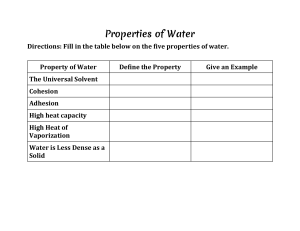
Available online at www.sciencedirect.com ScienceDirect Computer-aided molecular design of solvents for chemical separation processes Shiyang Chai1, Zhen Song2, Teng Zhou3,4, Lei Zhang1 and Zhiwen Qi2 Solvents are widely used in chemical industries, especially in various separation processes. As traditional trial-and-error solvent selection is time-consuming and expensive, modelbased methods for solvent selection/design become important for efficient and sustainable chemical manufacturing. A lot of contributions have been made in this area in the past few decades. This article first reviews the prediction methods for solvent properties, including single molecular properties and mixture properties. Then, the solution strategies of solvent design problems are summarized, including generate-and-test, deterministic optimization, and stochastic optimization methods. Next, latest progresses of computer-aided solventprocess design in separation processes including liquid–liquid extraction, extractive distillation, gas absorption, and crystallization are reviewed. Finally, several remaining challenges and possible future directions for solvent design in separation processes are pointed out. Addresses 1 Institute of Chemical Process Systems Engineering, School of Chemical Engineering, Dalian University of Technology, 116024 Dalian, China 2 School of Chemical Engineering, East China University of Science and Technology, 130 Meilong Road, 200237 Shanghai, China Process Systems Engineering, Max Planck Institute for Dynamics of Complex Technical Systems, Sandtorstr. 1, D-39106 Magdeburg, Germany 4 Process Systems Engineering, Otto-von-Guericke University Magdeburg, Universitätsplatz 2, D-39106 Magdeburg, Germany 3 Corresponding authors: Zhang, Lei (keleiz@dlut.edu.cn), Qi, Zhiwen (zwqi@ecust.edu.cn) Current Opinion in Chemical Engineering 2022, 35:100732 This review comes from a themed issue on Frontiers in chemical engineering; chemical product design – II Edited by Rafiqul Gani, Lei Zhang and Chrysanthos Gounaris For complete overview of the section, please refer to the article collection, “Frontiers in Chemical Engineering; Chemical Product Design – II” Available online 13th September 2021 https://doi.org/10.1016/j.coche.2021.100732 2211-3398/ã 2021 Elsevier Ltd. All rights reserved. Introduction Solvents are widely used in many chemical and related processes, such as reactions, separations, additives, and www.sciencedirect.com transportations. For example, as reaction media, solvents have a great influence on the reaction rate and selectivity for some specific reactions; as separation agents, solvents are indispensable in various processes, such as gas absorption, liquid–liquid extraction, extractive distillation, and crystallization [1,2]. Because of many factors that need to be considered, such as solvent properties (melting point, boiling point, flash point, viscosity, toxicity, safety and environmental impact, etc.) and process properties (vapor–liquid equilibrium, liquid–liquid equilibrium, solid–liquid equilibrium, etc.) [3,4], it is usually a difficult task to select suitable solvents in chemical separation processes. Knowing the important role of solvents in chemical processes, a careful solvent selection is often necessary to reduce process costs and improve product quality. However, it is obviously challenging to balance so many different aspects, such as solvent chemical/physical properties, mixing rules, phase equilibrium, mass/energy transfer, process design, economics, environmental impact, safety and health issues, and so on. Meanwhile, considering the large number of potential solvents, the trial-and-error method for solvent selection could be extremely time-consuming and expensive, and promising solvents may be missed if the selection is based on a fixed solvent database. To overcome this problem, modelbased solvent design/selection approaches are highly desirable, which can narrow down the huge solvent searching space and help focus the limited experimental resources on several promising candidates [5]. In model-based solvent design methods, property estimation models play an irreplaceable role as they connect the molecular structures with macroscopic properties. The performance of designed solvents depends largely on the accuracy of the used property estimation methods. Different property estimation methods have been developed for solvents, including Group Contribution (GC) [6], Quantum Mechanics (QM) [7], Machine Learning (ML) [8,9], and Molecular Dynamics (MD) [10]. Struebing et al. [11] used QM to calculate the reaction rate constant, and then established a surrogate model for the rate constant of the Menschutkin reaction. Liu et al. [12] proposed a novel ML-based atom contribution method to predict molecular surface charge density profiles (s-profiles). Wang et al. [13] developed ML models based on an extended experimental dataset to predict the toxicity of ionic liquids in leukemia rat cell line (IPC-81). To Current Opinion in Chemical Engineering 2022, 35:100732 2 Frontiers in chemical engineering; chemical product design achieve a higher accuracy, property prediction models are constantly being improved and developed. As a kind of model-based design method, Computer Aided Molecular Design (CAMD) is a powerful technique for pre-screening existing solvents and designing novel ones. In CAMD, a set of preselected building blocks of molecules (usually functional groups) are assembled by mathematical algorithms to generate promising molecules according to the objective functions and constraints (structural constraints, property constraints, process constraints, etc.) [14,15]. The mathematical formulation of CAMD is essentially a Mixed Integer Linear Programming (MILP) or Mixed Integrated Non-Linear Programming (MINLP) problem, and different solution approaches have been developed to solve such MILP/ MINLP problems (generally classified as enumerationbased generate-and-test, deterministic optimization methods, and stochastic optimization methods) [2]. Since CAMD methods were first proposed for the design of extractants for liquid–liquid extraction processes [16], it has been widely extended to extractive distillation [17– 19], liquid–liquid extraction [20–25], crystallization [4,12,26,27], reaction [28–30], CO2 capture [31–36], and many other fields [37–39]. The applications of CAMD have greatly freed researchers from extremely heavy experimental work. In this review paper, solvent design methods for chemical separation processes are specifically discussed, which are based on multi-scale simulation and optimization methods and tools, as shown in Figure 1. The topics cover the levels of solvent molecular structures, Quantitative Structure Property Relationship (QSPR), mixture properties, and separation processes. In Section ‘Methods for solvent property estimation’, methods of solvent property estimation are reviewed, which is a crucial step in solvent design. In Section ‘Computer-aided methods for separation solvent design’, the solution methods for the solvent design models are summarized. In Section ‘Solvent-process design’, several selected cases of solvent-process design problems for separation processes are discussed. In Section ‘Discussion and perspectives’, challenges and perspectives of separation solvent design are elaborated. Methods for solvent property estimation Property estimation models are the prerequisite for solvent design, which determine the range of application and the accuracy of the solvent design results. In this section, property estimation methods for single molecular and mixture properties are discussed. Single molecular properties The properties of a molecule are determined by its molecular structure. Conversely, with the molecular structure as the input, the properties could be estimated. The QSPR model addresses the relationship between molecular structure and molecular properties. The Group Contribution (GC) method is one of the most commonly used and effective methods to predict the properties of single molecules [14]. The GC methods are also known as the addition methods, which consider the properties of a molecule to be the summation of the property contributions of all the groups constituting the molecule [40]. Despite the popularity and effectiveness of first-order GC models in many cases, their accuracy is sometimes limited Figure 1 Quantitative Structure Property Relationship Property Prediction Multi-scale simulation and optimization GC method QM method Solvent group ML method Single molecular properties Mixture properties Phase equilibrium Process model Process simulation MD method Optimized solvent structure & process parameters CAMD Applications Liquid-liquid extraction Extractive distillation Gas absorption Crystallization Separation processes Current Opinion in Chemical Engineering Multi-scale simulation and optimization-based solvent design methodology. Current Opinion in Chemical Engineering 2022, 35:100732 www.sciencedirect.com Computer-aided molecular design of solvents for chemical separation processes Chai et al. due to the neglect of the interaction between different groups. To overcome this problem, more sophisticated GC models have been developed, such as high-order GC and Artificial Neural Network-Group Contribution (ANN-GC) methods [41]. Higher-order groups can be used to distinguish some structural isomers, for example, 2-methyl pentane and 3-methyl pentane, and therefore, the GC-based methods are able to distinguish between isomers like this. Nevertheless, for many stereoisomers (such as cis and trans isomers), their properties cannot be differentiated by GC-based methods, which need to be essentially determined by experimental measurement [40]. QM-based method overcomes the shortcomings of GC methods, that is, the missing molecular properties are obtained by ab initio calculation and do not depend on experimental data. Some QM-based property prediction methods have already been used in CAMD problems, such as the Conductor-like Screening Model (COSMO) based ones (e.g. COSMO-RS and COSMO-SAC) [42]. Recently, QM-based CAMD methods have been applied to the design of homogeneous catalysts, separation, and reaction solvents [43]. However, the application of QM in CAMD is limited by the calculation speed. One of the future directions of QM for CAMD can be integrating ML methods into semi-empirical QM methods to speed up the calculation of QM. Similar to QM, MD can also provide property estimation through its molecular scale simulation results, which can be used for product design or to establish QSPR models [44]. MD methods have been applied in fields such as gas adsorption and polymer design. Kupgan et al. [45] used MD to design polymers for CO2 capture. Liang et al. [37] proposed a new optimization-based Computer-Aided Polymer Design (CAPD) framework by combining MD and CAMD. However, similar to the QM methods, the computational speed limits its application in CAMD. The establishment of QSPR models based on MD simulation results is one of the future directions for chemical product design model. With the help of ML-based methods, it is possible to establish QSPR models to correlate molecular descriptors and target physical, chemical, and biological properties of molecules with higher computation speed and accuracy [8]. The steps of establishing QSPR models based on ML methods include data collection, data pre-processing and feature engineering, and model establishment [46], as shown in Figure 2. It is essential to establish a database before establishing ML models. The data can be obtained from experiments, databases and/or model simulation such as QM, MD, CFD, and so on. However, these data are not always valid when directly applied in the establishment of ML models. Experimental system errors, inconsistent order of magnitude and redundant data often lead to poor training results of ML models. Therefore, it is necessary to www.sciencedirect.com 3 employ data pre-processing and feature engineering methods to process the original data. After data collection, pre-processing and feature engineering, the features are generated and prepared for the establishment of ML model, which includes model selection, model training and validation process. The model selection depends on the application scenarios, which are generally categorized into supervised learning, unsupervised learning, and Reinforcement Learning (RL) [47]. Supervised learning [48] is employed to solve two problems of regression and classification, while unsupervised learning [49] is used to solve the problems such as clustering and association analysis. RL [50] is the process of training the model through sequences of state-action pairs, observing the rewards that result, and adapting the model predictions to those rewards using policy iteration or value iteration until it accurately predicts the best results. Su et al. [9] proposed a deep learning approach for QSPR modeling of critical properties, while Datta et al. [51] used decision trees and genetic programming to develop predictive models for reaction rate constants. Mixture properties Mixture properties include functional and equilibriumbased properties. As for functional properties, the prediction is based on the properties of pure components and a mixing rule for a given set of molecules and their compositions with a predefined phase identity. Liu et al. [52] summarized 26 mixture properties prediction models based on mixing rules, including molecular weight, density, and enthalpy. The equilibrium-based properties usually include vapor–liquid equilibrium (VLE), liquid–liquid equilibrium (LLE), solid–liquid equilibrium (SLE), and so on. The estimation of these properties requires the integration of pure component properties and thermodynamic property models. Mixture thermodynamic models used in CAMD include the UNIQUAC functional group activity coefficients (UNIFAC) methods [53], statistical associating fluid theory (SAFT) theory equation of state [54], COSMO-based activity coefficient methods [42], and so on. The UNIFAC methods, as a kind of GC methods, are widely used for estimating activity coefficient in phase equilibria calculation. However, due to the deficiency of GC methods mentioned in Section ‘Single molecular properties’, it is difficult to distinguish all isomers using UNIFAC methods. In addition, the UNIFAC methods require experimental data to obtain the binary interaction parameters between groups [55]. COSMO is a continuum solvation models based on first principles. Klamt et al. [56] proposed the first COSMO-based activity coefficient model, named as COSMO-RS (Conductor-like Screening Model for Real Solvents), which is suitable for the prediction of thermodynamic properties of fluid systems. Based on the principle similar to COSMO-RS, Lin and Sandler [57] proposed the COSMO-SAC model (Conductor-like Screening Model for Segment Activity Coefficient). With Current Opinion in Chemical Engineering 2022, 35:100732 4 Frontiers in chemical engineering; chemical product design Figure 2 Step 1: Data collection Experiments Step 2: Data pre-processing and feature engineering Databases Experimental system errors Model simulation Inconsistent order of magnitude (QM, MD, CFD, etc) Redundant data Step 3: Model establishment Supervised learning Unsupervised learning Reinforcemant learning Current Opinion in Chemical Engineering Flow chart of establishing QSPR models based on ML methods. COSMO-RS/SAC models, the activity coefficients of systems involving new molecules can be estimated directly and quickly without the need for molecular and group-specific parameters. Thus, COSMO-RS/SAC models are also widely used in many fields to predict thermodynamic properties [58,59]. In addition, MLbased methods are also used to predict the mixture properties. Zhang et al. [60] established the StructureOdor Relationship (SOR) model of aroma mixtures using the molecular surface charge density distribution as descriptors. process constraints including equipment design equations, mass balance equations, energy balance equations, and phase equilibrium equations, which can also be linear or nonlinear equations [61]. For different MINLP/MILP problems, the solution strategies for the solvent design problems are different, which can be classified as ‘generate-and-test’, deterministic optimization and stochastic optimization methods [2]. Generate and test method Computer-aided methods for separation solvent design The separation solvent design problem can be expressed as an MILP/MINLP model, as shown in Eqs. (1)–(4) [52,61]. Min=Max f obj ðX; N Þ ð1Þ Structural constraints : g 1 ðN Þ 0 ð2Þ Property constraints : g 2 ðN Þ 0 ð3Þ Process model and other constraints : g 3 ðX; N Þ 0 ð4Þ Here, X is a vector of continuous variables related to the process variables, N is a vector of integer variables related to the molecular structure of the molecule. Eq. (1) is the objective function, which could be economic benefits, product quality, social demand, sustainability, and so on [27]. Eq. (2) is the structural constraints, which includes the octet rule and structural complexity constraints [45]. Eq. (3) represents the property constraints of single molecules/mixtures, which can be linear or nonlinear, depending on the used property models. Eq. (4) is the Current Opinion in Chemical Engineering 2022, 35:100732 Gani and Brignole [16] proposed the ‘generation-andtest’ method for the design of liquid extraction solvents. The method is to generate all possible candidate molecules from a set of given groups, and then use constraints to test each candidate molecule. Finally, molecules that cannot meet any of the constraints are discarded, and the remaining molecules are sorted according to their performance. Because all candidate molecules generated by group combination need to be tested, the generate-andtest method becomes very time-consuming when the group number of the molecule increases. To overcome this problem, Gani et al. [62] improved the method: first, structural feasibility constraints are used to ensure that only chemically feasible molecules are generated; then, these molecules are screened through multi-level property constraints, and molecules that cannot meet the constraints will be eliminated. This method greatly reduces the molecular design space, which is very beneficial to the calculation efficiency of the generate-and-test method. However, once the number of given groups is too large, the method still faces the problem of combinatorial explosion. In addition, the traditional generate-and-test method cannot distinguish isomers. To solve this problem, Harper and Gani [63] proposed a multi-level CAMD framework: first, all combinations of first-order groups are determined by the generate-and-test method; then, by using higher-order groups, molecular modeling techniques or experimental data, all possible isomers of the candidate molecules obtained in the first step are further evaluated. The multi-level CAMD method has been www.sciencedirect.com Computer-aided molecular design of solvents for chemical separation processes Chai et al. implemented in the software ProCAMD (https://www. pseforspeed.com/icas/procamd/). Deterministic optimization methods The deterministic optimization methods are based on gradient-based algorithms for nonlinear programming (NLP) problems, and branch-and-bound type algorithms for integer variables to obtain the optimal molecules from the solutions of MILP or MINLP problems. For the CAMD problem formulated as an MILP, deterministic optimization methods can always guarantee global optimal solution. However, for nonconvex MINLP problem, the global optimal solution cannot be always guaranteed. Karunanithi et al. [4] proposed the decomposition algorithm to solve MINLP problems for product design. In this algorithm, the complex MINLP problem is decomposed into a set of MILP and NLP subproblems. First, the candidate molecules satisfying the structural constraints and linear property constraints are obtained by solving the MILP problem. Then, the nonlinear constraints are used to verify each candidate molecule. Finally, the molecules satisfying all constraints are retained and sorted according to the objective function value. The prerequisite for using the decomposition algorithm is that the constraints of the MINLP problem are independent of each other. Since the constraints of solvent properties (linear/nonlinear) in CAMD problem are usually independent, the decomposition algorithm is suitable for solving most solvent design problems. The gap between the solution obtained from decompositionbased algorithms and the truly optimal solution depends on the number of feasible solutions generated in the first step. If all feasible solutions are generated in the first step, the global optimality of the solution can be ensured [61]. Stochastic optimization methods The stochastic optimization methods are to find the optimal solution by using an adaptive search strategy. With this kind of methods, global or nearly global optimal solution can be obtained for convex and nonconvex problems. The commonly used stochastic optimization methods are genetic algorithm [64], tabu search algorithm [65], and so on. Genetic algorithm is a kind of heuristic algorithm based on natural selection. In CAMD problems, genetic algorithm usually searches in the space of feasible molecular structure. Each molecule in a generation is evaluated, and the best molecular characteristics are passed on to the next generation, and the process is continued until the algorithm converges. Venkatasubramanian et al. [66] first introduced genetic algorithms to the CAMD problem by encoding the molecular structure into a series of substituents and performing genetic manipulation on the individual. Van Dyk and Nieuwoudt [17] proposed a coding method based on the UNIFAC groups. Scheffczyk et al. [67] applied genetic algorithm to the design of liquid–liquid extractant based on the COSMO-RS model. Zhou et al. [68] combines genetic www.sciencedirect.com 5 algorithm (GA) with gradient-based deterministic algorithm to solve the continuous nonlinear optimization problem with fixed molecular variables. Tabu search is also a stochastic optimization method. In tabu search algorithm, a set of initial solutions (molecules) needs to be given first, and these solutions are appropriately changed through some operations. The operation is repeated as long as the changed molecules do not appear in the tabu list (that is, the list of molecules forbidden to be considered based on various factors). The forbidden factors in tabu list include frequency of occurrence (to ensure that the same molecules are not always visited), infeasible molecules, poor target properties, and other factors. Mcleese et al. [69] applied the tabu search algorithm to the design of ionic liquids in absorption refrigeration. The selection of solution strategy depends on the characteristics of specific problems. A brief list of references and main applications classified by solution strategy is summarized in Table 1. At present, the requirement for solution strategy is to minimize the computational cost while ensuring the global optimality of the solution. Solvent-process design The solvent design is closely related to its related process. Therefore, it is necessary to consider process models in the solvent design to obtain the overall optimal solvents. However, the existence of process models could make the solvent design problem very complex and highly nonlinear, which may bring about great challenges to the solution strategy. For different solvent design problems, the modeling and solution methods are also different. In this section, several selected applications of solventprocess design in separation processes are briefly discussed, which include liquid–liquid extraction, extractive distillation, gas absorption, and crystallization. These four areas are chosen because they have been extensively studied and can basically cover different types of solvent-process design problems in separation processes. Solvent-process design for liquid–liquid extraction The selection of extractant is an important issue in liquid–liquid extraction, which affects the quality and composition of extraction products and the performance of extraction operation [87]. A lot of work on the optimal selection and design of solvents for liquid–liquid extraction has been reported, as shown in Table 2. It can be seen from Table 2 that for liquid–liquid extraction processes, several important solvent properties such as Tm, Tb, toxicity and LLE need to be considered. Here, some selected studies and recent progress are reviewed. Gani and Brignole [16] first proposed a CAMD method and applied it to extractant design. Among them, g and g 1 are predicted by the UNIFAC method, and the extractants with good performance are determined using generate Current Opinion in Chemical Engineering 2022, 35:100732 6 Frontiers in chemical engineering; chemical product design Table 1 A brief list of references and main applications classified by solution strategy Solution strategy Applications Generate and Liquid–liquid test extraction Extractive distillation Gas absorption Crystallization Deterministic Liquid–liquid optimization extraction Extractive distillation Gas absorption Crystallization Reaction solvent Stochastic Liquid–liquid optimization extraction Gas absorption References Gani and Brignole [16], Shankar et al. [21], Song et al. [22], Gani et al. [62], Harper and Gani [63], Song et al. [70], Yang and Song [71], Chao et al. [72] Karunanithi et al. [4], Liu et al. [12], Karunanithi et al. [26], Chai et al. [27], Chong et al. [32], Liu et al. [52], Cheng and Wang [73], Xu et al. [74], Scilipoti et al. [75], Harini et al. [76], Lekutaiwan et al. [77], Cignitti et al. [78], Chen et al. [79], Ahmad et al. [80], Scilipoti et al. [81], Zhang et al. [82], Watsona et al. [83] Wang et al. [34], Venkatasubramanian et al. [66], Scheffczyk et al. [67], Zhou et al. [68], Mcleese et al. [69], Zhou et al. [84], Zhang et al. [85], Gebreslassie and Diwekar [86] and test method. Cheng and Wang [73] presented computer-aided process/solvent design to find a feasible biocompatible solvent for the extractive fermentation and separation process. In this model, the goals including the maximum production rate, extraction efficiency, and the limitation of solvent utilization were considered simultaneously. Among them, Tb, Tm, –log(LC50), and 4G were predicted by the GC methods, and g was predicted by the UNIFAC method. The process constraints were steadystate material balance and LLE. Finally, the established MINLP model was solved using Mixed-Integer Hybrid Differential Evolution (MIHDE), which belongs to the deterministic optimization algorithm. Shankar et al. [21] employed CAMD to design organic solvents for the liquid–liquid extraction of ephedrine from its aqueous solution. Three main solvent performance indicators (high ephedrine solubility, low solvent loss and high partition coefficient) were used to identify the solvents. Here, Tb, Tm, 4Hfus, h and –log(LC50) were predicted by the Joback-Reid method, which is a simple GC method; d was predicted by Albahari’s GC-based correlation, and g was predicted by the UNIFAC method. The SLE equations were considered as the process constraints. Finally, the optimal solvents were determined by the Exhaustive Direct Search (EDS), which belongs to the generationand-test method. The above two cases are examples for the design of single molecular extractant. Xu et al. [74] proposed a new blended extractant design and screening method to solve the problem of co-extraction of phenols, polycyclic Current Opinion in Chemical Engineering 2022, 35:100732 aromatic hydrocarbons (PAHs) and nitrogen heterocyclic compounds (NHCs) in coal chemical wastewater. First, two single-component extractant candidate sets were generated by database search based on property constraints (Tm, Tb, Psat, r, s, h and Mw). Then, the composition of the extractant mixture was determined by solving a series of NLP problems. The solution method was grid parallel computing method, which belongs to deterministic optimization algorithm. Among them, g of component i in the solution was calculated by the Dortmund UNIFAC model, and the process constraints include LLE and mass balance equations. In addition to traditional organic solvents, the design of ionic liquids (ILs) has also been reported considering the attractive physico-chemical properties of ILs for liquid– liquid extraction process, such as non-volatility, selective solubility for different compounds, and widely tunable character. For instance, Song et al. [22] proposed a systematic method combining phase equilibrium calculation, physical property prediction and process simulation to select suitable ILs as extraction solvents. The COSMO-RS model was employed to predict r and LLE data and the GC methods were used to estimate the key physical properties (Tm, Tb and h) of the prescreened ionic liquids. For the top IL candidates, the performance was further analyzed using Aspen Plus to determine the optimal solvents based on the process. Finally, the proposed method was applied to the extraction desulfurization process, and two most promising ILs were determined. As the design of ILs is often limited by the lack of GC thermodynamic methods, Song et al. [70] proposed an extended UNIFAC-IL model for the optimal design of ILs for the extractive desulfurization of fuel oils. The extended UNIFAC-IL model was based on 3653 experimental data of infinite dilution activity coefficient, covering seven conventional main groups and 20 IL main groups, and its high reliability was verified through a large experimental liquid–liquid equilibria database. Subsequently, the computer-aided design of ILs was performed by using the extended UNIFAC model for estimating g in the objective function (extraction performance indicator). The main solvent properties (Tm, h and Mw) were predicted by the GC methods. Finally, Aspen Plus was used to analyze the performance of the top candidate ILs. It can be seen from the above discussions that the design of novel extractants such as ILs has received more and more attention. For novel extractant design, one of the challenges is the lack of reliable predictive models for some physical properties. Therefore, the establishment of QSPR models for such properties is crucial for novel solvent design. Wang et al. [13] developed ML models based on an extended experimental dataset to predict the toxicity of ionic liquids, which provides a good reference for the use of data-driven machine learning methods to establish property prediction models for novel extractants. www.sciencedirect.com Computer-aided molecular design of solvents for chemical separation processes Chai et al. 7 Table 2 A brief list of solvent-process design for liquid–liquid extraction Reference Properties Property prediction method Process model Solution strategy Gani and Brignole [16] Cheng and Wang [73] g, g 1 UNIFAC GC LLE Steady-state material balance LLE Generate and test SLE Exhaustive Direct Search (EDS)b) Tm, Tb, –log(LC50), Shankar et al. [21] Xu et al. [74] Song et al. [22] 4G, g UNIFAC Joback-Reid method Tm, Tb, 4Hfus, –log(LC50), h, Albahari’s GC-based method d, g UNIFAC Database-based Tm, Tb, Psat, r, s, h, Mw, g method Dortmund UNIFAC GC Tm, h, r, Mw, g COSMO-RS Song et al. [70] UNIFAC-IL Tm, h, Mw, g COSMO-RS Yang and Song [71] Ss, Sd, Sp, Sl, Tm, Tb, g 1 Scilipoti et al. [75] Zhang et al. [85] Ss, Sd, Sp, Sl, g 1 Ss, Sd, Tm, h, g 1 Gebreslassie and Diwekar Ss, Sd, Tb, g 1 [86] Harini et al. [76] Sd, Sp, Sl, Kow, Tm, Td, g 1 GC UNIFAC UNIFAC GC-FFANN COSMO-SAC GC UNIFAC GC UNIFAC Mixed-Integer Hybrid Different Evolutiona) Mass conservation Grid parallel computing methoda) LLE Mass conservation Energy conservation LLE Mass conservation Energy conservation LLE Generate and test Generate and test LLE Generate and test LLE Deterministic optimization method LLE Stochastic optimization algorithm LLE Stochastic optimization algorithm LLE Deterministic optimization method g: activity coefficients; g 1: infinite dilution activity coefficient; Tm: melting point; Tb: boiling point; –log(LC50): toxicity; 4G: Gibbs free energy;4Hfus: latent heat of fusion; h: viscosity; d: solubility parameter; Psat: vapor pressure; r: density; s: surface tension; Mw: molecular weight; SS: solvent selectivity ; Sd: solute distribution coefficient; Sp: solvent power; Sl: solvent loss; Kow: octanol water partition coefficient; Td: thermal decomposition temperature. The superscript a) indicates the deterministic optimization method. The superscript b) indicates the generate and test method. FFANN: Feed Forward Artificial Neural Network. Solvent-process design for extractive distillation Extractive distillation is commonly used to separate systems of close boiling compounds or azeotropes, in which a solvent (also called entrainer) is introduced [88]. A lot of work on the optimal selection and design of solvents for extractive distillation has been reported, as shown in Table 3. It can be seen that several important solvent properties such as Tm, Tb and VLE need to be considered in extractive distillation processes. Here, some selected studies and recent progress are reviewed. Lek-utaiwan et al. [77] proposed a framework for extractive distillation that integrates solvent screening, experimental verification, VLE data analysis, and process design and optimization for separate close-boiling mixtures. First, CAMD was used for solvent screening, where d, Tb, and Tm were predicted by the GC methods, and g was predicted by UNIFAC method. Second, the VLE experiments were carried out for the top ranked solvents. In the situation that the results of the existing performance prediction models were inconsistent with the experimental data, the interaction parameters in the performance prediction model were refitted properly according to the www.sciencedirect.com experimental results before the design and optimization of the distillation column. Finally, Aspen Plus was used for the process design and optimization for the best solvents from the previous step. The methodology was successfully applied to an industrial case study of EB/ mixed-xylene separation. Cignitti et al. [78] used CAMD to screen entrainers for extractive distillation processes based on new thermodynamic criteria. In the proposed CAMD problem, Tb and 4Hvb were predicted by GC methods, and g was predicted by the UNIFAC method. The new thermodynamic criteria take into account of the thermodynamic properties of binary mixtures and the isovolatility curves of ternary mixtures. Then, with the goal of minimizing energy consumption, Aspen Plus was used for process optimization. Finally, the feasibility of the framework was verified by the entrainer design for acetone-methanol separation. Zhou et al. [18] established a multi-objective optimization-based CAMD method and used it to search for promising entrainers for extractive distillation processes. In this method, selectivity and capacity as two important solvent properties determining the efficiency of extractive distillation were optimized Current Opinion in Chemical Engineering 2022, 35:100732 8 Frontiers in chemical engineering; chemical product design Table 3 A brief list of solvent-process design for extractive distillation Reference Properties Lek-utaiwan et al. [77] Tm, Tb, d, g Cignitti et al. [78] 4Hvb, g Zhou et al. [18] GC Mass conservation Energy conservation VLE Mass conservation Energy conservation VLE Mass conservation Energy conservation VLE UNIFAC UNIFAC GC Tm, Tb, g, g 1 Chao et al. [72] Process model GC Tb, Chen et al. [79] Property prediction method 1 1 a1 i;l , Si;j , Sp, Mw, Tb, g 1 Sp, S1 i;j , Tm, h, g UNIFAC GC UNIFAC GC UNIFAC-IL Solution strategy Decomposition algorithm Decomposition algorithm Decomposition algorithm VLE Deterministic optimization method VLE Generate and test 1 4Hvb: vaporization enthalpy; a1 i;l :relative volatility at infinite dilution; Si;j : selectivity at infinite dilution. simultaneously. Solvent properties (Tb and Tm) were predicted by GC methods, and g and g 1 were predicted by UNIFAC method. Then, the Pareto-optimal solvents were further selected through rigorous thermodynamic calculation and analysis. Finally, the extractive distillation process was optimized for each remaining solvent to determine the best candidate solvent with the highest process performance. It can be seen from the above investigations that the modeling of the solvent-process design of extractive distillation is different for different problems. The selection of suitable entrainers is one of the most important issues for extractive distillation. There are also many other factors that need to be considered. Ma et al. [19] reviewed extractive distillation from six aspects (thermodynamic analysis, QSPR, solvent screening, process design, process intensification, and dynamic control) and emphasized the importance of QSPR in selecting suitable solvents in extractive distillation. Sun et al. [89] reviewed the key aspects (conceptual design, solvent selection and separation strategies) of extractive distillation, and elaborated the application of CAMD in solvent selection of extractive distillation. The challenge of solvent design for extractive distillation is similar to that of liquid–liquid extraction, that is, sometimes lack of reliable property predictive models. In addition, due to the high energy consumption and large investment of extractive distillation, it is also necessary to carry out technological innovation on the extractive distillation processes in addition to solvent design. Process intensification and dynamic control are helpful to reduce energy consumption and promote the development of extractive distillation in the direction of intelligence and security. Solvent-process design for gas absorption Because of the rapid economic growth all over the world, reducing CO2 emissions has become a global challenge. As one of the important applications of gas absorption, carbon capture has attracted the attention of many Current Opinion in Chemical Engineering 2022, 35:100732 researchers. Choosing/designing suitable solvents can significantly reduce the energy consumption of the carbon capture process and relieve the problem of global warming. A lot of work on the optimal selection and design of solvents for carbon capture has been reported, as shown in Table 4. It can be seen from Table 4 that for carbon capture processes, several important solvent properties such as Tm and VLE need to be considered. Here, some selected studies and recent progress are reviewed. Papadopoulos et al. [31] proposed a CAMD framework considering life cycle assessment (LCA), and safety, hazard and environmental (EHS) properties. It calculates a total of 11 sustainability-related indicators and integrates several impact categories. A design example of phase-change solvents for chemisorption-based postcombustion CO2 capture was introduced. The proposed framework identified verifiably useful phase-change solvents, which showed favorable performance compared with a reference CO2 capture solvent. Ahmad et al. [80] used CAMD method to design new alternative solvents for the post-combustion carbon capture in power plants. Solvent properties (d, r, m, Tb, Tm, s and Tf) were predicted by the GC methods, and g was predicted by the UNIFAC method. The process performance was evaluated by calculating the heat required for the solvent regeneration process. Finally, according to the specified target properties, 25 candidate solvents have been successfully generated from amine and alcohol functional groups. Compared with the traditional solvent (ethanolamine), the candidate solvent can significantly save up to 31.4% of energy requirement for the regeneration process, thus greatly reducing the cost of carbon capture. Scilipoti et al. [81] successfully applied the CAMD method based on the GC-EOS (GC Equation of State) to the solvent selection for a pre-combustion CO2 capture process. This work systematically studied the effects of molecular functional groups on solvent properties and successfully predicted the solvent properties (Sp, a and Sl) under the changes of pressure, temperature, solvent www.sciencedirect.com Computer-aided molecular design of solvents for chemical separation processes Chai et al. 9 Table 4 A brief list of solvent-process design for carbon capture Reference Properties Property prediction method Process model Solution strategy Wang et al. [34] m, Tm, g 1 GC COSMO-SAC GC VLE Stochastic optimization algorithm Ahmad et al. [80] d, r, m, s, Tm, Tb, Tf, g UNIFAC Scilipoti et al. [81] Sp, a, Sl Chong et al. [32] Zhang et al. [82] r, 4Hvap, Cp, h, g Tm, Tb, Mw, Mv, h, Cp, l Zhang et al. [93] Tm, Tb, Mw, Mv, h, Cp, r Tf : flash point; GC-EOS GC UNIFAC GC ANN-GC GC ANN-GC Mass conservation Energy conservation VLE Mass conservation Energy conservation VLE Decomposition algorithm Deterministic optimization methods VLE Decomposition algorithm VLE Deterministic optimization methods Mass conservation Energy conservation VLE Deterministic optimization methods 4Hvap: heat of vaporization; Cp: heat capacity; Mv: molecular volume; l: thermal conductivity. structure, and system composition. Finally, based on a hot flash solvent recovery scheme, the most promising candidate solvents were optimized for the absorption cycle to reduce the energy consumption. For the carbon capture process, in addition to choosing traditional organic solvents, Chong et al. [32] proposed a design method of ILs and IL mixtures to replace the traditional organic solvent (ethanolamine) used in carbon capture. To overcome the problem of missing property data of IL mixtures, this paper presented a method which can directly use the performance data of pure Ils and combine the existing performance prediction models and experimental data to predict the performance of IL mixtures. Here, r, 4Hvap, Cp and h of pure Ils were predicted by GC methods and g was predicted by the UNIFAC method. In addition to GC-based methods, there are also QM and ML-based methods for the prediction of the properties of solventprocess design for carbon capture. Wang et al. [34] proposed a systematic Computer-Aided Ionic Liquid Design (CAILD) method for CO2 capture. Solvent properties (m and Tm) were predicted by the GC methods, and g 1 was predicted by COSMO-SAC model. Finally, the established MINLP model was solved using stochastic optimization algorithm. Tatar et al. [90] used artificial intelligence-based methods to successfully predict the solubility of CO2 in 14 different ionic liquids. Sistla and Sridhar [91] explored the interaction between CO2 and Ils in the process of CO2 adsorption at the molecular level based on QM methods. Song et al. [92] used ML-GC model to predict the solubility of CO2 in ionic liquids (ILS) based on a database containing 10116 CO2 solubility data in Ils at different temperatures and pressures. Subsequently, Zhang et al. [82] determined the optimal IL solvents based on the ML-GC model established by Song et al. [92] with the maximum CO2 equilibrium solubility as the objective function. More recently, Zhang et al. [93] used this surrogate solubility model (ML-GC model established by Song et al. [92]) to replace the www.sciencedirect.com traditional thermodynamic model and performed an integrated IL and process design work. It was found that to use the ML-based solubility model can largely reduce the computational difficulty, making it possible to find the global optimum for the integrated design problem. Excitingly, the designed IL-based process can potentially save 14.8% cost compared to the industrial Selexol process. These works provide accurate property prediction models for the novel solvent design of carbon capture. In addition, the combination of adsorption and other separation technologies (for example adsorption and membrane) has attracted the attention of researchers. Hybrid technologies in the selection of carbon capture solvents may be helpful to find more economical solutions, which deserves further study [41]. Solvent-process design for crystallization Solution crystallization is an important separation unit operation in the pharmaceutical industry. Solvent is one of the most important factors affecting product quality (purity, yield, crystal form, crystal morphology, particle size distribution, etc.). A lot of work on the optimal selection and design of crystallization solvents has been reported, as shown in Table 5. It can be seen from Table 5 that for the crystallization processes, several important solvent properties such as Tm, Tb, Tf, toxicity and SLE need to be considered. Here, some selected studies and recent progress are reviewed. Karunanithi et al. [4,26] first used the decomposition algorithm to design crystallization solvents for ibuprofen and carboxylic acids. The objective function was the potential yield and the SLE was considered as process constraints, wherein the main physical properties (d, Tb, Tm, Tf, –log(LC50), dH and h) were predicted by the GC methods and g was predicted by the UNIFAC method. Among the considered physical properties, dH and h are used to constrain the crystal morphology. Because of the complexity of the crystallization process, the use of single solvent oftentimes cannot Current Opinion in Chemical Engineering 2022, 35:100732 10 Frontiers in chemical engineering; chemical product design Table 5 A brief list of solvent-process design for crystallization Properties Reference Karunanithi et al. [4 ,26] Watson et al. [83] Liu et al. [52] Chai et al. [27] Tm, Tb, d, Tf, g, –log(LC50), dH , h g, Mi Tm, Tb, d, Tf, g, –log(LC50), dH , h Mw, Tm, Tb, d, Tf, g, –log(LC50), h Liu et al. [12 ] Tm, Tb, d, Tf, g, –log(LC50), dH , h Property prediction method GC UNIFAC SAFT-g Mie equation of state GC COSMO-RS GC COSMO-SAC GC MLAC Process model Solution strategy SLE Decomposition algorithm SLE Deterministic optimization methods SLE Decomposition algorithm Mass conservation SLE Decomposition algorithm SLE Decomposition algorithm dH : hydrogen bond solubility parameter; Mi: miscibility. meet the requirements. Therefore, Watson et al. [83] proposed a general computer aided molecular/mixture design (CAMbD) for the crystallization process of pharmaceutical products. The proposed method can simultaneously identify the optimal process temperature, solvent and antisolvent molecules, and the composition of solvent mixture, wherein g was calculated from the SAFT-g Mie equation of state. The above crystallization solvent design examples considered the perspective of product performance but neglected the cost, pricing, and other economic factors. Chai et al. [27] proposed a Grand Product Design (GPD) model for the design of crystallization solvents, which contains objective function (single/ multi-objective) process submodel, property submodel, quality submodel, cost submodel, pricing submodel, economic submodel, and environmental submodel as well as other factors (such as company strategy, government policies and regulations). Finally, the GPD-model was successfully applied to the crystallization solvent design for 2-Mercaptobenzothiazole (MBT), for which the main solvent properties (Mw, Tb, Tm, Tf, –log(LC50), d, h) were predicted by GC methods and g was predicted by COSMO-SAC model. The process constraints include mass conservation and SLE. Finally, the established MINLP model was solved by the decomposition algorithm. Liu et al. [12] proposed an MLAC (Machine Leaning-based Atom Contribution) method to predict the charge density profiles and applied it to the crystallization solvent design of ibuprofen. This method balances the high computational cost of QM and the limited prediction accuracy of the GC method. Among them, the objective function and constraints refer to the work of Karunanithi et al. [4]. The difference is that g is predicted by the MLAC method. At present, the crystallization solvent-process design based on product purity and yield is relatively mature while the solvent-process design considering other product performances (such as crystal form, crystal habit and particle size distribution) is still in the exploratory stage. One possible research directions include the establishment of the QSPR relationship between solvent molecules and such product performances (crystal form, crystal habit, and particle size Current Opinion in Chemical Engineering 2022, 35:100732 distribution) by combining the results of molecular/macro simulation (through MD, CFD, population balance, etc.) with ML. Discussion and perspectives Although CAMD methods have already been widely applied in solvent design, it is still a research topic not fully developed and most solvents are still developed through experiment-based trial-and-error approaches. The systematic model and/or data-based methods and associated software tools should be able to make a major contribution to solvent design, and thereby, significantly reduce the design and development time and cost. The following four aspects are discussed and prospected: Different multidisciplinary methods and tools are used to solve complex problems in solvent design. For example, simplified models based on ML are used to solve the time-consuming problems of MD, QM and so on. The product design software contains a wealth of databases, property prediction models, solution strategies, and so on, which help to quickly screen/design all solvent molecules that meet the requirements. Data-driven based on machine learning is helpful to establish property prediction models that are difficult to obtain based on theoretical methods. High-throughput solvent design tool can quickly realize the experimental verification of the designed/ screened solvents. Manage the complexity of the multiscale multidisciplinary problems The solvent design problems for separation processes are multiscale and multidiscipline problems as molecular QSPR, molecular interaction, fluid dynamics, and separation processes are involved. Thus, different methods and tools from multidiscipline are needed. For example, for designing a crystallization solvent, quantum mechanics models are needed for structural optimization and obtaining missing force field parameters for MD simulation, MD models are used to predict the crystal www.sciencedirect.com Computer-aided molecular design of solvents for chemical separation processes Chai et al. 11 morphology and crystal growth rate, thermodynamic models are used to predict the solid–liquid equilibrium, population balance model and CFD simulation are needed for flow distribution and crystal size distribution, and finally process unit operation model is used for the design and optimization of the crystallization process with the optimal solvent. This problem is so complex that it is almost impossible to solve it in a fast and efficient way. One option of the solution strategy is using model reduction techniques. For example, use machine learning methods to replace the time-consuming MD calculation [94]. Apply solvent design tools to industrial cases Product design software is based on computer-aided molecular design method, which helps to quickly realize solvent screening/design. Kalakul et al. [95] presented a new version of the product design software tool ProCAPD. Compared with earlier versions, ProCAPD has improved software architecture, new and extended databases, models in the model library and new solution approaches. It can quickly and effectively solve wider ranges of CAMbD problems, as well as other CAMD and liquid formulation problems. Chai et al. [61] proposed a versatile modeling framework consisting of a collection of submodels (molecular structure, property, process, costing, pricing, economic analysis, quality, sustainability, environmental impact, and performance). The developed modeling framework has been incorporated into the software tool ProCAPD, along with an extended database and a library of product design templates. Although product design tools [63,96], have been developed, these tools focus on some specific types of products, mainly small molecules and mixtures. properties between compounds with very similar structures or even isomers, an appropriate molecular representation is critical. Recently, the adversarial autoencoder technique has been applied for molecular design where the ML models are trained on molecular descriptors, 3D structures or molecular graphs [99]. The data collection and processing steps are also important for ML-based solvent design. The data can come from experiments and/ or simulations such as DFT, MD, CFD, and so on. The consistent development of open-source databases and various property models is vital to accelerate future advances of this research field [44]. Moreover, raw molecular data often feature a large degree of noise and contain strongly correlated variables, which requires a careful data pre-treatment before model training can be performed. Finally, the selection of ML models is also important for achieving a good fitting performance, which is currently implemented using a heuristic-based or trialand-error strategy. High-throughput solvent design technology High throughput solvent design is the use of automated equipment to rapidly test thousands to millions of solvent samples in parallel. It utilizes robotics, liquid handlers, data processing, software, and sensitive detection systems. For instance, Gu et al. [100] used a robot-based high-throughput platform to quickly screen potential anti-solvents for different combinations of solvent and perovskite compositions. Because high-throughput technology typically aims to screen 100,000 or more samples per day, relatively simple and automation-compatible assay designs, robotic-assisted sample handling, and automated data processing are critical. Conclusions Data-driven solvent design based on machine learning In solvent design, the property estimation models and data need to be enlarged, which can be obtained from theory-based methods, data-driven methods or their hybrid. However, theoretical-methods are generally too complex and almost impossible to be implemented in a model-based solvent design. Therefore, with available methods and tools from data science, data-driven MLbased solvent design can be considered. Alshehri et al. [97] used machine learning and data science methods to address the shortcomings of the current GC-based models in fast and accurate estimation of 20 physicochemical properties. Chen et al. [98] established a TransformerCNN model to quickly predict the surface charge density profiles (s-profile) and cavity volumes (VCOSMO) of molecules by using the deep learning method. The model can predict s-profile and VCOSMO of millions of molecules in just a few minutes. Several similar studies have also been carried out [12,13,60,82,90–93]. In data-driven ML methods, the selection of molecular descriptors plays an essential role for property prediction as it determines the model performance. For the differentiation of www.sciencedirect.com This paper summarizes solvent-process design in chemical separation processes, including property prediction methods, solution strategies, some representative studies, challenges and future directions. Although a large progress has been made in computeraided solvent design, there are still challenges for the selection/design of the optimal solvents in different separation processes, as the requirements and properties involved vary for different problems. Property prediction models are the basis of solvent design, and these models need to be extended to more complex problems, such as reaction kinetics prediction, crystal morphology prediction, polymer properties prediction, design of cleaning agents, and additives in different products. One of the future directions is to consider data-driven ML-based solvent design. For solvent design tools, although several tools have been developed, the challenges are the needs of mature tools for industrial solvent design, which are easy to use and can accurately estimate all the solvent properties needed in industrial applications. In developing such tools, database, experiments, heuristic rules and Current Opinion in Chemical Engineering 2022, 35:100732 12 Frontiers in chemical engineering; chemical product design models including DFT (Density Functional Theory), MD, and so on. for the establishment of QSPR could be integrated to estimate more types of properties in a more accurate and efficient way. In high-throughput solvent design technology, better industry relevant models are needed, as in this process, the scale-down processes are used that they do not always hold true at commercial manufacturing scale or conditions. Another challenge is the increasing use for robotics and automation, which need the efforts from different disciplines. Conflict of interest statement z P, Caflisch A: Protein structure-based drug design: from 10. Sled docking to molecular dynamics. Curr Opin Struct Biol 2018, 48:93-102. 11. Stuebing H, Obermeier S, Siougkrou E, Adjiman CS, Galindo A: A QM-CAMD approach to solvent design for optimal reaction rates. Chem Eng Sci 2016, 159:69-83. 12. Liu Q, Zhang L, Tang K, Liu L, Du J, Meng Q, Gani R: Machine learning-based atom contribution method for the prediction of charge density profiles and solvent design. AIChE J 2021, 67: e17110 This paper proposed a novel ML-based atom contribution method to predict molecular surface charge density profiles (s-profiles). 13. Wang ZH, Zhen S, Zhou T: Machine learning for ionic liquid toxicity prediction. Process 2021, 9:65. Acknowledgements 14. Zhang L, Pang JQ, Zhuang Y, Liu LL, Du J, Yuan ZH: Integrated solvent-process design methodology based on COSMO-SAC and quantum mechanics for TMQ (2,2,4-trimethyl-1,2Hdihydroquinoline) production. Chem Eng Sci 2020, 226:115894. The financial support from National Nature Science Foundation of China (22078041, 21808025, 21776074 and 21861132019) and ‘the Fundamental Research Funds for the Central Universities (DUT20JC41)’ is acknowledged. 15. Zhang L, Mao HT, Liu LL, Du J, Gani R: A machine learning based computer-aided molecular design/screening methodology for fragrance molecules. Comput Chem Eng 2018, 115:295-308. Declaration of Competing Interest 16. Gani R, Brignole E: Molecular design of solvents for liquid extraction based on UNIFAC. Fluid Phase Equilib 1983, 13:331340. Nothing declared. The authors report no declarations of interest. References and recommended reading Papers of particular interest, published within the period of review, have been highlighted as: of special interest 1. Chen YQ, Koumaditi E, Gani R, Kontogeorgis GM, Woodley JM: Computer-aided design of ionic liquids for hybrid process schemes. Comput Chem Eng 2019, 13:106556. 2. Chemmangattuvalappil NG: Development of solvent design methodologies using computer-aided molecular design tools. Curr Opin Chem Eng 2020, 27:51-59 This article reviews the recent developments of solvent design using computer-aided molecular design (CAMD) tools. 3. Ten JY, Liew ZH, Oh XY, Hassim MH, Chemmangattuvalappil N: Computre-aided molecular design of optimal sustainable solvent for liquid-liquid extraction. Process Integr Optim Sustain 2021, 5:269-284. 4. Karunanithia AT, Acheniea LEK, Gani R: A computer-aided molecular design framework for crystallization solvent design. Chem Eng Sci 2006, 61:1247-1260 This article proposed the decomposition algorithm to solve MINLP problems for crystallization solvent design. 5. 6. 7. Liu QL, Zhang L, Tang K, Feng YX, Zhang JY, Zhuang Y, Liu LL, Du J: Computer-aided reaction solvent design considering inertness using group contribution-based reaction thermodynamic model. Chem Eng Res Des 2019, 152:123-133. Gmehling J: Present status and potential of group contribution methods for process development. J Chem Thermodyn 2009, 41:731-747. M, Adjiman CS: Solvent design using a Sheldon TJ, Folic quantum mechanical continuum solvation model. Ind Eng Chem Res 2006, 45:1128-1140. 8. Alshehri AS, Gani R, You FQ: Deep learning and knowledgebased methods for computer aided molecular design—toward a unified approach: state-of-the-art and future directions. Comput Chem Eng 2020, 141:107005 This article reviews recent progress, limitations, and opportunities in CAMD for both knowledge-based and deep learning-based approaches. 9. Su Y, Wang Z, Jin S, Shen W, Ren J, Eden MR: An architecture of deep learning in QSPR modeling for the prediction of critical properties using molecular signatures. AIChE J 2019, 65: e16678. Current Opinion in Chemical Engineering 2022, 35:100732 17. van Dyk B, Nieuwoudt I: Design of solvents for extractive distillation. Ind Eng Chem Res 2000, 39:1423-1429. 18. Zhou T, Song Z, Zhang X, Gani R, Sundmacher K: Optimal solvent design for extractive distillation processes: a multiobjective optimization-based hierarchical framework. Ind Eng Chem Res 2019, 58:5777-5786. 19. Ma YX, Cui PZ, Wang YK, Zhu ZY, Wang YL, Gao J: A review of extractive distillation from an azeotropic phenomenon for dynamic control. Chin J Chem Eng 2019, 27:1510-1522. 20. Khor SY, Liam KY, Loh WX, Tan CY, Ng LY, Hassim MH, Ng DKS, Chemmangattuvalappil NG: Computer aided molecular design for alternative sustainable solvent to extract oil from palm pressed fibre. Process Saf Environ 2017, 106:211-223. 21. Shankar KN, Adhikari J, Noronha SB: Computer-aided solvent selection and design for the efficient extraction of a pharmaceutical molecule. Can J Chem Eng 2019, 97:1605-1618. 22. Song Z, Zhou T, Qi Z, Sundmacher K: Systematic method for screening ionic liquids as extraction solvents exemplified by an extractive desulfurization process. ACS Sustain Chem Eng 2017, 5:3382-3389. 23. Lyu Z, Zhou T, Chen L, Ye Y, Sundmacher K, Qi Z: Reprint of: simulation based ionic liquid screening for benzenecyclohexane extractive separation. Chem Eng Sci 2014, 115:186-194. 24. Ten JY, Liew ZH, Oh XY, Hassim MH: Chemmangattuvalappil: computer-aided molecular design of optimal sustainable solvent for liquid-liquid extraction. Proc Integr Optim 2021, 5:269-284. 25. Wang J, Cheng H, Song Z, Chen L, Deng L, Qi Z: Carbon dioxide solubility in phosphonium-based deep eutectic solvents: an experimental and molecular dynamics study. Ind Eng Chem Res 2019, 58:17514-17523. 26. Karunanithi AT, Acquah C, Achenie L, Sithambaram S, Suib SL: Solvent design for crystallization of carboxylic acids. Comput Chem Eng 2009, 33:1014-1021. 27. Chai S, Liu Q, Liang X, Guo Y, Zhang S, Xu C, Du J, Yuan Z, Zhang L, Gani R: A grand product design model for crystallization solvent design. Comput Chem Eng 2020, 135:106764. 28. Zhou T, McBride K, Zhang X, Qi Z, Sundmacher K: Integrated solvent and process design exemplified for a Diels-Alder reaction. AIChE J 2015, 61:147-158. www.sciencedirect.com Computer-aided molecular design of solvents for chemical separation processes Chai et al. 13 29. Zhou T, Qi Z, Sundmacher K: Model-based method for the screening of solvents for chemical reactions. Chem Eng Sci 2014, 115:177-185. 45. Kupgan G, Abbott LJ, Hart KE, Colina CM: Modeling amorphous microporous polymers for CO2 capture and separations. Chem Rev 2018, 118:5488-5538. 30. Liu Q, Zhang L, Liu L, Du J, Meng Q, Gani R: Computer-aided reaction solvent design based on transition state theory and COSMO-SAC. Chem Eng Sci 2019, 202:300-317. 46. Liu Y, Zhao T, Ju W, Shi S: Materials discovery and design using machine learning. J Materiomics 2017, 3:159-177 This article reviews the typical modes and basic procedures for applying ML in materials science. 31. Papadopoulos AI, Shavalieva G, Papadokonstantakis S, Seferlis P, Perdomo FA, Galindo A, Jackson G, Adjiman CS: An approach for simultaneous computer-aided molecular design with holistic sustainability assessment: application to phasechange CO2 capture solvents. Comput Chem Eng 2020, 135:106769. 32. Chong FK, Eljack FT, Atilhan M, Foo DCY, Chemmangattuvalappil NG: A systematic visual methodology to design ionic liquids and ionic liquid mixtures: green solvent alternative for carbon capture. Comput Chem Eng 2016, 91:219232. 33. Papadokonstantakis S, Badr S, Hungerbühler K, Papadopoulos AI, Damartzis T, Seferlis P, Forte E, Chremos A, Galindo A, Jackson G, Adjiman CS: Toward sustainable solventbased postcombustion CO2 capture: from molecules to conceptual flowsheet design. Comput Aided Chem Eng 2015, 36:279-310. 34. Wang J, Song Z, Cheng H, Chen L, Deng L, Qi Z: Computer-aided design of ionic liquids as absorbent for gas separation exemplified by CO2 capture cases. ACS Sustain Chem Eng 2018, 6:12025-12035. 35. Song Z, Hu X, Wu H, Mei M, Linke S, Zhou T, Qi Z, Sundmacher K: Systemic screening of deep eutectic solvents as sustainable separation media exemplified by the CO2 capture process. ACS Sustain Chem Eng 2020, 8:8741-8751. 36. Wang J, Song Z, Cheng H, Chen L, Deng L, Qi Z: Multilevel screening of ionic liquid absorbents for simultaneous removal of CO2 and H2S from natural gas. Sep Purif Technol 2020, 248:117053. 37. Liang XY, Zhang X, Zhang L, Liu LL, Du J, Zhu XL, Ng KM: Computer-aided polymer design: integrating group contribution and molecular dynamics. Ind Eng Chem Res 2019, 58:15542-15552. 38. Jhamb S, Enekvist M, Liang X, Zhang X, Dam-Johansen K, Kontogeorgis GM: A review of computer-aided design of paints and coatings. Curr Opin Chem Eng 2019, 23:184-196. 39. Jonuzaj S, Cui J, Adjiman CS: Computer-aided design of optimal environmentally benign solvent-based adhesive products. Comput Chem Eng 2019, 130:106518. 47. Sah S: Machine Learning: A Review of Learning Types. 2020 http:// dx.doi.org/10.20944/preprints202007.0230.v1. 48. Fan C, Liu YC, Liu XY, Sun YJ, Wang JY: A study on semisupervised learning in enhancing performance of AHU unseen fault detection with limited labeled data. Sustain Cities Soc 2021, 70:102874. 49. Riazi A, Slovinsky P: Subaerial beach profiles classification: an unsupervised deep learning approach. Cont Shelf Res 2021, 226:104508. 50. Zhou SK, Le HN, Luu K, Nguyen HV, Ayache N: Deep reinforcement learning in medical imaging: a literature review. Med Image Anal 2021, 27:102193. 51. Datta S, Dev VA, Eden MR: Developing non-linear rate constant QSPR using decision trees and multi-gene genetic programming. Comput Chem Eng 2019, 127:150-157. 52. Liu QL, Zhang L, Liu LL, Du J, Tula AK, Eden M, Gani R: OptCAMD: an optimization-based framework and tool for molecular and mixture product design. Comput Chem Eng 2019, 124:285-301. 53. Fredenslund A, Jones RL, Prausnitz JM: Group-contribution estimation of activity coefficients in nonideal liquid mixtures. AIChE J 1975, 21:1086-1099. 54. Chapman WG, Gubbins KE, Jackson G, Radosz M: SAFT: equation-of-state solution model for associating fluids. Fluid Phase Equilib 1989, 52:31-38. 55. Chen G, Song Z, Qi Z, Sundmacher K: Neural recommender system for the activity coefficient prediction and UNIFAC model extension of ionic liquid-solute systems. AIChE J 2021, 67:e17171 This paper proposes a deep neural network based recommendation system (RS) for predicting the infinite dilution activity coefficient (g 1 ) and applying it to the extension of the UNIFAC model. 56. Klamt A: Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena. J Phys Chem 1995, 99:2224-2235. 57. Lin S, Sandler SI: A priori phase equilibrium prediction from a segment contribution solvation model. Ind Eng Chem Res 2002, 41:899-913. 40. Gani R: Group contribution-based property estimation methods: advances and perspectives. Curr Opin Chem Eng 2019, 23:184-196 This article reviews the advances and perspectives of properties prediction methods based on group contribution methods. 58. Peng D, Zhang J, Cheng H, Chen L, Qi Z: Computer-aided ionic liquid design for separation processes based on group contribution method and COSMO-SAC model. Chem Eng Sci 2017, 159:58-68. 41. Zhou T, McBride K, Linke S, Song Z, Sundmacher K: Computer aided solvent selection and design for efficient chemical processes. Curr Opin Chem Eng 2020, 27:35-44 This article reviews the challenges and perspectives of solvent selection and design for chemical processes. 59. Zhang J, Peng D, Song Z, Cheng H, Chen L, Qi Z: COSMOdescriptor based computer-aided ionic liquid design for separation processes. Part I: modified group contribution methodology for predicting surface charge density profile of ionic liquids. Chem Eng Sci 2017, 162:355-363. 42. Klamt A, Schüürmann G: COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc 1993, 2:799-805. 43. Gertig C, Leonhard K, Bardow A: Computer-aided molecular and processes design based on quantum chemistry: current status and future prospects. Curr Opin Chem Eng 2020, 27:8997 This article reviews the challenges and perspectives of computer-aided molecular and processes design based on quantum chemistry. 44. Zhang L, Mao HT, Liu QL, Gani R: Chemical product design recent advances and perspectives. Curr Opin Chem Eng 2020, 27:22-34 This article reviews the latest developments and perspectives of chemical product design. www.sciencedirect.com 60. Zhang L, Mao H, Zhuang Y, Wang L, Liu L, Dong Y, Du J, Xie W, Yuan Z: Odor prediction and aroma mixture design using machine learning model and molecular surface charge density profiles. Chem Eng Sci 2021, 245:116947 This paper establishes the Structure-Odor Relationship (SOR) model of aroma mixtures using the molecular surface charge density distribution as descriptors. 61. Chai SY, Zhang L, Du J, Tula AK, Gani R, Eden MR: A versatile modeling framework for integrated chemical product design. Ind Eng Chem Res 2020, 60:436-456 This paper proposes a versatile modeling framework for chemical product design, which consists of a collection of submodels (molecular structure, property, process, costing, pricing, economic, quality, sustainability, environmental, and performance). Current Opinion in Chemical Engineering 2022, 35:100732 14 Frontiers in chemical engineering; chemical product design 62. Gani R, Nielsen B, Fredenslund A: A group contribution approach to computer-aided molecular design. AIChE J 1991, 37:1318-1332. 82. Zhang X, Wang J, Song Z, Zhou T: Data-driven ionic liquid design for CO2 capture: molecular structure optimization and DFT verification. Ind Eng Chem Res 2021, 60:9992-10000. 63. Harper PM, Gani R: A multi-step and multi-level approach for computer aided molecular design. Comput Chem Eng 2000, 24:677-683. 83. Watson OL, Galindoa A, Jacksona G, Adjiman CS: Computeraided design of solvent blends for the cooling and anti-solvent crystallisation of ibuprofen. Comput Aided Chem Eng 2019, 46:949-954. 64. Maulik U, Bandyopadhyay S: Genetic algorithm-based clustering technique. Pattern Recogn 2000, 33:1455-1465. 65. Abdelaziz AY, Mohamed FM, Mekhamer SF, Badr MAL: Distribution system reconfiguration using a modified tabu search algorithm. Electr Pow Syst Res 2010, 80:943-953. 66. Venkatasubramanian V, Chan K, Caruthers JM: Computer-aided molecular design using genetic algorithms. Comput Chem Eng 1994, 18:833-844. 67. Scheffczyk J, Fleitmann L, Schwarz A, Lampe M, Bardow A, Leonhard K: COSMO-CAMD: a framework for optimizationbased computer-aided molecular design using COSMO-RS. Chem Eng Sci 2017, 159:84-92. 68. Zhou T, Zhou YG, Sundmacher K: A hybrid stochasticdeterministic optimization approach for integrated solvent and process design. Chem Eng Sci 2017, 159:207-216. 84. Zhou T, Wang J, McBride K, Sundmacher K: Optimal design of solvents for extractive reaction processes. AIChE J 2016, 62:3238-3249. 85. Zhang J, Qin L, Peng D, Cheng H, Chen L, Qi Z: COSMOdescriptor based computer-aided ionic liquid design for separation processes. Part II: task-specific design for extraction process. Chem Eng Sci 2017, 162:364-374. 86. Gebreslassie BH, Diwekar UM: Efficient ant colony optimization for computer aided molecular design: case study solvent selection problem. Comput Chem Eng 2015, 78:1-9. 87. Qin L, Zhang JN, Cheng HY, Chen LF, Qi ZW, Yuan WK: Selection of imidazolium-based ionic liquids for vitamin E extraction from deodorizer distillate. ACS Sustain Chem Eng 2016, 4:583590. 69. Mcleese SE, Eslick JC, Hoffmann NJ, Scurto AM, Camarda KV: Design of ionic liquids via computational molecular design. Comput Chem Eng 2010, 34:1476-1480. 88. Song Z, Li XX, Chao H, Mo F, Zhou T, Cheng HY, Chen LF, Qi ZW: Computer-aided ionic liquid design for alkane/cycloalkane extractive distillation process based on task-specifically fitted UNIFAC-IL model. Green Energy Environ 2019, 4:154-165. 70. Song Z, Zhang C, Qi Z, Zhou T, Sundmacher K: Computer-aided design of ionic liquids as solvents for extractive desulfurization. AIChE J 2018, 64:1013-1025. 89. Sun S, Lü L, Yang A, Wei S, Shen W: Extractive distillation: advances in conceptual design, solvent selection, and separation strategies. Chin J Chem Eng 2019, 27:1247-1256. 71. Yang XG, Song HH: Computer aided molecular design of solvents for separation processes. Chem Eng Technol 2006, 29:33-43. 90. Tatar A, Naseri S, Bahadori M, Hezave AZ, Kashiwao T, Bahadori A, Darvish H: Prediction of carbon dioxide solubility in ionic liquids using MLP and radial basis function (RBF) neural networks. J Taiwan Inst Chem E 2016, 60:151-164. 72. Chao H, Song Z, Cheng HY, Chen LF, Qi ZW: Computer-aided design and process evaluation of ionic liquids for n-hexanemethylcyclopentane extractive distillation. Sep Purif Technol 2018, 196:157-165. 73. Cheng HC, Wang FS: Computer-aided biocompatible solvent design for an integrated extractive fermentation-separation process. Chem Eng J 2010, 162:809-820. 74. Xu R, Zhao YH, Han QZ, Ning PG, Cao HB, Wen H: Computeraided blended extractant design and screening for coextracting phenolic, polycyclic aromatic hydrocarbons and nitrogen heterocyclic compounds pollutants from coal chemical wastewater. J Clean Prod 2020, 277:122334. 75. Scilipoti JA, Cismondi M, Andreatta AE, Brignole EA: Selection of solvents with A-UNIFAC applied to detoxification of aqueous solutions. Ind Eng Chem Res 2014, 53:17051-17058. 76. Harini M, Jain S, Adhikari J, Noronha SB, Rani KY: Design of an ionic liquid as a solvent for the extraction of a pharmaceutical intermediate. Sep Purif Technol 2015, 155:45-57. 77. Lek-utaiwan P, Suphanit B, Douglas PL, Mongkolsiri N: Design of extractive distillation for the separation of close-boiling mixtures: solvent selection and column optimization. Comput Chem Eng 2011, 35:1088-1100. 78. Cignitti S, Rodriguez-Donis I, Abildskov J, You X, Shcherbakova N, Gerbaud V: CAMD for entrainer screening of extractive distillation process based on new thermodynamic criteria. Chem Eng Res Des 2019, 147:721-733. 79. Chen BH, Lei ZG, Li QS, Li CY: Application of CAMD in separating hydrocarbons by extractive distillation. AIChE J 2005, 51:3114-3121. 80. Ahmad MZ, Hashim H, Mustaffa AA, Maarof H, Yunus NA: Design of energy efficient reactive solvents for post combustion CO2 capture using computer aided approach. J Clean Prod 2018, 176:704-715. 81. Scilipoti JA, Sánchez FA, Pereda S, Brignole EA: Molecular design of solvents for CO2 capture using a group contribution EOS. Fluid Phase Equilib 2019, 490:114-122. Current Opinion in Chemical Engineering 2022, 35:100732 91. Sistla YS, Sridhar V: Molecular understanding of carbon dioxide interactions with ionic liquids. J Mol Liq 2020, 325:115162. 92. Song Z, Shi H, Zhang X, Zhou T: Prediction of CO2 solubility in ionic liquids using machine learning methods. Chem Eng Sci 2020, 223:115752. 93. Zhang X, Ding X, Song Z, Zhou T, Sundmacher K: Integrated ionic liquid and rate-based absorption process design for gas separation: global optimization using hybrid models. AIChE J 2021:e17340 http://dx.doi.org/10.1002/aic.17340. 94. Deringer VL, Caro MA, Csanyi G: Machine learning interatomic potentials as emerging tools for materials science. Adv Mater 2019, 31:1902765. 95. Kalakul S, Zhang L, Fang Z, Choudhury HA, Intikhab S, Elbashir N, Eden MR, Gani R: Computer aided chemical product designProCAPD and tailor-made blended products. Comput Chem Eng 2018, 116:37-55. 96. Gani R, Hytoft G, Jaksland C, Jensen AK: An integrated computer aided system for integrated design of chemical processes. Comput Chem Eng 1997, 21:1135-1146. 97. Alshehri AS, Tula AK, Zhang L, Gani R, You FQ: A platform of machine learning-based next-generation property estimation methods for CAMD. Comput Aid Chem Eng 2021, 50:227-233. 98. Chen GZ, Song Z, Qi ZW: Transformer-convolutional neural network for surface charge density profile prediction: enabling high-throughput solvent screening with COSMOSAC. Chem Eng Sci 2021, 246:117002 http://dx.doi.org/10.1016/j. ces.2021.117002. 99. Polykovskiy D, Zhebrak A, Vetrov D, Ivanenkov Y, Aladinskiy V, Mamoshina P, Bozdaganyan M, Aliper A, Zhavoronkov A, Kadurin A: Entangled conditional adversarial autoencoder for de novo drug discovery. Mol Pharma 2018, 15:4398-4405. 100. Gu E, Tang XF, Langner S, Duchstein P, Zhao YC, Levgen L, Kalancha V, Stubhan T, Hauch J, Egelhaaf HJ et al.: Robot-based high-throughput screening of antisolvents for lead halide perovskites. Joule 2020, 4:1806-1822. www.sciencedirect.com