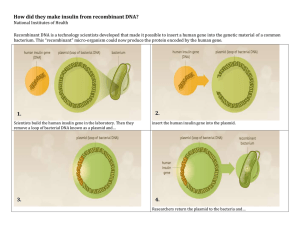
Unit 1 MODULE 2 CAPE BIOLOGY – ASPECTS OF GENETIC ENGINEERING (PART 1) Genetic Engineering (GE)/Recombinant DNA technology: ‘the transfer of a gene from one organism (donor) to another (recipient/host) e.g. gene coding for human insulin can be removed from human DNA and transferred to bacteria’; manipulation of genes (& thus proteins synthesized); genetic constitution ALTERED as a result – now is RECOMBINANT DNA (DNA from 2 different species); host organism = RECOMBINANT/TRANSGENIC/ GENETICALLY MODIFIED ORGANISM (GMO). Works thanks to universal nature of genetic code! 4.1 OUTLINE THE PRINCIPLES OF RESTRICTION ENZYME USE IN ‘CUTTING’ SECTIONS OF DNA AND LIGASE ENZYMES TO ‘PASTE’ DNA TOGETHER GENOME: total set of genes/genetic information in an organism Restriction enzymes (R.E.s) aka restriction endonucleases ➢ Naturally occurring enzymes found in bacterial cytoplasm. ➢ Play role in natural defences of bacteria; released on encounter with foreign DNA (especially of phage viruses that often infect bacteria). ➢ Destroy foreign DNA by cutting/cleaving it into smaller fragments [molecular scissors] How are restriction enzymes used? Essential - remove particular segments of DNA that can then be rejoined to another section of DNA. o Many different types exist – each specific o Each recognizes & cleaves DNA at SPECIFIC SITES; enzyme active site ‘recognises’ particular base sequences (4 – 8 bases long) in DNA molecule [recognition sites]. DNA sequences often palindromic (nucleotide sequences that read the same forward & backward) o Named for bacteria from which they are extracted e.g. EcoR1, HPaI o Some R.E.s make BLUNT ENDS – cutting across both strands at a single point o Others make STICKY ENDS - cutting at a slant/producing a staggered cut leaving short, single-stranded DNA overhanging. (Sticky ends are short lengths of unpaired bases at cut site). N.B. Sticky ends permit complementary base pairing which can result in ‘sticking’ cleaved ends together under the right conditions. DNA ligase needed to reseal fragments, linking sugar-phosphate backbones (ligation [molecular glue] N.B. Recent steps in GE include development of CRISPR-Cas9: advanced genome editing tool! Fast, cheap & precise! Lots of applications as permits user to edit parts of genome – removing, adding or altering parts of sequence. Cas9 = enzyme that acts like scissors, cutting the DNA strands at a particular location so as to remove or add DNA fragments. Guide RNA (gRNA) is a small length of RNA that can find & bind to DNA, thus bringing Cas9 to the desired location. When DNA is cut, cell’s repair machinery can be manipulated to introduce specific changes. May treat medical conditions with genetic components including cancer. N.B. Somatic cell use only! Gene editing in germline cells is currently illegal! 1 Diagram showing how the CRISPR-Cas9 editing tool works. Image credit: Genome Research Limited 2 4.2 EXPLAIN THE BASIC STEPS INVOLVED IN RECOMBINANT DNA TECHNOLOGY (refer to isolation & cloning of genes, vectors; using examples including insulin production) Recombinant DNA Technology: the transfer of a gene from one organism (donor) into another (recipient) such that the gene is expressed in its new host (host now said to be transgenic).e.g. gene coding for Factor VIII may be taken from human cells and transferred to bacteria such as E. coli. Involves essential steps: - Identifying and obtaining (isolating) desired gene & cloning it. Different options exist to do so: o Make synthetic DNA: work out DNA code if know amino acid sequence of protein desired, then make DNA in lab (Direct DNA synthesis). o Make DNA from mRNA (reverse transcription), i.e. use mRNA transcribed from desired gene to make DNA copy. o May isolate gene from entire genome by cutting total DNA into fragments using restriction enzymes, then identifying piece containing desired gene using gene probes (single strand of DNA carrying part of gene desired; labelled). Tedious method for big proteins! o Multiple copies of gene can be made using PCR (polymerase chain reaction) i.e. cloning the gene - Inserting gene into vector & cloning the vector - Inserting gene into organism via vector - Growing & culturing transformed cells to synthesise protein product N.B. VECTOR in GE = organism/structure that delivers/transfers gene into host cell (e.g. plasmid, virus, liposome) STEPS illustrated in INSULIN production: 1. Isolate gene coding for human insulin. Often use mRNA carrying code for insulin. [Insulin is a small protein so it would be problematic to isolate the gene from DNA in nucleus]. mRNA extracted from beta cells in pancreatic islets of Langerhans (mRNA will carry code complementary to exons of human insulin gene). mRNA then incubated with free DNA nucleotides and reverse transcriptase (enzyme from retroviruses) resulting in formation of complementary DNA (cDNA will initially be single stranded; converted to double stranded DNA using DNA polymerase & free DNA nucleotides). Outcome = insulin gene. ‘Sticky ends’ (short lengths of unpaired bases) may be added if needed by attaching single stranded guanine (G) nucleotides using enzymes (OR by adding non-coding DNA which can then be cut using particular restriction enzymes to produce sticky ends). N.B. the insulin gene obtained may then be CLONED to make many copies of cDNA molecules using polymerase chain reaction (PCR)*. 2. Insert gene into a vector. Often use viral DNA or bacterial plasmids – small circular pieces of DNA found in many bacteria that are also easily taken up. Plasmids can self-replicate & are often exchanged between bacteria. Access plasmids by treating bacteria with enzymes to dissolve cell walls – centrifuge to separate out from main (larger) bacterial DNA. Cut open plasmid at specific site using restriction enzyme; add sticky ends using cytosine nucleotides (OR use same restriction enzyme as in step 1, thus producing same sticky ends). Mix cut plasmid & cut human DNA (insulin genes); C and G bases pair (OR complementary sticky ends pair) as H bonds form; DNA ligase catalyses formation of covalent phosphodiester bonds so that gene is permanently added to plasmid (ligation occurs), creating Recombinant DNA (rDNA). Splicing! N.B. NOT all plasmids will take up insulin gene – some will just rejoin. 3. Insert gene into bacteria. Mix plasmids with bacterial culture, e.g. E. coli. (Usually treat with cold calcium chloride solution that increases permeability of plasma membranes making cells more likely to take up plasmids. Then heat treat or ‘zap’ with brief electrical shock). About 1% of bacteria take up plasmids with insulin gene (others take up none or take up plasmids that do not contain the insulin gene). Transformed bacteria have taken up the recombinant DNA. Separate these from others with a screening process** often using antibiotic resistance provided by other genes also carried on plasmid. Clone transformed bacteria to produce MANY genetically identical cells (clones), each with recombinant plasmid, & culture on large scale in industrial fermenters. Insulin gene will be expressed & secreted insulin can be extracted, purified & sold. 3 *PCR: biological photocopying, fully automated – allows many identical copies of DNA to be made from a small sample! 1st must denature DNA (by heating to 95°C) – the 2 strands SEPARATE (exposing bases). Primers (short single-stranded lengths of DNA, complementary to sequences at 3’ ends of DNA segment to be copied) added on cooling to about 65°C. Annealing occurs (primers attach to DNA indicating where DNA polymerase is to bind). Taq DNA polymerase action is promoted (raising temperature to 72°C), and nucleotides are added in a complementary base-pairing fashion (elongation phase). Having now copied the gene (forming 2 complete DNA molecules), the cycle can be repeated multiple times! (Carried out in thermocycler). **Screening process: FINDING THE GM BACTERIA (the 1%!) – sorting transformed from non-transformed! A. Choose a plasmid containing two resistance genes that will act as genetic markers (other genes whose effects are seen readily). One gives resistance to ampicillin and the other to tetracycline (both are antibiotics) B. Insert the human insulin gene in the middle of ONE of the resistance genes (often the one for tetracycline), using appropriate restriction enzymes C. Intact gene will still confer resistance to ampicillin; but spliced gene will NOT function, so bacteria would be susceptible to tetracycline D. Grow bacteria (which will be a mixture of transformed & non-transformed) on an agar plate containing ampicillin. Any bacteria that grow MUST have taken up the plasmid with the resistance gene (BUT we cannot be sure if the plasmid itself was transformed, i.e. has the insulin gene). Bacteria that are killed did NOT take up the plasmid at all. E. Take an imprint of the colonies growing on this first plate in process called replica plating. Transfer sample of colonies to new agar plate containing tetracycline. Retain first plate for later comparison. F. Any bacteria that are killed on this second plate (with tetracycline) are those that HAVE the recombinant plasmid as the resistance gene for tetracycline has been interrupted in the splicing process. Any that grow on this medium do NOT contain the recombinant plasmid (since they are resistant to tetracycline the gene must be working!). G. Compare the colony positions to those on the first plate – reject those that are growing on the tetracycline plate (the second) but note the positions of those absent on the second plate but present on the first as these must lack the functional gene for tetracycline resistance and must therefore contain the human insulin gene. Locate and culture these bacteria from the first plate. H. After the transformed bacteria have been identified & isolated, culture the GM bacteria on a large scale (i.e. clone them, reproduce asexually, by binary fission; also, plasmid replicates) using an industrial fermenter, providing necessary nutrients & oxygen. Recombinant bacteria will then secrete insulin – can be harvested, purified & sold. [REVIEW ADVANTAGES TO USING HUMULIN!] N.B. because of the risks involved with the use of antibiotic resistance genes (e.g. horizontal gene transfer), fluorescent markers are more commonly used today, or another marker gene that produces a harmless product that is not normally made by cells and is easily stained. N.N.B!! ☺ To get a donor gene to work, it is often necessary to insert the promoter gene also in order to control the expression of the desired gene (initiates transcription). Yeast cells may be genetically modified to produce insulin as they are eukaryotic and can carry out the necessary post-translational protein modification necessary to produce ‘mature insulin’ (they have the required organelles!). 4 Sources include but are not limited to: UCIE: A Level Science Applications Support Booklet: Biology CAPE BIOLOGY UNIT 1 5