Uploaded by
Muran Mujahed
Rheumatoid & Osteoarthritis Case Studies & Review
Muran Mjahed 1 Rheumatoid arthritis History A 34-year-old woman attends the rheumatology outpatient clinic with a 6-month history of painful hands. Previously fit and well, she developed mild pain in her metacarpophalangeal (MCP) joints which deteriorated over a few weeks to profound pain and marked stiffness affecting both hands and wrists, particularly in the morning. Her symptoms are now very intrusive. Her general practitioner has prescribed diclofenac with minimal benefit. She is otherwise well but is becoming isolated and depressed; she delayed going to see her GP because she was concerned that she had developed the same ‘rheumatism’ as her grandmother who was wheelchair-dependent. Examination This woman is thin and tearful. She has marked soft-tissue swelling of her wrists, MCPs and several proximal interphalangeal (PIP) joints in both her hands, and has reduced grip strength bilaterally. Apart from a non-tender nodule on her elbow, the rest of her examination is entirely normal History A 46-year-old man with rheumatoid arthritis attends an emergency review at his rheumatology department. His joint disease is well-controlled on methotrexate 20 mg/week. Over the last few weeks, however, he has become increasingly short of breath with a marked reduction in exercise tolerance. He has a dry, unproductive cough and, although he denies infective symptoms, he recalls feeling feverish at the beginning of his illness. He is a non-smoker and there is no relevant occupational history. Examination This man is mildly short of breath at rest (18 breaths per minute). He is not febrile. Oxygen saturation is 98 percent on room air, desaturating rapidly to 88 percent on exercise. Breath sounds are heard throughout both lung fields, with late inspiratory fine crackles bibasally. History A 54-year-old woman is reviewed in the rheumatology clinic. She was diagnosed with seropositive rheumatoid arthritis 14 years ago with prolonged early-morning stiffness, a small-joint arthropathy and a marked acute-phase response (elevated ESR and C-reactive protein). Despite aggressive therapy with disease-modifying agents, she has never achieved disease remission, continues to struggle with joint pains and stiffness and struggles with activities of daily living. Her livelihood is at risk as she is unable to type effectively. She has previously tried methotrexate but it produced marked nausea and deranged liver function tests at 15 mg/week and had to be stopped. Hydroxychloroquine and azathioprine were both ineffective. Current medications are leflunomide 20 mg once daily, prednisolone 4 mg once-daily, alendronate 70 mg weekly and calcichew D3 forte once-daily. Examination This woman has marked synovitis with tenderness and swelling of many of her metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints, her right wrist and left knee Chronic inflammatory autoimmune disease involving the synovium of joints The usual age of onset is 20 to 40 years; it is more common in women than in men Family history for RA is not unusual due to the presence of susceptibility genes such as HLA-DR. Signs and symptoms 1. Morning stiffness lasting >1 hour 2. Joints involved: hands (PIP, MCP) and wrists, knees, ankles, elbows, hips, and shoulders 3. Hand deformities: a. Ulnar deviation of the MCP joints b. Boutonnière deformities of the PIP joints Muran Mjahed 2 c. Swan-neck contractures of the fingers 4. Low-grade fever, weight loss and fatigue. 5. Subluxation and instability of cervical spine is common at C1-C2 (a life threatening complication of RA, radiographs should be performed prior to any surgery to prevent neurologic damage) 6. Cardiac involvement may include pericarditis, pericardial effusions, conduction abnormalities, and valvular incompetence 7. Pulmonary involvement; usually pleural effusions; interstitial fibrosis 8. Ocular involvement; episcleritis, anterior uveitis, keratoconjunctivitis sicca or scleritis with dry eyes. 9. Soft tissue swelling 10. Drying of mucous membranes; Sjögren xerostomia 11. Subcutaneous rheumatoid nodules over extensor surfaces and visceral organs ( the presence of rheumatoid nodules on examination indicates such seropositivity and a worse prognosis) 12. Nervous system involvement; mononeuritis multiplex 13. Blood: Anemia, Felty syndrome: triad of RA, neutropenia and splenomegaly. 14. Renal system: NSAID-induced nephropathy and Amyloid 15. Patients have an increased tendency to infections. Pathogenesis: Environmental triggers at mucosal surfaces→ PADs are induced→ arginine is converted into citrulline→ Modified proteins are presented to T cells after being processed by APCs; such as dendritic cells→ local and systemic production of antibodies directed against the altered peptides. ACPAs and cytokines→ increase in the circulation years before RA symptoms occur→ sudden formation of immune complexes→ increases vascular permeability in the synovium and activates synovial cells→ inflammatory mediators contribute to initiation and perpetuation of arthritis→ damage and destruction of cartilage Diagnosis: LABs 1. RF is associated with the severity of the disease- nonspecific (RF is found in other inflammatory conditions (e.g. Sjögren’s syndrome, cryoglobulinemia), certain infections and apparently healthy individuals) Helpful in determining prognosis. High titers → more severe disease 2. Anti Citrullinated peptide/protein antibodies (ACPA) 3. ↑ ESR, ↑ CRP 4. Anemia of chronic disease 5. ↑ anti CCP (RA associated with ILD) 6. Renal and liver functions 7. Immunoglobulins and protein electrophoresis RADIOGRAPHS Not required for a diagnosis of RA 1. Loss of juxta-articular bone mass 2. Narrowing of the joint space 3. Bony erosions at the margins of the joint. 4. Soft-tissue swelling 5. Osteopenia 6. Joint space narrowing evidence of joint destruction and ankylosis of bones on Xray in some patients. Synovial fluid analysis can be done to exclude infections or crystalline arthritis. Disease activity is measured by: disease activity score (DAS), which is a composite score of the clinical evidence of synovitis, the current inflammatory response and the patient’s own assessment of their health. Muran Mjahed 3 Treatment: Non pharmacological; the goal is to minimize the swelling, and exercise. Pharmacological; 1. NSAIDs 2. ↓dose corticosteroids 3. DMARDs (Methotrexate, alternatives: leflunomide, sulfasalazine, hydroxychloroquine, if not effective: anti-TNF (infliximab), Non-TNF biologics (abatacept, rituximab, tofacitinib)) - LFTs should be performed when prescribing methotrexate to prevent liver toxicity 4. Surgery (Synovectomy, Joint replacement surgery) Methotrexate side effects: GI upset, oral ulcers, mild alopecia, bone marrow suppression, pulmonary fibrosis. Methotrexate lung is an idiosyncratic reaction occurs more commonly in the first 2–3 years of treatment As TNF-a is associated with increased risk of increased malignancy, lymphoma and reactivation of tuberculosis. Diagnosis for methotrexate lung: - full blood count, elevated eosinophils, elevated CRP, pulmonary function test and CT of lungs Treatment of methotrexate lung: - Oral corticosteroids, with cessation of the drug, - Progressive form is hard to treat and called Hamman–Rich syndrome - Steroids and cyclophosphamide radiological evidence of active inflammation in the form of ground-glass shadowing Differential diagnosis: 1. Rheumatoid arthritis 2. Psoriatic arthritis in association with skin and typically nail disease 3. Post-infective arthritis (septic) 4. Systemic lupus erythematosus in the presence of pronounced extra-articular symptoms and a positive ANA 5. Parvovirus arthropathy, particularly in those with contact with small children Complications: 1. Cervical spine Subluxation 2. Nephrotic syndrome due to NSAIDs and amyloids 3. Osteopenia 4. Secondary osteoporosis due to steroid use Differential diagnosis for a breathless rheumatoid patient a. Rheumatoid lung: interstitial lung disease, generally a non-specific interstitial pneumonitis b. Drug reaction causing allergic alveolitis (methotrexate) c. Coexistent infection: bacterial or pneumocystis pneumonia d. Pleural effusion e. Bronchiolitis obliterans Osteoarthritis History An 80-year-old man presents with progressively worsening pain in his left groin and buttock radiating to the knee with no pins and needles. There is no history of pre-existing trauma or disease in that hip. He complains of pain made worse on walking and has difficulty in putting on shoes and tying shoe laces. He obtains only partial relief of the symptoms with analgesia and rest. Examination The patient walks with an antalgic gait. He has a fixed flexion deformity of 10 degrees. His range of motion in the left hip is restricted and painful in all directions. There is no neurovascular deficit in the leg. Muran Mjahed 4 History A 73-year-old woman presents with left knee pain. This pain has gradually deteriorated over the last three years. She reports early-morning stiffness in the knee. Her walking distance has reduced significantly and is limited due to pain. She obtains minimal relief with analgesia and rest. She has noticed some swelling in the knee and finds walking over uneven surfaces painful, and she finds going down stairs difficult. She has developed pain at night that keeps her awake. She has no history of trauma or infection. She reports no pain in her hips or back. Examination This woman has valgus (knock) knees with the left more affected than the right, associated with an effusion. She has an antalgic gait. She has marked tenderness over the lateral joint line and crepitus in the patellofemoral joint. She has flexion from 5 to 85 degrees with correctable valgus deformity degeneration of cartilage with hypertrophy of bone at the articular margins Any joint can be affected, but weight-bearing joints are most commonly involved (hips, knees, cervical, and lumbar spine) Usually affects people older than 60 years of age Modified risk factors - Obesity and excessive joint loading Occupation Smoking Bone density Physical activity Deposition diseases Hemophilia Non modified risk factor - Age Gender Genetic Joint injury (repeated microtrauma or macrotrauma) Altered joint anatomy Signs and symptoms: 1. Joint pain (often monoarticular) a. This is caused by movement of one joint surface against another due to cartilage loss b. Deep, dull progressive ache that is relieved with rest and worsened with activity 2. Morning stiffness for less than 30 minutes 3. Limited range of motion due to bony enlargement of joints 4. Crepitus may be present 5. Absence of erythema or warmth over the involved joints 6. Swelling indicates inflammation 7. Heberden and Bouchard nodes on fingers 8. If the spine is involved, nerve roots may become compressed and lead to radicular pain. Pathogenesis: Injury or damage to the cartilage→ pro inflammatory factor→ produce proteolytic enzymes→ responsible for the degradation and damage to the matrix→ chondrocytes multiply and form clusters→ bony lumps form called bone spurs. Damage to the matrix→ also causes thickening of the bone underneath the cartilage→ fluid-filled areas in the bone called bone cysts→ +inflammation of the joint’s synovium. Age-related matrix change → abnormal calcification referred to as chondrocalcinosis → from accumulation of calcium pyrophosphate dihydrate CPPD crystal- aka pseudogout Muran Mjahed 5 Diagnosis: LABS 1. Blood tests are normal RADIOGRAPHS 1. Plain radiographs: a. Joint space narrowing due to loss of cartilage b. Osteophytes; bony spurs c. Sclerosis of subchondral bony end plates adjacent to diseased cartilage d. Subchondral cysts on acetabular and femoral sides 2. MRI of the spine if indicated (neurologic findings, before surgery) Clinical signs on examination may include an antalgic gait, positive Trendelenburg sign, unequal leg length, muscle wasting, and a restriction of hip joint movements. Treatment: Nonpharmacologic treatment. 1. Avoid activities that involve excessive use of the joint. 2. Weight loss. 3. Physical therapy can be beneficial, mainly anaerobic. 4. Use canes or crutches to reduce weight on the joint. Pharmacologic treatment. 1. Acetaminophen 2. NSAIDs (GI bleeding is a concern with long-term use) 3. COX-2 inhibitors 4. Intra-articular injections of corticosteroids, 3-4 injections per year 5. Viscosupplementation; hyaluronic acid is injected into the knee joint Surgery for a serious disability. Nutritional products; glucosamine and chondroitin sulfate. Secondary causes of osteoarthritis: a. developmental dysplasia of the hip b. septic arthritis of the hip c. Perthes’ disease d. slipped capital femoral epiphysis e. rheumatoid arthritis f. seronegative arthropathy g. trauma h. crystal arthropathy i. avascular necrosis of the femoral head Osteoporosis History A 58-year-old woman presents to the emergency department with acute back pain. She stumbled off a step and, although she denies falling or receiving direct trauma to the spine, she developed acute and severe mid-thoracic pain immediately afterwards. Her past medical history is unremarkable. She admits to drinking up to 30 units of alcohol a week and smokes 30 cigarettes per day. Examination This woman is thin and in considerable pain. The pain is central with no radiation. There are no peripheral stigmata of systemic disease and cardiovascular, respiratory, abdominal and neurological examinations are normal. She has a marked kyphosis and has point tenderness over several thoracic vertebrae. Decreased bone mass/quality causes increased bone fragility and fracture risk Muran Mjahed 6 It is often difficult to differentiate between primary and secondary osteoporosis, and the two may coexist. It is best to attempt to identify any predisposing conditions and eliminate them if possible. Most patients are postmenopausal women due to decreased estrogen and elderly men Osteoporosis is a “silent” disease. It is asymptomatic until a fracture occurs An exercise program along with calcium and vitamin D supplementation is the mainstay of the therapy for prevention or treatment of osteoporosis. When taking history, ask the patient about hemoptysis and recent weight loss. 1. maternal family history of hip fracture 2. oestrogen deficiency (e.g. premature menopause, prolonged secondary amenorrhoea) 3. corticosteroid therapy with a prednisolone dose >7.5 mg/day for over 6 months 4. low BMI (<19 kg/m2), due to a combination of reduced estrogen levels and reduction in impact loading 5. smoking and excess alcohol 6. anorexia nervosa 7. prolonged immobilization. Bone mass or mineral density relies on a balance of osteoblastic and osteoclastic activity. Postmenopausal bone loss is the most common cause of osteoporosis, but secondary osteoporosis may occur in the context of a number of medical conditions: • postmenopausal oestrogen deficiency (most common cause) • amenorrhoea • iatrogenic (corticosteroid therapy) • endocrine (hyperparathyroidism, hyperthyroidism, Cushing’s disease, hypopituitarism, hypogonadism) • malabsorption (e.g. coeliac disease) • osteomalacia • multiple myeloma • inflammatory arthropathy (probably due to elevated IL-1 and/or TNF-a). Classification: 1. Primary osteoporosis (two types that are impractical clinically) a. Type I (most often in postmenopausal women 51 to 75 years of age), excess loss of trabecular bone; vertebral compression fractures and Colles fractures (a distal radius fracture) are common b. Type II (most often in men and women over 70 years of age), equal loss of both cortical and trabecular bone; fractures of femoral neck, proximal humerus, and pelvis most common 2. Secondary osteoporosis is when an obvious cause is present, such as excess steroid therapy/ Cushing syndrome, immobilization, hyperthyroidism, long-term heparin, hypogonadism in men, and vitamin D deficiency Risk factors: Modifiable risk factors - - Estrogen depletion (modified by giving HRT) a. Postmenopausal state, all women are estrogen deficient after menopause;however, osteoporosis does not develop in all women b. History of athletic amenorrhea, eating disorders, oligomenorrhea c. Early menopause Calcium deficiency/vitamin D deficiency Decreased physical activity or prolonged immobility Endocrine, hypogonadism in men (with low testosterone), hyperthyroidism Smoking and alcohol abuse Medications, corticosteroids, prolonged heparin use Non- modifiable risk factors - Female gender, women have a lower peak bone mass and smaller vertebral end plates (non modified) Decreased peak bone mass Heritable risk factors, family history, European or Asian ancestry, thin/slight build Signs and symptoms: 1. Vertebral body compression fractures ( middle and lower thoracic and upper lumbar spine) are the most common. Very rare in cervical spine a. Result in pain and deformity, including kyphosis Muran Mjahed 7 b. Severe back pain after minor trauma c. Restricted spinal movement, loss of height 2. Colles fracture (distal radius fracture) are usually due to fall on outstretched hand; more common in postmenopausal women 3. Hip fractures; femoral neck, intertrochanteric fractures 4. Increased incidence of long bone fractures (humerus, femur, tibia) Pathogenesis: consequence of genetic, hormonal, dietary, lifestyle and physical factors→ failure to attain optimal (peak) bone mass before age 30, or rate of bone resorption exceeds rate of bone formation after peak bone mass is attained→ decreased bone density→ increased susceptibility to fractures and osteoporosis Diagnosis: 1. DEXA (dual-energy x-ray absorptiometry) scan is the gold standard. a. Indications for bone mineral density measurement: i. All women 65 and older ii. Postmenopausal women <65 with one or more risk factors for fracture. iii. Men with risk factors for fracture. b. Sites are femoral neck and lumbar spine. Must be compared with the bone density of a healthy 30-year-old c. T-scores are used and can range from normal to osteopenia to osteoporosis.according 2. Rule out secondary causes—check calcium, phosphorus, alkaline phosphatase, TSH, vitamin D, free PTH, creatinine, CBC. 3. X-Ray may show wedge fractures of the involved bones Treatment: - Nonpharmacologic therapy 1. Diet—adequate calorie intake, avoid malnutrition 2. Supplemental elemental calcium and vitamin D 3. Exercise; weight-bearing exercise for 30 minutes, at least 3 times a week, to stimulate bone formation 4. Smoking cessation is critical—smoking accelerates bone loss 5. Eliminate or reduce alcohol intake 6. Hormone replacement therapy is used to prevent osteoporosis but has no beneficial effect in already presented diseases. - Pharmacologic therapy Indicated in the following patients: - Postmenopausal women with established osteoporosis or fragility fracture (hip or vertebral) - High-risk postmenopausal women with osteopenia 1. Bisphosphonates inhibit bone resorption and are first-line treatment a. They decrease osteoclastic activity and decrease the risk of fractures b. Oral bisphosphonates (alendronate, risedronate) are preferred in most patients c. Side effects include reflux, esophageal irritation, and ulceration d. If patient cannot tolerate oral bisphosphonates, use IV bisphosphonates (IV zoledronic acid) 2. PTH therapy or human recombinant PTH therapy - PTH is an effective drug that increases bone mineral density and reduces fracture risk. - Not first line therapy due to its cost - Indicated in patients with severe osteoporosis who cannot tolerate bisphosphonates, or who continue to fracture despite being on bisphosphonates for 1 year - Maximum duration of treatment is 24 months, because of concern for osteosarcomas, which have been observed in rats. - After stopping PTH, can restart bisphosphonates 3. Calcitonin (can be administered by nasal spray), only useful as short-term therapy Muran Mjahed 8 4. Estrogen–progestin therapy is no longer a first-line treatment in postmenopausal women because of increased risk of breast cancer, stroke, venous thromboembolism, and perhaps CAD Systemic Lupus Erythematosus History A 27-year-old Afro-Caribbean woman presents to the rheumatology clinic with a long history of painful hands and oral ulcers. She feels she ‘hasn’t been well’ for many years with various problems including chest pains, occasional rashes and hair loss. She recently visited her family in Jamaica and felt much worse, with a resurgence of all of her symptoms, particularly the rash. She is currently experiencing an attack of her typical central chest pain which is sharp and relieved by sitting forward. Examination This young woman is uncomfortable and slightly short of breath. Pulse is 115/min sinus rhythm, blood pressure 95/62 mmHg, and oxygen saturation 98 percent on room air. Her jugular venous pressure (JVP) is elevated at 2 cm and her heart sounds are quiet with no added sounds. Chest examination reveals stony dull bases with reduced vocal resonance and diminished breath sounds. History A 28-year-old woman presents with a 3-day history of a facial rash and swelling of her ankles. She has never had a rash before, has not changed her facial cream or been exposed to anything that might precipitate an allergic reaction. The swelling of the ankles coincided with the development of the rash. Initially it was mild with ‘sock-marks’ around her ankles, but now the swelling is more pronounced and has spread up to her knees. She is otherwise well and on no medication. Examination This young woman has a maculopapular rash across the bridge of her nose and cheeks with sparing of the nasolabial folds. There is pitting oedema of the lower limbs to just above the knee. She is hypertensive at 154/90 mmHg, but the remainder of the examination is normal. History A 38-year-old patient presents to the emergency department with acute pleuritic chest pain and breathlessness. She denies cough, infective symptoms or risk factors for deep venous thrombosis. She is otherwise well. Her medical history includes lupus erythematosus which is currently in remission, and a second-trimester miscarriage. Examination This woman is uncomfortable and short of breath at rest with a respiratory rate of 16 breaths per minute. Her pulse is 96/min sinus rhythm, blood pressure 115/68 mmHg, and oxygen saturation 90 percent on room air. She is not febrile. The remainder of her clinical examination is normal. → likely diagnosis for the current presentation is pulmonary embolism diagnosed by history of sudden-onset pleuritic chest pain and shortness of breath. The diagnosis is Antiphospholipid syndrome History A 22-year-old woman consults her general practitioner complaining of fever, joint pains and chest pain. The joint pains began 3 weeks previously and affected the small joints of her hands and wrists with moderate early-morning stiffness. The muscle pains are relatively non-specific and are not associated with any loss of power. Overall she has derived some benefit from treatment with regular non-steroidal anti-inflammatory drugs (NSAIDs). In the last 48 hours, however, she has also developed a low-grade fever and a sharp right sided chest pain on deep inspiration. She denies breathlessness or cough and has Muran Mjahed 9 no risk factors for pulmonary embolism. Her medical history includes acne, for which she has been taking minocycline for the past year. Examination This young woman is well, with a normal respiratory rate and oxygen saturations. Her temperature is 37.6°C. Although her joints are tender she has no objective evidence of synovitis. Respiratory examination reveals a pleural rub over the painful area in the right lower zone anteriorly but is otherwise normal. The GP was concerned she had developed systemic lupus erythematosus (SLE) and ordered some basic blood tests and an immunology screen. autoimmune disorder leading to inflammation and tissue damage in multiple organ systems. SLE is an idiopathic chronic inflammatory disease with genetic, environmental, and hormonal factors (estrogen). It is less common in children and usually occurs in females of reproductive years. African-American patients are more frequently affected than Caucasian patients a multisystem autoimmune condition characterized by antibodies directed against nuclear components. Types of SLE I. Spontaneous SLE II. Cutaneous lupus erythematosus (skin lesions without systemic disease) III. Drug-induced lupus (procainamide, isoniazid, phenytoin, hydralazine, quinine) IV. ANA-negative lupus—associated findings; 1. Arthritis, 2. Raynaud phenomenon, 3. subacute cutaneous lupus Environmental factors of SLE ➔ Ultraviolet light (UV- A, B) ➔ Hormones, e.g. oestrogens, prolactin ➔ Viral infections, e.g. EBV, CMV, retroviruses, parvovirus B19 ➔ Chemicals and heavy metals, e.g. silica and mercury ➔ Drugs, e.g. hydralazine, procainamide, isoniazid, quinidine, methyldopa, chlorpromazine, minocycline Minocycline induces anti-histone antibodies only in up to 15 per cent of cases, and up to three-quarters of patients generate a positive pANCA; drug should be stopped Signs and symptoms: 1. Constitutional symptoms: Fatigue, malaise, fever, weight loss. 2. Cutaneous: Butterfly or malar rash, photosensitivity, discoid lesions, oral or nasopharyngeal ulcers (mucosal involvement), alopecia, raynaud phenomenon. 3. Musculoskeletal: Arthralgias, arthritis (2 or more joints), myalgia. 4. Cardiac: Pericarditis, endocarditis (Libman–Sacks endocarditis), myocarditis 5. Pulmonary: Pleuritis (serosal involvement), pleural effusion, pneumonitis, pulmonary HTN. 6. Hematologic: Hemolytic anemia, leukopenia, lymphopenia, thrombocytopenia. 7. Renal: Proteinuria >0.5 g/day, glomerulonephritis, azotemia, pyuria, uremia, HTN. 8. Immunologic: Impaired immune response due to many factors, including autoantibodies to lymphocytes, abnormal T-cell function, and immunosuppressive medications; often associated with antiphospholipid syndrome 9. GI: Nausea and vomiting, dyspepsia, dysphagia, peptic ulcer disease 10. CNS: Seizures, psychosis, depression, headaches, TIA, cerebrovascular accident 11. Ocular manifestation; sicca syndrome (sjogren's syndrome), nonspecific conjunctivitis, retinal vasculitis, optic neuritis, cataract, and glaucoma. Pathogenesis: Systemic lupus erythematosus→ global loss of self-tolerance with activation of autoreactive T and B cells→ production of pathogenic autoantibodies and tissue injury→ activation of innate immune cells via Fc receptor (FcR)-mediated uptake of complexes→ tissue destruction and vasculitis→ production of symptoms Muran Mjahed 10 Cytokines in patients with SLE IL-2 IL-6 IL-10 IL-12 Other cytokines ↑Serum IL-15, IL-16, and IL-18 concentrations ↑IFN-γ mRNA expression in PBMCs ↑Serum IFN-γ Diagnosis: 1. First step in diagnosis must be checking the presence of inflammation by ESR, CRP, C3 and C4 in immune complexes 2. CBC, hemoglobin level… 3. The patient must have at least 4 criteria out of 11, or biopsy proven lupus nephritis with a positive ANA or anti-dsDNA. 4. Autoantibodies in lupus: a. ANA: Sensitive but not specific. b. Anti-ds DNA: very specific but not sensitive- during active disease. c. Anti-Smith: very specific but not sensitive. d. Antiphospholipid antibody positivity: as determined by 1. positive lupus anticoagulant (less specific), 2. high-titer anticardiolipin antibody level (gives false positive for syphilis), or 3. positive anti-β-2 glycoprotein. e. Antihistone Abs: are present in most cases of drug-induced lupus. If negative, drug-induced lupus can be excluded. f. Ro (SS-A) and La (SS-B) are found in. Associated with: ➔ Sjögren syndrome ➔ Subacute cutaneous SLE ➔ Neonatal lupus (with congenital heart block) ➔ Complement deficiency (C2 and C4) ➔ ANA-negative lupus 5. LABS and RADIOGRAPHS ➔ CBC shows anemia, leukopenia, lymphopenia or thrombocytopenia ➔ ↑ESR and CRP ➔ Urinalysis; in cases of lupus nephritis ➔ X-Rays 6. In cases of lupus nephritis; biopsy is indicated Antiphospholipid syndrome- APPS: ➔ Hypercoagulable state ➔ More prone to clots ➔ Deep vein thrombosis ➔ Hepatic vein thrombosis ➔ Stroke Lifelong anticoagulation therapy for APPS Diagnosis of PE associated with APPS: chest X-ray, CT–pulmonary angiogram, ECG to exclude complications - History of lupus, miscarriages, and thromcocytopenia are diagnostic - lupus anticoagulant or anticardiolipin antibody Extra clinical features: cardiac valvular disease, livedo reticularis, mild chronic thrombocytopenia, neurological disease such as chorea or transverse myelitis Antiphospholipid syndrome may occur independently of SLE. Estrogen containing contraceptive and HRT are contraindicated in women with APPS Muran Mjahed 11 Treatment: 1. 2. 3. 4. Avoid sun exposure because it can exacerbate cutaneous rashes NSAIDs; for less severe symptoms Either local or systemic corticosteroids; for acute exacerbations Systemic steroids for severe manifestations (Methotrexate and azathioprine) - Methotrexate side effects: GI upset, oral ulcers, mild alopecia, bone marrow suppression, pulmonary fibrosis. 5. Antimalarial agents such as hydroxychloroquine; best long-term treatment, associated with production of retinal toxicity. 6. Cytotoxic agents such as cyclophosphamide or mycophenolate mofetil; for systemic diseases and active glomerulonephritis 7. ACE Inhibitors to treat and control HTN if present with lupus nephritis 8. Diuretics to treat edema associated with lupus nephritis 9. Monitor the following and treat appropriately: a. Renal disease, which produces the most significant morbidity b. HTN c. Retinal toxicity Cyclophosphamide reduces fertility and this side-effect should be specifically discussed with patients of child-bearing age; mycophenolate mofetil is preferred. Periods of flare ups * prevention of flare ups is by avoiding sunlight ⤷ ⤴ *corticosteroids and immunosuppressants are used to limit severity Remission Conditions in Which ANAs Are Elevated 1. SLE 2. RA 3. Scleroderma 4. Sjögren syndrome 5. Mixed connective tissue disease 6. Polymyositis and dermatomyositis 7. Drug-induced lupus Associated conditions: 1. antiphospholipid syndrome 2. lupus nephritis - anti-DNA immune complexes deposition in glomeruli - nephritic or nephrotic syndrome - diffuse proliferative is the most common and most severe type 3. drug-induced lupus - typically positive for antinuclear and antihistone antibodies - typically without renal or neurologic involvement 4. Libman-Sacks endocarditis (LSE) - noninfectious endocarditis characterized by thrombi on the mitral or aortic valves (LSE in SLE) 5. Raynaud phenomenon 6. neonatal lupus erythematosus - associated with patients with anti-Ro or anti-La antibodies - neonates present with rashes and congenital heart block Complication: - Renal disease remains the most feared complication of lupus and all patients (including this one!) should have their blood pressure checked, urine examined for casts and serum creatinine checked regularly. Muran Mjahed 12 Scleroderma History A 36-year-old woman is referred by her general practitioner with cold, painful hands and shortness of breath on exertion. Over the last 6 weeks she has noticed her fingers and hands turning blue, then white and finally red and painful in episodic attacks, particularly in the cold. In addition the skin on her fingers feels taut, reducing her ability to make a fist. The shortness of breath and reduced exercise tolerance has developed insidiously over the same period but she has not had a cough or infective symptoms. She has no history of, or risk factors for, respiratory disease. Examination This woman is dyspnoeic on walking from the waiting room to the clinic; her oxygen saturations are initially 92 percent but increase to 99 percent with rest. She has no peripheral stigmata of respiratory disease. The skin over her fingers and hands is tight and bound-down but is intact with no evidence of ulcers or vasculitis. The thickened skin extends beyond her elbows and she also has restricted mouth opening. Her jugular venous pulse is elevated, with a parasternal heave and a loud second heart sound, but there is no peripheral oedema. In the respiratory system she has bibasal mid-to-late fine inspiratory crackles A chronic connective tissue disorder that can lead to widespread fibrosis. Skin is tight and shiny, with no wrinkles or folds. May lead to cancer in some cases. Scleroderma is more common in women. Average age of onset is 35 to 50 years. There are two types of scleroderma: Diffuse (20%) and limited (80%) Only diffuse scleroderma form has renal, lung, and heart involvement Signs and symptoms: 1. Constitutional symptoms: discomfort, fatigue, weight loss 2. Raynaud phenomenon a. Present in almost all patients; usually appears before other findings b. Caused by vasospasm and thickening of vessel walls in the digits c. Can lead to digital ischemia, with ulceration and infarction/gangrene d. Cold temperature and stress bring about color changes of fingers; blanching first, then cyanotic, and then red from reactive hyperemia 3. Cutaneous fibrosis- CREST syndrome a. Tightening of skin of the face and extremities (sclerodactyly refers to a clawlike appearance of the hand, and reduces the function of the hand) b. Can lead to contractures, disability, and disfigurement c. “puffy fingers” from interstitial non-pitting oedema d. Mucocutaneous telangiectasia: dilation of superficial capillaries mostly on face or oral mucosa e. Skin ulcerations due to breakdown of atrophic skin 4. GI involvement a. Occurs in most patients (both diffuse and limited) b. Findings include dysphagia/reflux from esophageal immobility (due to connective tissue disorder), delayed gastric emptying, constipation/diarrhea, abdominal distention, and pseudo-obstruction. Prolonged acid reflux may eventually lead to esophageal strictures c. GERD due to decreased motility in the lower ⅔ of the esophagus 5. Pulmonary involvement a. Most common cause of death from scleroderma b. Interstitial fibrosis and/or pulmonary HTN due to connective tissue disorder, affecting the vessels Muran Mjahed 13 - ILD has a restrictive pattern: FEV1 /FVC ratio is normal or increased. All lung volumes are low. Both FEV1 and FVC are reduced, but the ratio is often preserved c. On radiographs: honeycomb appearance or ground glass opacities in the lower lobes of the lung 6. Cardiac involvement a. pericardial effusions b. myocardial involvement that can lead to CHF ( in the case above; right sided heart failure), arrhythmias c. HTN and CAD 7. Renal involvement a. renal crisis; rapid malignant hypertension, occurs in patients with diffuse disease b. Glomerulonephritis with sclerotic kidneys c. Renal failure limited long standing SS associated with raynaud’s phenomenon → acro-osteolysis Patterns of limited disease occur as isolated fibrotic plaques (morphoea) or bands of affected skin (linear scleroderma, also known as ‘en coup de sabre’ when found on the face). Pathogenesis: Endothelial injury → intimal proliferation → vasoconstriction → hypoxia; ↑ROS, ↑PAH → angiogenesis → platelet aggregation → fibrosis and innate humoral immunity (macrophages, mast cells, cytokines…) 3 fundamental aspects of Systemic Sclerosis; 1. ↑ inflammation 2. ↑ vasoconstriction, and angiogenesis 3. ↑ fibroproliferation of vessel wall After endothelial cell dysfunction: ↑ET-1, ↑platelets, ↑adhesion molecules, ↑ IL33, ↓NO Diagnostic tests: 1. Almost all patients have elevated ANAs, (high sensitivity, low specificity). 2. Anticentromere antibodies are very specific for the limited form. 3. Anti-RNA polymerases 4. Anti Topoisomerase I Ab ( antiscleroderma-70 or anti-SCL 70 indicated severe disease) is very specific for the diffuse form. 5. Barium swallow (esophageal dysmotility) and pulmonary function tests are used to detect complications. 6. Nailfold capillaroscopy 7. Labs: a. CBC; anemia from malabsorption, iron deficiencyI or from bleeding in the GI. b. Serum creatinine; level indicates renal dysfunction c. CK; elevated in patients with myopathy or myositis d. Urinalysis with urine sediment; proteinuria and/or cellular casts e. Modified Rodnan Skin Score (mRSS); used to measure thickness of skin, pliability, and fixation to underlying structures. f. Durometer, Plicometer, High frequency Ultrasound Treatment: 1. No effective cure, and treatment is symptomatic depending on the organs involved 2. NSAIDs for musculoskeletal pains 3. H2 blockers or proton pump inhibitors for esophageal reflux, plus pro motility (metoclopramide) 4. Raynaud phenomenon; avoid cold and smoking, keep hands warm; if severe, use calcium-channel blockers; nifedipine 5. ACE inhibitors are used to prevent and treat renal hypertensive crisis 6. Diuretics for heart failure 7. For severe diffuse skin and organ involvement, systemic immunosuppression is indicated 8. Antibiotics for skin infection Muran Mjahed 14 Sjogren Syndrome History A 79-year-old man presents to his general practitioner with bilaterally swollen cheeks. He also complains of feeling ‘tired all the time’, weight loss and occasional night sweats. Although he admits to an irritating dry cough he denies frank breathlessness or change in exercise tolerance. His medical history includes hypertension, for which he takes ramipril. Examination This elderly man is slightly pale, with bilateral parotid enlargement and cervical History A 44-year-old woman presents to the rheumatology department with a long history of sore, stiff hands and dry, painful eyes. She delayed seeking medical attention as she was concerned she had developed rheumatoid arthritis. The pain in her hands affects principally her wrists, metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints with protracted early-morning stiffness. Her eyes feel ‘gritty’ much of the time, getting worse as the day progresses such that they are very painful by the end of the day. During her worst symptoms she complains of photophobia. She has recently noticed a reduction in saliva and finds it difficult to eat dry foods without ‘washing them down with water’. Her medical history is unremarkable and she is on no regular medication. Examination This woman is well with mild synovitis of her wrist, MCPs and PIPs. Her sclerae are minimally injected but there is no discharge and her acuity is normal. The remainder of her examination is normal. Sjögren syndrome is an autoimmune disease most common in women. Lymphocytes infiltrate and destroy the lacrimal and salivary glands A multiorgan disease (can also involve the skin, lungs, thyroid, vessels, and liver) Parotid enlargement differentials: Sjögren’s syndrome, sarcoidosis, lymphoma Sjögren’s syndrome is often associated with a polyclonal hypergammaglobulinemia and may develop lymphoma Types: Primary Sjögren syndrome: Dry eyes and dry mouth, along Secondary Sjögren syndrome: Dry eyes and dry mouth with lymphocytic infiltration of the minor salivary glands (on along with a connective tissue disease (RA, systemic sclerosis, histology) SLE, polymyositis) patients do not have another rheumatologic disease Signs and symptoms: 1. Dry eyes, burning, redness, blurred vision, keratoconjunctivitis sicca 2. Persistent dry mouth (Xerostomia) and tooth decay; with difficulty swallowing dry food, inability to speak continuously 3. Arthralgias, arthritis, fatigue, peripheral joint pain 4. Interstitial nephritis and vasculitis 5. Swelling of parotid and submandibular glands 6. Urticaria of limbs 7. Pulmonary involvement in some patients; chronic cough, dyspnoea, upper and lower respiratory infections. Radiographs show ground-glass pattern, thickened bronchial walls 8. Renal involvement characterized by interstitial renal disease, and glomerulonephritis 9. ⅓ of patients with primary SjS have thyroid disease. Muran Mjahed 15 10. Patients have increased risk of non-Hodgkin lymphoma. Malignancy is the most common cause of death Ask patients with sjogren if swallowing biscuits and bread is hard without water Pathogenesis: lymphocytic infiltrates → destruction of the salivary and lacrimal glands → systemic production of autoantibodies to the ribonucleoprotein particles Ro (SS-A) and La (SS-B) →The infiltrating cells; T- and B-cells, dendritic cells interfere with glandular function → resulting in dry eyes and dry mouth due to decreased secretions from the salivary glands. Diagnosis: 1. ANAs are present in most patients. Rheumatoid factor (RF) is also present in many patients with secondary disease. 2. Ro (SS-A) is present in 60% of patients, and La (SS-B) (which is more specific) is present in 40% of patients. - Patients with antibodies to Ro (SS-A) are at increased risk of having a child with neonatal SLE (with congenital heart block). 3. Schirmer test: Filter paper inserted in the eye to measure lacrimal gland output (degree of wetting in a specified time period) has high sensitivity and specificity. 4. Salivary gland biopsy (lip or parotid) is usually not performed, lymphocytic infiltrates. 5. Elevated ESR, CRP normal 6. Ultrasound / CT/ MRI of salivary glands in some cases 7. Biopsy of the salivary glands in severe cases anti-Ro and anti-La antibodies cross the placenta and cross-react with the developing conducting system in the fetal heart. As a result, patients of child-bearing age with Ro and La antibodies should be counseled regarding potential fetal heart-block Treatment: 1. Pilocarpine or Cevimeline (to enhance oral and ocular secretions) 2. Artificial tears for dry eyes 3. Good oral hygiene 4. NSAIDs, steroids for arthralgias, arthritis 5. Patients with secondary Sjögren syndrome must treat any connective tissue disorder Gout History A 56-year-old man presents to the emergency department with a 2-day history of an acutely painful, swollen and hot knee. He is otherwise well but felt feverish on the morning of his admission. There is no history of trauma and the system's inquiry is unremarkable. Past medical history includes left ventricular failure, hypertension and renal impairment. Current medications are aspirin, furosemide and ramipril. He lives alone, smoked 15 cigarettes per day and drinks 30 units of alcohol per week. Examination This man is uncomfortable but well. His temperature is 37.1°C, pulse 110/min and blood pressure 145/83 mmHg. General examination is unremarkable, but his right knee is warm with a tense effusion. Both passive and active movement are restricted by pain, and weight-bearing is uncomfortable. Other joints are normal Gout is an inflammatory monoarticular arthritis caused by the crystallization of monosodium urate in joints Hyperuricemia is a hallmark of the disease, but it is not specific to gout. Ninety percent of patients are men over 30 years of age. Women are not affected until after menopause due to decreased estrogen-mediated urinary urate excretion Risk factors: 1. Male >30 2. Females of postmenopausal years 3. Obesity 4. Genetic polymorphism 5. Kidney diseases 6. NSAID use Muran Mjahed 16 7. Purine rich foods 8. Alcohol intake 9. Thiazide and loop diuretics Risk factors for gout are under-excretion or overproduction of urate, which is a product of DNA breakdown and present in large amounts in certain foods and alcohol Signs and symptoms: 1. Intermittent acute monoarthritis especially the first metatarsophalangeal joint (MTP) 2. Desquamation of skin may occur 3. Erythema, pain, swelling, tenderness, and warmth 4. Osteopenia is characteristically absent. Stages of gout: 1. Asymptomatic hyperuricemia. a. Increased serum uric acid level in the absence of clinical findings of gout, may be present without symptoms for 10 to 20 years or longer. b. Should not be treated 2. Acute gouty arthritis. a. Initial attack usually involves sudden onset of exquisite pain. b. Pain often wakes the patient from sleep. c. Mostly affecting the big toe; the first metatarsophalangeal joint (podagra). d. Fever may be present. e. the patient may have desquamation of overlying skin when the attack is gone. 3. Intercritical gout. a. An asymptomatic period after the initial attack. b. Between two acute attacks c. Sixty percent of patients have a recurrence within 1 year. d. Few patients never have another attack of gout. e. Attacks tend to become polyarticular with increased severity over time. 4. Chronic tophaceous gout. a. Occurs in people who have had chronic poorly controlled gout b. Tophi: - Aggregations of urate crystals surrounded by giant cells in an inflammatory reaction. - Tophi causes deformity and destruction of hard and soft tissues and may be extra-articular. c. Usually on the extensor surface of forearms, elbows, knees, Achilles tendons, and pinna of the external ear. Pathogenesis: Increased production of uric acid (by: 1. Hypoxanthine-guanine phosphoribosyltransferase deficiency 2. Phosphoribosyl pyrophosphate synthetase overactivity 3. Increased cell turnover) OR Decreased excretion of uric acid (by: 1. Renal disease 2. NSAIDs 3. loop and thiazide diuretics 4. Acidosis) Accumulation of uric acid crystals in the synovial fluid→ PMNs initiate acute inflammation of gout→ IgGs coat monosodium urate crystals→ leading to the release of inflammatory mediators and proteolytic enzymes from the PMNs→ results in inflammation Crystal arthropathy is caused by either uric acid (gout) or pyrophosphate (pseudogout) crystals precipitating out into the synovial fluid, generating a brisk inflammatory response. Diagnosis: 1. Arthrocentesis to aspirate joint and analyze synovial fluid under polarized light microscopy; needle-shaped and negatively birefringent monosodium urate crystals. positively birefringent brick-shaped crystals in pyrophosphate disease 2. Serum uric acid is not helpful in diagnosis because it can be normal even during an acute gouty attack. Muran Mjahed 17 3. Blood test; during acute flares, uric acid is decreased in Blood 4. Radiographs: a. X Ray reveals punched-out erosions and sclerotic margins b. Ultrasound; double contour or margins of the affected joint c. CT Scan usually not performed Complications of Gout: 1. Nephrolithiasis 2. Degenerative arthritis Urate crystals may also deposit in the skin, causing painful discrete lumps known as tophi, or in the renal tract, causing urate nephropathy or lithiasis Treatment: 1. In all stages, avoid secondary causes of hyperuricemia. - Medications that increase uric acid levels (thiazide and loop diuretics) - Obesity - Reduce alcohol intake. a. Reduce dietary purine intake. Limit intake of seafood/red meat. 2. Acute gout. a. Bed rest is important. b. NSAIDs; indomethacin c. Avoid aspirin (aggravates the symptoms) and acetaminophen d. Colchicine i. An alternative for patients who cannot take or did not respond to NSAIDs. e. Corticosteroids i. Oral prednisone (7- to 10-day course) if patient does not respond to or cannot tolerate NSAIDs and colchicine. ii. Intra-articular corticosteroid injections (if only one joint is involved) 3. Prophylactic therapy. - at least two acute gouty attacks before initiating prophylactic therapy. - The presence of tophi is also an indication for prophylactic therapy. a. When giving prophylaxis, add either colchicine or an NSAID for 3 to 6 months to prevent an acute attack. b. Use of uricosuric drugs or allopurinol depends on how much uric acid is excreted in the urine in a 24-hour period. - Uricosuric drugs (probenecid, sulfinpyrazone) increase the renal excretion of uric acid, indicated when the 24-h urine uric acid is <800 mg/day, (underexcretion of urate from kidneys). - They are contraindicated if the patient has a history of renal stones. - Xanthine oxidase inhibitors (allopurinol, febuxostat) decrease uric acid synthesis and are indicated when the 24-h urine uric acid is >800 mg/day, (indicates overproduction.) - They are not contraindicated in kidney dysfunction. - Uricases (pegloticase, rasburicase) catalyze the conversion of urate into allantoin, a more soluble purine degradation product. They may cause infusion reactions. Differential diagnosis may include: 1. Septic arthritis 2. crystal arthritis - Gout - Pseudogout 3. Kidney impairment Muran Mjahed 18 Pseudogout (Calcium Pyrophosphate Deposition Disease) Calcium pyrophosphate crystals deposit in joints, leading to inflammation. a form of arthritis characterized by sudden, painful swelling in one or more of the joints Risk factors: 1. Deposition increases with age and with OA of the joints. 2. conditions that may increase crystal deposition include hemochromatosis, hyperparathyroidism, hypothyroidism, and Bartter syndrome (genetic defect in kidney function). Signs and symptoms: 1. Symptoms similar to gout but typically occur in larger joints - The most common joints affected are knees and wrists. 2. It is classically monoarticular, but can be polyarticular. Pathogenesis: abnormal formation and deposition of calcium pyrophosphate dihydrate (CPPD) crystals in the cartilage and the synovial fluid→ leading to a sudden attack of arthritis similar to gout. Diagnosis: 1. Joint aspirate is required for definitive diagnosis; positively birefringent, rod-shaped and rhomboidal crystals in synovial fluid (calcium pyrophosphate crystals) 2. Radiographs—chondrocalcinosis (cartilage calcification) Treatment: 1. Treat the underlying disorder (if identified) 2. Symptomatic management is similar to that for gout - First-line therapy includes NSAIDs - Colchicine for prophylaxis - Intra-articular steroid injections, with triamcinolone 3. Total joint replacement Polymyalgia rheumatica History A 72-year-old woman presents to her general practitioner with a few weeks’ history of pain and stiffness affecting her shoulders and hips. The pain began insidiously but is now severe; her shoulders and neck are worse and effected symmetrically Although she denies objective weakness, the pain is limiting her activities of daily living and frequently wakes her from sleep. The stiffness lasts several hours in the morning. Otherwise, she is entirely well and has no medical history of note. Examination This elderly woman is uncomfortable and finds it difficult to move from the chair to the examining couch. She is generally tender around the neck, shoulders and pelvic girdle. There is no evidence of synovitis but full range of movement is limited by periarticular rather than true joint pain Usually occurs in elderly patients, and it is more common in women. aching and stiffness of the shoulder, neck, and hip-girdle area that can occur with giant cell arteritis + flu-like symptoms - Self-limited disease (duration of 1 to 2 years). A significant minority of RA patients may present with a polymyalgia onset Signs and symptoms: 1. Bilateral hip and shoulder muscle pain Muran Mjahed 19 a. Often begins abruptly; but may be gradual b. Stiffness in shoulder and hip after a period of inactivity c. Pain occurs on movement; muscle strength is normal d. Profound morning stiffness is common 2. Constitutional symptoms are usually present: malaise, fever, depression, recent weight loss, and fatigue 3. Joint swelling a. some patients have synovitis in knees, wrists, or hand joints (similar to RA) b. Synovitis and tenosynovitis around the shoulder may lead to rotator cuff tendonitis or adhesive capsulitis c. Signs and symptoms of temporal arteritis might be present Pathogenesis: age-related immune alterations in genetically predisposed subjects (associated with HLA-DR4 allele) contribute to development of the disease. But the true cause is yet to be determined. Diagnosis: 1. Essentially a clinical diagnosis 2. ESR is usually elevated and aids in diagnosis a. Almost always >50, frequently >100 b. Correlates with disease activity 3. Ultrasound, MRI of shoulders and hips in some cases 4. Funduscopic examination to monitor patients with associated giant cell arteritis Treatment: 1. Low dose corticosteroids a. Response usually occurs within 1 to 7 days. b. Corticosteroids are not curative, but are effective in suppressing inflammation until the disease resolves itself. c. After 4 to 6 weeks, begin to diminish slowly. d. Most patients can stop corticosteroids within 2 years. A few patients have symptoms for up to 10 years. Differential diagnosis: 1. Polymyalgia rheumatica 2. Fibromyalgia (normal ESR, younger age at onset) 3. Infection 4. Rheumatoid arthritis 5. Hypothyroidism (elevated TSH, normal ESR and CRP) 6. Depression 7. Polymyositis Complications of PMR: - Giant cell arteritis - Blindness in untreated GCA Fibromyalgia History A 46-year-old woman is referred to the rheumatology department with a long history of muscle pain and fatigue. Her symptoms developed insidiously and she now complains of pain ‘all over her body’, poor sleep and exhaustion much of the time. She denies constitutional upset or specific symptoms but is finding it hard to work or care for her family. Her medical history is unremarkable and she takes no regular medications. Examination Muran Mjahed 20 This woman is well and has no peripheral stigmata of inflammatory disease. Although her joints are mildly tender, there is no evidence of synovial thickening or effusion. Palpation of a number of soft-tissue points is tender but her muscle strength is preserved. All systems examinations are normal. Occurs predominantly in adult women; due to imbalanced serotonin and dopamine hormones. Chronic nonprogressive course with waxing and waning in severity. noninflammatory syndrome of unknown etiology Secondary fibromyalgia is not uncommon in patients with inflammatory disease such as rheumatoid arthritis or SLE. Presence of multiple trigger points that are tender to palpation aid with diagnosis; patients may cry upon palpating these areas. a. Symmetrical. b. Eighteen characteristic locations have been identified, including occiput, neck, shoulder, ribs, elbows, buttocks, and knees.. Fibromyalgia may coexist with depression. Etiology is unknown—somatization is not a proven cause Signs and symptoms: 1. Stiffness, arthralgia, body, musculoskeletal aches, fatigue. a. Pain is constant and aching, and is aggravated by weather changes, stress, sleep deprivation, and cold temperature. It is worse in the morning. 2. Sleep patterns are disrupted, and sleep is unrefreshing. 3. Anxiety and depression (psychiatric symptoms). 4. Headaches. 5. Paraesthesias. 6. Cognitive dysfunction. 7. Raynaud’s phenomenon Tender points: occiput; trapezius, supraspinatus; gluteal; greater trochanter; low cervical; second rib; lateral epicondyle; knee Pathogenesis: Physical or emotional stress→ triggers central sensitization phenomenon→ dysfunction of neural-circuits→ transmission and processing of afferent nociceptive stimuli→ manifestation of pain at the level of the locomotor system. Fibromyalgia is not related to autoimmune response. Diagnosis: 1. history of widespread pain (in all quadrants of the body) for at least three months and when pain is caused by digital pressure in at least 11 out of 18 allogeneic points, called tender points. 2. no confirmatory tests for fibromyalgia, therefore, it is important to rule out: (differentials) a. Myofascial syndromes b. Rheumatoid disease c. Polymyalgia rheumatica d. Ankylosing spondylitis e. Spondyloarthropathy f. Chronic fatigue syndrome g. Lyme disease h. Hypothyroidism (thyroid disorders may lead to myalgias) i. Polymyositis j. Depression and somatization disorder k. Hypertrophic osteoarthropathy l. Hepatitis m. Endocrine disorders Muran Mjahed 21 Treatment: 3. Complete metabolic panels (ESR, CRP,CBC, TSH) should be performed routinely. 1. 2. 3. 4. 5. Activity and engaging in low intensity exercise. First-line treatment for fibromyalgia is amitriptyline. Local anesthetic at trigger points. Milnacipran and pregabalin (gabapentin)- SNRI or SSRI. Cognitive behavioral therapy (CBT). Ankylosing spondylitis History A 26-year-old man presents with a 6-month history of back and buttock pain and stiffness. His symptoms localize to his lumbar spine and right buttock and are particularly marked first thing in the morning, but start to settle within a couple of hours. The pain never spreads down the back of the leg and he has not noticed any change in sensation or loss of power. He is otherwise fit and well, with the exception of two episodes of an acutely painful red eye for which he received ‘eye drops’ from the ophthalmology service. He is self-employed with two children and smokes heavily. Examination This young man has dramatically reduced lumbar spinal movements in all planes but cervical movements are full and pain-free. His right hip is irritable with reduced range of movement, particularly external rotation. There is a small effusion of the left knee with evidence of Achilles tendinopathy and plantar fasciitis on the left foot. The remainder of his examination is normal →inflammatory spinal disease (spondyloarthropathy) and sacroiliitis evidence of peripheral arthropathy, tendinopathy, possible extra-articular symptoms in the form of previous ocular disease, and an elevated inflammatory response Strong association with HLA-B27 Three times more common in male than in female patients Bilateral sacroiliitis (inflammation of the sacroiliac joint) is a prerequisite for making the diagnosis Patients with back pain longer than 3 months, with age younger than 45 years at onset. It is characterized by “fusion” of the spine in an ascending manner (from lumbar to cervical spine) Early sacroiliitis in a patient with ankylosing spondylitis, indicated by prominent edema in the juxta articular bone marrow (asterisks), synovium and joint capsule (thin arrow), and interosseous ligaments (thick arrow) on a short tau inversion recovery (STIR) MRI Course: 1. There is a slow progression, but acute exacerbations are common. 2. Life expectancy is unchanged. 3. The first 10 years of the disease can give an indication of long-term severity. Signs and symptoms: 1. Low back pain and stiffness secondary to sacroiliitis. 2. limited motion in the lumbar spine. 3. Neck pain and limited motion in the cervical spine. 4. Enthesitis: inflammation at tendinous insertions into bone (usually occurs in the heels; achilles tendon, and supraspinatus tendon) 5. Low back pain and stiffness are worse in the morning and better as the day progresses. They improve with activity and a hot shower. Muran Mjahed 22 6. 7. 8. 9. 10. With extensive spinal involvement, the spine becomes brittle and is prone to fractures with minimal trauma. Chest pain and diminished chest expansion; due to thoracic spine involvement Shoulder and hip pain are most commonly in the peripheral joints Constitutional symptoms—fatigue, low-grade fever, weight loss Extra-articular manifestations a. Eye involvement ;acute anterior uveitis b. Cardiac (AV heart block and aortic insufficiency). c. Renal involvement. d. Pulmonary involvement. e. Nervous systems. 11. Loss of lumbar lordosis 12. Dactylitis: sausage like fingers (usually associated with Crohn's disease) Pathogenesis: HLA-B27 belongs to the MHC-I surface protein encoded by the MHC B gene on chromosome 6. HLA-B27→ peptide antigens to T immunocytes of the human body defense process→ inflammatory processes that are linked to AS→ inflammation, bone erosion and syndesmophyte (spur) formation. The inflammation leads to symptoms of pain and stiffness, while fibrosis and ossification at these sites generates signs of increasing spinal restriction. Diagnosis: 1. Schober’s test 2. Imaging studies of lumbar spine and pelvis (plain film, MRI, or CT) reveal sacroiliitis and bamboo spine. MRI highlights areas of high water content (joint inflammation and bone edema) and shows features of AS: the corners of the lumbar vertebrae are ‘shiny’ with edema and there is symmetrical inflammatory change at the sacroiliac joints with irregularity of the joint space. 3. Elevated ESR in some patients due to inflammation 4. HLA-B27 is not necessary for diagnosis, and may be positive in otherwise healthy persons. Complications of Ankylosing Spondylitis: 1. Restrictive lung disease 2. Cauda equina syndrome 3. Spine fracture with spinal cord injury 4. Osteoporosis 5. Spondylodiscitis Treatment: 1. Physical therapy; maintaining good posture, extension exercises. 2. NSAIDs; indomethacin. 3. Anti-TNF medications; etanercept, infliximab. 4. Surgery. 5. Patients who sustain minor trauma or complain of neck or back pain should be immobilized to prevent spinal cord injury until imaging studies prove the opposite. differential diagnosis of spondyloarthropathy: 1. Ankylosing spondylitis (prototypical spondyloarthritis), most common in males under 35 years of age 2. Psoriatic arthropathy 3. Enteropathic, associated with inflammatory bowel disease, but unrelated to disease activity 4. Reactive arthritis following genitourinary or gastrointestinal infection (in B27) Muran Mjahed 23 Reactive Arthritis/Reiter Syndrome History A 41-year-old man presents to the rheumatology department with bloody diarrhea, ulcers over his shins and a painful, stiff back. Although his back stiffness began more than 5 years ago, he chose to ignore it until recently when, in combination with his other symptoms, he began to struggle to cope at home. His back pain and stiffness are worse in the morning and tend to ease by lunchtime. It has, on occasion, been associated with a painful and swollen right knee. The diarrhea and leg ulcers began six weeks previously and he is awaiting a gastroenterology outpatient appointment. Examination This man has limited spinal movement in all planes with a Schober’s test reduced at 19 cm. His right hip is irritable and there is a moderate effusion of the right knee. There are two ulcers over his left shin with shiny yellow bases and red/blue overhanging edges. The remainder of the examination is normal. History A 52-year-old man presents with a longstanding history of weight loss, diarrhea and painful, stiff hips and knees. The arthritis developed first, affecting principally his hips and knees but tending to move around. A rheumatoid factor has been checked in the past and was negative, so he felt reassured. The diarrhea then became an issue over the last 6 months; he has intermittent abdominal pain and complains that the stools are foul-smelling and hard to flush away. There has never been any blood, mucus or slime. With continuing diarrhea he has lost weight (5 kg in 6 months), despite a good appetite. Examination There is evidence of recent weight loss and a mild effusion of the right knee. Otherwise, system examination is normal. History A 28-year-old man presents to the emergency department with an acutely painful and swollen right knee. The symptoms began 24 hours previously and have not responded to non-steroidal anti-inflammatory drugs (NSAIDs). He denies fever or constitutional upset. His medical history is unremarkable, except for a self-limiting but severe diarrhoeal illness 3 weeks previously. Examination This young man appears well but uncomfortable. His right eye has an injected sclera and there is a moderate effusion in his right knee which is warm and displays reduced range of movement. He has a pustular rash on both heels. The remainder of the examination is normal. Inflammatory arthritis that develops after certain enteric or urogenital infections. The term undifferentiated spondyloarthropathy is used when a patient has features of reactive arthritis but there is no evidence of previous infection and the classic findings of Reiter syndrome are absent. Reactive arthritis is asymmetric inflammatory oligoarthritis of lower extremities. Reiter syndrome is an example of reactive arthritis, but most patients do not express the typical symptoms, which are: arthritis, uveitis, and urethritis, so the term reactive arthritis is now used. Presence of rash might suggest psoriatic arthritis; but the combination of rash, ocular disease, self-limiting diarrhea and an acute monoarthropathy make reactive arthritis the most likely diagnosis Causative agents: 1. Enteric causes ; by shigella, salmonella, campylobacter, yersinia 2. Urogenital causes; by chlamydia trachomatis, HIV Muran Mjahed 24 Signs and symptoms: 1. Infection of GI or genitourinary tracts 1 to 4 weeks prior to the onset of symptoms. 2. Asymmetric arthritis that progresses sequentially from one joint to another 3. Joints are painful, with effusions and lack of mobility that persist or recur over a long-term period. 4. Fatigue, malaise, weight loss, and fever are common. 5. Eye inflammation. 6. Enthesitis may be present. 7. Dactylitis and swollen fingers and toes 8. Onychodystrophy; Reiter’s nails 9. Characteristic skin rash; Keratoderma blennorrhagicum or syphilitic skin lesion Seronegative arthritis may develop in up to 15 percent of patients with any form of inflammatory bowel disease, including ulcerative colitis (UC), Crohn’s disease or microscopic and collagenous colitis. The most common clinical presentations are a peripheral arthritis (commonly divided into type I and type II) and spondyloarthritis - Type I enteropathic arthritis is an asymmetrical oligoarthritis which follows disease activity in the bowel. ● Whipple’s disease is a systemic illness caused by the Gram-positive bacillus Tropheryma whippelii ● Other IBS: crohns, ulcerative colitis, celiac disease - Type II enteropathic arthritis is symmetrical and polyarticular; runs an independent course to bowel disease Pathogenesis: Recent infection→ triggers immune-mediated response→ T lymphocytes are activated by bacterial fragments→ activated cytotoxic-T cells then attack the synovium and other self-antigens anti-bacterial cytokine response is also impaired in reactive arthritis, resulting in the decreased elimination of the bacteria. It occurs mostly in HLA-B27–positive individuals The joint itself is not infected. Diagnosis: 1. CBC, leukocytosis, Thrombocytosis, Increased serum Ig (previous infection), normocytic anemia 2. Cultures; blood, urine, stool, throat to detect any active infection 3. HLA-B 27+ 4. Radiology; Sacroiliitis (if associated with AS), Enthesitis 5. MRI of the sacroiliac joints and lumbosacral spine 6. Send synovial fluid for analysis to rule out infection or crystals. 7. Endoscopy in case of enteropathic arthritis to exclude inflammatory bowel diseases. As for all patients with a malabsorptive history, the investigations should also assess nutritional status, and calcium, B12/iron/folate and vitamin D levels should be checked. Treatment: 1. NSAIDs are first-line therapy. 2. DMARDs; sulfasalazine and immunosuppressive agents; azathioprine. 3. Antibiotic use is controversial; Azithromycin, Ciprofloxacin, Doxycycline 4. Intra-Articular Glucocorticoids Septic arthritis History An 84-year-old woman with diabetes has been admitted to the emergency department with an acutely swollen and painful left knee. She has been unwell with a raised temperature and productive cough for the last week and for the last 24 hours has been unable to bear weight because of her knee pain. Examination Muran Mjahed 25 This elderly woman is unwell, sweaty and febrile. Her pulse is 108 bpm and blood pressure 98/60 mmHg. Oxygen saturation in room air is 92 per cent. Her left knee is held rigid in fixed flexion and is hot and red with a moderate effusion. Her respiratory rate is 22/min. There is decreased expansion on the right side, with dullness to percussion, increased vocal resonance and coarse crackles at the base. The remainder of her examination is normal. Chest X-ray Right basal consolidation Acute infectious arthritis occurs when microorganisms (usually bacteria) invade the joint space (not the bone itself), where they release endotoxins and trigger cytokine release and neutrophil infiltration. These inflammatory reactions ultimately lead to erosion and destruction of the join Risk factors for acute infectious arthritis. 1. Prior joint damage (e.g., rheumatoid arthritis). 2. Joint prosthesis 3. Concurrent or recent bacterial infection 4. immunocompromised (diabetes and old age) 5. extremes of age Signs and symptoms: 1. The joint is swollen, warm, and painful. a. The range of motion (active or passive) is very limited. b. An effusion can be palpated. 2. Constitutional symptoms such as fever, chills, and malaise are common. Pathogenesis: microorganisms penetrate the joint via the following mechanisms: a. Hematogenous spread—most common route. b. Contiguous spread from another locus of infection (e.g., osteomyelitis, abscess, or cellulitis). c. Traumatic injury to joints. d. Iatrogenic (e.g., from arthrocentesis, arthroscopy) Diagnosis: 1. Needle aspiration of synovial fluid 2. Blood cultures 3. Imaging (X-Ray, CT, MRI) 4. Joint aspirate (microscopy and culture) for possible septic arthritis Treatment: 1. Antibiotic to treat the infection 2. Drainage of effusion Differential diagnosis: - Septic arthritis - Osteoarthritis - Gout - Reactive arthritis Psoriatic arthritis History A 38-year-old man visits his general practitioner with a 2-month history of painful hands. It developed insidiously over a few weeks and affects primarily the distal and proximal interphalangeal joints in his hands, the whole of his third toe on the right foot and his right heel. The pain, stiffness and swelling are most marked in the morning but improve with exercise and ibuprofen. His past medical history is unremarkable. Systemic enquiry reveals a longstanding rash which affects his forearms and umbilicus for which he uses emollients. Examination There are plaques of scaly skin affecting his forearms, scalp and umbilicus. His nails are dystrophic with pitting and ridging and there is soft-tissue swelling and synovitis affecting the Muran Mjahed 26 distal and proximal interphalangeal joints. He has a ‘sausage toe’ on his right foot. His right Achilles tendon is swollen and tender in its distal third, with pain on palpation of the insertion into the calcaneus. Develops gradually in some patients with psoriasis (within 10 years). Usually asymmetric and polyarticular. Rash, nail and joint involvement are characteristic Are the normal inflammatory markers unusual? Psoriatic arthritis are part of group of disorders called seronegative spondyloarthropathy, like reactive arthritis; The seronegativity (i.e. RF and ACPA negative) is also very supportive Nail disease is very helpful in differentiating psoriatic arthritis from other forms of inflammatory arthropathy. Signs and symptoms: 1. Dactylitis and tendonitis 2. Predilection for DIP and PIP joints (MCP are less commonly affected. Distal and proximal polyarticular diseases). 3. Joint pain, stiffness and swelling. 4. Onycholysis and nail pitting 5. Upper extremities are more common to be involved than lower extremities; smaller joints are more common than large joints. 6. Oligoarthritis (particularly hips and knees) 7. Anterior uveitis 8. Sacroiliitis and/or spondyloarthropathy. Pathogenesis: Dendritic cells (DC)→ produce and secrete IL-23 and IL-12 → result in enthesitis → stimulate Th17→stimulate the production of inflammatory cytokines → IL6, IL17, IL22 → IL17 derives synovial fibroblasts, and macrophages to promote inflammatory cytokines (IL1B, IL16, TNF) that promote bone destruction → IL22 is responsible for production of new bones. Diagnosis: 1. Labs a. CRP / ESR; correlates with disease activity. b. HLA B-27 + c. Hyperuricemia. 2. Radiographs a. Periostitis b. Ankylosis c. Osteolysis d. Dactylitis no diagnostic test and often the acute phase is not as marked as in other inflammatory joint diseases. Radiology is unhelpful early on: the periarticular osteopenia of rheumatoid is characteristically absent and the classic osteolysis leading to ‘pencil-in-cup’ deformities is a late feature. Tendinopathy, enthesopathy, plantar fasciitis, sacroiliitis and spondyloarthropathy may be evident on MRI Treatment: 1. Initial treatment is NSAIDs, 2. Methotrexate or antiTNF agents for persistent arthritis. 3. Steroids are typically not used. Differential diagnosis of the rash and arthropathy: 1. Psoriatic arthritis 2. Systemic lupus erythematosus Muran Mjahed 27 3. Vasculitis 4. Sarcoidosis 5. Enteric arthropathy Juvenile arthritis History A six-year-old girl is brought to her general practitioner by her mother, with an 8-week history of a stiff and painful swollen right knee. There is no recall of trauma and she is otherwise well, with no recent history of infectious illness. Her medical history is unremarkable and her vaccinations are all up to date. A course of a non-steroidal antiinflammatory drug (NSAID) has been of minimal benefit. Examination The girl is uncomfortable getting undressed and on to the examination couch. There is no peripheral stigmata of inflammatory disease, but she has a moderate effusion of the right knee, which is warm and shows reduced flexion. Juvenile arthritis is a long-lasting, chronic disease. It is the most common form of arthritis in children. The three main types of juvenile arthritis are: 1. oligoarticular arthritis (<5 joints; subdivided into persistent and extended) 2. polyarticular arthritis (>5 joints; subdivided into RF positive and RF negative) 3. systemic-onset JIA 4. enthesitis-related arthritis (linked to the presence of HLAB27) 5. psoriatic arthritis Signs and symptom: 1. Painful joints in the morning that improve by afternoon, the first sign of the disease is a morning limp, caused by an affected knee. 2. Joint swelling and pain may also be noted, a child may feel irritable or tired and not want to play. 3. Sometimes, it causes lymph node swelling in the neck (cervical lymphadenopathy) or in other parts of the body. 4. Anterior uveitis with oligoarticular disease anterior uveitis may be asymptomatic, early referral to ophthalmology services for slit-lamp examination is recommended The risk of developing anterior uveitis is increased in patients with high ANA Diagnosis: 1. Children under 16 years 2. Swelling pain stiffness growth problem 3. Eye problem 4. ↑ESR, ↑ CRP 5. Positive ANA 6. Negative RF Treatment: 1. NSAID 2. DMARD; methotrexate 3. Corticosteroids 4. Surgery Muran Mjahed 28 Inflammatory Myopathies History A 72-year-old woman is admitted by the medical team with muscle weakness and pain. Her symptoms began insidiously over the last few weeks, but are now so severe she finds it hard to climb stairs or raise her arms above her head. Fine movements and grip strength are unaffected. She has also noticed a scaly rash over the back of her hands and her palms are becoming cracked and unsightly. Otherwise she is well, with no medical history of note. Systems review is unremarkable; she denies weight loss and in fact has noticed that her abdomen is mildly swollen. Examination This elderly woman has marked proximal weakness, graded 2 to 3 out of 5 at shoulder and hip in the absence of neurological features. She has a lilac discolouration over the back of her eyes, a scaly rash on the back of her fingers and papules over her metacarpophalangeal (MCP) joints. Her palms are fissured and cracked and nail-beds are ragged with dilated nailfold capillary loops. Cardiorespiratory examination is unremarkable. Abdominal examination reveals a non-tender pelvic mass and a small amount of ascites. Autoimmune condition that involves proximal muscle weakness and muscle inflammation. Classification: a. Polymyositis. b. Dermatomyositis. - The only difference between them both is that polymyositis does not involve the skin; however, skin is involved and develops skin rash in dermatomyositis. Females are more likely to be affected than males, 2:1 ratio, with age at onset 40-50 Dermatomyositis associated with malignancy often remits once the tumor is removed Weakness Causes: Genetic predisposition (HLA 8,1 PTPN22, STAT4, TRAF6) Environmental triggers (UV radiation, smoking, previous infection, previous lung disease, occupational exposure, medication, dietary supplements). Signs and symptoms: 1. Symmetrical proximal muscle weakness 2. Most severely affected muscle groups are the neck flexors, shoulder girdle, and pelvic girdle muscle. (in a majority of patients) 3. Distal extremity weakness is less frequent and typically less severe 4. Difficulty with: a. Walking up the stairs. b. Sitting up from a chair c. Reaching for items on shelves and cupboards. 5. Myalgia in a minority of patients. 6. Dysphagia; involvement of striated muscle in the upper GI tract in a minority of patients Associated findings include (other autoimmune conditions) a. Interstitial lung disease. b. CHF c. Conduction defects. d. Raynaud's phenomenon. e. Polyarthritis and arthralgias. f. SLE . 7. Features unique to dermatomyositis; skin findings which can occur at the same time with myalgia: Muran Mjahed 29 Pathogenesis: a. Diagnosis : Dermatomyositis: humoral immune mechanisms; involvement of B and CD4 cells leading to perifascicular vascular abnormalities, around the muscle. b. Polymyositis and inclusion body myositis: cell-mediated process; involvement of CD8 cytotoxic T cells; which cause damage to muscles directly. - Both cause damage and necrosis of muscles 1. 2. 3. 4. 5. 6. 7. 8. 9. Treatment: a. Heliotrope rash → a lilac discoloration of the back of the eyes b. Rash similar to malar rash in lupus. c. Gottron papules; papular, erythematous, scaly lesions over the knuckles (MCP, PIP, DIP)→ pathognomonic for dermatomyositis (diagnostic). d. V sign; rash on the face, neck, and anterior chest, similar to Shawl, but occurs on the anterior area of the neck e. Shawl sign; hyperpigmented rash on shoulders and upper back, elbows, and knees. - Both V and shawl signs occur due to exposure to sun f. Holster sign; hyperpigmented rash on the lateral thighs g. Periungual erythema with telangiectasia h. Vasculitis of GI (swelling of the abdomen), eyes, kidneys and lungs i. Calcinosis cutis: Subcutaneous calcifications in children; painful j. There is an increased incidence of malignancy in older adults (lung, breast, ovary, GI tract, and myeloproliferative disorders) Clinically; presence of rash on the face and palms CK level is significantly elevated due to muscle necrosis. LDH, aldolase, AST, ALT are also elevated (muscle waste products). ANA in majority of cases Antibodies (anti-Jo-1 antibodies, anti Mi, anti SRP)—abrupt onset of fever, cracked hands, Raynaud phenomenon, interstitial lung disease and fibrosis, arthritis EMG; abnormal in most patients CT and X-ray to exclude underlying malignancy T2 weighted MRI helpful in identifying areas of swelling and aids in taking biopsy Muscle biopsy a. Shows inflammation and muscle fiber fibrosis and necrosis. b. Red rimmed vacuoles with beta amyloid c. Dermatomyositis→perivascular and perimysial d. Polymyositis and inclusion body myositis→endomysial 1. Corticosteroids. 2. Steroid-sparing immunosuppressive agents: a. Methotrexate. b. Azathioprine. 3. Hydroxychloroquine. 4. Physical therapy. 5. vitamin D and calcium supplementation may be required to protect bones. Differential diagnosis: a. Inflammatory myopathy; Poly- and dermatomyositis b. Drug-induced myopathy; colchicine, alcohol c. Neuromuscular disease; myasthenia gravis d. Endocrine disease; Cushing’s and Addison’s disease e. Metabolic disease; Glycogen storage disorders f. Rheumatic disorders; Polymyalgia rheumatica, fibromyalgia Muran Mjahed 30 Giant cell arteritis History A 62-year-old man presents to his general practitioner with a headache. The pain focuses over the temporal areas, where his scalp also feels tender such that he finds it painful to lie on his side at night. Over the same period as his headache has developed, he has noticed pain and fatigue in his jaw when chewing his food. He is otherwise ‘reasonably well’ but has been experiencing pain and stiffness in his shoulders and hips which he has attributed to heavy gardening, and has lost a few kilograms in weight. His past medical history and systems enquiry is unremarkable and he takes no regular medication. Examination This man appears well but uncomfortable. He has marked tenderness to even light touch over his temples; his temporal arteries are tender and pulseless bilaterally. Fundoscopy and the rest of his physical examination are normal. Also called temporal arteritis. granulomatous vasculitis of unknown cause; it is T Cell mediated immune response. typical patient is >50 years of age; twice as common in women as men The temporal arteries are most frequently affected, but it may involve other arteries, such as the aorta or carotids. Carotid bruits, decreased pulses in the arms, and aortic regurgitation may also be observed. Associated with increased risk of aortic aneurysm and aortic dissection Microscopically there is inner media granulomas Keys to Diagnosing Temporal Arteritis - Age >50 years - New headache - Tender/palpable temporal artery - High ESR - Jaw claudication Signs and symptoms: 1. Constitutional symptoms of malaise, fatigue, weight loss, and low-grade fever 2. Headaches which may be severe 3. Visual impairment a. Caused by involvement of ophthalmic artery b. Optic neuritis; amaurosis fugax; may lead to blindness in up to 50% if not treated early and aggressively 4. Jaw pain with chewing, intermittent claudication of jaw when chewing 5. Tenderness over temporal artery; absent temporal pulse 6. Palpable nodules 7. Forty percent of patients also have polymyalgia rheumatica Pathogenesis: The pathophysiology is not well understood intimal hyperplasia and luminal obstruction leading to ischemic manifestations involving extracranial branches of carotid arteries and aorta involving all layers of the arterial wall Macrophages in the adventitia produce pro-inflammatory cytokines such as IL-1, IL-6 and tumor necrosis factor α. These cytokines promote arterial wall and systemic inflammation. ) Diagnosis: 1. Clinically 2. ESR elevated (high ESR may precipitate blindness) 3. Biopsy of the temporal artery. - A single negative biopsy does not exclude the diagnosis. 4. Angiography in cases of large vessels involvement 5. Examination of the ophthalmic artery (ciliary branch) Muran Mjahed 31 Diagnostic triad: anemia, fever, ESR On biopsy: temporal artery inflammation is segmental, granulomas will be visible Treatment: 1. Use high-dose steroids (prednisone) early to prevent blindness, before the biopsy results. a. If visual loss is present, admit the patient to the hospital for IV steroids; otherwise, start oral prednisone b. If the diagnosis is confirmed, continue treatment for at least 4 weeks, then taper gradually, but maintain steroid therapy for up to 2 to 3 years. Relapse is likely to occur if steroids are stopped prematurely 2. Follow up on ESR levels to monitor effectiveness of treatment. 3. Visual loss in one eye may be temporary or permanent; treated by steroids. - Methylprednisolone within 24 hours restores vision. 4. Bone protection with bisphosphonates and calcium/vitamin D supplementation Even if untreated, the disease is usually eventually self-limiting in most patients, although vision loss may be permanent Takayasu arteritis History A 24-year-old Asian woman presents to her general practitioner with dramatic weight loss and intermittent fever. Her symptoms have been present for several weeks, but over the last few days she has developed difficulty walking. Although previously very fit and active, she can now walk only a quarter of a mile before developing fatigue and cramping pain in her calves that forces her to stop. Once she has rested for a while, she is able to continue walking again, but the symptoms return. Examination This young woman is well but her GP is unable to detect a blood pressure from the left arm: the pulses are absent on that side and a subclavian bruit is clearly audible. The opposite arm is normal. Further examination of her vascular system reveals another bruit over her right femoral artery with decreased pulses distally on the same side. The rest of her pulses are present and the remainder of her physical examination is normal Suspect Takayasu arteritis in a young woman with: 1. Decreased/absent peripheral pulses 2. Discrepancies of BP (arm vs. leg) 3. Arterial bruits Most common in Asian women younger than 50 years of age. Granulomatous vasculitis of aortic arch and its major branches, leading to fibrosis and causing to stenosis or narrowing of vessels Signs and symptoms: 1. Constitutional symptoms; fever, night sweats, malaise, arthralgias, fatigue. 2. Pain and tenderness over involved vessels. 3. Absent pulses in carotid, radial, or ulnar arteries; aortic regurgitation may be present. 4. ischemia develops in areas supplied by involved vessels. 5. Severe complications include a. limb ischemia b. aortic aneurysms c. aortic regurgitation d. stroke e. secondary HTN due to renal artery stenosis. 6. Causes visual disturbances due to ocular involvement and hemorrhage of retinal arteries Muran Mjahed 32 7. Visual and neurological symptoms 8. Claudication Pathogenesis: The inflammatory lesions in Takayasu's arteritis originate in the vasa vasorum and are followed by cellular infiltration, mainly composed of T cells (gammadelta lymphocytes, cytotoxic T lymphocytes, T helper cells), but also of natural killer cells, dendritic cells, monocytes and granulocytes, invading the outer layer of the media and/or its neighboring adventitia. interleukin-6, interleukin-1 are released in large amounts by infiltrating inflammatory cells within damaged tissue, as well as by circulating inflammatory cells, and very likely help maintain the aberrant immune activation, by promoting endothelial cells activation and by inducing lymphocyte infiltration. Diagnosis: 1. Arteriogram. 2. Check for granuloma (may be present in some cases). 3. Elevated ESR and CRP. 4. CBC; Anemia, low hematocrit, thrombocytosis. 5. Elevated Serum Creatinine. 6. MRI and Urinalysis; Proteinuria, Hematuria if complicated Treatment: 1. High dose steroids; prednisone. 2. Treat HTN. 3. Cyclophosphamide is for those who do not achieve remission with standard therapy 4. Surgery or angioplasty may be required to recannulate stenosed vessels. Bypass grafting is sometimes necessary Polyarteritis nodosa (PAN ) History A 63-year-old man presents to his general practitioner with painful hands. His hands have been stiff and sore for several months but he denies frank swelling. The arthralgia is associated with a ‘blotchy rash’ over his trunk, profound fatigue and weight loss. In addition, he has experienced bouts of testicular pain and over the last 48 hours has developed central abdominal pain after each meal. Examination This man has evidence of recent weight loss and a blue/purple net-like rash over his abdomen and thighs. His joints are tender but there is no clinical evidence of synovitis. Genitourinary examination is unremarkable and he has mild abdominal tenderness to deep palpation but no organomegaly. The remainder of the examination is unremarkable. Symptoms involve the nervous system and GI tract. Can be associated with hepatitis B, HIV, and drug reactions. Necrosis is segmented, leading to “rosary sign” as a result of aneurysms. Affects young to middle-aged individuals. Has an overall poor prognosis. Signs and symptoms: 1. Early symptoms are fever, weakness, weight loss, myalgias, arthralgias, and abdominal pain (bowel angina). 2. HTN 3. Mononeuritis multiplex. 4. Livedo reticularis (purpura) 5. Glomerulonephritis is rare 6. Testicular pain 7. GI bleeding Muran Mjahed 33 8. Active hepatitis B (uncommon) 9. No pulmonary involvement (distinguish it from GPA) Pathogenesis: PMN invasion of all layers → fibrinoid necrosis → intimal proliferation → reduced luminal area→ ischemia, infarction, and aneurysms. Diagnosis: 1. biopsy of involved tissue or mesenteric angiography. 2. ESR is elevated, ↑BUN, and p-ANCA may be present. 3. Test for fecal occult blood. Treatment: 1. Start with corticosteroids. 2. If severe, add cyclophosphamide to prevent interstitial ischemia 3. Plasmapheresis for patients with HBV Kawasaki disease Affects children younger than 5 years of age. More prominent in children of asian background; Japan/China > Europe A self-limiting disease. Stages: 1. Acute; 1-2 weeks 2. Sub acute; 2-8 weeks 3. Chronic; longer than 8 weeks Cause is Infection/Genetic trigger 1. Signs and symptoms: 2. Symptoms mimic viral infection; misdiagnosed in most cases. 3. Can be fatal with coronary artery aneurysms (in cases of subacute, echocardiogram is required). 4. Thrombosis with myocardial infarction (if subacute CAD was not treated). 5. CRASH & BURN a. Conjunctivitis b. Rash c. Adenopathy; enlargement of cervical lymph nodes d. Strawberry tongue; Oral mucositis e. Hand/Foot Changes; Erythema, Edema f. Fever >5 days Differential diagnosis include: Scarlet fever (someone exposed to group A strep to become sick with strep throat or scarlet fever, illness usually begins with a fever and sore throat) Diagnosis: 1. clinical evaluation; no specific diagnostic technique 2. CBC; CRP/ESR 3. Urinalysis; may show pyuria /proteinuria 4. ECG/Echo; for cardiac abnormalities Treatment: 1. Refer to Pediatrician 2. High dose aspirin for Fever; (Reye’s disease may develop) 3. IVIG to Prevents coronary artery aneurysms 4. Disease is self-limiting Muran Mjahed 34 Granuloma with polyangiitis, GPA (Wegener Granulomatosis) History A 58-year-old woman is admitted to the emergency department with haemoptysis. She has been non-specifically unwell with recurrent low-grade fever, non-productive cough and weight loss for the last few weeks but is now ‘exhausted’ and short of breath on exertion. Her general practitioner had done some blood tests and commenced oral antibiotics with no benefit and she has been coughing up blood for 24 hours. Her medical history includes type 2 diabetes for which she takes metformin. She is a smoker with a 20-pack per year history and is teetotal. Examination This woman looks unwell and sounds nasally congested. She is not febrile. Pulse rate is 88/min, blood pressure 145/92 mmHg and oxygen saturations 95 percent on room air. There are fine inspiratory crackles in the mid and lower zones of the chest, but cardiovascular and abdominal examination is unremarkable. ANCA-associated Vasculitides (usually c-ANCA positive). Necrotizing granulomatous vasculitis. Occurs in older individuals, and predominantly in caucasians. Etiology is yet to be identified. Signs and symptoms: 1. Constitutional symptoms (fever, weight loss, fatigue) 2. Ear, Nose, Throat: a. Rhinosinusitis d. Epistaxis b. Oral and nasal ulcers e. c. Otorrhea Otitis media 3. Lower respiratory tract: a. Cough d. Dyspnea b. Hemoptysis c. Interstitial lung disease e. f. Pleuritic chest pain Pulmonary nodules 4. Renal system: a. Asymptomatic hematuria b. ↑ creatinine c. Glomerulonephritis b. Livedo reticularis c. Urticaria d. Secondary nephritic syndrome 5. Cutaneous: In half of patients. a. Leukocytoclastic angiitis d. Erythema nodosum 6. 7. 8. 9. 10. Eye involvement: conjunctivitis or scleritis. Arthralgias and myalgias. Heart involvement: pericarditis, myocarditis. Neurologic findings: sensory neuropathies Tracheal stenosis. Muran Mjahed 35 The classic triad of Wegener’s granulomatosis is the presence of upper and lower respiratory tract disease and renal impairment. Pathogenesis: anti-neutrophil-cytoplasmic-antibody (c-ANCA) associated vasculitides (AAV). The granulomas in GPA begins→ formation of neutrophilic microabscesses→ partial or total occlusion of blood vessels→ the granulomas in GPA are not well-formed→ consist of giant cells surrounded by plasma cells, lymphocytes, and dendritic cells→ that damage the submucosa and penetrate the surrounding tissues, cartilage, or bone→ necrosis and permanent deformities. Diagnosis: 1. CXR→ nodules and infiltrates 2. Urinary sediment → microscopic hematuria 3. Biopsy of artery or perivascular area → granulomatous formation (gold standard) or lung biopsy. 4. Labs → ↑ ESR, anemia, positive c-ANCA, proteinase a 3, thrombocytopenia. Treatment: 1. cyclophosphamide and corticosteroids combination. 2. Immunosuppressants. 3. oral trimethoprim/sulfamethoxazole for upper respiratory tract infection and kidney impairment 4. Consider renal transplantation if the patient has end-stage renal disease (ESRD). Prognosis is poor, patients die within a year Eosinophilic Granulomatosis With Polyangiitis (EGPA ) History A 45-year-old woman presents to the emergency department unable to lift her right foot. She was well the night before and noticed the problem immediately on waking in the morning; she came straight to hospital as she was frightened she was having a stroke. Her past medical history reveals poorly controlled adult-onset asthma for which she takes inhaled steroids and beta-agonist, and persistent rhinitis for which she uses nasal spray. She has no cardiovascular risk factors. She takes no oral medication and has no other relevant medical or family history. Her only foreign travel was a two-week holiday in Menorca 4 years ago. She does not smoke or drink. Examination This woman is well and not febrile. She walks with a high-stepping gait on the right side to avoid her toes dragging on the floor. She has weakness of dorsiflexion and eversion of the foot, with some reduced sensation over the top of the foot. There is a non-blanching rash over her legs which she reports has been present for a week and is gradually deteriorating. The remainder of her examination is normal. Known as Churg–Strauss Syndrome, and allergic granulomatosis. A multi system disorder, causing granulomatous inflammation of both, small and medium sized arteries. Key to diagnosis: hypereosinophilia and tissue infiltration of eosinophils Genetic predisposition plays a role in the formation of EGPA. Leukotriene receptor antagonists also play an important role in the development of EGPA. Patients may be first misdiagnosed with allergies. 5-year survival rate Stages: 1. Prodromal stage a. Occurs in 20s and 30s. b. Atopic disease with allergic rhinitis and asthma. 2. Eosinophilic stage a. Hypereosinophilia. b. Organs begin to become infiltrated with eosinophils. 3. Vasculitic stage Muran Mjahed 36 a. Occurs in 30s and 40s. b. Most of the signs and symptoms develop in this stage. c. Granulomatous inflammation of small and medium sized vessels. Classic triad of EGPA: 1. Asthma and allergic rhinitis 2. Eosinophilic lung disease similar to pneumonia 3. Systemic vasculitis a. Mononeuritis multiplex b. Peripheral neuropathy c. Peripheral eosinophilia Signs and symptoms: 1. Lung involvement: a. Athma, dyspnea b. Pleural effusion c. Pulmonary opacities on imaging 2. Ear, Nose, Throat a. Otitis media b. Recurrent sinusitis c. Allergic rhinitis d. Polyps in the nasal canal 3. Peripheral neuropathies in most patients 4. Eosinophilic gastroenteritis a. abdominal pain b. Melena or hematochezia c. Diarrhea 5. Skin lesions a. Subcutaneous nodules that are often tender b. Palpable purpura 6. Cardiac involvement which posses an increased risk of mortality a. Pericarditis b. Hypertension 7. Renal involvement: a. Hematuria b. Oliguria c. Kidney injury 8. Constitutional symptoms are most prominent during the Vasculitic stage. Pathogenesis: EGPA is typically considered a Th2-mediated disease and blood and tissue eosinophilia represent the cornerstone of diagnosis. Besides, ANCA are known for inducing endothelial injury and vascular inflammation by activating the circulating neutrophils. Diagnosis: 1. Eosinophilic count >1500/ microL. 2. p-ANCA positive in a majority of cases. 3. ↑IgE levels, MPO-ANCA 4. CXR to identify any pulmonary opacities 5. Biopsy of lung or skin tissue shows prominence of eosinophils. Treatment: 1. Systemic glucocorticoids 2. Cyclophosphamide and rituximab 3. Patients with life threatening diseases are treated with plasmapheresis. 4. Azathioprine and methotrexate for maintenance. Muran Mjahed 37 Microscopic Polyangiitis A small vessel disease, similar to GPA. The only difference is that microscopic polyangiitis does not involve the nose. Signs and symptoms: 1. Skin involvement →palpable purpura 2. Lungs → cough, dyspnea, hemoptysis, pulmonary fibrosis, pulmonary hypertension, but less commonly involved. 3. kidneys →glomerulonephritis Diagnosis: Can also be distinguished from GPA by biopsy, which shows necrotizing vasculitis without granulomas Treatment: 1. Systemic glucocorticoids 2. Cyclophosphamide and rituximab 3. Patients with life threatening diseases are treated with plasmapheresis. 4. Azathioprine and methotrexate for maintenance. Familial Mediterranean Fever History A 16-year-old Turkish boy is seen in the emergency department with abdominal pain and fever. His right knee is also exquisitely tender and slightly swollen. He has suffered recurrent attacks with a similar presentation since childhood, although normally they are more severe and also include pleurisy. He has been told that ‘it runs in the family’. The attacks resolve spontaneously and in between times he is entirely well. Examination This adolescent is very uncomfortable, with a temperature of 38.8°C. His pulse rate is 110/min, blood pressure 115/63 mmHg. He has marked abdominal tenderness with rebound and guarding; his bowel sounds are reduced. There is a mild and cool effusion of his right knee, which is extremely tender. The rest of his examination is normal. FMF is an autosomal recessively inherited periodic syndrome characterized by stereotyped attacks of fever and inflammatory features which last up to 4 days and then remit spontaneously The combination of recurrent serositis (peritonitis and pleurisy), fever and eastern Mediterranean origin raises the possibility of familial Mediterranean fever (FMF) as the underlying diagnosis The arthritis is classically monoarticular and symptoms often outweigh the signs, with extreme tenderness despite only mild effusions and a marked absence of increased temperature Signs and symptoms: 1. fever 2. peritonitis 3. arthritis 4. pleuritis 5. rash (erysipelas-like). Diagnosis: 1. Check for acute abdomen - CBC, inflammatory markers, amylase and lipase, lactate (severe disease) 2. Abdominal and chest X-ray 3. Genetic testing Muran Mjahed 38 Complications: - Amyloidosis a multisystem disease caused by the deposition of insoluble protein in the extracellular matrix: renal, cardiac and hepatic tissues are commonly affected. Primary amyloid (AL): deposition of immunoglobulin light chains and occurs in isolation or in the context of myeloma. Secondary or reactive amyloid (AA): deposition of amyloid A rather than light chains and may complicate any chronic infective, malignant or inflammatory process such as FMF. Treatment: Colchicine (diminishes the attacks for serositis and reduces the inflammation and pain) It is not only very effective at diminishing the attacks of serositis in FMF, but has also been shown to reduce the development of renal amyloidosis in these patients by up to two-thirds. Mononeuritis multiplex History A 64-year-old man is referred urgently by his general practitioner with acute neuropathy. Two days prior to presentation the patient awoke to find himself unable to lift his right foot and toes while walking, such that he kept tripping. Over the next 24 hours he found he had lost control of his left thumb and could not grip things properly. His problems were associated with altered sensation in his hand and foot. His past medical history is entirely unremarkable. Examination This man has a high steppage gait and inability to dorsiflex the right foot and walk on his heels only. Sensation is also diminished over the top of the foot and ankle. In the left hand, there is loss of abduction and opposition of the thumb and reduced sensation in the lateral three and a half digits. Mononeuritis multiplex can be caused by any of the medium or small vessel vasculitis Signs and symptoms: 1. livedo reticularis 2. back pain 3. testicular pain in polyarteritis nodosa 4. sinus or audiovestibular disease in Wegener’s granulomatosis 5. adult-onset asthma or nasal polyps in Churg–Strauss syndrome 6. abdominal pain and rectal bleeding in Henoch–Schönlein vasculitis. Diagnosis: - nerve biopsy and a common site for this procedure is the sural nerve Treatment: 1. potent immunosuppression - high-dose corticosteroids and cyclophosphamide 2. treatment of underlying infections such as hepatitis 3. Patients with foot drop will also require orthoses and physiotherapy to maximize physical function. Mixed Connective Tissue Disease (MCTD) History A 44-year-old woman presents to the rheumatology department with a constellation of symptoms. She was well until 8 months ago when she developed Raynaud’s phenomenon and joint pains. The latter responded to a non-steroidal anti-inflammatory drug (NSAID), so she ‘got on with normal life’. Over the last few months she has noticed the skin over her fingers becoming tighter. In recent weeks she has developed muscle weakness such that she finds it difficult to climb stairs or get out of a chair unaided. Muran Mjahed 39 Examination There is some skin tethering over the fingers, with reduction in the finger pulps. Although her joints are painful there is no objective evidence of synovitis. Proximal muscle power is reduced to 3/5. Respiratory examination reveals fine inspiratory crackles bibasally, but the remainder of her examination is normal. The general practitioner who referred her performed an ‘autoimmune screen’. Mixed connective tissue disease is an “overlap” syndrome with clinical features similar to those of SLE, RA, systemic sclerosis, and polymyositis. Findings consistent with each of these diseases do not necessarily occur simultaneously. It usually takes some time for a pattern to be identified and a diagnosis of mixed connective tissue disease to be made. Features seen in RA, SLE, myositis and scleroderma in the presence of anti-U1-RNP antibodies Signs and symptoms: 1. joint symptoms 2. Raynaud’s phenomenon 3. Sclerodactyly 4. proximal myopathy 5. Other frequent problems include esophageal dysmotility, serositis and lymphadenopathy Diagnosis: 1. presence of anti-U1-RNP Abs 2. High ANA and RF Treatment: - Depends on the predominant underlying disease. Cryoglobulinemia History A 78-year-old woman is referred from a leg-ulcer clinic to rheumatology for advice. She has been treated with compression bandaging for presumed venous ulcers for several months, with no perceptible benefit, and on a recent review was found to have a few scattered lesions of palpable purpura, an elevated ESR and a very high rheumatoid factor. Examination This woman has palpable purpura over her feet and toes, with a shallow painful ulcer on her left shin and one over her right first metatarsophalangeal (MTP) joint. The edges of the ulcers are punched out but not heaped up, and the bases are clean with no evidence of infection. There is no clinical evidence of synovitis and the remainder of the examination is normal. Caused by deposition of cryoglobulins, which are immunoglobulins that precipitate in cold temperatures. Some cryoglobulins have rheumatoid factor activity, giving rise to a very high RF titre in the absence of rheumatoid arthritis Classified into Type I, Type II, and Type III based on the type and clonality of immunoglobulins Causes: 1. Chronic HCV infection- most common cause 2. Chronic HBV infection 3. HIV infection 4. Other causes of liver disease (autoimmune hepatitis, primary biliary cirrhosis) Cryoglobulinemia exist in single and mixed forms a. A single homogeneous form (25 percent), also known as type I, can be essential or idiopathic, but is often associated with myeloma, lymphoma or Waldenström’s macroglobulinemia b. The mixed form (75 per cent) is further subdivided into mixed monoclonal (type II) and polyclonal (type III), and both types may form complexes with self IgG (i.e. have rheumatoid factor activity). Muran Mjahed 40 Although mixed cryoglobulinemia can be essential, type II is often associated with myeloma, lymphoma, Sjögren’s syndrome and infections (particularly hepatitis C). Type III is classically associated with autoimmune inflammatory disease (particularly RA and SLE) Signs and symptoms: 1. palpable purpura 2. renal disease 3. arthralgias/arthritis 4. peripheral neuropathy 5. Hypocomplementemia ( low serum levels of C3 and C4) 6. Pulmonary and CNS involvement are rare Palpable purpura may occur in conditions such as meningococcal sepsis and thrombocytopenic purpura, but one of the most common inflammatory causes of palpable purpura is vasculitis. The most common underlying conditions are Henoch–Schönlein purpura, leukocytoclastic vasculitis and cryoglobulinemia Complications: 1. Cutaneous with palpable purpura, petechiae, distal ulceration/necrosis and livedo 2. Raynaud’s phenomenon 3. Arthritis 4. Peripheral neuropathy 5. Renal disease (should check renal function and blood pressure, along with a urine dip checking for proteinuria or haematuria) 6. Liver disease Pathogenesis: Proposed mechanism is deposition of antigen-antibody complexes (such as HCV antibodies) in small vessels → complement activation and inflammation. Liver disease may contribute to decreased immune complex clearance,→ greater tissue deposition and disease activity. Diagnosis: 1. clinically 2. circulating cryoglobulins levels ( measuring can be challenging as significant quantities are lost when the blood cools to below 37°C) 3. biopsy →leukocytoclastic vasculitis (inflammation with polymorphonuclear leukocyte nuclear debris). 4. Patients should be tested for underlying HCV, HBV (serology), and HIV. Treatment: - depends on the extent of disease and the association with hepatitis C. - If not associated with hepatitis: 1. Cold avoidance 2. NSAIDs 3. Hydroxychloroquine 4. Corticosteroids and steroid-sparing agents such as azathioprine 5. Cytotoxic therapy (cyclophosphamide) or plasmapheresis is for organ or life-threatening disease - Hepatitis associated cryoglobulinemia 1. interferon-a with or without the antiviral ribavirin 2. immunosuppressive agents if the vasculitis is very severe - If asymptomatic: only monitoring is required - If mild to moderate: low dose corticosteroids - If moderate to severe: INF, ribavirin - medium dose corticosteroids - If severe: plasmapheresis Muran Mjahed 41 Buerger disease History A 17-year-old youth is referred urgently to the rheumatology department with a rapidly progressing rash over his buttocks and legs, and severe, cramping abdominal pain. The rash began as flat, red areas which became ‘bumpy’ and then developed into palpable dark red–purple lumps that failed to blanch on pressure. The abdominal pain occurs in spasms and does not appear to relate to food. His bowel habit is unchanged. Examination This youth is well but clearly very uncomfortable. His pulse rate is 84/min and blood pressure 117/62 mmHg. He has palpable purpura over his buttocks and the backs of his legs. His right ankle is mildly synovitic but otherwise his joints are normal and he has no other stigmata of inflammatory disease. Palpation of his abdomen reveals diffuse tenderness but no guarding or rebound. There is no organomegaly and normal bowel sounds are audible. Buerger disease is also known as Henoch–Schönlein purpura (HSP) and IgA nephropathy Purpura are the result of a spontaneous extravasation of blood from the capillaries into the skin. If small they are known as petechiae, when they are large they are termed ecchymoses. The combination of palpable purpura, abdominal pain, arthritis and renal disease is a classic presentation of Henoch–Schönlein purpura (HSP). HSP is frequently self-limiting small-vessel vasculitis that can affect any age; but the majority of cases present in children aged 2–10 years. Differential diagnosis for purpura include: a. Infection; meningococcal disease, infective endocarditis, septicaemia b. Thrombocytopenia; idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura c. Vascular defect; senile or steroid-induced purpura, vasculitis d. Coagulation defect; hemophilia e. Drugs; steroids, sulphonamides Signs and symptoms: 1. Petechiae 2. Abdominal pain 3. Renal involvement characterized by proteinuria and microscopic hematuria (glomerulonephritis) 4. Arthritis Diagnosis: 1. Urinalysis and urine dipstick for renal function 2. Skin biopsy ( leukocytoclastic vasculitis with IgA deposition in the affected blood vessels) 3. Kidney biopsy shows IgA deposition in glomeruli Treatment: 1. mild cases do not require a specific therapy 2. For arthritis; NSAIDs, steroid therapy 3. High-dose steroids and cytotoxics such as cyclophosphamide for renal disease 4. Plasma exchange or intravenous immunoglobulin for refractory cases Behçet syndrome History A 26-year-old Turkish man presents to his general practitioner with a painful aphthous ulcer on his scrotum. Although he has had oral ulcers on and off ‘for years’, this is the first episode in the genitourinary tract. He denies other genitourinary symptoms and is not sexually active. Four weeks previously he experienced a rash over the front of his shins: discrete red Muran Mjahed 42 tender lumps appeared and then faded spontaneously, going through color changes like a bruise. On systems review he mentions that his right knee is intermittently painful and swollen, but denies any other problems. Examination This young man has a deep ulcer on his scrotum, but his penis is normal. He has no oral ulcers. The sclera of his right eye is injected. He has a solitary brown slightly raised lesion on his left shin and a small effusion in the right knee. He has an erythematous lesion in the left antecubital fossa from blood donation 48 hours previously An autoimmune, multisystem vasculitic disease; cause is unknown. Both genders are affected equally but males develop more severe disease. Associated with HLA B51- MHC class 1 Often present in Middle Easterners, mediterranean and turkish individuals Signs and symptoms: 1. Painful, sterile oral and genital ulcerations (pathergy) 2. Arthritis (knees and ankles most common) 3. Eye involvement (uveitis, optic neuritis, iritis, conjunctivitis) 4. CNS involvement (meningoencephalitis, intracranial HTN) 5. Fever 6. Weight loss 7. Erythema nodosum and cutaneous manifestations 8. Pathologic lesions are systemic perivasculitis with early neutrophil infiltration and endothelial swelling Pathogenesis: Activated neutrophils with ↑Th1, Th17, cytotoxic CD8 and γ T cells Diagnosis: 1. biopsy of involved tissue (inflammation and necrosis) 2. Antibodies against: - Alpha enolase (endothelial cells) - Selenium binding proteins - Anti saccharomyces cerevisiae 3. No specific diagnostic test or laboratory finding Treatment: 1. Steroids for severe disease 2. Tacrolimus 3. Mucocutaneous disease can be treated with colchicine, dapsone or thalidomide 4. Cyclophosphamide is generally reserved for life- or sight-threatening disease Differential diagnosis: 1. Sarcoidosis 2. Reactive arthritis 3. Vasculitis 4. Crohn's disease 5. Systemic lupus erythematosus Rheumatic fever History A 16-year-old boy is referred to the medical team with abnormal involuntary facial movements. He was well until 8 weeks before admission when he developed a sore throat and febrile illness, associated with pain and swelling in several large joints. The symptoms began in his knees, before moving to his ankles and finally his elbows and responded only partially to Muran Mjahed 43 ibuprofen. However, he made a full recovery within 3 weeks. For the last 24 hours his parents have noticed emotional lability and involuntary rapid and purposeless movements, particularly in his face. He denies weakness or sensory loss. Examination This adolescent is well and not febrile. He has brief uncoordinated facial movements consistent with chorea. His skin, nails and mucosal surfaces are normal. Cardiovascular examination reveals a friction rub and pansystolic murmur in the mitral area. Respiratory and abdominal examinations are normal. There is a cool effusion in his left elbow. Acute rheumatic fever is an immunologically mediated systemic process that may progress to rheumatic heart disease. The arthritis associated with rheumatic fever is mild, nonerosive and self-limiting, tending to resolve within 2–4 weeks. Signs and symptoms: 1. Arthritis of large joints (knees, ankles, elbows and wrists) in most patients 2. Fever 3. Chorea 4. polyarthritis 5. Subcutaneous nodules Diagnosis of Acute Rheumatic Fever requires two major criteria or one major and two minor criteria 1. Major criteria a. Migratory polyarthritis b. Erythema marginatum c. Cardiac involvement (e.g., pericarditis, CHF, valve disease) d. Chorea e. Subcutaneous nodules 2. Minor criteria a. Fever b. Elevated erythrocyte sedimentation rate c. Polyarthralgias d. Prior history of rheumatic fever e. Prolonged PR interval f. Evidence of preceding streptococcal infection Diagnosis: 1. Echocardiogram (assess valvular insufficiency and to identify a pericardial effusion) 2. Based on Duckett Jones 3. Evidence of previous infection with GAS 4. elevated Antistreptolysin titre helps confirm the diagnosis 5. elevated ESR/CRP, a normocytic anemia, neutrophilia and hypoalbuminemia Treatment: 1. Treatment of GAS with penicillin; if resistant, treat with macrolides or cephalosporins 2. Give NSAIDs Prophylaxis with penicillin should be continued for 5 years or until 21 years of age, whichever is longer. Sarcoidosis History A 28-year-old African-American man presents to the rheumatology department with a 2-week history of painful swollen ankles and a rash over his shins. His rash is fluctuating with crops of raised tender lumps that fade spontaneously. His ankle pain initially responded to a non-steroidal anti-inflammatory drug (NSAID), but over the last few days has deteriorated such that he finds it hard to walk. His medical history is unremarkable, except for an acutely painful red eye several months Muran Mjahed 44 previously for which he saw an ophthalmologist and was given steroid eye-drops. He cannot recall the diagnosis he was given at the time. Systems review is unremarkable. Examination This man has synovitis affecting both ankles, with a mild effusion on the left and lesions suggestive of resolving erythema nodosum over his shins A chronic systemic granulomatous disease characterized by noncaseating granulomas, often involving multiple organ systems. Lungs are almost always involved. Etiology unknown. Occurs most often in the African American population, especially women Caused by exposure to smoking, amiodarone use… Signs and symptoms: 1. Constitutional symptoms; malaise, fever, anorexia, weight loss 2. Lungs: dry cough, dyspnea (especially with exercise), chest pain 3. Skin; erythema nodosum, plaques, subcutaneous nodules, maculopapular eruptions 4. Eyes; visual impairment, anterior uveitis, posterior uveitis, conjunctivitis 5. Heart; arrhythmias, conduction disturbances, such as heart block Sudden death 6. Musculoskeletal system; arthralgias and arthritis and bone lesions 7. Nervous system; Cranial nerve VII involvement (Bell palsy), optic nerve dysfunction, papilledema, peripheral neuropathy Pathogenesis: Stages of sarcoidosis: a. Stage 0: Normal b. Stage 1: Bilateral hilar lymphadenopathy c. Stage 2: Bilateral hilar lymphadenopathy and pulmonary infiltrates d. Stage 3: Pulmonary infiltrates and restrictive lung disease. Diagnosis: 1. Chest X-ray (Bilateral hilar adenopathy) 2. Skin anergy with tuberculin skin test 3. Angiotensin-converting enzyme (ACE) is elevated; secreted from the lungs 4. Hypercalciuria and hypercalcemia 5. transbronchial biopsy (noncaseating granulomas) Treatment: 1. NSAIDs 2. Low dose corticosteroids; high doses are required in the presence of arthritis 3. Intra-articular injection of steroid may abort an acute attack 4. Immunosuppression such as cyclophosphamide is reserved for neurological or life-threatening disease
 0
0

No more boring flashcards learning!
Learn languages, math, history, economics, chemistry and more with free StudyLib Extension!
- Distribute all flashcards reviewing into small sessions
- Get inspired with a daily photo
- Import sets from Anki, Quizlet, etc
- Add Active Recall to your learning and get higher grades!

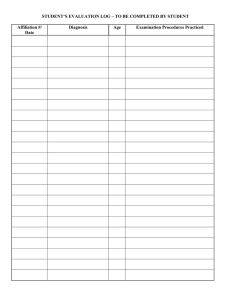
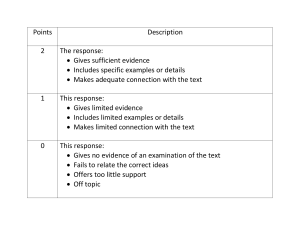

Add this document to collection(s)
You can add this document to your study collection(s)
Sign in Available only to authorized usersAdd this document to saved
You can add this document to your saved list
Sign in Available only to authorized users