrium constants for each of the separate reactions. For example, nitrogen dioxide, a major air pollutant, is formed from the reaction of nitrogen and oxygen gas at high temperatures from the following reaction; N2 g + 2O2 g > 2NO2 g with the equilibrium constant expression; Keq ¼ ½NO2 2 ½N2 ½O2 2 This overall reaction actually occurs in two very fast stepwise reactions: ½NO2 N2 g + O2 g > 2NO g with Keq ¼ ½N2 ½O2 and ½NO2 2 2NO g + O2 g >2NO2 g with Keq ¼ ½NO2 ½O2 222 7. CHEMICAL EQUILIBRIUM The overall reaction is the sum of the two stepwise reactions: N2(g) + O2(g) → ← 2NO(g) + 2NO(g) + O2(g) → ← 2NO2(g) = N2(g) + 2O2(g) → ← 2NO2(g) and the overall equilibrium constant expression for reaction is obtained as the product of the equilibrium constants of the two stepwise reactions: [NO]2 × [NO2]2 = [NO2]2 [N2][O2] [NO]2[O2] [N2][O2]2 In summary: • When two or more chemical equations can be summed to give a combined chemical reaction, the equilibrium constant for the overall chemical reaction is equal to the product of the equilibrium constants for each chemical reaction in the summation. 7.4 LE CHATELIER’S PRINCIPLE: DISTURBING A CHEMICAL EQUILIBRIUM When a reversible chemical reaction reaches chemical equilibrium under a particular set of conditions, there is no further change in the concentrations of reactants and products. But, if a change is made in the conditions under which the equilibrium was established, the equilibrium will be disturbed. The changes in conditions that will disturb a chemical equilibrium are: (1) (2) (3) (4) a a a a change change change change in in in in the the the the concentration of a reactant or product, temperature of the reaction, volume of a reaction in the gas phase, or pressure of a reaction in the gas phase. If a change is made in one of these conditions after the equilibrium is established, it is said that a stress is put on the equilibrium and a chemical change will occur which will establish a new chemical equilibrium under the new conditions. This is known as Le Chatelier’s principle, after the nineteenth century chemist Henry-Louis Le Chatelier, who studied the effects of changes in reaction conditions on a chemical equilibrium. The exact statement of Le Chatelier’s principle is; • When a chemical reaction at equilibrium is subjected to a change in reaction conditions, the position of the equilibrium will shift to counteract the effect of the change until a new equilibrium is established. The ways that a chemical reaction at equilibrium will respond when disturbed by a change in the original conditions can be predicted. These predicted responses and their 223 7.4 LE CHATELIER’S PRINCIPLE: DISTURBING A CHEMICAL EQUILIBRIUM TABLE 7.1 Conditions The Response of a Reversible Chemical Reaction at Equilibrium to a Change in Reaction Change in Condition Response Effect on Keq Increase concentration of reactant Formation of more products None Increase concentration of product Formation of more reactants None Decrease concentration of reactant Formation of more reactants None Decrease concentration of product Formation of more products None Increase in volume Increase in the total number of moles None Decrease in volume Decrease in the total number of moles None Increase in pressure Decrease in the total number of moles None Decrease in pressure Increase in the total number of moles None Increase in temperature Absorption of heat Change Decrease in temperature Release of heat Change Changes in volume and pressure affect GAS reactions only effects on the equilibrium constant are summarized in Table 7.1 for each of the changing conditions. If the concentration of a reactant or product in a reversible chemical reaction at equilibrium is changed, by the addition or the removal of either a reactant or a product, the equilibrium will be reestablished by adjusting the concentrations of reactants and products so that their ratio and the value of Keq will remain the same. For example, if the concentration of reactant A in the generic Eq. (3) of Section 7.1 is increased by the addition of A to the reaction system, the reaction will shift to consume the added reactant A and form more of the products C and D. This increases the value of the numerator of Keq and decreases the value of the denominator compensating for the increase in the value of the denominator caused by the added reactant. This response can be useful in an industrial process if it is necessary to convert the maximum possible amount of reactant B into the products C and D when reactant B is an expensive or rare material and reactant A is inexpensive and abundant. In this case, the addition of excess reactant A becomes a means of conserving the more expensive reactant B while producing as much product as possible. This response could also be useful in creating a greener process if reactant B were the most environmentally hazardous material in the reaction. If the concentration of one of the products is decreased, the reaction will proceed in a direction that will increase the product concentration and decrease the concentrations of the reactants. This shift in concentrations of reactants and products keeps their ratio constant and maintains the value of Keq. This is essentially what happens if one of the products of the reaction is removed from the reaction vessel as it is formed. If product C in reaction (3) is removed as it is formed, the reaction would shift to the right to produce more C and decrease the concentration of reactants in an attempt to keep the ratio of products to reactants constant and maintain the value of Keq. This can also be used in an industrial process as a 224 7. CHEMICAL EQUILIBRIUM means of forcing reversible reactions with small equilibrium constant values to produce as much product as possible. In a similar manner, if the concentration of one of the products is increased or one of the reactants is decreased, the reaction will shift to the left to consume added product or form more of the reactants until the ratio of the concentrations of products to reactants reproduces the value of Keq and chemical equilibrium is reestablished. In every case, the equilibrium concentrations of the reactants and products will have changed from their original values and the ratio of products to reactants will shift so that the value of the equilibrium constant remains the same and a new chemical equilibrium is established with new concentrations of reactants and products. EXAMPLE 7.4: DETERMINING THE EFFECT OF A CHANGE IN CONCENTRATION ON A CHEMICAL EQUILIBRIUM For the decomposition reaction of phosphorous pentachloride to phosphorous trichloride and chlorine gas, determine the direction of the response and new equilibrium concentrations with an addition of 0.2 mol of PCl5 to the equilibrium concentration of 0.058 M PCl5, 0.042 M PCl3, and 0.042 M Cl2 in a 1.00 L container. (Keq ¼ 0.030 M at 250°C) 1. Write the balanced chemical equation. PCl5 g > PCl3 g + Cl2 g 2. Write the equilibrium constant expression for the reaction. Keq ¼ ½PCl3 ½Cl2 ¼ 0:030M ½PCl5 3. Determine the direction of response. The addition of reactant will cause the equilibrium concentrations to shift to form more products. 4. Derive expressions for the new equilibrium concentrations. At equilibrium, an unknown amount (x) of the new concentration of PCl5 will be decomposed to form PCl3 and Cl2 ½PCl5 ¼ ð0:058 + 0:2Þ x M ½PCl3 ¼ 0:042 + x M ½Cl2 ¼ 0:042 + x M 5. Insert concentration values into the equilibrium constant expression and solve for x. Keq ¼ ½PCl3 ½Cl2 ð0:42 + xÞð0:42 + xÞ ¼ 0:030 M ¼ ½PCl5 ð0:78 xÞ The Keq expression takes the form of a quadratic equation: x2 + 1.14x 0.0576 ¼ 0 Solving the quadratic equation gives: x ¼ 0.039 M 7.4 LE CHATELIER’S PRINCIPLE: DISTURBING A CHEMICAL EQUILIBRIUM 225 EXAMPLE 7.4: DETERMINING THE EFFECT OF A CHANGE IN CONCENTRATION ON A CHEMICAL EQUILIBRIUM—CONT’D 6. Calculate the new equilibrium concentrations: ½PCl5 ¼ 0:258 0:039M ¼ 0:219M ½PCl3 ¼ 0:042 + 0:039M ¼ 0:081M ½Cl2 ¼ 0:042 + 0:039M ¼ 0:081M CASE STUDY: CHEMICAL EQUILIBRIUM AND PH INDICATORS Acid-base indicators (also known as pH indicators) are chemical substances which change color at a particular hydronium ion concentration. These indicators can be weak acids or weak bases, but most common indicators are weak acids. The generic chemical reaction of a weak acid pH indicator is; − + HInd(aq) + H2O(l) → ← Ind (aq) + H3O (aq) color A color B ð9Þ where HInd is the weak acid form of the indicator and Ind is the conjugate base form of the indicator. The equilibrium constant expression for the ionization of a weak acid indicator is also known as the acid ionization constant described in Section 5.2; KInd ¼ ½H3 O + ½Ind ½HInd The acid form and its conjugate base have different colors, as indicated in Eq. (9). When an indicator is added to a solution of unknown pH, the ratio of [Ind]/[HInd] determines the resulting color of the solution, which then indicates the hydronium ion concentration of the solution. If the indicator is added to a solution with a low pH, the hydronium ion concentration is high and so, according to LeChatelier’s Principle, the equilibrium position in Eq. (9) shifts to the left in favor of the unionized form of the indicator in order to maintain a constant value of KInd. With the higher concentration of HInd, the solution has the color A. But, if the indicator is added to a solution with a high pH, the hydronium ion concentration is low and so the equilibrium position in Eq. (9) shifts to the right in favor of the conjugate base. With the higher concentration of Ind, the solution has color B. A mixture of different indicators that show smooth color changes at a wide range of pH values is known as a universal indicator. The most common universal indicator is typically composed of bromthymol blue, thymol blue, methyl orange, and phenolphthalein. These indicators are complex weak organic acids with varying equilibrium constants. So, they each show a different color change at their turning point due to differences in their complicated organic structures. However, since there are several indicators present, many different colors are produced due to the mixture of each 226 7. CHEMICAL EQUILIBRIUM FIG. 7.4 The color changes of a universal indicator at different pH values. From Dejan Jovic DJ, Wikimedia Commons. indicator either in their HInd or Ind form. The colors of a universal indicator at different pH values are shown in Fig. 7.4. Many natural plants contain chemical species from the anthocyanin family, in a class called flavonoid compounds. Anthocyanins can be found in a variety of colored plants including red cabbage, geranium, poppy, rose, blueberry, blackcurrent, and rhubarb. The leaves of red cabbage, in particular, are usually colored dark red to purple, but can also be green or yellow. The anthocyanin in red cabbage is cyaniclin, which has a very complex organic structure with multiple weak acid and weak base sites that can act as indicators. This causes the color of the plant to change according to the pH of the soil. In acidic soils, the leaves grow more reddish, in neutral soils they will grow purplish, while in an alkaline soil the leaves are greenish yellow to yellow. When cyaniclin is extracted from the leaves of red cabbage, it can be used as a universal indicator as shown in Fig. 7.5. Cyaniclin is red in acidic solution (pH1–2), blue in neutral solution (pH 7–8), and yellow in basic solution (pH >10). Intermediate colors of purple and green are observed at intermediate pH values due to the overlap of the yellow, blue, and red forms. Unlike the multiple weak acid indicators used in the commercial universal indicators, this natural universal indicator has only one molecular species. The varied colors achieved from this one molecular species are possible because of the complex nature of cyaniclin, which has the ability to change its structure at the three different H3O+ concentrations. FIG. 7.5 Color changes at pH values (from left to right) equal to 1, 3, 5, 7, 8, 9, 10, 11, and 13 using natural cyaniclin extracted from red cabbage as a pH indicator. From IndikatorBlaukraut.JPG, Wikimedia Commons. 7.4 LE CHATELIER’S PRINCIPLE: DISTURBING A CHEMICAL EQUILIBRIUM 227 Although indicators can be used to determine the pH of a single solution, they are also commonly used in acid-base titrations to signal the completion of an acid-base reaction, as described in Section 5.6. When an acid of unknown concentration is titrated with a base of known concentration, the hydronium ion concentration in the solution at the beginning of the titration is high and the pH is low. According to LeChatelier’s Principle, the presence of the acid shifts the chemical equilibrium of the indicator to the left in favor of reactants and the weak acid indicator is almost entirely in the HInd form. As the titration proceeds, the addition of the base titrant reacts with hydronium ions, decreasing their concentration and shifting the indicator equilibrium to the right in favor of the products. The intensity of the color of HInd decreases and the intensity of the color of Ind increases. The point where the indicator will change from color A to color B should occur at the equivalence point of the titration if the indicator is chosen properly. This color change occurs at the point where; ½HInd ¼ ½Ind So, the pH of the solution at the color change is the pH at which half of the indicator is in its acid form and half is in the conjugate base form. Since [Hind] ¼ [Ind] at the point of color change the equilibrium expression at this point becomes; KInd ¼ ½H3 O + This means that the hydronium ion concentration of the solution at the color change is equal to the value of the equilibrium constant of the indicator. Also, the pH of the solution when the indicator changes color is equal to the pKInd of the indicator. The proper choice of an indicator for an acid-base titration requires a knowledge of the pH of the solution at the equivalence point. The equivalence point of the acid-base titration will have a pH dependent on the relative strengths of the acid and base used. This is because the final pH is controlled by the relative strengths of the conjugate acid and conjugate base after the reaction is complete. The titration of a strong acid with a strong base or a weak acid with a weak base will have an equivalence point near a pH of 7. Since both acid and base are of approximately the same strength, their conjugate acid and base will be about the same strength. However, the reaction of a strong acid with a weak base will have an equivalence point at a pH < 7, since the conjugate acid of the weak base will be a strong acid. Similarly, the reaction of a weak acid with a strong base will have an equivalence point at a pH > 7, since the conjugate base of the weak acid will be a strong base. When the products of a titration reaction are either a strong conjugate acid or a strong conjugate base, the exact pH at the equivalence point can be calculated from the equilibrium constant of the reaction of the strong conjugate acid or strong conjugate base with water as; HAðaqÞ + H2 OðlÞ ! H3 O + ðaqÞ + A ðaqÞ; Keq ¼ B ðaqÞ + H2 O ðlÞ ! BH + ðaqÞ + OH ðaqÞ; Keq ¼ ½H3 O + ½A ½HA ½BH + ½OH ½B The pH at the equivalence point is determined from the [H3O+] or [OH] as described in Chapter 5. An appropriate indicator for the titration can then be chosen by matching up the value of the calculated pH at the titration equivalence point with the value of the pKInd for the available indicators. 228 7. CHEMICAL EQUILIBRIUM In a gas phase reversible chemical reaction, if the volume of the container is changed, such as occurs in a piston, the pressure of the system is changed. According to the ideal gas law (P ¼ nRT/V), a decrease in volume results in an increase in pressure and an increase in volume results in a decrease in pressure. As far as the gas phase chemical reaction is concerned, decreasing the volume is the same as increasing the pressure and vice versa. When the volume or pressure of a reaction is changed, the concentration (amount/volume) of both reactants and products are also changed. A decrease in volume results in an increase in concentration and an increase in volume results in a decrease in concentration of both the reactants and the products. The reaction will shift to maintain the value of the ratio of the concentration of products to reactants and according to the value of the equilibrium constant. For a gas phase chemical reaction that has more moles of reactants than moles of products such as; ½AB A g + B g >AB g with Keq ¼ ½A½B when the reaction at equilibrium is compressed into a smaller volume, the concentrations of [A], [B], and [AB] are increased by the same amount. But since there are more reactants than products, the value of the denominator in the equilibrium expression is increased more than the value of the numerator, so the ratio [products]/[reactants] will be decreased. The reaction will shift in a direction that will bring the reaction to a new equilibrium in which the total concentration of reactants will be lowered and the concentration of product will be increased in order that Keq remains unchanged. This results in a decrease in the total number of moles, which results in a decrease in pressure. For a gas phase chemical reaction that has more moles of products than moles of reactants such as; ½C½D½E A g + B g >C g + D g + E g with Keq ¼ ½A½B when the reaction at equilibrium is compressed into a smaller volume, the value of the numerator in the equilibrium expression is increased more than the value of the denominator. The equilibrium will shift to the left lowering the total concentration of the products and increasing the concentration of reactants in order that Keq remains unchanged. This results in a decrease in the total number of moles, which results in a decrease in pressure. An increase in volume will have the opposite effect. Since in the larger volume the concentration of products and reactants has decreased, a new equilibrium can be achieved by increasing the total number of moles of reactants and products. This is accomplished by shifting the reaction in the direction of the largest number of moles. If the total number of moles of reactants is greater than the total number of moles of products, the reaction will shift to consume the products and produce more reactants, thus increasing the total number of moles. Similarly, if the total number of moles of products is greater than the total number of moles of reactants, the reaction will shift to consume the reactants and produce more products, reducing the total number of moles. For the special case where the number of moles of reactants and the number of moles of products in a gas phase reaction are equal; 7.4 LE CHATELIER’S PRINCIPLE: DISTURBING A CHEMICAL EQUILIBRIUM 229 ½C½D A g + B g > C g + D g with Keq ¼ ½A½B a change in pressure or volume affects the concentrations of reactants and products equally. So the value of the ratio of the concentrations of products to the concentrations of reactants and the value of Keq remains the same. In this case, there is no need for a change in the equilibrium concentrations to occur. In summary; • An increase in pressure of a gas phase reaction by decreasing the volume will result in a shift in the chemical equilibrium that decreases the total number of moles of gas molecules, effectively decreasing the total concentration and decreasing the pressure. • A decrease in pressure of a gas phase reaction by increasing the volume will result in a shift in the chemical equilibrium that increases the total number of moles of gas molecules, effectively increasing the total concentration and increasing the pressure. • If the total number of moles of products and reactants in a gas phase reaction is equal, a change in pressure caused by a change in volume will result in no shift in the chemical equilibrium. EXAMPLE 7.5: DETERMINING THE EFFECT OF A CHANGE IN VOLUME ON A CHEMICAL EQUILIBRIUM What is the direction of the response and new equilibrium conditions for the decomposition reaction of phosphorous pentachloride with initial concentrations of 0.058 M PCl5, 0.042 M PCl3, and 0.042 M Cl2 when the volume is increased from 1.00 to 2.00 L (Keq ¼ 0.030 M at 250°C). 1. Write the balanced chemical equation. PCl5 g >PCl3 g + Cl2 g 2. Write the equilibrium constant expression for the reaction. Keq ¼ ½PCl3 ½Cl2 ¼ 0:030M ½PCl5 3. Determine the equilibrium response to the new conditions. The increase in volume makes the concentrations of all reactants and products to decrease. Since there are more moles of products than reactants, the equilibrium will shift in favor of the products in order to increase the total number of moles. 4. Determine the new initial concentrations due to the change in volume. The increase in volume from 1.00 to 2.00 L will cause the concentrations of all reactants and products to decrease by a factor of 2 according to Avagodro’s law. (V1/n1 ¼ V2/n2). n2/n1 ¼ V2/V1 ¼ 2.00 L/1.00 L [PCl5] ¼ 0.058/2 M ¼ 0.029 M [PCl3] ¼ 0.042/2 M ¼ 0.021 M [Cl2] ¼ 0.042/2 M ¼ 0.021 M Continued 230 7. CHEMICAL EQUILIBRIUM EXAMPLE 7.5: DETERMINING THE EFFECT OF A CHANGE IN VOLUME ON A CHEMICAL EQUILIBRIUM—CONT’D 5. Derive expressions for the new equilibrium concentrations. At equilibrium, an unknown amount (x) of the reactant PCl5 will be decomposed to form products PCl3 and Cl2: ½PCl5 ¼ 0:029 x M ½PCl3 ¼ 0:021 + x M ½Cl2 ¼ 0:021 + x M 6. Insert concentration values into the equilibrium constant expression and solve for x. Keq ¼ ½PCl3 ½Cl2 ð0:021 + xÞð0:021 + xÞ ¼ 0:030M ¼ ½PCl5 ð0:029 xÞ The Keq expression takes the form of a quadratic equation: x2 + 0.072x 0.00043 ¼ 0 Solving the quadratic equation gives: x ¼ 0.0055 M 7. Calculate the new equilibrium concentrations. ½PCl5 ¼ 0:029 0:0055M ¼ 0:024M ½PCl3 ¼ 0:021 + 0:0055M ¼ 0:026M ½Cl2 ¼ 0:021 + 0:0055M ¼ 0:026M Determining the effect of changing the temperature on a chemical reaction at equilibrium is more difficult than determining the effect of changing the other conditions listed in Table 7.1. This is because the value of the equilibrium constant is temperature-dependent and a change in temperature will change the value of the equilibrium constant. However, a qualitative determination is possible by first determining whether heat is given off or absorbed during the reaction. This determines the general direction of the equilibrium shift and the change in Keq without a numerical determination of the equilibrium constant. A chemical reaction that releases energy in the form of heat (kJ/mol) during the reaction is known as an exothermic reaction. In this case, heat can be expressed as a product of the reaction in the chemical equation; A + B > C + D + heat ðkJ=molÞ (10) A chemical reaction that absorbs energy in the form of heat from its surroundings is known as an endothermic reaction. In endothermic reactions, heat can be expressed as a reactant in the chemical equation; A + B + heat ðkJ=molÞ >C + D (11) 231 7.4 LE CHATELIER’S PRINCIPLE: DISTURBING A CHEMICAL EQUILIBRIUM TABLE 7.2 Responses of a Chemical Reaction at Equilibrium to Changes in Temperature Reaction Temperature Equilibrium Shift Heat Keq Exothermic Decrease Increase products Released Increases Endothermic Decrease Increase reactants Released Decreases Exothermic Increase Increase reactants Absorbed Decreases Endothermic Increase Increase products Absorbed Increases For reversible reactions, if the forward reaction is exothermic, the back reaction would be endothermic by exactly the same amount of energy. The responses of a chemical reaction at equilibrium to changes in temperature are outlined in Table 7.2. If the temperature of a reversible reaction at equilibrium is decreased, according to Le Chatelier’s principle the position of the equilibrium will shift in such a way as to increase the temperature by the release of heat to the surroundings. If the reaction is exothermic, as in Eq. (10), heat can be released by shifting the equilibrium to the right increasing the concentration of products. So, the concentration of the products will increase over the concentrations of the reactants and the value of Keq will increase. If the reaction is endothermic as in reaction (11), the reverse reaction will be exothermic and heat can be released by shifting the equilibrium to the left increasing the concentration of reactants. In this case, the concentration of the reactants will increase over the concentrations of the products and the value of Keq will decrease. If the temperature of a reversible chemical reaction at equilibrium is increased, the position of the equilibrium will shift in such a way that the temperature is decreased by absorbing the extra heat. If the reaction is exothermic, the reverse reaction will be endothermic and heat is absorbed by shifting the equilibrium to the left increasing the concentration of reactants. The concentration of the reactants will increase over the concentrations of the products and the value of Keq will decrease. If the reaction is endothermic, heat is absorbed by shifting the equilibrium to the right increasing the concentration of the products. So, the concentration of the products will increase over the concentrations of the reactants and the value of Keq will increase. In summary: • If the temperature of reversible chemical reaction at equilibrium is increased, the equilibrium shifts in the direction that will absorb the extra heat, favoring the endothermic reaction. • If the temperature of a reversible chemical reaction at equilibrium is decreased, the equilibrium shifts in the direction that will release heat, favoring the exothermic reaction. • Changing the temperature of a reversible chemical reaction at equilibrium will change the value of the equilibrium constant. The direction of the change will depend on the direction of the temperature change and whether the reaction is exothermic or endothermic. 232 7. CHEMICAL EQUILIBRIUM 7.5 THE REACTION QUOTIENT The reaction quotient (Q) is the ratio of the product to reactant concentrations raised to the power of their stoichiometric coefficients for a reversible chemical reaction at any point in the reaction. The expression for the reaction quotient (Q) is identical to the expression for the equilibrium constant (Keq). For the generic reversible reaction (3); Q¼ ½Cc ½Dd (12) ½Aa ½Bb The rules for determining the reaction quotient expressions for reversible reactions are the same as those for determining the equilibrium constant expressions outlined in Section 7.2 with the exception of rule 1, which states that all concentrations must be equilibrium values. This is because the reaction quotient does not describe the ratio of product to reactant concentrations of a reversible reaction at equilibrium as with Keq. Instead, it describes the ratio of the product to reactant concentrations for a reversible reaction at any point in the reaction. The determination of the value of the reaction quotient from the concentrations of the reactants and products at any particular point in time helps to determine the direction the reaction is likely to proceed from that point. A comparison of the value of the reaction quotient (Q) with the value of the equilibrium constant Keq tells which way the reaction will shift in order to reach chemical equilibrium as demonstrated in Fig. 7.6. When Q is less than Keq, the ratio of [products] to [reactants] for Q is less than for Keq and there is an excess of reactants. This means that the reaction has not yet reached equilibrium. In order to reach equilibrium, more products must be formed from the available reactants. So the reaction proceeds to the right. But, when Q is greater than Keq, there are an excess of products. To achieve equilibrium, the reaction must proceed to the left using up the excess products and forming more reactants. Keq Q [Products] [Reactants] FIG. 7.6 A comparison of the values of Q and Keq at any point in a chemical reaction tells how the concentrations of reactants and products will change for the reaction to achieve equilibrium. Reactants Æ Products Equilibrium Products Æ Reactants 7.5 THE REACTION QUOTIENT 233 In summary; • Q < Keq—The ratio of products to reactants is less than that for the system at equilibrium. More products will be formed from the excess reactants for the reaction to reach equilibrium. • Q > Keq—The ratio of products to reactants is larger than that for the system at equilibrium. More products are present than there would be at equilibrium. The reaction must produce more reactants from the excess products for the reaction to reach equilibrium. • Q ¼ Keq—The reaction is already at equilibrium. There is no tendency to form more reactants or more products. EXAMPLE 7.6: DETERMINING THE REACTION QUOTIENT AND THE DIRECTION OF A REVERSIBLE CHEMICAL REACTION Determine the reaction quotient for the reaction of nitrogen and hydrogen to yield ammonia if the concentrations are: [N2] ¼ 0.60 M, [H2] ¼ 0.40 M, and [NH3] ¼ 0.06 M. Given that Keq ¼ 0.080 M2 at 500°C, what direction will the reaction proceed? 1. Write the balanced chemical equation. N2 + 3H2 > 2NH3 2. Write the reactant quotient expression for the reaction. Q¼ ½NH3 2 ½N2 ½H2 3 3. Insert concentration values into the reaction quotient expression and solve for Q. Q¼ ð0:06MÞ2 ð0:60MÞð0:40MÞ3 ¼ 0:094M2 4. Determine the direction that the reaction will proceed. If Q ¼ 0.094 M2 and Keq ¼ 0.080 M2, Q > Keq and the reaction will proceed to the left producing more reactants. CASE STUDY: CHEMICAL EQUILIBRIUM AND CARBON MONOXIDE POISONING Hemoglobin (abbreviated Hb) is a protein containing iron found in the red blood cells. It is responsible for transporting oxygen to the cells and returning carbon dioxide from the cells back to the lungs. Each hemoglobin molecule is made of four protein subunits as shown in Fig. 7.7A. Each subunit surrounds a central heme group containing iron(II) at the center. Each iron is tightly bonded to five nitrogen atoms in the hemoglobin subunit through covalent bonds shown in Fig. 7.7B. The axial position on the iron can reversibly bind to oxygen by a coordinate covalent bond completing the octahedral group of six ligands. Oxygen binds in an “end-on bent” geometry where one oxygen atom binds to Fe and the other juts out at an angle. When the iron is not bound to oxygen, a very 234 7. CHEMICAL EQUILIBRIUM FIG. 7.7 (A) A hemoglobin molecule consisting of four subunits each containing a heme group with an Fe+2 at the center, (B) The Fe+2, already tightly bound to five nitrogen atoms in the hemoglobin molecule, can reversibly bind to an oxygen molecule in the axial position. Modified from OpenStax College, Wikimedia Commons. O O Subunit 3 Subunit 1 N Fe+2 Heme Subunit 4 Subunit 2 N Fe N N weakly bonded water molecule fills the axial site. The iron in each subunit can bind to one oxygen molecule allowing each hemoglobin molecule to bind four oxygen molecules forming the species Hb(O2)4, known as oxyhemoglobin. The binding of hemoglobin to oxygen can be expressed as the reversible chemical reaction; HbðaqÞ + 4O2 g > HbðO2 Þ4 ðaqÞ with the equilibrium constant; Keq ¼ HbðO2 Þ4 ½Hb½O2 4 ¼ 3:6 106 M4 The formation of oxyhemoglobin in the lungs occurs at the equilibrium partial pressure of oxygen at 100 Torr. The transport of oxyhemoglobin from the lungs to active muscle tissue, where the oxygen partial pressure is 20 Torr, results in a reaction quotient that is larger than Keq. This shifts the equilibrium toward the reactants, releasing oxygen from the oxyhemoglobin into the oxygen-depleted tissue. At high altitudes, where the air pressure is lower and the concentration of oxygen in the air is less, the hemoglobin reaction with oxygen in the lungs is not at equilibrium (Q > Keq) and the equilibrium is shifted away from the oxyhemoglobin toward deoxyhemoglobin. Without adequate oxygen transported to the cells, this could cause light headedness and may make it necessary to increase the oxygen concentration by supplementing oxygen from a compressed gas tank reestablishing equilibrium in the lungs. Some people living at high altitudes can become accustomed to the effects of lower oxygen concentrations and their bodies become able to compensate for the lower oxygen concentration in the air by producing increased levels of blood hemoglobin. This higher hemoglobin concentration increases the value of the denominator in the Keq expression in the presence of lower oxygen concentration forcing the reaction to proceed in favor of the product oxyhemoglobin. Some other molecules also have the ability to bind to hemoglobin at iron(II) axial site. These include carbon monoxide, cyanide, sulfur monoxide, nitric oxide, hydrogen sulfide, and other sulfides. Carbon monoxide is especially dangerous since it is a colorless, odorless, and tasteless gas, not readily detectable. Carbon monoxide can also bind to hemoglobin in groups of four in a similar manner as oxygen forming the product carboxyhemoglobin. HbðaqÞ + 4CO g > HbðCOÞ4 ðaqÞ N IMPORTANT TERMS 235 The equilibrium constant for this reaction is 276 times greater than the equilibrium constant for the reaction between Hb and O2. HbðCOÞ4 Keq ¼ ¼ 9:95 108 M1 ½Hb½CO4 This very high equilibrium constant means that essentially all reactants are converted to the product Hb(CO)4 at equilibrium and the forward reaction is approached irreversibility. Due to this strong affinity of hemoglobin for carbon monoxide, the hemoglobin that has bonded with carbon monoxide can no longer bind to oxygen. Personal exposure to carbon monoxide is most often in the form of tobacco smoking, car exhaust, and incomplete combustion in home furnaces. Carbon monoxide in small quantities can cause headaches and dizziness, but larger concentrations can be fatal. Carbon monoxide poisoning causes acute symptoms such as nausea, weakness, angina, shortness of breath, loss of consciousness, seizures, and coma. Carbon monoxide levels in inhaled air of 0.02% can cause headache and nausea, while concentrations of 0.1% can cause unconsciousness. Carbon monoxide is a major product of tobacco combustion with concentrations of 1%–5% in cigarette smoke. It has been shown that in heavy smokers, up to 20% of the oxygen active hemoglobin sites are permanently blocked by CO. IMPORTANT TERMS Dynamic equilibrium the state of a chemical reaction where the forward and reverse reactions occur at the same rate so that the concentrations of reactants and products do not change. Endothermic reaction a chemical reaction that absorbs energy in the form of heat from its surroundings. Equilibrium constant (Keq) value of the chemical reaction quotient when the reaction has reached chemical equilibrium. Equilibrium constant expression the ratio of the concentrations of the products over the reactants in a chemical reaction at equilibrium raised to the power of their stoichiometric coefficients. Exothermic reaction a chemical reaction that releases energy in the form of heat during the reaction. Forward reaction a reversible reaction in which the reactants produce products. Heterogeneous reaction a chemical reaction that involves reactants and/or products that are present in more than one phase. Homogeneous reaction a chemical reaction that involves reactants and products that are all present in the same phase, usually gas phase or solution phase. Le Chatelier’s principle when a chemical reaction at equilibrium is subjected to change in reaction conditions, the position of the equilibrium will shift to counteract the effect of the change until a new equilibrium is established. pH indicator a chemical substance which changes color at a particular hydronium ion concentration. Qualitative dealing with the properties of a system without numerical measurements. Reaction quotient (Q) the ratio of the product to reactant concentrations raised to the power of their stoichiometric coefficients for a reversible chemical reaction at any point in the reaction. Reverse reaction a reversible reaction in which the products react to form the original reactants. Reversible reaction a chemical reaction where the reactants form products, which then react together to reform the reactants. 236 7. CHEMICAL EQUILIBRIUM STUDY QUESTIONS 7.1. 7.2. 7.3. 7.4. What determines a reversible chemical reaction? What is a dynamic equilibrium? When does a chemical reaction reach dynamic equilibrium? Write the equilibrium constant expression for the generic reversible chemical reaction: aA + bB > cC + dD 7.5. What is a homogeneous chemical reaction? 7.6. What is a heterogeneous chemical reaction? 7.7. What species are not included in the equilibrium constant expression? Why are they not included? 7.8. What is the molar concentration of water when used as a solvent? 7.9. What is the relationship between the value of the equilibrium constant for a chemical reaction and the value of the equilibrium constant for the reverse chemical reaction? 7.10. If a chemical reaction has a very large equilibrium constant, what is the size of the equilibrium constant of the reverse reaction? 7.11. What determines the units for the chemical equilibrium constant? 7.12. An equilibrium constant that is much greater than one means what? 7.13. An equilibrium constant that is much smaller than one means what? 7.14. When two or more chemical equations are summed to give a combined chemical reaction, what determines the equilibrium constant for the overall chemical reaction? 7.15. What is Le Chatelier’s principle? 7.16. What four reaction conditions will disturb an equilibrium if they are changed? 7.17. Which reaction condition changes the value of the equilibrium constant if it is changed? 7.18. What is the response of a chemical reaction at equilibrium if the concentration of a reactant is increased? 7.19. What is the response of a chemical reaction at equilibrium if the concentration of a product is increased? 7.20. What is the response of a chemical reaction at equilibrium if the concentration of a reactant is decreased? 7.21. What is the response of a chemical reaction at equilibrium if the concentration of a product is decreased? 7.22. What is the response of a chemical reaction at equilibrium if the volume is increased? 7.23. What is the response of a chemical reaction at equilibrium if the volume is decreased? 7.24. For what special case does a change in volume of a chemical reaction not affect the equilibrium? 7.25. What is an exothermic reaction? 7.26. What is an endothermic reaction? 7.27. If the temperature of a exothermic chemical reaction is increased, how is the equilibrium effected? How is Keq affected? 7.28. If the temperature of an endothermic reaction is increased, how is the equilibrium effected? How is the Keq affected? PROBLEMS 237 7.29. If the temperature of a exothermic chemical reaction is decreased, how is the equilibrium effected? How is Keq affected? 7.30. If the temperature of an endothermic reaction is decreased, how is the equilibrium effected? How is the Keq affected? 7.31. What is a pH indicator? 7.32. When a pH indicator changes color, the hydronium ion concentration is equal to what? 7.33. What is the reaction quotient? 7.34. If the reaction quotient is greater than Keq, in which direction will the reaction proceed? 7.35. If the reaction quotient is less than Keq, in which direction will the reaction proceed? PROBLEMS 7.36. Write the equilibrium constant expressions for the following reactions: (a) (b) (c) (d) (e) 2NO2(g) > 2NO(g) + O2(g), 2SO2(g) + O2(g) > 2SO3(g), CaCO3(s) > CaO(s) + O2(g), Cu(s) + 2Ag+(aq) > Cu2+(aq) + 2Ag(s), CO2(g) + H2(g) > CO(g) + H2O(l). 7.37. What is the equilibrium constant expression for the sum of the following two reactions: 2CO2(g) > 2CO(g) + O2(g) and 2H2(g) + O2(g) > 2H2O(g)? 7.38. If the equilibrium constant for the reaction: N2O4(g) > 2NO2(g) is 7.82 atm at 25°C, what is the equilibrium constant for the reaction: 2NO2(g) > N2O4(g) at 25°C? 7.39. What are the units for the equilibrium constants for the following reactions if the reactant and product concentrations are molar: (a) (b) (c) (d) (e) I2(g) + Cl2(g) > 2ICl(g), 3O2(g) > 2O2(g), N2(g) + 3H2(g) > 2NH3(g), CH4(g) + H2O(g) > CO(g) + 3H2O(g), 2HCl(g) > H2(g) + Cl2(g). 7.40. What are the units for the equilibrium constants for the reactions in Problem 7.39 if the reactant and product concentrations are in partial pressures? 7.41. If the equilibriunm constant for the reaction I2 + Cl2 > 2ICl is 81.9 at 25°C when the concentrations are in partial pressures (atm), what is the value of the equilibrium constant at the same temperature if the concentrations are given in molarity? 7.42. If the equilibrium constant for the reaction C(s) + CO2(g) > 2CO is 0.25 M at 700°C, what is the value of the equilibrium constant at 700°C when the concentrations are in partial pressures (atm)? 7.43. The equilibrium constant for the decomposition of dinitrogen monoxide gas to nitrogen and oxygen gas at 25°C is 7.3 1034 M. (a) Does nitrogen monoxide decompose at 25°C? (b)What is the value of K in atmospheres at 25°C? 238 7. CHEMICAL EQUILIBRIUM 7.44. For the following equilibrium constants, determine the relative concentrations of reactants to products: (a) Keq ¼ 14.5, (b) Keq ¼ 1.6 1021, (c) Keq ¼ 0.64, (d) Keq ¼ 1 103. 7.45. Determine the equilibrium constant for the following reaction; 2NH3(g) > N2(g) + 3H2(g) at 650°C, if the equilibrium concentrations are: [NH3] ¼ 1.42 M, [N2] ¼ 0.29 M, [H2] ¼ 0.87 M. 7.46. Determine the equilibrium constant for the following reaction: CO2(g) + H2(g) >CO + H2O(g), if [CO2] ¼ 0.095 M, [H2] ¼ 0.045 M, [CO] ¼ 0.0046 M, and [H2O] ¼ 0.0046 M. 7.47. Calculate the equilibrium constant for the reaction: 2NO(g) + 2H2(g) > N2(g) + 2H2O(g), if the following amounts are present in a 2 L flask at equilibrium: 0.124 mol NO, 0.024 mol H2, 0.038 mol N2, 0.276 mol H2O. 7.48. The equilibrium constant for the reaction 2IBr(g) > I2(g) + Br2(g) is 4.13 102 at a given temperature. What are the equilibrium concentrations of I2 and Br2 if the equilibrium concentration of IBr is 0.0124 M at the same temperature? 7.49. The equilibrium constant for the reaction HC2H3O2(aq) + H2O(l) > H3O+ + C2H3O2– is 1.80 105 at 25°C. What are the equilibrium concentrations of the products if the equilibrium concentration of HC2H3O2 is 0.10 M? 7.50. The equilibrium constant for the reaction of ammonia with water NH3(aq) + H2O(l) > NH4+(aq) + OH(aq) is 1.8 105 at 25°C. Calculate the equilibrium concentrations if the original solution contained only 0.200 M NH3. 7.51. The Keq for the reaction 2HI(g) > H(g) + I2(g) is 1.40 102 at a given temperature. Determine the equilibrium concentration of HI if the equilibrium concentrations of H2 and I2 are both 2.00 104 M at the same temperature. 7.52. The values of the equilibrium constants for the following reactions are: 2CO2(g) + H2O(g) > 2O2 + CH2CO(g); Keq ¼ 6.1 108 CH4(g) + 2O2(g) > CO2(g) + 2H2O(g); Keq ¼ 1.2 1014 What is the value of the equilibrium constant for the reaction: CH4(g) + CO2(g) > CH2CO(g) + H2O(g)? 7.53. The reaction 2CO(g) + O2(g) > 2CO2(g) is at equilibrium. Predict the response of the reaction for each of the following changes in reaction conditions: (a) the amount of O2 is increased, (b) the amount of CO2 is increased, (c) the amount of O2 is decreased. 7.54. The following reaction at equilibrium; N2(g) + 3H2(g) > 2NH3(g). Predict the response of the reaction for each of the following changes in reaction conditions: (a) an increase in the amount of N2, (b) an increase in the amount of NH3, (c) a decrease in the amount of N2, (d) a decrease in the amount of NH3. 7.55. After the reaction in Problem 7.54 has reestablished equilibrium, what will be the change in H2 concentration compared to the original concentration? 7.56. The reaction C(s) + H2O(g) > CO(g) + H2(g) has the following equilibrium concentrations: [H2O] ¼ 0.01 M, [CO] ¼ 0.04 M, [H2] ¼ 0.04 M. (a) What is the value of the equilibrium constant? (b) What are the new equilibrium concentrations if the concentration of H2 is increased to 0.06 M? PROBLEMS 239 7.57. For a weak acid pH indicator with a HInd form that is pink in color and an Ind form that is blue in color, what is the color of the indicator solution if: (a) HCl is added to the solution, (b) sodium hydroxide is added to the solution? 7.58. For the equilibrium reaction H2(g) + I2(g) > 2HI, what is the response of the reaction if the volume is decreased? 7.59. For the reversible reaction N2O4(g) > 2NO2(g), the initial equilibrium conditions are [N2O4] ¼ 3.48 atm, [NO2] ¼ 1.05 atm and Keq ¼ 0.316 atm. (a) Determine the direction of the response and (b) the new equilibrium concentrations when the volume is decreased from 1.00 to 0.50 L. 7.60. For the equilibrium reaction N2(g) + 3H2(g) > 2NH3(g), what is the response of the reaction to: (a) a decrease in volume, (b) a decrease in pressure, (c) an increase in volume? 7.61. After equilibrium is reestablished, how does each of the changes in conditions in Problem 7.60 affect the concentration of NH3? 7.62. The following reaction is endothermic; H2(g) + I2(g) > 2HI. If the temperature is decreased on the reaction at equilibrium, (a) which way will the reaction shift, (b) how will the concentration of HI change, (c) how will the value of the equilibrium constant change? 7.63. If the temperature of the reaction in Problem 7.63 is increased, (a) which way will the reaction shift, (b) how will the concentration of H2 change, (c) how will the value of the equilibrium constant change? 7.64. The reaction N2(g) + 3H2(g) > 2NH3(g) is exothermic. If the temperature is increased on the reaction at equilibrium, (a) which way will the reaction shift, (b) how will the concentration of NH3 change, (c) how will the value of the equilibrium constant change? 7.65. If the temperature of the reaction in Problem 7.65 is decreased, (a) which way will the reaction shift, (b) how will the concentration of N2 change, (c) how will the value of the equilibrium constant change? 7.66. The current conditions for the reaction; CO(g) + H2O(g) > H2(g) + CO2(g) are [CO] ¼ 0.15 M, [H2O] ¼ 0.25 M, [H2] ¼ 0.42 M, [CO2] ¼ 0.37 M, with an equilibrium constant of 5.10 at 527°C. (a) Calculate the reaction quotient for the reaction and (b) determine the direction that the reaction will proceed to attain equilibrium. 7.67. The reaction of rust with hydrogen gas to form iron and water vapor; Fe2O3(s) + 3H2(g) > 2Fe(s) + 3H2O(g) has an equilibrium constant of 0.064 M at 340°C. For the initial conditions of [H2] ¼ 0.45 M, [H2O] ¼ 0.37 M, determine (a) the reaction quotient and (b) the direction that the reaction will proceed. 7.68. The equilibrium constant for the reaction H2(g) + I2(g) > 2HI(g) is 51 at 448°C. If 0.02 mol of HI, 0.01 mol of H2, and 0.03 mol of I2 are placed in a 2 L container, (a) calculate the reaction quotient and (b) predict how the reaction will proceed. 7.69. The equilibrium constant for the reaction 2SO2(g) + O2(g) > 2SO3(g) is 0.15 M1 at 1220°C. If [SO2] ¼ 0.35 M, [O2] ¼ 0.20 M, and [SO3] ¼ 0.06 M, (a) determine the reaction quotient and (b) predict how the reaction will proceed. C H A P T E R 8 Thermodynamics and Energy Balance O U T L I N E 8.1 Chemical Thermodynamics 241 8.2 The First Law of Thermodynamics: Heat and Work 244 8.3 Enthalpy 250 8.4 Standard Enthalpies 255 8.5 Bond Enthalpy 259 8.6 The Second Law of Thermodynamics: Entropy 8.6.1 The Carnot Cycle 261 263 8.7 The Third Law of Thermodynamics: Entropy and Temperature 267 8.8 Gibbs Free Energy 270 8.9 Standard Gibb Free Energies and Chemical Equilibrium 272 Important Terms 275 Study Questions 277 Problems 278 8.1 CHEMICAL THERMODYNAMICS In Chapter 7, we examined the quantitative relationships for chemical reactions in different phases and developed the equilibrium expressions that determine the relationships between products and reactants. At this point, we have noted that some reactions have large equilibrium constants while others have small ones, but we have not yet developed an approach to be able to predict what the values of these equilibrium constants would be. Chemical thermodynamics is the examination of the fundamental properties of the chemical species involved in reversible chemical reactions and how these properties determine their behavior in the reactions. Thermodynamics makes use of the bulk properties of the materials to evaluate their physical and chemical behavior. It is a macroscopic approach and so does not rely upon the atomic nature of the chemical species being studied. Chemical thermodynamics relates heat and work to chemical reactions as well as the changes of physical states of the chemical species. It is confined by the laws of thermodynamics and thus involves making General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00008-4 241 # 2018 Elsevier Inc. All rights reserved. 242 8. THERMODYNAMICS AND ENERGY BALANCE FIG. 8.1 Josiah Willard Gibbs (left), father of chemical thermodynamics (1839–1903), and the Gibbs medal (right). Photo Willard Gibbs by Serge Lachinov, Wikimedia Commons; Photo courtesy of Chicago Section, The American Chemical Society and the photographer, Josh Kurutz. use of measurements of bulk thermodynamic properties as well as the use of mathematical relationships between the properties to allow predictions for chemical reactions. An important aspect of chemical thermodynamics is thermochemistry, which is the study of the heat evolved from chemical reactions. For example, thermochemistry can be applied to the study of the energy released from combustion reactions. One of the founders of chemical thermodynamics and thermochemistry is Josiah Willard Gibbs (Fig. 8.1). Dr. Gibbs was the first doctoral graduate in engineering from Yale University in 1863 and is considered to be the only American scientist whose discoveries are as fundamental in nature as those of Newton. His work in the field of chemistry has been celebrated by the Chicago Section of the American Chemical Society by establishing the Gibbs Medal, with the purpose: “To publicly recognize eminent chemists who, through years of application and devotion, have brought to the world developments that enable everyone to live more comfortably and to understand this world better” (Chicago Section, American Chemical Society). Gibb’s work focused on making use of the laws of thermodynamics to be able to predict if a chemical reaction will occur. This involved determining how much energy would be required for the reaction to occur or how much energy would be released from the reaction if the reaction was spontaneous. The concept of spontaneity, when a chemical reaction occurs without being driven by some outside force, is at the heart of chemical thermodynamics. However, chemical thermodynamics only determines if the reaction can occur. It does not predict the time scale on which the reaction will occur, which is determined by the kinetics of the reaction. The kinetics of chemical reactions will be discussed in Chapter 9. Thermodynamics is based on three laws and the concept of thermal equilibrium. The first law of thermodynamics is the application of conservation of energy to the system. The second 8.1 CHEMICAL THERMODYNAMICS 243 law addresses the importance of order or disorder in the system, and the third law addresses the order of the system at a temperature of absolute zero. We will further define these laws and examine them in detail as they relate to chemical reactions and processes in Sections 8.2, 8.6, and 8.8. The concept of thermal equilibrium describes the condition between two systems where no heat flows between them when they are connected by a path that can transfer heat. If two systems are in thermal equilibrium with a third system, then they are in thermal equilibrium with each other. A system that is in thermal equilibrium is aid to be adiabatic, where heat neither enters nor leaves the system. In order to describe these chemical systems in a bulk sense, we will first need to define a few terms. The most basic is the definition of a system, which is the part of the universe that is being studied that consists of a number of elements or bodies. It can be small or very large, but has to be part of the universe. The regions outside of the system, essentially the rest of the universe, are considered to be the surroundings. A system can be a closed system or an open system. A closed system is one where mass cannot be exchanged between the system and the surroundings. An open system is one that can readily exchange matter with its surroundings. So, an open system can be influenced by its surroundings. So, experiments that measure thermodynamic properties need to take the surroundings into consideration. Experimental designs require that the interaction with the surroundings be limited and so the system being studied should be as close to a closed system as possible. A system is described by its properties. These properties can be either intensive properties or extensive properties. Intensive properties are those that describe the bulk or physical properties of the system. They do not depend upon how much matter is present and do not change when material is added or taken away. Examples of intensive properties include temperature, density, color, boiling point, melting point, and hardness. Extensive properties are those that depend on how much material is present. They change when matter is added to or taken away from the system. Examples of extensive properties include mass, volume, weight, and length. Chemical thermodynamics is the study of the equilibrium states of systems. Since chemical thermodynamics is a bulk approach to describing systems, it uses the macroscopic properties such as the pressure, temperature, volume, density, and chemical composition to define the state of the system. Equilibrium states are those states of a system where these macroscopic properties do not change. The macroscopic properties that define the equilibrium state are called state functions. State functions depend only on the initial and final states of a system and are not dependent on the manner in which the state changed from the initial to the final state. Examples of state functions are: pressure, temperature, volume, mass, chemical composition, and energy. A common example of a state function is where you sit in your classroom or workplace, assuming that you sit in the same chair every day. That position is defined by a specific longitude and latitude. If you move to your home and sit at your dining table or in a specific chair in the living room, that new position is also defined by a specific longitude and latitude. The exact difference between the two longitudes and latitudes is defined by an exact distance between the two seats. That distance is a state function. However, the exact distance you travel between the two seats may actually vary tremendously each day as you travel a different route to get to the seat in your home. The distance travelled to and from the two seats will almost never be exactly the same and therefore the distance along the exact route 244 8. THERMODYNAMICS AND ENERGY BALANCE taken from one seat to the other seat is not a state function. State functions in chemical thermodynamics are the same. It does not matter how the chemical or physical changes occur in a system, it only matters what the initial and final equilibrium states of the system are. Changes in state functions describe a change in state of the system. Since the change in the state functions are only dependent upon the initial and final states of a system and are not dependent upon the process that occurs to get from the initial state to the final state, the difference between the values of the state functions for the initial and final states defines the change in state of the system. For example, the change in state for one mole of an ideal gas in an initial equilibrium state described by P1, T1, and V1 going to a final equilibrium state described by P2, T2, and V2 is defined as the differences between the values of the state functions of the initial state and the final state. The change in state is then defined by; ΔP ¼ P2 P1 ΔT ¼ T2 T1 ΔV ¼ V2 V1 : State functions are related to each other by equations of state, mathematical relationships between state functions that describe the equilibrium state of a system under a given set of physical conditions. For example, as described in Chapter 6, the state functions pressure, temperature, volume, and amount of an ideal gas are related by the equation of state called the ideal gas law; P ¼ nRT/V. So, if the temperature and volume is known for one mole of an ideal gas, the pressure is fixed. This is a simple equation of state relating the state functions, temperature, pressure, volume, and amount of an ideal gas. Other equations of state may be more complex, but the basic concept is the same. 8.2 THE FIRST LAW OF THERMODYNAMICS: HEAT AND WORK Heat (q) is defined as the amount of thermal energy transferred from a system to its surroundings or from one system to another due to the fact that they are at different temperatures. Heat is measured in units of energy, usually calories or joules. Heat will always flow spontaneously from a hotter system to a colder system. Although the process of transferring heat from one system to another changes the internal energy of both systems, it is not a property of the state of the system and so is not a state function. Temperature is a measure of the average thermal energy of a system. When a substance absorbs heat, one of two things can happen, the temperature of the substance can increase or the substance can undergo a phase change. In the absence of a phase change, the change in temperature of the substance is proportional to the amount of heat absorbed. The more heat added, the larger the temperature change. The specific heat capacity (C) of a substance is defined as the amount of energy in the form of heat that must be added to one gram of a substance to increase the temperature by one degree Kelvin. The specific heat capacity, in units of J • g1 • K1 or cal • g1 • K1, is essentially a ratio of the heat added to a substance of mass “m” to the resulting change in temperature of that substance; C ¼ q=mΔT (1) 8.2 THE FIRST LAW OF THERMODYNAMICS: HEAT AND WORK 245 The relationship between the heat gained or lost by a specific mass of a substance to the resulting temperature changes of that substance from Eq. (1) is; q ¼ CmΔT (2) The heat capacity of a substance can also be expressed on a molar basis. The heat capacity defined in this way is the molar heat capacity, the amount of energy in the form of heat that must be added to one mole of a substance to increase the temperature by one degree Kelvin. So, the molar heat capacity is measured in units of J • mol1 • K1 or cal • mol1 • K1. EXAMPLE 8.1: DETERMINING THE HEAT REQUIRED TO RAISE THE TEMPERATURE OF A SUBSTANCE Given the specific heat capacity of water is 1.00 cal • g1 • K1, how much heat is needed to raise the temperature of 0.5 kg of water from 0°C to 100°C? 1. Determine △ T. 373:15K 273:15K ¼ 100 K 2. Determine q from the specific heat capacity. q ¼ C m △T ¼ 1:00cal g1 K1 500g 100K q ¼ 50:0kcal Specific heat capacities can be determined for elements, molecules, and materials. They are particularly useful in engineering applications since they reflect the ability of materials to store or transfer heat. The specific heat capacities for some solid materials are listed in Table 8.1. All of these, including ice, are lower than that for liquid water. Beeswax, with a specific heat capacity of 0.84 cal • kg1 • K1, has the highest heat capacity of the solids listed in Table 8.1. The very high heat capacity of beeswax acts to store heat during the day and release it slowly at night, maintaining a more uniform temperature in the hive when ambient temperature changes can be rapid and significant. So, it may seem that bees are very good engineers when selecting the building material for their hives. The ability of materials to absorb or release heat is commonly referred to in the building industry as thermal mass (Cth). Thermal mass is defined as the ability of a material to absorb and store heat. It is related to specific heat capacity as Cth ¼ m C, where “m” is the mass of the material used. The heat absorbed by a material is given by; q ¼ Cth ΔT (3) in units of J • K1 or cal • K1. Higher density materials, such as concrete, bricks, and tiles, require more heat to raise their temperature than lower density materials, such as wood. So, higher density materials have a higher thermal mass and low density materials have a lower thermal mass. Building materials with an ideal thermal mass are those that have both high density and a high specific heat capacity. Another process that exchanges energy between systems is work. Work is the transfer of energy by any means other than heat. Physics defines work as the force required to move an object a certain distance. Work (w) is calculated by multiplying the force (F) by the distance 246 8. THERMODYNAMICS AND ENERGY BALANCE TABLE 8.1 Specific Heat Capacities Under Conditions of Constant Pressure (Cp) in Units of cal • kg1 • K1 and Densities in Units of g • cm3 for Some Solid Building Materials Building Material Heat Capacity (CP) Density (g • cm23) Aluminum 0.22 2.7 Asphalt 0.22 2.3 Beeswax 0.84 1.0 Brick 0.20 1.9 Concrete 0.21 1.7 Cork 0.45 0.2 Fiberboard 0.55 0.8 Glass, pyrex 0.18 2.2 Glass, silica 0.20 2.2 Marble 0.21 2.7 Paper 0.33 0.9 Paraffin 0.70 0.9 Plaster 0.23 0.9 Plastics 0.35 1.0 Ice 0.49 0.9 Wood, oak 0.48 0.8 Wood, pine 0.60 0.5 moved (d), where the force is equal to mass of the object (m) multiplied by the acceleration (a) of the movement; w ¼ F d ¼ ma d (4) Heat and work are both processes that transfer energy from one system to another or from a system to the surroundings. So, they both change the internal energy of a system. Although heat and work are related to each other, work can be totally converted into heat, but heat cannot be totally converted to work. CASE STUDY: WORK AND THE INTERNAL COMBUSTION ENGINE An important form of work in chemical thermodynamics is accomplished by pressure and volume changes in the gas phase. This is the kind of work done by the combustion of fuels in an internal combustion engine, which moves the pistons in the cylinders of an automobile. The work done by the pistons is transferred to the drive train, which turns the wheels of the automobile and moves its mass over the distance driven. A diagram of a simple piston is shown in Fig. 8.2. Combustion of the 8.2 THE FIRST LAW OF THERMODYNAMICS: HEAT AND WORK 247 FIG. 8.2 Diagram of a piston which moves from position (A) to position (B) after the combustion of gasoline in the cylinder. The volumes V1 and V2 are the internal volumes of the cylinder before and after the expansion. The Area of the piston is given by A. gasoline in the piston increases the amount of gas molecules resulting in the upward movement of the piston inside the cylinder from position (A) to position (B). The distance traveled by the piston is equal to h2 h1 and the work done by the piston is given by; w ¼ F d ¼ F ðh2 h1 Þ The force (F), which is constant over the piston area (A), is equal to the initial pressure of the gas in the cylinder before combustion. This is equal to the external pressure (Pext) on the piston. The external pressure is defined as the force per unit area or Pext ¼ F/A as described in Section 6.2. So, F ¼ Pext A and the work done by the piston is equal to; w ¼ Pext A ðh2 h1 Þ Since the change in the volume of the cylinder is equal to the area of the piston multiplied by the height of the cylinder below the piston: ΔV ¼ V2 V1 ¼ Ah2 Ah1 ¼ A(h2 h2). The work done by the piston then becomes; w ¼ Pext Aðh2 h1 Þ ¼ Pext ΔV So, the work accomplished by the change in volume of the cylinder created by the combustion of gasoline is directly related to the external pressure on the piston. If the external pressure is changed, the amount of work accomplished by the piston will change, even though the change in volume inside the cylinder remains the same. This means that the work done in an internal combustion engine depends upon the conditions of the cylinder operation. Along with the volume change produced from the combustion process in the cylinder, there is also a change in temperature of the gases in the cylinder, which produces heat. So, the energy released from the combustion reaction inside the cylinder produces both work and heat. Heat flow happens when there is a difference in temperature between a system and its surroundings, in this case between the cylinder and the surroundings. To maintain a reasonable temperature in the engine, the heat needs to flow from the hot cylinder to cooler surroundings. Initially, heat is removed from the cylinder during its exhaust cycle and is released as hot gases in the exhaust system of the car. Additional heat is removed by using a coolant, which flows through the engine to carry away the heat and maintain a constant engine temperature. This coolant takes up the heat from the combustion reaction in the cylinder and moves it to the radiator, maintaining cylinder and engine temperature within reasonable limits. 248 8. THERMODYNAMICS AND ENERGY BALANCE The first law of thermodynamics, also known as the law of conservation of energy, states; • Energy cannot be created or destroyed, it can only be transformed from one form to another form. Since energy cannot be created or destroyed, any change in the internal energy of a system must result in a corresponding change in the energy of the system’s surroundings. The internal energy of a system is the energy contained within the system and can be viewed as the energy required to create the system. The value of internal energy of a system (E) is a property of the system at equilibrium and is therefore a state function. The two processes that can change the internal energy of a system are heat and work and the change in the internal energy (ΔE) of the system is equal to the sum of these two processes; ΔE ¼ q + w (5) Heat can move from the system to the surroundings ( q) or from the surroundings to the system (+ q). Similarly, work can be done by the system on the surroundings ( w) or by the surroundings on the system (+ w). These sign conventions used for work and heat are related to the internal energy change of the system (ΔE). When work or heat is positive, energy has been supplied to the system and the internal energy of the system increases. When work or heat is negative, the system has lost energy and the internal energy of the system decreases. In summary: • • • • q is positive—heat is absorbed by the system q is negative—heat is released from the system w is positive—work is done on the system w is negative—work is done by the system Unfortunately, there is more than one form of the equation relating internal energy with heat and work. Eq. (5) is the form adopted by the International Union of Pure and Applied Chemists (IUPAC) and is used exclusively by chemists. This is the form used in this text. Some engineering applications often use the form ΔE ¼ q w. In this form, “w” is defined as work done by the system on the surroundings and is designated as positive (+ w). Either form is consistent with the first law of thermodynamics and the results obtained by either form are the same. The difference between the two lies in the sign conventions for work. Notice, however, that once the value of work is substituted into either equation, they become equivalent. For example, work done by the system gives: ΔE ¼ q + w ¼ q + ðwÞ ¼ q w for Eq: ð5Þ, and ΔE ¼ q w ¼ q ð +wÞ ¼ q w for the alternate form: EXAMPLE 8.2: DETERMINING THE CHANGE IN INTERNAL ENERGY OF A SYSTEM What is the change in internal energy of a system that releases 100 J of energy to the surroundings in the form of heat and has 200 J of work done on it? 8.2 THE FIRST LAW OF THERMODYNAMICS: HEAT AND WORK 249 EXAMPLE 8.2: DETERMINING THE CHANGE IN INTERNAL ENERGY OF A SYSTEM— CONT’D 1. Determine the signs for heat and work. Heat is released from the system so it is negative. Work is done on the system so it is positive. 2. Substitute the values into the equation for change in internal energy of a system. ΔE ¼ q + w ¼ 100J + 200J ¼ 100J Work in chemical reactions is normally achieved by way of a change in pressure and volume, as in gas phase reactions. Since work is defined as the product of force times a change in position, if there is no change in position no work is being accomplished. For many gas phase chemical reactions that take place in a closed container at constant volume, ΔV ¼ 0 and no work is done. If no work is done, the change in internal energy of the system becomes; ΔEv ¼ qv (6) where the “v” subscript indicates that the system is at constant volume. When chemical reactions give off energy in the form of heat, the internal energy difference between the initial and final states is lower for the products than for the reactants; qv and ΔEv are both negative and the reaction is exothermic. When chemical reactions take up energy in the form of heat, the difference between the initial and final states is lower for the reactants than for the products; both qv and ΔEv are positive and the reaction is endothermic. The specific heat capacity of a substance under conditions of constant volume (Cv) is related to the change in internal energy as; Cv ¼ qv =mΔT ¼ ΔEv =mΔT (7) Since the heat released or taken up by a chemical reaction at constant volume is equal to the change in the internal energy of the system, we can simply determine the amount of heat given off or taken up to determine the change in internal energy for a chemical reaction at constant volume. This can be accomplished by measuring the change in temperature of a substance, such as water, with a known heat capacity that is in direct contact with the reaction system. The SI unit for energy is the Joule, which is defined as 1 kg • m2 • s2. However, a Joule is a lot of energy compared to the energy of a single chemical bond, which is approximately 1018 J. An alternate unit of energy more appropriate to chemical reactions is the thermal unit of a calorie (cal), also known as the gram calorie or small calorie, which is defined as the amount of energy that it takes to heat one gram of water at one degree Celsius at one atmosphere pressure. A kilocalorie (kcal), also known as the large calorie (Cal), is the unit commonly used to measure the energy content in food. It is therefore sometimes referred to as the food calorie. The kilocalorie is defined in terms of the kilogram rather than the gram and is equal to 1000 small calories. The calorie is equal to 4.184 J and the kilocalorie is equal to 4.184 kJ. Other units commonly used in engineering are the British Thermal Unit (BTU), which is the amount of heat needed to raise one pound of water one degree F at one atmosphere pressure. A BTU is equivalent of 1055.06 J. The unit of choice for chemists is the kcal, which will be used in this textbook. 250 8. THERMODYNAMICS AND ENERGY BALANCE 8.3 ENTHALPY For practical reasons, most chemical reactions are typically not carried out at constant volume. They are usually carried out in systems open to the atmosphere and so they are at constant pressure, typically one atmosphere. The change in energy of a system at constant pressure, when the only work done by the system is pressure-volume work, is given by; ΔEp ¼ qp + wp ¼ qp PΔV qp ¼ ΔEp + PΔV This expression for qp is dependent only on the three state functions: internal energy, pressure, and volume. It is therefore equivalent to a new state function, which is called enthalpy (H) and is defined by; H ¼ E + PV (8) Enthalpy is the total thermal energy of the system. It is equal to the internal energy of the system plus the product of pressure and volume. In a physical sense, it can be viewed as the energy required to create the system at equilibrium (internal energy) plus the amount of energy required to create space for the system by expanding its volume against a constant pressure (PV). This new state function allows us to be able to predict the changes in energy for chemical reactions at constant pressure. Although the value of enthalpy for an equilibrium state cannot be calculated directly, the change in enthalpy (ΔH) between equilibrium states can be calculated and is given by; ΔH ¼ ΔE + ΔðPV Þ ¼ q + w + ΔðPV Þ (9) For chemical reactions at constant pressure, where the only work done by the system is pressure-volume work, wp is equal to PΔV and the change in enthalpy becomes; ΔHp ¼ qp PΔV + PΔV ΔHp ¼ qp (10) So, for the condition of constant pressure, the change in enthalpy is simply equal to the heat transferred to or from the system (qp). Similar to ΔEv, ΔHp will also be negative for exothermic reactions as the heat released at constant pressure will move to the surroundings from the system, and ΔHp will be positive for endothermic reactions when heat must be supplied to the system for the reaction to occur. Heat capacities are also most important under conditions of constant pressure since most materials and chemical species are used under these conditions. The specific heat capacity of a substance under conditions of constant pressure (Cp) is related to the change in internal energy as; Cp ¼ qp =mΔT ¼ ΔHp =mΔT (11) Changes in enthalpy are also associated with the phase transitions discussed in Chapter 1. Since phase transitions are caused by heat added to or released from a system at constant pressure, they are accompanied with a change in enthalpy according to Eq. (10). The enthalpy changes that occur in each phase transition are related to each other as shown in Fig. 8.3. The 8.3 ENTHALPY 251 FIG. 8.3 The relationships between changes in enthalpy for different phase transitions. The length of the arrows represents the relative amount of heat required for each phase transition. Transitions with arrows pointing up are endothermic. Transitions with arrows pointing down are exothermic. length of the arrows in Fig. 8.3 represents the relative amount of heat required for each phase transition. The phase transitions with arrows pointing up require heat input and are endothermic processes. The phase transitions with arrows pointing down release heat and are exothermic processes. As shown in Fig. 8.3, the change in enthalpy for any phase transition is equal but opposite in sign of the reverse transition (e.g., Δ Hvap ¼ Δ Hcnd). Also, the enthalpy changes for linked transitions must balance. For example, when going from the solid to the gas phase, the enthalpy change of the one step process (sublimation) must be equal to the sum of the enthalpy changes of the two step processes (fusion and vaporization). ΔHfus + ΔHvap ¼ ΔHsub ΔHfrz + ΔHcnd ¼ ΔHdep One important case for enthalpy changes associated with phase transitions is that for water. Changes in the phases of water are well-studied and have many uses ranging from refrigeration and cooking to steam engines. The changes in temperature of water as heat is added and it going through the phase changes of ice to liquid water and liquid water to steam are shown in Fig. 8.4. The amount of heat required for the ice to reach the melting point is determined from the specific heat capacity of ice at constant pressure (Cp) and Eq. (11) as; q ¼ m Cp ΔT. When the ice reaches the melting point and both ice and liquid water are present, the temperature remains constant at 273.15 K until all of the ice is melted. This absorption of heat with no change in temperature is due to the enthalpy of fusion, also called the heat of fusion, of ice. The heat of fusion (ΔHfus) is defined as the change in enthalpy due to adding heat to a substance in order to change its state from a solid to a liquid at constant pressure. The amount of heat required is dependent on the mass of ice and is determined by; q ¼ m ΔHfus (12) After all the ice is melted, the liquid water will take up heat according to Eq. (11) where Cp is now the specific heat capacity of water. The temperature will increase until the boiling point is reached. Once the boiling point is reached, the temperature will remain constant at 373.15 K 252 8. THERMODYNAMICS AND ENERGY BALANCE Temperature (K) (q = m × cp(g) × ΔT ) (q = m × ΔHvap) 373.15 (q = m × cp(l ) × ΔT ) 273.15 (q = m × ΔHfus) (q = m × cp(s) × ΔT ) Ice Water Steam Time (s) FIG. 8.4 The changes in the temperature of water as it goes through two phase changes (ice to water and water to steam). The determination of the heat required for each transition is given in parentheses. until all of the liquid has been converted to steam. The absorption of heat at the boiling point with no change in temperature is due to the enthalpy of vaporization, also called the heat of vaporization, for water. The heat of vaporization (ΔHvap) is defined as the change in enthalpy due to adding heat to a substance in order to change its state from a liquid to a gas at constant pressure. The amount of heat required is again dependent on the mass of water and is determined by; q ¼ m ΔHvap (13) Once all the water is converted to steam, it will then take up heat according to Eq. (11) where Cp is the specific heat capacity of steam. The temperature of the steam will continue to increase as heat is absorbed. The ability of a substance to take up heat without a change in temperature during a phase change is very useful for chemists and engineers. Chemists can use ice baths to maintain a constant temperature for chemical reactions. A mixture of ice and liquid water is placed in contact with an exothermic chemical reaction. The ice-water bath will absorb the heat released by the exothermic reaction and still remain at a constant temperature until all the ice is melted. If the bath is large enough and sufficient ice is present, a significant amount of heat can be absorbed without a rise in temperature. CASE STUDY: MEASURING HEAT—THE CALORIMETER Understanding changes in energy is important in understanding chemical reactions and the phase changes of materials, but how can it be measured accurately? According to Eq. (6), the change in internal energy is equal to the amount of heat transferred at constant volume, and according to Eq. (10), the change in enthalpy is equal to the amount of heat transferred at constant pressure. So, these changes in internal energy can be measured by designing the experiment to maintain constant pressure or constant volume and assure that heat is not lost to the surroundings. A device that is used to measure the heat released or absorbed during a chemical reaction or phase change is called a calorimeter. The process of making this measurement is called calorimetry. To determine the heats of reactions, known amounts of materials are placed into a calorimeter and the temperatures of the 8.3 ENTHALPY 253 FIG. 8.5 An engraving of the constant pressure ice calorimeter made by Marie Lavoisier, wife of Antoine Lavoisier. The calorimeter was used by Lavoisier and Laplace in the 1780s to determine the heat of fusion of ice for the first time. initial and final states are measured. If the heat capacity of the calorimeter is known, the amount of heat absorbed or released can be determined directly. Errors arise from the fact that some heat is lost to the surroundings. There is also some heat loss in making the actual measurements with a thermometer or thermocouple, which takes up a small amount of heat in order to make the measurement. The early chemists Antoine Lavoisier and Pierre-Simon Laplace used a constant pressure ice calorimeter shown in Fig. 8.5 to measure the heat of fusion of ice for the first time. The calorimeter had three concentric sheet iron chambers. The inner chamber (M), which held the sample, was surrounded by a middle chamber (A) and an outer chamber (B), both of which were filled with ice. The ice-filled outer chamber served as a means of isolating the inner chamber from the surroundings. The ice-filled middle chamber absorbs the heat given off by the sample in the inner chamber. The heat melts the ice in the middle chamber and the amount of heat is calculated from the amount of ice that melts and the specific heat capacity of ice. For the measurement of the heat of fusion of ice, a mixture of hot water and ice cubes was placed in the inner chamber and the temperature monitored until it leveled off (Δ T). The mass of ice melted (mi) was determined from the change in volume of the water in the inner chamber and the density of water (mi ¼ ρw ΔVw). The heat lost by the hot water (q) was then determined by Eq. (7) and the heat of fusion of ice was determined by; ΔHfus ¼ q=mi 254 8. THERMODYNAMICS AND ENERGY BALANCE FIG. 8.6 Schematic diagram of a bomb calorimeter used to determine the heat released during combustion reactions. Other experiments were conducted to determine the heat capacities of selected materials. In these experiments, the inner chamber contained a sample that was heated to a given temperature. The water from the melted ice in the middle chamber was drained off at D and weighed. The amount of heat lost by the sample is then determined from the heat capacity of ice and the specific heat capacity of the sample was determined from Eq. (7). A modern constant volume calorimeter that is used to measure the heat released during a combustion reaction, called a bomb calorimeter, is shown in Fig. 8.6. In a bomb calorimeter, the sample is placed in a steel bomb, a container designed to withstand high pressure. The bomb contains an electrical ignition coil and is filled with oxygen gas at high pressure (typically 30 atm). It is surrounded by a known amount of water. The entire system is in an insulated container that isolates it from the surroundings. The temperature of the water is measured before ignition and after complete combustion of the sample (ΔT). The heat released during the combustion reaction (qcomb) is determined from the change in temperature of the water and the heat capacity of the bomb calorimeter [Cv(cal)], which includes the steel bomb, the water, and the insulated container. The heat capacity of the calorimeter is determined using a measured amount of a substance with a known heat of combustion. The qcomb of unknown samples can then be determined by; qcomb ¼ Cv ðcalÞ ΔTw It is interesting to note that the calorie content in foods was initially determined by using a bomb calorimeter and reported as kilocalories. It was later realized that the energy obtained from food in body metabolism is not the same as the heat given off during a combustion reaction. The calorie content of food is now determined based upon biochemical uptake processes. These processes do not make use of fiber in the food and so give a lower energy content than was originally obtained using the original bomb calorimeter. The system used to determine the metabolically available energy in food today is the Atwater system, which is based on theoretical calculations rather than direct measurements. 8.4 STANDARD ENTHALPIES 255 8.4 STANDARD ENTHALPIES Standard enthalpies (ΔH°) are enthalpy changes that occur in chemical reactions with all materials in their standard states. The standard state of an element or a compound is defined as the most stable form of the physical state that exists at 1 atmosphere pressure and 298 K. For the chemical reaction that results in the formation of a molecule directly from its elements, the standard enthalpy is called the standard enthalpy of formation or standard heat of formation (ΔH°f). Other types of standard enthalpies include the standard enthalpy of neutralization (ΔH°neut), which is the change in enthalpy that occurs when an acid and a base undergo a neutralization reaction to form one mole of water and a salt, and standard enthalpy of combustion (ΔH°comb), which is the enthalpy change that occurs when one mole of the compound is burned completely in oxygen. Here we will focus mainly on the standard enthalpies of formation. Standard enthalpies of formation are very useful in determining whether a compound is stable at the standard room temperature of 298 K. They are also useful in determining the enthalpies of formation for unknown reactions or reactions that are not easy to measure quantitatively. The standard enthalpy of formation is defined as the change in enthalpy that occurs during the formation of one mole of a compound from its elements, with all substances in their standard states. So, the chemical equation describing the formation must be written with all species in their standard states and the stoichiometry of the reaction must be set so that that one mole of product is produced. For example, the reaction of solid carbon with oxygen to form carbon monoxide is normally written as; 2CðsÞ + O2 g ! 2CO g However, the stoichiometry of this equation shows two moles of carbon monoxide being produced. The equation for the standard enthalpy of formation can be obtained by dividing the normal chemical equation by 2 resulting in the equation; CðsÞ + ½O2 g ! CO g The use of the stoichiometric coefficient of ½ for O2 may seem strange but the reaction cannot be written as; CðsÞ + O g ! CO g because the single oxygen atom is not the standard state of the element oxygen. The stable form of oxygen at 298 K and one atmosphere is O2(g). So, in order to yield one mole of CO and keep all species in their standard states, oxygen must be written as ½O2. Some standard enthalpies of formation are presented in Table 8.2 for selected molecules and atoms. The standard enthalpy of formation for any element in its standard state is defined as zero. This is because the formation of the element in its standard state from the element in its standard state involves no reaction. For example, in the formation of elemental carbon from itself; C(s) ! C(s), no change has occurred, no heat is released or absorbed and so ΔH°f ¼ 0. The enthalpy of formation for the atoms in column 3 of Table 8.2 reflects the heat required to change the element in its standard state to a single atom in the gas phase. For example, the standard state for hydrogen, oxygen, and the halogens is the diatomic form. The standard enthalpy of formation given in Table 8.2 is for the reaction; 256 8. THERMODYNAMICS AND ENERGY BALANCE TABLE 8.2 Standard Enthalpies of Formation (ΔH°f) in kcal • mol1 for Some Selected Molecules and Atoms Molecule Phase H2O Gas HCl ΔH°f Atom Phase ΔH°f 57.8 H Gas 52.1 Gas 22.1 O Gas 59.6 CO Gas 26.4 C Gas 171.3 CO2 Gas 93.9 N Gas 113.0 CH4 Gas 17.8 S Gas 66.3 NO Gas 21.8 F Gas 19.0 NO2 Gas 7.9 Cl Gas 29.0 N2O Gas 19.5 Br Gas 26.7 NH3 Gas 11.0 I Gas 25.5 O3 Gas 34.1 SO2 Gas 70.9 H2S Gas 4.8 H2O Liquid 68.3 H2O2 Liquid 44.9 CH3OH Liquid 57.2 Al2O3 Solid 400.5 BaSO4 Solid 352.1 CaO Solid 153.9 CaCO3 Solid 288.6 CuO Solid 37.6 Cu2O Solid 40.3 SiO2 Solid 205.0 ZnO Solid 83.2 ½O2 ! O Also, the standard state for both carbon and sulfur is the solid form, so the standard enthalpy of formation for a single atom of carbon the gas phase is for the reaction; CðsÞ ! C g The standard enthalpies of formation are related to the stability of compounds under standard conditions. Generally, the more heat released in the formation, the more stable the compound. For example, Al2O3(s), known as alumina, has a standard enthalpy of formation of 380 kcal • mol1. This means that a large amount of heat is released in forming alumina. So, alumina is a very stable compound. In contrast, NO(g), with a heat of formation 8.4 STANDARD ENTHALPIES 257 of +21.6 kcal • mol1, implies that the formation of NO requires energy. So, nitric oxide is not very stable and tends to react with oxygen to form NO2(g). The heat of formation for NO2(g) (7.9 kcal • mol1) is also positive, but less than that for NO(g) by 13.9 kcal • mol1. So, NO2(g) is more stable than NO(g). Both of these gases will not return to their elemental forms once formed, but they are reactive and much less stable than alumina. Some compounds with a very high enthalpy of formation are very unstable and can be explosive. Mercury(II) fulminate [Hg(CNO)2(s)], with a enthalpy of formation of +64.1 kcal • mol1, is used in blasting caps or percussion caps as it will react explosively releasing a large amount of CO(g), CO2(g), and N2(g) along with mercury upon decomposition. The standard enthalpies of formation can be used to calculate the enthalpy change for a chemical reaction under standard conditions as; ΔH° rxn ¼ ΣnΔH° fðproductsÞ ΣnΔH° fðreactantsÞ (14) where “n” is the stoichiometric coefficient for each product and reactant in the balanced chemical equation for the reaction. EXAMPLE 8.3: DETERMINING THE STANDARD ENTHALPY OF REACTION Determine the standard enthalpy of reaction for the complete combustion of methane. 1. Write the balanced chemical equation for the reaction with all species in their standard states. CH4 + 2O2 ! CO2 + 2H2 O 2. Determine the sum of the enthalpies of formation for the reactants. CH4 ¼ 17:8kcal mol1 O2 ¼ 0kcal mol1 ðan element in its standard state is defined as 0Þ 17:8 + 2ð0Þ ¼ 17:8kcal mol1 3. Determine the sum of the enthalpies of formation for the products. CO2 ¼ 93:9kcal mol1 H2 O ¼ 57:8kcal mol1 93:9 + 2ð57:8Þ ¼ 209:5kcal mol1 4. Subtract the enthalpy for the reactants from the enthalpy for the products. ΔH° rxn ¼ 209:5kcal mol1 17:8kcal mol1 ¼ 191:7kcal mol1 The known standard enthalpies of formation can also be used to determine unknown standard enthalpies of formation for other compounds that are difficult to measure directly. Consider the reaction of carbon with oxygen to form carbon dioxide; CðsÞ + O2 g ! CO2 g The standard enthalpy of this reaction can be measured in a calorimeter, but the reaction tends to produce carbon monoxide along with the carbon dioxide. So, the result would not 258 8. THERMODYNAMICS AND ENERGY BALANCE be applicable to the formation of CO2 alone. The standard enthalpy of formation for carbon dioxide can be determined by taking advantage of the fact that enthalpy is a state function. Since state functions depend only on the initial and final states and not on the manner in which the state changed from the initial to the final state, it does not matter if carbon dioxide is formed in one step or two. So, the value of the enthalpy of formation of CO2(g) directly from C(s) is the same as the value when forming it in two steps: (1) forming CO from C(s), (2) forming CO2 from the combustion of CO with oxygen. So, the standard enthalpy of formation for CO2 can then be obtained by summing the standard enthalpy of formation for CO (step 1) and the standard enthalpy of combustion of CO (step 2); 1) C(s) + 2) CO(g) ½O2(g) ½O2(g) CO(g) CO2(g) ________________________________________________________ C(s) O2 CO2 ΔHof = 26.4 kcal mol ΔHocomb = 1 67.6 kcal mol 1 _________________________________________________________ ΔHof = 94.0 kcal mol 1 This summation approach to the determination of standard enthalpies is known as Hess’s Law. Hess’s law allows for the calculation of the enthalpy change for a chemical reaction when it cannot be measured directly. The exact statement of Hess’s law, also known as the law of heat summation, is; • The total enthalpy change during a chemical reaction is the same whether the reaction takes place in one step or in several steps. EXAMPLE 8.4: DETERMINING THE STANDARD ENTHALPY OF FORMATION FROM HESS’S LAW Determine the standard heat of formation for methane (CH4) using Hess’s law, given the following standard enthalpies for the reactions: (1) C(s) + O2(g) ! CO2(g) ΔH° f ¼ 94:0 kcal mol1 (2) H2(g) + ½O2(g) ! H2O(l) ΔH° f ¼ 68:3 kcal mol1 (3) CH4(g) + 2O2(g) ! CO2(g) + 2H2O(l) ΔHcomb ¼ 212.7 kcal • mol1 1. Write the chemical equation for the formation of methane from the elements in standard states. 2CðsÞ + 2H2 g ! CH4 2. Determine how the given equations can be summed to give the chemical equation for the formation of methane. (a) Equation (1) has carbon as a reactant. (b) Equation (2) has hydrogen as a reactant, but since the formation of one mole of methane requires two moles of hydrogen, the equation needs to be multiplied by 2; 2H2 g + O2 g ! 2H2 OðlÞ (c) Equation (3) has methane as a reactant instead of a product so it must be reversed; CO2 g + 2H2 OðlÞ ! CH4 g + 2O2 g 8.5 BOND ENTHALPY 259 EXAMPLE 8.4: DETERMINING THE STANDARD ENTHALPY OF FORMATION FROM HESS’S LAW— CONT’D 3. Sum the three equations. CðsÞ + O2 g ! CO2 g 2H2 g + O2 g ! 2H2 OðlÞ CO2 g + 2H2 OðlÞ ! CH4 g + 2O2 g CðsÞ + 2O2 g + 2H2 g + CO2 g + 2H2 OðlÞ ! CO2 g + 2H2 OðlÞ + CH4 g + 2O2 g Since 2O2(g), CO2(g), and 2H2O(l) appear on both sides of the equation, they can be eliminated to give; CðsÞ + 2H2 g ! CH4 4. Calculate the standard enthalpy of formation by summing these three reactions to give the equation for the standard enthalpy of formation of methane. (a) ΔH°f for equation (1) is unchanged ¼ 94.0 kcal • mol1 (b) ΔH°f for equation (2) is multiplied by two since the equation was multiplied by two ¼ 2(68.3) ¼ 136.6 kcal • mol1 (c) The sign of ΔH°comb for equation (3) is reversed because the equation is reversed ¼ +212.7 kcal • mol1 ΔH° f ¼ 94:0kcal mol1 136:6kcal mol1 + 212:7kcal mol1 ¼ 17:9 kcal mol1 : 8.5 BOND ENTHALPY Bond enthalpy (ΔHb), commonly called bond energy, is the measure of the strength of a chemical bond. It is defined as the average value of the gas phase bond dissociation energies for all bonds of the same type in the same chemical species. Bond dissociation enthalpy (ΔHbd), commonly known as bond dissociation energy, is the standard energy required to break one specific bond in a molecule in the gas phase. The bond dissociation energy of a specific chemical bond in a molecule depends on the molecular environment surrounding the bond. The bond energy values usually given for a particular kind of chemical bond are values averaged over different environments. Water is a good example of how these two related energy terms differ from one another. The bond dissociation energy of the first OdH bond is 120 kcal • mol1 at 298 K. This value is the bond dissociation energy (ΔH°bd) for the HOdH bond. But, the bond dissociation energy for the second OdH bond is 101 kcal • mol1 at 298 K. This value is the bond dissociation energy for the OdH bond. Thus, the bond energy (ΔH°b) for the OdH bonds in the water molecule is the average of the bond dissociation energies for the two OdH bonds. So, the bond energy for water is the sum of the two ΔHbd values for the OdH bonds divided by the number of bonds, or; 260 8. THERMODYNAMICS AND ENERGY BALANCE TABLE 8.3 Bond Energies (Bond Enthalpies) in kcal • mol1 at 298 K for Selected Bonds Bond Bond Energy Bond Bond Energy Bond Bond Energy CdH 98.7 HdF 135.5 C]C 146.8 CdC 83.2 HdCl 103.0 C^C 200.5 CdN 70.0 HdBr 87.5 C]O 213.0 CdO 85.6 HdI 71.5 C]S 114.0 CdF 115.9 OdH 110.7 N]N 99.9 CdCl 78.4 OdO 34.9 N]O 145.1 CdBr 66.0 SdH 81.0 N^N 224.9 CdI 57.4 SdS 63.6 O]O 118.3 CdS 61.9 FdF 37.0 NdH 81.0 CldCl 57.8 NdN 39.0 BrdBr 46.1 NdO 48.0 IdI 36.1 HdH 104.2 ΔHb ¼ ΣΔHbd =number of bonds ¼ 120kcal mol1 + 101kcal mol1 =2bonds ¼ 110:5kcal mol1 Table 8.3 gives some bond energies for selected types of chemical bonds. Bond energies are always positive as it requires energy to break a bond. The sign of the energy required to form a bond is always negative because energy is released when bonds are formed. The energy required to form a specific bond is the negative value of the bond energy for the same bond. The change in enthalpy during a chemical reaction (ΔHrxn) can be estimated from bond energies by adding up all the bond energies for the bonds broken during the reaction and subtracting all the bond energies for all the bonds formed during the reaction; ΔHrxn ¼ ΣΔHðbonds brokenÞ ΣΔHðbonds formedÞ (15) EXAMPLE 8.5: DETERMINING THE ENTHALPY OF REACTION FROM BOND ENERGIES Calculate the enthalpy of reaction for the reaction of methane with chlorine gas to form carbon tetrachloride (CCl4) and hydrochloric acid. 1. Write the balanced chemical equation for the reaction: CH4 g + 4Cl2 g ! CCl4 g + 4HCl g 8.6 THE SECOND LAW OF THERMODYNAMICS: ENTROPY 261 EXAMPLE 8.5: DETERMINING THE ENTHALPY OF REACTION FROM BOND ENERGIES— CONT’D 2. Determine the number of bonds broken and the number of bonds formed during the reaction. Bonds broken ¼ 4 CdH bonds and 4 CldCl bonds Bonds formed ¼ 4 CdCl bonds and 4 HdCl bonds 3. Determine the bond energies for the bonds broken in the reaction. 4 CdH bonds ¼ 4 98.7 ¼ 394.8 kcal • mol1 4 CldCl bonds ¼ 4 57.8 ¼ 231.2 kcal • mol1 394.8 kcal • mol1 + 231.2 kcal • mol1 ¼ 626.0 kcal • mol1 4. Determine the bond energies for the bonds formed in the reaction. 4 CdCl bonds ¼ 4 78.4 ¼ 313.6 kcal • mol1 4 HdCl bonds ¼ 4 103.0 ¼ 412.0 kcal • mol1 313.6 kcal • mol1 + 412.0 kcal • mol1 ¼ 725.6 kcal • mol1 5. Subtract the sum of the bond energies of the bonds formed from the sum of the bond energies of the bonds broken. ΔHrxn ¼ ΣΔHðbonds brokenÞ ΣΔHðbonds formedÞ ¼ 626:0kcal mol1 725:6kcal mol1 ¼ 99:6kcal mol1 8.6 THE SECOND LAW OF THERMODYNAMICS: ENTROPY The primary use of thermodynamics and thermochemistry in the fields of chemistry and engineering is in determining whether a chemical reaction will occur, under what conditions it will occur, and if it will be spontaneous. The early chemists Julius Thomsen and Marcellin Berthelot proposed that the release of heat was the driving force of a chemical reaction. According to this concept, known as the Thomsen–Berthelot principle, only exothermic reactions would occur spontaneously. This means that only reactions with ΔEv or ΔHp would be spontaneous. It is true that a number of reactions or phase changes will happen quickly or spontaneously when the ΔHp is negative. For example, water will spontaneously freeze to form ice at a temperature of 263.15 K. This phase change releases 1.3 kcal • mol1 of energy (ΔHp of 1.3 kcal • mol1). However, some reactions or phase changes will still proceed quickly and can be considered to be spontaneous even though the ΔHp is positive. An example of this is the evaporation of liquid water to form water vapor at a low pressure of 0.013 atm and a temperature of 298.15 K. This change in state requires about 10 kcal • mol1 of energy, for a positive ΔHp. Since ΔHp is positive, it would be assumed under the Thomsen–Berthelot principle that the liquid water should remain in the liquid phase, which it does not! As chemists and engineers further studied chemical reactions and examined other systems, it was discovered that neither ΔEv nor ΔHp alone could determine absolutely if a reaction would occur spontaneously. So, the Thomsen–Berthelot principle was disproved. The second law of thermodynamics was developed by Rudolf Clausius as a means of predicting spontaneous reactions. While the first law of thermodynamics is concerned with 262 8. THERMODYNAMICS AND ENERGY BALANCE the total amount of energy in a system and its surroundings, the second law of thermodynamics is concerned with the quality of that energy. The second law of thermodynamics, as developed by Clausius, states that as energy is transferred from one system to another or from one form to another, it is dispersed from a localized state to a more spread out state. In other words, it is transformed from an ordered state to a more disordered state. This process is described by a state function called entropy (S) with units of cal • mol1 • K1. Entropy is the measure of the dispersal of energy or how much energy is spread out in a process and how widely spread out it becomes. The modern statement of the second law of thermodynamics, commonly known as the law of increased entropy, is; • The entropy of any isolated system always increases. An isolated system is a system that cannot exchange energy (heat or work) or mass with its surroundings. Consider entropy in nature as the tendency for the system to become more random. For example, if you take a pack of playing cards that are neatly stacked in order and throw them up into the air, the cards will not tend to fall neatly back into the stack of 52 cards, but will fall into a disordered pile of cards all over the room. For the case of chemical reactions or phase changes, entropy is considered to be made up of all of the possible positions of atoms and molecules that make up the chemical system under specific conditions. An example of this would be the phase changes of water. When one mole of water in the solid state changes to water in the liquid state and then to water in the gaseous state, the entropy of the system is increased at the molecular level. The hydrogen bonding, which acts to give order to the system, is increasingly disrupted at each phase change and the molecules are allowed to move more freely between more possible positions, so the entropy of the system increases. Entropy is directly related to the heat added to the system and, as such, is related to the extent of molecular movement. Entropy is also related to the temperature of a system as long as it is not undergoing a phase change as shown in Fig. 8.7. As the temperature of the solid increases, heat is transferred to the solid, the movement of the atoms or molecules that make up the solid increases, the molecules become more spread out, disorder increases, and so entropy increases. When the melting point of the solid is reached, there is a rapid increase of temperature as heat is transferred to a system undergoing two phase changes: (1) from a solid to a liquid, (2) from a liquid to a gas. Entropy (cal mol-1 k-1) FIG. 8.7 Change in entropy (ΔS) as a function 0 ΔSvap ΔSfus Tmp Tbp Temperature (K) 8.6 THE SECOND LAW OF THERMODYNAMICS: ENTROPY 263 in entropy with no change in temperature until all the solid is converted to the liquid. During the melting phase, disorder is increased by changing the more ordered and compact solid into a liquid where the molecules become more mobile and can occupy more random sites. This rapid increase in entropy as the solid melts is known as the entropy of fusion (ΔSfus). When all the solid is converted to the liquid form, the temperature begins to rise again. As more heat is transferred to the liquid, the molecules begin to move more rapidly and acquire even more random positions. The entropy increases with temperature until the boiling point is reached. At that point, there is another sharp rise in entropy with no change in temperature due to vaporization of the liquid (ΔSvap). This continues until all of the liquid has been converted to the gas. Again, as the temperature of the gas is increased, heat is transferred to the system and the gas molecules move more rapidly causing an increase in disorder. 8.6.1 The Carnot Cycle Rudolf Clausius and Lord Kelvin developed the concept of entropy after careful examination of work done by Nicholas Leonard Sadi Carnot, a military man who was trained in physics and engineering. Carnot proposed the first successful theory of how to maximize the efficiency of a heat engine, an idealized system that converts thermal energy and chemical energy to mechanical energy. A heat engine acts by transferring energy from a warm area to a cool area and, in the process, converting some of the energy to mechanical work. Carnot showed that the efficiency of the heat engine is a function only of the two temperatures of the reservoirs between which it operates. This concept of an idealized heat engine was successfully applied to the essential features of a real world steam engine. It allowed for exact calculations to be made, which avoided complications that arose from the crude features of the steam engines of the time. The heat engine shown in Fig. 8.8 was designed to exchange heat between two reservoirs that were at two different temperatures, one at a high temperature (TH) and one at a low temperature (TC). The working material (WM), which was usually a gas, absorbs the heat (qh) from the hot reservoir, causing it to expand. The expansion of the working material causes it to do work on the surroundings and can be converted to mechanical work such as moving a piston. The working material then transfers the heat (qc) to the cold reservoir causing it to compress back to its initial size. If the heat engine is allowed to undergo four steps that complete a cycle and return the material to its initial state, the cycle is known as the Carnot cycle. FIG. 8.8 Schematic diagram of a heat engine showing the working mateTH qh WM qc Piston TC rial (WM), hot reservoir (TH), and cold reservoir (Tc). The work created by the expansion of the WM is converted to mechanical work by moving a piston. 264 8. THERMODYNAMICS AND ENERGY BALANCE The Carnot cycle includes two reversible isothermal steps, which take place with no change in temperature, and two reversible adiabatic steps, which take place with no heat exchange. A reversible adiabatic process is also an isentropic process, which takes place with no chance in entropy. The four steps involved in the Carnot cycle are: (1) A reversible, isothermal expansion of the working material at temperature TH. (2) A reversible adiabatic (isentropic) expansion of the working material where the temperature of the working material drops to TC. (3) A reversible isothermal compression of the working material at the temperature TC. (4) A reversible adiabatic (isentropic) compression of the working material to increase the temperature back to TH and return it to its initial starting state. The change in temperature of the working material as a function of the change in volume is shown in Fig. 8.9. During step (1), the working material absorbs heat (qh) from the hot reservoir and expands from V1 to V2 without a change in temperature. The expansion of the working material is caused by the absorption of heat accompanied by an increase in entropy, resulting in the working material doing work on the surroundings. During step (2), the working material continues to expand to V3, doing work on the surroundings and losing an amount of internal energy equal to the work that is done with no loss of heat (ΔE ¼ w). The expansion causes the working material to cool to the temperature TC and the entropy remains unchanged. At step (3), heat (qc) is transferred from the working material to the low temperature reservoir. The entropy decreases by the same amount that was increased in step (1). During step (4), the surroundings do work on the working material, compressing it and causing the temperature to rise to TH. The entropy remains unchanged and the working material returns to the initial state before step (1). The amount of work produced by the heat engine at the end of the cycle is equal to the net heat transferred during the process; w ¼ qh qc (16) The efficiency (η) of this ideal heat engine, the fraction of the heat energy extracted from the hot reservoir and converted to mechanical work, is equal to; FIG. 8.9 The change in temperature as a function of volume for the Carnot cycle. (Step 1) is an isothermal expansion at TH. (Step 2) is an adiabatic expansion with the temperature dropping from temperature TH to TC. (Step 3) is an isothermal compression at TC. (Step 4) is an adiabatic compression raising the temperature back to initial conditions and at TH. 8.6 THE SECOND LAW OF THERMODYNAMICS: ENTROPY η¼ 265 w qh qc qc ¼ ¼1 qh qh qh The ratio of the heat lost (qc) to the heat absorbed (qh) is equal to the ratio of the temperature of the cold reservoir (Tc) to the temperature of the hot reservoir (Th); η¼1 TC TH (17) Then, the efficiency of a Carnot heat engine is dependent only on the temperatures of the two reservoirs. The Carnot cycle is useful in studying real engines because the efficiency can be derived simply from the temperatures of the heat reservoirs. But, since the second law of thermodynamics states that not all heat supplied in a heat engine can be used to do work, the Carnot efficiency represents an upper limit to the fraction of heat that can be used. So, the efficiency of a real engine will always be less than the efficiency of a Carnot engine operating between the same temperatures. The wider the range between the temperatures TC and TH, the more efficient the engine. The value of TC is limited by the type of heat sink used. For example, some common heat sinks for a real engine might be the atmosphere, ocean, river, or whatever is available. Normally, the lowest temperature range for commonly available heat sinks is 283.15–293.15 K (10 20°C). The maximum value of TH is limited by the thermal strength of the available materials. CASE STUDY: CHOICE OF WORKING MATERIALS—GASES FOR REFRIGERATION Each step in the Carnot cycle for a heat engine is reversible. This means that the entire cycle can be reversed. When the Carnot cycle for a heat engine is reversed, it becomes the Carnot refrigeration cycle. The result is that the directions of the heat and work interactions on the working material are reversed. Heat is absorbed from the low temperature reservoir, lost to the high temperature reservoir, and work is done on the working material by the surroundings. The high temperature reservoir acts as a cooling device or refrigeration apparatus and, as work is done on the working material by the surroundings, the work is used to move the heat from the low temperature reservoir to the high temperature reservoir. The determination of efficiency is the same for both forward and reverse cycles. Most refrigerators use a working fluid that is a liquid when compressed and a gas when heated. This is because liquids will take up heat when they are vaporized and gases will give up heat when compressed. Fig. 8.10 shows the basic working parts of a refrigeration system. Step (1) is the reversible adiabatic compression of the refrigerant. The working fluid enters the compressor as a gas, which is compressed slowly from TC to TH. Step (2) is the reversible isothermal compression of the refrigerant. The gas enters the condenser and is condensed to a liquid releasing heat (qh) to the heat exchanger at constant temperature TH. The heat exchanger, which links the system to the surroundings, is usually a set of coils that are in contact with a series of metal veins. This allows good heat exchange with the outside air. Step (3) is the reversible adiabatic expansion of the liquid phase refrigerant. The liquid enters an expansion valve, which allows it to expand slowly to reform the gas. The temperature decreases from TH to TC at constant entropy. Step (4) is the reversible isothermal expansion. The gas enters the evaporator where it absorbs heat (qc) at constant temperature TC. 266 8. THERMODYNAMICS AND ENERGY BALANCE FIG. 8.10 Basic working parts of a refrigerator including the steps of the Carnot refrigeration cycle: (Step 1) the reversible adiabatic compression of the refrigerant, (Step 2) the reversible isothermal compression of the refrigerant, (Step 3) the reversible adiabatic expansion of the liquid phase refrigerant, and (Step 4) the reversible isothermal expansion. The Carnot refrigeration cycle is the same principle that is used in air conditioners to cool the outside air. The working fluids first used in these systems were small alcohols, ammonia, sulfur dioxide, chloroform (CH3Cl), and methyl formate (CH3OC]OdOH). All of these compounds had substantial safety concerns as they are all fairly toxic and leakage of the working fluids from the closed refrigeration systems was a health hazard. Also, some of the gases were flammable with the added risk of fire hazards. During the 1920s, General Motors (GM) began to look into finding safer and more cost-effective materials that could be used in refrigeration units to replace the toxic fluids being used at the time. Thomas Midgely, Jr., a trained mechanical engineer leading a group of scientists at the GM research laboratories, developed synthetic procedures to generate chlorofluorocarbons (CFCs), which were to be used as working fluids in refrigeration systems. Dichlorodifluoromethane (CCl2F2) and other CFCs were widely accepted because they were chemically inert, had very low flammabilities, and very low toxicities. Midgely received the Priestly Medal from the American Chemical Society for his work and was elected President of the Society just before his accidental death in 1944. Later in the mid-1970s, Dr. F. Sherwood Rowland and Dr. Mario Molina, working at the University of California, Irvine, proposed that the CFCs could cause damage to the stratospheric ozone layer. The chemistry behind this process will be covered in detail in Chapter 9. Since stratospheric ozone is a natural filter of harmful ultraviolet solar radiation, destruction of the stratospheric ozone could result in cancer causing UV radiation reaching the earth’s surface. After ozone depletion was observed over Antarctica by NASA, the CFCs were banned for use by all countries and were replaced with the more chemically reactive hydrochlorofluorocarbons or HCFCs. Though safer, the HCFCs are still problematic with regard to ozone destruction and the search for better refrigeration fluids and techniques is still ongoing. The CFCs were originally developed to generate a safer working fluid for use in refrigeration and air conditioning. At the time they were considered to be a major advance in safety, but would later prove to potentially lead to a global catastrophe through the destruction of stratospheric ozone. This example emphasizes the importance of understanding how chemicals behave and how they can react in our environment before they are put into widespread use. Chemicals and materials that 8.7 THE THIRD LAW OF THERMODYNAMICS: ENTROPY AND TEMPERATURE 267 seem to be a perfect solution to a complex problem when considering the system at hand may prove to be far more hazardous when considering the effects on the surroundings. An increased knowledge of the impacts of chemicals on the environment is essential for both professional engineers and chemists and will become increasingly important in the future as we face the problems of decreasing resources, environmental pollution, and climate change. These environmental problems may serve to amplify the overall impacts of materials and chemicals chosen to solve complicated engineering problems and improve our way of life. 8.7 THE THIRD LAW OF THERMODYNAMICS: ENTROPY AND TEMPERATURE As with internal energy (E) and enthalpy (H), in chemical thermodynamics only changes in entropy (ΔS) are measured. However, absolute values of entropy can be defined with respect to a reference state. This reference state is described by the third law of thermodynamics. An early statement of the third law, given by G.N. Lewis in 1923, is the entropy of each element in a perfect crystal is zero at the temperature of absolute zero. The location and orientation of each element in a perfect crystal is exact and, as the energy of the crystal is reduced, the vibrations of each element in the crystal are reduced. These vibrations cease at absolute zero. The third law provides the reference point for the determination of the entropy of a system at any other temperature above absolute zero. The entropy determined relative to this zero point is then the absolute entropy of the system under a specific set of conditions. A more modern statement of the third law of thermodynamics is; • The entropy of any pure substance in its equilibrium state approaches zero at a temperature of absolute zero. The entropy of the pure substance under any set of conditions is the entropy gained when converting the substance from 0 K to that set of conditions. Since increasing the temperature requires adding heat and adding heat increases entropy, all substances have positive entropy at temperatures above absolute zero. Standard molar entropy (S°) is defined as the entropy gained when converting one mole of a substance from a perfect crystal at 0 K1 atm and a specified temperature. Some standard molar entropies are given in Table 8.4 and some generalities can be observed these values: • For a given substance, there is a large change in standard molar entropy with a phase change. • Gases have larger standard molar entropies than liquids. • Liquids have larger standard molar entropies than solids. • Larger and more complex molecules have larger standard molar entropies than smaller molecules. 268 8. THERMODYNAMICS AND ENERGY BALANCE TABLE 8.4 Standard Molar Entropies (cal • mol1 • K1) of Selected Elements and Compounds at 298 K Species (phase) Entropy Species (phase) Entropy Al(s) 6.8 H2O(s) 9.7 Al(l) 8.4 H2O(l) 16.8 AlCl3(s) 26.1 H2O(g) 45.1 Al2O3(s) 12.2 I2(s) 27.7 Ba(s) 14.9 I2(g) 62.3 BaCl2(s) 29.6 HI(g) 49.4 BaO(s) 17.2 Fe(s) 6.5 Br2(g) 58.7 FeCl2(s) 28.2 Br2(l) 36.4 Hg(l) 18.1 HBr(g) 47.5 HgCl2(s) 34.9 C(s)—graphite 1.4 N2(g) 45.8 C(s)—diamond 0.6 NO(g) 50.4 C(g) 37.8 NO2(g) 57.4 CCl4(l) 51.7 N2O(g) 52.6 CCl4(g) 74.1 NH3(g) 46.1 CH4(g) 44.5 O2(g) 49.0 CH3OH(l) 30.3 O3(g) 57.1 CH3OH(g) 57.3 S(s) 7.7 CO(g) 47.3 S(g) 40.1 CO2(g) 51.1 H2S(g) 49.2 Ca(s) 9.9 SO2(g) 59.3 Ca(g) 37.0 Zn(s) 9.9 CaO(s) 9.6 ZnO(s) 10.5 CaCO3(s) 21.9 ZnS(s) 57.7 CaCl2(s) 25.9 ZnCl2(s) 26.6 Cl2(g) 53.3 Zn2SiO4(s) 31.4 HCl(g) 44.7 He(g) 30.1 F2(g) 48.5 Ne(g) 35.0 HF(g) 41.5 Ar(g) 37.0 H2(g) 31.2 Kr(g) 39.2 D2(g) 34.6 Xe(g) 40.5 8.7 THE THIRD LAW OF THERMODYNAMICS: ENTROPY AND TEMPERATURE 269 The first three of these general observations are reasonable as we consider that entropy can be viewed on a molecular level as an increase in molecular motion accompanied by an increase in the number of possible positions of the molecules. A molecular species in the gas phase allows for the molecules to have more motion and therefore more possible molecular arrangements resulting in larger standard molar entropy values than the same species in the liquid phase. Compare: H2O(g) (S° ¼ 45.1 cal • mol1 • K1) with H2O(l) (S° of 16.8 cal • mol1 • K1). The same is true for molecular species in the liquid phase compared to the same species in the solid phase. Compare: H2O(s) (S° ¼ 9.7 cal • mol1 • K mol1) with H2O(l) (S° ¼ 16.8 cal • mol1 • K1). The liquid phase has more possible molecular arrangements, more molecular motion, and higher standard molar entropy values than the solid phase. The standard molar entropy difference between the gas and liquid phases is not as large as with the liquid and solid phases. This is an indication that the gas phase has the largest molecular motion and the largest number of possible molecular arrangements of all three phases. During a phase change, the large change in entropy is associated with the conversion of the molecular species from a more ordered system to a less ordered system as shown in Fig. 8.7. The standard entropy change during the phase change from ice to liquid water (ΔS°fus) is 5.3 cal • mol1 • K1 and the standard entropy change during the phase change from liquid water to water vapor (ΔS°vap) is 26.1 cal • mol1 • K1. The last observation listed from Table 8.4 is that larger molecules have larger standard molar entropies than smaller molecules. There is a general trend of increasing standard molar entropies with increasing molecular mass for similar types of molecular species. Note the trend of the standard molar entropies (cal • mol1 • K1) for the gaseous hydrogen halides: HF(41.5) < HCl(44.6) < HBr(47.4) < HI(49.3). Also consider the standard molar entropy trend for zinc and its compounds: Zn(9.9) < ZnO(10.5) < ZnCl2(26.6) < Zn2SiO4(31.4). Polyatomic molecules with more atoms tend to have larger standard molar entropies. These trends are all consistent with the idea that larger molecules have a larger range of motion than smaller molecules. The internal states of larger atoms and molecules have the ability to vibrate and bend along bonds axes, which adds to the number of possible states of the molecule and so to increased disorder and increased entropy. The general observations of standard molar entropies can be used to predict the general direction of the change in entropy for chemical reactions or phase changes. For example, in the decomposition of AlCl3 into its elements; 2AlCl3 ðsÞ ! 2AlðsÞ + 3Cl2 g an increase in entropy is predicted because 3 moles of chlorine gas is produced in the products from a solid reactant. But, the standard molar entropies can be used to calculate entropy changes during a chemical reaction under standard conditions (ΔS°rxn) as; ΔS° rxn ¼ ΣnΔS° ðproductÞ ΣnΔS° ðreactantÞ (18) The entropy of both products and reactants are multiplied by their stoichiometric coefficients (n), summed, and entropies of reactants subtracted from the entropies of the products. 270 8. THERMODYNAMICS AND ENERGY BALANCE EXAMPLE 8.6: DETERMINING THE CHANGE IN ENTROPY DURING A CHEMICAL REACTION Determine the standard molar entropy change during the reaction of ammonia with N2O gas to give nitrogen gas and liquid water. 1. Write the balanced chemical equation for the reaction. 2NH3 g + 3N2 O g ! 4N2 g + 3H2 OðlÞ 2. Determine the standard molar entropies for the reactants. 2NH3 g ¼ 2 46:1 ¼ 92:2cal mol1 K1 3N2 O g ¼ 3 52:6 ¼ 157:8cal mol1 K1 3. Determine the standard molar entropies for the products. 4N2 g ¼ 4 45:8 ¼ 183:2cal mol1 K1 3H2 OðlÞ ¼ 3 16:8 ¼ 50:4cal mol1 K1 4. Subtract the sum of the standard molar entropies for the reactants from the sum of the standard molar entropies for the products. ΔS° rxn ¼ ΣnΔS° ðproductÞ ΣnΔS° ðreactantÞ ¼ 233:6 250 ¼ 16:4cal mol1 K1 8.8 GIBBS FREE ENERGY The second law of thermodynamics states that the entropy of an isolated system always increases. Completely isolated systems are an ideal concept and not readily applicable to chemical processes. But, since the universe (system + surroundings) is the ultimate isolated system, the second law of thermodynamics predicts that for a nonisolated chemical reaction or process to be spontaneous, the entropy of the universe must increase (Δ Ssystem + Δ Ssurroundings > 0). While this approach is useful, it requires the calculation of the change in entropy of both the system and the surroundings. It would be more useful if there were a means of determining whether a chemical reaction or process is spontaneous by addressing the system alone without considering the surroundings. This is exactly what Josiah Willard Gibbs accomplished when he developed a new state function, which has been given his name; Gibbs free energy (G) also known as Gibbs energy or the Gibbs function. The term “free” was used to mean that it represents the energy available (or free) to do useful work. Gibbs free energy is defined as being equal to the enthalpy of a system less the entropy times the temperature of the system; G ¼ H TS and the change in Gibbs free energy for a system at constant temperature and pressure is; ΔG ¼ ΔH TΔS (19) 271 8.8 GIBBS FREE ENERGY Gibbs free energy is the maximum work that may be performed by a system at a constant temperature and pressure. The change in the Gibbs free energy for a chemical reaction (ΔGrxn ¼ ΔHrxn TΔSrxn) determines whether the reaction is thermodynamically possible or not. If Δ G of the initial state (reactants) is greater than the Δ G of the final state (products), Δ G(rxn) < 0 and the reaction will take place spontaneously. If Δ G of the initial state is greater than Δ G of the final state or Δ G(rxn) > 0, the reaction will not take place unless energy is added to the system and it will not be spontaneous. But, since the Δ G for the reverse reaction is the same as the value for the forward reaction, but with an opposite sign; ΔGforward ¼ ΔGreverse the reverse reaction will be spontaneous. In addition, once the Gibbs free energy reaches its minimum possible value, Δ G(rxn) ¼ 0 and the state of chemical equilibrium is reached. In summary: • If Δ G(rxn) < 0, the forward reaction will be spontaneous. • If Δ G(rxn) > 0, the reverse reaction will be spontaneous. • If Δ G(rxn) ¼ 0, the reaction is at equilibrium and there will be no change in the concentrations of reactants or products. According to Eq. (19), the factors that affect the value of Δ G of a chemical reaction are: Δ H, Δ S, and the temperature. Assuming Δ H and Δ S are independent of temperature, the values of these three factors will determine the sign of Δ G°(rxn) and the spontaneous direction of the reaction. The five possible combinations among these three factors are listed in Table 8.5. If Δ H is < 0 (exothermic reaction) and Δ S is > 0, Δ G (ΔH TΔS) will be <0 and the forward reaction will be spontaneous at all temperatures. If Δ H is > 0 (endothermic reaction) and Δ S is <0, Δ G will be >0 and the reverse reaction will be spontaneous at all temperatures. Under conditions where Δ H and Δ S are either both positive or both negative, Δ G will be <0 for a limited range of temperatures. If they are both positive, Δ G is <0 only at high temperatures and if they are both negative, Δ G is <0 only at low temperatures. For these cases, the exact temperatures where Δ G will be <0 and the reaction spontaneous must be calculated from Eq. (19). TABLE 8.5 The Effect of the Signs of ΔH and ΔS on the Sign of Δ G for a Chemical Reaction ΔH ΔS ΔG Spontaneous Direction Temperature Range <0 >0 <0 Forward All temperatures >0 <0 >0 Reverse All temperatures >0 >0 >0 Reverse Low temperatures >0 >0 <0 Forward High temperatures <0 <0 <0 Forward Low temperatures <0 <0 >0 Reverse High temperatures 272 8. THERMODYNAMICS AND ENERGY BALANCE The change in Gibbs free energy also applies to the conditions under which phase changes will occur. In a phase transition such as vaporization, both phases (liquid and gas) coexist at the vaporization temperature (Tvap) and the process is at equilibrium (Δ G ¼ 0). Under these conditions; ΔGvap ¼ ΔHvap Tvap ΔSvap ¼ 0 ΔHvap ¼ Tvap ΔSvap Tvap ¼ ΔHvap =ΔSvap For water, the values for ΔHvap is 9711 cal • mol1 and ΔSvap is 26 cal • mol1 • K at a pressure of one atmosphere. This determines the temperature of water at the boiling point; Tvap ¼ ΔHvap =ΔSvap ¼ 9711cal mol1 = 26cal mol1 K ¼ 373:5K: Since ΔH ¼ TΔS at the equilibrium temperature of 373.5 K, at temperatures higher than 373.5 K, TΔS > ΔH and Δ G < 0. So, at temperatures higher than 373.5 K, water will spontaneously go from a liquid to a vapor! In other words, above 373.5 K water will only exist as vapor. At temperatures less than 373.5 K (TΔS < ΔH and ΔG > 0), the vaporization phase change will not be spontaneous. However, the reverse process (condensation) will occur spontaneously; ΔHcond ¼ ΔHvap ¼ 9711cal mol1 ΔGcond ¼ 9711calmol1 T 26cal mol1 K So, Δ G < 0 and the condensation of water vapor will be spontaneous at temperatures below 373 K. As with standard molar enthalpies of formation, the standard molar Gibbs free energy of formation (Δ G°f) of any compound is the change in Gibbs free energy that occurs during the formation of one mole of the compound from its elements with all substances in their standard states. Also, for any element in its standard state, the standard molar Gibbs free energy of formation is defined as zero. Some standard molar Gibbs free energies of formation are given in Table 8.6. The change in Gibbs free energy during a chemical reaction under standard conditions can be calculated in the same manner as with the standard enthalpy of reaction, ΔH°rxn, and the standard entropy of reaction, ΔS°rxn; X X ΔG° ðrxnÞ ¼ nΔG° f ðproductsÞ nΔG° fðreactantsÞ (20) The standard molar Gibbs free energies of both products and reactants are multiplied by their stoichiometric coefficients (n), summed, and the summation for the reactants is subtracted from the summation for the products. 8.9 STANDARD GIBB FREE ENERGIES AND CHEMICAL EQUILIBRIUM Fig. 8.11 represents the change in Gibbs free energy during the course of a spontaneous chemical reaction. At the beginning of the reaction, the Gibbs free energy decreases and 8.9 STANDARD GIBB FREE ENERGIES AND CHEMICAL EQUILIBRIUM TABLE 8.6 and Atoms 273 Standard Gibbs Free Energy of Formation (Δ G°f) in kcal • mol1 for Selected Compounds Molecule Phase H2O Gas HCl ΔG°f Atom Phase ΔG°f 54.6 H Gas 48.6 Gas 22.8 O Gas 55.4 CO Gas 32.8 C Gas 160.4 CO2 Gas 94.3 N Gas 108.9 CH4 Gas 12.1 S Gas 56.6 NO Gas 20.9 F Gas 14.9 NO2 Gas 12.3 Cl Gas 25.2 N2O Gas 24.8 Br Gas 19.7 NH3 Gas 3.9 I Gas 16.8 O3 Gas 39.0 SO2 Gas 71.7 H2S Gas 4.8 H2O Liquid 56.7 H2O2 Liquid 28.8 CH3OH Liquid 39.8 Al2O3 Solid 378.2 BaSO4 Solid 325.6 CaO Solid 144.2 CaCO3 Solid 269.9 CuO Solid 31.0 Cu2O Solid 34.9 SiO2 Solid 205.0 ZnO Solid 83.2 the slope of the curve in Fig. 8.11 is negative (Δ G < 0), indicating that the forward reaction is spontaneous. The Gibbs free energy reaches a minimum when the reaction reaches equilibrium. At this point, the slope of the curve is zero (Δ G ¼ 0). Beyond the point of equilibrium, the slope of the curve is positive (Δ G > 0) indicating that further progression toward products is not spontaneous. The reaction quotient (Q) can be used to describe the relationship between the reaction equilibrium and Gibbs free energy of the reaction. In Fig. 8.11, when Δ G < 0, Q < Keq and when ΔG > 0, Q > Keq. At the point where the reaction reaches equilibrium, there is no further change in the concentrations of reactants and products, Q ¼ Keq, and Gibbs free energy reaches a minimum (Δ G ¼ 0). 274 8. THERMODYNAMICS AND ENERGY BALANCE Gibbs free energy (G) Q < Keq ΔG < 0 ΔG° < 0 Q > Keq ΔG > 0 Equilibrium: Q = Keq G is minimum ΔG = 0 FIG. 8.11 0 1 Reactants only Products only Extent of reaction The Gibbs free energy changes during the course of a spontaneous chemical reaction. The relationship between the Gibbs free energy of a chemical reaction at any moment in time (ΔG) and the standard molar Gibbs free energy of a chemical reaction (ΔG°) is given by; ΔG ¼ ΔG° + RT ln Q 1 1 (21) 1 1 where R is the ideal gas constant (8.314 J • mol • K or 1.987 cal • mol • K ), T is the temperature in Kelvin, and ln Q is the natural log of the reaction quotient. This means that at a given temperature, Δ G at any point in a chemical reaction is given by the values of Δ G° and Q. For a reversible chemical reaction at equilibrium, Δ G ¼ 0 and Q ¼ Keq; ΔG° + RT ln Keq ¼ 0 ΔG° ¼ RT ln Keq ln Keq ¼ ðΔG∘ Þ=RT Keq ¼ eΔG=RT (22) (23) Eqs. (22), (23) provide a means of determining the standard free energy change for a chemical reaction from the equilibrium constant for the reaction experimentally. They also allow for the estimation of the equilibrium constant from tabulated values of Δ G°. IMPORTANT TERMS 275 EXAMPLE 8.7: DETERMINING THE EQUILIBRIUM CONSTANT FROM THE STANDARD FREE ENERGY OF A CHEMICAL REACTION What is the equilibrium constant for the formation of 1 mole of ammonia from its elements with all substances in their standard states if the standard Gibbs free energy of formation of is 3.9 kcal • mol1? 1. Determine Δ G°/RT. The units of ΔG° and R must be the same. Δ∘ =RT ¼ 3900cal mol1 = 1:987cal mol1 K1 298K ¼ ð3900Þ=ð592Þ ¼ 6:59 2. Determine Keq ¼ eΔ G/RT. Keq ¼ eΔG=RT ¼ e6:59 ¼ 728 According to Eq. (2), when Δ G° is negative, ln Keq is greater than 0, Keq is greater than 1, and the concentration of the products is greater than the concentration of the reactants at equilibrium. The more negative the value of Δ G°, the larger Keq, and the reaction is said to be “product favored.” Conversely, when Δ G° is positive, ln Keq is less than 0, Keq is less than 1, and the concentration of the reactants is greater than the concentration of the products at equilibrium. The more positive the value of Δ G°, the smaller Keq, and the reaction is said to be “reactant favored.” Very large negative values of ΔG° means that Keq will be very large and the reaction will essentially go close to completion! Very large positive for ΔG° indicates that Keq will be very small and few products will be formed. When ΔG° is equal to zero, Keq is equal to one and the concentrations of products will equal the concentrations of reactants. In summary: • When Δ G° is <0, Keq > 1, reaction is product favored. • When Δ G° is >0, Keq < 1, reaction is reactant favored. • When Δ G° ¼ 0, Keq ¼ 1. IMPORTANT TERMS Adiabatic a process that takes place with no heat exchange. Bomb calorimeter a constant volume calorimeter that is used to measure the heat released during a combustion reaction. Bond energy the average value of the gas phase bond dissociation energies for all bonds of the same type in the same chemical species. Bond dissociation energy the standard energy required to break one specific bond in a molecule in the gas phase. British thermal unit (BTU) the amount of heat needed to raise one pound of water one degree Fahrenheit at one atmosphere pressure. Calorie (cal) amount of energy required to raise one gram of water one degree Celsius at one atmosphere pressure. Calorimeter a device used to measure the heat released or absorbed from a chemical reaction or phase change. 276 8. THERMODYNAMICS AND ENERGY BALANCE Carnot cycle a four step heat engine cycle, consisting of two reversible isothermal steps and two reversible adiabatic steps. Chemical thermodynamics the examination of the fundamental properties of the chemical species involved in reversible chemical reactions and how these properties determine their behavior in the reactions. Closed system one where mass cannot be exchanged between the system and the surroundings. Engine efficiency the fraction of the heat energy extracted from the hot reservoir that is converted to mechanical work. Enthalpy (H) the total thermal energy of a system. Entropy (S) is the measure of the dispersal of energy or how much energy is spread out in a process and how widely spread out it becomes. Equations of state mathematical relationships between state functions that describe the equilibrium state of a system under a given set of physical conditions Equilibrium states the states of a system where the macroscopic properties do not change. Extensive properties bulk properties that depend on the amount of matter present. First law of thermodynamics energy cannot be created or destroyed, it can only be transformed from one form to another. Gibbs free energy (G) the maximum work that may be performed by a system at a constant temperature and pressure. It is equal to the sum of the enthalpy minus the temperature in Kelvin times the entropy of the system. Heat (q) the amount of thermal energy transferred from a system to its surroundings or from one system to another due to the fact that they are at different temperatures. Heat engine an idealized system that converts thermal energy and chemical energy to mechanical energy. Heat of fusion (ΔHfus) the change in enthalpy due to adding heat to a substance in order to change its state from a solid to a liquid at constant pressure. Heat of vaporization (ΔHvap) the change in enthalpy due to adding heat to a substance in order to change its state from a liquid to a gas constant pressure. Hess’s Law the total enthalpy change during a chemical reaction is the same whether the reaction takes place in one step or several steps. Intensive properties bulk properties that do not depend upon how much matter is present. Isentropic a reversible adiabatic process, which takes place with no chance in entropy. Isolated system a system that cannot exchange energy (heat or work) or mass with its surroundings. Isothermal a process that takes place with no change in temperature. Molar heat capacity the amount of heat needed to increase the temperature of one mole of a substance one degree Kelvin. Open system a system that can readily exchange matter with its surroundings. Second law of thermodynamics the entropy of any isolated system always increases. Specific heat capacity the amount of heat needed to increase the temperature of one gram of a substance one degree Kelvin. Spontaneity when a chemical reaction occurs without being driven by some outside force. Standard enthalpy of formation (ΔH°f) the change in enthalpy that occurs during the formation of one mole of a compound from its elements, with all substances in their standard states. Standard free energy of formation (Δ G°f) change in Gibbs free energy that occurs during the formation of one mole of the compound from its elements with all substances in their standard states. Standard molar entropy (S°) the entropy gained when converting one mole of a substance from a perfect crystal at 0 K to 1 atm and a specified temperature. Standard state an element or compound in the most stable form of the physical state that exists at 1 atm and 298 K. State functions macroscopic properties that define the equilibrium state of a system, which depend only on the initial and final states of a system and not on the way the state changed from the initial to the final state. Surroundings everything outside of the system, essentially the universe. System the part of the universe being studied. Thermal equilibrium the condition between two systems where no heat flows between them when they are connected by a path which can transfer heat. Thermal mass the ability of a material to absorb and store heat. Third law of thermodynamics the entropy of any pure substance in its equilibrium state approaches zero at a temperature of absolute zero. Work (w) the force required to move an object a certain distance. STUDY QUESTIONS 277 STUDY QUESTIONS 8.1 8.2 8.3 8.4 8.5 8.6 8.7 8.8 8.9 8.10 8.11 8.12 8.13 8.14 8.15 8.16 8.17 8.18 8.19 8.20 8.21 8.22 8.23 8.24 8.25 8.26 8.27 8.28 8.29 8.30 8.31 8.32 8.33 8.34 8.35 8.36 8.37 What determines that a system is in thermal equilibrium? What is another term that describes a system that is in thermal equilibrium? What is a system? What are the surroundings? How does an open system differ from a closed system? What is an intensive property of a system? What is an extensive property of a system? What determines an equilibrium state of a system. What is a state function? What is an equation of state? Give an example for the equation of state for one mole of an ideal gas. What is heat? What is the specific heat capacity of a substance? What are the two process that can exchange energy between systems? How is work calculated? What is the statement of the first law of thermodynamics? What is the sign of q if heat is absorbed by the system? What is the sign of w if work is done by the system? How is the specific heat capacity under conditions of constant volume (Cv) related to the internal energy of a system? What is enthalpy? How is the heat capacity at constant pressure (Cp) related to the change in enthalpy of a system? How is the change in enthalpy related to heat flow at constant pressure? What is the name of the device used to determine the heat absorbed or released during a chemical reaction? What is the standard state of an element or compound? What is the standard state for the following elements: (a) oxygen, (b) helium, (c) mercury, (d) carbon, (e) nitrogen. What is the standard enthalpy of formation? What is the value of ΔH°f for elements in their standard state? What is Hess’s Law? What is the difference between bond energy and bond dissociation energy? What state function is related to the second law of thermodynamics? What is an isothermal process? What is an isentropic process? What determines the efficiency of a heat engine? The efficiency of a Carnot heat engine is dependent only on what two values? What is the third law of thermodynamics? What is the standard molar entropy? Which physical phase has the largest number of possible molecular arrangements? List the following in the order of increasing standard molar entropy: BaO, Ba, Ba(NO3)2, BaCl2. 278 8.38 8.39 8.40 8.41 8.42 8.43 8.44 8.45 8.46 8.47 8. THERMODYNAMICS AND ENERGY BALANCE What is Gibbs free energy? How is the Gibbs Free Energy defined in terms of enthalpy and entropy? How is the change in Gibbs free energy defined at constant temperature and pressure? What is the value of ΔG for a reaction at equilibrium? What is the value of Δ G for a spontaneous reaction? What is the relationship between Δ G of the forward reaction and Δ G of the reverse reaction? What is the relationship between the standard free energy and the equilibrium constant for a reaction at equilibrium? What value of ΔG° results in an equilibrium reaction constant that is greater than one? What value of ΔG° results in an equilibrium reaction constant that is less than one? What is the value of ΔG° when K ¼ 1? PROBLEMS 8.48 Given that the specific heat capacity of metallic lead is 0.0310 kcal • kg1 • K1, how much heat would be needed to increase the temperature of a lead brick weighing 10.0 kg from 298 to 348 K? 8.49 If a system has 12 kcal of heat added to it and it does 20 kcal of work, what is the change in its internal energy? 8.50 What is the change in internal energy if 145 kcal of work are done on a system and 240 kcal of heat is produced? 8.51 If a system does 10.0 kcal of work and the internal energy of the system is changed by 3.5 kcal what was the transfer of heat during the process? 8.52 A substance requires 500 cal to heat 10 g from 298 to 323 K. What is the material’s specific heat capacity? 8.53 A 200 g bar of silver metal is found to have a thermal mass of 0.010 kcal K1. What is the specific heat capacity for silver? 8.54 The molar heat capacity at constant pressure for CO(g) is 6.97 cal • mol1 • K1. How much heat is required to raise one mole of CO(g) from 298 to 398 K? 8.55 The molar heat capacity at constant pressure for gaseous ethane (C2H6) has been determined to be 12.71 cal • mol1 • K1. How much heat energy is required to raise two moles of ethane from 298 to 333 K? 8.56 Sodium hydroxide gives off heat when it is dissolved in water. Calorimetry measurements have determined that the ΔH for this process is 10 kcal mol1. How much heat is released when 6 moles of sodium hydroxide are dissolved into a liter of water to make up a 6 molar solution? 8.57 In making up one liter of the 6 molar sodium hydroxide solution in Problem 8.56, if the water is originally at 298 K, what would be the temperature of the solution if no heat was lost to the surroundings? (Cp for water is 1 kcal kg1 K1) 8.58 The enthalpy of combustion of octane which is used as the standard for gasoline engines is 1.3 103 kcal • mol1. How much heat is liberated in combusting a liter of octane if its density is 0.700 g • mL1 and its molecular weight is 114.2 g • mol1? PROBLEMS 279 8.59 Ethanol is being proposed as an alternative to gasoline. The enthalpy of combustion of ethanol is 295 kcal • mol1. The density of ethanol is 0.789 g • mL1. The molecular weight of ethanol is 46.07 g • mol1. How much heat is released from combustion of one liter of ethanol? 8.60 Compare the heat released from combustion of one liter of octane (gasoline) in Problem 8.58 to the heat released from combustion of one liter of ethanol Problem 8.59. Which has the higher energy content? How much more of the lower energy fuel would you have to combust to get the same amount of energy output as the higher energy containing fuel? 8.61 According to Table 8.2, Which is the more stable compound: H2S or SO2? 8.62 According to Table 8.2, Which is the more unstable compound: CaO or CaCO3? 8.63 If the enthalpy of vaporization for a molecule is 12 kcal • mol1, what is the enthalpy for condensation? 8.64 The ΔH of vaporization for ice at 298 K is 10.5 kcal • mol1. The Δ H of fusion for ice at 273 K is 1.4 kcal • mol1. What is the Δ H of sublimation for ice? 8.65 The enthalpy of sublimation of a solid is determined experimentally to be 7.5 kcal • mol1. What is the enthalpy of deposition for the substance? 8.66 Determine the standard enthalpy of reaction for the reaction: O3(g) + NO(g) ! NO2(g) + O2(g) using the standard enthalpies of formation in Table 8.2. 8.67 Calculate the standard enthalpy of reaction for the complete combustion of methanol (CH3OH) using the standard enthalpies of formation in Table 8.2. 8.68 What is the standard enthalpy of reaction for the formation of water from hydrogen and oxygen using the standard enthalpies of formation in Table 8.2. 8.69 Use Hess’s Law to determine the ΔH°f for calcium hydroxide Ca(OH)2(s) using the following reactions: H2(g) + ½O2(g) ! H2O(l) ΔHf ¼ 68.3 kcal • mol1 Ca(s) + ½O2(g) ! CaO(s) ΔHf ¼ 151.8 kcal • mol1 CaO(s) + H2O(l) ! Ca(OH)2(s) ΔHrxn ¼ 15.3 kcal • mol1 8.70 Calculate the standard enthalpy of reaction for the reaction: SO2(g) + ½O2(g) ! SO3(g) using Hess’s law given that the standard enthalpies of formation of SO2(g) is 71.0 kcal • mol1 and SO3(g) is 94.6 kcal • mol1. 8.71 Calculate the standard enthalpy for the reaction: 4NH3(g) + 5O2(g) ! 4NO(g) + 6H2O(g), given the following data: N2(g) + O2(g) ! 2NO(g) ΔHrxn ¼ 43.1 kcal • mol1 N2(g) + 3H2(g) ! 2NH3(g) ΔHrxn ¼ 21.9 kcal • mol1 2H2(g) + O2(g) ! 2H2O(g) ΔHrxn ¼ 115.6 kcal • mol1 8.72 Estimate the standard enthalpy of formation of CCl4 using bond energies from Table 8.3. 8.73 Estimate the standard enthalpy of formation CH2Cl2 using bond energies from Table 8.3. 8.74 Estimate the standard enthalpy of formation of CBr3H using bond energies from Table 8.3. 8.75 Using bond energies from Table 8.3, determine the Δ H° for the reaction between hydrogen and oxygen gas to yield water. (Hint: What is the bond order of O2?) 280 8. THERMODYNAMICS AND ENERGY BALANCE 8.76 Calculate the enthalpy of reaction between hydrogen and iodine gas to form hydrogen iodide gas using bond energy values from Table 8.3. 8.77 A heat engine takes in 167 cal of heat from a high temperature reservoir and rejects 120 cal of heat to a lower temperature reservoir. How much work does the engine do in each cycle? 8.78 Calculate the efficiency of a Carnot engine operating between temperatures of 400°C and 25°C. 8.79 Determine the change in entropy during the reaction: O3(g) + NO(g) ! NO2(g) + O2(g) using the standard molar entropies in Table 8.4. 8.80 What is the change in entropy for the decomposition of solid calcium carbonate to solid calcium oxide and carbon dioxide gas? 8.81 Methane gas reacts with water vapor to produce carbon monoxide and hydrogen with a ΔH° of 49.2 kcal • mol1 and a ΔS° of 51.4 cal • mol1 • K1. Calculate ΔG° for this reaction at 25°C. 8.82 The main industrial process for the manufacture of ammonia from atmospheric nitrogen and hydrogen gas, called the Haber process, has a ΔH of 22.2 kcal • mol1 and a ΔS of 47.3 cal • mol1 • K1. At what temperature will this reaction be spontaneous? 8.83 A plastic compound is recycled from the polymer to the monomer in a reversible reaction at a certain temperature. Knowing ΔH and ΔS for the reaction, write the equation you would use to determine the temperature at which this reaction occurs. 8.84 Given the following data, which reactions will be spontaneous at all temperatures? Which will not be spontaneous at any temperature? (a) (b) (c) (d) ΔH°rxn ¼ 59 kcal • mol1, ΔS°rxn ¼ 85 cal • mol1 • K1 ΔH°rxn ¼ 21 kcal • mol1; ΔS°rxn ¼ 27 cal • mol1 • K1 ΔH°rxn ¼ 25 kcal • mol1; ΔS°rxn ¼ 5 cal • mol1 • K1 ΔH°rxn ¼ 25 kcal • mol1; ΔS°rxn ¼ 42 cal • mol1 • K1 8.85 Calculate the ΔSvap for liquid water at 373 K if ΔHvap ¼ 9.7 • kcal • mol1. 8.86 Calculate the ΔSvap for liquid Cl2 at its boiling point of 238.5 K if ΔHvap ¼ 4.9 kcal • mol1. 8.87 Calculate the ΔHvap for molten PbCl2 in kcal mol1 at the boiling point of 1145 K given ΔSvap ¼ 21.7 cal • K1 • mol1. 8.88 Given the ΔHvap for benzene (C6H6) is 7.35 kcal • mol1 and ΔSvap ¼ 20.8 cal • K1 • mol1, what is its boiling point in K? 8.89 Given that the ΔHvap for chloroform (CH3Cl) is 7.02 kcal • mol1 and ΔSvap ¼ 21.0 cal • K1 • mol1, what is its boiling point in K? 8.90 Ethanol (C2H5OH) boils at 351 K, its ΔSvap is 21.0 cal • K1 • mol1, what is its ΔHvap in kcal • mol1? 8.91 Determine the standard Gibbs free energy change for the decomposition of solid calcium carbonate to solid calcium oxide and carbon dioxide gas using the Gibbs free energy of formation. 8.92 Determine the standard Gibbs free energy change for the reaction of carbon monoxide gas with oxygen gas to give carbon dioxide gas using the Gibbs free energy of formation. 8.93 Calculate the ΔG°rxn for the complete combustion of one mole of methane in oxygen gas using the standard Gibbs free energy of formation. PROBLEMS 281 8.94 Calculate ΔG°rxn for the gas phase reaction of O3(g) + NO(g) ! NO2(g) + O2(g) using the Gibbs free energies of formation in Table 8.6. 8.95 What is the equilibrium constant for the reaction in Problem 8.94 at 298 K? Are reactants or products favored? 8.96 The equilibrium constant for the reaction: NH3 ðaqÞ + H2 OðlÞ ! NH4 + ðaqÞ + OH ðaqÞ is 1.8 105 at 25°C. Calculate Δ G° for this reaction at this temperature. 8.97 Calculate Keq for the reaction: MgCO3(s) ! MgO(s) + CO2(g) at 298 K given that Δ G°rxn ¼ 119 kcal • mol1. C H A P T E R 9 Kinetics and the Rate of Chemical Reactions O U T L I N E 9.1 Reaction Rate 283 9.6 Reaction Mechanisms 303 9.2 Rate Laws 285 9.7 Chain Reaction Mechanisms 306 9.3 Integrated Rate Laws 289 Important Terms 310 9.4 Half-Life 294 Study Questions 311 9.5 Collision Theory 296 Problems 313 9.1 REACTION RATE Chapters 7 and 8 have dealt with equilibrium reactions and how the laws of thermodynamics can be used to determine whether the reactions will occur spontaneously under a given set of conditions. In addition, the use of equilibrium constants has allowed for the determination of whether products or reactants were favored at equilibrium. So far, these determinations have only taken into account the relative stabilities of the reactants and products and have not considered the rates of the chemical reactions or the pathways that the reactions follow in going from reactants to products. This is because chemical thermodynamics deals only with why chemical reactions occur. It gives us an understanding of the nature of the chemical equilibrium, but it does not address how fast the reaction will reach equilibrium. In some cases, thermodynamics will predict that a reaction should occur spontaneously, but the rate at which the reaction occurs is so slow that for all practical purposes it is not seen to occur at all. One example of this is the transition of carbon from graphite to General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00009-6 283 # 2018 Elsevier Inc. All rights reserved. 284 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS diamond. Although this transition has a negative free energy and so is thermodynamically favorable at 25°C and 1 atm, the reaction is so slow that it is never observed. This chapter will deal with the topic of chemical kinetics, which is the study of the rates and mechanisms of chemical reactions. It examines the dependence of the speed of a chemical reaction on the temperature and the reactant concentrations in order to explain how the reaction rate, defined as the change in concentration as a function of time, can often vary widely for different chemical reactions. While chemical thermodynamics predicts if a chemical reaction will occur, chemical kinetics predicts how fast a reaction will occur. So, it can be said that thermodynamics guides while kinetics decides if a reaction will occur in a time frame that will be useful. In addition, by determining the effects of temperature and concentrations on the reaction rate, chemical kinetics can provide a means of controlling and optimizing the speed of a chemical reaction. Chemical kinetics uses an experimental approach to study the rates and mechanisms of chemical reactions. It accomplishes this through careful measurements of the reactant and/or product concentrations as a function of time. For the general chemical reaction between reactants A and B producing the products C and D; aA + bB ! cC + dD the changes in the concentrations of [A], [B], [C], and [D] are measured as a function of time and are described mathematically in the form of a derivative of each concentration with respect to time. The reactants are represented as negative derivatives because their concentrations decrease with time and the products are represented as positive derivatives because their concentrations increase with time: Reactant concentration change ¼ d½A=dt and d½B=dt Product concentration change ¼ d½C=dt and d½D=dt The change in the concentrations between the reactants A and B and the products C and D will depend upon the stoichiometric coefficients: a, b, c, and d. For the simple case of a ¼ b ¼ c ¼ d ¼ 1, the concentrations of the reactants decrease at the same rate and the concentrations of the products increase at the same rate. The rate of the reaction can be described by the change in concentration of any one of the reactants or products as; Reaction rate ¼ d½A d½B d½C d½D ¼ ¼ ¼ dt dt dt dt For reactions with stoichiometric coefficients not equal to 1, the rate of change of each species is divided by its stoichiometric coefficient as; Reaction rate ¼ 1 d½A 1 d½B 1 d½C 1 d½D ¼ ¼ ¼ a dt b dt c dt d dt (1) Since the reaction rate can be measured in terms of any one reactant or product, all that is needed to obtain the reaction rate is the change in concentration of one of the reaction species along with its stoichiometric coefficient in the balanced chemical equation. 9.2 RATE LAWS 285 EXAMPLE 9.1: DETERMINING RATES OF REACTION FROM EXPERIMENTAL DATA Determine the rate of formation of nitric oxide from the reaction of 1 mole of nitrogen gas with oxygen if the nitrogen is found to drop to ½ mole in 0.25 s. 1. Write the balanced chemical equation. N2 g + O2 g ! 2NO g 2. Determine the rate of loss of nitrogen gas. d½N2 =dt ¼ 0:5mol=0:25s ¼ 2:0 mol s1 3. Determine the rate of formation of NO. According to the chemical equation, 2 moles of NO are formed for each mole of N2 lost. d½N2 =dt ¼ ½d½NO=dt ¼ 2 2mol s1 ¼ 4mol s1 9.2 RATE LAWS Normally, the reaction rate changes when the concentrations of the reactants change. This means that the reaction rate is not constant throughout a chemical reaction. How these concentration changes affect the reaction rate is determined experimentally by measuring the reaction rates as the concentrations of the reactants are varied. The result is a proportionality between the reaction rate and the concentration of reactants. This proportionality is called a rate law or rate equation. A rate law is an experimentally determined equation that relates the reaction rate with the concentration of the reactants. The proportionality coefficient is called the rate constant (k). The rate law will usually fit an equation of the form; Reaction rate ¼ dA ¼ k½Am ½Bn dt (2) The value of the rate constant is independent of concentration. However, it varies with temperature, usually increasing as temperature increases. It should also be emphasized that both the rate constant and the exponents “m” and “n” must be derived experimentally and that the rate law exponents have no relationship to the reaction’s stoichiometric coefficients. The reaction order with respect to one particular reactant is equal to the value of the exponent of that reactant’s concentration. In the general rate law (Eq. 2), the reaction order with respect to the reactant A is the value of “m” and the reaction order with respect to reactant B is the value of “n.” The overall reaction order is defined as the value of the sum of all exponents in the rate law or m + n for the general rate law (Eq. 2). In other words, the order of the reaction described by the rate Eq. (2) is m + n, while the reaction is mth order in reactant A and nth order in reactant B. The rate constant for a reaction of the order m + n has units of; mol1ðm + nÞ Lðm + nÞ1 s1 286 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS The reaction order can be determined from measurements of reaction rate for different concentrations of reactants. But, since the reaction rate changes as the concentration of the reactants change, the reaction rate is not constant throughout a chemical reaction. In order to determine reaction order, the concentrations of the reactants must be held constant so that the rate of reaction is held constant. One method of achieving this is called the “method of initial rates.” The initial rate of a reaction is the rate of the reaction at the instant the reactants are first mixed and so it corresponds to the initial concentrations of the reactants. This initial rate is determined graphically from experimental measurements made over a short time interval. The reaction order can then be determined by varying the initial concentrations of the reactants and analyzing the resulting changes in the initial rates. For example, if the concentration of reactant A is doubled, the initial rate will also double if the reaction is first order in reactant A (m ¼ 1). But, if the reaction is second order in reactant A (m ¼ 2), the initial rate will quadruple. EXAMPLE 9.2: DETERMINING REACTION ORDER FROM EXPERIMENTAL MEASUREMENTS OF REACTION RATE WITH CHANGING REACTANT CONCENTRATIONS Determine the reaction order for each reactant, the overall reaction order, and the rate law for the reaction of nitrogen oxide with bromine gas from the following initial rate measurements: Experiment (1) (2) (3) [NO] (M) 0.2 0.4 0.2 [Br2] (M) 0.2 0.2 0.1 Rate of Reaction (M • s21) 4.0 104 1.6 103 2.0 104 1. Determine the reaction order for NO. Comparing experiments 1 and 2: the NO concentration is increased by a factor of 2 and the Br2 is constant. The reaction rate increases by a factor of 4. This means that the reaction is second order in NO. 2. Determine the reaction order for Br2. Comparing experiments 1 and 3: the concentration of NO is constant and the concentration of Br2 is decreased by a factor of 2. The reaction rate is decreased by a factor of 2. So, the reaction is first order in Br2. 3. Determine the overall rate order. Since the reaction is second order in NO and first order in Br2, the overall reaction rate is third order: 2 + 1 ¼ 3. 4. Determine the rate law. Reaction rate ¼ k[A]m[B]n where m ¼ 2 and n ¼ 1 Reaction rate ¼ k[NO]2 [Br2]. A zero order reaction is one with a rate law in which the sum of the exponents is equal to zero ([A]0). Since [A]0 ¼ 1, the rate law for a zero order reaction is equal only to the rate constant; Reaction rate ¼ d½A ¼k dt (3) 9.2 RATE LAWS 287 The rate constant for a zero order reaction is in units of concentration per time (mol • L1 • s1 or M • s1). The rate of a zero order reaction is independent of the concentration of the reactant or reactants. Changing the concentration of a reactant has no effect on the speed of the reaction. This type of reaction is extremely rare in chemistry. For those reactions that have been observed, the rate is usually limited by something other than the concentration of the reactants. One example of a zero order reaction is a chemical reaction that occurs on a metal surface which has a limited number of reaction sites such as the decomposition of N2O on a hot platinum surface. 2N2 O g + 2N2 g ! O2 g The N2O molecules that can react under these conditions are limited to those that are attached to the surface of the solid platinum. Once all of the limited surface sites are occupied, the gas phase N2O molecules cannot react until some of the adsorbed molecules decompose and free up a surface site. So, the rate is dependent on the number of sites on the hot platinum surface, but not on the gas phase concentration of N2O. The overall order of the reaction is zero because it is not dependent on the concentration of the reactant. Another example of a zero order reaction is enzyme reactions in living organisms. These reactions begin with the attachment of the reactant to an active site on the enzyme, forming an enzyme-reactant complex. If the number of active enzyme sites is limited compared to the concentration of reactant molecules, the reaction is independent of reactant concentration and so is zero order. A first order chemical reaction is one with a rate law in which the sum of the exponents is equal to one. The reaction rate is then proportional to the concentration of one reactant. Even though other reactants may be present, their concentrations will not affect the reaction rate and so each of these other reactants will be zero order. The rate law for a first order reaction is given by; Reaction rate ¼ d½A ¼ k½A dt (4) Since the reaction rate is equal to the rate constant multiplied by the concentration of one reactant, the units of the rate constant are in inverse time (s1). A second order chemical reaction has a rate law in which the sum of the exponents is equal to two. There are two possible combinations that result in this sum of exponents. The reaction rate can be proportional to the square of the concentration of one reactant or it can be proportional to the product of the concentrations of two reactants. So, the rate law for a second order reaction can then take two forms: d½A ¼ k½A2 dt d½A Case ð2Þ reaction rate ¼ ¼ k½A½B dt Case ð1Þ reaction rate ¼ (5) While the overall order of the reaction is second order, the reaction is said to be second order in reactant A in case 1 and first order in both reactants A and B in case 2. The reaction rate constant for a second order reaction will have the units of concentration per time (L • mol1 • s1 or M1 • s1). The second order reaction is the one most commonly observed in chemical reactions. 288 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS A third order reaction has a rate law in which the sum of the exponents is equal to three. This can be obtained by three possible combinations. The reaction rate can be proportional to the cube of the concentration of one of the reactants, the product of the square of the concentration of one reactant and the concentration of one other reactant, or the product of the concentrations of three reactants: d½A ¼ k½A3 dt d½A ¼ k½A2 ½B Case ð2Þ reaction rate ¼ dt d½A ¼ k½A½B½C Case ð3Þ reaction rate ¼ dt Case ð1Þ reaction rate ¼ (6) The reaction is said to be third order in reactant A in case 1, second order in reactant A and first order in reactant B in case 2, and first order in each of the reactants A, B, and C, in case 3. The rate constants for third order reactions are in units of L2 • mol2 • s1 or M2 • s1. EXAMPLE 9.3: DETERMINING THE RATE LAW FROM EXPERIMENTAL MEASUREMENTS Determine the rate law and the rate constant for the reaction of carbon monoxide gas with nitrogen dioxide to give carbon dioxide and nitrogen monoxide from the following measurements: Experiment (1) (2) (3) [CO] (M) 5.10 104 5.10 104 1.02 103 [NO2](M) 0.350 104 0.700 104 0.350 104 Rate (M/h) 3.4 108 6.8 108 6.8 108 1. Determine the reaction order for NO2. In experiments 1 and 2: the CO concentration is constant and the concentration of NO2 is doubled. The reaction rate also doubled. This means that the reaction is first order in NO2. 2. Determine the reaction order for CO. In experiments 2 and 3: the concentration of NO2 is constant and the concentration of CO is doubled. The reaction rate is also doubled. The reaction is first order in CO. 3. Determine the reaction rate law. Since the reaction is first order in CO and first order in NO2, the reaction rate law is: Reaction rate ¼ k½CO½NO2 4. Determine the value of the rate constant. Rate ¼ k½CO½NO2 k¼ 3:4 108 M h1 Rate ¼ 1:9M1 h1 ¼ ½CO½NO2 5:10 104 M 0:350 104 M 289 9.3 INTEGRATED RATE LAWS In Summary: • • • • Zero order: First order: Second order: Third order: Rate ¼ k Rate ¼ k[A] Rate ¼ k[A]2; k[A][B] Rate ¼ k[A]3; k[A]2[B]; k[A][B][C] Units Units Units Units of of of of k ¼ M • s1 k ¼ s1 k ¼ M1 • s1 k ¼ M2 • s1 9.3 INTEGRATED RATE LAWS The rate laws outlined in Section 9.2 are in the form of differential equations that express the reaction rate as a function of a change in the concentration of reactants over time (d[A]/dt). These differential rate laws describe how the rate of a reaction varies with the reactant concentrations and are used to determine the reaction rate at a specific reaction concentration. By integrating these differential rate laws, we obtain the integrated forms of the rate laws, which express the reaction rate as a function of the initial reactant concentration [A]0 and a concentration after an amount of time has passed [A]t. The integrated rate law allows us to find: the concentration of reactant at any time after the beginning of the reaction, the reaction order, and the reaction rate constant. In order to determine these values, experimental measurements of concentration versus time are displayed in graphical form in four different ways: Graph Graph Graph Graph (1) (2) (3) (4) [A]t vs time ln[A]t vs time 1/[A]t vs time 1/[A]2t vs time According to the integrated rate laws, only one of these four graphs will be linear (Figs. 9.1 and 9.3) and this will determine the reaction order as well as the integrated rate law. Once the reaction order is known, the rate constant can be determined from the slope of the linear plot. Eq. (3) gives the differential rate law of a zero order reaction as d[A]/dt ¼ k, which can be rearranged to give; d[A] ¼ k dt. Integrating this differential rate equation over the limits of A0 to At and t0 to t yields an expression that describes the decrease in concentration of reactant A as a function of time; Zero order First order of time for a zero order reaction (left) and ln[A]t as a function of time for a first order reaction (right). In [A]0 Slope = –k Time (s) In [A]t [A]t [A]0 FIG. 9.1 The plot of [A]t as a function Slope = –k Time (s) 290 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS ð ð d½A ¼ k dt (7) ½At ¼ ½A0 kt In Eq. (7), [A]t is the concentration of reactant A at a particular time t, [A]0 is the initial concentration of reactant A, and time t0 ¼ 0 s. The integrated rate law of the zero order reaction is in the form of the equation for a straight line (y ¼ mx + b) where the slope is k and the y intercept is [A]0. So, the plot of [A]t as a function of time (Fig. 9.1, left) is a straight line with a slope of k. This means that if experimental measurements plotted as the concentration of reactant A versus time (Graph 1) are in the form of a straight line, the reaction is zero order. For zero order reactions, all other graphs will not be linear but will result in a curved shape. The differential rate law for a first order reaction; d[A]/dt ¼ k[A] can be rearranged to d[A]/[A] ¼ k dt. Integration of this expression over the same limits (A0 ! A and t0 ! t) gives: ð ð d½A ¼ k dt ½A ½At ¼ ½A0 ekt (8) ln ½At ¼ ln ½A0 kt (9) Eq. (9) is the most useful for determining the reaction order because it is also in the form of an equation for a straight line with a slope of k and an intercept of ln[A]0 as shown in Fig. 9.1, right. So, a linear result for Graph 2 is the criteria for a first order reaction. An example of a first order reaction is the decomposition of hydrogen peroxide to form hydrogen and oxygen gas; H2 O2 g ! H2 g + O2 g with a differential rate law of : d½H2 O2 =dt ¼ k½H2 O2 and the integrated rate law of : ln ½H2 O2 t ¼ ln ½H2 O2 0 kt: The graph of [H2O2]t versus time does not yield a straight line as shown in Fig. 9.2, left. This is because there is not a linear decrease in [H2O2] with time. Instead, the concentration of H2O2 is a logarithmic function with time and the graph of ln[H2O2] versus time (Graph 2) results in a straight line with a negative slope ( k). FIG. 9.2 The plot of [H2O2]t as a function In [H2O2]t In [H2O2]0 [H2O2]t of time (left) and ln[H2O2]t as a function of time (right). The linear plot for ln[H2O2]t versus time shows that the reaction is first order. Time (s) Slope = –k Time (s) 9.3 INTEGRATED RATE LAWS 291 EXAMPLE 9.4: DETERMINING THE CONCENTRATION OF REACTANT LEFT AFTER A SPECIFIED TIME The rate constant for the first order decomposition of an organic compound CxHy is 8.7 103 s1. How much of the organic will be left after 1 min if the initial concentration is 0.10 M? Using the exponential form of the integrated first order rate law: ½At ¼ ½A0 ekt ¼ ð0:01MÞeð0:0087Þð60Þ ¼ ð0:01Þð0:593Þ ¼ 5:93 103 M As noted in Section 9.2, the differential rate law for a second order reaction can take two forms, one where the reaction is second order in one reactant (case 1) or where the reaction is first order in each of two reactants (case 2). The differential rate law for case 1 is d[A]/dt ¼ k [A]2, which can be rearranged to d[A]/[A]2 ¼ k dt. Integration of this equation using the same limits as before gives: ð ð ½A ¼ k dt ½A2 (10) 1 1 ¼ + kt ½At ½A0 This again is in the form of an equation for a straight line with a slope of + k and a y intercept of 1/[A]0. So, if a plot of 1/[A]t versus time is a straight line as shown in Fig. 9.3, left, the reaction is second order. An example of a second order reaction is the decomposition of nitrogen dioxide into nitrogen monoxide and oxygen; 2NO2 g ! 2NO g + O2 g with a differential rate law of : d½NO2 =dtÞ ¼ k½NO2 2 and an integrated rate law of : 1=½NO2 t ¼ 1=½NO2 0 + kt Slope = k Third order 1/(2[A]t2) 1/[A]t Second order Slope = k 1/(2[A]02) 1/[A]0 Time (s) Time (s) FIG. 9.3 The plot of 1/[A]t as a function of time for a second order reaction (left) and 1/(2[A]2t ) as a function of time for a third order reaction (right). 292 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS FIG. 9.4 The plot of [NO2]t as a function of time (left), ln[NO2]t as a function of time (center), and 1/[NO2]t as a function of time (right).The linear plot for 1/ [NO2] versus time shows that the reaction is second order. The graph of [NO2]t versus time does not yield a straight line as shown in Fig. 9.4, left, neither does the graph of ln[NO2]t versus time (Fig. 9.4, center). Both of these plots results in a curved line. Since, as the integrated rate law shows, the inverse of the concentration of NO2 is a linear function with time, the graph of 1/[NO2]t versus time results in a straight line with a positive slope (+k) and an intercept of 1/[NO2]0. For the type of second order reaction where the reaction is first order in each of two reactants (case 2), the differential rate equation can be simplified if the initial concentrations of the two reactants are set to be equal [A]0 ¼ [B]0 and the stoichiometry is 1:1. For this special case, the differential rate equation; d[A]/dt ¼ k[A][B] ¼ k[A][A] ¼ k[A]2 becomes the same as for case 1 and the integrated rate law is the same as Eq. (9). However, for situations where the stoichiometry is not 1:1 and [A]0 6¼ [B]0, the integrated rate equations become much more complicated and will not be covered in this text. The differential rate law for a third order reaction has three possible cases: one where the reaction is third order in one reactant (case 1), one where the reaction is first order in one reactant and second order in one reactant (case 2), and one where the reaction is first order in three reactants (case 3). The differential rate law for (case 1) is d[A]/dt ¼ k[A]3, which can be rearranged to d[A]/[A]3 ¼ k dt. Integration of this equation using the same limits as before gives: ð ð d½A ¼ k dt ½A3 (11) 1 1 ¼ + 2 kt ½At 2 ½A0 2 This rate law is in the form of an equation for a straight line with a slope of +2k and a y intercept of 1/[A]20. So, if a plot of 1/[A]2t versus time is a straight line as shown in Fig. 9.3, right, the reaction is third order. The differential rate equation for case 3; d[A]/dt ¼ k[A][B][C] can easily be reduced to the differential rate equation for case 1 if the experiment is set up so that all reactant concentrations are equal ([A] ¼ [B] ¼ [C]) and the stoichiometry is 1:1. For this special situation, Eq. (11) represents the integrated rate equation for case 3 and a plot of 1=½At 2 versus time is a straight line. For case 3 reactions with stoichiometry that is not 1:1 and for reactions that fall into case 2, the integrated rate equations become much more complicated. However, third order reactions are quite rare in chemistry except for some gas phase reactions such as: 2NO + O2 ! 2NO2 2NO + Cl2 ! 2NOCl 9.3 INTEGRATED RATE LAWS 293 The reaction rates for this type of case 2 reactions are commonly determined using special experimental techniques. EXAMPLE 9.5: DETERMINING REACTION ORDER GRAPHICALLY Determine the overall order of this reaction, the differential and integrated rate laws, and estimate the rate constant with units from the experimental data for the decomposition of dinitrogen pentoxide to nitrogen dioxide and oxygen presented in graphical form below: 1. Determine the overall order of the reaction. The overall order of the reaction is determined by the graph that gives a linear fit to the data. The graph of ln[N2O5] is linear so the reaction is first order. 2. Determine the reaction rate laws. The first order differential rate law is; d[A]/dt ¼ k[A] The first order integrated rate law is; ln[A]t ¼ ln[A]0 kt 3. Estimate the rate constant for the reaction. Since the raw data is not given, estimate the slope of the line in plot 2 from the graph. The concentrations for each point in the graph can be estimated from graph 1. For the highest concentration: (0.04 M), ln[N2O5] is 3.2 at 0 s. For the lowest concentration: (0.01 M), ln [N2O5] at 700 s is 4.4. k ¼ slope ¼ Δy=Δx ¼ y2 y1 =x2 x1 ¼ ½ð4:4Þ ð3:2Þ=ð700 0Þ ¼ 1:2=700 k ¼ 1:7 103 M1 s1 294 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS 9.4 HALF-LIFE For many reactions, the concept of half life (t½) is very useful. It is a means of describing the speed of a reaction without the need to measure the time required for the reaction to reach equilibrium. Half life is defined as the amount of time required for the reactant concentration to fall to half (50%) of its original value. The half life of a reaction can be determined from the integrated rate law by substituting ½[A0] for [A]t and t½ for t then solving for t½. In order to determine the value of the half life, you need to know: (1) the order of the reaction, (2) the value of the rate constant, and (3) in most cases the initial reactant concentration. Half life is most often used for first order reactions. Recall from Section 9.3 that the integrated rate law for a first order reaction follows an exponential decay. So, according to Eq. (8), at t ¼ t½: ½A½ 1 kt½ ¼ ¼e ½A0 2 ln 0:5 ¼ kt½ t½ ¼ (12) 0:693 k So, the half life of a first order reaction is a constant and does not depend on the initial reactant concentration ([A]0). This means that the time required for 1 mol of a reactant to be reduced to 0.5 mol is the same time required for 100 mol to be reduced to 50 mol. Also, the rate constant and the half life of a first order reaction are inversely related by the value 0.693 (ln 0.5). Since the units for the rate constant for a first order reaction are s1, the half life is given in seconds (s). After the reaction has reached its half life, a second half life can be determined by setting the initial concentration equal to ½[A]0. The value of the concentration of the reactant at the end of the second half life would then be ¼[A]0. Fig. 9.5 shows the result of a series of half lives taken in succession for a first order reaction following an exponential decay. After n half lives, the initial reactant concentration of a first order reaction has fallen by a factor of 1/2n or by a percentage of 1/2n 100. So, after four half lives, the reactant concentration has fallen to 1/128 or 6.25% of its original value. After 10 half lives, the reactant concentration will fall to 1/1024 or 0.1% of the original value. At this point, the reaction is considered to approach equilibrium. 1 Fraction of [A]0 FIG. 9.5 The percent decrease in concentration of reactant A during a first order reaction after 1–4 half lives. 0.8 0.6 50% 0.4 25% 0.2 12.5% 6.25% 0 t½ = 0 1 2 3 4 Half-life 5 6 7 8 295 9.4 HALF-LIFE So, the time required to achieve 10 half lives provides a good estimate of the time required for a first order reaction to reach equilibrium. EXAMPLE 9.6: DETERMINING THE REACTION TIME FROM THE REACTION HALF LIFE How much time will it take for the concentration of a reactant in a first order reaction to fall from 8 to 0.5 M if the half life is of 10 s? 1. Determine the concentration reduction factor. 0:5 M=8 M ¼ 1=16 2. Determine the number of half lives. After n half lives, the initial reactant concentration is reduced by 1/2n 1/2n ¼ 1/16 n ¼ 4 half lives 3. Determine the time elapsed. 4 half lives ¼ 4 10s ¼ 40s The half life for a second order reaction is obtained from the integrated rate law (Eq. 10). At t ¼ t½ and [A]0 ¼ ½[A]0; 1 1 ¼ + kt½ ½½A0 ½A0 (13) t½ ¼ 1=k½A0 So, the half life for a second order reaction is not a constant, but depends on the initial concentration of the reactant. This is also true for zero order and third order reactions (see Table 9.1) and is the reason that the concept of half life is much more useful for a first order reaction than for reactions of other orders. TABLE 9.1 Summary of Rate Parameters for Zero, First, Second, and Third Order Reactions Zero Order First Order Second Order Third Order Differential rate law Rate ¼ k Rate ¼ k[A] Rate ¼ k[A] Rate ¼ k[A]3 Integrated rate law [A]t ¼ [A]0 kt ln[A]t ¼ ln[A]0 kt 1=½At ¼ 1=½A0 + kt 1=½A2t ¼ 1=½A20 + 2kt Linear plot [A]t vs t ln[A]t vs t 1=½At vs t 1=½A2t vs t Rate constant units M • s1 s1 M1 • s1 M2 • s1 Half life ½A0 2k 0:693 k 1=k½A0 3=2½A20 2 296 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS 9.5 COLLISION THEORY Rate constants and reaction rates can vary widely for different chemical reactions. Some reactions such as the rusting of an iron bar can take days, weeks, or even years, while others such as the reaction of sulfur, carbon, and potassium nitrate in gunpowder happen immediately. So far, we have only discussed reaction rates and have not addressed how these rates may be affected by different reaction conditions. The rate of a reaction can be affected by concentration of the reactants, the temperature of the reaction, and the presence of a catalyst. The effect of each of these factors on reaction rate can be explained by collision theory. Collision theory states that, in order for molecules to react, they must collide with each other. But, a collision between reactant molecules is not enough to cause a reaction. These collisions must also be energetic enough to be able to break and reform molecular bonds. In addition, the reactant atoms must be oriented in such a way that they can easily rearrange to form the products. A molecular collision that results in a chemical reaction between reactants is called an effective collision. So, according to collision theory the rate of a chemical reaction is equal to the frequency of the effective collisions. Consider the gas phase reaction between reactants A and B, which collide to form the product A B. A+B!AB The number of collisions between reactants A and B is dependent upon the number of molecules of A and B and so the number of collisions is proportional to the concentration of each reactant. If the concentration of either A or B is doubled, the frequency of collisions between A and B will double. As the frequency of collisions is increased, the frequency of effective collisions is also increased. So, increasing the concentration of the reactants will have the effect of increasing the reaction rate. For conditions of one atmosphere pressure and a temperature of 273 K, the collision frequency between the gas reactants A and B has been estimated to be 2 108 mol • L1 • s1. If every collision between A and B was effective, this would be a very fast reaction rate where the two gases would almost completely react in a billionth of a second (109 s) after mixing. While there are a few gas reactions that approach this rate, most chemical reaction rates are much slower. Common rates for second order chemical reactions are in the range of 102 to 103 mol • L1 • s1. A comparison of this reaction rate with the collision frequency of 108 mol • L1 • s1 shows that most collisions are not effective! In order for a collision to be effective, A and B must collide with enough energy to break the chemical bonds in the reactants and form new bonds in the product. To accomplish this, the reactants must have enough kinetic energy and collide with enough force to break bonds. The minimum kinetic energy that a molecule must have so that a collision between reactants will result in a chemical reaction is called the activation energy (Ea). The kinetic-molecular theory of gases, described in Chapter 6, explains the macroscopic properties of gases as arising from their molecular motion. According to the kinetic-molecular theory, the temperature of the gas is directly proportional to the average kinetic energy (KEav) of the atoms or molecules; 3 (14) KEav ¼ kT 2 297 9.5 COLLISION THEORY FIG. 9.6 The kinetic energy distribution, known as a Maxwell-Boltzmann distribution, for reactant molecules at temperatures of 300 and 400 K. The number of molecules achieving the activation energy (Ea) is represented in blue area for a temperature of 300 K and blue + red area for a temperature of 400 K. where k is the Boltzmann constant (1.381 1023 J • K1). A higher temperature results in a higher average kinetic energy among the reactant molecules. This gives more of the molecules sufficient energy to undergo effective collisions, even though the average kinetic energy is still below the activation energy. This is because the reactant molecules have a distribution of kinetic energies, which is dependent on the temperature. The distribution of molecular kinetic energy for temperatures of 300 and 400 K is shown in Fig. 9.6. The colored areas in the figure indicate the relative number of molecules that have enough energy to be able to react at each temperature. Notice that the number of molecules reaching or exceeding the activation energy (Ea) increases significantly at the higher temperature (400 K) compared to the lower temperature (300 K), although the average kinetic energy is still below the activation energy for both temperatures. Even if the reactant molecules collide with sufficient kinetic energy, they also need to line up with one another in such a manner that chemical bonds can easily break and reform to produce products. The lower the probability of achieving the correct orientation, the smaller the reaction rate. Consider the gas phase reaction of a chlorine atom with a hydrogen molecule to form hydrogen chloride and a hydrogen atom; Cl g + H2 g ! HCl g + H g This reaction can be described by a reaction coordinate diagram shown in Fig. 9.7. In this diagram, the horizontal axis is the progress of the reaction along the reaction pathway, called the reaction coordinate, and the vertical axis is the potential energy of the reaction system. If the chlorine atom and the hydrogen molecule collide with enough force to react, they also must be oriented so that the HdH bond can break while the HdCl bond forms. The Cl atom could collide with the H2 molecule either end-on or side-on and these two orientations would have different probabilities of reaction. The most favorable orientation for this reaction would be an end-on collision forming a linear transition state, an intermediate formed during the transition from reactants to products in a chemical reaction. Cl g + H2 g ! Cl⋯H⋯H‡ g ! HCl g + H g 298 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS Potential energy FIG. 9.7 The reaction coordinate diagram for the reaction of a Cl atom with an H2 molecule. The formation of the transition state (Cl⋯ H⋯ H‡) is the point of highest potential energy. (Cl¨¨H¨¨H‡) HCl + H Ea forward Cl + H2 Ea reverse ΔE Reaction coordinate The transition state (Cl⋯ H⋯ H‡) has the highest potential energy on the reaction pathway, as shown in Fig. 9.7. The reaction is endothermic in the forward direction since the products are of higher energy than the reactants. The overall change in energy during the reaction (ΔE) is given by the difference in energy of the reactants and products. Note that the activation energy for the forward reaction is higher than the activation energy for the reverse reaction (Ea forward > Ea reverse). For an exothermic reaction, the Ea of the forward reaction would be smaller than the Ea for the reverse reaction (Ea forward < Ea reverse). Collision theory was developed to take into account the effects on reaction rate of the number of molecular collisions, the kinetic energy, and the molecular orientation. The quantitative basis of the relationship between these factors and the reaction rate is described by the Arrhenius equation; k ¼ AeEa=RT (15) where “k” is the reaction rate constant, “Ea” is the activation energy, “R” is the ideal gas constant (8.314 103 kJ • K1 • mol1), and “T” is the temperature in Kelvin. The parameter “A” is called the frequency factor. It is related to the number of collisions with the correct orientation for reaction. The factor e Ea/RT is the fraction of molecules with the minimum amount of energy required for reaction. The frequency factor (molecular orientation) has a very small temperature dependence and can be considered to be independent of temperature. The exponential factor is strongly temperature-dependent. Taking the natural logarithm of Eq. (15) gives: ln k ¼ ln A Ea =RT ln k ¼ ln A ðEa =RÞð1=TÞ (16) Eq. (16) is in the form of a straight line with a slope of Ea/R and an intercept of ln A. If the reaction rate constants are measured experimentally at different temperatures and plotted as ln k versus 1/T, the graph is known as an Arrhenius plot. The activation energy of the reaction can be obtained from the slope of the Arrhenius plot as; Slope ¼ Ea =R Ea ¼ ðR slopeÞ: 9.5 COLLISION THEORY FIG. 9.8 −16.0 An Arrhenius plot for the reaction of an oxygen atom (O) with the organic molecule acrolein (C3H4O) in the gas phase. −16.5 In (k) 299 −17.0 −17.5 −18.0 2.2 2.4 2.6 2.8 3 3.2 3.4 3.6 1/T (K−1) An Arrhenius plot for the reaction of an oxygen atom with the organic molecule acrolein (C3H4O) in the gas phase is shown in Fig. 9.8. This reaction has an activation energy of 2.4 kcal • mol1and and frequency factor of 1.4 1010 mol1 • s1. This very high frequency factor shows that the reaction has a high frequency of effective collisions. EXAMPLE 9.7: DETERMINING THE ACTIVATION ENERGY FROM THE ARRHENIUS EQUATION Determine the activation energy of a reaction that has a rate constant of 1.6 105 s1 at 600 K and 6.36 103 s1 at 700 K. 1. Determine the slope of the line. Even though the straight line is determined by only 2 points, the slope can be determined as: Slope ¼ ðy2 y1 Þ=ðx2 x1 Þ ¼ ð ln k2 = ln k1 Þ=ð1=T2 1=T1 Þ ¼ ln 6:36 103 =1:6 105 =ð1=700 1=600Þ ¼ ln ð398Þ= 2:38 104 ¼ 2:51 104 2. Determine the activation energy. Ea ¼ ðR slopeÞ ¼ 8:314 103 kJ K1 mol1 2:51 104 K ¼ 209kJ mol1 The reaction rate can also be affected by the addition of a catalyst. A catalyst is a substance that increases the rate of a chemical reaction, but is chemically unchanged at the end of the reaction. Catalysts do not act as reactants and so are not consumed during the reaction. Instead, they only provide the reactants with an alternate reaction pathway, which has a lower activation energy than the original reaction pathway. Fig. 9.9 shows the activation energy of the chemical reaction with no catalyst (Ea) is higher than the activation energy with the catalyst present (Eacat). Originally, only the number of molecules represented by the dark blue area in the graph is able to achieve effective collisions. After the addition of a catalyst, the additional number of molecules represented by the area in light blue can also achieve 300 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS FIG. 9.9 Arrhenius plot for a chemical reaction showing the activation energies with a catalyst (Eacat) and without a catalyst (Ea). The area in dark blue is the number of molecules reacting without a catalyst and the area in dark blue + the area in light blue signify the number of molecules reacting with a catalyst. effective collisions. This added number of molecules reacting to form products results in an increase in reaction rate. Different reactions use different catalysts and they are all extremely important in industry. Because they speed up chemical reactions, they make industrial processes much more efficient and economical. If the catalyst is in the same phase as the reactants, it is known as a homogeneous catalyst, if it is in a different phase than the reactants, it is known as a heterogeneous catalyst. The heterogeneous catalyst is preferred in industry because it is easily separated from the product and expensive catalysts can usually be fully recovered. The most common examples of chemical processes involving heterogeneous catalysts in industry involve gas phase reactants passed over the surface of a solid catalyst. The catalyst is often a metal, a metal oxide, or a zeolite (aluminosilicates). Some important examples of heterogeneous catalysts used widely in industry are: an iron catalyst used in the formation of ammonia from nitrogen and hydrogen (the Haber process), a nickel catalyst used in the hydrogenation of unsaturated fats into solid fats such as margarine, a vanadium catalyst used in the formation of sulfuric acid from sulfur dioxide and oxygen, and a platinum catalyst used in the formation of nitric acid from ammonia and oxygen. Both sulfuric and nitric acid are then used in the production of fertilizers. CASE STUDY: CATALYTIC CONVERTERS Automobiles are a major source of air pollutants in urban as well as rural areas. Over 60 million cars were produced worldwide in the year 2012, or about 165,000 cars per day. This is projected to increase to 1300 million by 2030. All automobiles that use gasoline as fuel emit carbon monoxide, unburned hydrocarbons, and nitrogen oxides from the combustion chamber. The carbon monoxide and hydrocarbons are produced because the combustion of the fuel is incomplete. Nitrogen oxides (NO and NO2) are produced from the reaction of nitrogen and oxygen in the air at the high temperatures in the engine. 9.5 COLLISION THEORY 301 FIG. 9.10 The platinum catalyst inside of a catalytic converter embedded on the surface of a honeycomb ceramic substrate Photo by RedBurn, Wikimedia Commons. The catalytic converter was developed to convert these pollutants in the exhaust to nontoxic products in order to comply with EPA’s motor vehicle emission standards. Over 90% of today’s new cars are equipped with catalytic converters. These devices contain a platinum catalyst embedded on a honeycomb solid ceramic support as shown in Fig. 9.10. They are integrated in the car’s exhaust manifold where the entire exhaust stream can flow through the screen like solid catalyst. This structure maximizes the contact of the exhaust stream with the catalyst and decreases the amount required of the very expensive precious metal. The carbon monoxide and unburned hydrocarbons can react with oxygen to form carbon dioxide and water vapor; 2CO g + O2 g ! 2CO2 g Cx Hy g + O2 g ! xCO2 g + y=2H2 O g but these reactions have a high activation energy in the gas phase and would not occur alone in the exhaust stream. The addition of the high surface precious metal catalyst greatly lowers the activation energy of these reactions and increases the reaction rate so that they can occur as the exhaust flows through the catalytic converter. The reaction mechanism in the catalytic converter involves the binding of the reactants to the surface of the metal catalyst by van der Waals forces (a process called adsorption) followed by reaction with oxygen to form CO2 and H2O and desorption of the products back into the exhaust stream. The mechanism for the homogeneous reaction of carbon monoxide with oxygen to give carbon dioxide in the gas phase proceeds by way of a bimolecular transition state as; CO g + O2 g ! O─C⋯O─O‡ ! CO2 g + O g The heterogeneous mechanism in the presence of the platinum catalyst involves the adsorption of both carbon monoxide molecules and oxygen molecules on the surface of the metal catalyst. The oxygen molecules dissociate into oxygen atoms, which are held strongly onto the platinum surface. The reactants then migrate toward each other where an oxygen atom combines with an adsorbed carbon monoxide molecule to form a carbon dioxide molecule, which is then desorbed from the surface of the catalyst into the gas phase. 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS FIG. 9.11 Reaction coordinate diagram for the reaction of carbon monoxide with oxygen to form carbon dioxide with (blue) and without (red) a platinum catalyst. The designation (ad) indicates that the species is adsorbed on the surface of the catalyst. Ea without catalyst Potential energy 302 Ea with catalyst Ea adsorption Ea desorption CO(g), O2(g) CO(ad), O2(ad) CO2(ad) CO2(g) Reaction Coordinate CO g >COðadÞ O2 g >O2 ðadÞ ! 2OðadÞ COðadÞ + OðadÞ ! CO2 ðadÞ CO2 ðadÞ ! CO2 g Each of these steps has an activation energy associated with it, as shown in Fig. 9.11. However, they are all dramatically lower than the activation energy required in the gas phase reaction without the catalyst present. This is because of the altered reaction pathway involving the decomposition of the oxygen molecules on the surface of the catalyst before reacting with the carbon monoxide molecules. This pathway eliminates the formation of the transition state (OdC⋯ OdO‡) providing a lower activation energy than the gas phase reaction involving the formation of the intermediate. Also, the immobilization of the reactants on the surface of the catalyst increases their probability of energetic contact with each other. The catalyst in a catalytic converter can be poisoned by some atmospheric pollutants. Poisoning refers to the partial or total deactivation of a catalyst caused by exposure to a range of chemical compounds that bind strongly to the catalyst’s surface preventing the reactants access to the reactive sites. One of the strongest catalyst poison is lead. This is why vehicles equipped with catalytic converters can only use unleaded fuel. Some other common catalyst poisons include sulfur, manganese, and silicon. Manganese comes primarily from the gasoline additive MMT (Methylcyclopentadienyl manganese tricarbonyl) used to increase octane rating after the ban of tetra ethyl lead. Silicon can enter the exhaust stream when there is a leak in the engine that allows coolant to enter the combustion chamber. Reaction rates can also be affected by the use of different isotopes of the same element in a chemical reaction. For example, a chemical bond involving a deuterium bonded to a carbon atom (CdD) is stronger than the same chemical bond with a hydrogen bonded to a carbon atom (CdH). This difference in bond strength is due to the difference in the mass of the isotopes (H ¼ 1.00 amu, D ¼ 2.01 amu). It takes more energy to break the CdD bond than 9.6 REACTION MECHANISMS 303 the CdH bond by about 1.2 kcal • mol1. So, a reaction that involves the breaking of a CdD bond will have a higher activation energy than the same reaction involving the breaking of a CdH bond. This means that the rate of a chemical reaction that involves the breaking of a CdH bond is faster than same reaction with a CdD bond. This change in the rate of a chemical reaction when one of the atoms of the reactants is substituted with one of its isotopes is called the kinetic isotope effect. It can be measured as the ratio of the rate constant of the reaction with the light isotope (kL) to the rate constant of the reaction with the heavy isotope (kH); Kinetic isotope effect ¼ kL= kH : (17) For the reactions involving the substitution of a deuterium for a hydrogen, the kinetic isotope effect would be measured as; kHyd/kDeu. Although the kinetic isotope effects are stronger with the lighter elements, chemical reactions that involve the isotopes of heavier elements will also result in kinetic isotope effects. For example, some plants preferentially use 13CO2 instead of 12CO2 from the air in their photosynthesis reactions. This preference can be used to identify the sources of plant-derived materials by measuring the ratio of [13C]/[12C]. But, since the isotopes of the heavier elements are much closer in weight than the isotopes of the lighter elements, the isotope effects involving the heavier elements like carbon are much smaller than those involving the lighter elements. 9.6 REACTION MECHANISMS In addition to determining reaction rate, chemical kinetics is a very useful tool in determining how chemical reactions occur. Although the stoichiometric equation shows a chemical reaction taking place in one step, many chemical reactions actually take place in a sequence of steps. For example, consider the reaction of N2O5 decomposing to form NO2 and O2. The balanced chemical equation is written as; 2N2O5(g) ! 4NO2(g) + O2(g) However, this reaction actually undergoes a number of steps in the reaction process. (1) (2) (3) (4) (5) N2O5(g) + N2O5(g) ! N2O5*(g) + N2O5(g) N2O5*(g) ! NO2(g) + NO3(g) NO2(g) + NO3(g) ! N2O5(g) NO2(g) + NO3(g) ! NO(g) + NO2(g) + O2(g) NO(g) + NO3(g) ! 2NO2(g) The collision of two N2O5 gas molecules generates an energetic N2O5* molecule in step 1. The superscript “*“means that the N2O5 molecule has gained extra energy from the collision. This highly energetic molecule is capable of dissociation into NO2 and NO3 in step 2 without requiring another collision. But the reverse reaction also occurs to reform the N2O5 in step 3. This forward and reverse reaction pair (steps 2 and 3) is the equivalent of the equilibrium reaction; N2 O5 g >NO2 g + NO3 g 304 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS The reaction between NO2 and NO3 can also give NO along with the products NO2 and O2 in step 4. The NO then reacts with NO3 to give NO2 in step 5. Each of these individual reactions (1–5) is elementary reaction step, which expresses how the molecules actually react with each other on the molecular level during the overall chemical reaction to give the products NO2 and NO3. The sequence of elementary reaction steps that describes the overall chemical reaction on a molecular level is the reaction mechanism. Each elementary reaction step is classified by its molecularity, which is the number of molecules, atoms, or ions that come together to react in that particular step. The molecularity is equal to the sum of the stoichiometric coefficients of all the reactants in the elementary reaction step. An elementary reaction can be classified as unimolecular, bimolecular, or termolecular depending on how many molecules, atoms, or ions are involved. A unimolecular reaction is one where there is only one reactant species. Step 2 of the chemical mechanism for the decomposition of N2O5 is a unimolecular reaction step, since there is only one reactant (N2O5*). The reverse reaction that reforms N2O5 in step 3 is a bimolecular reaction step since it involves two reactant molecules (NO2 and NO3). A termolecular reaction step is one that involves three reactant molecules. An example of a termolecular reaction is two nitric oxide molecules reacting with molecular oxygen to form nitrogen dioxide; 2NO g + O2 g ! 2NO2 g The simultaneous collision of three reactants with the proper energy and orientation to cause a reaction has a low probability of occurring unless one of the reactants is in high concentration. Because of this, most termolecular reactions involve the reaction of two reactants and a third molecule called a third body, which is in high concentration. This third body does not take part in the chemical reaction. Its function is to absorb the excess energy produced when the chemical bond is formed in the product and so acts to stabilize the product molecule. An example of this type of termolecular reaction is the formation of ozone from the reaction of an oxygen atom with an oxygen molecule in air; (18) O2 g + O∗ g + M g ! O3 g + M g where “M” is the third body, which can be N2, Ar, or another O2 molecule. The third body molecule is unchanged in the reaction. It only acts as a means of stabilizing the ozone molecule formed from the energetic oxygen atom combining with the molecular oxygen. Since the concentration of the third body is not changed in the reaction, the reaction rate appears to be second order overall; rate ¼ k½O½O2 ½M ¼ k0 ½O½O2 where k0 ¼ k[M] is a constant. The rate laws for each elementary step in a reaction mechanism are written the same as with a single step reaction, according to the rate constant of the elementary step, the concentration of the reactants, and the stoichiometry of the elementary step. The exact rate of the overall reaction can be determined from the set of simultaneous rate equations for the individual steps of the mechanism. This is a very complicated determination. But, it can be simplified by identifying the slowest step in the mechanism, which limits the rate of 9.6 REACTION MECHANISMS 305 the overall reaction. The entire series of elementary reaction steps, no matter how complicated, cannot proceed faster than the slowest step. This slow step in the reaction mechanism is called the rate limiting step or rate determining step. The rate law for the overall reaction is determined by the stoichiometry of the rate determining step. The correct rate determining step in a reaction mechanism is identified by comparing the rate law for each elementary step with the experimentally determined rate law for the overall reaction. For example, consider the reaction; 2NO2 g + F2 g ! 2NO2 F g One would assume from the stoichiometry that this reaction is termolecular with a rate law of; Rate ¼ ½ d½NO2 =dt ¼ k½NO2 2 ½F2 However, the rate law is found experimentally to be second order overall and first order in NO2; ½ d½NO2 =dt ¼ kex ½NO2 ½F2 where “kex” is the experimentally determined rate constant. The contradiction between the stoichiometry and the experimental results indicates that the reaction occurs in more than one step with a rate determining step that is second order. One possibility for the reaction mechanism is one that proceeds in two steps with the first step being the reaction of NO2 with F2 to give one molecule of NO2F and a fluorine atom. The second step would be the reaction of the fluorine atom with a second molecule of NO2 to produce the second molecule of NO2F. NO2 g + F2 g ! NO2 F g + F g ðk1 is slowÞ NO2 g + F g ! NO2 F g ðk2 is fastÞ If the first elementary step is slow and the second elementary step is fast, the overall rate equation would be; rate ¼ k1[NO2][F2], as observed experimentally. So, step 1 is the rate determining step and kex ¼ k1. CASE STUDY: THE MECHANISM OF THE REACTION OF NITRIC OXIDE WITH MOLECULAR OXYGEN TO FORM NITROGEN DIOXIDE Nitric oxide is formed in large amounts during combustion processes at high temperatures and pressures. Exhaust gas contains more than 99% NO as it is released from the tailpipe. As the exhaust gas cools, the nitric oxide reacts with atmospheric oxygen to form nitrogen dioxide by the following reaction; 2NO g + O2 g ! 2NO2 g This reaction is of potential concern due to the fact that the nitrogen dioxide is a brown gas, which can absorb sunlight. This light absorption causes the NO2 to decompose to form NO and an oxygen atom, a process known as photolysis. The oxygen atom can then form ozone in the atmosphere by reaction with O2, creating poorer air quality. 306 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS According to the stoichiometry, the rate of the reaction of NO with O2 to form NO2 in the atmosphere should be third order involving two NO molecules and one O2 molecule. Experimental measurements have confirmed that the reaction is second order in NO and first order in O2 with a rate law of; Rate ¼ ½ d½NO=dt ¼ k½NO2 ½O2 One mechanism for this reaction is the one step termolecular reaction involving two NO molecules and one O2 molecule according to the stoichiometric equation. This one step mechanism would obey the observed rate law. However, another possible mechanism that also obeys the observed rate law would involve two elementary steps; NO g + NO g >N2 O2 g ðfastÞ N2 O2 g + O2 g ! 2NO2 g ðslowÞ The first step is the reaction of two NO molecules with each other to form an N2O2 dimer (ONdNO) in a fast equilibrium reaction. The second step is the slow, rate determining reaction of N2O2 with O2 to form NO2. Both mechanisms have the same rate law and obey the experimental evidence. But the two step mechanism may be more plausible since it is known that the NO dimer (N2O2) can be formed at low temperatures. As yet, it cannot be proven experimentally which mechanism is correct. So, there are two possible mechanisms for the reaction of NO with O2 that can explain the observed rate law. How important is this reaction in converting NO to NO2 in the atmosphere once the exhaust is released in air that contains 21% oxygen? The concentrations of pollutant gases emitted from an automobile exhaust tail pipe are usually in the region of a few ppm and they are rapidly diluted as they mix with the atmosphere. The reaction rate is strongly dependent on the concentration of NO because of the [NO]2 term in the rate law. For example, if a lecture bottle of NO is released into the air at 13% of one atmosphere (130,000 ppm), 85% of the NO will be converted to NO2 in 15 s. However, the concentration of NO in car exhaust is in the range of 0.1 ppm. This amount of NO would take 226 days to be 85% converted to NO2 because the square of a small concentration in the rate law results in a very small reaction rate and a long reactant lifetime. But it has been observed that the lifetime of NO in the atmosphere is not long. This means that the reaction of NO with O2 is not the main route for NO oxidation in the atmosphere! This reaction will only be important if the concentrations of NO become high enough so that the square of the [NO] results in a faster reaction rate. 9.7 CHAIN REACTION MECHANISMS Many chemical reaction mechanisms involve chain reactions, chemical reactions in which the products themselves act as reactants, restarting the reaction cycle. In many cases, these reactive products take the form of a molecule, atom, or ion with unpaired valence electrons, known as a free radical. In the chain reaction, the free radical reactant is regenerated as a product in one of the elementary steps of the mechanism. It then serves as a reactant to continue to promote the reaction. The unpaired electrons on the free radicals make them very 9.7 CHAIN REACTION MECHANISMS 307 reactive so chain reactions can occur at a very fast reaction rate. Important examples of free radical chain reactions are the formation of organic polymers, which will be discussed in detail in Chapter 13. An example of a chain reaction is the mechanism of the production of NO2 from NO in the atmosphere. It was determined that the direct reaction of NO with molecular oxygen could not be major contributor because the low concentrations of atmospheric NO would result in a very slow rate constant for the reaction. The reaction actually occurs in the atmosphere by reaction of NO with the hydroperoxyl radical (HO2) in a chain reaction mechanism: (1) OH·(g) + CO(g) (2) H(g) + O2(g) CO2(g) + H(g) HO2·(g) (3) HO2·(g) + NO(g) NO2(g) + OH·(g) The chain propagating species in this chain reaction is the hydroxyl radical (OH). In step 1, the hydroxyl radical reacts with carbon monoxide to give carbon dioxide and a hydrogen atom. In step 2, the hydrogen atom reacts with O2 to give HO2. In step 3, NO is converted to NO2 by reaction with HO2 and the hydroxyl radical is regenerated as a product. The hydroxyl radical produced in step 3 can then react with a second molecule of CO in step 1 restarting the reaction chain. Chain reaction mechanisms involve three main types of reactions: (1) an initiation reaction, (2) one or more propagation reactions, and (3) one or more termination reactions. An initiation reaction is the first elementary step in a chain reaction that initially creates the chain propagating species. Propagation reactions are elementary reaction steps that continuously regenerate the chain propagating species. Termination reactions are elementary reaction steps in which the chain propagating species is converted irreversibly into a nonreactive species without the formation of a new chain propagating species. For example, the initiation reaction in the chain reaction mechanism of the conversion of NO to NO2 in the atmosphere is the initial generation of hydroxyl radical, which is formed primarily from the photolysis of ozone in the presence of water vapor; O3 ! O∗ + O2 O∗ + H2 O ! 2OH The propagation reactions are the three elementary reaction steps (1), (2), and (3), which outline the consumption and reformation of the hydroxyl radical in the production of NO2. The termination reactions are reactions that convert OH into a nonreactive species such as: OH g + NO2 g ! HNO3 g OH g + OH g ! H2 O2 g Both of these reactions consume the hydroxyl radical, effectively stopping the recycling of the chain reaction. However, the free radical concentrations in any free radical chain reaction are very small. So, the probability of two free radicals reacting with each other is low. This means that free radical chain reactions having only this type of termination step will have a relatively long lifetime before the reaction stops. 308 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS CASE STUDY: STRATOSPHERIC OZONE DEPLETION BY CHLOROFLUOROCARBONS Our atmosphere consists of layers, as shown in Fig. 9.12. In these layers, the pressure decreases with increasing altitude going from sea level to outer space. The lowest level of the atmosphere, with the highest pressure, is called the troposphere, which is separated from the stratosphere by the tropopause. Generally, the temperature drops along with the pressure with increasing altitude in the troposphere until you reach the tropopause. In the tropopause, the temperature remains constant. It then increases with altitude in the stratosphere, remains constant in the stratopause, and then decreases again with altitude in the mesosphere. Located in the lower stratosphere, at about 20–30 km, is the stratospheric ozone layer. The ozone is formed from the photolysis of molecular oxygen. The ultraviolet radiation in the stratosphere is energetic enough to break the O]O bond and form oxygen atoms. The oxygen atoms then react with oxygen molecules to form ozone in a third body reaction, as described in Section 9.6. O2 g + O∗ g + M g ! O3 g + M g This gas phase reaction is considered to be pressure-dependent, since the partial pressures of the reactants are related to their concentrations by the ideal gas law. So, as the pressure decreases with altitude in the stratosphere, the concentration of the reactants decreases and the rate of formation of ozone decreases. This is why there is more ozone in the lower portion of the stratosphere creating the stratospheric ozone layer. In 1974, Drs. F. Sherwood Rowland and Mario Molina became concerned that the widespread use of CFCs might lead to a decrease in stratospheric ozone. As discussed in Chapter 8, CFCs were used as refrigeration working fluids. At this time, they were also being used as spray can propellants because of their inflammability and low toxicity. Rowland and Molina suggested that because of FIG. 9.12 The location of the layers of the atmosphere as a function of temperature. The stratospheric ozone layer is shown in green. 9.7 CHAIN REACTION MECHANISMS 309 their low atmospheric reactivities, the concentration of the CFCs could build up in the troposphere, and over time, would likely begin to mix into the stratosphere. The high levels of ultraviolet radiation in the stratosphere can break the CdCl bonds in the CFCs, producing highly reactive chlorine free radicals (Cl), which then react with ozone in a chain reaction mechanism: (1) CFC + UV Cl· initiation (2) Cl·(g) + O3(g) ClO·(g) + O2(g) propagation (3) ClO·(g) + O*(g) Cl·(g) + O2(g) propagation (4) 2ClO·(g) + UV 2Cl·(g) + O2(g) propagation When the chlorine free radicals react with ozone, they produce another highly reactive free radical, ClO (step 2). The ClOreacts with oxygen atoms, produced in the stratosphere from the photolysis of ozone, reforming the chlorine free radicals in step 3. Alternatively, the ClO free radicals can undergo photolysis to reform chlorine free radicals in step 4. While reaction (2) leads to the direct loss of ozone in the stratosphere, reaction (3) also contributes to ozone loss by taking up oxygen atoms before they can react with molecular oxygen to reform ozone. The propagation steps (3) and (4) both result in the production chlorine radicals, which then act as reactants in step (2) to continue the chain reaction. In this manner, one chlorine free radical can destroy thousands of ozone molecules before one of the following termination reactions stops the process: (1) Cl(g) + CH4(g) ! HCl(g) + CH3(g) (termination) (2) ClO(g) + NO2(g) ! ClNO3(g) (termination) (3) 2Cl(g) ! Cl2(g) (termination) The lifetime of HCl is a few weeks and the lifetime of ClNO3 is about a day. So, eventually each of these termination species will react to give back the free radicals and continue the chain reaction. Ozone is a strong absorber of radiation in the ultraviolet region called the UV–B (280 –315 nm). The addition of the CFCs into the atmosphere could lead to a lowering of the ozone concentrations in the stratosphere and an increase in the UV–B radiation reaching the surface of the earth. This radiation can be damaging to most terrestrial organisms and some shallow sea environments. The potential of a depletion of stratospheric ozone resulting in higher levels of UV–B radiation reaching ground level became a major concern. Ground-based measurements of total column ozone taken in the Antarctic in the late 1970s indicated that there was a significant lowering of stratospheric ozone in the Antarctic region. This observed ozone depletion was reported as the development of a so-called “ozone hole” over Antarctica. The cause of this observed ozone depletion was highly debated. Was it in fact due to the reaction of chlorine free radicals produced from the photolysis of CFCs or was it due only to the special meteorological conditions in the region? Because of its location at the southern pole, the Antarctic region has a dramatic change in the photoperiod during the year resulting in a very strong change in atmospheric photochemistry. But, it also has a meteorology that isolates it from the rest of the atmosphere in winter. October is spring in the southern hemisphere, the season when the photoperiod begins to lengthen and atmospheric photochemistry begins to become important. The question was answered when atmospheric concentrations of the predicted reaction intermediate ClO were measured by satellite. A comparison of the O3 concentrations with ClO concentrations obtained from satellite measurements is shown in Fig. 9.13 for one of the highest ozone depletion events that occurred in March of 2011 in the Arctic. The fact that ClO was found in parts 310 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS FIG. 9.13 Comparison of ozone and ClO concentrations obtained from satellite measurements in March of 2011 over the Arctic. Image: NASA/JPL-Caltech. per billion (ppb) concentrations in the stratosphere over Antarctica confirms the chain reaction mechanism of Cl as the cause of the O3 depletion in this same area. This shows how a proposed chemical mechanism can be confirmed by product analysis. The confirmation of the chlorine free radical reaction mechanism and the recognition of the potential damage caused by the stratospheric ozone depletion from the use of CFCs led to the passing of the Montreal Protocol in 1989. In this agreement, the countries of the world began to phase out the use of CFCs to protect the Earth’s ozone layer. It is interesting to note that CFCs are also very strong greenhouse gases, so reducing their release into the atmosphere also had an important impact in the area of climate change. For their work, Drs. Rowland and Molina shared the Nobel Prize in Chemistry in 1995 with Paul Crutzen, who predicted that a number of trace gases could be mixed into the stratosphere from the troposphere through strong thunderstorm activity and that this mixing could affect stratospheric ozone chemistry. IMPORTANT TERMS Activation energy (Ea) the minimum kinetic energy that a molecule must have so that a collision between reactants will result in a chemical reaction Arrhenius equation the quantitative basis of the relationship between the activation energy, temperature, molecular orientation, and the reaction rate Arrhenius plot a plot of the natural logarithm of the observed rate constant as a function of 1/T in Kelvin. Bimolecular reaction an elementary reaction that involves two molecules, atoms, or ions coming together to form products. Catalyst a substance that increases the rate of a chemical reaction, but is chemically unchanged at the end of the reaction. Chain reactions a reaction that regenerates a key reactant in the reaction mechanism. Chemical kinetics the study of the rates and mechanisms of chemical reactions. Collision theory a theory used to predict the rates of chemical reactions based on the assumption that, for a reaction to occur, it is necessary for the reacting species to collide with one another. Effective collision a molecular collision that results in a chemical reaction between reactants. STUDY QUESTIONS 311 Elementary reaction steps simple chemical reactions that expresses how molecules actually react with each other on the molecular level during an overall chemical reaction. Exponential decay the decrease in any quantity “N” according to; N(t) ¼ N0 e λt. First order a chemical reaction whose rate is proportional to the concentration of one reactant. Frequency factor the parameter “A” in the Arrhenius equation which is related to the number of collisions with the correct orientation for reaction Half life the amount of time required for the reactant concentration to fall to half of its original value. Initiation reaction the first elementary step in a chain reaction that initially creates the chain propagating species. Integrated rate law an experimentally derived equation that expresses the reaction rate as a function of the initial reactant concentration [A]0 and the reactant concentration after an amount of time has passed [A]t. Kinetic isotope effect change in the rate of a chemical reaction when one of the atoms of the reactants is substituted with one of its isotopes. Molecularity the number of molecules, ions, or atoms that come together to react in an elementary reaction step. Photolysis the decomposition of molecules after the absorption of light. Propagation reactions elementary reaction steps in a chain reaction that continuously regenerate the chain propagating species. Rate constant a proportionality coefficient relating the rate of a chemical reaction to the concentration of reactant or reactants. Rate determining step the elementary step in a reaction mechanism with the slowest reaction rate. Rate law an experimentally derived equation that relates the rate of a chemical reaction with the concentration of the reactants. Reaction mechanism the sequence of elementary reaction steps that describes how the molecules actually react with each other on the molecular level during the overall chemical reaction to give the products. Reaction order the sum of the values of all the exponents on all the concentration terms in the rate law. Reaction rate the change in concentration of a reactant or product as a function of time. Second order a chemical reaction whose rate is proportional to the square of the concentration of one reactant or the product of the concentrations of two reactants. Termination reactions elementary reaction steps in a chain reaction in which the chain propagating species is converted irreversibly into a nonreactive species without the formation of a new chain propagating species. Termolecular reaction an elementary reaction that involves three molecules, atoms, or ions coming together to form products. Third order a chemical reaction whose rate is proportional to the cube of one of the reactants, the product of the square of the concentration of one reactant and the concentration of one other reactant, or the product of three reactants. Transition state an intermediate formed during the transition from reactants to products in a chemical reaction. Unimolecular reaction an elementary reaction that involves only one molecule, atom, or ion as a reactant. Zero order a chemical reaction whose rate is proportional only to the rate constant and not to the concentration of reactants. STUDY QUESTIONS 9.1 9.2 9.3 9.4 9.5 9.6 What topics does chemical kinetics cover? How is the rate of a chemical reaction determined? What is a rate law? Is the rate law for a chemical reaction determined theoretically or experimentally? What is a rate constant? If the rate of a chemical reaction between reactants A and B has the rate law: reaction rate ¼ [A]m[B]n; What is the overall reaction order? What is the order in [A]? What is the order in [B]? 9.7 How is a zero order reaction rate related to concentration of the reactants? 9.8 How is a first order reaction related to the concentration of reactants? 312 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS 9.9 What are the two possible rate laws for a second order reaction involving two reactants? 9.10 What are the three possible rate laws for a third order reaction involving three reactants? 9.11 What would the overall reaction order be for a chemical reaction with a rate law of: rate ¼ k[A][B][C]? What is the order in each of the individual reactants, A, B, and C? 9.12 If a reaction is second order what are the units of the rate constant? 9.13 What are the units of the rate constant for a first order reaction? 9.14 For a reaction that is third order overall, what are the units of the rate constant? 9.15 What is an integrated rate law? 9.16 For a chemical reaction involving reactant A going to products, what is the integrated rate law for a zero order reaction? What is the integrated rate law for a first order reaction? 9.17 What is the integrated rate law for a second order chemical reaction that is second order in one reactant? 9.18 What is the integrated rate law for a third order chemical reaction that is third order in one reactant? 9.19 In order to determine the order of a chemical reaction from experimental measurements of concentration versus time, four graphs are used. These are: (1) [A]t versus time, (2) ln [A]t versus time, (3) 1/[A]t versus time, and (4) 1/[A]2t versus time. Which of these are linear for a second order reaction? Which is linear for a first order reaction? 9.20 How would you determine graphically if a chemical reaction is zero order? 9.21 What is the order of a chemical reaction that has an integrated rate law that follows an exponential decay? Give this rate law in its exponential form. 9.22 Define half life for a chemical reactant. 9.23 What is the equation that describes the half life for a first order chemical reaction? 9.24 Is the half life for a first order chemical reaction dependent or independent on the starting concentration? 9.25 What is the equation for the half life of a second order chemical reaction? 9.26 Is the half life for a second order chemical reaction dependent upon the initial reactant concentration? 9.27 According to collision theory, what two properties must a collision between reactant molecules have in order to result in a chemical reactants? 9.28 What is a molecular collision that results in a chemical reaction called? 9.29 What is the activation energy? 9.30 If a reaction coordinate diagram for a chemical reaction shows that the reactants are at a higher potential energy than the products, what type of reaction is the forward reaction? What type of reaction is the reverse reaction? 9.31 What is the intermediate formed during the transition from reactants to products in a chemical reaction called in collision theory? 9.32 What is the Arrhenius equation? Give its mathematical formula. 9.33 What term in the Arrhenius equation is related to the number of molecular collisions with the correct orientation for reaction? What is it called? 9.34 What term in the Arrhenius equation is related to the number of collisions with sufficient energy for reaction? 9.35 How can the Arrhenius equation be used to determine Ea from rate constant and temperature data? PROBLEMS 313 9.36 What is a catalyst? 9.37 How does a catalyst speed up a reaction? 9.38 When a hydrogen atom is replaced by a deuterium atom in an organic molecule, the rate of a chemical reaction that involves the breaking the CdH bond is found to be faster than rate of the reaction with a CdD bond. What is this effect called? 9.39 What is a reaction mechanism? 9.40 What is the molecularity of a reaction? 9.41 What is the difference between a unimolecular, biomolecular, and termolecular reaction? 9.42 Is it likely that a reaction might be tetramolecular or higher? Why? 9.43 For a two step reaction that involves a fast elementary step followed by a slow elementary step, what is the rate determining step? 9.44 What is photolysis? 9.45 Describe a chain reaction. 9.46 What are the three types of reactions in a chain reaction mechanism? 9.47 What is the chain propagating species in the chain reaction mechanism of the destruction of ozone in the stratosphere by CFCs? PROBLEMS 9.48 Experimental studies of the decomposition of N2O5(g) to give NO2(g) and +O2(g) have shown that the reactant concentration falls from 0.012 to 0.010 M in 100 s. What is the rate of reaction? 9.49 Studies of the decomposition of nitrogen dioxide to nitrogen monoxide and oxygen have reported a decrease in nitrogen dioxide from 0.01 to 0.008 M in 50 s. What is this reaction rate? 9.50 For a general reaction 3A + 2B ! 2C, how would the rate of disappearance of reactant A be related to the disappearance of B? Be related to the appearance of the product C? 9.51 Gas phase methane (CH4) reacts with oxygen to give carbon dioxide and water. Which reactant will have the slower loss rate? Which product will have the faster rate of production? 9.52 For the general reaction A + B ! 2C, if the concentration of A falls from 1 to 0.6 M after 10 s what is the rate of the loss of A? What is the rate of formation of C? 9.53 Determine the reaction order for each reactant and the overall order for the chemical reaction of reactants A and B from the following experimental measurements of reaction rate at different starting concentrations: 1. [A] ¼ 0.1 M, [B] ¼ 0.1 M, rate ¼ 1 104 M • s1 2. [A] ¼ 0.1 M, [B] ¼ 0.2 M, rate ¼ 1 104 M • s1 3. [A] ¼ 0.3 M, [B] ¼ 0.1 M, rate ¼ 3 104 M • s1 9.54 Determine the reaction order for each reactant and the overall order for the reaction of nitrogen oxide with hydrogen from the following experimental measurements of reaction rate at different starting concentrations: 314 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS 1. [NO] ¼ 0.001 M, [H2] ¼ 0.004 M, rate ¼ 0.002 M • s1 2. [NO] ¼ 0.002 M, [H2] ¼ 0.004 M, rate ¼ 0.008 M • s1 3. [NO] ¼ 0.004 M, [H2] ¼ 0.002 M, rate ¼ 0.016 M • s1 9.55 Ozone is known to thermally decay to molecular oxygen by the following reaction: 2O3(g) ! 3O2(g). The rate of production of O2 is determined to be 0.125 M • s1 over the first 12 s of the reaction. How many moles of O2 were formed during that amount of time? 9.56 The reaction, A + B + C ! 3D, is found experimentally to follow the rate law; rate ¼ k[A][B][C]. What is the order of the reaction for each of the reactants? What is the overall order of the reaction? 9.57 The gas phase reaction of nitrogen monoxide and oxygen to give nitrogen dioxide follows the rate law; rate ¼ k [NO]2[O2]. If the pressure of NO is tripled how much faster will the rate be? If the pressure of NO is halved how will this affect the rate of reaction? If O2 is doubled how will this affect the overall rate? 9.58 A study of the gas phase reaction of NO with Cl2 gives the following data: [NO] (M) 0.200 0.400 0.200 [Cl2] (M) 0.200 0.200 0.100 Rate (M • s1) 1.0 104 2.0 104 5.0 105 What is the rate law for this reaction? What is the value of the rate constant? 9.59 The reaction A + B ! C + D is third order. If the concentration of A is doubled, the rate is increased by a factor of 2. What is the effect on the rate if the concentration of B is cut in half? What is the rate law for this reaction? 9.60 A reaction is studied by measuring the decrease in reactant concentration in molar units using a stopwatch to obtain time in seconds. What will be the units of the rate constant if it is a third order reaction? A second order reaction? 9.61 An experimental study of the reaction of bromine gas with nitric oxide to form NOBr showed that the reaction rate increased by a factor of 8 if the concentrations of both reactants were doubled. But, if the concentration of bromine is doubled and the concentration of NO is held constant, the reaction rate is only doubled. What is the rate law for this reaction? What are the units of the rate constant? 9.62 Determine the rate law, overall reaction order, and the value of the rate constant for the reaction A2 + B2 ! 2AB given the following data: 1. [A2] ¼ 0.001 M, [B2] ¼ 0.001 M, rate ¼ 0.01 M/s 2. [A2] ¼ 0.001 M, [B2] ¼ 0.002 M, rate ¼ 0.02 M/s 3. [A2] ¼ 0.002 M, [B2] ¼ 0.002 M, rate ¼ 0.08 M/s 9.63 Determine the rate law, overall reaction order, and the value of the rate constant for the reaction A + B ! C given the following data: 1. [A] ¼ 0.01 M, [B] ¼ 0.02 M, rate ¼ 0.02 M/s 2. [A] ¼ 0.04 M, [B] ¼ 0.04 M, rate ¼ 0.08 M/s 3. [A] ¼ 0.04 M, [B] ¼ 0.04 M, rate ¼ 0.08 M/s 315 PROBLEMS 9.64 The decomposition of dinitrogen pentoxide gas at 318.15 K is measured as the decrease of N2O5 concentration in partial pressures (mm): The data are shown graphically: Time (s) 0 600 1200 2400 3600 4800 6000 7200 [N2O5](mm) 348 247 195 105 58 33 18 10 1/[N2O5] 0.00287 0.00405 0.00541 0.00952 0.01724 0.0303 0.05556 0.1 ln[N2O5] 5.8522 5.5093 5.22036 4.65396 4.06044 3.49651 2.89037 2.30259 What is the order for this reaction? What is the value of the rate constant? 9.65 Using the data presented in Problem 9.64, what is the concentration of N2O5 remaining after 150 min? 9.66 It has been suggested that a reaction A + B ! C + D is third order. What is the most useful way to plot the measurements of the concentration of A as the reaction proceeds in order to determine if the reaction is actually third order? 9.67 The second order gas phase decomposition hydrogen iodide gas has a rate constant of 30 M • min1. What is the concentration after 3 min if the initial concentration is 0.01 M? 9.68 The rate constant for the reaction: A ! B + C is 6.20 103 M • s1. If the initial concentration of A is 0.050 M, what is its concentration after 500 s? 9.69 Three different reactions are found to have the following rate constants: (1) k ¼ 2.3 M1 • s1, (2) k ¼ 1.8 M • s1, (3) k ¼ 0.75 • s1, which reaction is zero order? 9.70 The decomposition of an organic molecule with the formula C4H6 at 326 °C is measured as the decrease of C4H6: The data are shown graphically: 316 9. KINETICS AND THE RATE OF CHEMICAL REACTIONS Time (s) 0 900 1800 3600 6000 [C4H6](M) 0.017 0.014 0.012 0.010 0.007 1/[C4H6] 58.1 69.9 81.3 105 137 ln[C4H6] 4.06 4.25 4.40 4.65 4.92 Determine the overall reaction order, the rate law, and the rate constant for the reaction. 9.71 A first order reaction has a rate constant of 6.93 s1. What is the half life for the reaction? 9.72 If a first order reaction has a half life of 115.5 s, what is the first order rate constant for the reaction? 9.73 A first order reaction starts out with 1 M concentration in the reactant A. If the half life is 30 s, what will be the concentration of A after the reaction has gone 30 s? 60 s? 90 s? 9.74 What percentage of a first order reactant A will remain after 40 s, if the half life is 10 s? 9.75 The reaction rate for the decomposition of N2O5 to form NO2 and O2 was studied as a function of temperature. The first order reaction rate constants were found to be: T (K) 273 298 308 318 328 338 What is the Ea for this reaction? k (s1) 7.9 107 3.5 105 1.4 104 5.0 104 1.5 103 4.9 103 PROBLEMS 317 9.76 The rate constant for the reaction H2(g) + I2(g) ! 2HI(g) is 5.4 104 M1 • s1 at 326°C and 2.8 102 M1 • s1 at 410°C. What is the activation energy for this reaction? 9.77 A reaction has the stoichiometry of A + 2B + C ! 2D. Experimentally, the rate of the reaction is found to be first order in A and first order in B. What is the overall order of the reaction? 9.78 What is the molecularity of the elementary reaction step: Cl + O3 ! ClO + O2? 9.79 The gas phase reduction of NO by H2 is proposed to begin by the following reaction step: H2 + 2NO ! N2O + H2O. What is the molecularity of this reaction step? 9.80 The proposed mechanism for the formation of phosgene from carbon monoxide and chlorine gas is: 1. Cl2 + M ! 2Cl + M (fast) 2. Cl + CO + M ! ClCO + M (fast) 3. ClCO + Cl2 ! Cl2CO + Cl (slow) What is the rate law for this mechanism? 9.81 The mechanism for the reaction; Br2 + H2 ! 2HBr is: 1. 2. 3. 4. 5. Br2 ! 2Br Br + H2 ! HBr + H H + Br2 ! HBr + Br H + HBr ! H2 + Br 2Br ! Br2 9.82 What is the propagation species for this reaction? What is the initiation step? What is the termination step? C H A P T E R 10 Oxidation-Reduction Reactions and Electrochemistry O U T L I N E 10.1 Oxidation-Reduction Reactions 319 10.6 Electrolysis 339 10.2 The Galvanic Cell 323 10.7 Batteries 342 10.3 Balancing Oxidation-Reduction Equations 325 10.8 Fuel Cells 347 10.4 Standard Cell Potentials 327 Important Terms 350 Study Questions 351 Problems 352 10.5 Reactions at Nonstandard Conditions: The Nernst Equation 334 10.1 OXIDATION-REDUCTION REACTIONS Many common chemical reactions, such as the acid-base reactions, discussed in Chapter 5, are based on the transfer of a proton (H+) between reactants. Another common type of chemical reactions involves the transfer of electrons between reactants. The branch of chemistry that studies this type of reactions is electrochemistry, the study of chemical processes that cause electrons to move. These chemical reactions that involve the transfer of electrons from one reactant to another are called oxidation-reduction reactions, or redox reactions for short. Historically, the study of these reactions focused on reactions involving oxygen as a reactant, which formed an oxide product. One common example of this type of reaction is the reaction of zinc metal with oxygen to form zinc oxide; 2Zn0 ðsÞ + O2 g ! 2ZnOðsÞ General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00010-2 319 # 2018 Elsevier Inc. All rights reserved. 320 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY In this reaction, the zinc metal is oxidized by the oxidizing species oxygen. This oxidation of zinc involves the transfer of two electrons from the Zn0 to an oxygen atom forming Zn2+ and O2. As the study of oxidation-reduction reactions progressed, they were expanded to include other oxidizing species besides oxygen, and ultimately, it was generalized to include all reactions involving the transfer of electrons between reactants. Oxidation is now defined as the loss of electrons from a reactant in a chemical reaction with an oxidizing agent. The oxidizing agent gains the electrons from the species being oxidized. The process of gaining electrons is called reduction and the species that loses the electrons is the reducing agent. In the reaction of zinc metal with oxygen, the zinc metal loses two electrons and is oxidized. The oxygen gains two electrons and is reduced. So, in this reaction, oxygen is the oxidizing agent and the zinc metal is the reducing agent. Another example of an oxidation-reduction reaction that is commonly used in batteries is the reaction between zinc metal and copper(II) ion; This reaction involves the transfer of two electrons from the zinc metal to the copper(II) ion to give the products Zn2+ ion and copper metal. In this reaction, it is quite clear that the chemical species that is oxidized (Zn0) loses two electrons and the chemical species that is reduced (Cu2 + ) gains those two electrons. Since the copper(II) ion accepts the electrons from the zinc metal, it is the oxidizing agent, and since the zinc metal gives up the electrons to the copper, it is the reducing agent. So, in an oxidation-reduction reaction, the reducing agent is oxidized (loses electrons) and the oxidizing agent is reduced (gains electrons). A summary of these principles is shown in Fig. 10.1. The charges on the species in an oxidation-reduction reaction are called oxidation states, or oxidation numbers, since they tell how many electrons have been gained or lost from the neutral state of the elements. Oxidation states allow the oxidation-reduction reactions to be described in terms of the electrons lost or gained. A positive oxidation state shows the total FIG. 10.1 The concepts of oxidation-reduction as they apply to the reaction of zinc metal with copper(II) ion. 10.1 OXIDATION-REDUCTION REACTIONS 321 number of electrons that have been removed from an atom and a negative oxidation state shows the total number of electrons that have been added to an atom. Oxidation results in an increase in oxidation state and reduction results in a decrease in oxidation state. There are some simple rules for determining the oxidation states of atoms. These are: 1. 2. 3. 4. The oxidation state of all atoms in their elemental form is zero. The oxidation state of a monatomic ion is equal to its charge. The sum of the oxidation states for all atoms in a compound is zero. The sum of the oxidation states for all atoms in a polyatomic ion is equal to the net charge on the ion. 5. The oxidation state of an oxygen atom in a polyatomic molecule is 2 in all compounds except for peroxides, (H2O2, Na2O2, etc.) where the oxidation state is 1. 6. The oxidation state of a hydrogen atom in a polyatomic molecule is +1 for all compounds except metal hydrides where the oxidation state is 1. For simple monatomic ions, the oxidation state is simply the charge associated with the ionic form, as they are listed in Chapter 3. For example, sodium chloride dissolves in water to give Na+ and Cl ions. The oxidation states of the sodium and chloride ions are then +1 and 1, respectively. Similarly, the iron chlorides, Fe(II)Cl2 and Fe(III)Cl3, are made up of the ions Fe2+, Fe3+, and Cl and their oxidation states of the two iron ions are +2 and +3, and the oxidation state of the chloride ion is 1. In polyatomic ions with mixed ionic and covalent bonding, the oxidation states of the atoms can still be determined according to the above rules, even though in reality the covalently bonded electrons are shared between atoms. For example, according to the rules, the oxidation states of the hydrogen and oxygen in the polyatomic ion HPO32 are: H ¼ +1 and O ¼ 2. Since the overall charge on the polyatomic ion is 2, the oxidation state of the phosphorous must be + 3: +1 + (3 2) + 3 ¼ 2. Recall from Chapter 3 that there are a number of groups in the periodic table where all the elements have the same ionic forms. For example, all of the alkali metals in group 1 of the periodic table have the same 1+ ionic form. So, all the alkali metals will have a +1 oxidation state. Similarly, the alkaline earth metals of group 2 all have a 2+ ionic form and they will have a +2 oxidation state. The next to last group (group 17) in the periodic table contains the halogens that all form ions with a 1 charge and they all will have an oxidation state of 1. This periodic trend can be used to determine the oxidation states for ions that cannot otherwise be determined by using the list of rules. EXAMPLE 10.1: DETERMINING OXIDATION STATES Determine the oxidation states of the atoms in the following compounds: (a) NO, (b) NaH, (c) BaO2, (d) CS2, and (e) (NH4)2MoO4. (a) The oxidation states in NO. NO is not a peroxide, so the oxidation state of oxygen is 2. Since NO has no overall charge, the oxidation state of nitrogen has to be +2. (b) The oxidation states in NaH. NaH is a metal hydride, so H will have an oxidation state of 1. Continued 322 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY EXAMPLE 10.1: DETERMINING OXIDATION STATES— CONT’D The net charge is zero, so Na will have an oxidation state of +1. (c) The oxidation states in BaO2. BaO2 is a peroxide, so O will have an oxidation state of 1. The net charge is zero, since there are two oxygens with an oxidation state of 1: 2 1 ¼ 2 and Ba will have an oxidation state of +2. (d) The oxidation states in CS2. This is a case where the rules do not give us a clear place to start. Referring to the periodic table in Chapter 3: S tends to form 2 ions. Since the overall charge is zero, C must have an oxidation state of + 4: (2 2) + 4 ¼ 0 (e) The oxidation states in (NH4)2MoO4. In the NH4+ ion, H has an oxidation state of +1. Since the overall charge on the ion is 1 +, N must have an oxidation state of 3. Since there are two NH4+ ions, the overall charge on the MoO4 must be 2. has an oxidation state of 2 Mo must have an oxidation state of +6 to give the overall charge of 2; (4 2) + 6 ¼ 2 Oxidations states are useful in determining if a chemical reaction is in fact an oxidationreduction reaction. The oxidation states of each atom in the reactants are determined and compared to the oxidation states of the same atoms in the products. If there is a difference in the oxidation states of the atoms in the reactants and the products, then the reaction is an oxidation-reduction process. If there is no difference in the oxidation states of the atoms in the reactants and products, an oxidation-reduction does not take place during the reaction and the chemical reaction is not of the oxidation-reduction type. EXAMPLE 10.2: DETERMINING IF A CHEMICAL REACTION IS THE OXIDATION-REDUCTION TYPE Is the reaction: BrO + NO 2 ! Br + NO3 an oxidation-reduction reaction? If so, how many electrons are transferred? 1. Determine the oxidation states in the reactants. The oxidation state of O in both reactants is 2 The oxidation state of Br must be +1 since the overall charge in BrO is 1 The oxidation state of N must be + 3 since the overall charge in NO2 is 1 . 2. Determine the oxidation states of the products. The oxidation state of Br is the same as the charge on the ion; 1 . In NO3, the oxidation state of O is 2 and the oxygen state of N must be +5 since the overall charge is 1 : (3 2) +5 ¼ 1. 3. Determine if there is a change in oxidation state from reactants to products. Br in the reactants is +1 and in the products is 1. N in the reactants is +3 and in the products is +5. There is a change in the oxidation states of Br and N. 10.2 THE GALVANIC CELL 323 EXAMPLE 10.2: DETERMINING IF A CHEMICAL REACTION IS THE OXIDATION-REDUCTION TYPE— CONT’D This is an oxidation-reduction reaction! 4. Determine the number of electrons transferred. Br goes from a +1 oxidation state to a 1 oxidation state, a gain of two electrons (reduction). N goes from a +3 oxidation state to a +5 oxidation state, a loss of two electrons (oxidation). Two electrons are transferred from the N to the Br during the reaction. 10.2 THE GALVANIC CELL Oxidation-reduction reactions are very common in everyday life. They are the reactions in combustion and explosives, in photosynthesis, in the spoiling of fruit, and in the corrosion of metal surfaces. Oxidation-reduction reactions are also very useful in that they can occur with the reactants separated in physical space as long as they are connected by some medium that allows for the flow of electrons. This flow of electrons through an external medium can be harnessed and used to do work. One of the first scientists to report this phenomenon was Luigi Galvani, who observed that when a zinc metal strip and a copper metal strip were each applied to the opposite ends of a nerve in a frog leg soaked in brine, the leg would move. Not understanding the chemistry, he called this phenomenon “animal electricity.” What was actually happening was an oxidation-reduction reaction between the zinc metal and hydrogen ions in the brine. The frog nerve served as a conduit for the electrons to travel from the zinc strip to the copper strip at the other end causing the muscle to contract. This concept was further developed by Alessandro Volta who, in an attempt to prove that the results of Galvani’s experiment had nothing to do with living tissue, constructed a stack of alternating copper and zinc discs separated by cardboard spacers soaked in salt water. This device, called a voltaic pile, produced a steady flow of electricity and proved that an electrical current could be generated chemically. This discovery paved the way for the development of electrical batteries. Simple devices that derive electrical energy from spontaneous oxidation-reduction reactions are called galvanic cells after Luigi Giovani, sometimes called voltaic cells after Alessandro Volta. An example of a galvanic cell, based on the Zn0/Cu2+ reaction discussed in Section 10.1, is shown in Fig. 10.2. This cell consists of two containers, one with an aqueous solution of zinc sulfate and one with an aqueous solution of copper sulfate. A strip of zinc metal is placed in the container with the zinc ions and a strip of copper metal is placed in the container with the copper ions. Each of the metal strips is known as an electrode, a conductor through which a current enters or leaves a nonmetallic medium in a galvanic cell. The metal electrodes are connected to a voltmeter, which allows the two cells to be in electrical contact with each other while providing a means of measuring the electrical current. Electrons can then flow from the zinc electrode, as it dissolves to form Zn2+(aq) ions, through the voltmeter to the copper electrode where they are taken up by the Cu2+(aq) ions in solution forming Cu0(s). As the Cu0(s) is formed, it is deposited on the Cu electrode. So, as the reaction proceeds, the Zn electrode loses mass and the Cu electrode gains mass. The two containers are connected to each other by a device called a salt bridge, which provides electrical contact between the two aqueous solutions. The salt bridge is filled with an electrolyte, an electrical conducting medium such as KCl(aq) or NH4NO3(aq). 324 FIG. 10.2 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY The design of a simple galvanic cell based on the zinc-copper oxidation-reduction reaction. The electrolyte conducts electricity due to its ability to dissociate into ions in aqueous solution. The electrolyte solution is held in the salt bridge by dissolving it into a gelatinous material and plugging the ends with glass wool. The purpose of the salt bridge is to allow for the movement of ions between the two solutions in order to prevent any net charge build up in either container as the reaction proceeds. As Zn2+(aq) ions are produced in the left container, the overall positive charge increases. Similarly, as Cu2+(aq) ions are removed from the solution in the right container, the overall negative charge increases. If the ionic charges are allowed to continue to increase in each container, the charge difference would prevent the electrons from moving from the left electrode to the right electrode and the reaction would stop. The salt bridge allows for ions to diffuse from one container to the other to keep from accumulating a net charge that would stop the flow of electrons. Each of the two containers in Fig. 10.2 with their electrode and electrolyte solution is called a half cell because it is one half of the galvanic cell. The half cell on the left contains the oxidation reaction and the half cell on the right contains the reduction reaction. Each of these reactions is called a half cell reaction. The two half cell reactions are written as: Zn0 ðsÞ ! Zn2 + ðaqÞ + 2e Cu2 + ðaqÞ + 2e ! Cu0 ðsÞ Adding the two half cell reactions gives the net oxidation-reduction reaction: Zn0 ðsÞ ! Zn2 + ðaqÞ + 2e ðoxidationÞ Cu2 + ðaqÞ + 2e ! Cu0 ðsÞ ðreductionÞ Zn0 ðsÞ + Cu +2 ðaqÞ ! Zn2 + ðaqÞ + Cu0 ðsÞ The two electrodes in each half cell are called the anode and the cathode. The anode is the electrode in the container where the oxidation reaction occurs and the cathode is the electrode in the container where the reduction reaction occurs. The electrical current flows from the anode to the cathode in the galvanic cell. In Fig. 10.2, the anode is the Zn0(s) electrode and the cathode is the Cu0(s) electrode. 325 10.3 BALANCING OXIDATION-REDUCTION EQUATIONS 10.3 BALANCING OXIDATION-REDUCTION EQUATIONS The first step in balancing the chemical equation for an oxidation-reduction reaction is to balance the number of electrons lost in the oxidation reaction with the number of electrons gained in the reduction reaction. For the zinc/copper system discussed in Section 10.2, both oxidation and reduction reactions involve two electrons, so the stoichiometric coefficients of both the copper and zinc species are one. Consider the reaction of copper metal with silver ions forming the products silver metal and copper(II) ions; Cu0 ðsÞ + Ag + ðaqÞ ! Cu2 + ðaqÞ + Ag0 ðsÞ The oxidation state of copper goes from zero to +2 and the oxidation state of the silver goes from +1 to zero. In order to balance the overall chemical equation for this reaction, we must first balance the total transfer of electrons. This is done by balancing the half cell reactions. The reaction of copper metal with silver ions involves the following half cell reactions: Cu0 ðsÞ ! Cu2 + ðaqÞ + 2e ðoxidationÞ Ag + ðaqÞ + 1e ! Ag0 ðsÞ ðreductionÞ Since Cu0(s) loses two electrons and Ag+(aq) accepts only one electron, there needs to be two Ag+(aq) ions for each molecule of Cu0(s). So, the reduction half reaction involving silver must be multiplied by two in order to balance the electron transfer with the oxidation half reaction. 2Ag + ðaqÞ + 2e ! 2Ag0 ðsÞ reduction, gain of 2e Cu0 ðsÞ ! Cu2 + ðaqÞ + 2e ðoxidation, loss of 2e Þ The balanced oxidation-reduction equation then becomes; Cu0 ðsÞ + 2Ag + ðaqÞ ! 2Ag0 ðsÞ + Cu2 + ðaqÞ EXAMPLE 10.3: BALANCING OXIDATION-REDUCTION CHEMICAL EQUATIONS Balance the chemical equation for the reaction of iron metal with Cu+(aq) to form Fe3+(aq) and copper metal. 1. Determine the oxidation and reduction half reactions. Fe0 ! Fe3 + + 3e oxidation Cu + + 1e ! Cu0 reduction 2. Balance the electrons in the half reactions. Fe0 loses three electrons and Cu+ gains one electron. So, the electrons must be balanced by multiplying the reduction half reaction by 3: Continued 326 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY EXAMPLE 10.3: BALANCING OXIDATION-REDUCTION CHEMICAL EQUATIONS— CONT’D Fe0 ! Fe3+ + 3e 3Cu+ + 3e!3Cu0 3. Add the two half reactions. Fe0 ! Fe3 + + 3e + 3Cu + + 3e ! 3Cu0 Fe0 ðsÞ + 3Cu + ðaqÞ + 3e ! Fe3 + ðaqÞ + 3CuðsÞ + 3e The balanced chemical equation for the oxidation-reduction reaction is: Fe0 ðsÞ + 3Cu + ðaqÞ + 3e ! Fe3 + ðaqÞ + 3CuðsÞ + 3e CASE STUDY: THE THERMITE REACTION The oxidation-reduction reaction of a metal oxide with metallic aluminum (Al0(s)), to form alumina (Al2O3(s)), and a molten metal is called the thermite reaction. It was originally discovered in 1893 and patented in 1895 by the chemist Hans Goldschmidt and so it is sometimes called the “Goldschmidt Reaction.” What Goldschmidt discovered was that aluminum metal would react with metal oxides very rapidly, and sometimes violently, when given an initial burst of heat. The reactions were quite exothermic once started and would produce a molten metal that could be used for welding. Fig. 10.3 shows a picture of Hans Goldschmidt and the practical application of the thermite reaction for welding railroad rails together. FIG. 10.3 Hans Goldschmidt (left) inventor of the thermite reaction and the thermite reaction being used to weld railroad rails (right). Photograph by Skatebiker (left), Wikimedia commons. 10.4 STANDARD CELL POTENTIALS 327 Although many metal oxides will react in the thermite reaction, the most commonly used is Fe2O3(s). The reaction of Al0(s) with Fe2O3(s) is usually carried out by mixing the metal oxide and aluminum in powder form and placing them into a ceramic pot or on top of a sand pile to absorb the heat given off by the reaction. This reaction produces molten iron very quickly and since the Al2O3(s) produced is light and floats to the top of the liquid iron, the iron can easily be separated by drawing it off at the bottom of the pot. This method of making pure molten iron quickly had great advantages for producing clean welds. Once ignition temperature is reached, the reactions are extremely exothermic and, in the case of iron oxide, can reach temperatures in excess of 2700 K. Since the reaction makes use of the oxygen in the metal oxide, it does not require oxygen in the air to proceed. This allows it to be used for welding underwater and other anoxic environments. At one time, the thermite reaction was used in chemistry laboratory demonstrations to impress prospective students. This is no longer done since the highly exothermic thermite reaction can be dangerous and presents a potential fire and burn hazard to casual observers. So, why is the thermite reaction classified as an oxidation-reduction reaction? The Fe2O3(s) is reduced to molten iron by reaction with metallic Al0(s), while the aluminum metal is oxidized to Al2O3(s). In order to balance the chemical equation for this reaction, we first need to determine the oxidation states of the iron in Fe2O3(s) and the aluminum in Al3O3. From the oxidation-reduction rules, oxygen has an oxidation state of 2. Since there are three oxygen atoms in both reactant and product molecules, the total oxidation state of oxygen in either compound is 6. So, the two iron atoms in Fe2O3 must have a total oxidation state of +6 to give a net oxidation state of zero. This means that each iron atom has an oxidation state of +3. In the reduction half reaction, the Fe3+ atoms are reduced to metallic iron (Fe0), which requires a gain of three electrons; Fe3 + + 3e ! Fe0 ðlÞ ðreductionÞ In the oxidation half reaction, the total oxidation state of 2Al in Al2O3 must be +6 in order to give a net oxidation state of zero and each Al has an oxidation state of +3. The oxidation half reaction requires a loss of three electrons; Al0 ðsÞ ! Al3 + + 3e ðoxidationÞ The balanced chemical equation for the thermite reaction is; Fe2 O3 ðsÞ + 2Al0 ðsÞ ! Al2 O3 ðsÞ + 2Fe0 ðlÞ which involves the total transfer of six electrons from the Al0(s) to the Fe3+. In this reaction, the Fe2O3(s) is the oxidizing agent and Al0(s) is the reducing agent. 10.4 STANDARD CELL POTENTIALS The flow of electrons from the anode to the cathode in a galvanic cell is driven by the difference in electrical potential energy (measured in volts) between the two electrodes. The electrical current will flow from the electrode with a higher electrical potential energy to the electrode with a lower electrical potential energy. The electrical potential energy 328 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY difference between two half cells of a galvanic cell is called the cell potential. The cell potential depends on the chemical species in the half cells, the concentrations of the solutes, the partial pressures of the gases, and the temperature. Because the cell potential varies with the cell conditions, standard state conditions are defined in order to be able to directly compare the cell potentials of different galvanic cells. A cell potential measured under the standard state conditions is called the standard cell potential (E0). These standard state conditions are: • • • • all reactants and products in their standard states, gases at 1 atmosphere pressure, solutes at 1 M concentration, temperature at 298 K unless otherwise specified. It is impossible to measure the potential of a single half cell since there would be no oxidation-reduction reaction taking place and no flow of electrons. The cell potential can only be measured as a difference in the potential energy between two half cells. In order to determine half cell potentials that can be used for a comparison of different half reactions, one half cell must be chosen to serve as a standard. The half cell that was chosen as the standard against which all other half cells are measured is the standard hydrogen electrode (SHE). The SHE is composed of hydrogen gas at 1 atm, bubbled over an inert platinum metal electrode, submerged in a 1 M acid solution. The SHE half cell is arbitrarily assigned a standard cell potential of 0 V. So, when the potential of a half cell under standard conditions is measured against the SHE, the difference between the potential of the half cell and the potential of the SHE (0 V) is equal to the standard half cell potential of the companion half cell. The SHE can function as either an anode or a cathode by the following half cell reactions: (1) H2 g ! 2e + 2H + g ðanodeÞ (2) 2H + ðaqÞ + 2e ! H2 g ðcathodeÞ depending on the reaction with which the half cell is paired. In a galvanic cell, the electrons always flow from the anode to the cathode and whether a half cell is operating as an anode or a cathode can be identified by determining the direction of the electron flow. This is done by using a voltmeter with the positive terminal connected to the SHE. If the measurement is positive, the SHE acts as the cathode by reaction (1) and the other half cell is the anode. If the reading is negative, the SHE acts as the anode by reaction (2) and the other half cell is the cathode. In Fig. 10.4, the SHE is paired with the copper half cell used in in Fig. 10.2. In both figures, the Cu0(s) electrode is the cathode. The Zn0(s) electrode is the anode in Fig. 10.2, while the SHE is the anode in Fig. 10.4. At the zinc anode, Zn0(s) loses two electrons to form Zn2+(aq) and at the SHE anode, H2(g) loses two electrons to form 2H+(aq). At the copper cathode in both examples, the Cu2+(aq) ions gain two electrons to form Cu0(s) metal. In Fig. 10.4, the electrical current moves from left to right, from oxidation at the SHE anode to reduction at the copper cathode, and the voltage measured is +0.34 V when all chemical species are in standard conditions. This is the standard half cell potential for the Cu0/Cu2+ half cell; 10.4 STANDARD CELL POTENTIALS 329 FIG. 10.4 A simple galvanic cell using a standard hydrogen electrode (SHE) as the anode and a copper electrode as the cathode. Cu2 + ðaqÞ + 2e ! Cu0 ðsÞ E0 ¼ + 0:34 V If the Cu0/Cu2+ half cell Fig. 10.4 is replaced with the Zn0/Zn2+ from Fig. 10.2, the electron flow moves in the opposite direction, from right to left. For this case, the reduction reaction occurs at the SHE, which is now the cathode, and the oxidation reaction occurs at the zinc electrode, which is now the anode. The voltage is measured at 0.76 V when all chemical species are in standard conditions, which is the standard half cell potential for Zn0/Zn2+ half cell; Zn2 + ðaqÞ + 2e ! Zn0 ðsÞ E0 ¼ 0:76 V The standard half cell potentials for a number of half cell reactions can be determined by measuring the voltage of each metal/ion half cell under standard conditions against the SHE. Table 10.1 lists some important standard half cell potentials. A more complete list of half cell potentials is given in Appendix III. Notice that all of the half cell reactions listed are written in the form of the reduction reaction. This is a commonly used convention for reporting half cell potentials. So, the standard half cell potentials given for each half cell reaction represent the potential for the reduction half cell and are called standard reduction potentials. To obtain the standard half cell potential for the oxidation half cell, the sign of the value reported in Table 10.1 or Appendix III must be reversed. The standard half cell potentials then become the standard oxidation potentials. The standard reduction potential and the standard oxidation potential for a half cell are related to each by a factor of 1. The half cell reactions in Table 10.1 are listed in order of increasing reduction potential. The sign of the reduction potentials (+ or ) is the sign of the half cell potential when combined with the SHE. A negative sign indicates that the half cell reaction is an oxidation and a positive sign indicates that the half cell reaction is a reduction when operated with the SHE. All the half cell reactions listed above the SHE have a negative standard reduction potential and all TABLE 10.1 Some standard half cell reduction potentials Half Cell Reaction Oxidizing Agent Reducing Agent E0 (V) Li+(aq) + 1e ! Li0(s) 3.04 2+ ! Ba (s) 2.91 + ! Na (s) 2.71 0 Ba (aq) + 2e 0 Na (aq) + 1e ! Mg (s) 2.37 3+ ! Al (s) 1.66 2+ ! Zn (s) 0.76 3+ ! Cr (s) 0.74 2+ 2+ 0 Mg (aq) + 2e 0 Al (aq) + 3e 0 Zn (aq) + 2e 0 Cr (aq) + 3e ! Fe (s) 0.44 2+ ! Cd (s) 0.40 +2 ! Co (s) 0.28 2+ ! Ni (s) 0.25 2+ ! Sn (s) 0.14 2+ ! Pb (s) 0.13 3+ ! Fe (s) 0.04 + Fe (aq) + 2e 0 0 Cd (aq) + 2e 0 Co (aq) + 2e Ni (aq) + 2e 0 0 Sn (aq) + 2e 0 Pb (aq) + 2e Fe (aq) + 3e 0 ! H2(g) 0.00 (defined) 4+ ! Ge (s) 0.12 4+ ! Sn (aq) 0.15 3+ ! Bi (s) 0.31 2H (aq) + 2e Ge (aq) + 4e 0 2+ Sn (aq) + 2e 0 Bi (aq) + 3e Cu (aq) + 2e ! Cu (s) Fe(CN)63(aq) + 1e ! Fe(CN)6 (aq) 0.36 ! Cu (s) 0.52 2+ 0 + 0 Cu (aq) + 1e I2(g) + 2e 0.34 4 ! 2I (aq) 0.54 ! H2O2(aq) 0.70 3+ ! Tl (s) 0.72 3+ ! Fe (aq) 0.77 + ! Ag (s) 0.80 ! Pd (s) 0.92 Pt (aq) + 2e ! Pt (s) 1.19 Cr2O72(aq) + 14H+(aq) + 6e ! 2Cr (aq) + 7H2O(l) 1.33 + O2(g) + 2H (aq) + 2e 0 Tl (aq) + 3e Fe (aq) + 1e 2+ 0 Ag (aq) + 1e 2+ 0 Pd (aq) + 2e 2+ 0 3+ Cl2(g) + 2e ! 2Cl (aq) 1.36 MnO4(aq) + 8H+(aq) + 5e ! Mn (aq) + 4H2O(l) 1.51 ! Au (s) 1.52 ! Au (s) 1.83 ! O2(g) + 7H2O(l) 2.08 3+ 0 Au (aq) + 3e + Au (aq) + 1e + O3(g) + 2H (aq) + 2e F2(g) + 2e 2+ 0 ! 2F (aq) 2.65 10.4 STANDARD CELL POTENTIALS 331 the half cell reactions listed below the SHE have positive standard reduction potential. The relative values of the standard reduction potentials are an indication of the strength of the oxidizing agents in the half cell reaction. The species with the most positive standard reduction potentials (e.g., F2(g)) are the strongest oxidizing agents. The species with the most negative standard reduction potentials (e.g., Li+) are the weakest oxidizing agents. Put differently, the more negative the standard reduction potential, the less likely the half reaction will proceed as a reduction when paired with a half cell other than the SHE and the more likely that it will occur as an oxidation. Since the standard cell potential for a galvanic cell is the measured difference between the standard potential of the two half cells, it is determined as the difference between the standard reduction potentials of the cathode (E0cathode) and the anode (E0anode); E0 cell ¼ E0 cathode E0 anode (3) For example, the standard cell potential for the zinc/copper galvanic cell shown in Fig. 10.2 will be the difference between the reduction potential of the Cu0/Cu2+ half cell (cathode) and the reduction potential of the Zn0/Zn2+ half cell (anode)as; E0 cell ¼ E0 cathode E0 anode ¼ 0:34 ð0:76Þ ¼ 1:10V For a combination of any two half cell reactions, the half cell that will function as the anode (oxidation) will be the one with the lowest reduction potential. Fig. 10.5 shows the standard reduction potentials of some selected metal/ion half cell pairs from Table 10.1 plotted on a horizontal axis with the SHE at 0 V. For any combination of two half cells, the metal/ion pair that lies to the left on the horizontal axis (more negative value) will always function as the anode and the one that lies to the right will always function as the cathode. The standard cell potential (E0cell) for the pair of half cells will be the positive value of the difference between the half cell reduction potentials. FIG. 10.5 Standard reduction potentials for selected metal/ion pairs compared to the standard hydrogen electrode (SHE) defined as 0 V. 332 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY EXAMPLE 10.4: DETERMINING THE OXIDATION-REDUCTION REACTION AND THE STANDARD CELL POTENTIAL OF A GALVANIC CELL Determine the oxidation-reduction reaction and calculate the standard cell potential in a galvanic cell composed of the aqueous ion/metal electrode pair: Zn/Zn2+ and Ag0/Ag+. 1. Determine which half cell will function as the anode and which will function as the cathode. The reduction half reactions for each half cell are: Zn2+(aq) + 2e ! Zn0(s) standard reduction potential ¼ 0.76 V Ag+(aq) + 1e ! Ag0(s) standard reduction potential ¼ +0.80 V The standard reduction potential of the Zn2+/Zn half cell is more negative than the standard reduction potential of the Ag0/Ag+ half cell. Zinc will be the cathode and silver will be the anode. 2. Balance the half reactions. Zn2+(aq) + 2e ! Zn0(s): gain of two electrons Ag+(aq) ! Ag0(s) + 1e: loss of one electron 2Ag+(aq) ! 2Ag0(s) + 2e: loss of two electrons 3. Write the balanced oxidation-reduction reaction for the galvanic cell. Zn0(s) + 2Ag+(aq) ! Zn2+ + 2Ag0(s) 4. Determine the standard cell potential for the galvanic cell. E0 cell ¼ E0 cathode E0 anode ¼ 0:80V ð0:76VÞ ¼ 1:56V Note that while the silver half reaction is multiplied by 2 to get the balanced oxidation-reduction equation, the standard reduction potential for the half cell reaction is not changed when calculating the potential for the half cell pair. CASE STUDY: PREVENTING CORROSION BY USING SACRIFICIAL MAGNESIUM AND ZINC BLOCKS Corrosion of metal surfaces is a major problem in the building and in shipping industries. Many of the common metals used in structural materials, such as iron and copper, are prone to oxidation reactions that will convert the metals to their oxides when exposed to moist air. Iron metal “rusts” very easily and copper or bronze statues will become covered with a green copper oxide coating over time. The oxidation of these metals involves an oxidation-reduction reaction with oxygen in the presence of water. For example, the oxidation-reduction half reactions for the corrosion of iron are: Fe0 ðsÞ ! Fe2 + ðaqÞ + 2e ðoxidationÞ ðreductionÞ O2 g + 2H2 OðlÞ + 4e ! 4OH ðaqÞ (4) (5) The iron metal is oxidized to iron(II) ions and oxygen from the air is reduced to hydroxide ions in the presence of water. Two iron atoms are oxidized for every one oxygen molecule that is reduced giving the balanced oxidation-reduction equation as; 10.4 STANDARD CELL POTENTIALS 333 2FeðsÞ + O2 g + 2H2 OðlÞ ! 2Fe2 + ðaqÞ + 4OH ðaqÞ The Fe2+ and OH ions combine to form Fe(OH)2(s), which reacts further with oxygen and water to form rust (Fe2O3). The more negative the value of the standard reduction potential for the metal, the stronger the oxidation potential and the more likely the metal will become oxidized. Several methods are available to protect these important structural metals from oxidation. They can be covered with an inert material such as paint, a plastic coating, or a less reactive metal, which isolates the metal from the moist atmosphere and prevents the oxidation-reduction reactions (4) and (5) from occurring. For example, iron is often galvanized to prevent rust. This involves dipping the iron parts in a bath of molten zinc to achieve a protective coating. Another way to prevent the oxidation of important metals is to electrically connect them to a “sacrificial” metal that is more readily oxidized. The metals commonly used for this purpose are those that have a half cell reduction potential with a fairly large negative value. One of these common metals with a large negative standard reduction potential is magnesium. Mg2 + ðaqÞ + 2e ! Mg0 ðsÞ standard reduction potential ¼ 2:37V A block of magnesium is connected to the metal that is to be protected using a joint that conducts electrons. This creates a galvanic cell with the more easily oxidized metal as the anode and the less easily oxidized metal as the cathode. The magnesium anode is oxidized and the electrons generated from the oxidation travel from the magnesium to the new cathode where the reduction reaction (reaction 5) takes place. This process is called cathodic protection since it uses the more easily oxidized metal as an anode, making the protected metal the cathode of a galvanic cell. This prevents oxidation of the protected metal as long as the electron flow is supported. The sacrificial metal continues to lose mass as it oxidizes to the ionic form until eventually it ceases to function and must be replaced. Another metal that is used for cathodic protection is zinc. Zn2 + ðaqÞ + 2e ¼ Zn0 ðsÞ standard reduction potential ¼ 0:76V For the galvanizing process discussed above, the zinc not only protects as a coating by isolating the protected metals from the oxidizing agents in the air, but also functions in a similar manner as a cathodic protector if the coating becomes scratched. However, true cathodic protection requires that the anode is separated from the metal to be protected and connected only by an electrically conductive material to support the flow of electrons. In this arrangement, any area of the structural metal can be protected from oxidation. But, when a metal is galvanized, the anode and cathode are in physical contact. This means that the flow of electrons is localized and only exposed areas of the protected metal that are very close to the zinc coating are protected. Applications of true cathodic protection include: pipelines, storage tanks such as home water heaters, oil platforms, and metal reinforcement bars in concrete structures. Zinc is commonly attached to the undersides of navy ships to protect the steel hulls from oxidation as shown in Fig. 10.6. The bolts used to fasten the bars to the hull serve as the electrical connection between the zinc anode and the steel cathode. Since the ships are regularly removed from the water for inspections and maintenance, it is easy to replace the zinc anodes when they become consumed. 334 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY FIG. 10.6 Large naval vessel using white zinc blocks as anodes for cathodic protection to reduce corrosion of the hull. Photo by US Gov., Wikimedia commons. 10.5 REACTIONS AT NONSTANDARD CONDITIONS: THE NERNST EQUATION The standard reduction potentials and the standard cell potentials calculated from them only hold for half cells under standard conditions. These standard conditions are fixed concentrations and temperatures. But what if a galvanic cell is run under conditions that are not standard? If the concentrations of the aqueous ions in the half cells are something other than 1 M, then the cell potential will change. For the general oxidation-reduction reaction; aA + bB ! cC + dD the observed cell potential at 298.15 K is given by: Ecell ¼ E0 cell 0:059 ½Cc ½Dd log ne ½Aa ½Bb Ecell ¼ E0 cell 0:059 log Q ne (6) where ne is the number of electrons transferred in the oxidation-reduction reaction, Q is the reaction quotient, and E0cell is the standard cell potential. Eq. (6), developed by the German chemist Walther Nernst, is called the Nernst equation. The Nernst equation relates the cell potential of a galvanic cell to the standard cell potential and the concentrations of the chemical species in the oxidation-reduction reaction. 10.5 REACTIONS AT NONSTANDARD CONDITIONS: THE NERNST EQUATION 335 EXAMPLE 10.5: DETERMINING THE POTENTIAL OF A GALVANIC CELL WITH NONSTANDARD CONCENTRATIONS What is the potential of a galvanic cell with the half reactions: Cd2+ + 2e! Cd and Pb + 2e ! Pb, at 25oC with the concentrations of Cd2+ at 0.02 M and Pb2+ at 0.20 M? 2+ 1. Determine the overall cell reaction. Since both half reactions involve the transfer of two electrons, the overall reaction is: Pb2 + ðaqÞ + CdðsÞ ! Cd2 + ðaqÞ + PbðsÞ 2. Determine the cell potential under standard conditions. E0 of Cd2 + ¼ 0:40; E0 of Pb2 + is 0:13: Cadmium will be the anode since it has the lowest reduction potential and lead will be the cathode. The standard cell potential is: E0 cell ¼ E0 cathode E0 anode ¼ 0:13 ð0:40Þ ¼ 0:27 V 3. Determine the cell potential with the nonstandard concentrations. 0:059 0:059 log Q ¼ 0:27 V log ð0:02MÞ=ð0:20MÞ ne 2 Ecell ¼ 0:27 V 0:03 log ð0:10Þ ¼ 0:27 + 0:03 ¼ 0:3 V Ecell ¼ E0 cell Notice that, as with the equilibrium constant expression, contributions from the solid electrodes are not included in the reaction quotient. The potential of a galvanic cell at nonstandard conditions is also dependent on the temperature. The variation of cell potential with temperature is determined by the expanded form of the Nernst equation: Ecell ¼ E0 cell ðRT=ne FÞ ln Q ¼ E0 cell ð2:303 RT=ne FÞ log Q (7) where R is the gas constant (8.314 J/K• mol), T is the temperature, and F is the faraday constant. The Faraday constant is the magnitude of electric charge carried by one mole of electrons. The amount of charge carried by one electron is 1.602 1019 C, so the amount of charge carried by one mole of electrons is: F ¼ 1:602 1019 C=e 6:022 1023 e =mol (8) F ¼ 96 485 C=mol At 298 K, 2.303 RT/F ¼ 0.059 and Eq. (9) reduces to Eq. (6). A galvanic cell that has both half cells with the same composition will have a standard cell potential of zero: E0 cathode ¼ E0 anode ; E0 cell ¼ E0 cathode E0 anode ¼ 0. This only holds for standard conditions of 1 M concentrations. The Nernst equation predicts that if the concentrations of 336 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY the ions are different in the two half cells (Q 6¼ 0), there will still be a voltage generated (Ecell 6¼ 0). Ecell ¼ 0:059 log Q (9) This is due to the driving force of the oxidation-reduction reaction trying to reach equilibrium. The same reaction occurs in both half cells, but in opposite directions. The cell with the lower concentration becomes the anode, increasing the concentration of the ions, and the cell with the higher concentration becomes the cathode, decreasing the concentration of the ions. This type of cell system is called a concentration cell, a form of galvanic cell where both half cells have the same composition with differing concentrations. But, since an order of magnitude concentration difference in the half cells only produces less than 60 mV, these cells are not typically used to generate electrical energy. Instead, concentration cells are more commonly used in chemical methods of analysis. Because the cell potential is determined by a difference in concentration of one chemical species, the measurement of cell potential can be used to determine the concentration of a chemical species in one half cell if the concentration in the other half cell is known. The concentration of the unknown solution is then determined from the Nernst equation, the standard cell potential (E0cell), and the measured cell potential (Ecell). EXAMPLE 10.6: DETERMINING THE CELL POTENTIAL OF A CONCENTRATION CELL What is the cell potential of a galvanic cell composed of two Ag0/Ag+ half cells, one with a silver ion concentration of 0.01 M and the other with a silver ion concentration of 1 M. 1. Determine the oxidation-reduction reaction. The cell with the lower concentration (0.01 M) becomes the anode: Ag0 ðsÞ ! Ag + ðaqÞ ½0:01M + 1e ðanodeÞ The cell with the higher concentration becomes the cathode: Ag+(aq) [1.0 M] + 1e ! Ag0(s) (cathode) The chemical equation for the oxidation-reduction reaction is: Ag0(s) + Ag+[1.0 M] ! Ag+[0.01 M] + Ag0(s) 2. Determine the cell potential. E¼ 0:059 log Q ne where: E0 ¼ 0, ne ¼ 1, Q ¼ [0.01 M]/[1.0 M] E ¼ E0 0:059 log Ag + 0:01M = Ag + 1:0M ¼ 0 V ½ð0:059Þð2Þ ¼ 0:118 V 10.5 REACTIONS AT NONSTANDARD CONDITIONS: THE NERNST EQUATION 337 CASE STUDY: THE PH METER Solution pH can be determined by the color of a chemical indicator, as described in Chapter 7. But, these measurements are only approximate determinations of solution pH and a more quantitative method is required when more accurate measurements of pH are needed. Solution pH can be determined accurately by using the principles of a concentration cell based on the H2(g)/H+(aq) half reactions. The potential generated between two half cells with different H+(aq) concentrations is measured to determine the difference in the H+(aq) concentrations electrochemically. This was originally accomplished by using two hydrogen electrodes as shown in Fig. 10.7. Both electrodes were constructed like the SHE, but one was submerged in a solution of unknown pH (the sensing electrode) and the second was submerged in a solution of 1 M H+(aq) (the reference electrode). Since all the unknown solutions will have a hydrogen ion concentration less than 1 M, the sensing electrode functions as the cathode and the reference electrode functions as the anode. 2H + ðaqÞ ½1M + 2e ! H2 g ðanodeÞ H2 g ! 2H + ðaqÞ ½?M ðcathodeÞ 2H + ðaqÞ ½1M ! 2H + ðaqÞ ½?M ðoxidation reductionÞ The expression for the Nernst equation for this cell is; 0:059 log Q ne 0:059 ½H + 2 0:059 log ½H + ¼ log 2 ¼0 1:0M 2 2 1 Ecell ¼ 0:059 pH Ecell pH ¼ 0:059 Ecell ¼ E0 cell (10) So, the measurement of the cell potential with this apparatus gives a measurement of the pH of the unknown solution directly! FIG. 10.7 solution pH. A hydrogen ion concentration cell constructed of two hydrogen ion electrodes for the measurement of 338 FIG. 10.8 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY The glass electrode and reference electrode used for the measurement of pH in a modern pH meter. Although the hydrogen ion concentration cell has many advantages for the direct measurement of solution pH, it is expensive and difficult to set up and operate. Because of these difficulties, the hydrogen electrode is used only rarely today. The modern pH meter is a special type of concentration cell that uses a glass sensor electrode coupled with a reference electrode as shown in Fig. 10.8. The glass electrode is composed of a silver wire coated with solid silver chloride and submerged in a 1.0 M HCl solution. The tip of the electrode is made of a very thin semipermeable glass membrane that is sensitive to differences in H+ concentrations. The reference electrode is a silver-silver chloride wire in a saturated KCl solution. The two electrodes are both placed in the unknown solution and connected by a salt bridge. The thin membrane at the tip of the glass electrode binds H+ from both sides of the membrane, resulting in a H+ concentration differential across the membrane. Both sides of the glass are charged by the adsorbed protons and the difference in the charge across the glass membrane creates a potential difference. The difference in potential across the glass membrane is measured by the potential difference between the reference and indicator electrodes. This potential is described by the Nernst equation and is directly proportional to the pH difference between the solutions on both sides of the glass membrane. Ecell ¼ E0 cell + 0:059 log ½H + inside ½H + outside ¼ E0 cell + 0:059 pHoutside pHinside ¼ E0 cell + 0:059pHoutside 10.6 ELECTROLYSIS 339 The principles behind the operation of the concentration cell are important in Engineering because they can control corrosion. Concentration cell corrosion occurs when two or more areas of a metal surface are in contact with different concentrations of the same ions or gases. The two areas of the metal will then be at different electrical potentials due to the differences in concentration, forming a concentration cell. Concentration cell corrosion is one of the main forms of localized corrosion causing many structural failures. It occurs most often in critical applications, such as with pipes buried in soil where the properties of the soil vary. Concentration cell responsible for corrosion falls into two major categories; ion concentration cells and oxygen concentration cells. Ion concentration cell corrosion occurs when one part of a metal surface is exposed to a lower concentration of ions than other parts of the surface. The area exposed to the lower concentration functions as an anode and the area exposed to higher concentrations functions as a cathode. So, the part of the metal surface that is exposed to the lower concentrations of ions corrodes faster in an attempt to increase the local ion concentration. The difference in speed of the corrosion is proportional to the difference in potential of the metal surfaces, which is proportional to the differences in ion concentrations surrounding the metal. Oxygen concentration cells occur when different areas of a metal surface are exposed to different oxygen levels. Although corrosion occurs whenever oxygen has access to a damp metal surface, the corrosion process occurs more rapidly in areas where the concentration of oxygen is the lowest. This is because in the areas of higher oxygen concentrations the equilibrium in Eq. (5) will lie to the right decreasing the concentration of oxygen and increasing the concentration of hydroxide ion, while the areas of the metal exposed to smaller oxygen concentrations will increase corrosion according to Eq. (4). When a metal structure is partially covered with dirt, the subsurface area will corrode faster than the area exposed to open air because of the difference in oxygen concentrations. For the same reasons, metal parts partly submerged in water will corrode faster below the water level than above. Also, because the rate of diffusion of air into water may produce a decreasing oxygen concentration with depth, corrosion of underwater metal structures will occur faster in the oxygen-depleted areas at lower depths. This is especially a problem for offshore structures in the Gulf of Mexico, which suffers from a very large oxygen-depleted “dead zone” in the summer caused by pollution runoff. 10.6 ELECTROLYSIS So far we have only considered oxidation-reduction reactions that occur spontaneously to generate an electric current. The opposite process, which uses an external electric current to drive a nonspontaneous oxidation-reduction reaction, is called electrolysis. An electrolytic cell is very similar to a galvanic cell. Both have a cathode, an anode, an electrolyte, and consistent flow of electrons from the anode to the cathode. However, where a galvanic cell converts chemical energy to electrical energy, an electrolytic cell converts electrical energy into chemical energy. Electrolysis is essentially the exchange of atoms and ions by the removal or addition of electrons from the external electrical circuit. To drive the reaction, the potential applied from 340 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY the external source must be higher than the largest potential of the half reactions. Electrolysis is carried out on either a molten salt or on an aqueous solution because the sample must be in the form of ions in order for an electric current to flow. The products of electrolysis are often in a different physical state than the reactants and can be removed by physical separation processes. Consider an electrolysis cell consisting of two silver electrodes placed into a solution of silver nitrate and connected to the opposite sides of a voltage source. Even though both half cells have the same cell potential, the electrical current that is passed through the solution forces oxidation to occur at one electrode, the anode, and reduction to occur at the other electrode, the cathode. Ag0 ðsÞ ! Ag + + 1e anodeoxidation Ag + + 1e ! Ag0 ðsÞ cathodereduction As the reaction proceeds, the silver anode dissolves causing an increase in Ag+ ions in the solution. At the same time, Ag0 is deposited at the cathode decreasing the concentration of Ag+ ions in solution and increasing the mass of the cathode. If this electrolysis cell was constructed with a silver anode and a copper strip as the cathode, as shown in Fig. 10.9, the silver would be deposited on the copper strip causing a silver coating to form. The electrode potential required for this to happen must be higher than 0.80 V, the standard reduction potential of silver. In 1883, Michael Faraday proposed two laws of electrolysis that predicted the behavior of electrolytic reactions in a quantitative manner; 1. The mass of a substance produced by electrolysis is directly proportional to the quantity of electricity used. 2. When the same quantity of electricity is used to produce different substances, the mass of substance produced is directly proportional to the equivalent weights of the substances. FIG. 10.9 An electrolytic cell used for electroplating silver on copper. The anode is silver metal and the cathode is a copper strip. During the process, the copper strip becomes coated with a layer of silver. 10.6 ELECTROLYSIS 341 The first law means that if a current (I) is applied to an electrolytic cell for a specific amount of time (t), the mass of substance produced (m) is equal to; m ¼ kðI ÞðtÞ ¼ k q where “k” is a proportionality constant and q ¼ I t is the total charge passed in the system. According to the second law, the mass of the substance is also proportional to the atomic mass of the substance “ma.” Combining the two laws gives; m ¼ ðI ÞðtÞðma Þ (11) ne F where ne is the number of moles of electrons transferred in the electrolytic reaction, and the proportionality constant becomes the Faraday constant F ¼ (96,485 C/mol) described in Section 10.5. Although the SI unit of charge is the coulomb, the faraday (F) is also a unit of charge that is often used in electrochemistry. It is related to the Faraday constant and is equal to 96,485 C. In electroplating, the faraday is equal to the number of moles of electrons (ne) required to produce 1 mol of metal in an electrolysis cell times the number of moles of metal produced; Charge in faradays ¼ ðne Þ ðmoles of metalÞ EXAMPLE 10.7: DETERMINING THE AMOUNT OF CHARGE NEEDED TO DEPOSIT A GIVEN MASS OF METAL Determine the amount of charge in faradays required to electroplate 5.0 g of copper onto a metal key using an aqueous solution of Cu(NO3)2 and a Cu electrode. How many minutes will it take to complete the process if the current is 1 A? 1. Determine the number of moles of electrons required to produce one mole of copper. Cu0 ðsÞ ! Cu2 + + 2e oxidation Cu2 + ðaqÞ + 2e ! Cu0 ðsÞ reduction 2 moles of electrons are required to produce 1 mole of Cu. 2. Determine the number of moles of copper produced. atomic weight ¼ 63.54 g mol1 5 g Cu/63.54 ¼ 0.079 mol Cu 3. Determine the charge in faradays. F ¼ ne moles Cu ¼ 2 0:079 mol ¼ 0:158F 4. Determine the time required in minutes. q ¼ I t; where I is measured in amperes, t is measured in seconds, and q is measured in coulombs. Convert q to coulombs using the Faraday constant: q ¼ 0.158 F 96,485 C/F ¼ 15,345 C. t ¼ q/I ¼ 15,345 C/1 A ¼ 15,345 s ¼ 15,345/60 min ¼ 255.8 min 342 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY Electrolysis is very important in industry for refining metals such as copper and aluminum. It is also commonly used in electroplating to produce a thin coating of one metal on top of another, such as plating gold on a silver or brass base to reduce cost for the manufacture of jewelry. It also is used for plating parts of vehicles with nickel and chromium to protect them from corrosion. Silver plating of electrical parts made of brass or copper to improve electrical connections while reducing cost is also a common practice because silver is a better electrical conductor than the base metals. 10.7 BATTERIES A battery is an electrical device consisting of one or more galvanic cells, with external connections provided to allow the battery to power electrical devices. Batteries, like galvanic cells, are composed of an anode, a cathode, and an electrolyte, which provide a conductive medium for ions and electrons to move between the electrodes. When the electrolyte is a liquid, usually an aqueous salt, the battery is called a wet cell and when it is in the form of a paste or a solid the battery is called a dry cell. Each cell has an electromotive force (emf), equal to the cell potential, which is a measure of its ability to drive the electric current from the interior of the cell to the exterior of the cell. The net emf of the cell (εcell) is the equal to the difference between the emfs (or reduction potentials) of the half cells (ε2 ε1). So: εcell ¼ ε2 ε1 ¼ E0 cathode E0 anode ¼ Ecell : Batteries are classified into two categories: primary batteries and secondary batteries. Primary batteries cannot be returned to their original state by recharging. Secondary batteries, or rechargeable batteries, can be returned to their original state by the application of an external electric current, which reverses the oxidation-reduction half cell reactions. The first primary battery was developed in 1866 by Georges Leclanche and is called a Leclanche cell after its inventor. A diagram of the original wet cell design of the Leclanche cell is shown in Fig. 10.10. The cell uses a zinc anode and a cathode made up of a mixture of carbon Carbon and MnO2 cathode Zinc anode NH4Cl electrolyte AR SJ AS GL FIG. 10.10 The original design of the Lelanche battery (left) and the modern version of the zinc-carbon battery (right). Modified from Mcy jerry, Wikimedia Commons. 10.7 BATTERIES 343 and manganese dioxide wrapped in a porous material. The electrolyte is an aqueous solution of ammonium chloride. The reactions occurring at the two electrodes in the Leclanche cell are: Zn0 ðsÞ ! Zn2 + ðaqÞ + 2e ðanodeÞ 2NH4 + ðaqÞ + 2e ! 2NH3 g + H2 g ðcathodeÞ The zinc is oxidized at the anode to Zn2+ and the ammonium ion in the electrolyte is reduced at the cathode to form ammonia and hydrogen gases. These gases must be absorbed in order to avoid an increase in gas pressure in the sealed cell. Two further reactions accomplish this: ammonia reacts with zinc and chloride ions in the electrolyte to form solid zinc ammonium chloride and hydrogen reacts with manganese dioxide in the cathode to form solid dimanganese trioxide and water: 2NH3 g + ZnCl2 ðaqÞ ! ZnðNH3 Þ2 Cl2 ðsÞ 2MnO2 ðsÞ + H2 g ! Mn2 O3 ðsÞ + H2 OðlÞ The Lelanche battery was later modified to function as a dry cell battery by substituting an ammonium chloride paste for the aqueous ammonium chloride solution. This form is still used today in the modern zinc-carbon battery shown in Fig. 10.10. The zinc can that contains the battery components also functions as the anode and the carbon-MnO2 cathode is wrapped around a central carbon core in the modern design. The oxidation-reduction reactions remain the same as in the original Lelanche cell. While the Lelanche battery was used in early telegraph operations and electric bells, modern zinc-carbon batteries today are used in wall clocks, remote controllers, and other devices that require a small electrical charge over a long period of time. A similar type of primary dry cell battery is the zinc-alkaline battery. The battery is still composed of a zinc anode and a MnO2 cathode, but the electrolyte is potassium hydroxide. The reaction at the anode is the oxidation of zinc metal to zinc(II) ion, which then reacts with the hydroxide ion in the electrolyte to form zinc oxide and water. The reaction at the cathode in the presence of the potassium hydroxide electrolyte is now the reduction of Mn4+ to Mn3+, which reacts with water to yield manganese(III) oxide and the reformation of the hydroxide ion. Zn0 ðsÞ + 2OH ðaqÞ ! ZnOðsÞ + H2 OðlÞ + 2e ðanodeÞ 2MnO2 ðsÞ + H2 OðlÞ + 2e ! Mn2 O3 ðsÞ + 2OH ðaqÞ ðcathodeÞ The capacity of a zinc-alkaline battery is three to five times greater than the zinc-carbon battery because of the construction. The MnO2 is purer and denser and the space taken up by the internal components including the electrodes and the electrolyte is less than the zinc-carbon battery resulting in a smaller battery design. Zinc-alkaline batteries are found in all common AAA, AA, C, and D size cells. Some zinc-alkaline batteries are designed to be recharged and thus sometimes cause rupture of the battery case and leaking of the electrolyte, which can cause corrosion of equipment. Nevertheless, attempts at recharging zinc-alkaline batteries a limited number of times (10 or less) have been successful and 344 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY chargers are commercially available. Still, since their materials are inexpensive, most of these batteries are viewed as disposable and they are not usually recycled after use. One of the first commercially available secondary batteries is the lead-acid battery, which is still in use today. This battery, invented by Gaston Plante in 1859, is the oldest type of wet cell battery. A single cell, shown in Fig. 10.11A, consists of a lead anode, a lead dioxide cathode, and a sulfuric acid electrolyte. An insulating separator between the anode and cathode prevents short circuiting through physical contact of the electrodes, which would be most commonly caused by the accumulation of solids in the electrolyte. Although a single cell produces only about 2.1 V, common lead-acid batteries used to start engines are constructed of six single cells in series (Fig. 10.11B), producing a fully charged output voltage of 12.6 V. As the battery is discharged the lead anode is oxidized to Pb2+, which combines with the SO2 4 in the electrolyte forming solid lead sulfate. The lead sulfate then precipitates out on the anode surface. The two electrons produced in the oxidation reaction move through the electrolyte to the cathode where the lead dioxide is reduced to Pb2+ ions, which also form PbSO4(s) that precipitates onto the cathode surface. Pb0 ðsÞ + HSO4 ðaqÞ ! PbSO4 ðsÞ + H + ðaqÞ + 2e ðanodeÞ PbO2 ðsÞ + HSO4 ðaqÞ + 3H + ðaqÞ + 2e ! PbSO4 ðsÞ + 2H2 OðlÞ ðcathodeÞ The lead-acid battery can be recharged by passing an electric current through it to return the half cells to their original state. During the recharge, the PbSO4(s) coating on the electrodes is converted back to Pb0 and PbO2(s) by the reverse reaction; 2PbSO4 ðsÞ + 2H2 OðlÞ ! Pb0 ðsÞ + PbO2 ðsÞ + 2H + ðaqÞ + 2H2 SO4 ðaqÞ In an automobile, the battery produces a large initial current to start the car engine, discharging the battery. The battery is recharged during normal driving by an alternator that FIG. 10.11 A single cell of a lead-acid battery (A) and 6 cells connected in parallel (B) to form the common 12 V battery used to start engines. 10.7 BATTERIES 345 drives an electric current through the battery terminals. The recharging process is possible as long as the PbSO4(s) remains coated on the electrode surfaces. After time, the coatings will flake off the electrodes and fall to the bottom of the battery case. At this point the current forced through the battery by the alternator cannot reach the solid PbSO4 to return it to its original forms and the battery cannot be recharged. One issue with the lead-acid battery is that, as the battery is discharged, H2SO4 is consumed at both electrodes. This decreases the sulfuric acid concentration of the electrolyte and reduces the cell potential. Also, overcharging the battery will cause the water to undergo electrolysis producing hydrogen and oxygen gases, which leads to a decrease in water content and an increase in acid concentration causing an increase in electrode corrosion. Older leadacid batteries provided ports for the addition of water to compensate for any loss through electrolysis. Newer sealed batteries may suffer from bulging of the case due to the gas buildup. Other drawbacks to this type of battery are that they are large and heavy and the toxic lead is an environmental hazard. Because of the high toxicity of the lead components, lead-acid batteries are required to be recycled at a certified recycling facility. Another secondary rechargeable battery, invented by Waldemer Jungner in 1899, is the nickel-cadmium (NiCad) battery. This battery is composed of a nickel oxide hydroxide cathode, a cadmium metal anode, and a potassium hydroxide electrolyte. During discharge, the cadmium is oxidized to Cd2+ at the anode and Ni3+ is reduced to Ni+ at the cathode. Cd0 ðsÞ + 2OH ðaqÞ ! CdðOHÞ2 ðsÞ + 2e ðanodeÞ 2NiOðOHÞðsÞ + 2H2 OðlÞ + 2e ! 2NiðOHÞ2 ðsÞ + 2OH ðaqÞ ðcathodeÞ Nickel-cadmium rechargeable batteries produce a nearly constant voltage at 1.2 V and were once used widely in portable devices, but the high cost and disposal restrictions because of the high toxicity of cadmium have reduced their use. Other batteries, such as the rechargeable nickel-metal hydride or lithium-ion batteries, have replaced them because of the toxicity and environmental problems related to cadmium. In the nickel-metal hydride battery (NiMH), the nickel oxide hydroxide becomes the anode and the cathode is constructed of a hydrogen-absorbing metal alloy composed of a mixture of metals with the stoichiometric formulas AB2 or AB5. The most common composition is the AB5, where A is actually a mixture of rare earth metals and B is nickel, cobalt, manganese, or aluminum. The AB2 form is a higher capacity cathode where A is titanium or vanadium and B is zirconium or nickel. Whatever the stoichiometric form they all serve the same purpose, the reversible formation of a metal hydride (MH). NiðOHÞ2 ðsÞ + OH ðaqÞ ! NiOðOHÞðsÞ + H2 OðlÞ + e ðanodeÞ H2 OðlÞ + MðsÞ + e ! OH + H2 OðlÞ + MH ðcathodeÞ This construction results in a rechargeable battery with all the advantages of the nickelcadmium battery without the high cost and toxicity of the cadmium. The lithium-ion battery was first marketed in 1991 and is now the most commonly used rechargeable battery for many applications from laptop computers to electric cars. The lithium-ion battery is constructed with an anode made of a mixture of graphitic carbon and lithium and a cathode of a transition metal oxide, usually cobalt oxide. Since lithium 346 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY is very easily oxidized with an oxidation potential of 3.04, lithium reacts readily with water. Because of this, the lithium-ion battery cannot use an aqueous electrolyte. Instead, the electrolyte is a lithium salt in an organic solvent. A porous membrane separator prevents short circuiting while allowing the flow of ions. The structure of both electrodes is in the form of layered plates and the mechanism for the oxidation-reduction reaction involves the intercalation of lithium between the plates of the electrodes as shown in Fig. 10.12. Intercalation is the reversible insertion of an ion, atom, or molecule into a chemical species with a layered structure. During discharge, the lithium ions move from the open space between the plates of the graphitic carbon electrode through the electrolyte into the open spaces between the plates of the cobalt oxide electrodes. During the recharging cycle, the lithium ions move in the reverse direction back into the carbon anode. The half reactions of the lithium-ion battery are; LiC6 ðsÞ ! C6 ðsÞ + Li + + e ðanodeÞ CoO2 ðsÞ + Li + + e ! LiCoO2 ðsÞ ðcathodeÞ where C6 indicates the structure of graphite, which will be further discussed in Chapter 11. The lithium is oxidized at the anode to lithium ion, which then travels through the electrolyte to the cathode. The cobalt is reduced at the cathode (Co4+ to Co5+) and combines with the lithium ion to form the mixed metal oxide. During the recharging cycle, cobalt is oxidized back from Co5+ to Co4+ releasing the lithium ions. Lithium-ion batteries provide lightweight, high energy density power sources for a variety of devices. To power larger devices, such as electric cars, many cells must be connected in a FIG. 10.12 The construction of a lithium-ion battery where the lithium ions (blue circles) reside between the plates of a carbon graphite electrode. During discharge, they move from inside the carbon anode through the organic electrolyte and into layers in the CoO2 cathode, passing through a porous membrane separator. 10.8 FUEL CELLS 347 parallel circuit. While a mobile phone uses a single cell battery, an 85 kW battery in an electric car uses 7104 lithium-ion cells. Battery lifetimes typically consist of 1000 charge-recharge cycles. Some batteries, constructed with a lithium titanium oxide anode instead of lithiumcarbon, have resulted in more than 4000 recharge cycles. Lithium-ion batteries are generally categorized as nontoxic waste since the components are less toxic than other types of batteries. These components, which include iron, copper, nickel, cobalt, and carbon, are considered safe for disposal in both incinerators and landfills and so the batteries can be discarded in household waste. 10.8 FUEL CELLS A fuel cell is an electrochemical device that produces a continuous electrical current from the chemical oxidation of positively charged hydrogen ions with an oxidant, usually oxygen. The general structure of a hydrogen-oxygen fuel cell is shown in Fig. 10.13. The hydrogen ions are produced at the anode from hydrogen gas or another hydrogen containing fuel. The hydrogen ions then travel through the electrolyte to the cathode where they react with oxygen to form water. Fuel cells are another application of oxidation-reduction reactions, but they are different from batteries in that they require a continuous source of fuel and oxygen to sustain the chemical reaction. Batteries, on the other hand, derive their electric current from the materials inside the battery. Fuel cells can produce electricity continuously as long as the reactants are supplied while batteries cease to function once the reactive component of the anode is depleted. The main difference between the different types of fuel cells is the type of electrolyte used and they are classified according to the electrolyte system. For example, a proton exchange FIG. 10.13 Schematic diagram of a hydrogen-oxygen gas fuel cell. Modified from R. Dervisoglu, Wikimedia Commons. 348 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY membrane fuel cell (PEMFC) uses a proton exchange membrane electrolyte, which is a hydrated polymeric proton exchange membrane (PEM). It is a specially treated material that enables it to conduct positively charged ions, while blocking the flow of electrons. The best known of these membranes is called Nafion, a porous organic complex polymer. These types of polymers will be discussed further in Chapter 13. Another type of fuel cell uses phosphoric acid as the electrolyte. Because it is an acid, phosphoric acid will transport the H+ ions instead of electrons. This device is called a phosphoric acid fuel cell (PAFC). Solid acid fuel cells (SAFCs) have also been developed that use CsHSO4(s) and CsH2PO4(s) as electrolytes. Other fuel cells include the alkaline fuel cell (AFC) that uses a concentrated NaOH or KOH electrolyte. The AFCs use porous carbon electrodes impregnated with a catalyst, usually a Pt metal, to assist in the decomposition of hydrogen. The half reactions of the hydrogen-oxygen fuel cell are: 2H2 g ! 4H + + 4e ðanodeÞ O2 g + 4H + + 4e ! 2H2 O g ðcathodeÞ When the H2 contacts the anode and is oxidized to form H+ and electrons, the protons go directly through to the membrane to the cathode. Since the electrons cannot pass through the electrolyte, they must travel through an external circuit to the cathode. Along the way, they are used to perform useful work, like lighting a bulb or driving a motor. The oxygen, usually from air, enters at the cathode where it reacts with H+ ions and electrons supplied by the cathode to produce water as a waste product. The water exits the cell with the unused gases. One important fuel cell system that has been developed for high temperature applications makes use of a solid oxide ceramic electrolyte, called a solid oxide fuel cell (SOFC). The solid electrolyte is different from all the other electrolytes in use as it is capable of transporting negative oxygen ions from the cathode to the anode while blocking the flow of electrons. The chemical reactions at the anode and cathode of an SOFC are: 2H2 g + 2O2 ! 2H2 O g + 4e ðanodeÞ O2 g + 4e ! 2O2 ðcathodeÞ Since the cathode in the SOFC generates the O2 charge carrier, the ion mobility in the fuel cell runs in the opposite direction of other fuel cells, as shown in Fig. 10.14. The ceramics used in SOFCs do not become ionically active until they reach very high temperatures of 1100–1400 K. These high operating temperatures result in extremely high electrical efficiencies (>60%). The high temperatures are also capable of converting other fuels into hydrogen gas, so the solid oxide fuel cell has the potential of using a variety of hydrocarbons other than hydrogen gas as a primary fuel. The advantages of high temperature operation contribute to better economics, but the very high temperatures also create more engineering challenges. While the SOFCs have been used for many applications, including powering satellites, a single cell only produces a voltage of about 0.7 V, so many applications require cells to be stacked or placed in series to generate sufficient voltage. Since SOFC technology has no moving parts or corrosive liquid electrolytes, they are expected to lead to stationary electricity generation systems that are highly reliable and require low maintenance. The SOFC exhaust 10.8 FUEL CELLS 349 FIG. 10.14 Schematic diagram of a solid oxide fuel cell (SOFC). Modified from Sakurambo, Wikimedia Commons. gases exit the cell at a temperature between 800oC and 850oC and this heat could be recovered to increase energy efficiency for a total system efficiency of up to 85%. A hybrid of both fuel cell and battery concepts is the zinc-air battery. These primary batteries are composed of a zinc anode, an aqueous KOH electrolyte, and a moist porous carbon cathode that has access to air through a hole in the battery. Atmospheric oxygen is reduced at the cathode, but is not contained in the electrode. Instead, it is taken up from the surrounding air as it is needed. This battery is like a fuel cell in that it makes use of O2(g) in the air for operation. It is also like a battery in that it is limited in its lifetime by the amount of zinc metal in the anode. The chemical reactions involved in the zinc-air battery cell are: Zn0 ðsÞ + 4OH ! ZnðOHÞ4 2 + 2e ðanodeÞ ZnðOHÞ4 2 ! ZnOðsÞ + H2 OðlÞ + 2OH O2 g + H2 OðlÞ + 4e ! 4OH ðcathodeÞ Oxygen from the air reacts at the cathode with water already present in the porous cathode producing hydroxide ions, which migrate to the anode. Zinc is oxidized to Zn2+ ions at the anode releasing electrons that travel to the cathode and the zinc ions combine with the hydroxide ions to form the complex ion Zn(OH)42. The tetrahydroxozincate ion decomposes to zinc oxide, water, and hydroxide ion. The zinc oxide is an insoluble and irreversible product that limits the life of the battery. The zinc-air battery produces 1.35–1.4 V in most available cells and is used in a number of common applications including “button” batteries used in watches and hearing aids. Larger battery styles constructed to optimize the amounts of zinc and air input are used as cylindrical 350 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY cells for safety lamps at road and rail construction sites or as power sources for electric fences. When sealed, the battery has an excellent shelf life with a self discharge rate of only two percent per year. Zinc-air batteries also store more energy per gram than any other primary battery. However, they are sensitive to extreme temperatures, humidity, and carbon dioxide concentrations. High levels of carbon dioxide can cause the formation of carbonates which reduces electrical conductivity. IMPORTANT TERMS Anode the electrode in a galvanic cell where the oxidation reaction occurs. Battery an electrical device consisting of one or more galvanic cells, with external connections provided to allow the battery to power electrical devices. Cathode the electrode in a galvanic cell where the reduction reaction occurs. Cathodic protection a method of protecting structural metal components by using a more easily oxidized metal as an anode, making the protected metal as the cathode of a galvanic cell. Cell potential the electrical potential energy difference between two half cells of a galvanic cell. Concentration cell a form of galvanic cell where both half cells have the same composition with differing concentrations. Dry cell a battery that uses an electrolyte in the form of a paste or a solid. Electrochemistry the study of chemical processes that cause electrons to move. Electrode a solid conductor through which an electrical current enters or leaves a galvanic cell. Electrolysis a process which uses an external electrical current to drive a nonspontaneous oxidation-reduction reaction Electrolyte a substance that can conduct electricity due to its ability to dissociate into ions when dissolved. Electromotive force (emf) a measure of the ability of a half cell to drive the electric current from the interior to the exterior of the cell. Electroplating the application of electrolysis to produce a thin coating of one metal on top of another Faraday constant (F) the amount of electric charge produced by one mole of electrons. Fuel cell an electrochemical device that produces a continuous electrical current from the chemical oxidation of a fuel with oxygen or another oxidizing agent. Galvanic cell a device that derives electrical energy from spontaneous oxidation-reduction reactions. Half cell one half of a galvanic cell consisting of an electrode and an electrolyte. Intercalation the reversible insertion of an ion or molecule into a chemical species with a layered structure. Nernst equation a mathematical equation that relates the cell potential of a galvanic cell to the standard cell potential and the concentrations of the chemical species in the oxidation-reduction reaction. Oxidation the loss of electrons from a reactant in an oxidation-reduction reaction. Oxidation state a number assigned to an atom that represents the number of electrons lost or gained by that atom. Oxidizing agent the reactant in an oxidation-reduction reaction that accepts electrons from a reactant being oxidized. Oxidation-reduction (redox) a chemical reaction that involves the loss of electrons by one reactant and the gain of electrons by another reactant. Primary battery a battery that cannot be returned to its original state by recharging Reducing agent the reactant in an oxidation-reduction reaction that loses electrons. Reduction the gain of electrons by a reactant in an oxidation-reduction reaction. Salt bridge a device used to connect the half cells of a galvanic cell in order to provide electrical contact between the two cells. Secondary battery a battery that can be returned to its original state by the application of an external electric current that reverses the oxidation-reduction reactions. Standard cell potential (E0) a cell potential measured under standard state conditions. STUDY QUESTIONS 351 Standard half cell potential the difference between the potential of the half cell and the potential of the standard hydrogen electrode. Standard hydrogen electrode (SHE) the half cell that is used as a reference, with an assigned potential of 0 V, against which all other half cell potentials are measured. Standard oxidation potentials the standard half cell potential for the oxidation half cell. Standard reduction potentials the standard half cell potential for the reduction half cell. Wet cell a battery that uses a liquid electrolyte, usually an aqueous salt. STUDY QUESTIONS 10.1 10.2 10.3 10.4 10.5 10.6 10.7 10.8 10.9 10.10 10.11 10.12 10.13 10.14 10.15 10.16 10.17 10.18 10.19 10.20 10.21 10.22 10.23 10.24 10.25 10.26 10.27 What is an oxidation-reduction reaction? What is oxidation? What is reduction? What is an oxidizing agent? What is a reducing agent? What is the oxidation state of an atom? What is the value of the oxidation state of any atom in its elemental form? What is the value of the sum of the oxidation states for all atoms in a compound? What is the oxidation state of a hydrogen atom in a metal hydride? What is the oxidation state of an oxygen atom in a peroxide? What is a galvanic cell? What is another name for these types of cells? What is an electrode? What is the purpose of a salt bridge? What is an electrolyte? At which electrode does the oxidation reaction occur? At which electrode does the reduction reaction occur? What is the half cell reaction that occurs at a zinc anode in a zinc-copper galvanic cell?? What is the half cell reaction that occurs at a copper cathode in a zinc-copper galvanic cell? What are standard cell potentials? In what form are standard half cell potential reactions always written? How are the standard reduction potential and the standard oxidation potential for a half cell related? What electrode system are all half cell potentials determined against? What E0 is assigned to this electrode? When zinc or magnesium metal is used as a sacrificial metal to protect a structural metal from corrosion, what term is used to describe this process? If the system in Question 10.23 is considered as a galvanic cell, what electrode would the zinc be? What is the mathematical equation that relates the cell potential of a galvanic cell to the standard cell potential and the concentrations of the chemical species in the oxidation-reduction reaction? A galvanic cell that is composed of two half cells of the same composition but different concentrations is called what? What is electrolysis? What is the process called where electrolysis is used to produce a thin coating of one metal on top of another metal? 352 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY 10.28 10.29 10.30 10.31 10.32 10.33 10.34 10.35 10.36 10.37 10.38 10.39 What is the Faraday constant? What equation is used to determine pH by using a pH meter? What is the difference between a wet cell and a dry cell battery? What is emf? What is the difference between a primary and a secondary battery? A secondary battery is also known as what? What rechargeable battery is used today for starting automobile and other engines? What is the mobile species in a lithium-ion battery? What is a fuel cell? How does a battery differ from a fuel cell? How are they the same? How do the PAFC, SAFC, and AFC differ? How is the operation of the SOFC different from other fuel cells? What is the charge carrier? 10.40 How are “button batteries” like a fuel cell? How are they like a normal battery? PROBLEMS 10.41 Identify the species that is oxidized and the species that is reduced in the following reactions: (a) Cr+ + Sn4+ ! Cr3+ + Sn2+; (b) 3Hg2+ + 2Fe0(s) ! 3Hg2 + 2Fe3+, (c) 2As0(s) + 3Cl2(g) ! 2AsCl3. 10.42 In Problem 10.41, what is the oxidizing agent? What is the reducing agent? 10.43 Identify the species that is oxidized and the species that is reduced in the following reactions: (a) Zn0(s) + Fe2+(aq) ! Zn2+(aq) + Fe0(s), (b) Zn0(s) + Cu2+(aq) ! Zn2+(aq) + Cu0(s), (c) Fe0(s) + Cu2+(aq) ! Fe2+(aq) + Cu0(s). 10.44 In the reaction: Fe2O3(s) + 3CO(g) ! 2Fe0(s) + 3CO2(g), what is the oxidizing agent and what species is being oxidized? 10.45 For the following reaction involving different chlorine species: 2Cl ðaqÞ + 4H + ðaqÞ + 2ClO3 ðaqÞ ! 2ClO2 g + 2H2 OðlÞ + Cl2 g Which of the chlorine containing species is the oxidant and which is being oxidized? 10.46 What are the oxidation states of the atoms in the following compounds: (a) CO, (b) H2O, (c) CO2, and (d) CaH2? 10.47 What are the oxidation states of the atoms in the following complex ions: (a) CO32, (b) Cd(OH)42, (c) CrO2, (d) MoO42, and (e) HSO3? 10.48 Which of the following are oxidation-reduction reactions: (a) ClO + NO2 ! Cl + NO3 (b) Ag + S ! Ag2S, (c) 2Na + FeCl2 ! 2NaCl + Fe, and (d) 2H2 + O2 ! 2H2O? 10.49 What are the anode and cathode reactions for a galvanic cell that is based on the following reaction: Ni(s) + 2Fe3+ ! Ni2+ + 2Fe2+? 10.50 For a galvanic cell composed of a nickel electrode submerged in Ni(NO3)2(aq) and an aluminum electrode submerged in Al(NO3)3(aq). (a) Write the equations for the anode and cathode half reactions, and (b) write the overall cell equation 10.51 (a) Write a balanced equation for the reaction of HCl(aq) with Mg0(s) to form Mg2+(aq), Cl(aq), and H2(g). (b) What are the two half reactions for this balanced net reaction? (c) How many electrons are transferred in this reaction? PROBLEMS 353 10.52 In order to convert Ni0(s) to NiCl2(aq), (a) what kind of redox agent would be needed? (b) How many electrons are transferred? (c) What is the half reaction for this process? 10.53 (a) Balance the following oxidation-reduction equation: Cr0(s) + Cl2(g) ! CrCl3(s). (b) How many electrons are transferred in this reaction? 10.54 What are the half reactions for the reaction in Problem 10.49? 10.55 For the galvanic cell that uses the reduction half reaction: Cl2 + 2e ! 2Cl, what cathode would be used in this half cell? 10.56 If a galvanic cell is constructed with a Zn0(s) electrode in a solution of Zn2+(aq) ions and an Al0(s) electrode in a solution of Al3+(aq) ions, (a) which metal will oxidize? (b) What is the standard potential of the galvanic cell? 10.57 If a galvanic cell is constructed with a magnesium electrode and a copper electrode, (a) which metal would be the anode and which the cathode? (b) What would be the expected E0 for the cell? 10.58 If the following two half cell reactions are up in a galvanic cell, (a) what voltage would be obtained? Cu2 + ðaqÞ + 2e ! Cu0 ðsÞ Cl2 g + 2e ! 2Cl ðaqÞ (b) Which reaction would occur at the anode and which would occur at the cathode? 10.59 If a galvanic cell is set up using electrodes of Mg0(s) and Ag0(s) and electrolytes of Mg2+(aq) and Ag+(aq), (a) which electrode will be the cathode and which will be the anode? (b) How many electrons are transferred in the reaction? (c) What is the balanced chemical equation for the cell? (d) What is the standard potential of the cell? 10.60 Given the following oxidation-reduction reaction: Cu2+(aq) + Ba(s) ! Cu(s) + Ba2+(aq) (a) Determine the half reactions and their standard reduction potentials. (b) Which half reaction would occur at the anode and which would occur at the cathode of a galvanic cell? (c) Determine the E0cell for the galvanic cell. 10.61 Given the following half reactions: Al3+(aq) ! Al(s) + 3e and Sn2+(aq) + 2e ! Sn(s), (a) What is the complete balanced oxidation-reduction equation for a galvanic cell? (b) What is the standard cell potential? 10.62 What would be the cell potential for a zinc/nickel galvanic cell if the electrolytes were composed of (a) a Zn2+(aq) ion concentration of 0.02 M and a Ni2+(aq) concentration of 0.2 M, (b) a Zn2+(aq) ion concentration of 1.0 M and a Ni2+(aq) concentration of 1.0 M? 10.63 Determine the cell potential of a galvanic cell based on the following reduction half reactions: Cd2+ + 2e ! Cd and Pb2+ + 2e ! Pb where [Cd2+] ¼ 0.020 M and [Pb2+] ¼ 0.200 M. 10.64 What is the potential of the galvanic cell: Cu(s) + 2Fe3+(aq) ! Cu2+(aq) + 2Fe2+(aq) with electrolyte concentrations of: [Fe3+] ¼ 1.0 104 M; [Cu2+] ¼ 0.25 M; [Fe2+] ¼ 0.20 M? 10.65 What is the predicted voltage output of a silver/silver ion concentration cell with the following electrolyte concentrations: (a) an anode concentration of 0.001 M and a cathode concentration of 1.0 M? (b) an anode concentration of 0.1 M and a cathode concentration of 1.0 M? 354 10. OXIDATION-REDUCTION REACTIONS AND ELECTROCHEMISTRY 10.66 Determine the amount of charge (in faradays) required to plate out 25 g of Cu0(s) onto an electrode using a solution of Cu2+(aq) ions? 10.67 If 0.400 F of charge is passed through an electrolysis cell with a cathode reaction of: Ag+(aq) + 1e ! Ag0(s), how many grams of silver would plate out on the electrode? 10.68 How many moles of Zn2+(aq) will plate out on an electrode surface as Zn0(s) if 1.3 F of charge is passed through the electrolysis system? How many grams of zinc? 10.69 If five amps of electric current is passed through an electrolysis system for 1 h, how many moles of Ag+(aq) can be electroplated onto the electrode surface, as Ag0(s)? How many grams? 10.70 If the electroplating of Cu2+(aq) continues for 2 h with 10 A of current, how many grams of Cu0(s) is plated onto the electrode? C H A P T E R 11 Solids O U T L I N E 11.1 Crystalline Solids 356 11.6 Amorphous Solids 380 11.2 Ionic Solids 361 Important Terms 382 11.3 Molecular Solids 369 Study Questions 383 11.4 Atomic Solids 372 Problems 385 11.5 Metallic Solids 374 In previous chapters, the focus has been on the structure and behavior of individual molecules or small groups of molecules. The properties of the macromolecular solids are dependent on this structure and behavior of the individual molecules that make up the solids. Chapter 1 describes solids as rigid and maintaining a fixed shape and volume under pressure. Solids behave in this way because they are composed of extended arrays of atoms, ions, or molecules held together by strong chemical bonds. It is the behavior and properties of the array of chemical bonds that give solids their distinct properties. The classification of solid materials is shown in Fig. 11.1. As outlined in Chapter 1, there are two main types of solids: crystalline and amorphous. Crystalline solids have a long range, highly ordered three-dimensional arrangement of atoms, molecules, or ions, whereas amorphous solids are characterized by a lack of a long range ordered geometrical arrangement. Amorphous solids are divided into two groups: microcrystalline solids and pseudo solids. Pseudo solids have properties belonging to all solids, but also have some fluid properties. They are sometimes considered not to be true solids. There are four types of crystalline solids: ionic solids, molecular solids, atomic solids, and metallic solids. In addition, molecular solids are classified according to the types of forces holding the molecules together. The three types of molecular solids are: nonpolar molecular solids, polar molecular solids, and hydrogen-bonded molecular solids. Atomic solids are also divided into two groups: those that are entirely made of three-dimensional covalently bonded atoms (covalent) and those General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00011-4 355 # 2018 Elsevier Inc. All rights reserved. 356 11. SOLIDS Solids Amorphous Microcrystalline lonic Crystalline Pseudo solids Atomic Molecular Covalent Nonpolar FIG. 11.1 Polar Metalline Mixed bonding H–bonded Classification of solid materials according to their structure and properties. that are made up of two-dimensional covalently bonded networks that are held together by weaker intermolecular forces (mixed bonding). 11.1 CRYSTALLINE SOLIDS Some examples of crystalline solids are shown in Fig. 11.2. The process by which a crystalline solid is formed is known as crystallization. Crystals can be formed by precipitating from a highly concentrated solution, by freezing of a liquid mixture, or by deposition from a gas phase. Crystallization is a chemical separation technique commonly used in industrial process that will be further discussed in Chapter 12. As described in Chapter 1, a crystalline solid is a solid material with the constituent species (atoms, ions, or molecules) arranged in a highly ordered microscopic structure that extends in all directions. Because crystalline solids have a highly ordered internal structure that extends throughout the material, the geometric shape of the macroscopic form of a crystalline solid is dependent on the microscopic structure. The geometric shapes of these macroscopic crystals consist of flat surfaces, or faces, with sharp angles oriented in different directions. The faces intersect at angles that reflect the internal arrangement of the component species. When a crystalline solid is exposed to X-rays, the constituent species inside the crystal cause the X-ray beam to diffract into many directions. A three-dimensional picture of the microscopic arrangement of the crystalline solid can be obtained by measuring the angles and intensities of the diffracted X-ray beams, called a diffraction pattern. By using this technique, called X-ray crystallography, the positions of the constituent species, the chemical bonds, and the extent of any disorder in the solid can be determined. The microscopic structure of a crystalline solid is made up of simple repeating units called the unit cell. The unit cell is defined by its lattice points, which are simply the geometric 357 11.1 CRYSTALLINE SOLIDS FeS2 CaSo4 CaCo3 SiO2 SbS2 FIG. 11.2 Some examples of crystalline solids. From Rob Lavinsky, iRocks.com, Wikimedia Commons. points that define the shape of the unit cell and are also the positions of the constituent species. When the unit cell is repeated in three dimensions, the resulting network is called the crystal lattice, the symmetrical three-dimensional arrangement of the constituent species inside the crystalline solid. The defining properties of a unit cell are: • The unit cell is the simplest repeating unit in the crystal lattice. • The opposite faces of a unit cell are parallel. • The edge of the unit cell is connected to equivalent points in adjacent unit cells. There are seven classes of unit cells shown in Fig. 11.3. These seven classes are: cubic, tetragonal, orthorhombic, monoclinic, triclinic, rhombic, and hexagonal. Each unit cell class is in the shape of a hexahedron and each is differentiated by its shape, the length of its sides (a, b, and c), and the angles between the edges (α, β, and γ). The shapes of the first three unit cell classes (cubic, tetragonal, and orthorhombic) are based on a simple rectangular prism with 90 degrees angles at all the corners. They differ by the length of the sides. The shapes of the next four unit cell classes (monoclinic, triclinic, rhombic, and hexagonal) take the form of a hexahedron with varying angles between the adjoining edges. They differ in both the length of the sides and the size of the angles between the edges. The hexagonal unit cell gets its name from the fact that it is ⅓ of a hexagonal prism. Each unit cell class can exist in different forms shown in Fig. 11.4. These are: simple, bodycentered, face-centered, and side-centered. The simple forms have the constituent species (atoms, ions, or molecules) only at the corners of the unit cell. Body-centered forms have an additional species at the center of the unit cell. Face-centered forms have additional species 358 11. SOLIDS c a b Cubic Tetragonal Orthorhombic a=b=c a = b = g = 90 degrees a=b≠c a = b = g = 90 degrees a≠b≠c a = b = g = 90 degrees g a b Monoclinic a≠b≠c a = g = 90 degrees b > 90 degrees Triclinic a≠b≠c a ≠ b ≠ g ≠ 90 degrees Rhombic a=b=c a = b = g ≠ 90 degrees Hexagonal a=b=c a = b = 90 degrees g = 120 degrees FIG. 11.3 The seven unit cell classes. The lengths of the sides are designated as a, b, and c. The size of the angles between the sides are α, β, and γ. FIG. 11.4 The four forms of unit cells. Simple Body centered Face centered Side centered at the center of each face of the unit cell and side-centered forms (also called base-centered) have additional species at the center of two opposing sides of the unit cell. The different unit cell forms allowed for each of the unit cell classes are listed in Table 11.1. The number of constituent species in a unit cell depends on how many of the atoms (ions, molecules) in the unit cell belong to more than one unit cell and this depends on the form of the unit cell. All simple forms have one constituent species at each of the eight corners 11.1 CRYSTALLINE SOLIDS 359 TABLE 11.1 The Types of Forms Allowed for Each of the Seven Unit Cell Classes Unit Cell Class Forms Cubic Simple, face-centered, body-centered Tetragonal Simple, body-centered Orthorhombic Simple, face-centered, body-centered, side-centered Monoclinic Simple, side-centered Triclinic Simple Rhombic Simple Hexagonal Simple FIG. 11.5 The number of unit cells that share the defining constituent unit in the simple cubic, body-centered cubic, and facecentered cubic unit cells. Simple cubic Body centered cubic Face centered cubic (Fig. 11.4). But each of these is shared by eight unit cells as shown in Fig. 11.5. So, the total number of atoms in one unit cell of the simple form is: 8 atoms/8 unit cells ¼ 1 atom. A body-centered unit cell form has all the species of the simple form and one extra species at the center of the cell, which is not shared by any other unit cell. So, the number of atoms in one unit cell of the body-centered form is: 1 atom + 1 atom ¼ 2 atoms. A face-centered unit cell has all the species of the simple form and six extra species at the center of each face of the unit cell. Each of those in the center of the face is shared by two unit cells. So, the number of atoms in one unit cell of the face-centered form is; 1 atom + 6 atoms/2 unit cells ¼ 4 atoms. The side-centered unit cell is similar to the face-centered form except it has only two extra species at the center of two opposing sides that are each shared by two unit cells. The number of atoms in one unit cell of the side-centered form is: 1 atom + 2 atoms/2 unit cells ¼ 2 atoms. In summary, if the constituent species were atoms, the number of atoms for each form of unit cell would be: Simple ¼ 8 atoms 8 unit cells ¼ 1 atom: Body-centered ¼ ð8 atoms 8 unit cellsÞ + 1 atom ¼ 2 atoms: Face-centered ¼ ð8 atoms 8 unit cellsÞ + ð6 atoms 2 unit cellsÞ ¼ 4 atoms: Side-centered ¼ ð8 atoms 8unit cellsÞ + ð2 atoms 2 unit cellsÞ ¼ 2 atoms: 360 11. SOLIDS TABLE 11.2 The Formulas Used to Calculate the Volumes of the Different Types of Unit Cells, Which are Needed for the Determination of Solid Density Unit Cell Volume Cubic a b c ¼ a3 Tetragonal a b c ¼ a2 c Orthorhombic abc Monoclinic a b c sin(β) Triclinic a b c [(1cos2αcos2β cos2γ) + 2(cos(α) cos(β) cos(γ))]½ Rhombic a b c (sin 60°) ¼ a3 sin(60°) Hexagonal a b c (sin 60°) ¼ a2 c sin(60°) The letters a, b, and c refer to Fig. 11.3. The density of a crystalline solid, in g/cm3, can be determined from the density of the unit cell. This is obtained by dividing the mass of the constituent units (atoms, ions, or molecules) by the volume of the unit cell. The mass of the constituent units in the unit cell (Mcell) is determined by the molar mass of the species divided by the Avogadro’s number (NA) and multiplied by the number of constituent species in the unit cell (n). ρ¼ Mcell molar mass NA n ¼ Vc Vc (1) Table 11.2 lists the method of calculating the volumes for different types of unit cells. As long as all the angles of the cell are 90 degrees (cubic, tetragonal, and orthorhombic), the volume is calculated as the volume of a rectangle: V ¼ length (a) width (b) height (c). The formulas for calculating the volumes of the four types of unit cells that have angles other than 90 degrees include sine and cosine factors. This text will only consider the first three unit cell types, which are the most commonly encountered in simple chemical elements. EXAMPLE 11.1: DETERMINING THE DENSITY OF A CRYSTALLINE SOLID FROM THE CRYSTAL STRUCTURE Krypton freezes at 157.4°C into a face-centered cubic crystal structure with an edge length of 559 pm. What is the density of the unit cell of solid krypton? 1. Determine the number of krypton atoms in a unit cell. The face-centered cubic form has ð8 atoms 8 unit cellsÞ + ð6 atoms 2 unit cellsÞ ¼ 4 atoms of krypton: 2. Determine the volume of the unit cell. The volume units should be in cm3 so : 197pm ¼ 1:97 108 cm 3 The volume of a cubic unit cell is equal to : a3 ¼ 5:59 108 ¼ 1:747 1022 cm3 11.2 IONIC SOLIDS 361 EXAMPLE 11.1: DETERMINING THE DENSITY OF A CRYSTALLINE SOLID FROM THE CRYSTAL STRUCTURE— CONT’D 3. Calculate the mass of one krypton atom. Mass of one atom of krypton ¼ molar mass=NA ¼ 83:80 g=mol=6:02 1023 atoms=g ¼ 1:392 1022 g=atom 4. Calculate the mass of krypton in the unit cell. Mcell ¼ 1:392 1022 g=atom 4 atoms=unit cell ¼ 5:568 1022 g 5. Determine the density from Eq. (1). ρ ¼ ðMcell Þ=ðVc Þ ¼ 5:568 1022 g = 1:747 1022 cm3 ¼ 3:19 g=cm3 11.2 IONIC SOLIDS Ionic solids consist of positive and negative ions held together by electrostatic attractions, as discussed in Section 3.2. But, they do not exist as simple pairs of positive and negative ions as represented by their chemical formulas. Instead, ionic solids exist as millions of ions arranged in a crystal lattice where each cation and anion has multiple nearest neighbors. The ions are typically packed into an arrangement that maximizes the attractions between ions of opposite charge, while minimizing the repulsions between ions of like charge. This is achieved by packing as many cations around each anion, and as many anions around each cation, as possible. The number of nearest neighbor ions of opposite charge surrounding any ion is called the coordination number for that ion. The coordination number for any ion in an ionic solid is limited by the ratio of cations to anions in the chemical formula. For example, in a sodium chloride crystal there are six chloride anions around each sodium cation. Since the ratio of sodium ions to chloride ions in the chemical formula is 1:1, there must also be six sodium cations around each chloride anion. So, in a crystal of sodium chloride, both Na+ and Cl have a coordination number of 6. But, in a crystal of aluminum oxide (Al2O3), each Al3+ cation is surrounded by six O2 anions giving it a coordination number of 6. In order to maintain the ratio of cations to anions of 2:3 dictated by the chemical formula, there must be four Al3+ ions around each O2 ion, with a coordination number of 4. The most energetically stable arrangement of ions in an ionic solid is generally those in which there is a minimum of empty space between the ions. The most tightly packed and space-efficient arrangement of an ionic crystal lattice is known as a close packed structure. The close packed lattice structures are the face-centered forms, the most common is facecentered cubic. The anions in close packed structures, which are typically larger than cations, occupy the lattice points of the unit cell, while the smaller cations occupy the voids between them. There are two types of voids in close packed structures: octahedral voids that are surrounded by six anions, tetrahedral voids that are surrounded by four anions. So, a cation in an octahedral void has a coordination number of 6, a cation in a tetrahedral void has a coordination number of 4. 362 11. SOLIDS In close packed structures, there are the same number of octahedral voids as there are anions in the unit cell (# octahedral voids ¼ # anions); there are twice as many tetrahedral voids as there are anions (# tetrahedral voids ¼ 2 # anions). So, an ionic solid of a close packed structure that has cations in the tetrahedral voids can have a maximum cation/anion ratio of 2/1 when all of the tetrahedral holes are filled. If the cation/anion ratio is <2, some of the tetrahedral voids are empty. Similarly, an ionic solid of a close packed structure with cations in the octahedral voids will have a maximum cation/anion ratio of 1/1 when all of the octahedral voids are filled. If the cation to anion ratio is <1, some of the octahedral voids are empty. EXAMPLE 11.2: DETERMINING THE EMPIRICAL FORMULA OF AN IONIC SOLID FROM CRYSTAL STRUCTURE Determine the empirical formula of an unknown ionic solid AnBm if it is known to crystallize in a close packed crystal structure with ½ of the tetrahedral voids filled. 1. The anions (B) occupy the lattice points of the unit cell, while the cations (A) occupy the tetrahedral voids. 2. Since there are 2 tetrahedral voids for every anion and ½ of these voids are occupied by cations. number of anions ¼ 1=ð½ 2Þ ¼ 1=1: number of cations 3. Empirical formula ¼ AB Any ionic solid will adopt the crystal structure that will provide it with the greatest stability. This is generally the structure that allows the maximum number of oppositely charged ions to bond without requiring ions of the same charge to come in close contact. The unit cell type and the placement of the ions will depend on the size of the ions. The unit cell for sodium chloride is shown in Fig. 11.6A. The large chloride anions (green) make up a close packed face-centered cubic unit cell structure, while the smaller sodium ions (blue) are located in the octahedral voids between the larger anions. The placement of the smaller sodium ions in the voids of the face-centered cubic structure of chloride anions minimizes the unoccupied spaces in the crystal lattice and so maximizes its stability. The six ion coordination surrounding the sodium ion is shown by dotted lines in the center of the unit cell in Fig. 11.6A. This six ion coordination forms an octahedral structure with FIG. 11.6 The unit cell for the crystal lattice of sodium chloride (A). The larger chloride anions are in green and the smaller sodium cations are in blue. The six ion coordination surrounding the central sodium ion is outlined with dotted lines in (A). The six ion coordination for all sodium and chloride ions in the expanded crystal lattice is demonstrated by dotted lines in (B) for one chloride ion (upper left) and one sodium ion (middle right). (A) (B) 11.2 IONIC SOLIDS 363 the six chloride ions at the corners of the octahedron. As the unit cell is repeated in three dimensions to form the extended crystal lattice, each chloride and sodium ion has this same octahedral structure with a coordination number of 6. This is shown by the dotted lines in Fig. 11.6B for one sodium cation and one chloride anion. The energy released when a crystal lattice is formed is known as the lattice energy. For any ionic solid, the lattice energy is a constant that measures how tightly the ions are held together. This lattice energy is equal to the sum of the interaction of all ions with all other ions in the crystal lattice and includes both attractions between ions of opposite charge and repulsion between ions of the same charge. The energy of these interactions can be calculated by Coulomb’s law in a similar manner as for the calculation of the strength of a single ionic bond (Section 3.2). The lattice energy (E) is directly proportional to the number of ion pairs in the lattice (n) and the ionic charges (q1 and q2) multiplied by a unitless constant (A), which is dependent on the crystal structure type and the arrangement of ions in the crystal lattice. The lattice energy is inversely proportional to the sum of the ionic radii (r1 + r2), which is equal to ionic bond length. E ¼ kn q1 q2 A r1 + r2 (2) It is impossible to measure the enthalpy change of converting the gaseous ions into a solid crystal. But lattice energies can be determined from experimental measurements by using a thermodynamic cycle developed by Max Born and Fritz Haber called the Born-Haber cycle. This cycle is an application of Hess’s law described in Section 8.4. Recall that according to Hess’s law, the enthalpy change in a chemical reaction is the same whether it occurs in one step or several steps. So, the determination of the lattice enthalpy of an ionic solid can be determined by comparing the standard enthalpy of formation of an ionic solid directly from its elements in the standard form to its formation from a series of stepwise reactions that include the enthalpy of formation of the crystal lattice. It is possible to write a series of chemical reactions that sum to the standard enthalpy of formation for any ionic solid. This series of stepwise reactions is the Born-Haber cycle. Hess’s law is then applied to the multistep series of reactions and the standard enthalpy of formation to obtain the lattice energy of the ionic solid. In general, constructing a Born-Haber cycle for the determination of the lattice energy of an ionic solid involves the following steps: (1) (2) (3) (4) Convert Convert Convert Convert any diatomic species into the monatomic form. (enthalpy of dissociation) the nongaseous elements into their gaseous states. (enthalpy of sublimation) the atomic species into their ionic forms. (ionization energy; electron affinity) the separate ions into the solid form. (lattice energy) The sum of these 4 reaction steps gives the same enthalpy as the formation of the ionic solid from the elements in their standard states. (5) Convert the elements in their standard forms to the solid crystal. (enthalpy of formation) Lattice energy ¼ enthalpy of formation enthalpy of dissociation + enthalpy of sublimation + Σ ionization energies + Σ electron affinities 364 11. SOLIDS For example, a Born-Haber cycle for sodium chloride includes the following five steps: (1) (2) (3) (4) (5) ½ Cl2(g) ! Cl(g) Na(s) ! Na(g) Cl(g) + e ! Cl Na(g) ! Na+ + e Na+(g) + Cl(g) ! NaCl(s) ΔH° ¼ + 121.3 kJ/mol ΔH° ¼ + 107.3 kJ/mol ΔH° ¼ 349 kJ/mol ΔH° ¼ + 496 kJ/mol ΔH° ¼ ? Enthalpy of dissociation Enthalpy of sublimation Electron affinity Ionization energy Lattice energy The sum of these five reaction steps is the same as the reaction for the standard enthalpy of formation of sodium chloride; ΔH°f ¼ – 411.12 kJ/mol Na(s) + ½Cl2(g) ! NaCl(s) Enthalpy of formation According to Hess’s law: ΔHf° ¼ ΔH1° + ΔH2° + ΔH3° + ΔH4° + ΔH5° So: ΔH5° ¼ ΔHf° ΔH1° + ΔH2° + ΔH3° + ΔH4° ¼ 411:1 ð121:3 + 107:3 349 + 496Þ ¼ 411:1 375:6 ¼ 786:7kJ=mol EXAMPLE 11.3: DETERMINING THE LATTICE ENERGY OF AN IONIC SOLID USING THE BORN-HABER CYCLE Calculate the lattice energy for CaCl2 given the following information: Enthalpy of sublimation 1st ionization energy 2nd ionization energy Dissociation energy Electron affinity Enthalpy of formation Ca(s) ! Ca(g) Ca(g) ! Ca+(g) + e Ca+(g) ! Ca2+(g) + e Cl2(g) ! 2Cl(g) Cl(g) + e ! Cl(g) Ca(s) + Cl2 ! CaCl(s) ΔH° ¼ 178.2 kJ/mol ΔH° ¼ 590 kJ/mol ΔHf° ¼ 1145 kJ/mol ΔH° ¼ 244 kJ/mol ΔH° ¼ 349 kJ/mol ΔHf° ¼ – 795.8 kJ/mol step (1) Convert Cl2 into the monatomic form. ΔH° ¼ dissociation energy for Cl2 g ¼ 244kJ=mol step (2) Convert all the elements into their gaseous states. Since Cl is already a gas: ΔH°sub ¼ enthalpy of sublimation for CaðsÞ ¼ 178:2kJ=mol: step (3) Convert all the atomic species into their ionic forms. Since 2 electrons are added to Cl, the electron affinity must be multiplied by 2: 365 11.2 IONIC SOLIDS EXAMPLE 11.3: DETERMINING THE LATTICE ENERGY OF AN IONIC SOLID USING THE BORN-HABER CYCLE— CONT’D ° ΔHel aff for Cl ¼ 349 2 ¼ 698kJ=mol ° ΣΔHion E for Ca ¼ 590 + 1145 ¼ 1735kJ=mol step (4) Convert the separate ions into the solid form. ΔH° ¼ ? step (5) Convert the elements in their standard forms to the solid crystal. ΔHf° ¼ 795:8kJ=mol Determine lattice energy: Lattice energy ¼ enthalpy of formation enthalpy of dissociation + enthalpy of sublimation +Σ ionization energies + Σ electron affinities ¼ 795:8 ð244 + 178:2 698 + 1735Þ ¼ 2255 kJ=mol Recall from Section 3.2 that the strength of an ionic bond can be predicted by using Coulomb’s law. The magnitude of the bond strength depends on the magnitude of the product of the ionic charges and the bond length. Larger charges will have stronger interactions and a stronger ionic bond. Also, smaller ions will be able to get closer together and have smaller bond lengths and stronger bonds. As with the ionic bond strength, the stronger the bonds between the ions the more stable the crystal lattice. So, the lattice energy of an ionic crystal follows the same trends as the ionic bond strength. Smaller ions and larger ionic charges will result in stronger crystal lattices and higher lattice energies. Similarly, larger ions and smaller ionic charges result in weaker crystal lattices and lower lattice energies. EXAMPLE 11.4: DETERMINING THE RELATIVE STRENGTH OF THE LATTICE ENERGY OF IONIC SOLIDS List the following ionic solids in the order of increasing lattice energy: Na F, Na Cl, Ca Cl, Na Br, Sc Cl According to Coulomb’s Law, the lattice energy is directly proportional to the product of the ion charges (q1 q2) and inversely proportional to the bond length (r1 + r2). 1. First determine the product of the ionic charges for each ion pair. Na F ¼ + 1 1 ¼ 1 Na Cl ¼ + 1 1 ¼ 1 Ca Cl ¼ + 2 1 ¼ 2 Continued 366 11. SOLIDS EXAMPLE 11.4: DETERMINING THE RELATIVE STRENGTH OF THE LATTICE ENERGY OF IONIC SOLIDS— CONT’D Na Br ¼ + 1 1 ¼ 1 Sc Cl ¼ + 3 1 ¼ 3 2. According to the charges of the ions, the order of increasing lattice energy is: Na Br,Na Cl,Na F < Ca Cl < Sc Cl Since Na-F, Na-Cl, and Na-Br have the same ionic charge product and the same cation, the bond strength is determined by the size of the anion which is: Br > Cl– > F. So, in the order of increasing lattice energy: Na – Br < Na – Cl < Na – F < Ca – Cl < Sc – Cl. Because of the strong bonding between cations and anions and the rigid structure of the crystal lattice, ionic solids have high melting and boiling points. Melting of an ionic solid requires that the ions break out of the rigid crystal structure and move about independently in a liquid state. The ionic bonds are very strong and require a lot of energy to break. This causes ionic solids to have very high melting points. The melting points are dependent on the lattice energy and the ionic bond strength and so can be predicted by Coulomb’s law. In general, the larger the charges on the ions, the stronger the ionic bonds and the higher the melting point. For example, NaF composed of singly charged ions melts at 996°C, while MgO composed of doubly charged ions melts at 2852°C. Also, for solids composed of ions of similar charge and different sizes, the smaller the ionic radius the stronger the ionic bond and the higher the melting point. Compare the melting point of NaI at 661°C to that of NaF at 996°C. EXAMPLE 11.5: DETERMINING THE RELATIVE MAGNITUDE OF MELTING POINTS FOR IONIC SOLIDS List the following ionic solids in the order of increasing melting points: LiF, LiBr,MgBr2 , LiI,FeBr3 The melting points of ionic solids increase as the strength of the lattice energy increases according to Coulomb’s Law. 1. Determine the product of the ionic charges for each ion pair. Li–F ¼ +1 1 ¼ 1 Li–Br ¼ +1 1 ¼ 1 Mg–Br ¼ +2 1 ¼ 2 Li–I ¼ +1 1 ¼ 1 Fe–Br ¼ +3 1 ¼ 3 So, according to the charges of the ions: FeBr3 > MgBr2 > LiF, LiBr, LiI. 2. Determine the relative melting points for solids with the same ionic product by comparing the bond length (r1 + r2). LiF, LiBr, and LiI have the same ionic product and the same cation. The length of the bond with Li is dependent on the relative size of the anion, which is: 11.2 IONIC SOLIDS 367 EXAMPLE 11.5: DETERMINING THE RELATIVE MAGNITUDE OF MELTING POINTS FOR IONIC SOLIDS— CONT’D I > Br > F So, the order of increasing melting points is the same as the order of increasing lattice energy: LiI < LiBr < LiF < MgBr2 < FeBr3 Ionic solids are also hard and brittle. They are hard because the positive and negative ions are strongly attracted to each other and are difficult to separate by the application of an external compressive force. However, they can also be brittle. When pressure is applied to an ionic solid, the ions of like charge can be forced close enough for the ionic repulsion to cause the solid to break. In addition, when an outside force causes one plane of ions to shift slightly, ions of like charge are brought closer together. The ionic repulsion can cause the solid to split along the axis parallel to the planes of the ions producing new faces in the macroscopic crystal that intersect at the same angles as those in the original crystal. This process is called cleaving. Ionic solids are poor conductors of both heat and electricity. Electric conductivity requires the movement of charged particles, either electrons or ions. Since the ions in an ionic solid are held tightly in their lattice positions, ionic solids cannot conduct electricity. However, when melted, the ions are free to move relative to each other. Cations can move in one direction while anions move in the opposite direction and so are free to carry an electric current. Ionic solids become very good electrical conductors when molten. CASE STUDY: FAST ION CONDUCTORS Silver iodide is an ionic solid with the chemical formula AgI. At room temperature, the crystal lattice of silver iodide is a close packed face-centered cubic unit cell structure. This is similar to the crystal structure for sodium chloride shown in Fig. 11.6 with the larger iodide anions occupying the lattice points. But unlike sodium chloride, the silver cations occupy the tetrahedral voids in the lattice structure instead of the octahedral voids. There are eight tetrahedral voids in a face-centered cubic unit cell and the silver cations occupy four of these available tetrahedral voids. In this structure, both the iodide ions and the silver ions have a coordination number of 4. Like all ionic solids, silver iodide is essentially an insulator (about 105 Ω1 cm1) at room temperature. However, at a temperature of 420 K, solid silver iodide shows an abrupt increase in conductivity of over three orders of magnitude as shown in red in Fig. 11.7. The conductivity increases only slightly as temperature is increased above 420 K and actually decreases by 10% on melting at 829 K. This anomalous behavior of silver iodide can be compared with that of the typical ionic solid sodium chloride, which has a conductivity of less than 108 Ω1 cm1 at room temperature. The conductivity of sodium chloride, shown in blue in Fig. 11.7, increases slowly with temperature up to about 105 Ω1 cm1 just below the melting point. This low level increase in the conductivity of sodium chloride just below the melting point is due to small amounts of ionic impurities in the crystal lattice which can become mobile in the lattice structure as the temperature rises. At the melting point 368 11. SOLIDS FIG. 11.7 Conductivity as a function of temperature for sodium chloride (blue) and silver iodide (red). The melting points for each compound are indicated by a dotted line. FIG. 11.8 The available tetrahedral voids in the close packed face-centered cubic (left) and the open body-centered cubic (right) unit cells. of 1074 K, the conductivity of sodium chloride increases abruptly by five orders of magnitude to a value of 3 Ω1 cm1. The large increase in the conductivity of silver iodide at a temperature below its melting point is associated with a solid-solid phase transition shown in Fig. 11.8. At 420 K, the silver iodide crystal structure changes from the close packed face-centered cubic to a more open body-centered cubic form. The iodide ions still occupy the lattice points and the silver ions still occupy tetrahedral voids. But, since the high temperature body-centered cubic form is not a close packed structure, it has many more voids than the close packed face-centered cubic form. The voids include 12 tetrahedral voids (24 voids shared with 2 cells), 6 octahedral voids, and an additional 24 tetragonal voids. The high temperature coupled with the more open structure of the body-centered form allows the 2 silver ions per unit cell to move rapidly between any of these 42 voids. This gives the silver ions a very high mobility in the crystal structure, resulting in the anomalously high electrical conductivity. So, the high conductivity of the solid silver iodide in the high temperature body-centered cubic lattice is a result of the large cationic radius of the iodide anion, which is restricted to the lattice points maintaining the rigid solid structure, and the high mobility of the small silver ion, which is allowed to move freely between the 42 voids in the open structure. This phenomenon is referred to as fast ion conduction. 11.3 MOLECULAR SOLIDS 369 This high temperature phase of silver iodide was the first fast ion conductor ever discovered. Fast ion conductors function as solid electrolytes. They possess the rigidity of solids with a disorder on the atomic scale similar to a liquid. This has sometimes been referred to as a “molten sublattice.” Other fast ion conductors are the silver halides: AgBr and AgCl, the copper halides: CuI, CuBr, and CuCl, and silver compounds from anions in group 16 of the periodic table: Ag2S, Ag2Se, and Ag2Te. Fast ion conductors are used in solid state supercapacitors, batteries, solid oxide fuel cells, and in chemical sensors as ion carriers. 11.3 MOLECULAR SOLIDS Molecular solids are composed of atoms or molecules held together by intermolecular forces. The constituent species of molecular solids are molecules except in the solidified noble gases, where the units are atoms. While the bonding within the molecules is covalent and strong, the forces which hold the molecules together in the crystal lattice are the weak intermolecular forces discussed in Section 3.8. These include: dipole-dipole forces, London dispersion forces, and hydrogen bonding. There are three types of molecular solids, which are classified according to the type of intermolecular force involved: nonpolar molecular solids, polar molecular solids, and hydrogen-bonded molecular solids. Molecular solids form crystals where the molecules tend to pack in a way that maximizes the intermolecular forces. So, the way that the molecules are arranged in the crystal lattice depends upon the shape of the molecules and the types of intermolecular forces involved. Nonpolar molecular solids are composed of atoms of the noble gases, nonpolar diatomic molecules, or nonpolar organic molecules. The attractive force that holds them together in the solid form is the weak London dispersion forces. Because the forces between the molecules are very weak, very few nonpolar molecular solids exist in solid form at room temperature and pressure. The solid form can only be observed at very low temperatures and they frequently sublime, going directly from the solid phase to the gas phase without passing through the liquid phase. In nonpolar molecular solids, the London dispersion force between the molecules (F) is directly proportional to the inverse seventh power of the distance (r) between the centers of the molecules; F¼ k r7 (3) Since the attractive force declines rapidly with distance, the molecules must pack as closely as possible to maintain the solid structure. So, the atomic or diatomic nonpolar molecular solids form more simple crystal structures since their molecular size is small. The noble gases all form face-centered cubic crystal structures at low temperatures. Diatomic hydrogen forms a simple hexagonal lattice structure and the diatomic molecules (H2 and the halogens) form monoclinic lattice structures. Nonpolar organic molecules vary widely in their solid forms due to the wide variation in molecular shapes and sizes. In crystalline methane (CH4), the molecules are arranged in a face-centered cubic structure. Carbon tetrachloride forms a 370 11. SOLIDS monoclinic crystal structure in order to accommodate the larger chlorine atoms. At the other extreme, larger hydrocarbons such as pentane (C5H7) have only one molecule per unit cell. For large organic molecules with more irregular shapes, the London forces depend on the rotational orientation of the molecules. So, in order to maximize the intermolecular force, the molecules in the crystal may have very unusual arrangements. Polar molecular solids are formed by polar molecules held together by dipole-dipole forces, which guide the formation of the crystal lattice. The orientation of the dipole-dipole forces is a major driving force behind the crystal structure of the polar molecular solid. The molecules will arrange so that the partially positive area of one molecule (δ+) interacts with the partially negative area of another molecule (δ). This interaction may be accomplished with more than one arrangement of molecules. For example, solid formaldehyde (CH2O) will prefer to adopt the following three configurations: The magnitude of the attractive force between two molecules depends on the dipole moment of each molecule (μ1, μ2), the distance between the dipoles (r), and the orientation of the two molecules. For the case where the orientation is end to end, the dipole-dipole force is given by; μ μ (4) F ¼ k 1 3 2 r The dipole-dipole force is directly proportional to the inverse third power of the distance between the centers of the molecules. So, it does not fall off as rapidly with distance as the London dispersion force. The polar molecular solids are capable of forming crystal structures where the molecules can be somewhat farther apart than in the nonpolar molecular solids. For example, sulfur dioxide forms an orthorhombic crystal structure with only four molecules per unit cell. Most molecular solids actually fall in the third category, hydrogen-bonded solids, involving the interaction of a δ+ hydrogen on one molecule with a δ oxygen, nitrogen, or fluorine on an adjacent molecule. Hydrogen bonds are the strongest intermolecular force due to the very strong dipole moment in molecules with hydrogen covalently bonded to a very electronegative atom (O, F, or N). These solids are the hardest of all the molecular solids with the highest melting points. Many hydrogen-bonded solids remain in the solid form at room temperature and pressure. The most common hydrogen-bonded solid is ice, discussed in detail in Section 3.8. Ice, with a hexagonal crystal lattice where each water molecule is hydrogen-bonded to four other water molecules, has a more open structure than liquid water with a partially ordered 11.3 MOLECULAR SOLIDS 371 tetrahedral structure. Recall that this is why water expands as it freezes. Other examples of hydrogen-bonded molecular solids are the organic acids and amino acids. Organic acids all have the functional group dCO2H, which will be discussed in detail in Chapter 13. The hydrogen bonding occurs between the δ+ hydrogen on the dCO2H and the δ of the C]O on an adjacent molecule. Two simple organic acids: formic acid (CO2H) and acetic acid (CH3CO2H) both have orthorhombic crystal structures with each molecule linked to two others through hydrogen bonds. The properties of molecular solids are dictated by the type and strength of the intermolecular forces between the molecules. While the melting points of ionic solids range from 1000°C and higher, the melting points of molecular solids are less than 300°C. For the most part, molecular solids are gases, liquids, or low melting point solids because the intermolecular forces of attraction are weak compared to ionic or covalently bonded solids. The value of the melting and boiling points will directly depend on the strength of the intermolecular forces, increasing in the order of: nonpolar molecular solids < polar molecular solids < hydrogen-bonded molecular solids. In the nonpolar solids, larger molecules will have larger London dispersion forces and higher boiling and melting points than smaller molecules. In both polar solids and hydrogen-bonded solids, the more polar molecules have stronger dipoles and stronger attractive forces resulting in higher boiling and melting points. EXAMPLE 11.6: DETERMINING THE RELATIVE MAGNITUDE OF MELTING POINTS OF MOLECULAR SOLIDS List the following molecular solids in the order of increasing melting points: H2 O, CO2 ,CCl4 , H2 S,C2 H6 : 1. Determine the type of molecular solid. H2O is a hydrogen-bonded molecular solid. CO2 is a nonpolar molecular solid. CCl4 is a nonpolar molecular solid. H2S is a polar molecular solid. Evan though it contains hydrogen, the electronegative atom must be either oxygen, nitrogen, or fluorine to be hydrogen-bonded. C2H6 is a nonpolar molecular solid. 2. Determine the order of increasing melting points. Nonpolar < polar < hydrogen-bonded So: CO2, CCl4, C2H6 < H2S < H2O. The melting points of the nonpolar solids range according to molecular size: CO2 < CCl4 < C2 H6 So, the order of increasing melting points is: CO2 < CCl4 < C2 H6 < H2 S < H2 O Most molecular solids are soft, but can also be ductile or brittle. While brittle solids fracture immediately when enough stress is applied, ductile solids undergo a period of 372 11. SOLIDS deformation followed by fracture. The intermolecular forces in many molecular solids are directional, leading to mechanical properties that vary with the direction of the applied stress. Molecular solids are usually insulators because the localization of electrons in the covalent bonds of the molecules prevents their movement in the crystal lattice. However, if an alkali metal is intercolated into the crystal lattice voids, the valence electrons on the alkali metal can be easily ionized allowing them to move freely in the crystal lattice. This gives the molecular solid electrical conductivity it would not otherwise have without significantly changing the other properties. 11.4 ATOMIC SOLIDS Atomic solids, also called network solids, are composed of atoms connected by covalent bonds in a continuous network. There are two types of atomic solids: those that are totally composed of an extended three-dimensional covalent network and those that are composed of two-dimensional covalent networks, which are held together by weaker van der Waals forces (mixed bonding). In the three-dimensional networks, the covalent bonding extends throughout the solid and the result is a macroscopic interlocking covalent lattice structure. Any distortion of the crystal geometry can only occur through breaking the strong covalent bonds. In the two-dimensional networks, distortion of the three-dimensional geometry is more easily accomplished by disturbing the weaker intermolecular forces holding the two-dimensional planes together. The most common examples of atomic solids are the allotropes of carbon. These allotropes, different solid forms of carbon, are shown in Fig. 11.9. The two most common allotropes of carbon are diamond and graphite. In the diamond structure, each carbon atom is covalently FIG. 11.9 Four allotropes of carbon. Modified from diamond and graphite.jpeg; Torsten Brandmueller; and Rao010384, Wikimedia Commons. Diamond Graphite 0.1 5n m Fullerene Nanotube 11.4 ATOMIC SOLIDS 373 bonded to four other carbon atoms in a tetrahedral arrangement to create a three-dimensional extended crystal lattice. The unit cell of diamond is a face-centered cubic structure with four additional carbon atoms inserted into four of the tetrahedral holes. In graphite, each carbon is bonded to three other carbons forming a two-dimensional planar network of six member rings. These two-dimensional arrays of carbon atoms are stacked one on top of the other to form a three-dimensional solid. The two-dimensional layers are held together by London dispersion forces to form a hexagonal unit cell structure. As a result of the combination of covalent bonding and London forces, graphite exhibits properties typical of both covalent and molecular solids. Until the1980s, carbon was thought to exist only in these two allotropes. Then in 1985, a third form of carbon, C60, was discovered in interstellar dust. This new form of carbon was called buckminsterfullerene, or fullerene for short, after the American architect R. Buckminster Fuller who is famous for constructing geodesic domes showing a close similarity to the structure of C60. Fullerene is a sphere composed of six-member and five-member carbon rings. The C60 macromolecules are held together in a bulk structure by London forces, in a manner similar to graphite, with a face-centered cubic unit cell. Fullerene actually represents an entire class of carbon atomic solids. Distorted spheres containing more than 60 carbon atoms have been found along with long tubes of the same structure. The cylindrically shaped fullerenes are called carbon nanotubes because, although they can be very long, they are typically only a few atoms in circumference. Another type of carbon nanotube has been constructed from single two-dimensional sheets of graphite, known as graphene. They are usually only a few nanometers wide, but can range from less than a micrometer to several millimeters in length. They also can be held together in a hexagonal bulk structure by London forces. The unique molecular structure of carbon nanotubes gives them some extraordinary macroscopic properties including: high electrical and heat conductivity and high tensile strength. Graphene nanplatelets, small stacks of graphene sheets, have been developed that are better conductors of electricity than any other material at room temperature and are of great interest in the areas of electronics. Other substances can also form allotropes similar to those of carbon. For example, silicon dioxide forms a covalently bonded three-dimensional network similar to diamond, called quartz. It also forms a layered two-dimensional structure similar to graphite, called mica. Boron nitride (BN) also forms a three-dimensional cubic crystal structure similar to diamond (cBN) and a two-dimensional hexagonal form that corresponds to graphite (hBN). It can also form nanotubes with a structure similar to carbon nanotubes. Since the three-dimensional and two-dimensional forms of atomic solids are made up of different bonding structures, they have different properties. A comparison of some important properties between the allotropes of carbon, boron nitride, and silicon dioxide are shown in Table 11.3. The three-dimensional forms, diamond, cBN and quartz, all have high Mohs (scratch hardness) and Vickers (deformation hardness) hardness values due to the strong covalent bonds throughout the structure while graphite, hBN, and mica have low hardness values due to the weak van der Waals forces holding the two-dimensional sheets together. The strength values generally follow the same patterns except for tensile strength of boron nitride which is higher than expected in the hexagonal form. The electrical conductivity is generally low and the resistance is generally high for all atomic solids because the electrons are localized in the covalent bonds. This lack of movement 374 11. SOLIDS TABLE 11.3 A Comparison of Some Important Properties Between the Allotropes of Carbon, Boron Nitride, and Silicon Dioxide Diamond cBN Quartz Graphite hBN Mica Hardness (Mohs) 10 9.5–10 7 1.5 2 2–2.5 Hardness (Vickers) 8600 5000 1400 7–11 15–24 2 Tensile strength (MPa) 60,000 50 150 5 171 2.4 Compressive strength (MPa) 110,000 47,000 1100 96 24 230 16 10 1013 – 105 – – 720 14 24 30 53 3027 1715 3730 3000 1275 10 Electrical conductivity (S/m) – – Thermal conductivity (W/m • K) 2200 Melting point (C) 3550 10 17 4 10 10 Resistance (Ω • m) 11 14 in the solid structure does not allow for the flow of electricity. The one exception to this is graphite. The carbon atoms in graphite are covalently bonded to three other carbons in sp2 hybrid bonds. The fourth available valence electron is not involved in bonding and becomes “delocalized.” Delocalized electrons are electrons that are not associated with a single atom or a covalent bond. Since the delocalized electrons in graphite are not involved in bonding, they are free to move in the graphene plane making graphite electrically conductive. This bonding structure will be discussed in more detail in Chapter 13. However, the electrons can only move within the two-dimensional plane and so graphite does not conduct electricity in a direction at right angles to the covalent plane. In summary, due to the very strong covalent bonds in the three-dimensional arrays, they have very high melting and boiling points, they are solids at room temperature, they are extremely hard, and they do not conduct electricity. However, most are good thermal conductors. The strong covalent bonds give them their hardness and high melting points while the lack of delocalized electrons account for the lack of electrical conductivity. The atoms in the three-dimensional crystals lie along several different planes and, when cut along these planes, light is reflected by them causing the cut crystals to have a sparkle typical of diamond and zirconia (ZrO2). The two-dimensional arrays held together by weak London forces also have high melting and boiling points, but they are soft because the weakly bonded layers will slide over each other. They also lack electrical conductivity, except for graphite, and have low thermal conductivities. 11.5 METALLIC SOLIDS Metallic solids, such as crystals of copper, iron, and zinc, are composed of metal atoms held together by a unique type of bonding known as metallic bonding. The metallic crystal structures are primarily body-centered cubic, face-centered cubic, and hexagonal. Some of the larger elements in groups 13 through 15 of the periodic table include tetragonal (In, Sn), orthorhombic (Ga), and monoclinic (Bi) structures. The bonding that holds the atoms together 11.5 METALLIC SOLIDS 375 in the crystal structure is a variation of covalent bonding where electrons are shared over multiple atoms. As described in Chapter 3, electrons in a covalent bond reside in molecular orbitals formed from the overlap of the atomic orbitals. In metallic bonding, all of the valence atomic orbitals on all of the atoms in the metallic solid overlap to give a vast number of molecular orbitals that extend over the entire solid. For example, the metals in group 1 of the periodic table have a single valence electron in the outer s subshell. In a metallic solid of the group 1 metals, the valence electrons are shared with the nearest neighbors in a molecular orbital in a manner similar to the formation of a covalent bond in an atomic solid. However, unlike in a traditional covalent bond where the electrons are shared between two atoms, in a metallic solid the electrons can also be shared with the nearest neighbors, the second closest neighbors, their closest neighbors, and so on. In this manner, the electrons in these molecular orbitals are free to move from atom to atom across the entire solid and so each electron becomes separated from its parent atom. The large array of overlapping molecular orbitals in the metallic solid is known as the conduction band and the electrons in this band are said to be delocalized. The metallic solid is held together by the strong forces of attraction between the positively charged atoms and the delocalized electrons. This metallic solid structure is often described as a uniform array of metal atoms in a “sea” of delocalized electrons as shown in Fig. 11.10. Although all metallic solids are good electrical conductors due to the mobility of the delocalized electrons, the electrical conductivities of each metal vary according to their ability to delocalize the valance electrons. Fig. 11.11 (top) shows the average electrical conductivity for metals in each group of the periodic table. They range from 1.8 106 to 2.1 107 S/m in groups 1 through 10. The values are higher for groups with unfilled subshells and lower for groups with filled or half-filled subshells. This is because the valence electrons in metals with closed shells are held more tightly to the parent atom and are less likely to become delocalized. The group 11 metals (Cu, Ag, Au) have the highest electrical conductivity at 5.5 107 S/m. With a valence electronic configuration of d10s1, the s1 electron is very easily delocalized in the group 11 metals. Although the metals in group 1 also have a valence electronic configuration of s1, the electrons are more tightly held to the positively charged atoms than metals in group 11 due to their smaller size, resulting in a lower electrical conductivity for the metals in group 1. The electrical conductivities of the metals after group 11 decrease from group 12 through 16 as the electronic shells become filled. FIG. 11.10 Metallic bonding formed by metallic solids in group 1 of the periodic table. From Muskid, Wikimedia Commons. 376 11. SOLIDS FIG. 11.11 Electrical conductivity (S/m 107) and thermal conductivity (W/m • K) for the average of the metals in each group of the periodic table. Metallic solids are also good thermal conductors because the atoms are closely packed together and the delocalized electrons can carry kinetic energy through the solid. Fig. 11.11 (bottom) compares the average thermal conductivity to the average electrical conductivity for each group of the periodic table. The thermal conductivity follows the same periodic trend as the electrical conductivity for the same reasons. Because thermal conductivity also depends on the ability of the valence electrons to move through the solid, it increases with the increased tendency of the valence electrons to become delocalized. The thermal conductivity of the metals in groups 1 through 10 range from 16 to 134 W/(m • K). The metals in group 11 have the highest thermal conductivity at 383 W/(m • K) and the values decrease from group 12 to group 16. Metallic solids also have high melting points because of the strong attraction between closely packed positive metal ions and the delocalized electrons. However, the strength of a metallic bond can vary widely and so the energy required to break the bond also varies widely. For example, cesium melts at 28.4°C, while tungsten melts at 3422°C. Fig. 11.12 (top) shows the average melting points for metals in each group of the periodic table. Metallic bonds tend to be weakest for elements that have either nearly empty or nearly full valence subshells and strongest for elements with half-filled valence shells. So, the melting points of the metals increase going across the periodic table from group 1 (82°C) to group 6 377 11.5 METALLIC SOLIDS FIG. 11.12 Melting Point (°C) and Mohs Hardness for the average of the metals in each group of the periodic table. (2651°C), which has two half-filled valence subshells (d5s1). The melting points then generally decrease from group 7 to the metalloids. Other properties related to the strength of the metallic bonds, such as boiling points, tensile strength, and hardness, have similar periodic trends as melting point. For example, the average Mohs hardness for the metals in each group of the periodic table is also shown in Fig. 11.12 (bottom). The hardness of the metals increase going across the periodic table from group 1 (0.4) to group 6 (7.2) following the same trend as the melting point. The hardness values also generally decrease after group 6 going across from group 7 to the metalloids. Metallic alloys are nonstoichiometric homogeneous mixtures of metals. There are two types of alloys. The first is a substitutional alloy where some of the metal atoms in the crystal lattice of the host metal are replaced by other atoms of the same size. One example of a substitutional alloy is brass, where one third of the atoms in a crystal of copper are replaced with zinc atoms. Pewter is also a substitutional alloy where copper (7%), bismuth (6%), and antimony (2%) replace atoms of tin in the crystal lattice. The second type of alloy is an interstitial alloy where atoms of one or more different metals reside in the voids in the crystal lattice of the host metal. An important example of an interstitial alloy is steel, where carbon occupies the voids in an iron crystal lattice. The higher the percentage of carbon, the stronger the steel. Some steels can be both substitutional and interstitial. In these alloys, other metals such as 378 11. SOLIDS chromium and vanadium substitute for the iron atoms in the lattice structure, while carbon remains in the voids. These types of steel are used for special applications such as cutlery and bicycle frames. CASE STUDY: SEMICONDUCTORS The metalloids that lie on the border between metals and nonmetals in the periodic table exist as solid crystalline structures with properties between atomic and metallic solids. For example, silicon and germanium have electrical conductivities much lower than the metals, but much higher than the atomic solids such as diamond. These solids are known as semiconductors because they possess electrical conductivities that lie between the metals and other types of solids. These intermediate electrical conductivity values are due to the unique structure of the molecular orbitals in the solid. Both silicon and germanium (s2p2) have four valence electrons which occupy four equivalent sp3 hybridized orbitals. These atomic orbitals overlap with atomic orbitals of other atoms in the crystal to give four molecular orbitals, which are split into two groups of different closely spaced energies as shown in Fig. 11.13. The lower energy molecular orbitals are called the valence band where the electrons reside in traditional covalent bonds between two atoms. The higher energy molecular orbitals form the conduction band where electrons can become delocalize due to the extended overlap of the molecular orbitals with surrounding atoms. The energy region between the valence band and the conduction band is known as the band gap (Eg). In silicon, the band gap is equal to 1.94 1019 J and in germanium it is 1.06 1019 J. Electrons can’t exist in the band gap since it does not consist of molecular orbitals. Electrons can only reside in the molecular orbitals of the valence band or the conduction band. The electrons in the valence band must gain energy equivalent to the band gap in order to reach the conduction band. According to the Boltzmann distribution of kinetic energies discussed in Section 9.5, only a few electrons have enough energy to make the transition from the valence band into the conduction band at room temperature. This small number of electrons in the conduction band gives silicon and germanium higher electrical conductivities than the atomic solids, but lower electrical conductivities than the metallic solids. One way to increase the conductivity of a semiconductor is to increase the temperature (see Fig. 9.6). The increase in temperature provides extra kinetic energy, which increases the number of electrons capable of reaching the conduction band. Another way of increasing the number of electrons reaching the conduction band is by a process called doping. Doping is the deliberate FIG. 11.13 The four sp3 hybridized valence atomic orbitals in a silicon atom combine in the silicon crystal to give molecular orbitals in two closely spaced energy bands: the valence band, which is almost completely filled at room temperature, and the conduction band, which is almost empty. 379 11.5 METALLIC SOLIDS introduction of impurities into the crystal structure of a semiconductor in order to alter the electrical properties of the pure substance. For the case of silicon, this process involves the addition of a group 15 element, such as arsenic or antimony, which is substituted for the silicon atoms (group 14) in the crystal lattice. The group 15 elements (s2p3) have five valence electrons. This introduces one more electron into the silicon crystal than is needed for bonding. Since there is an additional negative charge due to the addition of the group 15 element, a semiconductor with this type of doping is called an n-type semiconductor. Less energy is required to promote these extra electrons into the conduction band than is required to promote the silicon electrons that are involved in bonding, which results in more electrons in the conduction band. The increase in the number of electrons in the conduction band results in an increase in electrical conductivity of the semiconductor. Silicon can also be doped by adding a group 13 element, such as gallium, to the crystal lattice. Group 13 elements (s2p1) have only 3 valence electrons. This results in one less electron and one unoccupied molecular orbital in the valence band. This unoccupied molecular orbital is commonly referred to as a “hole.” The electrons in the valence band can move to fill the hole when an external voltage is applied to the crystal as shown in Fig. 11.14. As the electrons move to fill the vacant orbitals, they leave behind a silicon atom with a positive charge. So, a semiconductor with this type of doping is called a p-type semiconductor. An alternate way to look at this process is that as the electrons move to fill the holes, they leave behind an unoccupied orbital creating another hole. This makes it appear that the holes are moving in the opposite direction as the electrons. This view of moving holes is a common way of describing electrical conductance of p-type semiconductors. But remember that the species that is actually moving is the valence electron. Both n- and p-type semiconductors are used extensively in electronics as rectifiers, transistors, and light emitting diodes. When a p-type semiconductor and an n-type semiconductor are brought in contact with each other, a p-n junction is formed. Some of the excess electrons in the n-type semiconductor FIG. 11.14 n-type semiconductor +− Conductor band − − − − − − − − − − − − − − − − − − − − Valence band p-type semiconductor + − Conductor band − − − − Valence band − − − − − − − − In an n-type semiconductor, the dopant has an extra electron which can more easily be promoted into the conduction band, resulting in an increased electrical conductance. In a p-type semiconductor, the dopant has one less electron than is needed for bonding. This results in an unoccupied molecular orbital or “hole” in the crystal lattice. The electrons in the valence band will move to occupy the holes when an external voltage is applied, making it appear that the holes are moving in the opposite direction. 380 11. SOLIDS migrate across the junction to fill the holes in the p-type semiconductor. The result is a buildup of positive charge in the n-type semiconductor and a buildup of negative charge in the p-type semiconductor. This charge buildup is called the junction potential, which prevents the further movement of electrons. Electrons will continue to move if an external potential is applied to the p-n junction, with the negative pole connected to the n-type material and the positive pole connected to the p-type material. This creates a device called a diode where current can flow in one direction, but not in the other direction. These p-n junctions serve as the basis of electronic gates and integrated circuits. 11.6 AMORPHOUS SOLIDS Some examples of amorphous solids are shown in Fig. 11.15. As described in Chapter 1, an amorphous solid is a solid material that lacks the highly ordered microscopic structure extending in all directions typical of a crystalline solid. This can be due to defects in the crystal lattice of a normally crystalline material or it can be due to materials consisting of very large molecules that cannot form ordered structures. One of the most defining properties of an amorphous solid is that they tend to melt slowly over a wide temperature range rather than having a well-defined melting point like a crystalline solid. This is primarily because many amorphous solids are composed of different types of bonding between the atoms or molecules, which require different amounts of energy to break the bonds. But it can also be because the solid is composed of molecules with the same structure but varying molecular weights, as with the polymers discussed in Chapter 13. Microcrysalline SiO2 Plastic FIG. 11.15 “Mouldavite” Glass BN ceramic Gel SiO2 Some examples of amorphous solids. From H. Raab, Takis Iazos, Materialscientist, and Petra Klawikowski; Wikimedia Commons. 11.6 AMORPHOUS SOLIDS 381 Amorphous solids are divided into two groups: microcrystalline solids and pseudo solids. As with crystalline solids, the atoms or molecules in microcrystalline solids are close together with little freedom to move. They have bonding similar to crystalline solids, but the crystalline structure contains many defects that disrupt any formation of an extended regular lattice structure. These defects take the form of lattice points that have missing atoms or atoms that are located at sites other than their regular positions. Because of these many defects, microcrystalline solids have an irregular bonding pattern and do not exhibit any kind of long range regular structure. But they do show a regular lattice arrangement over small regions, known as a short range or microcrystalline order. Any kind of crystalline solid can also exist as a microcrystalline amorphous solid. Because the formation of the highly ordered crystal lattice from an unordered liquid solution cannot occur rapidly, the crystallization process must be allowed to proceed slowly for the crystalline solid to form. If the conditions change too rapidly, crystalline order may begin to form, but it will become disrupted before the ordered form continues beyond a microscopic range. Microcrystalline solids also include the ceramics and cements. Hardened Portland Cement (calcium silica hydrate) has a short range ordered geometry consisting of layers of very long chains of silica molecules combined with layers of calcium oxide. These crystalline areas are broken by areas where the silica and calcium oxide layers mix creating voids in the structure. It is these voids that provide some flexibility to the building materials constructed of Portland cement. When under stress, the cement has the flexibility to stretch or compress instead of breaking like an ordered crystalline structure. Ceramics are mixtures of several compounds and are composed of microcrystalline regions cemented together by glassy structures. They are commonly composed of both covalent and ionic bonding. This strong bonding gives ceramics their high hardness, high compressive strength, high melting points, low thermal expansion, and good chemical resistance. But it also causes them to have a low ductility and low tensile strength. Since the valence electrons are held in the strong covalent or ionic bonds, most ceramics are poor conductors of electricity and heat. The microcrystalline structure of ceramics is very important because slight differences in this structure can strongly influence the properties of individual ceramic pieces. Although ceramics generally do not deform before they fail under stress, failure is caused by weak points in the bonding in the microcrystalline regions. These weak points are not consistent from sample to sample, so processing must be strictly controlled to reduce the inconsistencies in the microcrystalline regions in order to minimize the failure rates. Some special types of amorphous solids are the pseudo solids: glasses, organic polymers, gels, and thin films. They are commonly not considered to be true solids because they are capable of flowing like a fluid under an applied shear stress. Both glasses and organic polymers are formed of long irregularly shaped structures that can easily become tangled and disordered. They both exhibit a glass transition, a reversible transition from a hard and relatively brittle glassy state into a viscous or rubbery state as the temperature is increased. Like liquids, these disorganized solids are capable of flow. Over long periods of time, the molecules making up a glass will slowly shift in order to settle into a more stable, crystalline like formation. The closer to the glass transition temperature, the more rapidly the molecules move and the faster they can shift. The farther from the glass transition temperature, the slower the molecules move, the slower they shift, and the more solid the substance appears. 382 11. SOLIDS TABLE 11.4 Summary of Structures and Properties of Solid Materials Electrical Conductivity Melting Point Other Ionic bonds Poor as solid good as liquid High Hard, brittle Molecules London, dipoledipole, hydrogen bond Poor in solid and liquid Low Soft, ductile Atomic Atoms Covalent bond Poor (exception graphite) High 3d form is hard, 2d form is soft Metallic Metal cations Metallic bond Good in solid and liquid Varies Malleable, ductile, hardness varies Amorphous Covalent bonds of varying strengths Covalent bond Poor Wide range Irregular fracture Type Structural Units Internal Forces Ionic Cations and anions Molecular Although pure silica (quartz) will form a glass if cooled quickly, its high melting point (900°C) makes it difficult to shape and so is not suitable for making containers. However, it is widely used in UV-visible spectroscopy due to its good transparency in this wavelength region. The most common glass used in windows is composed of SiO2 with Na+ and Ca2+ ions distributed throughout a covalently bonded OdSidO network. This ionic-covalent structure is a giant polyanion called soda lime glass. This glass has a lower melting point and is easier to shape than pure silica. It also has a low thermal conductivity and a high coefficient of thermal expansion. This means that internal stresses are created when exposed to extreme heat or cold, which may cause it to shatter. These internal stresses can be eliminated by a process called annealing. This is done by slowly heating the glass to a temperature just below the glass transition and maintaining the temperature for a period of time before allowing it to cool slowly. This allows for a short range diffusion of atoms in the glass to occur relieving the stresses. Another important class of amorphous solids is organic polymers that can be molded into shapes when hot and hardened upon cooling. Some examples are Bakelite (the first plastic), poly ethylene, polypropylene, polystyrene, and polytetrafluroethylene (Teflon). Each of these is composed of long chain organic molecules that cannot form any ordered structure. Further discussion of organic polymers is left to Chapter 13. A summary of the structures and properties of solid materials is given in Table 11.4. IMPORTANT TERMS Allotropes two or more different forms of an element in the same physical state. Annealing a process used to relieve internal stresses in a glass by slowly heating the glass to a temperature just below the softening point, maintaining the temperature for a period of time, and allowing it to cool slowly. Atomic solids solids composed of atoms connected by covalent bonds. Band gap (Eg) the energy region between the valence band and the conductance band in a semiconductor. Born-Haber cycle an application of Hess’s law used to calculate lattice energies. Cleaving causing an ionic solid to split along the axis parallel to the planes of the ions by an application of an outside force. STUDY QUESTIONS 383 Close packed structure the most tightly packed and space-efficient arrangement of ions in a crystal lattice. Conduction band a group of molecular orbitals of the same energy in a crystal lattice through which electrons are free to move easily. Coordination number the number of nearest neighbor ions of opposite charge surrounding any ion in a crystal structure. Crystal lattice the simplest repeating unit in a crystalline solid. Crystalline solid a solid material with the constituent species arranged in a highly ordered microscopic structure that extends in all directions. Crystallization the process by which a crystalline solid is formed. Delocalized electrons electrons in a molecule, ion, or solid metal that are not associated with a single atom or a covalent bond. Diode a device constructed of a p-n junction that allows the flow of electrical current only in one direction. Doping the deliberate introduction of impurities into the crystal structure of a semiconductor in order to alter the electrical properties of the pure substance. Fast ion conductors ionic solids in which the ions are highly mobile. Glass transition a reversible transition in an amorphous solid from a hard and relatively brittle glassy state into a viscous or rubbery state as the temperature is increased. Interstitial alloy a metallic alloy where atoms of one or more different metals reside in the voids in the crystal lattice of a host metal. Ionic solids solids composed of positive and negative ions held together by electrostatic attractions. Junction potential the charge buildup at a p-n junction caused by the migration of electrons into the p-type material. Lattice energy the energy released when a crystal lattice is formed. Lattice points the geometric points that define the shape of the unit cell, which are also the positions of the constituent species. Metallic alloys nonstoichiometric homogeneous mixtures of metals. Metallic solids solids composed of metal atoms that are held together by metallic bonds. Microcrystalline solid a solid material with the constituent species arranged in an ordered microscopic structure over a limited range. Molecular solids solids composed of atoms or molecules held together by London dispersion forces, dipole-dipole forces, or hydrogen bonds. n-type semiconductor a semiconductor formed by the addition of an element with one more valence electron than the host element creating an excess of electrons in the crystal lattice. Octahedral voids spaces in a close packed ionic crystal structure that are surrounded by six ions of like charge. p-n junction a boundary between a p-type semiconductor and an n-type semiconductor. p-type semiconductor a semiconductor formed by the addition of an element with one less valence electron than the host element creating a deficit of electrons and vacant molecular orbitals in the crystal lattice. Pseudo solids amorphous solids that are capable of flowing like a fluid under large stresses. Semiconductor a crystalline solid that possess electrical conductivities that lie between the metals and other types of solids. Substitutional alloy a metallic alloy where some of the host metal atoms in the crystal lattice are replaced by other atoms of the same size. Tetrahedral voids spaces in a close packed ionic crystal structure that are surrounded by four ions of like charge. Unit cell the symmetrical three-dimensional arrangement of the constituent species inside a crystalline solid. Valence band a region of molecular orbitals in a crystalline solid where the electrons reside in traditional covalent bonds between two atoms. X-ray crystallography a technique used to determine the structure of a crystalline solid, by diffraction of X-rays. STUDY QUESTIONS 11.1. What are the two types of amorphous solids? 11.2. What are the four types of crystalline solids? 11.3. What is the difference between amorphous and crystalline solids? 384 11. SOLIDS 11.4. What technique can give a three-dimensional picture of the microscopic arrangement of the crystalline solid? 11.5. What is a unit cell? 11.6. What are the lattice points of a unit cell? 11.7. When the unit cell is repeated in three dimensions, the resulting network of ions is called what? 11.8. What are the three defining properties of a unit cell? 11.9. Name the seven classes of unit cells. 11.10. What type of unit cell is in the shape of a rectangle with all sides (length, width, and height) of equal length and all angles equal to 90 degrees? 11.11. What type of unit cell is in the shape of a rectangle with all sides (length, width, and height) of different lengths and all angles equal to 90 degrees? 11.12. What type of unit cell is in the shape of a hexahedron with all sides (length, width, and height) of different length and all angles not equal to 90 degrees? 11.13. What type of unit cell is in the shape of a hexahedron with all sides (length, width, and height) of different lengths and only two angles equal to 90 degrees? 11.14. The different types of unit cells can exist in different forms. What are the possible four forms? 11.15. Which type of unit cell can exist in all four forms? 11.16. An atom at a lattice point in a simple cubic unit cell is shared by how many unit cells? 11.17. The atom located on a face a face-centered cubic unit cell is shared by how many unit cells? 11.18. How many atoms are there in a single unit cell of the following type: (a) face-centered cubic, (b) side-centered cubic, (c) simple rhombic, (d) side-centered monoclinic, (e) body-centered orthorhombic? 11.19. What is the number of nearest neighbor ions of opposite charge surrounding an ion in an ionic solid called? 11.20. What is a close packed crystal structure? 11.21. A tetrahedral void in a unit cell is surrounded by how many ions? 11.22. An octahedral void in a unit cell is surrounded by how many ions of like charge? 11.23. What determines which crystal structure an ionic compound will adopt? 11.24. The energy released when an ionic crystal is formed is called what? 11.25. What is a Born-Haber cycle? 11.26. What are the five steps in a Born-Haber cycle? 11.27. How is lattice energy and ionic bond strength related? 11.28. What is cleaving in an ionic crystal? 11.29. (a) What important property does a fast ion conductor possess? (b) What structural anomaly gives rise to this property? 11.30. What are the forces that hold molecular solids together? 11.31. What are the three types of molecular solids and what is the connecting force for each? 11.32. What force connects the atoms in an atomic solid? 11.33. What are allotropes? 11.34. Name four allotropes of carbon. 11.35. What type of bonding do metallic solids have? PROBLEMS 11.36. 11.37. 11.38. 11.39. 11.40. 11.41. 11.42. 11.43. 11.44. 11.45. 11.46. 11.47. 11.48. 11.49. 11.50. 11.51. 11.52. 11.53. 385 What feature is responsible for the high electrical conductivity of metals? Why are metal good thermal conductors? Why do metallic solids have high melting points? What is a metallic alloy? (a) What is the difference between a substitutional alloy and an interstitial alloy? (b) Give an example of each. What is the conduction band in a semiconductor? What is the valence band? What is the space between the conduction band and the valence band called? Why does a silicon and germanium have a higher than expected electrical conductivity? What is meant by doping a semiconductor? What is an n-type semiconductor? What is a p-type semiconductor? What is a p-n junction? What is the junction potential? What is the function of a diode? (a) What is the difference between the melting points of crystalline solids and those of an amorphous solid? (b) What causes this difference? What is a microcrystalline solid? (a) Why are glasses and organic polymers considered to be amorphous solids? (b) Why are they pseudo solids? How does annealing help prevent glass from shattering? PROBLEMS 11.54. Identify the following solids as crystalline or amorphous: (a) Al2O3, (b) CH2O, (c) Teflon, (d) Al, (e) steel, (f) CadSidH2O. 11.55. Solid platinum has a face-centered cubic unit cell with an edge length of 392.0 pm. What is the density of platinum? 11.56. Nickel crystallizes in a face-centered cubic crystal lattice. The density of a unit cell is 8.908 g/cm3. What is the length of the unit cell edge in pm? 11.57. Metallic iron crystallizes in a type of cubic unit cell. The unit cell edge length is 287 pm and the density of iron is 7.87 g/cm3. (a) How many iron atoms are there within one unit cell? (b) What is the type of unit cell? 11.58. The ionic compound CuO2 has a crystal structure of body-centered cubic. If the coordination number of O2 is 4, what is the coordination number of Cu+? 11.59. The crystal structure of PuO2 is face-centered cubic with all the tetrahedral voids occupied by O2 ions. What is the coordination number of the Pu4+ ions? 11.60. The ionic solid AnBm crystallizes in a close packed array with B ions in two thirds of the octahedral voids. What is the empirical formula? 11.61. What is the coordination number of A in Problem 11.60? 11.62. Cadmium sulfide crystallizes with cadmium occupying ½ of the tetrahedral voids in a closest packed array of sulfide ions. (a) What is the empirical formula of cadmium 386 11.63. 11.64. 11.65. 11.66. 11.67. 11.68. 11.69. 11.70. 11.71. 11.72. 11.73. 11.74. 11.75. 11.76. 11.77. 11.78. 11.79. 11. SOLIDS sulfide? (b) What is the coordination number of cadmium? (c) What is the coordination number of sulfide? What is the empirical formula of cobalt oxide, which crystallizes with cobalt ions occupying ⅛ of the tetrahedral voids and ½ of the octahedral voids in a close packed array of oxide ions? What is the empirical formula of titanium in titanium oxide, which exists in a close packed array with titanium ions in ½ of the octahedral voids? What is the percent by mass of titanium? Calculate the lattice energy for lithium fluoride given the following information: The enthalpy of sublimation for solid lithium is 161 kJ/mol, the first ionization energy of lithium is 520 kJ/mol, the F2 bond dissociation energy is 77 kJ/mol, the electron affinity of fluorine is 328 kJ/mol, and the enthalpy of formation for solid lithium fluoride is 617 kJ/mol. Calculate the lattice energy for RbCl given the following information: The electron affinity of Cl is 349 kJ/mol, the first ionization energy of Rb is 403 kJ/mol, the bond dissociation energy of Cl2 is 242 kJ/mol, the enthalpy of sublimation of Rb is 86.5 kJ/mol, and the enthalpy of formation of RbCl(s) is 430.5 kJ/mol. Calculate the lattice energy for MgCl2 given the following information: The electron affinity of Cl is 349 kJ/mol, the first ionization energy of Mg is 738 kJ/mol, the second ionization energy of Mg ¼ 1451 kJ/mol, the bond dissociation energy of Cl2 is 122 kJ/mol, the enthalpy of sublimation of Mg is 148 kJ/mol, and the enthalpy of formation of RbCl(s) ¼ 643 kJ/mol. What information is needed to construct a Born-Haber cycle for the calculation of the lattice energy of an ionic solid MX2 composed of a metal M with a charge of +2 and a nonmetal X with a charge of 1 that is diatomic in its standard state? List the following ionic solids in order of increasing lattice energy: NaF, CaO, CsI. Which ionic solid has the largest lattice energy: MgF2, CaF2, or ZrO2? List the following ionic solids in order of decreasing lattice energy: KCl, LiCl, NaCl. List the following ionic solids in order of increasing lattice energy: KCl, KI, KF, KBr. List the following ionic solids in order of decreasing melting point: NaBr, NaF, NaCl, NaI. Which will have the higher melting point: (a) KCl or CaCl2, (b) NaCl or CaO? (c) Mg(OH)2 or MgO, (d) Al(OH)3 or MgO? Solid CaCl2 has a simple cubic structure with an edge length of 4.77 108 cm and a density of 6.80 g/cm3. How many formula units are there in one unit cell? (Hint: the number of anions to cations in the unit cell must obey the empirical formula.) Magnesium oxide has a face-centered cubic unit cell with oxide ions at the lattice points and magnesium ions in the octahedral holes. What is the density of magnesium oxide if the edge length of the unit cell is 422 pm? What is the intermolecular force in each of the following molecular solids: (a)F2, (b) NO, (c) PCl3, (d) H2SO4, (e) Kr, (f) XeF? List the following molecular solids in order of increasing melting point: C6H6, SO2, NH3, N2, CHCl3. Which will have the higher melting point: HCl or HF? Why? PROBLEMS 387 11.80. Identify the type of bonding in the crystal lattice composed of each of the following constituent species: (a) SiO2, (b) CH4, (c) Cr, (d) Na2S, (e) CaO, (f) C, (g) HF. 11.81. What type of solids is each of the following: (a) diamond, (b) SO2, (c) SiO2, (d) glass, (e) Ag, (f) C2H2. 11.82. What type of constituent species occupies the lattice points in each of the following: (a) SiC, (b) HBr, (c) Cu, (d) I2, (e) NH4ClO3, (f) SiO2? 11.83. What kind of bonds must be broken in order to melt the following solids: (a) quartz crystals, (b) graphene, (c) amorphous quartz, (d) solid argon, (e) ice, (f) lithium chloride? 11.84. A crystalline solid has a melting point of 776°C and a boiling point of 1500°C. It is a poor conductor of electricity but is a good conductor in the liquid form. What is the type of solid? 11.85. A crystalline solid has a melting point of 157°C and a boiling point of 153°C. It is a poor conductor of electricity in both solid and liquid form. What is the type of solid? 11.86. A crystalline solid has a melting point of 660°C and a boiling point of 2470°C. It is a good conductor of electricity in both the solid and liquid form. What is the type of solid? 11.87. List the following chlorine containing compounds in order of increasing melting points: Cl2, NaCl, and CCl4. 11.88. Which will have the higher melting point: (a) CuBr2 or Br2, (b) CO2 or SiO2, (c) S or Al, (d) CsBr or CaF2 (e) H2O or H2? (f) glass or graphite? 11.89. Identify each of the following as a p-type or n-type semiconductor: (a) aluminum doped silicon, (b) antimony doped germanium, (c) boron doped silicon, (d) gallium doped germanium. C H A P T E R 12 Solution Chemistry O U T L I N E 12.1 Solution Composition 390 12.2 Dissolution 393 12.3 The Effect of Pressure on Solubility 396 12.8 Colligative Properties 12.8.1 Vapor Pressure Depression 12.8.2 Boiling Point Elevation 12.8.3 Freezing Point Depression 12.8.4 Osmotic Pressure 408 408 409 410 411 12.4 The Effects of Temperature on Solubility 399 Important Terms 414 12.5 Solubility of Ionic Solids 403 Study Questions 415 12.6 Complexing Agents 405 Problems 416 12.7 Surfactants 406 Recall from Chapter 1 that a solution is a special case of a homogeneous mixture of two or more substances in a single phase, usually a liquid phase, whose components are distributed uniformly on the microscopic scale. The major component is called the solvent, which is commonly a liquid. The minor component is called the solute, which can be a solid, liquid, or gas. The defining property of a solution is that of dissolution. The solute and solvent must interact either physically or chemically in order for a homogeneous mixture to be classified as a solution. The solute will not settle out of the mixture, nor can it be removed by mechanical methods. They can, however, be separated by physical methods involving a phase change of one of the components of the mixture or by chemical methods based on the differences in the chemical properties of the pure substances. Important chemical methods of separation include recrystallization and solvent extraction, which will be discussed in Section 12.4. Although the chemical properties of the solvent and solute are not changed by dissolution, the physical properties of both are altered. So, when a solid solute is dissolved in a liquid solvent, both solute and solvent exist in the liquid phase and the solute then takes on the physical phase of the solvent. The important physical properties of the solvent that are changed by the presence of the solute are colligative properties, properties that depend upon the General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00012-6 389 # 2018 Elsevier Inc. All rights reserved. 390 12. SOLUTION CHEMISTRY concentration of solute molecules or ions, but not upon the identity of the solute. Colligative properties include freezing point depression, boiling point elevation, vapor pressure lowering, and osmotic pressure. These will be discussed in Section 12.8. 12.1 SOLUTION COMPOSITION The properties and behavior of solutions depend on their composition. Although the composition of a solution can vary from one solution to the next, since the mixture is homogeneous, composition remains the same throughout a single solution. A small sample taken from anywhere in the solution will always have the same composition. The terms unsaturated, saturated, and supersaturated are used to relatively compare the composition of solutions with respect to the quantity of solute in a given quantity of solvent. A saturated solution is a solution that contains the maximum possible amount of solute under its current conditions. Any solution containing less than the maximum amount of solute is unsaturated. A solution that is more concentrated than a saturated solution is a supersaturated solution. The process of obtaining a supersaturated solution will be discussed in Section 12.4. The quantitative composition of a solution is expressed as the concentration of solute in the solvent. This can be done in a number of ways, as listed in Table 12.1: based on mass (mass percent), on volume (volume percent), on mass solute per volume of solvent (mass volume percent, parts per million, parts per billion, parts per trillion), on amount (mole fraction, mole percent), on amount of solute per volume of solvent (molarity), or on amount of solute per mass of solvent (molality). The unit of concentration most closely tied to the SI units and that most commonly used by chemists is molarity (mol/L), which was described in Section 4.8. Mass percent and volume percent are both unitless values that are usually used for more concentrated solutions such as: 30% hydrogen peroxide (w/w) ¼ 30 g H2O2/100 g solution TABLE 12.1 Methods of Determining the Concentration of a Solution Concentration Unit Definition Mass percent % (m/m) or (w/w) (Grams solute/grams solution) 100 Volume percent % (v/v) (mL of solute/mL of solution) 100 Mass/volume percent % (m/v) (Grams of solute/mL of solution) 100 Parts per million ppm Milligrams of solute/liters of solution Parts per billion ppb Micrograms of solute/liters of solution Parts per trillion ppt Nanograms of solute/liters of solution Mole fraction χ Moles of solute/moles of solution Mole percent % (χ) χ 100 Molarity M Moles of solute/liters of solution Molality M Moles of solute/kilograms of solvent Normality N Gram equivalent weight/liters of solution 12.1 SOLUTION COMPOSITION 391 or 99.8% (w/w) “glacial” acetic acid ¼ 99.8 g CH3CO2H/100 g solution. Mass/volume percent, which measures the mass (or weight) of solute in grams and the volume of solution in mL, is used because the units are more readily obtainable. It is easier to measure the mass of a solid solute than the volume and it is easier to measure the volume of a liquid solvent than the mass. An example where mass/volume percent is used routinely is for medical saline solutions, which contain 0.9 g of NaCl for every 100 mL of solution or 0.9% (w/v) sodium chloride. More dilute solutions are often expressed as parts per million (ppm), parts per billion (ppb), or parts per trillion (ppt). These concentration units are most often used in environmental chemistry where the solute concentrations in water are very small. In order to use these concentration units, the values for solute and solvent must be in the same units. So, a solution with 1 g of solute in 1 106 mL of solution can be converted to grams of solution by using the density of the solution. Because the solution is so dilute, the density of the solution can be approximated by the density of the solvent and when the solvent is water, the density of solution is 1 g/mL. 1 106 mL solution 1g=mL ¼ 1 106 g solution So, 1 g of solute in 1 106 mL of solution is 1 part solute in 106 parts of solution or; 1 g/(1 106 g) ¼ 1 mg/1000 g. For aqueous solutions: 1000 g ¼ 1 L and for aqueous solutions: 1 ppm ¼ 1 mg=L 1 ppb ¼ 1 μg=L 1 ppt ¼ 1 ng=L The concept of mole fraction was introduced in Chapter 6 in the discussion of gas mixtures and Dalton’s law (Section 6.7). For a solution with only one solute, the mole fraction of solute is defined as the amount of solute (n1) divided by the total amount of solute + solvent (n1 + n2); χ1 ¼ n1 n1 + n2 (1) Mole percent is simply mole fraction multiplied by 100. EXAMPLE 12.1: DETERMINING THE MOLE FRACTION OF A SOLUTION What are the mole fractions of ethanol and water in a 100 proof bottle of vodka? Proof ¼ 2 % (m/m). 1. Determine the mass of ethanol and water. 100 proof ¼ 100/2 ¼ 50% (m/m) The solution is 50% (m/m) ethanol in water 50% (m/m) ¼ 50 g ethanol in 100 g of solution Mass of ethanol ¼ 50 g Mass of water ¼ 100 g 50 g ¼ 50 g 2. Determine the number of moles of ethanol. n1 ¼ 50 g/(46.1 g/mol) ¼ 1.08 mol ethanol Continued 392 12. SOLUTION CHEMISTRY EXAMPLE 12.1: DETERMINING THE MOLE FRACTION OF A SOLUTION— CONT’D 3. Determine the number of moles of water: n2 ¼ 50 g/(18.0 g/mol) ¼ 2.78 mol 4. Determine the mole fraction of ethanol. n1 1:08 1:08 χ1 ¼ ¼ ¼ ¼ 0:28 n1 + n2 1:08 + 2:78 3:86 5. Determine the mole fraction of water. n2 2:78 2:78 ¼ ¼ ¼ 0:72 χ2 ¼ n1 + n2 1:08 + 2:78 3:86 Notice that the sum of the mole fractions of the components of the solution equals 1.00: 0:28 + 0:72 ¼ 1:00 The expressions for molarity and molality are similar. They differ only in the units of the denominators. While molarity is defined as; Moles of solute Liter of solution (2) Moles of solute Kilogram of solvent (3) M¼ molality is defined as; m¼ Molality becomes important in more concentrated solutions where the amount of solvent must be known accurately, such as with the determination of colligative properties (Section 12.8). Also, since liquids expand as temperature increases, their volumes vary with temperature and so the molarities of solutions also vary with temperature. Since molality does not include a volume measurement and mass does not change with temperature, molality is independent of temperature. Normality is defined as; N¼ Gram equivalent weight Liter of solution (4) where the gram equivalent weight is the mass of one equivalent (eq) of an ion in a compound, which is the number of moles of an ion dissolved in a solution multiplied by the ionic charge. For example, if 1 mol of NaCl and 1 mol of CaCl2 are both dissolved in the same solution, there is 1 equivalent of Na+, 2 equivalents of Ca2+, and 3 equivalents of Cl in the solution. A given amount of 1+ ions provides the same amount of equivalents, while the same amount of 2 + ions provides twice the amount of equivalents. The amount of a substance in equivalents is often very small, so it is often described in terms of milliequivalents (meq). This method of determining ionic concentrations is no longer widely used except for the measurement of ions in biological fluids. But, it is also of use in titrations involving acids with more than one hydrogen ion or bases with more than one hydroxide ion. For example, the 12.2 DISSOLUTION 393 determination of the unknown concentration of H2SO4 by titration with NaOH using Eq. (12) of Section 5.6 would not give the correct result if the concentrations were given in molarity. Because sulfuric acid has 2 equivalents of H+ and NaOH has only one equivalent of OH, the concentration must be in normality in order to correct for the difference in equivalents and give the correct result by Eq. (12). 12.2 DISSOLUTION Solutions are formed by mixing two or more pure substances. The molecules of these pure substances interact on a molecular level as they are mixed to create the solution. When a solute is introduced into a solvent, the attractive forces between the molecules or ions in the solute and the molecules in the solvent are broken up and replaced by attractive forces between the solute and solvent. For a molecular solid or liquid solute, the intermolecular forces between the solute molecules and the solvent molecules disrupts the intermolecular forces between the solute molecules causing the solute molecules to separate from each other and become surrounded by solvent molecules that carry them into the solution. This process is called dissolution or solvation, as described in Chapter 4. For ionic solids, the attractive forces between the solvent molecules and ions are so strong that the ionic bonding holding the crystal lattice together is disrupted causing the individual ions to become separated from each other. The separate ions then become surrounded by solvent molecules which carry them into solution. Since the cations and anions of an ionic solute separate when the solute dissolves, the process is referred to as dissociation. The dissociation of ionic compounds in solution to give separate anions and cations creates an electrolyte. If the ionic compound dissociates completely, it is a strong electrolyte, but if some of the cations and anions remain associated with each other in solution, it is a weak electrolyte. Strong electrolytes are strong acids and bases and weak electrolytes are weak acids and bases as described in Chapter 5 (see Table 5.1). There is a rule among chemists that “like dissolves like.” This means that the solute and solvent must have similar chemical structures and similar intermolecular forces in order for a solution to form. Conversely, solutes and solvents that have different intermolecular forces will not form a solution when mixed. Nonpolar organic hydrocarbons are attracted only by London forces. They are soluble in nonpolar solvents that are also attracted by London forces. Polar molecules are attracted by dipole-dipole forces and are soluble in polar solvents that are also attracted by dipole-dipole forces. Water is often called “the universal solvent” because it can form a solution with more substances than any other liquid. This is because water, a polar molecule, is attracted by both dipole-dipole and hydrogen bonding forces. So, it can form solutions with solutes that are attracted to either one or both of these types of forces. This allows the water molecule to become attracted to many different types of molecules. It is especially important in vital biochemical and transportation functions in living organisms. Solubility is the relative ability of a solute to dissolve in a particular solvent and form a solution. It is measured as the maximum concentration of solute that can be obtained under a particular set of conditions. The solubility of a substance depends on the type of solvent, the temperature of the solution and, in the case of a gaseous solute, the pressure. The solubility of 394 12. SOLUTION CHEMISTRY a substance in a particular solvent is measured as the concentration of the solute in a saturated solution. It is determined by the strength of the intermolecular forces between solvent molecules, the strength of the attractive forces between the solute molecules, and the strength of the attractive forces between the solvent and solute. Solubility also depends on the presence of other species that may interact with the solute, such as surfactants or complexing agents discussed in Sections 12.6 and 12.7, or on the excess or deficiency of a common ion in the solution, known as the common ion effect discussed in Section 12.5. The change in enthalpy when a solution is formed from the pure substances is called the enthalpy of solution (ΔHsoln), which is the enthalpy change associated with the dissolution of a solute in a solvent at constant pressure. The net enthalpy change when a solution forms involves three processes as shown in Fig. 12.1. (1) Separation of the solvent molecules (ΔH1). (2) Separation of the solute molecules (ΔH2). (3) Joining the separated solvent molecules and the separated solute molecules together to form a solution (ΔH3). The enthalpy of solution is the sum of these three processes: ΔHsoln ¼ ΔH1 + ΔH2 + ΔH3 (5) Both process (1) and process (2) involve the disruption of attractive forces within the molecules or ions and require energy added to the system. So, both ΔH1 and ΔH2 are positive values. Process (3) involves the formation of attractive forces between the solute and solvent molecules, which releases energy. So, ΔH3 is a negative value. The final value of ΔHsoln can be either positive (an endothermic process) or negative (an exothermic process), depending on how much energy must be absorbed during processes (1) and (2) compared to how much energy is released in process (3). This depends on the difference in strength of the intermolecular forces in the solute and solvent as pure substances compared to the attractive forces between them as a solution. If ΔH1 + ΔH2 > ΔH3 the forces of attraction within the pure substances are greater than the forces of attraction in the solution, and if ΔH1 + ΔH2 < ΔH3, the forces of attraction in the solution are greater than the forces of attraction within the pure substances. So, dissolution will occur if the enthalpy of solvation (ΔH3) is more negative than the sum of the enthalpies of separation (ΔH1 + ΔH2). FIG. 12.1 Enthalpy changes during solution formation. If ΔHsoln < 0, the dissolution will be exothermic (left) and if ΔHsoln > 0, the dissolution will be endothermic. 12.2 DISSOLUTION 395 If ΔH1 + ΔH2 ≫ ΔH3, the solution will not form and the result will be a heterogeneous mixture. If the differences in enthalpy are small, as with nonpolar solutes, entropy changes must be considered to predict solubility. In reality, the enthalpy changes for each process are difficult if not impossible to determine for each process separately. The enthalpy of solution is commonly measured by a calorimeter similar to that described in Section 8.3. The calorimeter, consisting of an insulated container at constant pressure, contains the solvent. A solute is added to the solvent and the temperature of the solvent is measured before and after dissolution. The heat released or absorbed during the dissolution process (qsoln) is determined in kJ/mol from the change in temperature (ΔT), the mass of solution (m), and the specific heat capacity of the solution (Cv) as; ΔHsoln ¼ qsoln ¼ ðC m ΔTÞ (6) EXAMPLE 12.2: DETERMINING THE ENTROPY OF SOLUTION BY CALORIMETER The change in temperature when 5.19 g of Na2CO3 is dissolved in 75.0 g of water was determined to be +3.8°C by calorimetry. What is the enthalpy of solution? Assume that the calorimeter does not absorb any heat and that the specific heat capacity of the solution is the same as that of water (4.18 J • g1 • K1). (1) Determine the mass of the solution. m ¼ 75.0 g + 5.19 g ¼ 80.19 g (2) Determine the heat transferred. qsoln ¼ ðm C ΔT Þ ¼ 80:19g 4:18J=g K 3:8K ¼ 1:27kJ (3) Determine the amount of heat released per mole of solute. 1:27kJ 1:27kJ ¼ ΔHsoln ¼ 0:049mol 5:19g=105:0g=mol ¼ 25:9kJ=mol CASE STUDY: HOT AND COLD PACKS A practical application of the chemistry of dissolution is in hot and cold packs that are commonly used in sports medicine to relieve swelling from injuries such as muscle and joint sprains. They are constructed of a large pouch containing a dry ionic sold and an inner pouch filled with water. The pack is activated by breaking the seal that separates the inner pouch from the outer pouch and shaking the pack vigorously. This action mixes the dry chemical with the water causing it to dissolve. The enthalpy of solution resulting from this dissolution can be either positive (cold pack) or negative (hot pack) depending on the solute used. A commercial hot pack contains about 120 mL of water in the inner pouch and a fixed amount of calcium chloride with a particle size of 8 mesh for quick dissolution in the outer pouch. When the 396 12. SOLUTION CHEMISTRY FIG. 12.2 The change in water temperature upon dissolution of CaCl2 and NH4NO3 assuming the initial temperature of the water is 25°C. inner pouch is crushed and the water and CaCl2 are shaken together, the temperature of the pack rises due to the exothermic heat of solution according to the equation; CaCl2 ðsÞ ! Ca2 + ðaqÞ + 2Cl ðaqÞ + 82:8 kJ The rise in temperature of the pack is dependent on the amount of calcium chloride used as shown in Fig. 12.2. Assuming a beginning temperature of the water solution of 25°C, the rise in temperature of the solution can vary from 32°C for 5 g of CaCl2 to 91°C for 70 g of CaCl2. Commercial hot packs are available in either of two temperature ranges, 49–54°C (120–130°F) and 66–71°C (150–160°F). Each of these packs would require a mass of 21 g and 39 g of CsCl2. The packs are at their hottest immediately after mixing and tend to cool as they lose heat to the surroundings. Heat is transferred from the exothermic solution to the body area in contact with the pack. Many common cold packs use ammonium nitrate, which absorbs heat from the surroundings when it dissolves according to the equation; NH4 NO3 ðsÞ + 25:7 kJ ! NH +4 ðaqÞ + NO3 ðaqÞ The cold pack is constructed in the same fashion as the hot pack with the dry ammonium nitrate in the outer pack. As the ammonium nitrate dissolves, it absorbs energy from the surroundings and the temperature of the water in the pack decreases as shown in Fig. 12.2. The decrease in temperature is again dependent on the mass of NH4NO3. With an initial temperature of 25°C, the resulting temperature of solution can vary from 22°C for 5 g of NH4NO3 to 3°C for 70 g of NH4NO3. Commercial hot packs are usually in the temperature range of 3°C to 1°C (19°F to 17°F). Each of these packs would require a mass of 21 g and 39 g of CsCl2. The packs are at their hottest immediately after mixing and tend to cool as they lose heat to the surroundings. Heat is transferred from the body area in contact with the pack to the endothermic solution, causing the area of body in contact with the pack to feel cold. 12.3 THE EFFECT OF PRESSURE ON SOLUBILITY Changes in pressure do not affect the solubility of liquids or solids in liquids. They do, however, affect the solubility of gases in liquids. When a gas is in contact with a liquid, a dynamic equilibrium is established between the gas and the liquid. So, the rate that the 12.3 THE EFFECT OF PRESSURE ON SOLUBILITY 397 gas molecules enter the liquid equals the rate that they leave the liquid. If the partial pressure of the gas is increased, the gas molecules contact the surface of the liquid more often causing the rate that they enter the liquid to increase and so the concentration of gas in the solvent increases. A new dynamic equilibrium is established when the concentration of the gas dissolved in the solvent is high enough that the rate of gas molecules leaving the liquid again equals the rate of gas molecules entering the liquid. The solubility of the gas in the solvent is increased. The solubility of a gas solute in a liquid solvent is directly proportional to the gas pressure as defined by Henry’s law. Henry’s law is one of the gas laws formulated by William Henry in 1803. The exact statement of Henry’s law is; • At a constant temperature, the amount of a given gas that dissolves in a given type and volume of liquid is directly proportional to the partial pressure of that gas in equilibrium with the liquid. For aqueous solutions, this means that the concentration of a gas in the water solvent is directly proportional to the pressure or partial pressure of the gas above the water. Caq ¼ kH Pg (7) where Caq is the concentration of the gas in the water, Pg is the pressure (or partial pressure) of the gas above the water, and kH is the Henry’s law constant, which is dependent on the identity of the gas and the temperature. Since the dynamic equilibrium between the gas above the water and the gas in the water will result in the concentration of gas in the aqueous phase obtaining the solubility limit under the given conditions of pressure and temperature, Eq. (7) can also be written as; Saq ¼ kH Pg where Saq is the solubility of the gas in water under the specified temperature and pressure. The values of the Henry’s law constants for some common gases in aqueous solution at 298 K are listed in Table 12.2. Column 1 of Table 12.2 corresponds to the Henry’s law constants obtained from Eq. (7). But, Henry’s law is often used in forms other than the one in Eq. (7), resulting in Henry’s law constants with different units and different values. Three of the more common alternate forms of Henry’s law are also listed in Table 12.2 along with the Henry’s law constants derived from them. The four forms of Henry’s law shown in Table 12.2 can be divided into two groups depending on the expressions for the Henry’s law constant. Those expressions that have the aqueous phase concentration of the gas in the numerator and the gas phase concentration of the gas in the denominator (Caq/Pg and Caq/Cg) result in a value for kH that represents the Henry’s law solubility constant. The value of the solubility constant increases as the solubility of the gas (Sg) increases. The kH expressions that have the gas phase concentration of the gas in the numerator and the aqueous phase concentration of the gas in the denominator (Pg/Caq and Pg/χ aq) result in a value that represents the Henry’s law volatility constant. The value of the volatility constant decreases as the solubility of the gas increases. The expressions for the solubility constant and the volatility constant and their values are the inverse of each other as long as the concentration units remain the same. But, the two different groups of the mathematical forms of Henry’s law, also differ in the units that can be chosen to describe the gas concentration in the two phases. The forms listed in Table 12.2 use units of molarity 398 12. SOLUTION CHEMISTRY TABLE 12.2 Henry’s Law Constants for Some Common Gases in Water at 298 K for Four Different Forms of Henry’s Law Henry’s Law Equation Caq 5 kHPg χ aq ¼ kHg Saq 5 kHCg kH Equation Caq ¼ kHg C kH ¼ Paqg P kH ¼ Caqg P kH ¼ χ g aq kH ¼ Units of kH molg/(Lsoln • atm) (Lsoln • atm)/molg (atm • molsoln)/molg Dimensionless P P Caq Cg GASES O2 1.3 103 769.2 4.259 104 3.180 102 N2 6.1 104 1639.3 9.077 104 1.492 102 CO2 3.4 102 29.4 0.163 104 0.8317 4 4 CO 9.5 10 1052.6 5.828 10 2.324 102 H2 7.8 104 1282.1 7.099 104 1.907 102 He 3.7 104 2702.7 14.97 104 0.905 102 Ne 4.5 104 2222.2 12.30 104 1.101 102 Ar 1.4 103 714.3 3.955 104 3.425 102 Caq is the concentration of the gas in water (molg/Lsoln), Χaq ¼ mole fraction of the gas in water (moles of gas/moles of solution), Cg ¼ the concentration of the gas in the gas phase (molg/Lgas), Pg ¼ partial pressure of the gas (atmospheres). (molg/Lsoln) and mole fraction to describe the aqueous phase, while the gas phase is described as a partial pressure in units of atmospheres or concentration in units of molarity. Since all values of kH obtained from these different forms of Henry’s law are often referred to simply as “the Henry’s law constants,” you must be very careful when using Henry’s law constants to be sure which version of the Henry’s law equation they are derived from. When given a value for the Henry’s law constant, the units of the constant will indicate which form of Henry’s law has been used to determine the constant. All versions of Henry’s law will give the same result as long as the form of the Henry’s law equation used corresponds to the value of kH. EXAMPLE 12.3: DETERMINING THE CONCENTRATION OF A GAS IN WATER USING HENRY’S LAW How many grams of carbon dioxide gas is dissolved in a 1 L bottle of carbonated water at a pressure of 2.4 atm and 25°C if the Henry’s law constant for CO2 is 29.4 atm/M? 1. Determine the proper form of Henry’s law. The units of the kH given are atm/M ¼ atm/(mol/L) ¼ (atm • L)/mol So, the form of kH is partial pressure of gas/molarity of solution kH ¼ Pg/Caq The proper form of Henry’s law is: Caq ¼ Pg/kH 12.4 THE EFFECTS OF TEMPERATURE ON SOLUBILITY 399 EXAMPLE 12.3: DETERMINING THE CONCENTRATION OF A GAS IN WATER USING HENRY’S LAW— CONT’D 2. Determine the concentration of CO2 in water at 2.4 atm. Caq ¼ Pg =kH ¼ 2:4atm=29:4ðLsoln atmÞ=molg ¼ 0:082mol=L 3. Determine the number of grams dissolved in 1 L. (0.082 mol/L)(44.0 g/mol) ¼ 3.61 g of CO2 12.4 THE EFFECTS OF TEMPERATURE ON SOLUBILITY The aqueous solubility of most solid and liquid solutes in liquid solvents increases with increasing temperature. As the temperature of the solvent increases, heat is added to the system and the average kinetic energy of the molecules increases. The increased molecular motion allows the solvent molecules to more easily disrupt the attractive forces holding the solute molecules together. In addition, the increased molecular motion in the solute also acts to destabilize the attractive forces. Both of these temperature effects on kinetic energy result in an increase in solute solubility. However, the solubility of some solutes, such as sodium chloride, is fairly independent of temperature and a few, such as LiSO4 and Ce2(SO4)3, become less soluble in water as temperature increases as shown in Fig. 12.3. The trends of solubility with increasing temperature can be predicted by using Le Chatelier’s principle described in Section 7.4. For most solutes, the enthalpy of solution is positive, the dissolution process is endothermic, and heat is required for dissolution. Solute + liquid solvent + energy > solvated solute ΔH > 0 (8) The addition of heat with increasing temperature promotes dissolution by providing needed energy. So, when the temperature of an endothermic dissolution increases, the dissolution equilibrium will shift towards the products (solvated solute molecules) and the solubility of the solute will increase. But, for solutes with negative enthalpy of solution, heat is released during the dissolution process and the dissolution process is exothermic. Solute + liquid solvent > solvated solute + energy ΔH < 0 FIG. 12.3 80 NH4CI Solubility 60 KCI NaCI 40 Ba(NO3)2 20 Ce2(SO4)3 0 0 20 40 60 80 Temperature (°C) 100 (9) Temperature dependence of the solubility (g of solute in 100 g of water) of some ionic compounds in water. 400 12. SOLUTION CHEMISTRY The addition of more heat to the solution actually inhibits dissolution since excess heat is already being produced. The addition of extra heat will cause the dissolution equilibrium to shift towards the reactants. This causes dissolution to decrease in an attempt to reduce the amount of heat in solution. The result is a decrease in solubility of the solute with increasing in temperature. CASE STUDY: PRODUCT PURIFICATION Industrial chemists and engineers take advantage of the variations in compound solubility to purify industrial products once they are produced. Frequently, the desired product is contained in a mixture with undesired impurities. One important method for the separation of the desired compound from impurities is solvent extraction, also known as liquid-liquid extraction. Solvent extraction is a method of separation of compounds based on their relative solubilities in two different immiscible liquids. If two solvents do not mix in any proportions but remain in contact with each other as separate layers, they are said to be immiscible. In contrast, liquids that can be mixed together to form a homogeneous mixture in any proportions are said to be miscible. Often water is used as the first solvent because as “the universal solvent” it dissolves more chemical species than any other solvent. The second solvent is usually an organic solvent because most organic liquids are immiscible with water. Choice of the second solvent depends on the solubility of the desired compound. The solvent extraction procedure is shown schematically in Fig. 12.4. During solvent extraction, the entire mixture is dissolved in the first solvent, usually water, and a second immiscible solvent is added to the solution. The two solvent layers are mixed vigorously to maximize the surface area between them and increase the transfer of the soluble compound into the second solvent. The mixture is then allowed to settle reforming the separate layers. The desired compound is transferred to the second solvent leaving the impurities in the water solvent. It is collected by decanting and evaporating the second solvent. So, the desired compound is separated from impurities by extracting the compound from one solvent into a second solvent leaving the impurities behind in the first solvent. In order for the process to be selective, the desired compound must be more soluble in the second solvent than in the first solvent and the impurities must be insoluble in the second solvent. The process may have some problems if the desired compound and the impurities have similar chemical properties affecting their relative solubilities in both solvents. In this case, the separation will not be complete and further separation methods must be used to purify the desired product. FIG. 12.4 The method of solvent extraction for the purification of a desired compound (green) from an impure mixture. The mixture is dissolved in an aqueous solution and an immiscible solvent is added. The desired compound is transferred into the second solvent by agitation leaving the impurities (black) in the aqueous solvent. 12.4 THE EFFECTS OF TEMPERATURE ON SOLUBILITY 401 Solvent extraction was developed in the 1920s for use in the petrochemical industry because of the need to separate heat-sensitive hydrocarbon mixtures according to chemical type (e.g., aliphatic, aromatic) instead of using methods that require heating the mixture, which risk degrading the heatsensitive product. The technique was soon applied to the pharmaceutical industry for the separation of antibiotics from fermenter broth. Solvent extraction is also used in nuclear reprocessing and ore processing. In these applications, the separation of the solute can be enhanced by adding a chemical complexing agent to the solvent. The complexing agent aids in transferring the desired compound into the second solvent. This process will be discussed further in Section 12.6. Solvent extraction is used for essential oil extraction used in flavorings, fragrances, and food products. Most industrial extractors operate continuously with countercurrent flow of the two liquid phases providing a wellagitated dispersion of drops. The mixture is then sent to settling tanks for phase disengagement. Another useful method of compound purification is recrystallization, a procedure for the purification of an impure compound based on the principle that the solubility of most solids increases with increased temperature. Recrystallization is the most common method for purifying organic compounds and is responsible for purifying approximately 70% of all solid materials produced by industrial processes. During recrystallization, a mixture of the desired compound along with the impurities is dissolved in a small volume of an appropriate solvent at a temperature at or near its boiling point. At this high temperature, the solubility of the mixture is greatly increased and a much smaller volume of solvent is required to create a saturated solution. When the temperature of the solution is slowly decreased, the solubility of each of the compounds in the solution decreases. Each of the separate substances in the mixture will reach their solubility limit at different temperatures and will begin to form crystals at different times. The crystallization of a solid is not the same process as precipitation of a solid. In crystallization, the solid crystal lattice forms in a slow selective process that excludes the impurities resulting in a pure compound. The crystallization process consists of two major events: nucleation and crystal growth. Nucleation occurs as the solute molecules or atoms begin to come together in clusters as the solubility decreases. Nucleation is followed by crystal growth where the size of the nuclei increases and the solute molecules begin to arrange in a way that defines the crystal lattice structure. In contrast, precipitation (described in Section 4.7) is a rapid formation of an insoluble solid product. This rapid formation of the solid produces an amorphous form containing many trapped impurities within the microcrystalline structure. For this reason, chemical reactions that produce an insoluble solid by precipitation must always include a final recrystallization step to yield the pure compound. There are four steps in the recrystallization process: (1) (2) (3) (4) Identify a suitable solvent. Dissolve the impure mixture in a minimum volume of hot solvent. Slowly reduce the temperature to crystallize the desired compound from solution. Filter the hot solution to isolate the purified compound. The most important of these steps is; step (1) the identification of a suitable solvent. The desired solvent properties are: the desired compound must be highly soluble at high temperature, the desired compound must be only slightly soluble at a lower temperature, the unwanted impurities should be very soluble at the crystallization temperature of the desired compound, and the solvent should be volatile enough to be easily removed from the desired compound by evaporation after filtration. Industrially, the crystallization of pure crystalline solids from solutions is accomplished in large quantities in crystallizers. Crystallizers operate at low temperatures and low energy consumption 402 12. SOLUTION CHEMISTRY with very high product purity. The operational conditions of the crystallizers determine the product purity, filterability, and handling characteristics. So, effective operation requires a thorough knowledge of the chemical steps involved in the process, including the creation of supersaturation, nucleation, and crystal growth. Some examples of the use of industrial crystallizers include: the purification of potassium chloride from fertilizer quality material, the recovery of iron sulfate from steel pickling baths, and the recovery of food grade caffeine from decaffeination waste streams. One class of solutes that becomes less soluble in liquids with increasing temperature is the gases. Attractive intermolecular interactions in the gas phase are essentially zero. When a gas dissolves in a liquid, intermolecular interactions are formed between the gas and solvent molecules releasing energy. So, the dissolution of most gases in liquids is an exothermic process that follows reaction (9). Adding heat to the solution accompanied by a rise in temperature provides thermal energy that overcomes the intermolecular forces between the gas and the solvent molecules decreasing the solubility of the gas. The added thermal energy also causes an increase in kinetic energy in the solution. The higher kinetic energy causes more motion in the gas and solvent molecules, which disrupts the intermolecular interactions decreasing the solubility of the gas. So, nearly every gas becomes less soluble in liquid solution with increasing temperature. The decrease in gas solubility with temperature has both practical and environmental applications. The formation of limescale also called “boiler scale” in the giant boilers used to supply hot water or steam for industrial applications was discussed as a consequence of precipitation reactions in Section 4.7. Both the uptake of Ca+ and Mg2+ ions from geological substrates and their precipitation as carbonate compounds in the boiler as limescale are driven by the aqueous concentration of CO2 in the following reactions: CO2 g + H2 OðlÞ > CO2 ðaqÞ + energy CaCO3 ðsÞ + CO2 ðaqÞ + H2 OðlÞ > Ca2 + ðaqÞ + 2HCO3 ðaqÞ In the cold waters of springs and lakes where the water is originally obtained, the CO2(aq) concentrations are high and the second equilibrium lies in favor of the formation of the dissolved ions: Ca2+(aq) and HCO3 ðaqÞ. But, in the boiler where the temperatures are extremely high, the CO2(aq) concentrations decrease forcing the second equilibrium to the left in favor of the formation of the solid carbonates known as limescale. In environmental applications, lake or river water that is used to cool an industrial reactor or power plant is returned to the environment at a higher temperature than normal. This increase in temperature, known as thermal pollution, decreases the solubility of oxygen in the water according to Fig. 12.5. Fish and other aquatic organisms that need dissolved oxygen to live can literally suffocate if the oxygen concentration is too low. Federal data sets of the discharge temperatures of power plant cooling systems from 2001 to 2005 reported that maximum reported temperature discharges averaged 37°C, 10°C higher than the maximum intake water temperatures during summer months (Environ. Res. Lett. Doi:10.1088/ 1748-9326/8/3/035005). Some plants reported discharge temperatures higher than 5°C. This increase in temperature is large enough to decrease the oxygen solubility in water from the intake levels of 8 mg/L to a discharge concentration of 6.8 mg/L. 403 12.5 SOLUBILITY OF IONIC SOLIDS FIG. 12.5 The variation of oxygen solubility in water with decreasing temperature. 12.5 SOLUBILITY OF IONIC SOLIDS Some general guidelines were presented for predicting the solubilities of ionic compounds in Section 4.7. The solubility product constant (Ksp), an equilibrium constant that describes the equilibrium between an ionic solid and its ions in a saturated aqueous solution, can be used to predict the aqueous solubilities of ionic compounds more quantitatively. The general chemical equation for the dissolution of an ionic salt with the formula AxBy is; Aa Bb ðsÞ > aAx + ðaqÞ + bBy (10) The general expression for the solubility product constant for this dissociation is; a Ksp ¼ ½Ax + ½By b (11) EXAMPLE 12.4: DETERMINING THE K S P FROM SOLUBILITY DATA Calculate the solubility product constant for lead(II) chloride if 50.0 mL of a saturated solution contains 0.2207 g of lead(II) chloride. 1. Write the chemical equation and the Ksp expression for the dissolution of lead(II) chloride. Chemical equation: PbCl2(s) ! Pb2+(aq) + 2Cl(aq) Solubility product constant: Ksp ¼ [Pb2+][Cl]2 2. Determine the concentration of PbCl2. 0:2207 g ¼ 7:936 104 mol PbCl 278:1g=mol 7:936 104 mol ¼ 0:0159M PbCl2 0:050L Continued 404 12. SOLUTION CHEMISTRY EXAMPLE 12.4: DETERMINING THE K S P FROM SOLUBILITY DATA— CONT’D 3. Determine the concentrations of the ions in solution. Since, after dissociation, there are two chloride ions for every lead ion: [Pb2+] ¼ 0.159 M [Cl–] ¼ 2 0.159 M ¼ 0.0318 M 4. Calculate the Ksp. Ksp ¼ [Pb2+][Cl]2 ¼ (0.159)(0.038)2 ¼ 1.61 105 Since the solubility product constant is an equilibrium constant of a dissolution reaction, a solubility product quotient (Qsp), similar to the reaction quotient described in Section 7.5, would be the value of the expression for the Ksp in Eq. (11) with concentrations at any point in the reaction. If the product of the ionic concentrations in Eq. (11) is less than the value of the solubility product constant, the solution is unsaturated. If there is sufficient solid available, more will dissolve to achieve saturation. If the product of the ionic concentrations is greater than the value of the solubility product constant, the solution is above saturation and a precipitate will form to reduce the concentrations to achieve the solubility limit of the ionic solid. In summary: • Qsp < Ksp: The ion concentration product is less than the solubility product constant and the solution is unsaturated. More solid will dissolve to achieve saturation. • Qsp > Keq: The ion concentration product is larger than the solubility product constant and the solution is supersaturated. A precipitate will form to reduce the ion concentrations to the solubility limit. • Qsp ¼ Keq: The reaction is saturated. There is no tendency for a precipitate to form or for more dissolution to occur. The value of the solubility product constant depends only on temperature for a given ionic solid. The magnitude of Ksp indicates the degree to which a compound dissolves in water, the higher the solubility product constant, the more soluble the compound. The relative solubility of ionic compounds can be determined by comparing the values of the solubility product constants only if the compounds give equivalent numbers of ions upon dissociation. For example, you can compare the relative solubilities of all the compounds with a 1 to 1 ion ratio (e.g., LiF, AgBr, BaSO4) or all the compounds with a 1 to 2 ion ratio (e.g., PbCl2, CaF2, Li2CO3), but you can’t compare compounds that have different numbers of ions upon dissociation (e.g., LiF with PbCl2). Solubility product constants can only be used for slightly soluble ionic solids that result in a dilute solution of ions. Although all ionic solids have some limit to their solubilities, those with solubilities greater than about 0.01 mol/L can’t be treated by simple equilibrium constants (Ksp) because they tend to form ion pairs in concentrated solutions. This ion pair formation complicates their behavior and causes the effective concentrations to differ from that determined from the solubility directly. Because of this, Ksp is only used for ionic compounds with solubilities less than about 0.01 mol/L. 12.6 COMPLEXING AGENTS 405 The solubility of a slightly soluble ionic solid can be significantly affected by the presence of a highly soluble ionic solid in the same solution if they both share a common ion. For example, given the general dissolution reaction in Eq. (10), if a highly soluble solid AaCc is added to the solution, the value of the Ksp for compound AaBb would remain the same, but the value of [A]a in solution would be higher. According to Le Chatelier’s principle, the increase in concentration of species Ax+(aq) in solution would cause the solubility equilibrium in Eq. (10) to shift to the left to remove the excess Ax+(aq), decreasing the solubility of compound AaBb. This is called the common ion effect. EXAMPLE 12.5: DETERMINING THE SOLUBILITY OF A SLIGHTLY SOLUBLE IONIC SOLID IN A SOLUTION WITH A COMMON ION Given that the Ksp of silver chloride is 1.77 107, what is the solubility of silver chloride in a solution of 0.010 M sodium chloride? 1. Write the chemical equation and the Ksp expression for the dissolution of silver chloride. Chemical equation: AgCl(s) > Ag+(aq) + Cl(aq) Solubility product constant: Ksp ¼ [Ag+][Cl] 2. Determine the ion concentrations in solution. The concentrations of ions from the dissolution of AgCl are [Ag+] ¼ [Cl] ¼ x So, the total concentrations of the ions in solution after dissolution of AgCl is: [Cl] ¼ 0.010 M + x [Ag+] ¼ x 3. Determine the solubility of AgCl from the Ks. Ksp ¼ [Ag+][Cl] ¼ (x) (0.010 + x) ¼ x2 + 0.010x The Ksp expression takes the form of a quadratic equation. However, since AgCl is only slightly soluble under pure conditions: 0.010 + x 0.01 Ksp ¼ [Ag+][Cl–] ¼ (x) (0.010) ¼ 0.010x 0:01x ¼ 1:77 107 x ¼ Ag + ¼ ½Cl ¼ 1:77 108 M 12.6 COMPLEXING AGENTS The Ksp of an ionic compound describes the concentrations of ions in equilibrium with an ionic solid. But, if some of the cations become associated with anions rather than existing as separate ions, the predictions of the total solubility of the compound would differ substantially from the actual solubility. As described in Section 5.1, a metal cation reacts with a species containing a lone pair of electrons in a Lewis acid-base interaction forming a coordination complex. Metal ions in aqueous solution are surrounded by a shell of usually four or six water molecules. This hydrated metal ion is a kind of weak complex ion. Anions, which are a stronger Lewis base than water, can replace the waters of hydration. The formation of this stronger complex ion can affect the solubility of the metal ion. The solubility of an ionic compound may increase dramatically if when a Lewis base is added. 406 12. SOLUTION CHEMISTRY The formation of a complex between a metal ion (M) and an anionic ligand (L) is a substitution reaction of the ligand for waters of hydration. The general equation and the equilibrium constant for this reaction is; MðH2 OÞn + L > ½ML + H2 O M x Ly Keq ¼ MðH2 OÞn ½L The equilibrium constant for the formation of the complex ion is called the formation constant (Kf) or stability constant. The equilibrium constant expression for Kf has the same general form as any other equilibrium constant expression. Omitting the waters of hydration because of the high concentration of the solvent, the simplified reaction gives; xM + yL > ML Kf ¼ ½ML ½Mx ½Ly If the Kf is large, the complexation equilibrium lies in favor of the complex product. The complexation of the free cation acts to increase the solubility of the solid because the free cation can no longer take place in the solubility equilibrium in Eq. (10). According to Le Chatelier’s principle, the decrease in concentration of species Ax+(aq) in solution upon complexation with L would cause the solubility equilibrium in Eq. (10) to shift to the right to increase the concentration of species Ax+(aq), increasing the solubility of compound AaBb. For example, AgBr is a sparingly soluble ionic solid with a Ksp of 5.4 1013. However, silver forms a very stable complex with thiosulfate (S2 O3 2 ) with a Kf of 2.9 1013. The magnitude of the Kf indicates that nearly all the Ag+ ions in solution will be immediately complexed by thiosulfate. The effect of the addition of thiosulfate on the solubility of AgBr can be determined by treating the dissolution and the complexation as occurring in two stepwise reactions as described in Section 7.3: AgBrðsÞ>Ag + ðaqÞ + Br ðaqÞ Ksp1 ¼ 5:4 1013 3 Ag + ðaqÞ + 2S2 O3 2 ðaqÞ> AgðS2 O3 Þ2 ðaqÞ Kf ¼ 2:9 1013 3 AgBrðsÞ + 2S2 O3 2 ðaqÞ> AgðS2 O3 Þ2 ðaqÞ + Br ðaqÞ Ksp2 ¼ Ksp Kf ¼ 15 Comparing Ksp1 with Ksp2 shows that the formation of the complex ion increases the solubility of AgBr by approximately; Ksp2/Ksp1 or 3 1013. This increased solubility of AgBr is used in black and white photography during film processing to wash away excess silver bromide that was not converted to silver metal during exposure. 12.7 SURFACTANTS Surfactants (short for surface active agents) are compounds that lower the surface tension between two liquids or the interfacial tension between a liquid and a solid. They are commonly used as detergents, foaming agents, or wetting agents. Surfactants are composed of both a hydrophobic group and a hydrophilic group, which makes them both water soluble 12.7 SURFACTANTS 407 and oil soluble. Since the hydrophobic group is usually composed of a long chain hydrocarbon, it is referred to as the “tail” of the molecule. The smaller hydrophilic group is composed of a polar or ionic group and is referred to as the “head” of the molecule. Surfactants will diffuse in water and become adsorbed at the interface between air and water where the hydrophobic tail may extend out of the bulk water phase into the air disrupting the surface tension of the water. It can also become adsorbed at the interface between oil and water where the hydrophobic tail extends into the oil phase, while the hydrophilic head group remains in the water phase. In the aqueous phase, surfactants form aggregates called micelles as shown in Fig. 12.6, where the hydrophobic tails group together toward the center of the aggregate and the hydrophilic heads remain in contact with the water solution. Surfactants are classified according to the group used as the hydrophilic head as shown in Fig. 12.7. These are: nonionic, anionic, cationic, or amphoteric. Anionic surfactants contain anionic heads such as; sulfate (─SO4 2 ), sulfonate (─SO3 ), phosphate (─PO4 3 ), and carboxylate (─CO2 ), with the most common being carboxylate. Anionic surfactants are the most widely used for laundry detergents, dishwashing liquids, and shampoos. As a detergent, FIG. 12.6 A surfactant micelle where the hydrophobic tails group together leaving the hydrophilic heads exposed to the aqueous solution. From Super Manu, Wikimedia Commons. FIG. 12.7 Schematic representation of surfactant molecules classified according to the nature of their hydrophilic head groups. 408 12. SOLUTION CHEMISTRY the hydrophobic tails of the surfactant molecules are dissolved into the oily dirt and the hydrophilic head is dissolved in the water. This acts to carry the hydrophobic material into the water solution and prevents it from redepositing on the clean surface. Cationic surfactants contain cationic heads such as ammonium ion (─NH4 + ). They are used as fabric softeners with anionic surfactants, helping them to break down the interface between the oily dirt or stain and the water solution. Amphoteric surfactants have the ability to change charge from cationic to anionic with a change in solution pH because the head group contains both a negative and positive charge. Almost all of the amphoteric surfactants contain a quaternary ammonium ion with a carboxylate, sulfate, or sulfonate. Quaternary ammonium ions are of the structure NR4 + , where R is an alkyl or aryl organic group and the four R groups can be the same or different. These will be further discussed in Chapter 13. Amphoteric surfactants are very mild and are used in shampoos and other cosmetics. They are promoted in advertising campaigns as being “pH balanced” only because they have the ability to respond to the pH of a solution, not because their pH is equivalent to biological pH. Nonionic surfactants do not contain charged ions in the head group, but contain hydrophilic polar long chain alcohols. They are often used together with anionic surfactants. Their advantage is that they do not interact with calcium and magnesium ions in hard water as will the anionic or amphoteric surfactants. 12.8 COLLIGATIVE PROPERTIES Colligative properties of solutions are properties that depend upon the concentration of solute molecules or ions, but not upon the identity of the solute. In liquid solutions, the solute molecules displace some solvent molecules and so reduce the concentration of solvent. Colligative properties depend only on the ratio of solute to solvent molecules and not on the properties of the solvent or solute so that the colligative properties are independent of the nature of the solute. The colligative properties of solutions are: vapor pressure depression, boiling point elevation, freezing point depression, and osmotic pressure. 12.8.1 Vapor Pressure Depression When a nonvolatile solute is added to a solvent, the vapor pressure of the solvent above the solution is lower than the vapor pressure above the pure solvent. This occurs because the presence of the solute molecules at the surface of the solution reduces the surface area available for the solvent to escape the solution into the gas phase. So, the reduction in vapor pressure of the solvent is proportional to the number of solute particles (molecules or ions) in solution. This relationship is described by Raoult’s law, which states that the vapor pressure of the solvent (Psolv) is directly proportional to the mole fraction of solvent (χ solv) in the solution; Psolv ¼ χ solv Po solv where P o solv is the vapor pressure of the pure solvent. So according to Raoult’s law; (12) 12.8 COLLIGATIVE PROPERTIES • when χ solv ¼ 1, • when χ solv < 1, 409 Psolv ¼ Po solv Psolv < Po solv Remember that although the chemical nature of the solute does not affect the vapor pressure depression, the number of solute species does. The mole fraction of the solvent must include all the species in solution. If the solute is a molecule, like sucrose, then the mole fraction of solvent is equal to nsolv/(nsolv + nsucrose), but if the solute is an ionic solid, like sodium chloride, the mole fraction of solvent is equal to nsolv/(nsolv + 2nNaCl). EXAMPLE 12.6: DETERMINING THE VAPOR PRESSURE OF A SOLUTION What is the vapor pressure of an aqueous solution containing 200 g of sucrose in 500 mL of water given that the vapor pressure of pure water is 23.8 Torr? The molar mass of sucrose is 342.30 g/mol. 1. Determine the number of moles of water and sucrose. 500 mL H2O ¼ 500 g 500 g/18.01 g/mol ¼ 27.76 mol H2O 200 g/342.30 g/mol ¼ 0.584 mol sucrose 2. Determine the mole fraction of the solvent. χ solv ¼ 27.76/(27.76 + 0.584) ¼ 0.98 3. Determine the vapor pressure of water. Psolv ¼ χ solv Posolv ¼ 0:98 23:8Torr ¼ 23:4Torr 12.8.2 Boiling Point Elevation The normal boiling point of any liquid is the point where its vapor pressure reaches 1 atm. Because of the vapor pressure depression, when the solvent contains a nonvolatile solute, the solution will have a vapor pressure less than 1 atm at the normal boiling temperature. So, in order to reach a vapor pressure of 1 atm, the solution must be raised to a temperature higher than the normal boiling point. The boiling point elevation is calculated from a form of Raoult’s law except that the amount of solute particles is expressed as a molality of solute instead of a mole fraction of solvent. Molality is used for the concentration of solute instead of molarity because it is not affected by changes in temperature. The increase in boiling point of the solution (ΔTsoln) is directly proportional to the concentration (in molality) of the nonvolatile solute in a solvent; ΔTsoln ¼ Kb msolute (13) where Kb is the molal boiling point elevation constant of the solvent and msolute is the molal concentration of the solute species. The constant Kb is proportional to the heat of vaporization of the solvent, which varies depending on the strength of the intermolecular interactions between the solvent molecules. So, Kb has a specific value depending on the identity of the solvent. 410 12. SOLUTION CHEMISTRY EXAMPLE 12.7: DETERMINING THE BOILING POINT ELEVATION OF A SOLUTION What is the boiling point of a solution containing 96.0 g of sodium chloride in 380 mL of water? Kb(H2O) ¼ 0.52°C/m. 1. Determine the molality of the solute species. (380 mL)(1 g/mL) ¼ 380 g ¼ 0.380 kg H2O (96.0 g/58.44 g/mol)/0.380 kg ¼ 4.23 m Since NaCl dissociates into two ions, the solute species molality is: 4.23 2 ¼ 8.46 m 2. Calculate ΔT. ΔTsoln ¼ Kb msolute ¼ 0:52° C=m ð8:46mÞ ¼ 4:40° C 3. Calculate the new boiling point. 100°C + 4.4°C ¼ 104.4°C 12.8.3 Freezing Point Depression The freezing point of a pure liquid is the temperature at which the molecules begin to cluster to form a crystal lattice. Since the freezing point is also the melting point, at this temperature there is a dynamic equilibrium where the rate of freezing equals the rate of melting. While some of the solvent molecules cluster together to form a pure solvent crystal lattice, the liquid solution becomes more concentrated. According to Le Chatelier’s principle, the dynamic equilibrium will tend to shift in the direction of melting to correct the concentration difference between the pure solid and the solution. So, the rate of freezing proceeds slower than the rate of melting, and in order for the dynamic equilibrium to be reestablished, the freezing must occur at a lower temperature for the solution than for the pure solvent. The freezing point depression of a solution is proportional to the molality of the solute species in the same way as for the boiling point elevation; ΔTsoln ¼ Kf msolute (14) where Kb is the molal freezing point depression constant of the solvent, which depends on the strength of the intermolecular interactions between the solvent molecules and so depends on the identity of the solvent. Freezing point depression is used in many everyday applications. For example, salting of roadways takes advantage of this effect to lower the freezing point of ice in cold weather so that it will form at lower than normal temperatures. The maximum depression of the freezing point using NaCl is about 18°C (0°F), so if the ambient temperature is expected to drop below this limit calcium chloride can be used instead. Since CaCl2 dissolves to give three ions instead of two, it will result in a maximum freezing point depression of 27°C. Another everyday practical application of freezing point depression as well as boiling point elevation is the use of ethylene glycol, a nonvolatile alcohol, in automobile cooling systems. Ethylene glycol lowers the freezing point of the water-ethylene glycol solution so that it will not freeze in winter months in most climates. It also raises the boiling point of the coolant mixture to prevent engine overheating in hot weather. 411 12.8 COLLIGATIVE PROPERTIES 12.8.4 Osmotic Pressure Osmosis is the process in which a liquid passes through a membrane whose pores are large enough to permit the passage of solvent molecules, but are too small for the larger solute molecules to pass through. Such a membrane is known as a semipermeable membrane. Normally, in an aqueous solution the transport of solvent through a semipermeable membrane obeys Le Chatelier’s principle, flowing from an area of low solute concentration (high solvent concentration) to an area of high solute concentration (lower solvent concentration) as shown in Fig. 12.8. Pressure must be applied to the area of higher concentration to prevent the flow of water entering. Osmotic pressure is the minimum pressure which needs to be applied to the solution to prevent the inward flow of water across the semipermeable membrane. It is also defined as the measure of the tendency of a solution to take in water by osmosis. In Fig. 12.8, sucrose (common table sugar) is added to the left side of a U-tube, the water in the right side travels through the semipermeable membrane into the left side by osmosis. This causes the liquid level in the left side of the tube to rise. A dynamic equilibrium is achieved when the pressure in the left side of the tube equals the osmotic pressure of the solution in the right side of the tube. At this point, the rate of the solvent molecules passing through the membrane is the same in both directions. The height of the water column remains unchanged and the osmotic pressure of the solution can be calculated from the height of the liquid in the left side of the tube above the height of the liquid in the right side of the tube in much the same way that atmospheric pressure is measured by Torricelli’s barometer shown in Fig. 6.2 of Section 6.1. The osmotic pressure (Π) of a solution is calculated as; Π ¼MRT (15) where M is the molar concentration of the dissolved species, R is the ideal gas constant, and T is the temperature in Kelvin. Theoretically, since all the colligative properties are related to the concentration of solute in solution, the molar mass of the solute can be obtained by measuring one of the colligative properties of the solution, the mass of the solute, and the mass or volume of the solvent. But, measurements of vapor pressure depression and boiling point elevation FIG. 12.8 The flow of water through a semipermeable membrane by osmosis after the addition of sucrose (common table sugar) to the left side of a U-tube. The transport of water through the membrane from the side containing water into the side containing the sucrose solution results in an elevation of the liquid level in the side containing the sucrose solution. The difference in heights of the liquid column between the two sides of the tube is a measure of the osmotic pressure of the solution. Modified from KDS4444, Wikimedia Commons. 412 12. SOLUTION CHEMISTRY are not very sensitive to changes in the solute concentration and are not usually used for molar mass determinations. The colligative properties more commonly used are measurements of freezing point depression for solvents with a large Kf and osmotic pressure. Osmotic pressure is the most sensitive to changes in solution concentration and can be used to determine the molar mass of molecules with a low water solubility such as large biomolecules. EXAMPLE 12.8: DETERMINING MOLAR MASS FROM OSMOTIC PRESSURE What is the molar mass of an unknown polymer that has an osmotic pressure of 1.25 102 atm at 25°C for a solution of 2.50 g in 150 mL? 1. Calculate the molarity of the polymer. Since the polymer is not an ionic solid, the concentration of the species in solution is equal to the molarity of the polymer. M ¼ Π=RT ¼ 1:25 102 =ð0:082atm=mol KÞð298KÞ ¼ 5:11 104 mol=L 2. Calculate the amount of polymer. n ¼ 5:11 104 mol=L ð0:150LÞ ¼ 7:67 105 mol 3. Calculate the molar mass of the polymer. M ¼ 2:50g = 7:65 105 mol ¼ 3:26 104 g=mol Although most semipermeable membranes are artificially manufactured, one naturally occurring semipermeable membrane is the cell membrane. The interior of a biological cell contains different solutes and the resulting osmotic pressure is important in controlling the distribution and balance of solutes throughout the organism. When a cell is placed in a solution with a smaller concentration of solvents, water will flow across the cell membrane increasing the pressure within the cell. This is the main mechanism for the use of high concentrations of salt or sugar to preserve food. The addition of a 20% salt solution creates a solute-rich environment where osmotic pressure draws water out of microorganisms desiccating them. Salt is mainly used in meat curing. Sugar is used to preserve fruits, either in syrup with fruit such as apples, pears, peaches, apricots, plums or in a crystallized form where the fruit is cooked in sugar to the point of crystallization and the product is stored dry. This also creates a high solute environment that draws water from the cells of microorganisms preventing food spoilage. Honey was used to destroy microorganisms and limit decay as part of the mummification process in ancient Egypt. But, the growth of molds and fungi is not suppressed as efficiently by this process as the growth of bacteria due to the presence of a thick cell wall. CASE STUDY: REVERSE OSMOSIS Reverse osmosis (RO) uses a semipermeable membrane to remove ions, molecules, and large particles from water. In reverse osmosis, an external pressure is applied to the unpurified water solution that exceeds the osmotic pressure of the solution. This application of a high external pressure 413 12.8 COLLIGATIVE PROPERTIES to the solution side of the membrane reverses the natural flow of solvent, which would normally occur from the pure solvent into the solution. So, the solvent is forced from a region of high solute concentration through the semipermeable membrane to a region of low solute concentration by applying a pressure in excess of the osmotic pressure. The largest and most important application of reverse osmosis is the separation of pure water from seawater. The seawater is pressurized against one surface of the membrane, causing transport of the water across the membrane to obtain purified drinking water from the low pressure side. The membranes used for reverse osmosis have a dense layer in a polymer matrix where the separation occurs. In most cases, the membrane is designed to allow only water to pass through this dense layer, while preventing the passage of solutes. Since seawater has an osmotic pressure of about 27 bar (390 psi), this process requires a pressure of about 40–82 bar (600–1200 psi) on the high concentration side of the membrane. Although known for its use in desalination of seawater, since the early 1970s, it has also been used to purify fresh water for medical, industrial, and domestic applications. As discussed in Chapter 1, particle filtration removes particles of 1 μm or larger. Microfiltration removes particles of 50 nm or larger, while ultrafiltration removes particles of roughly 3 nm or larger and nanofiltration removes particles of 1 nm or larger. Reverse osmosis is used in series with membrane filtration removing particles larger than 0.1 nm. Water purification systems commonly include a number of steps shown in Fig. 12.9. These usually include four steps: (1) a sediment filter to remove large particles, (2) one or more activated carbon filters to remove organic chemicals and chlorine, (3) a reverse osmosis filter composed of a thin film composite membrane. Additionally, a second carbon filter can be added after the reverse osmosis membrane to capture those chemicals not removed by the membrane. The carbon filter is also a type of chemical filtration that will be Post carbon filter Clean faucet water Flow restrictor RO membrane 2 Particulate filter 3 Carbon filter Storage tank Drain water Carbon filter Auto shut off valve Cntaminated feed water 1 FIG. 12.9 A reverse osmosis system used for the desalination of seawater. The three main steps are: (1) carbon filtration, (2) particle filtration, and (3) reverse osmosis. An additional postcarbon filter is often used after the reverse osmosis membrane to assure that all dissolved organics have been removed. Modified from Johnlessdominic, Wikimedia Commons. 414 12. SOLUTION CHEMISTRY discussed in more detail in Chapter 15. A UV lamp is sometimes used for sterilization of any microbes that may escape filtering or be introduced after filtration. In practice, a small amount of living bacteria can pass through reverse osmosis membranes through minor imperfections or can bypass the membrane entirely through tiny leaks in surrounding seals. Reverse osmosis is also used in the food industry. It is more economical for concentrating fruit juices than processes that involve heating, which may degrade proteins and enzymes in the juices. It is used extensively in the dairy industry for the production of whey protein powders and for the concentration of milk to reduce shipping costs. Although once avoided in the wine industry, it is also now widely understood and an estimated 60 reverse osmosis machines are in use in France. Maple syrup producers started using reverse osmosis in 1946 to dewater the sap before boiling it down to syrup. This allows about 75%–90% of the water to be removed reducing energy consumption and exposure of the syrup to high temperatures. IMPORTANT TERMS Colligative properties properties of a solution that depend upon the concentration of solute molecules or ions, but not upon the identity of the solute. Common ion effect the decrease of solubility of a slightly soluble ionic solid by the presence in the same solution of a highly soluble ionic solid containing one of the same ions. Dissociation the separation of cations and anions of an ionic solute when the solute dissolves in a solvent. Dissolution the process by which gases, liquids, or solids become dissolved in a liquid solvent Enthalpy of solution (ΔHsoln) the enthalpy change associated with the dissolution of a solute in a solvent at constant pressure. Equivalent (eq) the number of moles of an ion dissolved in a solution multiplied by the ionic charge. Formation constant the equilibrium constant for the formation of a complex ion from its components in solution. Gram equivalent weight the mass of one equivalent of an ion in a compound. Henry’s law the amount of gas dissolved in a liquid solvent is directly proportional to the gas pressure above the liquid. Immiscible two liquids that do not mix in any proportions but remain in contact with each other as separate layers. Infinite dilution a solution that has a large enough excess of solvent so that adding more will not cause heat to be released or absorbed. Miscible liquids that can be mixed together to form a homogeneous mixture in any proportions. Osmosis the process in which a liquid passes through a membrane with pores small enough to permit the flow of the small solvent molecules but too small for the larger solute molecules to pass through. Raoult’s law the vapor pressure of the solvent is directly proportional to the mole fraction of solvent in the solution. Recrystallization a procedure for purifying an impure compound based on the principle that the solubility of most solids increases with increased temperature. Reverse osmosis (RO) the application of a pressure higher than the osmotic pressure to the solution side of a semipermeable membrane to cause the solvent to flow from the solution into the pure solvent. Saturated solution a solution that contains the maximum possible amount of solute under its current conditions. Semipermeable membrane a type of membrane that will allow certain molecules or ions to pass through but not others. Solubility the relative ability of a solute to dissolve in a particular solvent and form a solution measured as the maximum concentration of solute that can be obtained under a particular set of conditions. Solubility product constant (Ksp) an equilibrium constant that describes the equilibrium between an ionic solid and its ions in an aqueous solution. STUDY QUESTIONS 415 Solvent extraction a method of separation of compounds based on their relative solubilities in two different immiscible liquids Supersaturated solution a solution that is more concentrated than a saturated solution. Surfactants compounds that lower the surface tension between two liquids or the interfacial tension between a liquid and a solid. Unsaturated solution a solution that contains less than the maximum amount of solute under its current conditions. STUDY QUESTIONS 12.1 What is a saturated solution? 12.2 What is a supersaturated solution? 12.3 What are the unitless concentration values usually used for very concentrated solutions? 12.4 What are the concentration units commonly used for very dilute environmental solutions? 12.5 What are the common chemical equivalents in g/L for (a) 1 ppm, (b) 1 ppb, (c) 1 ppt in molarity? 12.6 How do you define mole fraction of a solute in solution? 12.7 What is the difference between molarity and molality? 12.8 When is it important to use molality instead of molarity? 12.9 How is normality defined? 12.10 What is solvation? 12.11 What is the measure of a solute’s solubility? 12.12 What molecular properties determine a solute’s solubility? 12.13 What is the relative magnitude of the enthalpy of solution for (a) an exothermic dissolution, (b) an endothermic dissolution? 12.14 The enthalpy of solution is commonly measured by what experimental technique? 12.15 What is the definition of Henry’s law for aqueous solutions? 12.16 How does Henry’s law solubility constant vary from the Henry’s law volatility constant? 12.17 How does increasing temperature affect an endothermic dissolution process? 12.18 How does increasing temperature affect an exothermic dissolution process? 12.19 What is solvent extraction? 12.20 What is meant by solvents being miscible? 12.21 (a) What is the most important step in recrystallization? (b) Why? 12.22 What one class of solutes becomes less soluble in liquids with increasing temperature? 12.23 What is the solubility product constant? 12.24 What is the relationship between Ksp and Q in a saturated solution? 12.25 What is the relationship between Ksp and Q when a precipitate forms in solution? 12.26 What is the common ion effect? 12.27 (a) What is a formation constant? (b) What is another name for a formation constant? 12.28 What is a surfactant? 12.29 What are micelles? 12.30 What are colligative properties of solutions? 12.31 Name four colligative properties of solutions. 416 12.32 12.33 12.34 12.35 12.36 12. SOLUTION CHEMISTRY What What What What What is is is is is Raoult’s law? osmosis? a semipermeable membrane? osmotic pressure? reverse osmosis? PROBLEMS 12.37 What is the percent composition by mass of a sugar solution composed of 4 g of sugar in 350 mL of water at 80°C. (Density of water at 80°C ¼ 0.975 g/mL). 12.38 What volume of alcohol is present in 250 mL of wine that contains 12.5% alcohol by volume? 12.39 What is a 300 ppm aqueous solution expressed as a % (m/m)? 12.40 What mass of silver nitrate is required to prepare 300 mL of an aqueous solution containing 0.1 ppm silver? 12.41 A sample of 300.0 g of drinking water is found to contain 38 mg of lead. What the lead concentration in ppb? 12.42 What is the mole fraction of NaCl in a solution of 0.100 mol of NaCl in 100.0 g of water? 12.43 What is the mole fraction of sugar in a 1.62 molal aqueous solution? 12.44 An aqueous solution contains 83 g of sodium hydroxide in 750 mL of water at 25°C. What is the concentration of the solution in (a) molality, (b) % (m/m), (c) mole fraction, (d) molarity? (Hint: assume that the density of the solution is equal to the density of water.) 12.45 If the enthalpy of solution of lithium iodide is 63 kJ/mol, how much heat will be evolved or absorbed if 25.0 g of lithium iodide is dissolved in water? 12.46 Will the dissolution in Problem 12.45 be exothermic or endothermic? 12.47 The enthalpy of solution of NaOH is 42 kJ/mol. What will be the final temperature of a 7.0 M solution of NaOH prepared in 500 mL of water at 21°C? 12.48 When 1.5 g of ammonium nitrate is dissolved in water, the enthalpy change is 0.335 kJ. What is the enthalpy of solution for ammonium nitrate? 12.49 What is the Henry’s law constant for neon dissolved in water given if the concentration of neon is 23.5 mL/L at STP? 12.50 What is the concentration of O2 in water in g/kg water if the water is in equilibrium with air at 25°C and 1 bar? The mole fraction of O2 in air is 0.21 and the Henry’s law constant for O2 is 1.3 103 mol/kg • atm. 12.51 How many grams of oxygen can be dissolved in 1 L of water at 20°C and 2.00 atm if the Henry’s Law constant for oxygen at 20°C is 1.38 103 M/atm? 12.52 Determine the Ksp of silver bromide, given that its molar solubility is 5.71 107 mol/L. 12.53 What is the molar solubility of AgCN? (Ksp ¼ 5.97 1017) 12.54 What is the balanced chemical equation for the dissociation of copper(II) ferrocyanide, Cu2[Fe(CN)6], in water? 12.55 What is the Ksp expression for the dissociation in Problem 12.53? 12.56 What is the molar solubility of the copper ion in Problem 12.53? 12.57 Calculate the Ksp for Mg3(PO4)2, given that its molar solubility is 3.57 106 mol/L. PROBLEMS 417 12.58 Will a precipitate of CaSO4 form in a solution with a Ca2+ concentration of 0.0025 M and a SO4 concentration of 0.030 M? Ksp(CaSO4) ¼ 2.4 105. 12.59 What is the solubility of AgI in a 0.274 M solution of NaI. Ksp of AgI ¼ 8.52 1017 12.60 What is the solubility of AgCl in 0.0300 M CaCl2 solution? Ksp ¼ 1.77 1010 12.61 What is the concentration of ferrocyanide ion, FeðCNÞ6 4 , in a 0.65 M solution of K4Fe(CN)6? Kf ¼ 1 1035. 12.62 What is the concentration of mercury(II) iodide (HgI2) in (a) pure water, (b) a 3.0 M solution of NaI? Assume that the only species that exists in the presence of excess I is HgI4 2 . Ksp(HgI2) ¼ 2.9 1029; Kf (HgI4)2 ¼ 6.8 1029 12.63 What is the Kf for (HgI4)2 in Problem 12.62? 12.64 What is the increase in solubility for HgI2 caused by the addition of excess I in Problem 12.62? 12.65 What is the vapor pressure of an aqueous solution that has a solute mole fraction of 0.100? The vapor pressure of water is 25.76 mm Hg at 25°C. 12.66 When 10.00 g of an unknown nonvolatile substance is dissolved in 100.0 g of benzene (vapor pressure ¼ 0.125 atm), the vapor pressure of the solution is 0.12 atm. Calculate the mole fraction of solute in the solution. 12.67 What is the vapor pressure of a solution composed of 0.340 mol of a nonvolatile nonelectrolyte in 3.00 mol of water (vapor pressure of 23.8)? 12.68 What is the boiling point of a solution prepared by adding 96.0 g of sodium acetate to 383 mL of water (boiling point constant of 0.52°C/m)? 12.69 What is the molecular weight of an organic compound that produces a boiling point elevation of 0.49°C when 0.64 g of the compound is dissolved in 36.0 g of CCl4? 12.70 Calculate the molar mass of a 5.00 g sample of a polymer dissolved in 16.0 g of carbon tetrachloride (boiling point ¼ 76.50°C) if the boiling point of the solution is 77.85°C. (The boiling point constant of CCl4 ¼ 5.03°C/m). 12.71 Determine the molal freezing point depression constant of benzene (freezing point ¼ 5.5°C) if 60 g of naphthalene dissolved in 20.0 g of benzene results in a freezing point of 2.8°C. 12.72 What is the freezing point of a solution formed by adding 31.65 g NaCl to 220.0 mL of water (ρ ¼ 0.994 g/mol; Kf ¼ 1.86°C • kg/mol) at 34°C? 12.73 If 2.00 g of an unknown compound reduces the freezing point of 75.00 g of benzene from 5.53°C to 4.90°C, what is the molar mass of the compound? 12.74 A solution with 35.0 g of a biopolymer in 1.00 L of solution has an osmotic pressure of 10.0 mm Hg at 25.0°C. What is the molar mass of the biopolymer? 12.75 What is the osmotic pressure in atmospheres of a saline solution prepared by dissolving 0.923 g of NaCl in 100.0 mL of solution? 12.76 What is the molar mass of an unknown organic compound if 0.97 g/L of an aqueous solution has an osmotic pressure of 62.9 Torr, at 25°C. 12.77 If a 4% starch solution and a 10% starch solution are separated by a semipermeable membrane, which starch solution will decrease in volume as osmosis occurs? 12.78 If a 0.1% (m/v) albumin solution and a 2% (m/v) albumin solution are separated by a semipermeable membrane, (a) which solution will have the higher osmotic pressure, (b) which solution will lose water? C H A P T E R 13 The Chemistry of Carbon O U T L I N E 13.1 Carbon Bonding and Hybridization 419 13.5 Polymer Chemistry 445 13.2 Organic Hydrocarbons: Alkanes, Alkynes, Alkenes, and Aromatics 425 Important Terms 452 Study Questions 454 Problems 456 13.3 Organic Functional Groups 431 13.4 Organic Reaction Mechanisms 440 13.1 CARBON BONDING AND HYBRIDIZATION Carbon is a unique element in that it is the only element that has a separate area of chemical study, organic chemistry. As the chemistry of carbon is very important in the chemistry of living organisms, organic chemistry originally obtained its name from the study of carbon compounds that were found in plants and animals. For many years, it was thought that only living organisms could produce organic compounds. We now know that this is not true and that organic molecules can be synthesized in the laboratory using inorganic reactants. Today, organic chemistry involves the study of organic compounds and their chemical reactions. As discussed in Chapter 1, organic compounds are those compounds that contain one or more atoms of carbon covalently bonded to atoms of other nonmetal elements. Due to its ability to also covalently bond to atoms other than carbon including: nitrogen, oxygen, sulfur, hydrogen, and the halogens, carbon can make simple as well as extremely complex molecular structures. The basic reason for the importance of carbon relative to the other elements is due to its special bonding and hybridization. Recall that a carbon atom has an electronic structure of 1s22s22p2, so it needs to share four electrons with other atoms in order to obtain the stable electronic configuration of neon (1s22s22p6). Because of this, stable carbon molecules will always have four bonds per carbon atom. These bonds can be sigma (σ) bonds or pi (π) bonds, but the number of bonds to carbon will always add up to four, no more and no less! These four bonds General Chemistry for Engineers https://doi.org/10.1016/B978-0-12-810425-5.00013-8 419 # 2018 Elsevier Inc. All rights reserved. 420 13. THE CHEMISTRY OF CARBON to carbon always include from two to four hybridized orbitals. Recall from Chapter 3 that the 2s and the 2p valence atomic orbitals of carbon are close in energy making it possible for them to interact. This interaction results in s-p atomic orbital mixing, called hybridization. Orbital mixing on the carbon atom can include: the 2s orbital and one 2p orbital (sp hybridization), the 2s orbital and two 2p orbitals (sp2 hybridization), or the 2s orbital and three 2p orbitals (sp3 hybridization). In summary: • sp hybridization results from the mixing of the 2s atomic orbital with one 2p atomic orbital on the central carbon atom resulting in two hybrid orbitals separated by 180 degrees with a linear geometry. • sp2 hybridization results from the mixing of the 2s atomic orbital with two 2p atomic orbitals on the central carbon atom resulting in three hybrid orbitals separated by 120 degrees with a trigonal planar geometry. • sp3 hybridization results from the mixing of the 2s atomic orbital with the three 2p atomic orbitals on the central carbon atom resulting in four hybrid orbitals separated by 109.5 degrees with a tetrahedral geometry. The shape and geometries of these three kinds of hybridized electronic orbitals around a central carbon atom are shown in Fig. 13.1. These hybrid orbitals of carbon are directed towards the bond axis leading to better orbital overlap and stronger bonds. The number of hybrid orbitals formed from each type of hybridization will be equal to the number of s and p atomic orbitals from which they were combined. So, sp hybridization results in two hybrid orbitals, sp2 hybridization results in three hybrid orbitals, and sp3 hybridization results in four hybrid orbitals. Since the sp3 hybridization results in four orbitals with one of carbon’s four valence electrons occupying each hybrid orbital, no other orbitals are needed to complete the necessary four bonds to carbon. But sp2 and sp hybridization result in less than the required four bonding orbitals for carbon. This means that unhybridized 2p orbitals must be utilized in bonding to make up the required number of four bonds to carbon. According to Section 3.6, the overlap of px or py orbitals forms π bonds, while the overlap of pz orbitals forms a σ bond. Since the hybridized orbitals are always aligned along the bond axis (σ bonds), the p orbital bonding utilizes the px and py orbitals forming π bonds. So, the carbon bonding with sp3 hybridized orbitals are all σ bonds, the carbon bonding with sp2 hybridized orbitals include three σ bonds and one π bond, and the carbon bonding with sp. hybridized orbitals include two σ bonds and two π bonds. Organic compounds usually have multiple carbon atoms linked together with these types of bonds and, in most cases, the carbon atoms are also bonded to hydrogen atoms. In the simplest of the organic molecules, methane (CH4), the carbon orbitals are sp3 hybridized with each of the four valence electrons occupying one of the sp3 hybrid orbitals. The central carbon atom forms four σ bonds to four hydrogen atoms sharing one of its four valence electrons with FIG. 13.1 The electron group geometries for the sp, sp2, and sp3 hybridized carbon orbitals. Modified from Jfmelero, Wikimedia Commons. 13.1 CARBON BONDING AND HYBRIDIZATION 421 each hydrogen atom. Each hydrogen atom has one 1s1 valence electron. The spherical 1s orbital of hydrogen overlaps with the sp3 hybridized orbital of carbon to form a σ bond, as shown in Fig. 13.2. Each of the hydrogen atoms shares its one valence electron with carbon giving carbon the stable valence electron configuration of neon. Because all four CdH σ bonds are directed along the bond axis, the geometry of methane is tetrahedral. When a carbon atom has sp2 or sp hybridized orbitals, the number of hybridized orbitals forms less than the required number of four bonds to carbon. So, the carbon atom must also form bonds with unhybridized 2p orbitals. This occurs when carbon forms bonds with atoms other than hydrogen such as: another carbon atom, an oxygen atom, or a nitrogen atom. These bonds are made up of one σ bond, involving the hybridized orbital, and one or two π bonds. For example, the two carbon atoms in ethene (CH2CH2) are sp2 hybridized allowing each carbon atom to form three σ bonds, one to the adjacent carbon atom and two to hydrogen atoms. This leaves one 2p orbital on each carbon available to form one π bond between the two carbon atoms, resulting in one σ bond and one π bond between the two carbon atoms. The two carbon atoms share two electron pairs resulting in a carbon-carbon double bond (C]C) as described in Section 3.3. The remaining two sp2 orbitals on each carbon bond to two hydrogen atoms giving the molecular structure shown in Fig. 13.3. Because all the bonds lie in the same plane, the geometry of the sp2 hybridized ethene molecule is trigonal planar. The carbon atoms in ethyne (C2H2: common name acetylene) are sp hybridized allowing each carbon atom to form two σ bonds, one to the adjacent carbon atom and one to a hydrogen atom. One hybridized sp orbital on one carbon can overlap with a sp hybridized orbital on the adjacent carbon to form a σ bond between the two carbons and have two 2p orbitals still remaining that are not hybridized. These two 2p orbitals on each carbon overlap to form two π bonds between the two carbon atoms. This leads to a triple bond between the carbon atoms (C^C) made up of one σ bond and two π bonds as shown in Fig. 13.4. Each carbon atom then has one remaining sp orbital that can bond with a hydrogen atom to form a carbonhydrogen σ bond (CdH). The ethyne structure then becomes; HdC^CdH with the two FIG. 13.2 The sp3-s carbon-hydrogen bonds in methane showing the shared valence electrons in each molecular orbital. Modified from Jfmelero, Wikimedia Commons. FIG. 13.3 The chemical bonding in ethene showing the one sp2 orbital and one 2p orbital (A) forming the double bond (B). 422 13. THE CHEMISTRY OF CARBON FIG. 13.4 The chemical bonding in ethyne showing the one sp hybrid orbital and two 2p orbitals (A) that form the triple bond (B). carbon atoms sharing three electron pairs with each other. Each carbon atom then shares one electron pair with a hydrogen atom giving it the stable valence configuration of neon. The resulting geometry of the sp. hybridized ethyne molecule is linear. In an organic molecule containing oxygen, such as formaldehyde (CH2O), the carbon atom bonded to oxygen is sp2 hybridized allowing it to form three σ bonds and one π bond. Oxygen has an electronic configuration of [He]2s22p4, so it needs to share two electrons to obtain the stable valence configuration of neon. It does this by sharing two of its valence electrons with carbon forming a double bond between the carbon and oxygen, which consists of one σ bond and one π bond. The remaining two sp2 orbitals on the carbon atom bond to two hydrogen atoms and the remaining four electrons on the oxygen atom exist as nonbonding pairs in the two remaining sp2 hybrid orbitals. Because it is sp2 hybridized, the formaldehyde molecule has a trigonal planar geometry like that of ethene. The structures of formaldehyde and ethene are compared in Fig. 13.5. The carbon atoms in both of these molecules are sp2 hybridized with the necessary four bonds to carbon being: three hybrid σ bonds and one π bond. In formaldehyde, the oxygen atom is also sp2 hybridized, but it only forms two bonds, one σ hybrid bond and one π bond. The other four valence electrons of oxygen occupy sp2 hybridized orbitals as lone pair electron not available for bonding. So the electronic group structure of formaldehyde is the same as that of ethene with an oxygen atom substituted for a carbon atom and the nonbonding electron pairs of oxygen in place of the two hydrogen atoms bonded to carbon. Carbon can also bond to oxygen atoms when they are sp3 hybridized. In this case, the simplest example would be methanol (CH3OH) with a molecular formula similar to that of methane. The structure of methanol would be the same structure as methane shown in Fig. 13.2 with one hydrogen atom in methane replaced by a dOH group. Since an oxygen atom needs two electrons to form a stable electronic structure, they can do this with a carbon atom by either forming a double bond through sp2 hybridization, as in formaldehyde, or by forming one single σ bond with carbon and one single σ bond with a hydrogen atom through sp3 hybridization, as in methanol. In organic molecules containing nitrogen, such as hydrogen cyanide (HCN), the carbon atom bonded to nitrogen is sp hybridized similar to ethyne. Nitrogen has five valence FIG. 13.5 A comparison of the chemical bonding in ethene (C2H4) and formaldehyde (CH2O). 13.1 CARBON BONDING AND HYBRIDIZATION 423 FIG. 13.6 A comparison of the chemical bonding in ethane (HC^CH) and hydrogen cyanide (HC^N). electrons and needs three more electrons to get to the stable valence configuration of neon. This can be achieved if each nitrogen atom shares three electron pairs with carbon in one σ bond and two π bonds. The resulting bond between carbon and nitrogen is a triple bond like that in ethyne. The nitrogen atom in hydrogen cyanide is also sp hybridized with one hybrid orbital taking part in the triple bond. The second hybrid orbital contains a lone pair of electrons that is not available for bonding. A comparison of the bonding in ethyne with that in hydrogen cyanide is shown in Fig. 13.6. The structure of hydrogen cyanide is the same as that of ethyne with one carbon of the ethyne molecule replaced with a nitrogen atom and the hydrogen replaced with the nonbonding pair of electrons. Carbon atoms can also bond with nitrogen atoms that are sp2 or sp3 hybridized. If the nitrogen atom is sp2 hybridized, with three hybrid atomic orbitals, it will usually form a double bond with carbon (one σ bond and one π bond) and one σ bond with a hydrogen atom. The lone pair of electrons remains in the remaining sp2 hybrid orbital. If the nitrogen atom is sp3 hybridized, with four available hybrid orbitals, it will usually form one single σ bond with the carbon atom and two σ bonds with two hydrogen atoms leaving the last hybrid orbital with the nonbonding pair of electrons. The carbon in these types of molecules is also sp3 hybridized and the structure is similar to methane shown in Fig. 13.2 with one of the methane hydrogens replaced with an dNH2 group. The main thing to remember when determining the hybridization of carbon atoms in molecules is that the number of π bonds to carbon will determine the type of hybridization. Zero π bonds means that the carbon atom is sp3 hybridized, one π bond means that it is sp2 hybridized, and two π bonds means that it is sp hybridized. The number of hybrid orbitals plus the number of π bonds must always equal four. Another way to say this is that if the carbon atom has only single bonds (four σ bonds), then it is sp3 hybridized. If the carbon has one double bond and three single bonds (one π bond and three σ bonds), it is sp2 hybridized, and if it has a one triple bond and one single bond (two π bonds and two σ bonds), then it is sp hybridized. EXAMPLE 13.1: DETERMINING THE HYBRIDIZATION AND GEOMETRIES OF ORGANIC MOLECULES What is the geometry of each carbon in the following: (a) CH3CH2CH3, (b) CH2]CHCH3, (c) CH^CCH3, (d) CH3CH2CH]O? (a) CH3CH2CH3 Count the number and type of bonds to each carbon: Carbon #1 has 4 σ bonds (3 to hydrogens and 1 to carbon) Carbon #2 has 4 σ bonds (2 to hydrogens and 2 to carbons) Carbon # 3 has 4 σ bonds (3 to hydrogens and 1 to carbon) Continued 424 13. THE CHEMISTRY OF CARBON EXAMPLE 13.1: DETERMINING THE HYBRIDIZATION AND GEOMETRIES OF ORGANIC MOLECULES— CONT’D Determine the hybridization: Every carbon has 4 σ bonds and there are no π bonds, so all carbons are sp3 hybridized Determine the geometry: The geometry is tetrahedral. (b) CH2]CHCH3 Count the number and type of bonds to each carbon: Carbon #1 has 3 σ bonds (2 to hydrogens and 1 to carbon) and 1 π bond (in the double bond). Carbon #2 has 3 σ bonds (1 to hydrogen and 2 to carbons) and 1 π bond (in the double bond). Carbon # 3 has 4 σ bonds (3 to hydrogens and 1 to carbon). Determine the hybridization: Carbon # 1 and # 2 has 1 π bond and so they are sp2 hybridized. Carbon # 3 has 4 σ bonds and no π bonds and so is sp3 hybridized. Determine the geometry: The geometry of carbon # 1 and 2 is trigonal planar. The geometry of carbon # 3 is tetrahedral. (c) CH^CCH3 Count the number and type of bonds to each carbon: Carbon #1 has 2 σ bonds (1 to hydrogen and 1 to carbon) and 2 π bonds (in the triple bond). Carbon #2 has 2 σ bonds (both to carbons) and 2 π bonds (in the triple bond). Carbon # 3 has 4 σ bonds (3 to hydrogens and 1 to carbon). Determine the hybridization: Carbon # 1 and # 2 have 2 π bonds and so are sp hybridized. Carbon # 3 has 4 σ bonds and no π bonds and so is sp3 hybridized. Determine the geometry: The geometry of carbon # 1 and # 2 is linear. The geometry of carbon # 3 is tetrahedral. (d) CH3CH2CH]O Count the number and type of bonds to each carbon: Carbon #1 has 4 σ bonds (3 to hydrogens and 1 to carbon). Carbon #2 has 4 σ bonds (2 to hydrogens and 2 to carbons). Carbon # 3 has 3 σ bonds (1 to carbon, 1 to hydrogen, and 1 to oxygen) and 1 π bond (in the double bond). Determine the hybridization: Carbon # 1 and # 2 have no π bonds and so are sp3 hybridized. Carbon # 3 has 1 π bond and so is sp2 hybridized. Determine the geometry: The geometry of carbon # 1 and # 2 is trigonal planar. The geometry of carbon # 3 is tetrahedral. 13.2 ORGANIC HYDROCARBONS: ALKANES, ALKYNES, ALKENES, AND AROMATICS 425 13.2 ORGANIC HYDROCARBONS: ALKANES, ALKYNES, ALKENES, AND AROMATICS Hydrocarbons are compounds that contain only hydrogen and carbon atoms. There are three basic types of hydrocarbon structures: straight chain hydrocarbons, branched hydrocarbons, and cyclic hydrocarbons. The straight chain hydrocarbons form one continuous chain of carbon atoms bonded together in a straight line where each carbon atom is bonded to no more than two other carbon atoms. Branched hydrocarbons have one or more of the carbon atoms in the chain bonded to three or four carbon atoms creating a branch in the straight carbon chain. Cyclic hydrocarbons are hydrocarbons in which the carbon chain joins to itself forming a ring. The simplest of the hydrocarbons are the alkanes. Alkanes are a class of hydrocarbons that contain only sp3 hybridized carbon atoms. They have the general formula CnH2n+2 and tetrahedral geometries. The alkanes are sometimes referred to as saturated hydrocarbons, meaning that all carbon bonds in the molecule are sp3 hybridized and all the bonds are σ single bonds. Each carbon is bonded to the maximum number of four neighboring carbon or hydrogen atoms. They are nonpolar and very chemically stable since they contain only CdC and CdH bonds. The larger alkanes are sometimes referred to as paraffin hydrocarbons because they appear waxy in the solid phase. Petroleum and natural gas are primarily composed of mixtures of alkanes. The simple alkanes are known as normal alkanes or straight chain alkanes. They are named according to the number of carbon atoms in the molecule. The names of the first 10 al