- No category
Philippine Pediatric Society Diplomate Exam Reviewer 2020
advertisement
PHILIPPINE PEDIATRIC SOCIETY Diplomate Exam Reviewer 2020 (topics were lifted from the HAB checklist) EMERGENCY MEDICINE ------------------------------------------------------------------------------------------------------------------------------------------------------------DERMATOLOGY ҉ Skin Disorders ----------------------------------------------------҉ Skin and Soft Tissue Infections ---------------------------------------OPHTHALMOLOGY ҉ Eye Pain and Discharge ----------------------------------------------------OTOLOGY ҉ Ear Pain ----------------------------------------------------NEONATOLOGY ҉ Neonatal Fever ----------------------------------------------------҉ Neonatal Jaundice ----------------------------------------------------҉ Respiratory Distress in the Newborn ---------------------------------------҉ Delayed Meconium Passage ---------------------------------------҉ Abdominal Distention ----------------------------------------------------҉ Newborn Dysmorphology ---------------------------------------҉ Malformations ----------------------------------------------------҉ Metabolic Disorders ----------------------------------------------------҉ Chronic Child ----------------------------------------------------ENDOCRINOLOGY ҉ Weight Gain ----------------------------------------------------҉ Diabetic Ketoacidosis ----------------------------------------------------҉ Endocrine Disorders ----------------------------------------------------҉ Short Stature ----------------------------------------------------GASTROENTEROLOGY ҉ Diarrhea ----------------------------------------------------҉ Constipation ----------------------------------------------------҉ Abdominal Pain ----------------------------------------------------҉ Prolonged Jaundice ----------------------------------------------------҉ Shock ҉ Pediatric Poisonings ҉ Animal Bites 2 4 6 7 10 12 14 17 19 21 25 26 27 29 33 49 53 55 57 63 66 69 71 73 NEUROLOGY / DEVELOPMENTAL PEDIATRICS ҉ Traumatic Brain Injury ----------------------------------------------------҉ Headache ----------------------------------------------------҉ Seizures ----------------------------------------------------҉ Raised Intracranial Pressure ---------------------------------------҉ Children with Special Needs ---------------------------------------HEMATOLOGY – ONCOLOGY ҉ Anemia ----------------------------------------------------҉ Cancer ----------------------------------------------------NEPHROLOGY ҉ Dysuria ----------------------------------------------------҉ Renal Disorders ----------------------------------------------------҉ Genital Disorders ----------------------------------------------------CARDIOLOGY ҉ Hypertension ----------------------------------------------------҉ Chest Pain ----------------------------------------------------҉ Heart Murmur ----------------------------------------------------҉ Arrhythmia ----------------------------------------------------҉ Heart Failure ----------------------------------------------------PULMONOLOGY / ALLERGOLOGY ҉ Sore throat ----------------------------------------------------҉ Cough ----------------------------------------------------҉ Colds ----------------------------------------------------҉ Allergic Disorders ----------------------------------------------------RHEUMATOLOGY / ORTHOPEDICS ҉ Collagen and Vascular Disorders ---------------------------------------҉ Limping Child ----------------------------------------------------҉ Musculoskeletal Disorders ---------------------------------------INFECTIOUS DISEASE ҉ Fever ----------------------------------------------------҉ Fever and Rash ----------------------------------------------------҉ Common Viral Illnesses ----------------------------------------------------҉ Common Bacterial Infections ---------------------------------------҉ Common Bacterial Causes of Nosocomial Infections -------------҉ Fungal Infections ----------------------------------------------------҉ Parasitic Infections ----------------------------------------------------- 74 75 79 83 85 89 92 98 100 116 120 124 129 135 138 141 143 155 156 158 168 171 173 183 188 209 231 233 235 Complex ▪ Acute process characterized by the body’s inability to deliver adequate oxygen to meet the metabolic demands of vital organs and tissues COMPENSATED SHOCK ▪ Compensatory mechanisms attempt to maintain BP by increasing CO and SVR ▪ Body attempts to optimize oxygen delivery to the tissues by increasing oxygen extraction and redistributing blood flow to the brain, heart, and kidneys at the expense of the skin and GIT DECOMPENSATED SHOCK ▪ Hypotension and tissue damage develop if treatment is not initiated or is inadequate CLINICAL MANIFESTATIONS DIAGNOSIS TREATMENT COMPLICATIONS / PROGNOSIS ▪ May initially manifest only as tachycardia ± tachypnea ▪ Progression leads to ↓ UO, poor peripheral perfusion, respiratory distress or failure, alteration of mental status, and low BP ▪ Hypotension – late finding, not a criterion for the diagnosis of shock ▪ Clinical diagnosis based on thorough history and physical examination (see Nelson’s 21st Ed., Fig. 88.1 and Fig. 88.2 for the algorithms for timesensitive, goal-directed stepwise management of hemodynamic support in Newborns and Infants and Children, respectively) ▪ After the 1sthr of therapy and attempts at early reversal of shock, focus on goaldirected end points should continue in the PICU (see Nelson’s 21st Ed., Table 88.6 for Hemodynamic Variables in Different Shock States) (see Nelson’s 21st Ed., Table 88.4 for Pathophysiology of Shock) HYPOVOLEMIC ▪ Most common cause of shock in children worldwide ▪ Due to ↓ preload secondary to internal or external losses Etiologies: Blood loss, plasma loss, water/electrolyte loss ▪ Cardiac pump failure secondary to poor myocardial function → systolic and diastolic dysfunction CARDIOGENIC DISTRIBUTIVE Etiologies: CHD, arrhythmias cardiomyopathies, ischemia, ▪ Abnormalities of vasomotor tone from loss of venous & arterial capacitance → maldistribution of blood flow away from vital organs & a compensatory ↑ in CO Etiologies: Anaphylaxis, neurologic (loss of sympathetic vascular tone 2O to SCI or brainstem injury), drugs ▪ ↓ CO 2O to direct impediment to right- or leftsided heart outflow or restriction of all cardiac chambers OBSTRUCTIVE Etiologies: tension pneumothorax, pericardial tamponade, pulmonary embolism, anterior mediastinal masses, critical coarctation of the aorta PPS Oral Exam Reviewer 2020 ▪ Often manifests initially as orthostatic hypotension ▪ Dry mucous membranes, dry axillae, poor skin turgor ▪ ↓ UO ▪ Tachypnea ▪ Cool extremities, delayed CRT, poor peripheral and/or central pulses ▪ Declining mental status ▪ ↓ UO ▪ Manifests initially as peripheral vasodilation and ↑ but inadequate CO ▪ Manifests as inadequate CO because of a physical restriction of forward blood flow (see Nelson’s 21st Ed., Table 88.2 for Criteria for Organ Dysfunction and Table 88.3 for Signs of Decreased Perfusion) Hematologic Abnormalities ▪ Thrombocytopenia, prolonged PT and aPTT, ↓ serum fibrinogen level, ↑ fibrin split products, and anemia ▪ ↑ Neutrophil counts and ↑ immature forms, vacuolation of neutrophils, toxic granulations and Döhle bodies can be seen with infection ▪ Neutropenia or leukopenia may be an ominous sign of overwhelming sepsis Hallmark of uncompensated shock: imbalance between oxygen delivery and oxygen consumption Falling mixed venous oxygen saturation (SVO2) reflects an increasing O2 extraction ratio and ↓ in O2 delivery relative to consumption ▪ Measured from a pulmonary arterial catheter ▪ Manifested clinically by ↑ lactic acid production caused by anaerobic metabolism and a compensatory ↑ in tissue oxygen extraction ▪ Initial assessment: airway, breathing and circulation ▪ Establish IV/IO access → rapid bolus of 20mL/kg of isotonic fluid; titrated to normalize HR, UO, CRT and mental status (may be repeated up to 60-80 mL/kg) ▪ If shock remains refractory, vasopressor therapy should be instituted (see Nelson’s 21st Ed., Table 88.13 and Table 88.14 for the Cardiovascular Drug Treatment of Shock and Vasodilators/Afterload Reducers in Treatment of Shock, respectively) ▪ Monitor and correct electrolytes ▪ Treat the hypoglycemia ▪ Hypocalcemia, which may contribute to myocardial dysfunction, should be treated with a goal of normalizing iCa concentration ▪ Adrenal function is an important consideration ● HAA replacement may be beneficial ● Corticosteroids may be considered in patients with shock that is unresponsive to fluid resuscitation and catecholamines (see Nelson’s 21st Ed., Table 88.8 for GoalDirected Therapy of Organ System Dysfunction in Shock) ▪ Hgb should be maintained at 10 g/dL, SVO2 at >70%, and cardiac index at 3.3-6.0 L/min/m2 ▪ Blood lactate levels and calculation of base deficit – useful markers for the adequacy of oxygen delivery ▪ Respiratory support – used as clinically appropriate ▪ Smaller boluses of fluids (5-10mL/kg) – replace deficits and maintain preload ▪ Early initiation of myocardial support with epinephrine or dopamine to improve CO is important ▪ Early consideration should be given to administration of an inodilator (e.g. milrinone) → may improve systolic function and ↓ SVR without causing a significant ↑ in HR ▪ Dobutamine or other vasodilating agents (e.g. nitroprusside) may be considered ▪ May benefit from early initiation of a vasoconstrictive agent to increase SVR ▪ Phenylephrine or vasopressin – to ↑ SVR in patients with SCI and spinal shock ▪ Epinephrine – for patients with anaphylaxis ▪ Fluid resuscitation may be briefly temporizing in maintaining CO but the primary insult must be immediately addressed ● Pericardiocentesis for pericardial effusion ● Pleurocentesis or CTT for pneumothorax ● Thrombectomy/thrombolysis for PE ● Initiation of a prostaglandin infusion for ductus-dependent cardiac lesions Batch Clingy ETIOLOGY / INCIDENCE / PATHOGENESIS 2 ▪ Encompasses multiple forms of shock (e.g. hypovolemic, distributive, cardiogenic) ▪ Hypovolemia from intravascular fluid losses occurs through capillary leak ▪ Cardiogenic shock results from the myocardialdepressant effects of sepsis ▪ Distributive shock is the result of decreased SVR SEPTIC ▪ In the early stages, CO ↑ to maintain adequate oxygen delivery and meet the greater metabolic demands of the organs and tissues (warm shock) ▪ As septic shock progresses, CO ↓ in response to the effects of numerous inflammatory mediators → compensatory ↑ in SVR (cold shock) Systemic inflammatory response syndrome (SIRS) ▪ Inflammatory cascade that is initiated by the host response to an infectious or noninfectious trigger Sepsis ▪ SIRS 2o to a suspected or proven infectious etiology Severe sepsis ▪ Sepsis + organ dysfunction Septic shock ▪ Severe sepsis + persistence of hypoperfusion or hypotension despite adequate fluid resuscitation or a requirement for vasoactive agents (see Nelson’s 21st Ed., Table 88.7 for the International Consensus Definition for Pediatric Sepsis) Select recommendations from the 2020 Surviving Sepsis Campaign for Children ▪ If lactate levels can be rapidly obtained, measure blood lactate – provide a valuable indirect marker of tissue hypoperfusion ▪ In children with septic shock, start antimicrobial therapy as soon as possible, within 1 hour of recognition ▪ In children with sepsis-associated organ dysfunction but without shock, start antimicrobial therapy as soon as possible after appropriate evaluation, within 3 hours of recognition ▪ Use empiric broad-spectrum therapy with one or more antimicrobials to cover all likely pathogens ● Once pathogens and sensitivities are available, narrow antimicrobial coverage ● If no pathogen is identified, narrow or stop empiric therapy according to clinical presentation, site of infection, host risk factors and adequacy of clinical improvement ▪ Use balanced/buffered crystalloids, rather than 0.9% saline for the initial resuscitation of children with septic shock ▪ Use either epinephrine or norepinephrine rather than dopamine in children with septic shock ● Epinephrine is associated with a lower risk of mortality and more organ failure-free days among survivors compared to dopamine ▪ SS suggest against using IV hydrocortisone to treat septic shock if fluid resuscitation and vasopressor therapy are able to restore hemodynamic stability ▪ SS suggest that either IV hydrocortisone or NO hydrocortisone may be used if adequate fluid resuscitation and vasopressor therapy are not able to restore hemodynamic stability Mortality for children with sepsis ranges from 4% to as high as 50%, depending on illness severity, risk factors, and geographic location Batch Clingy (see Nelson’s 21st Ed., Fig 88.4 for the hypothetical pathophysiology of the septic process) ▪ Alterations in temperature (hyperthermia or hypothermia) ▪ Tachycardia ▪ Tachypnea PPS Oral Exam Reviewer 2020 3 Complex ▪ >90% occur at home ▪ Most involve a single substance ▪ Ingestion accounts for majority of exposures, minority via dermal, inhalational, and ophthalmic routes ▪ 40% involve non drug items (ex. cosmetics, personal care items, cleaning solutions, plants, foreign bodies) ▪ Pharmaceutical preparations account for remainder of exposures, and analgesic, topical preparations, vitamins, and antihistamines are the most reported ▪ Common in <6yo, less common in 6-12yo, 2nd peak occurs in adolescence SIGN ODOR Bitter almonds Acetone Rotten eggs Wintergreen Garlic OCULAR SIGNS Miosis Mydriasis Nystagmus Lacrimation Retinal hyperemia CUTANEOUS SIGNS Diaphoresis Alopecia Erythema Cyanosis (unresponsive to oxygen) ORAL SIGNS Oral burns Gum lines GI SIGNS Diarrhea PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS Common Agents Toxic to <6yo in small doses ▪ Aliphatic hydrocarbons (gasoline, kerosene, lamp oil) – acute lung injury ▪ Antimalarials (Chloroquine, quinine) – seizures, dysrhythmias ▪ Benzocaine – methemoglobinemia ▪ B-adrenergic receptor blockers – bradycardia, hypotension ▪ Ca channel blockers – bradycardia, hypotension, hyperglycemia ▪ Camphor – seizures ▪ Caustics (pH <2 or >12) – airway, esophageal, gastric burns ▪ Clonidine – lethargy, bradycardia, hypotension ▪ Hypoglycemics (sulfonylureas) – hypoglycemia, seizures ▪ Laundry detergent packets – airway issues, respiratory distress, altered mental status ▪ MAO inhibitors – hypertension → delayed CV collapse ▪ Methylsalicylate – tachypnea, metabolic acidosis, seizures ▪ Opioids – CNS depression, respiratory depression ▪ Organophosphate pesticides – cholinergic crisis ▪ Phenothiazines – seizures, dysrhythmias ▪ Theophylline – seizures, dysrhythmias ▪ Tricyclic antidepressants – CNS depression, seizures, dysrhythmia, hypotension DIAGNOSIS APPROACH ▪ Stabilization ▪ Rapid assessment of ABC and mental state ▪ If with altered mental status, serum dextrose concentration should be obtained early, and naloxone should be considered ▪ Targeted history (intentional? Witnessed? Household stress? Brand? Generic? Timing? Symptoms – time and progression?) ▪ Past medical and Developmental history ▪ Social history ▪ Targeted PE ▪ Labs: basic chemistry panel (electrolytes, renal function, glucose), anion gap (especially if with acidosis), ALT AST, INR (especially for acetaminophen overdose), serum creatine kinase (for rhabdomyolysis), serum osmolality (surrogate marker for toxic alcohol exposure), urine pregnancy test (mandatory for all postpubertal females) ▪ Additional lab tests based on presumed poison (ex. Quantitative blood concentrations) ▪ Additional testing: ECG, CXR TOXIDROMES – recognized poisoning syndromes suggestive of toxicity from specific or class of substances TOXIN LABORATORY CLUES IN TOXICOLOGIC DIAGNOSIS Cyanide Isopropyl alcohol, methanol, paraldehyde, salicylates Hydrogen sulfide, sulfur dioxide, methyl mercaptans Methylsalicylate Arsenic, thallium, organophosphates, selenium Opioids, organophosphates, clonidine, phenothiazides, sedative-hypnotics, olanzapine Anticholinergics, sympathomimetics, post-anoxic encephalopathy, opiate withdrawal, cathinones, MDMA Anticonvulsants, sedative-hypnotics, alcohols, PCP, ketamine, dextromethorphan Organophosphates, irritant gas or vapors Methanol Cholinergics, sympathomimetics, withdrawal syndromes Thallium, arsenic Boric acid, elemental mercury, cyanide, carbon monoxide, disulfiram, scombroid, anticholinergics, vancomycin Methemoglobinemia, amiodarone, silver Corrosives, oxalate-containing plants Lead, mercury, arsenic, bismuth Antimicrobials, arsenic, iron, boric acid, cholinergics, colchicine, opioid withdrawal Anion gap metabolic acidosis (Mnemonic = MUDPILES CAT) ▪ Methanol, metformin ▪ Uremia ▪ DKA ▪ Propylene glycol ▪ Isoniazid, iron, massive ibuprofen ▪ Lactic acidosis ▪ Ethylene glycol ▪ Salicylates ▪ Cellular asphyxiants ▪ Alcoholic ketoacidosis ▪ Tylenol Elevated Osmolar Gap ▪ Alcohols: ethanol, isopropyl, methanol, ethylene glycol Hypoglycemia (Mnemonic = HOBBIES) ▪ Hypoglycemics oral: sulfonylureas, meglitinides ▪ Other: quinine, unripe ackee fruit ▪ Beta Blockers ▪ Insulin ▪ Ethanol ▪ Salicylates Hyperglycemia ▪ Salicylates (early) ▪ Ca channel blockers ▪ Caffeine TREATMENT ▪ Poison control center ▪ Majority of exposures in <6yo can be managed without direct medical intervention → product is not inherently toxic, or quantity is not sufficient to produce clinically relevant toxic effects ▪ Substances highly toxic to toddlers in small doses: carbon monoxide, batteries, and analgesics (mainly opioids) POISON PREVENTION EDUCATION – integral part of all well child visit starting 6mos ▪ Exposures in adolescence is usually intentional (suicide, misuse, or abuse of substances) → more severe toxicity MANAGEMENT ▪ Supportive care ▪ Decontamination ▪ Syrup of Ipecac – contains 2 emetic alkaloids that work both in CNS and GI to produce vomiting (but AAP, AACT and AAPCC published statements in favor of abandoning use of ipecac) ▪ Gastric lavage – under best circumstances only removes a fraction of gastric contents hence in most clinical scenarios is no longer recommended ▪ Single dose activated charcoal – most likely effective when given within 1 hour of ingestion ● Substances poorly adsorbed by activated charcoal > alcohols, caustics, cyanide, heavy metals, hydrocarbons, iron, lithium ● Cathartics have been used in conjunction w/ activated charcoal to prevent constipation and accelerate evacuation of charcoal-toxin complex ▪ Whole-Bowel Irrigation ▪ Directed therapy: Antidotal therapy, intralipid emulsion therapy ▪ Enhanced elimination: urinary alkalinization, hemodialysis, multidose activated charcoal COMMON ANTIDOTES POISON Acetaminophen Anticholinergics Benzodiazepines B-blockers Ca channel blockers Carbon monoxide ANTIDOTE N-acetylcysteine Physostigmine Flumazenil Glucagon Insulin Calcium salts Oxygen Batch Clingy ETIOLOGY / INCIDENCE 4 Hematemesis Constipation CARDIAC SIGNS Tachycardia Bradycardia Hypertension Hypotension RESPIRATORY SIGNS Depressed respirations Tachypnea CNS SIGNS Ataxia Coma Seizures Delirium/psychosis Sympathomimetics, anticholinergics, antidepressants, antipsychotics, methylxanthines, salicylates, cellular asphyxiants, withdrawal, serotonin syndrome, neuroleptic malignant syndrome, MDMA, cathinones B-blockers, Ca channel blockers, digoxin, clonidine, organophosphates, opioids, sedative-hypnotics Sympathomimetics, anticholinergics, MAO inhibitors, serotonin syndrome, neuroleptic malignant syndrome, clonidine withdrawal B-blockers, Ca channel blockers, cyclic antidepressants, iron, antipsychotics, barbiturates, clonidine, opioids, arsenic, amatoxin mushrooms, cellular asphyxiants, snake evenomation Cyanide Hypocalcemia ▪ Ethylene glycol ▪ Fluoride Rhabdomyolysis ▪ Neuroleptic malignant syndrome ▪ Statins ▪ Mushrooms (Tricholoma equestre) ▪ Any toxin causing prolonged immobilization (ex. Opioids, antipsychotics) or excessive muscle activity or seizures (ex. Sympathomimetics) Opioids, sedative-hypnotics, alcohol, clonidine, barbiturates Salicylates, sympathomimetics, caffeine, metabolic acidosis, carbon monoxide, hydrocarbon aspiration Alcohols, anticonvulsants, sedative-hypnotics, lithium, dextromethorphan, carbon monoxide, inhalants Opioids, sedative-hypnotics, anticonvulsants, antidepressants, antipsychotics, ethanol, anticholinergics, clonidine, GHB, alcohols, salicylates, barbiturates Sympathomimetics, anticholinergics, antidepressants, cholinergics, isoniazid, camphor, lindane, salicylates, nicotine, tramadol, water hemlock, withdrawal Sympathomimetics, anticholinergics, LSD, PCP, hallucinogens, lithium, dextromethorphan, steroids, withdrawal, MDMA, cathinones Radiopaque substance on KUB (Mnemonic = CHIPPED) ▪ Chloral hydrate, calcium carbonate ▪ Heavy metals (lead, zinc, barium, arsenic, lithium, bismuth) ▪ Iron ▪ Phenothiazines ▪ Play-Doh, potassium chloride ▪ Enteric-coated pills ▪ Dental amalgam, drug packets Digitalis Ethylene glycol, methanol Iron Isoniazid Lead and other heavy metals Methemoglobinemia Opioids Organophosphates Salicylates Sulfonylureas Tricyclic antidepressants Hydroxocobalamin Digoxin-specific Fab antibodies Fomepizole Deferoxamine Pyridoxine BAL (Dimercaprol) Calcium disodium EDTA Dimercapto-succinic acid Methylene blue 1% solution Naloxone Atropine Pralidoxime Na bicarbonate Octreotide and dextrose Na bicarbonate Lead, arsenic, mercury, organophosphates, nicotine Batch Clingy Peripheral neuropathy Arsenic, iron, caustics, NSAIDs, salicylates Lead PPS Oral Exam Reviewer 2020 5 Complex DISEASE ETIOLOGY/INCIDENCE Dog Bites 80-90% 3 categories: abrasions, puncture wounds, lacerations with or without an associated avulsion PPS Recommendations on Animal Bites ▪ Vaccinate pet dogs every year ▪ Families with pet dogs should be given rabies vaccine as preexposure prophylaxis ▪ First aid for bites: wash wound with soap and water for 10 mins → antiseptic (povidone iodine or alcohol) ▪ PREEXPOSURE PROPHYLAXIS: The Rabies Act of 2007 ● Rabies immunization for children aged 5-14 years living in highly endemic areas ● 3 doses: days 0, 7 and 21 or 28 ANIMAL AND HUMAN BITES ▪ POSTEXPOSURE PROPHYLAXIS: ● 5 intramuscular doses (days 0, 3, 7, 14 and 28) OR ● 8 intradermal doses (2 doses of 0.1mL each intradermally on both deltoids on days 0, 3, 7, and 28 or 30) ● The doses beyond d10 may be discontinued if the dog is still alive after 10-14 days DIAGNOSIS Attention to: ▪ Thorough history and PE ▪ Circumstances surrounding bite event ▪ History of drug allergies ▪ Immunization status of child (tetanus) and animal (rabies) PE: Type, size and depth of injury, foreign material in wound, status of underlying structures, location of injury, range of motion of affected area Radiograph if bone or joint was penetrated or fractured Aerobic and anaerobic cultures from infected wound TREATMENT ▪ Irrigation – copious amount of normal saline ▪ Debridement ▪ Wound closure (1o wound closure preferred) Antibiotic therapy for: ▪ Moderate to severe injuries <8h old ▪ Bone or joint space penetration ▪ Deep hand wounds ▪ Immunocompromised patients ▪ Wounds adjacent to a prosthetic joint ▪ Wounds close to genital area ▪ Coverage should include Pasteurella, Eikenella, Staph, Strep and anaerobes ▪ First choice: Co-amoxiclav ▪ No alternative tx for penicillin-allergic patients but can consider: Clindamycin, Doxycycline, Moxifloxacin, and macrolides in pregnant but poor coverage for anaerobes ▪ If IV is necessary: Ampisul, Cefoxitin, Ertapenem, Moxifloxacin ▪ Provide rabies & tetanus vaccination ▪ Elevation (esp. if edema present) ▪ Immobilization (ex. Use splint for hands) ▪ Follow-up (within 48hrs or sooner if worsening) ▪ Reporting COMPLICATION Infection is most common: ▪ Dog bite wounds (80%) ▪ Cat bite wounds (>50%) ▪ Rodent bite (rats 50%, other mammals 25%) ▪ Human bite (high risk) Batch Clingy ▪ There is no single shot rabies vaccine that will offer sufficient protection ▪ Bites and scratches by animal’s paw can transmit rabies, same is true for licks on open wounds and mucus membranes ▪ Rabies vaccination is not routinely given for rat bites because rats, mice, hamsters, guinea pigs and members of the rodent family are not significant carriers of rabies. However, all are tetanus prone. ▪ If a child has been previously vaccinated from a previous dog bite and was bitten again, he will still need rabies vaccine. Since previous vaccine has been completed, he will require only 2 booster doses (on day he was bitten and 3 days later) ▪ Unless a child is infected by the rabies virus, he cannot transmit rabies. Playmates do not need rabies vaccine. CLINICAL MANIFESTATIONS ▪ Dog bites: Crush injury to tissues ▪ Cat and rat bites: Puncture wounds ▪ Human bites: occlusion injury (upper and lower teeth come together on a body par) or a clenched-fist injury (when fist strikes the teeth of another individual) PPS Oral Exam Reviewer 2020 6 Complex ETIOLOGY/INCIDENCE/ PATHOGENESIS INFANTILE HEMANGIOMA ▪ Most common tumor of infancy (5% of NB) ▪ Present at birth or more commonly in the 1 st or 2nd week of life ▪ RF: prematurity, LBW, female, white ▪ Enlarge then spontaneously involute ▪ Most are mixed (superficial + deep components) ▪ Rapid phase of expansion → stationary period → spontaneous involution → regression (anticipated when the lesion develops pale gray areas centrally) → small cosmetic defects (telangiectasia, hypopigmentation, fibrofatty deposits, scar) ▪ 60% reach maximal involution by 5yo, 90-95% by 9yo HEMANGIOMA ▪ Vascular tumors ▪ Exhibit endothelial cell hyperplasia and proliferation Superficial IH ▪ 1st or 2nd month of life; Solitary or multiple ▪ On any area of the body, mostly on the face, scalp, back, anterior chest ▪ May have patterns of facial involvement – frontotemporal, maxillary, mandibular, frontonasal ▪ Erythematous or blue mark or area of pallor → fine telangiectatic pattern → bright, red, protuberant, compressible, sharply demarcated ▪ Burrowing and release of toxic or antigenic substances by the female mite Sarcoptes scabei var. hominis ▪ Most at risk: children and sexual partners of affected individuals ▪ Transmitted rarely by fomites because isolated mite dies within 2-3 days ▪ Gravid female impregnates on the skin surface → exudes a keratolytic substance → burrows into the s. corneum → forms a shallow well within 30 mins → extends this tract by 0.5-5mm/day along the boundary wall with the s. granulosum → deposits 10-25 oval eggs daily → completed egg laying in 4-5wks → female dies within the burrow → eggs hatch in 3-5 days → release of larvae → move to the skin surface → molt into nymph → mature in 23wks → mate → invades the skin PPS Oral Exam Reviewer 2020 LABORATORY FINDINGS/DIAGNOSIS ▪ GLUT-1 – immunohistochemical marker specific for IH COMPLICATIONS ▪ Impairment of a vital function (e.g. on the eyelid interfering with vision) ▪ Ulceration, secondary infection, permanent disfigurement Deep IH ▪ More diffuse, less defined ▪ Cystic, firm, or compressible ▪ Overlying skin may appear normal in color or have a bluish hue MULTIFOCAL INFANTILE HEMANGIOMA ▪ Occur in the skin and/or visceral organs CONGENITAL HEMANGIOMA ▪ Present at birth SCABIES CLINICAL MANIFESTATIONS ▪ GLUT 1 positive ▪ If with >5 cutaneous IH, do abdominal PE and liver UTZ to detect liver IH ▪ Red or blue hued with telangiectasia ▪ May have a ring of pallor ▪ Does not undergo further growth ● Non-involuting CH – stay stable ● Rapidly involuting CH – rapidly decrease in size → fibrofatty residual tissue ● Partially involuting CH – decrease in size to a certain point then remain stable instead of resolving completely ▪ Intense pruritus, particularly at night ▪ 1st sign of infestation: 1-2mm red papules, some excoriated, crusted, or scaling ▪ Classic lesion: threadlike burrows – may not be seen in infants (bullae and pustules); virtually pathognomonic of human scabies ▪ Wheals, papules, vesicles, superimposed eczematous dermatitis ▪ Infants: diffuse eczematous eruption in the scalp, neck, face ▪ Young children: palms, soles, and scalp ▪ Older children and adults: interdigital spaces, wrist flexors, anterior axillary folds, ankles, buttocks, umbilicus, belt line, groin, genitals (men), areola (women); Head, neck, palms, and soles are generally spared Nodular Scabies ▪ less common variant ▪ red-brown nodules in covered areas (axilla, groin, genitals) TREATMENT ▪ Expectant observation if without extensive growth/severe disfigurement ▪ Topical Timolol solution 1 drop BID ▪ Oral propranolol 1-3mkday – 1st line treatment for disfiguring, life- or vision-threatening, or ulcerated IH ● If <8wks old, inpatient initiation of treatment – start at 1mkday TID with HR & BP monitoring 1&2h after each dose → increase to 2mkday TID ● If OP treatment (has good social support & access to hospital), start at 1mkday. If tolerated for 3-7days, increase to 1.5mkday → 2mkday. ● Risks: Hypoglycemia, bradycardia, hypotension, GERD, hyperK, bronchospasm/wheezing, worsening of existing disease ▪ Systemic corticosteroids – if unable to tolerate propranolol or has not responded after weeks of treatment ▪ Intralesional CS injection – rapid involution of IH; risks: ulceration, tissue atrophy, blindness (if used near the orbit) ▪ Systemic propranolol ▪ Good prognosis, low morbidity DDx: Multifocal lymphangioendotheliomatosis ▪ Vascular tumors in the skin and visceral organs ▪ GLUT 1 negative ▪ (+) Severe thrombocytopenia and GI bleeding ▪ High mortality rate ▪ GLUT 1 negative DIFFERENTIAL DIAGNOSIS ▪ Papulovesicular – popular urticaria, canine scabies, chickenpox, viral exanthems, drug eruptions, dermatitis herpetiformis, folliculitis ▪ Eczematous – atopic dermatitis, seborrheic dermatitis ▪ Nodular scabies – urticaria pigmentosa, Langerhans cell histiocytosis COMPLICATIONS If untreated → eczematous dermatitis, impetigo, ecthyma, folliculitis, furunculosis, cellulitis, lymphangitis, id reaction ▪ Permethrin 5% cream – treatment of choice ● Apply to entire body from the neck down, leave on for 812hrs, reapply after 1 week ● In infants <2yo, apply also in the scalp if with lesions above the neck ▪ Sulfur ointment 5-10%, Crotamiton 10% lotion or cream ▪ Ivermectin – 200mcg/kg/dose PO for 2 doses 2wks apart; for severe infestations or in immunocompromised patients ▪ Treat the whole family ▪ Wash clothing, bed linens, and towels in hot water & dry using high heat ▪ If other items cannot be washed, dry clean or store in bags for 37 days OUTCOME ▪ Transmission of mites is unlikely >24h after treatment ▪ If pruritus persists for days to weeks, may be alleviated by topical Batch Clingy DISEASE 7 PEDICULOSIS ▪ Thickened and dystrophic nails ▪ Generalized lymohadenopathy ▪ Eosinophilia CANINE SCABIES ▪ Caused by S. scabei var canis (dog mite) ▪ Acquired by cuddling an infested puppy ▪ Sudden onset, usually 1-10 days after exposure ▪ Secondary to the development of a hypersensitivity reaction to mite antigens ▪ Most common in infants and young children (<5yo) ▪ Caused by phage group 2 staphylococci strains 71 and 55 ▪ Foci of infection: nasopharynx, umbilicus, urinary tract, superficial abrasion, conjunctivae, blood ▪ Hematogenous spread in the absence of specific antitoxin Ab of staphylococcal epidermolytic or exfoliative toxins A or B ▪ Tiny papules, vesicles, wheals, and excoriated eczematous plaques in the arms, chest, abdomen ▪ Burrows are not present – the mite infrequently inhabits the human s. corneum ▪ (+) Pruritus ▪ Localized bullous impetigo to generalized cutaneous involvement with systemic illness ▪ Rash may be preceded by malaise, fever, irritability, exquisite tenderness of the skin ▪ Scarlatiniform erythema – accentuated in the flexural areas ▪ Inflamed, occasionally purulent, conjunctivae ▪ Circumoral erythema; Radial crusting and fissuring around the eyes, mouth, nose ▪ Nikolsky sign - denudation of skin with gentle tangential pressure ▪ Brightly erythematous skin → desquamative phase after 25days → healing without scarring in 10-14 days ▪ Pharyngitis, conjunctivitis, superficial erosions of the lips, intraoral mucosal surfaces are spared ▪ Massive orthokeratosis and parakeratosis with numerous interspersed mites ▪ Psoriasiform epidermal hyperplasia, foci of spongiosis, neutrophilic abscesses ▪ Self-limited since humans are not a suitable host ▪ Bathe and change clothes ▪ Remove or treat the infested animal ▪ Symptomatic therapy for the pruritus Skin Biopsy: ▪ Subcorneal, granular layer split ▪ Absence of an inflammatory infiltrate ▪ Semisynthetic antistaphylococcal penicillin (e.g. nafcillin) ▪ First-generation cephalosporin (e.g. cefazolin) ▪ Clindamycin – used in addition to other agents ▪ Vancomycin – if considering MRSA ▪ Always clean and gently moisten the skin ▪ Emollients – lubrication, decreases discomfort ▪ Recovery is usually rapid COMPLICATIONS ▪ Excessive fluid loss, electrolyte imbalance ▪ Faulty temperature regulation ▪ Pneumonia, septicemia, cellulitis ▪ Head and pubic lice deposit 3-10eggs/day on the human host ▪ Body lice lay eggs in or near the seams of clothing, the ova or nits are glued to hairs or fibers of clothing but not directly on the body ▪ Laying of eggs → ova hatch in 1-2wks → mature in 1wk → larvae die unless a meal is obtained w/in 24h & every few days thereafter → nymphs and adult lice feed on human blood, inject their salivary juice into the host, deposit fecal matter on the skin ▪ Hallmark: pruritus Pediculosis humanus corporis ▪ Small, intensely pruritic, red macule or papule with a central ▪ Improved hygiene ▪ Body/clothing lice; vector of human disease (typhus, trench hemorrhagic punctum ▪ Hot water laundering of all infested clothing and bedding fever, relapsing fever) ▪ Shoulders, trunk, or buttocks ▪ Uniform temp of 65oC for 15-30mins kills all eggs and lice ▪ Vagabond’s skin – if chronic; lichenified, scaling, hyperpigmented plaques, often on the trunk Pediculosis humanus capitis ▪ Translucent 0.5mm eggs near the proximal portion of the hair ▪ Malathion 0.5% in isopropanol ▪ Head lice shaft that are adherent to one side of the shaft and cannot be ● Treatment of choice for resistance to pyrethroids; apply to ▪ Most common form of lice to affect children between 3-12 yo moved along or knocked off the hair shaft with the fingers dry hair until hair and scalp are wet, leave on for 12h. Reapply ▪ Modes of transmission: fomites, head-to-head contact, and ▪ Lice are not always visible, but nits are detectable on the hair after 7-9days if necessary. shared combs, brushes, towels most commonly in the occipital region and above the ears ● Not for use in neonates and infants ▪ Secondary pyoderma (due to trauma from scratching) → hair ▪ Spinosad/Benzyl alcohol lotion – if >6mos old loss ▪ Ivermectin – for difficult-to-treat head lice ▪ Matting together of the hair ▪ Cervical and occipital lymphadenopathy ▪ Treat all household members at the same time ▪ Dermatitis on the neck and pinnae ▪ Remove nits with a fine-toothed comb after application of a ▪ Id reaction – erythematous patches and plaques on the trunk damp towel to the scalp for 30mins ▪ Launder clothing and bed linens in very hot (>130oF) water then dry for at least 10mins at the highest setting ▪ Discard brushes and combs or coat with a pediculicide for 15mins PPS Oral Exam Reviewer 2020 Batch Clingy STAPHYLOCOCCAL SCALDED SKIN SYNDROME (Ritter Disease) NORWEGIAN (CRUSTED) SCABIES ▪ Highly contagious ▪ Occurs in those who are cognitively and physically debilitated, has Down syndrome, poor cutaneous sensation (e.g. leprosy, spina bifida), severe systemic illness (leukemia, DM), and immunosuppressed (HIV) ▪ Mites inhabit the crusts and exfoliating scales of the skin and scalp corticosteroids. ▪ If >2wks still with pruritus & new lesions, reexamine for mites ▪ Scrupulous isolation measures ▪ Removal of the thick scales ▪ Repeated and careful application of permethrin 5% cream ▪ Ivermectin (200-250mcg/kg) x 1 dose in refractory patients 8 Phthirus pubis ▪ Pubic or crab lice ▪ Modes of transmission: skin-to-skin, sexual contact ▪ Common in adolescents MOLLUSCUM CONTAGIOSUM ▪ Poxvirus – has three types, type 1 causes most infections ▪ Acquired by direct contact or from fomites ▪ Spread by autoinoculation ▪ Otherwise well children aged 2-6yrs and those who are immunosuppressed ▪ 80% of cases ▪ Most common precipitant: DRUGS – sulfonamides, NSAIDs, antibiotics, anticonvulsants ▪ Affected BSA <10% ▪ Drug-specific CD8 cytotoxic T cells, perforin/granzyme B, and granulysin trigger keratinocyte apoptosis → expanded enactment of apoptosis STEVEN- JOHNSON SYNDROME ▪ Moderate to severe pruritus ▪ Secondary pyoderma from scratching ▪ Shallow excoriations ▪ Maculae cerulae – steel-gray spots <1cm in diameter on the pubic area, chest, abdomen, thighs ▪ Oval translucent nits firmly attached to the hair shaft ▪ Grittiness when the fingers are run through infested hair ▪ Examine the trunk, thighs, axilla, beard, and eyelashes too since pubic lice may wander or be transferred to other sites on fomites ▪ Incubation period: ≥2 weeks ▪ 1-5mm discrete, pearly, skin-colored, smooth, dome-shaped, papules sometimes with mild surrounding erythema or an eczematous dermatitis ▪ Has a central umbilication from which a plug of cheese material can be expressed ▪ Anywhere in the body, commonly in the face, eyelids, neck, axillae, thighs ▪ In adolescents, may be found in clusters on the genitals or in the groin (may be associated with venereal dse if sexually active) ▪ Lesions may be preceded by a flulike URT illness ▪ Erythematous macules that rapidly and variably develop central necrosis → vesicles, bullae, areas of denudation on the face, trunk, extremities ▪ Accompanied by involvement of ≥2 mucosal surfaces – eyes, oral cavity, upper airway or esophagus, GIT, anogenital mucosa ▪ Burning sensation, edema, and erythema of the lips and buccal mucosa → bullae, ulceration, hemorrhagic crusting ▪ Often, severe pain from mucosal ulceration ▪ Minimal to absent skin tenderness ▪ Disseminated cutaneous bullae and erosions → increased insensible fluid loss → high risk for bacterial superinfection and sepsis then thoroughly clean in boiling water ▪ If object cannot be washed, seal it in a plastic bag for 48 hours ▪ 10-min application of a pyrethrin preparation. Retreat after 7-10 days if necessary. ▪ Petrolatum 3-5x/day for 8-10 days in on the eye lashes ▪ Thoroughly launder or dry clean clothing, towels, and bed linens ▪ Self-limited ▪ Average attack lasts 6-9 months but can persist for years, can spread to distant sites, and may be transmitted to others ▪ Avoid shared baths and towels ▪ Immunotherapy with either candida or trichophyton antigen every 4 weeks until resolution ▪ Liquid nitrogen cryotherapy ▪ Leukocytosis ▪ Elevated ESR ▪ Occasionally, increased LFTs and decreased serum albumin SJS-TEN Overlap Syndrome ▪ 10-30% BSA Toxic Epidermal Necrolysis ▪ >30% BSA ▪ Most severe disorder of the spectrum ▪ Widespread blister formation & morbilliform confluent erythema with skin tenderness ▪ Nikolsky sign in areas of erythema Other DDx: ▪ Urticaria, M. pneumoniae-associated mucositis, drug rash w/ eosinophilia & systemic symptoms (DRESS) syndrome, viral exanthems (e.g. KD) ▪ Mgt is supportive and symptomatic ▪ Discontinue offending drug as soon as possible ▪ Ophthalmologic exam ▪ Cryopreserved amniotic membrane – applied to the ocular surface during the acute phase to limit destruction & prevent long term sequelae ▪ Topical steroid for the eyes ▪ Mouthwash and glycerin swabs – oral lesions ▪ Topical (oral) anesthetics before eating for pain relief ▪ Saline or burrow solution compresses – for denuded skin lesions ▪ Observe vaginal lesions closely ▪ Complete healing may take 4-6 weeks ▪ Some may require ICU admission ▪ Systemic antibiotics for documented urinary/cutaneous infections & for suspected bacteremia (S. aureus/P. aeruginosa) ▪ IVIg 1.5-2g/kg/day for 3 days Batch Clingy COMPLICATIONS ▪ Corneal ulceration, anterior uveitis, panophthalmitis ▪ Bronchitis, pneumonitis, myocarditis, hepatitis, enterocolitis ▪ Polyarthritis, hematuria, ATN → renal failure ▪ Strictures of the esophagus, bronchi, vagina, urethra, anus PPS Oral Exam Reviewer 2020 9 Acute ETIOLOGY/INCIDENCE/ PATHOGENESIS ▪ Infection and inflammation of loose connective tissue, limited dermal involvement, sparing of the epidermis ▪ Secondary to a break in the skin for previous trauma, surgery, or an underlying skin lesion ▪ Most common etiologic agents: S. aureus, S. pyogenes (GAS) ▪ In immunocompromised, may be caused by P. aeruginosa, Aeromonas hydrophila, Enterobacteriaciae, Legionella spp., etc CLINICAL MANIFESTATIONS ▪ Localized area of edema, warmth, erythema, and tenderness ▪ Indistinct lateral margins ▪ May produce pitting when pressure is applied ▪ Regional adenopathy and constitutional SSx (fever, chills, malaise) ▪ S. aureus – more localized and may suppurate ▪ S. pyogenes – spread more rapidly, may be associated with lymphangitis LABORATORY FINDINGS/DIAGNOSIS ▪ In the neonate, do blood CS, LP ▪ Do blood CS if: ● < 1 year old ● (+) Signs of systemic toxicity ● An adequate examination cannot be carried out ● Immunocompromised ▪ Aspirate from the point of maximum inflammation ▪ Ultrasonography DIFFERENTIAL DIAGNOSIS ▪ Skeeter Syndrome – swelling disproportionate to erythema, has pruritus, no tenderness ▪ Cold Panniculitis – erythematous, nontender swelling after cold exposure CELLULITIS COMPLICATIONS ▪ Subcutaneous abscess, bacteremia, osteomyelitis, septic arthritis, thrombophlebitis, endocarditis, necrotizing fasciitis ABSCESS ▪ S. aureus – penetrates abraded perifollicular skin NECROTIZING FASCIITIS FURUNCLE (Boil) ▪ Risk Factors: obesity, hyperhidrosis, maceration, friction, preexisting dermatitis ▪ More common in those with low serum iron levels, diabetes, malnutrition, HIV infection, or other immunodeficiency states ▪ Recurrent furunculosis ● Carriage of S. aureus in the nares, axillae, or perineum, or close contact with someone who is a carrier CARBUNCLE ▪ Infection of a group of contiguous follicles accompanied by inflammatory changes in surrounding tissues ▪ Occur in any location, most common on the lower abdomen, buttocks, and legs ▪ Subcutaneous tissue infection, involves the deep layer of superficial fascia ▪ Polymicrobial, approx. 4 differential organisms ▪ Most common: S. aureus, Streptococcal sp., Klebsiella sp., E. coli, anaerobes ▪ Occur anywhere on the body, mostly perineum and trunk ▪ Most common in the immunocompromised – DM, neoplasia, post-surgery, on IV drugs, on immunosuppressive treatment (steroids) ▪ Can occur in healthy people – after minor puncture wounds, blunt trauma, surgery (abdomen, GIT, GUT, perineum) ▪ Skin changes may appear over 24 – 48hrs as nutrient vessels are thrombosed and cutaneous ischemia develops ▪ Acute onset of local or tense, edema, erythema, tenderness, heat ▪ Fever, pain, & tenderness disproportionate to cutaneous signs ▪ (+/-) Lymphangitis and lymphadenitis ▪ Bullae formation (straw-colored → bluish → hemorrhagic fluid) ▪ Darkening of affected tissues (red → purple → blue) ▪ Skin anesthesia ▪ Frank tissue gangrene and slough ▪ Ominous signs of advanced disease: vesicles, bullae, PPS Oral Exam Reviewer 2020 Infants and children >2mo with mild to moderate infections: ▪ PO as OP with a penicillinase-resistant penicillin (dicloxacillin) or a 1st generation cephalosporin (cefalexin), or clindamycin → shift to IV if no improvement or with significant progression in the 1st 2448h of therapy Infants and children >2mo with signs of systemic infection: ▪ Clindamycin or 1st gen cephalosporin (cefazolin) Unimmunized patients: ▪ Include a 3rd gen cephalosporin (cefepime, ceftriaxone, cefotaxime) or a β-lactam/ β-lactamase inhibitor combination (AmpiSul) to cover for H. influenzae type b & S. pneumoniae ▪ Once erythema, warmth, edema, and fever have significantly decreased, complete a 5- to 7-day total course of treatment ▪ Regular bathing with antimicrobial soaps (chlorhexidine) ▪ Wear loose-fitting clothing ▪ Frequent hot, moist compress – facilitate drainage of lesions ▪ Large lesions should be drained by a small incision ▪ Systemic antibiotics – for carbuncles or large/numerous furuncles ▪ Deep-seated, tender, erythematous, perifollicular nodule → central necrosis and suppuration → rupture, discharge of a central core of necrotic tissue and destruction of the follicle → scar formation ▪ Sites of predilection: hair-bearing areas on the face, neck, axillae, buttocks, groin ▪ Intense pain if the lesion is in an area where the skin is relatively fixed (i.e. EAC, nasal cartilage) ▪ May be accompanied by fever, leukocytosis, and bacteremia ABSCESS ▪ Community-acquired MRSA abscesses can complicate folliculitis ▪ Infection can be spread by crowding conditions, shared personal hygiene items, and a compromised skin barrier TREATMENT Neonates: ▪ IV antibiotics – β-lactamase stable antistaphylococcal antibiotic (nafcillin), Cefazolin, Vancomycin, + aminoglycoside (gentamicin) or a 3rd generation cephalosporin (cefotaxime) ▪ Mupirocin intranasally BID for 5d + Clorhexidine (in lieu of soap) or Diluted bleach baths (1tsp/gallon of water or ¼ cup per ¼ tub) QD for 5d ● To reduce colonization (and hence reinfection) in patients and in family members ▪ Definitive diagnosis is made by surgical exploration – gray necrotic fascia and subcutaneous tissue with little resistance on blunt probing ▪ CT and MRI – aid in delineating the extent and tissue planes of involvement ▪ Frozen section incisional biopsy COMPLICATIONS ▪ Compartment Syndrome – tight edema, pain on motion, loss of distal sensation and pulses; 6P’s (pain, poikilothermia, paresthesia, paralysis, pulselessness, pallor) ▪ Recolonization often occurs within 3mos of decolonization attempt ▪ Incision and drainage + oral antibiotics (7-10 days) ▪ Antibiotics (w/ MRSA coverage is recommended): ● Clindamycin 10-30mkday ● TMP-SMX 8-12mkday based on TMP content ● If >8yo, may give Doxycycline ▪ Remove all devitalized tissues, repeat exploration within 24-36h to confirm that no necrotic tissue remains ▪ Empiric antibiotic therapy ● Gram (+) – vancomycin, linezolid, or daptomycin ● Gram (-) – piperacillin-tazobactam ● May add ceftriaxone/metronidazole to cover mixed aerobicanaerobic organisms ▪ Penicillin + clindamycin – for GAS or Clostridium spp. ▪ Doxycycline + Cipro/Ceftaz – V. vulnificus ▪ Continue antibiotics for at least 5 days after resolution of SSx Batch Clingy DISEASE 10 ▪ Rare acute GAS infection of the deeper layers of the skin and the underlying connective tissue ERYSIPELAS IMPETIGO (Pyoderma) ▪ Most common form of extrapulmonary TB in children ▪ Occur within 6-9months of initial infection by MTB ▪ Tonsillar, anterior cervical, submandibular, supraclavicular nodes – extension of a 1o lesion of the ULF or abdomen ▪ Inguinal, epitrochlear, axillary LNs – from regional lymphadenitis associated with TB of the skin or skeletal system ▪ Discrete, nontender, firm, but not hard LN that gradually enlarge → multiple LNs are infected → matted nodes ▪ Nodes feel fixed to underlying or overlying tissue ▪ Often unilateral → bilateral 2o to crossover drainage patterns of lymphatic vessels in the chest and lower neck ▪ Reactive TST ▪ Normal CXR in 70% of cases ▪ Acute onset, rapid enlargement, tenderness, and fluctuance of LN, with high fever ▪ Ruptured node → draining sinus tract ▪ Typical duration of treatment is 4 weeks ▪ Culture thru needle aspirate of the advancing margin of the inflamed area DIFFERENTIAL DIAGNOSIS ▪ HSV, VZV, Tinea corporis, kerion, arthropod bites, scabies, pediculosis capitis DIFFERENTIAL DIAGNOSIS ▪ Neonates: Epidermolysis bullosa, bullous mastocytosis, herpetic infection, early SSSS ▪ Older Children: Allergic contact dermatitis, burns, erythema multiforme, linear IgA dermatosis, pemphigus, bullous pemphigoid ▪ Definitive diagnosis requires histologic or bacteriologic confirmation best accomplished by fine needle aspiration for culture, stain, and histology ▪ If FNA is unsuccessful, do excisional biopsy ▪ Mupirocin 2% – apply 2-3x/day for 10-14days, localized staph infection ▪ Cephalexin 25-50mkday 3-4x/day for 7days: ● Streptococcal infection ● Widespread staph infection ● Lesions near the mouth, where topical medicationss may be licked off ● Evidence of deep involvement – cellulitis, furunculosis, abscess, suppurative lymphadenitis ▪ Do a blood CS ▪ If MRSA, give clindamycin, doxycycline, or TMP-SMX COMPLICATIONS ▪ Bacteremia with subsequent osteomyelitis, septic arthritis, pneumonia, septicemia ▪ Cellulitis – nonbullous impetigo ▪ Lymphangitis, suppurative lymphadenitis, guttate psoriasis, scarlet fever – streptococcal disease ▪ Acute PSGN – nephritogenic strains of GABHS; common in children 3-7 years old 18-21 days after the skin infection ▪ Can resolve if left untreated but often progresses to caseation and necrosis ▪ Anti-Koch’s therapy, same regimen as PTB (2HRZE, 4HR) ▪ Responds well to anti-Koch’s but LN do not return to normal size for months or years DIFFERENTIAL DIAGNOSIS ▪ Infection caused by nontuberculous mycobacteria ▪ Cat scratch disease, tularemia, brucellosis, toxoplasmosis, pyogenic infection ▪ Non-infectious causes: Tumor, brachial cleft cyst, cystic hygroma Batch Clingy SCROFULA ▪ Most common skin infection in children throughout the world NONBULLOUS IMPETIGO ▪ >70% of cases ▪ Predominant organism: S. aureus; may be caused by GABHS ▪ Staph spread from nose to normal skin then infect the sk ▪ GABHS colonizes the skin 10d before the dev’t of impetigo ▪ May be spread to other parts of the body by fingers, clothing, towels BULLOUS IMPETIGO ▪ Always caused by S. aureus strains that produce exfoliative toxins (ETA, ETB, ETD) ▪ Toxins blister the superficial epidermis ▪ Infants and young children ecchymoses, crepitus, anesthesia, necrosis ▪ May be accompanied by significant systemic toxicity – shock, organ failure, death ▪ Swollen, red, very tender skin ▪ May have superficial blebs ▪ Occasional reddish streaks of lymphangitis projecting out from the margins of the lesion ▪ Abrupt onset with SSx of systemic infection (high fever) ▪ Lesions begin on the face or extremities that have been traumatized (insect bites, abrasions, lacerations, chickenpox, scabies, pediculosis, burns) ▪ Tiny vesicle/pustule → honey-colored crusted plaque ▪ Lesions have little to no pain or surrounding erythema ▪ Occasional pruritus ▪ Regional adenopathy (90%) ▪ Leukocytosis (50%) ▪ Flaccid, transparent bullae on the face, buttocks, trunk, perineum, extremities ▪ Bullae rupture easily → Narrow rim of scale at the edge of shallow, moist erosion ▪ (-) Adenopathy and surrounding erythema Neonatal Bullous Impetigo – can begin in the diaper area PPS Oral Exam Reviewer 2020 11 Acute ETIOLOGY/INCIDENCE/ PATHOGENESIS OPHTHALMIA NEONATORUM ▪ Infants <4mos of age ▪ Most common eye disease of newborns ▪ Acquired during vaginal delivery ▪ Etiologic Agents: ● N. gonorrhea – incubation period: 2-5 days ● C. trachomatis – incubation period: 5-14 days ● P. aeruginosa – acquired in the nursery, potentially serious ▪ May be caused by silver nitrate instillation (mild, self-limited) ▪ Potentially blinding CLINICAL MANIFESTATIONS ▪ Redness and chemosis (swelling) of the conjunctiva, edema of the eyelids, discharge (may be purulent) ▪ Inflammation caused by silver nitrate occurs within 6-12h after birth, clears by 24-48h LABORATORY FINDINGS/ DIFFERENTIAL DIAGNOSIS ▪ Conjunctivitis appearing after 48h should be evaluated for an infectious cause – gram stain and culture N. gonorrhea ▪ Mild inflammation, serosanguineous discharge → thick and purulent in 24hrs → edema of the eyelids, marked chemosis ▪ Gram negative diplococci C. trachomatis ▪ Mild inflammation to severe swelling of the eyelids, copious purulent discharge ▪ Mainly involves the tarsal conjunctivae, rarely affects the cornea CONJUNCTIVITIS ACUTE PURULENT CONJUNCTIVITIS ▪ EA: nontypeable H. influenzae (60-80% associated with ipsilateral OM), pneumoccocci (20%), staphylococci (5-10%) P. aeruginosa ▪ Days 5-18 of edema, erythema of the lids, purulent discharge, pannus formation, endophthalmitis, sepsis, shock, death ▪ Generalized (in 50-75%) conjunctival hyperemia, edema, mucopurulent exudate, glued eyes, & ocular pain and discomfort ▪ Little or no pruritus or periauricular LN enlargement VIRAL CONJUNCTIVITIS ▪ Occur more often in the summer and in older children (>5yo) ▪ Adenovirus – sometimes with pharyngitis/pneumonia ▪ Enterovirus – often hemorrhagic ▪ Watery discharge ▪ Follicular changes (small aggregates of lymphocytes) in the palpebral conjunctiva ▪ Often unilateral, often with periauricular nodes EPIDEMIC KERATOCONJUNCTIVITIS ▪ Adenovirus serotypes 8, 19, 37 ▪ Transmitted by direct contact ▪ Foreign body sensation beneath the lids, itching, burning ▪ Edema (chemosis), photophobia, large oval follicles within the conjunctiva ▪ Preauricular adenopathy, pseudomembrane on the conjunctival surface ▪ Subepithelial corneal infiltrates → blurring of vision (usually disappear but may permanently reduce VA) ▪ May have associated URTI and pharyngitis ▪ Fibrin-rich exudate forms on the conjunctival surface and permeates the epithelium ▪ Membrane is removed with difficulty and leaves raw bleeding areas ▪ The layer of fibrin-rich exudate is superficial and can be stripped easily, leaving the surface smooth MEMBRANOUS CONJUNCTIVITIS ▪ Classic: Diphtheria PSEUDOMEMBRANOUS CONJUNCTIVITIS ▪ Occurs in various bacterial and viral infections – staphylococcal, pneumococcal, streptococcal, or chlamydial conjunctivitis; epidemic keratoconjunctivitis, vernal conjunctivitis, SJS PPS Oral Exam Reviewer 2020 Differential Diagnosis: ▪ Dacryocystitis – caused by NLDO with lacrimal sac distention ▪ Intracytoplasmic inclusions in Giemsa-stained epithelial cells scraped from the tarsal conjunctivae (immunofluorescent staining, chlamydial antigen/DNA) ▪ Hyperacute Bacterial Conjunctivitis ● Caused by gonococcal or meningococcal infection ● Treat with systemic antimicrobials TREATMENT/ COMPLICATIONS/PREVENTION PREVENTION ▪ 0.5% erythromycin or 1% silver nitrate drops at birth ▪ 2% povidone iodine may be given especially in developing countries ▪ Prenatal screening and treatment of maternal gonorrhea ● If untreated, give infant Ceftriaxone 50mg/kg x 1dose IV/IM (max 125mg, decrease dose If preterm) ● If mother’s isolate is penicillin sensitive, give infant Penicillin 50,000 units ▪ Ceftriaxone 25-50mkday x 1dose IV/IM (not to exceed 125mg) ▪ Eye irrigation with saline q10-30mins increasing to 2h intervals until the purulent discharge has cleared COMPLICATIONS ▪ Corneal ulceration and perforation, iridocyclitis, anterior synechiae, panophthalmitis ▪ Erythromycin 50mkday QID PO for 2weeks – cures conjunctivitis and prevents chlamydial pneumonia COMPLICATION Chlamydial Pneumonia (10-20%) ▪ Systemic antibiotics + aminoglycoside ▪ Local saline irrigation ▪ Gentamicin ophthalmic ointment ▪ Warm compress ▪ Topical antibiotics (Table 644.2): ● Aminoglycosides – gentamicin, tobramycin ● Quinolones – ciprofloxacin, ofloxacin, moxifloxacin ▪ Self-limited ▪ Acute Hemorrhagic Conjunctivitis ● Enterovirus CA 24 or 70 ● Red, swollen, painful eyes with a hemorrhagic watery discharge ▪ Prevention is key ▪ Pharyngoconjunctival Fever ● High fever, pharyngitis, bilateral conjunctivitis, periauricular lymphadenopathy ● Highly contagious Batch Clingy DISEASE 12 ALLERGIC CONJUNCTIVITIS ▪ Seasonal (spring – summer) ▪ Intense itching, clear watery discharge, chemosis VERNAL CONJUNCTIVITIS ▪ Begins in the prepubertal years ▪ May recur for many years ▪ Extreme itching and tearing ▪ Large, flattened, cobblestone-like papillary lesions of the palpebral conjunctivae ▪ Stringy exudate, milky conjunctival pseudomembrane ▪ Horner – Trantas Dots – small elevated lesions of the bulbar conjunctivae adjacent to the limbus ▪ Hallmarks: lymphadenopathy and conjunctivitis PARINAUD OCULOGLANDULAR SYNDROME ▪ Bartonella henselae, transmitted from cat to cat by fleas (kittens are more likely than adult cats to be infected) ▪ Humans can become infected when scratched by a cat or may pass from a cat’s saliva to its fur during grooming CHEMICAL CONJUNCTIVITIS ▪ Occurs when an irritating substance enters the conjunctival sac – household cleaning substances, sprays, smoke, smog, metal halide bulbs, industrial pollutants ▪ Alkalis – linger in the conjunctival tissues and continue to inflict damage for hours or days ▪ Acid – precipitate the proteins in tissues; produce their effect immediately ▪ Inflammation of the tissues of the orbit ▪ Mean age: 6.8 years (1wk – 16yo) ▪ Boys > girls (2:1) ▪ EA: GAS, anaerobes, S. aureus, S. pneumoniae, Haemophilus sp. ▪ Direct infection of the orbit from a wound ▪ Hematogenous seeding of organisms during bacteremia ▪ Often, direct extension or venous spread of infection from contiguous sites – lids, conjunctiva, globe, lacrimal gland, nasolacrimal sac, paranasal (ethmoid) sinuses ▪ Spread of infection to the orbit from the sinuses is more prevalent in children due to: ● Thinner bony septa and sinus wall ● Greater porosity of bones ● Open suture lines ● Larger vascular foramina ▪ Spread of infection is facilitated by the venous and lymphatic communication between the sinuses and surrounding structures which allow flow in either direction, facilitating retrograde thrombophlebitis ▪ Self-limited ▪ Thorough and copious irrigation COMPLICATION Extensive tissue damage and loss of the eye – especially if alkali ▪ TRIAD: proptosis, pain limited eye movement, decreased VA ▪ Chemosis, inflammation, and swelling of the eyelids ▪ Feels ill, febrile, appear toxic ▪ Leukocytosis ▪ Increased suspicion for intracranial extension if with HA, vomiting, or focal neurologic deficit ▪ CT Scan with contrast of the orbit and paranasal sinuses ▪ Additional brain imaging if indicated ▪ Lumbar puncture – if with meningitis presentation provided there is no sign of ↑ICP or FND DIFFERENTIAL DIAGNOSIS Idiopathic orbital inflammation, myositis, sarcoidosis, granulomatous vasculitis, leukemia, lymphoma, histiocytic disorders, rhabdomyosarcoma, ruptured dermoid cyst, orbital trauma, orbital foreign body, primary/metastatic tumor in the orbit ▪ Systemic antibiotic therapy: Ampicillin Sulbactam or Clindamycin + Ceftriaxone, Cefepime (or Cefotaxime) ▪ If with suspicion of intracranial extension, Vancomycin + Cefotaxime (or Ceftriaxone) + Metronidazole ▪ If without signs of improvement or if with signs of progression, consider sinus drainage ▪ Orbital or subperiosteal abscess may require urgent drainage of the orbit ▪ If <9yo with medial subperiosteal abscess, treat with IV antibiotics then monitor q6 for signs of visual deterioration or pupillary abnormalities. If with development of such, do drainage COMPLICATIONS ▪ Visual loss secondary to ↑ orbital pressure that causes retinal artery occlusion/optic neuritis ▪ Cavernous sinus thrombosis ▪ Meningitis, epidural/subdural empyema, brain abscess ▪ Optic atrophy, exposure keratitis, retinal/choroidal ischemia Batch Clingy ORBITAL CELLULITIS ▪ Smear of the exudate – (+) eosinophils ▪ Cold compress ▪ Topical antihistamine drops for symptomatic relief ▪ Topical mast cell stabilizers or prostaglandin inhibitors ▪ Topical corticosteroid therapy ▪ Cold compress ▪ Topical mast cell stabilizers or prostaglandin inhibitors – when long-term control is needed PPS Oral Exam Reviewer 2020 13 Acute ETIOLOGY/INCIDENCE/ PATHOGENESIS ▪ Most common cause of acquired hearing loss in children ▪ 1st 2yrs of life (most common at 6-20mos of age) – due to less well-developed immunologic defenses and less favorable eustachian tubal factors ▪ Boys > girls Risk Factors: ▪ Poverty – crowding, limited hygienic facilities, suboptimal nutritional facilities, limited access to medical care, limited resources to buy meds ▪ Tobacco smoke exposure ▪ Repeated exposure to other children ▪ Season – cold weather months ▪ Congenital anomalies ● Unrepaired palatal clefts, submucous cleft palate, other craniofacial anomalies, Down syndrome ● Due to deficiency in eustachian tube function (muscular changes, tubal compliance, defective velopharyngeal valving) → disturbed aerodynamic and hydrodynamic relationships in the nasopharynx and proximal portions of the tubes ▪ Pacifier use, bottle feeding in the recumbent position ▪ HIV infection – high risk of recurrent OM ▪ Breastmilk has a protective effect OTITIS MEDIA Pathogenesis ▪ Hypertrophied NP adenoid tissue/tumor or inflammatory edema of the tubal mucosa (2o to a viral URTI) → eustachian tube obstruction → inflammatory process – secretory metaplasia, compromise of the mucociliary transport system, effusion of liquid into the tympanic cavity → decreased eustachian tube function (ventilation, protection, clearance) ● Protection and clearance – if the eustachian tube is patulous or excessively compliant, it may fail to protect the ME from reflux of infective NP secretions ● Mucociliary clearance – if impaired, contributes to persistent infection ▪ Progressive ↓ in tubal wall compliance + maturation of the innate immune system → ↓ occurrence of OM as they grow older CLINICAL MANIFESTATIONS TM EXAMINATION ▪ Otoscopy ▪ Clearing the EAC ● Children’s ears are “self-cleaning” d/t squamous migration of ear canal skin ● Cleaning with cotton-tipped swabs worsens cerumen impaction ● If obscured by cerumen, remove using an ear curette or gentle suction. If child is old enough to cooperate (≥5yo), lavage then mechanically remove TM FINDINGS ▪ Pars tensa – lower 2/3 of the TM ● Normally, pearly gray, slightly concave ● Abnormal: Full/bulging or extreme retraction ▪ Pars flaccida – more vascular in nature ▪ Normally, the TM is translucent with some degree of opacity in the 1st few MOL ▪ If with later opacification/abnormal whiteness, (+) scarring or underlying effusion ▪ Erythema – if alone and not intense, may be from crying/vascular flushing ▪ If with a persistent focal white area – congenital cholesteatoma ▪ Mobility ● Most sensitive and specific in determining the presence or absence of MEE ● Total absence of mobility – TM perforation after an ↑ in ME pressure associated with effusion ● Substantial impairment of mobility – more common finding in MEE if (-) perforation ▪ Bulging TM – most specific finding of AOM (97%) but less specific (51%) Signs of MEE (≥2 of the following) ▪ White, yellow, amber, or (rarely) bluish TM ▪ Opacification not caused by scarring ▪ Decreased or absent mobility LABORATORY FINDINGS/DIAGNOSIS ▪ Always distinguish between AOM & OME – dictates treatment Tympanometry (Acoustic Immittance Testing) ▪ Offers objective evidence of the presence/absence of MEE but cannot distinguish the effusion of OME from AOM ▪ May supplement PE of difficult to examine patients ▪ Helps confirm, refine, or clarify questionable otoscopic findings ▪ Type A curve ● Steep gradient, sharp-angled peak, ME air pressure that approximates atmospheric pressure ● Normal ME status ▪ Type B curve (flat) ● (+) ME abnormality causing ↓ TM compliance ▪ Type C curve ● Intermediate findings – shallow peak, obtuse-angled peak, or (-) ME air pressure peak ● May or may not be associated with MEE ● Nondiagnostic/equivocal with respect to OM ● Suggest eustachian tube dysfunction & some ongoing ME pathology Recurrent AOM ▪ 3x in 6mos or 4x in 12mos Conjunctivitis – Associated OM ▪ Young children and infants ▪ Often present in multiple family members ▪ Simultaneous appearance of purulent and erythematous conjunctivitis – 2o to nontypeable H. influenzae in most children ▪ Topical ocular antibiotics are ineffective Asymptomatic Purulent OM ▪ No overt signs + an obvious purulent MEE and bulging TM on otoscopy ▪ Treat with antibiotics PREVENTION ▪ Avoid exposure to individuals with respiratory infection ▪ Appropriate vaccination – pneumococcal & influenza ▪ Breast milk feeding ▪ Avoid environmental tobacco smoke TREATMENT ▪ Most crucial aspect of OM treatment: accurate diagnosis ▪ Most episodes of OM will spontaneously resolve ▪ Assess the pain and treat it – Acetaminophen/Ibuprofen Bacterial Resistance ▪ Greatest risk of resistance – <2yo, in regular contact with large groups of children, or recently received antimicrobial treatment INTRATEMPORAL COMPLICATIONS ▪ Infectious Dermatitis ● Contamination from purulent discharge from the ME → infection of the skin of the EAC ● Erythematous, edematous, tender skin ● Mgt: proper hygiene + systemic antibiotics + otic drops ▪ TM Perforation ▪ Chronic Suppurative OM (CSOM) ● Persistent ME infection + discharge through a TM perforation ● EA: P. aeruginosa, S. aureus ● Treatment is guided by microbiologic results ● If without cholesteatoma, parenteral antibiotics + assiduous aural cleansing ● Tympanomastoidectomy for refractory cases ▪ Mastoiditis ▪ Facial Paralysis ● Uncommon, often resolves after IV antibiotics & myringotomy ● If present, requires urgent attention. Prolonged infection → permanent facial paralysis ▪ Cholesteatoma – cyst like growth lined by keratinized, stratified squamous epithelium containing desquamated epithelium and/or keratin Acquired Cholesteatoma – 2o to long-standing chronic OM ● Progressive extension → bony resorption → extend into the mastoid cavity → extend intracranially ● Often found in the superior part of the TM (pars flaccida) ● Otoscopy: area of TM retraction/perforation with white, caseous debris persistently overlying the area ● Treatment: tympanomastoid surgery Congenital Cholesteatoma ● 2o to epithelial inflammation in the ME space during otologic development in utero ● Often in the anterior-superior quadrant of the TM ● Discrete, white opacity in the ME space ● Tx: surgical resection ▪ Labyrinthitis ● Spread of infection from the ME and/or mastoid to the inner ear ● Usual source: cholesteatoma or CSOM ● Vertigo, tinnitus, nausea, vomiting, hearing loss, nystagmus, clumsiness INTRACRANIAL COMPLICATIONS ▪ Meningitis, epidural/subdural abscess, focal encephalitis, brain abscess, sigmoid sinus thrombosis, otitic hydrocephalus ▪ Develop through direct extension, hematogenous spread, or thrombophlebitis ▪ ME infection + any systemic symptom (high grade fever, HA, extreme lethargy) or meningismus or any CNS sign PPS Oral Exam Reviewer 2020 Batch Clingy DISEASE 14 ▪ If suspected, do imaging, LP, ME exudate culture ▪ Myringotomy with tympanostomy tube placement ▪ Otitic hydrocephalus ● Form of idiopathic intracranial HTN or pseudotumor cerebri ● ↑ICP without ventricular dilation occurring in association with acute/chronic OM or mastoiditis ● MRI confirms the diagnosis ● Tx: Antibiotics + acetazolamide/furosemide + mastoidectomy, repeated LP, lumboperitoneal shunt, VP shunt ● If untreated, loss of vision 2o to optic atrophy ACUTE OTITIS MEDIA ▪ EA: nontypeable H. influenzae, S. pneumoniae, M. catarrhalis, GAS, S. aureus, gram-negative organisms, Alloicoccus otitidis ▪ S. aureus & gram-negative organisms – most common in neonates and very young hospitalized infants ▪ Viral pathogens ● Rhinovirus, RSV, influenza, adenovirus, parainfluenza ● Sets the stage for bacterial invasion (if with respiratory infection), amplifies the inflammatory process, interferes with resolution of the bacterial infection; still uncertain whether viruses alone can cause AOM ▪ Complication of bronchiolitis ▪ In young children, ear pain manifested as irritability, change in sleeping/eating habits, holding/tugging at the ear ▪ Fever may be present & is occasionally the only sign ▪ May have systemic symptoms & URTI symptoms ▪ Purulent otorrhea of recent onset ▪ Bullous myringitis – sign of AOM, not an etiologically discrete entity TM Findings ▪ Malleus may be obscured ▪ TM may resemble a bagel without a hole but with a central depression ▪ TM may be obscured or have a cobblestone appearance ▪ AOM Diagnosis (based on the AAP 2013 guidelines): 1) Moderate to severe bulging of the TM or new onset otorrhea not caused by otitis externa 2) Mild bulging of the TM + recent (<48h) onset of ear pain or intense TM erythema A diagnosis of AOM should not be made in a child without MEE. ▪ To support a diagnosis of AOM instead of OME in a child with MEE: 1) Distinct fullness/bulging of the TM may be present ± erythema, or at a minimum, 2) MEE + ear pain that appears clinically important Batch Clingy ▪ See alternative antibiotics in table 658.3 ▪ Duration of treatment: 10days, longer if very young or have severe episodes of whose previous OM has been problematic ▪ Indications for Myringotomy (3rd line treatment for those who failed 2 courses of antibiotics) ● Severe, refractory pain ● Hyperpyrexia ● Complications of AOM – facial paralysis, mastoiditis, labyrinthitis, CNS infection, immunologic compromise from any source ▪ Tympanostomy Tube Placement ● 3eps AOM in 6mos or 4eps in 1yr with 1ep in the preceding 6mos ● If otorrhea occurs post-op, maintain ear canal dry & give ototopical treatment – cipro/dexa, ofloxacin; have excellent coverage of even the most resistant strains + S. aureus & P. aeruginosa PPS Oral Exam Reviewer 2020 15 OTITIS MEDIA WITH EFFUSION ▪ 30% of cases have the same etiologic agents as AOM ▪ Often not accompanied by overt complaints of the child but can be accompanied by hearing loss manifested as changes in speech patterns (often goes undetected if unilateral/mild) ▪ Balance difficulties or disequilibrium ▪ Older children may complain of mild discomfort or sense of fullness in the ear COMPLICATIONS ▪ Predisposition to recurring AOM ▪ Pain; Disturbance of balance; Tinnitus ▪ Pathologic MEE changes ▪TM atelectasis, retraction pocket formation ▪ Adhesive OM; Cholesteatoma formation ▪ Ossicular discontinuity ▪ Conductive and sensorineural hearing loss TM Findings ▪ Absent or slight bulging of the TM ▪ Absent or slight erythema ▪ EA: P. aeruginosa (60%), S. aureus, Enterobacter aerogenes, P. mirabilis, K. pneumoniae, streptococci, CoNS, diphtheroids, fungi (Candida, Aspergillus) ▪ Result from chronic irritation and maceration from excessive moisture in the canal ▪ Other causes: cerumen impaction with trapping of water, trauma, loss of protective cerumen OTITIS EXTERNA (Swimmer’s ear) Necrotizing (malignant) otitis externa ▪ Invasive infection of the temporal bone and skull base ▪ Facial paralysis, other cranial nerve abnormalities, vertigo, SNHL ▪ Seen in the immunocompromised and severely malnourished DIFFERENTIAL DIAGNOSIS ▪ Furunculosis – occur in the lateral hair-bearing part of the ear canal; localized swelling of the canal limited to one quadrant ▪ Otitis Media ▪ Mastoiditis – postauricular fold is obliterated, tenderness is noted over the mastoid and not on movement of the auricle ▪ Fig 657.2 (Flowchart for managing AOE) PREVENTION ▪ Instillation of dilute alcohol or 2% acetic acid immediately after swimming or bathing ▪ Use of hair dryer to clear moisture after swimming ▪ Removed in the office setting without general anesthesia ▪ Gentle irrigation of the ear canal with body temperature water or saline for very small objects (only if the TM is intact) ▪ Large, deeply embedded foreign bodies with associated canal swelling are best removed by an ENT ▪ Disk batteries – remove immediately; leaks a basic fluid that can cause severe tissue destruction ▪ Insects – kill first with mineral oil or lidocaine prior to removal Batch Clingy FOREIGN BODY ▪ Predominant symptom: acute rapid onset (within 48h) of ear pain, often severe, accentuated by manipulation of the pinna or by pressure on the tragus and by jaw motion ▪ Severity of the pain may be disproportionate to the degree of the inflammation – skin of the external ear canal is tightly adhered to the underlying perichondrium and periosteum ▪ Itching – precursor of pain; characteristic of chronic inflammation of the canal/resolving AOE ▪ Conductive hearing loss secondary to 1) edema of the skin & TM, 2) serous/purulent secretions, or 3) canal skin thickening associated with chronic external otitis ▪ Prominent signs of acute disease: ● Edema of the ear canal ● Erythema ● Thick, clumpy otorrhea ▪ TM cannot be adequately visualized since the canal is often tender and swollen (concentric swelling) ▪ If TM is seen, looks normal or opaque ▪ TM mobility may be normal or reduced (if thickened) ▪ Palpable and tender periauricular lymph nodes ▪ Erythema and swelling of the pinna and periauricular skin ▪ Postauricular fold is preserved ▪ Assess for any baseline sensory, physical, cognitive, or behavioral factors risk w/c may signify risk of learning problems (see table 658.4) ▪ Most cases resolve without treatment within 3 months ▪ If MEE persists >3mos, do an age-appropriate hearing test and consider ENT referral ▪ If doing expectant management, examine every 3-6mos until ● (-) Effusion ● Significant hearing loss is identified ● Structural abnormalities of the eardrum/ME is suspected ▪ Antimicrobials – limited to those with associated bacterial URTI or untreated ME infection ▪ Myringotomy + Insertion of Tympanostomy Tubes ● Nearly uniformly reverse the CHL associated with OME ▪ Adenoidectomy ● reduce the risk of recurrence in older children who underwent tube insertion and who continues to have OM after tube extrusion ▪ Otic drops containing: ● Acetic acid with or without HAA ● Neomycin – against gram-positive organisms & some gram-negative organisms (Proteus sp.) ● Polymyxin – against gram-negative bacilli (Pseudomonas sp) ● Quinolone (Ciprofloxacin) with or without HAA ▪ Otic drops should fill the canal and the patient should remain in place for 3-5mins ▪ Oral analgesics (Ibuprofen, Acetaminophen) – for severe pain PPS Oral Exam Reviewer 2020 16 – Fanaroff Acute ETIOLOGY / INCIDENCE / PATHOGENESIS EARLY ONSET (≤ 72 HOL) ▪ Vertical transmission – through ascending amniotic fluid infection or through acquisition of bacterial flora from the mother’s anogenital tract during vaginal delivery ▪ T. pallidum & L. monocytogenes - cross placenta ▪ GBS, E. coli – most common in preterm ▪ Others: S. aureus, viridans group Streptococci, Entero-cocci, GAS, Listeria monocytogenes, Haemophilus, and other enteric Gramnegative organisms, including Klebsiella, Enterobacter, Citrobacter, Acinetobacter, and Pseudomonas SEPSIS ▪ Incidence: 0.77 per 1000 live births SIRS secondary to infection SIRS (≥2 of the ff) ▪ Fever ▪ Hypothermia ▪ Tachycardia ▪ Tachypnea ▪ Hypervent ▪ Abnormally high/low WBC CLINICAL MANIFESTATIONS ▪ Variable, nonspecific ▪ Hyper- or hypothermia, lethargy, or irritability, hypotonia, respiratory distress, cyanosis, apnea, feeding difficulties, poor perfusion, bleeding problems, abdominal distention Metabolic changes Hyper- or hypoglycemia Acidosis Jaundice ▪ Meningismus is uncommon – full fontanels, irritability, lethargy, and seizures Risk Factors ▪ Delivery ▪ Maternal GBS colonization ▪ Maternal fever ▪ Chorioamnionitis, PROM (>18 hours) ▪ Inadequate intrapartum antibiotic administration before delivery ▪ Prematurity and lower birth weight LATE ONSET (>72 HOL – 7th DOL) ▪ Through the infant’s environment ▪ Gut microbiome often involved URINARY TRACT INFECTION ▪ Uncircumcised at risk: enhanced bacterial adherence to foreskin and increased bacterial colonization in the urogenital tract ▪ Neonate: via hematogenous spread/ascending infection often associated with anatomic abnormalities – VUR, ectopic ureter, duplicated collecting system, renal dysplasia, and causes of obstruction, such as posterior urethral valves and uretero-pelvic junction stricture ▪ Additional RF: indwelling urinary catheter E. coli PPS Oral Exam Reviewer 2020 TREATMENT ▪ Ampicillin and aminoglycoside – cover for GBS, E. coli, Listeria WBC count w/ differential ▪ 6th-12thHOL – more likely to reflect pathologic inflammatory response ▪ Leukopenia, high percentage of immature to total WBC (>0.2) ▪ Empiric use of 3rd gen cephalosporins is not recommended d/t resistance and increased risk for invasive candidiasis BUT if gram (-) meningitis is suspected, add cefotaxime CRP ▪ ↑ within 6-8hrs after infection ▪ Peaks after 24hrs ▪ Increases sensitivity over the next 10-12hrs after onset ▪ Preterm have lower CRP values and response Procalcitonin ▪ Peak at 6-8hrs following infection though there is physiologic ↑ in the 1st 24hrs after birth ▪ Not affected by AOG ▪ Better sensitivity but less specific than CRP ▪ Ceftriaxone has potential to displace bilirubin, increased risk for kernicterus, associated w/ biliary sludging Duration: ▪ 10 days – bacteremia without focus ▪ 10-14 days – early preterm ▪ 10-14 days – gram (-) infection ▪ 14-21 days – uncomplicated GBS meningitis ▪ 21 days or 2w beyond 1st negative CSF culture w/c ever is longer – gram (-) meningitis ▪ Ampicillin & Aminoglycoside or Cefotaxime Urine culture ▪ Low yield in early onset sepsis since UTI in NB is primarily caused by renal seeding during bacteremia ▪ Do in late onset sepsis via suprapubic aspiration/sterile catheterization ▪ Extreme prematurity ● Greatest risk factor ● More impaired innate and adaptive immune function hence susceptible Other Risk Factors ▪ Use of H2 blockers and PPIs ▪ GIT pathology ▪ Indwelling foreign body, dependent on parenteral nutrition ▪ CoNS and S. aureus, invasive candidiasis, GBS, E. coli ▪ Term: 0.1 to 1%, preterm: 2%, ▪ Neonate: M>F (rare in the 1st 3 DOL) ▪ 1st 6mos: males decrease, females increase ▪ 1yo: F>M DIAGNOSIS ▪ 1ml blood culture – gold standard ▪ Lumbar puncture ▪ Nonspecific: fever, lethargy, failure to thrive, poor feeding, abdominal distention, vomiting, tachypnea, and cyanosis ▪ Preterm similar to that of term infants, although may also have hypoxia or apnea with bradycardia ▪ Urine culture by catheterization >1000 CFU/mL is meaningful ▪ Suprapubic bladder aspiration Any growth of a urinary pathogen ▪ Blood cultures should be obtained ▪ Urinalysis lacks specificity and sensitivity for dx of UTI in neonates and NOT RECOMMENDED ▪ 3rd gen cephalosporin – for suspected gram (-) meningitis ▪ Vancomycin (instead of ampicillin) – if preterm/infants with complex problems, surgeries, indwelling catheters ▪ Carbapenems – considered if previously given 3rd gen cephalosporin ▪ Empiric antifungal therapy – for invasive candidiasis, severe sepsis unresponsive to antibiotics, chronic TPN, unexplained cytopenias ▪ Empiric: Ampicillin + Aminoglycoside ▪ Vancomycin + Aminoglycoside – for hospitalized infants w/ late onset infection ▪ Ciprofloxacin: for multidrug-resistant, Gram-negative disease or orally when alternative oral antibiotics are not available ▪ Repeat urine CS after 2-3d of treatment if (+) highly resistant pathogen / with known anatomical abnormality ▪ If still (+) evaluate for potential reservoir of infection Duration: 7-14 days ▪ Give IV antibiotics for the entire course if with delayed clinical response, prematurity, bacteremia, or presence COMPLICATIONS / PROGNOSIS ▪ Harm associated with >5d of empiric antibiotics – ↑ risk for NEC, candidemia, mortality among preterm ▪ Early antibiotic exposure may permanently alter the microbiome & predispose to obesity PREVENTION ▪ Universal antenatal screening for GBS colonization ▪ Intrapartum antibiotic prophylaxis PREVENTION ▪ Infection control in the postnatal environment ▪ Hand hygiene ▪ Proper management of central venous catheters ▪ Appropriate use of antibiotics ▪ Limited use of H2 blockers and PPIs ▪ All neonates with UTI should undergo imaging evaluation d/t high prevalence of urinary tract abnormalities ▪ If prenatal ultrasound data are not available, do a renal ultrasound once clinically stable ▪ If renal damage is suggested by ultrasound, do renal cortical scintigraphy to assess for renal scarring ▪ Voiding cystourethrogram ● Detects VUR, done 3-6 weeks after completion of antibiotics Batch Clingy DISEASE 17 ▪ Most common ▪ Can invade & replicate w/in uroepithelial cells Adhesin – ↑ adhesion to urogenital tract cells Fimbrae – promotes persistence of infection Others: ▪ Enterobacteriaceae: Klebsiella, Proteus, Enterobacter, Citrobacter, Salmonella, and Serratia ▪ Gram-positive organisms – less frequent although Enterococci, S. aureus, and coagulase-negative Staphylococci are also known to cause UTI in newborns ▪ More common in the 1st month of life than any other age ▪ 0.3 per 1000 live births in developed countries ▪ 0.8 - 6.1 per 1000 in developing countries Early onset ▪ Within 1st WOL, usually within 72 HOL ▪ Vertically transmitted ▪ Associated w/ complications of labor & delivery ▪ GBS, E. coli ▪ Others: Klebsiella, Enterobacter, Citrobacter, and Serratia species (formation of brain abscess), Listeria monocytogenes (rhombencephalitis) MENINGITIS Late onset ▪ After 1st wk of life ▪ Community/ nosocomial transmission ▪ Nosocomial pathogens: CoNS, Candida, and resistant Gramnegative organisms, particularly Pseudomonas more prominent of anatomic abnormalities ▪ Older infants w/ uncomplicated disease may be switched to oral antibiotics after demonstration of clinical improvement ▪ Nonspecific, similar w/ sepsis ▪ Temp instability (60%) Common neurologic symptoms: Irritability, emesis, lethargy, poor tone, seizures, full but not bulging fontanels, without meningeal signs Other symptoms: Poor feeding, respiratory distress, apnea, diarrhea ▪ Blood culture ▪ CBC with differential ▪ Urine culture if >6DOL ▪ Lumbar puncture for CSF GS/CS, cell count with differential, glucose, and protein (in order of priority) ▪ If CSF findings not definitive in infant whose clinical picture is otherwise suspicious, a repeat LP at 24-48 hours later will still show pleocytosis if true meningeal inflammation is present even with use of antibiotics ▪ Ampicillin + aminoglycoside – early onset/ GBS LONG-TERM OUTCOME ▪ Renal scarring ▪ Subsequent hypertension and CKD ▪ Cefotaxime/higher gen cephalosporin in addition to ampicillin – Gram (-) Neurologic sequelae: Developmental delay, seizures, hydrocephalus, cerebral palsy, blindness, and hearing loss. ▪ Aminoglycoside – not used as monotherapy due to poor CNS penetration 21%-38% – mild deficits 24%-29% – severe neurologic sequelae ▪ If improved & CSF is sterilized, treatment can be narrowed to ampicillin or penicillin monotherapy to continue for 14 days after 1st negative culture Predictors of poor neurologic outcomes: ▪ Seizures >72 hours ▪ Presence of coma ▪ Hypotension requiring inotropes ▪ Leukopenia (<5.0) ▪ Abnormal EEG ▪ If LP is traumatic, not recommended to adjust CSF WBC as this does not improve diagnostic utility for neonates ▪ For gram (-) infection – 3rd gen (cefotaxime) for 21 days or for 14 days after the 1st (-) CSF culture. Add aminoglycoside until CSF sterilization, which usually takes longer for gram (-) meningitis ▪ Consider repeat LP if with complicated clinical course, including seizures, abnormal neuroimaging, prolonged (+) CSF cultures, slow clinical response to treatment ▪ Enterobacteriaceae family with inducible β-lactamase resistance (e.g., Citrobacter, Enterobacter, Serratia) and for Pseudomonas – 4th gen cephalosporin (cefepime) or a carbapenem (meropenem) + aminoglycoside ▪ Listeria monocytogenes, Enterococcus (not susceptible to Ceph) – Ampicillin + amino-glycoside until CSF sterilization, followed by ampicillin monotherapy x 14 days after 1st (-) culture Batch Clingy ▪ CoNS (preterm, foreign body in the CNS) - vancomycin x 14-21 days after CSF sterilization, with removal of any FB if feasible. PPS Oral Exam Reviewer 2020 18 Acute DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS Risk Factors for ↑ indirect bilirubin: Maternal age, race (Chinese, Japanese, Korean, Native American), maternal DM, prematurity, drugs (vitamin K3, novobiocin), altitude, polycythemia, male sex, trisomy 21, cutaneous bruising, blood extravasation (cephalhematoma), oxytocin induction, breastfeeding, weight loss (dehydration or caloric deprivation), delayed bowel movement, family history of, or a sibling who had, physiologic jaundice CLINICAL MANIFESTATIONS / PHYSICAL EXAM ▪ Indirect bilirubin in umbilical cord serum is 1-3 mg/dL ● Rises at a rate of <5mg/dL/day → visible jaundice on D2-3 of life ● D2-4 of life – peaks at 5-6 mg/dL ● D5-7 of life – decrease to <2mg/dL ▪ Breakdown of fetal RBCs + transient limitation in the conjugation of bilirubin by the immature neonatal liver → (↑) bilirubin production DIAGNOSIS ▪ Exclude known causes of jaundice based on the history, clinical findings, and laboratory data Pathologic Jaundice ▪ Jaundice on the 1st 24-36 hrs after birth ▪ Rising faster than 5 mg/dL/day ▪ >12 mg/ dL in a full-term infant (especially in the absence of risk factors) or 10-14 mg/dL in a preterm infant ▪ Persists after 10-14 days after birth, or direct bilirubin fraction is >2 mg/dL at any time TREATMENT / COMPLICATIONS / PROGNOSIS Phototherapy ▪ Bilirubin absorbs light maximally in the blue range (420-470 nm) ▪ Reversible photoisomerization reaction ● Toxic native unconjugated 4Z,15Z-bilirubin → unconjugated configurational isomer, 4Z,15E-bilirubin, which can be excreted in bile without conjugation ▪ Lumirubin – irreversible structural isomer converted from native bilirubin that can be excreted by the kidneys in the unconjugated state ▪ Contraindication: porphyria ▪ AE: loose stools, erythematous macular rash, purpuric rash associated with transient porphyrinemia, overheating, dehydration (increased insensible water loss, diarrhea), hypothermia from exposure, bronze baby syndrome (direct hyperbilirubinemia) IVIG ● 0.5-1.0 g/kg/dose; repeat in 12 hrs ● Reduces the need for exchange transfusion in hemolytic disease PHYSIOLOGIC JAUNDICE Metalloporphyrin Sn-mesoporphyrin (SnMP) ▪ Competitive enzymatic inhibition of the rate-limiting conversion of hemeprotein to biliverdin (an intermediate metabolite in the production of unconjugated bilirubin) by heme-oxygenase Single IM dose if ABO/G6PD jaundice anticipated ▪ AE: transient erythema if the infant is receiving phototherapy BREASTFEEDING JAUNDICE ABO HEMOLYTIC DISEASE BREASTMILK JAUNDICE ▪ 2% of breastfed infants ▪ β-glucoronidase → deconjugation of bilirubin, ↑ enterohepatic circulation ▪ Free fatty acids, pregnanediol → interfere w/ bilirubin conjugation BREASTFEEDING JAUNDICE ▪ Dehydration hemoconcentrates bilirubin ▪ Fewer bowel movement → ↑ enterohepatic circulation ▪ Jaundice after 7th DOL with maximum concentration of 1030 mg/dL at 2nd-3rd wk of life ▪ 20% of cases at risk for ABO mismatch but only 1-10% develops hemolysis because naturally occurring maternal antibodies against ABO antigens almost exclusively IgM and do not cross the placenta ▪ Jaundice in the 1st 24hrs ▪ (-) Pallor and hepatosplenomegaly ▪ Rarely, development of hydrops fetalis or kernicterus ▪ Jaundice on the 1st week after birth ▪ Hyperbilirubinemia - in 10–20% of affected, unconjugated serum bilirubin may reach ≥20mg/dL ▪ Mild anemia and reticulocytosis ▪ PBS: polychromasia, nucleated RBCs, spherocytes Complication: Rarely, kernicterus ▪ Frequent breastfeeding (>10x/day) ▪ Rooming-in with night feeding ▪ Ongoing lactation support ▪ Supplementation with formula or EBM if intake inadequate, weight loss is excessive, (+) dehydration ▪ Phototherapy ▪ IVIG/Exchange transfusion – if severe ▪ pRBC transfusion – may be required at several weeks of age because of hyporegenerative or slowly progressive anemia ▪ Post discharge monitoring of hemoglobin or hematocrit is essential Batch Clingy ▪ Some group O mothers produce IgG Ab against blood group A or B antigens → can cross the placenta → immune-mediated hemolysis Double Volume Exchange Transfusion if intensive phototherapy fails ▪ AE: metabolic acidosis, electrolyte abnormalities, hypoglycemia, hypocalcemia, thrombocytopenia, volume overload, arrhythmias, NEC, infection, graft-versus-host disease, death ▪ If BF is continued, bilirubin gradually decreases but may persist for 3-10wks at lower levels ▪ If BF is discontinued, falls rapidly reaching normal within few days ▪ Phototherapy may be beneficial PPS Oral Exam Reviewer 2020 19 ▪ No G6PD → no NADPH → no reduced glutathione → cells susceptible to oxidative stress → damage induced to cell membrane of RBC → hemolysis ▪ X- linked G6PD DEFICIENCY *see anemia* ▪ Other substances noted in neonates: Naphthalene, herbal medications, henna application, menthol containing umbilical potions ▪ Falling hemoglobin and hematocrit ▪ Increasing reticulocyte count ▪ Hyperbilirubinemia ▪ Parenteral education ▪ Avoidance of known triggers ▪ Phototherapy ▪ Exchange transfusion G6PD testing performed several weeks after acute hemolysis ▪ May give false normal result because older RBCs, depleted in G6PD enzyme activity may be destroyed, leaving younger cells with higher enzyme activity intact G6PD-A – Africa, southern Europe, African americans G6PD Mediterranean – mediterranean countries, middle east, India G6PD Canton – Asia ▪ Hemolysis or impaired conjugation → decreased excretion of bilirubin → ↑ total bilirubin ▪ Neonatal erythrocytes are particularly susceptible to cell injury and Heinz body formation in response to oxidative stress ▪ DIC from sepsis → hemolysis as erythrocytes traverse the deposition of fibrin within the microvasculature ▪ Conjugated hyperbilirubinemia from hepatitis secondary to bacterial, viral, fungal and protozoal infection Batch Clingy SEPSIS *see neonatal fever* G6PD ▪ Major part in stabilization of the RBC membrane against oxidative damage ▪ Catalyzes the 1st step in the HMP pathway, oxidizing glucose6-phosphate to 6-phosphogluconolactone, thereby reducing nicotinamide adenine dinucleotide phosphate (NADP) to NADPH (essential for the regeneration of reduced glutathione from oxidized glutathione, a substance that plays an integral part in the body’s antioxidative mechanisms) ▪ Pathway also instrumental in stimulating catalase, another important antioxidant ▪ Ingestion of or contact w/ fava beans (favism) → severe hemolytic episodes, jaundice and anemia PPS Oral Exam Reviewer 2020 20 Complex ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Occurs in premature, 60-80% in <28 wks AOG ▪ Risk Factors: maternal DM, multiple births, CS delivery, precipitous delivery, asphyxia, cold stress, maternal hx of prev affected infants ▪ Decreased risk: chronic or pregnancy HTN, maternal heroin use, prolonged ROM, antenatal corticosteroid prophylaxis ▪ SURFACTANT DEFICIENCY 2o to decreased production and secretion Surfactant: ● Appears in amniotic fluid bet 28-32wks, mature after 35w ● Contains dipalmitoyl phosphatidylcholine (lecithin), phosphatidylglycerol, apoproteins (SP-A, B, C, D), cholesterol ● Synthesized and stored in type II alveolar cells ● Reduce surface tension, help maintain alveolar stability at end expiration RESPIRATORY DISTRESS SYNDROME ▪ Absence of pulmonary surfactant → ↑ alveolar surface tension → atelectasis-impaired ability to attain adequate FRC ▪ Atelectasis, volutrauma, ischemic injury, oxygen toxicity →injury to epithelial and endothelial cells → effusion of proteinaceous material and cellular debris into the alveolar spaces forming hyaline membranes → impaired oxygenation ▪ Alveolar atelectasis, hyaline membrane formation, interstitial edema → less compliant lungs→ greater pressure required to expand alveoli and small airways ▪ Highly compliant chest walls in preterms → less resistance to natural tendency of lungs to collapse ▪ Perfused but not ventilated alveoli → atelectasis → hypoxia ▪ Decreased lung compliance, small tidal volumes, ↑ physiologic dead space, and insufficient alveolar ventilation → hypercapnia. ▪ Hypercapnia + hypoxia, + acidosis = pulmonary arterial vasoconstriction with ↑ R to L shunting through the foramen ovale and ductus arteriosus and within the lung itself TRANSIENT TACHYPNEA OF THE NEWBORN ▪ 3- 6 per 1000 infants ▪ Most common etiology of tachypnea in NB ▪ Self limited Risk Factors: twin, maternal asthma, late prematurity, precipitous delivery, GDM, CS without labor ▪ Delayed clearance of fetal lung fluid PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS ▪ Within minutes of birth, tachypnea, prominent expiratory grunting, intercostal and subcostal retractions, nasal flaring, cyanosis ▪ Breath sounds – normal or ↓ with a harsh tubular quality, fine crackles on deep inspiration Untreated RDS: progressive worsening of cyanosis and dyspnea, mixed respiratory metabolic acidosis, edema, ileus, oliguria, hypotension, respiratory failure ▪ Apnea & irregular respirations - ominous signs ▪ Signs peak within 3 days after which improvement is gradual DIAGNOSIS ▪ CXR: low lung volumes, diffuse, fine reticular granularity of the parenchyma (ground-glass appearance), air bronchograms ▪ ABG: hyoxemia, hypercapnia, metabolic acidosis ▪ 2DED: if no response to surfactant ▪ Lung profile (lecithin:sphingo- myelin ratio and phosphatidyl- glycerol determination) on a tracheal aspirate – to check surfactant deficiency for atypical RDS PREVENTION ▪ Avoidance of unnecessary or poorly timed early (<39w GA) CS or induction of labor ▪ Appropriate mgt of high-risk pregnancy and labor (antenatal corticosteroids) ▪ Prediction of pulmonary immaturity w/ possible in utero acceleration of maturation Signs of improvement: spontaneous diuresis, improved blood gas at lower inspired O2 levels and/or lower ventilator support ▪ Surfactant: for symptomatic prophylactic (immediately after birth or early rescue (during the 1st few hrs), repeated dosing q612h for 2-4 doses ▪ iNO ▪ Early (1st 10 days) low dose steroid – 1mkd HAA BID x 7d or 0.5mkd x 3d, reduces BPD risk ▪ Dopamine/HAA for hypotension ▪ Ampicillin +aminoglycoside Signs of Respiratory Failure ▪ pH <7.2 ▪ PaCO2 >60mmHg ▪ SaO2 <90% at O2 40-70% and nCPAP of 5-10 ▪ Persistent/severe apnea ▪ Tachypnea, retractions, expiratory grunting, cyanosis relieved by minimal O2 (<40%) ▪ Clear BS TREATMENT ▪ Steroids – give in those with preterm labor between 24-36wks AOG and likely to deliver within 1wk ▪ Maintain PaO2 50-70mmHg ▪ If SaO2 <90% at FiO2 >40-70% apply nCPAP at 5-10cmH2O – reduces collapse of alveoli and improves FRC and VQ match; early use reduces need for MV ▪ INSURE ● Intubate ● Surfactant replacement, then ● Extubate back to nCPAP once the infant is stable (usually within minutes to <1hr) ▪ Mech Vent: if with signs of respiratory failure, use high rate (>60/min) and low tidal volume strategy (4-6mL/kg) ▪ Diagnosis of exclusion CXR: prominent perihilar pulmonary vascular markings, fluid in intralobar fissures and rarely, small pleural effusion ▪ Supportive ▪ Salbutamol ● ↑ expression of ENaC and Na-K-ATPase ● Facilitate fluid clearance ● When given early, improve oxygenation, shorten duration of O2 therapy, expedite COMPLICATIONS / PROGNOSIS COMPLICATIONS OF SURFACTANT ▪ Transient hypoxia, hypercapnia, bradycardia, hypotension, blockage of ETT, pulmonary hemorrhage COMPLICATIONS OF INTUBATION ▪ Pulmonary air leaks, asphyxia (obstruction/ dislodgment of the tube), bradycardia during intubation or suctioning, subglottic stenosis, bleeding trauma, posterior pharyngeal pseudodiverticula, need for tracheostomy, ulceration of the nares (pressure from the tube), permanent narrowing of the nostril (tissue damage and scarring from irritation / infection around the tube), erosion of the palate, avulsion of a vocal cord, laryngeal ulcer, papilloma of a vocal cord, persistent hoarseness, stridor, edema of the larynx. Risks with umbilical arterial catheterization ▪ Vascular embolization, thrombosis, spasm, vascular perforation, ischemic or chemical necrosis of abdominal viscera, infection, accidental hemorrhage, HTN, impairment of circulation to a leg with subsequent gangrene COMPLICATIONS ▪ Hypercapnia, acidosis, RF — uncommon but when it occurs, resolves rapidly (<12-24h) Rapid Recovery Batch Clingy DISEASE 21 ▪ Ineffective expression of ENaC & Na-K-ATPase → slow absorption of fetal lung fluid → ↓ pulmonary compliance and impeded gas exchange ▪ Meconium stained – 10-15% of birth, common in term/post term ▪ 5% requires MV, 3-5% die ▪ Fetal distress and hypoxia occur before passage of meconium recovery ▪ Airway obstruction → respiratory distress within 1st hours, tachypnea (may persist many days or several weeks), retractions, grunting ▪ Partial obstruction → pneumo-mediastinum, pneumothorax, or both CXR: patchy infiltrates, coarse streaking of both lungs, increased AP diameter, flattening of diaphragm ▪ Normal CXR with severe hypoxemia and no cardiac malformation → Pulmonary HTN ▪ Supportive, standard mgt for RD ▪ Exogenous surfactant and/or iNO with MAS and hypoxemic RF or pulmonary HTN requiring MV → ↓ need for ECMO ▪ If no signs of sepsis – no role for antibiotics PROGNOSIS ▪ Depends on extent of CNS injury from asphyxia and presence of associated problems (Pulmonary HTN) ▪ Mortality higher than nonstained ▪ Residual lung problems rare – symptomatic cough, wheezing, persistent hyperinflation for up to 5-10yrs ▪ Overdistention of chest is prominent PREVENTION ▪ Rapid identification of fetal distress and initiation of prompt delivery ▪ Improves within 72 hours but if it requires assisted ventilation, high risk for mortality MECONIUM ASPIRATION SYNDROME PNEUMONIA Fanaroff Congenital Pneumonia ▪ Acquired in utero – aspiration of infected amniotic fluid, ascending infection thru intact/ruptured membranes, hematogenous spread thru the placenta ▪ Presents immediately after delivery ▪ Bacteria: GBS, E. coli (esp preterm), Klebsiella, Enterobacter, GAS, Staphylococcus, and L. monocytogenes, anaerobes such as Bacteroides (occasionally) ▪ Viral: Herpes simplex, adenovirus, enterovirus, mumps, rubella, an unusual presentation of congenital CMV, syphilis and toxoplasmosis ▪ 70% with disseminated candidal infections ▪ Risk Factors: prematurity, PROM, GBS colonization, chorioamnionitis, intrapartum maternal fever PPS Oral Exam Reviewer 2020 ▪ Nonspecific ▪ Temperature instability, lethargy, apnea, tachycardia, metabolic acidosis, abdominal distention, poor feeding, neurologic depression ▪ Respiratory distress – tachypnea, retractions, cough (unusual, <1/3 of neonates) ▪ Delayed diagnosis and treatment → PPHN and septic shock ▪ CXR: nonspecific, unilateral or bilateral streaky densities, confluent mottled opacified areas, or a diffusely granular appearance with air bronchograms ▪ Blood CS, CBC, ↑CRP, neutropenia, immature WBCs, thrombocytopenia ▪ Tracheal GS/CS within the 1st 8 hours of life VAP CRITERIA ▪ Increased ventilator support ▪ Temperature instability ▪ New onset of purulent tracheal secretions or a change in the character of sputum ▪ Leukopenia or leukocytosis ▪ Symptoms of respiratory distress ▪ Bradycardia or tachycardia ▪ New changes in chest radiograph that persist in at least two consecutive films ▪ Ampicillin + Gentamicin ▪ Complete for 10-14 days if uncomplicated pneumonia SUPPORTIVE CARE ▪ Circulatory support – fluids and inotropes, ▪ Mechanical ventilation and O2 – correct hypoxemia ▪ Correct acidosis, hypoglycemia, and electrolyte imbalance ▪ Parenteral nutrition w/ amino acids, carbohydrates, and lipids – adequate nutrition and prevent protein catabolism & amino and fatty acid deficiency. ▪ Feeding should be started ASAP to supply adequate calories. In the absence of respiratory distress or abdominal pathology with concern for aspiration, gavage feedings can be started Batch Clingy EARLY PNEUMONIA (1st 3-7 days) ▪ Aspiration of infected amniotic fluid or bacteria colonizing the birth canal during labor 22 LATE PNEUMONIA (After 7 days) ▪ Ventilator associated pneumonia, nosocomial infection, hematogenous spread ▪ Bacteria: coagulase-negative staphylococci and Staphylococcus aureus, S. pyogenes, S. pneumoniae, E. coli, Klebsiella, Serratia, Enterobacter cloacae, Pseudomonas, Bacillus cereus, and Citro-bacter ▪ Chlamydia trachomatis acquired during labor may present as late as 2-4 wks of life due to long incubation period ▪ Viral: RSV, adenovirus, enteroviruses, parainfluenza, rhinovi-ruses, and influenza viruses ▪ Nonspecific ▪ New-onset or increased apnea, respiratory distress, ↑ oxygen requirement or mechanical support ▪ Abdominal distension or feeding intolerance ▪ Temperature instability ▪ Hyperglycemia ▪ Cardiovascular instability ▪ Vancomycin + Gentamicin or ▪ Vancomycin + Cefotaxime ▪ Nafcillin + Gentamicin – to ↓ vancomycin resistance ▪ If Pseudomonas: aminoglycoside + antipseudomonal β-lactam, (ceftazi-dime, cefepime, or piperacillin-tazobactam) ▪ Uncomplicated pneumonia: 7-10d ▪ Prolonged use of antibiotics and corticosteroids increase risk of candidal pneumonia, especially in ELBW Alveolar overdistention ▪ PPV during resuscitation ▪ Ball valve phenomenon (aspiration, bronchial/bronchiolar obstruction AIR LEAK SYNDROME Spontaneous rupture ▪ Lobar emphysema, congenital lung cyst, pneumatocele Pneumothorax with pulmonary hypoplasia ▪ 1st few hours after birth ▪ ↓ Alveolar surface area and poorly compliant lungs Associated with: ▪ ↓ Amniotic fluid volume – potter syndrome, renal agenesis, renal dysplasia, chronic amniotic fluid leak ▪ ↓ Fetal breathing movement – oligohydramnios, neuromuscular disease ▪ Pulmonary space occupying lesions – diaphragmatic hernia, pleural effusion, chylothorax ▪ Thoracic abnormalities – thoracic dystrophies Pulmonary interstitial emphysema ▪ Gas from ruptured alveolus escapes into the interstitial spaces of lung where it tracks along small conducting airways and dissects along the peribronchial and perivascular connective tissue sheaths to the hilum of the lung ▪ If volume of escaped air great enough, it may collect to the: ● Mediastinal space – pneumomediastinum ● Rupture into pleural space – pneumothorax ● Subcutaneous tissue – subcutaneous emphysema ● Peritoneal cavity – pneumoperitoneum ● Pericardial sac – pneumopericardium PPS Oral Exam Reviewer 2020 Asymptomatic Pneumothorax ▪ Hyperresonance, ↓ ipsilateral sounds ± tachypnea breath Symptomatic Pneumothorax ▪ Respiratory distress, high RR to severe dyspnea, tachypnea, cyanosis, irritability, restlessness, apnea ▪ PE: chest asymmetry with ↑ AP diameter, hyperresonance, ↓ or absent BS, heart and cardiac apex displaced on contralateral side, diaphragm displaced downward, as is the liver causing abdominal distention Pneumomediastinum ▪ Usually asymptomatic ▪ If great, may cause bulging of mid thoracic area, distended neck veins, low BP (tamponade of systemic and pulmonary veins), subcutaneous emphysema (pathognomonic) Pulmonary Interstitial Emphysema ▪ May precede or occur independently with pneumothorax → ↑ PA-aO2 and intrapulmonary shunting → ↓ compliance, hypercapnia, and hypoxemia → ↑ respiratory distress Pneumothorax ▪ Edge of collapsed lung standing out in relied against the pneumothorax Pneumomediastinum ▪ Hyperlucency around the heart border and between the sternum and the heart border ▪ Transillumination – affected side of the thorax transmits excessive light Pulmonary hypoplasia ▪ Signs of uterine compression ▪ Small thorax on CXR ▪ Severe hypoxia and hypercapnia ▪ Signs of primary disease –hypotonia, diaphragmatic hernia, Potter syndrome Pneumopericardium ▪ Sudden shock w/ tachycardia, muffled heart sounds, poor pulses suggesting tamponade ▪ Close observation – small pneumothorax ▪ Frequent small feedings – prevent gastric dilation and minimize crying ▪ 100% O2 – accelerate resorption of free pleural air into blood by reducing the nitrogen tension in the blood and producing a resultant nitrogen pressure gradient from the trapped gas in the blood ▪ Needle thoracentesis – emergency compression for severe distress. Immediately or after catheter aspiration, insert chest tube and attach to underwater seal drainage. ▪ If air leak is ongoing, do continuous suction (-5 to -20cm H2O) ▪ Surfactant – pneumothorax reduces incidence of ▪ Pneumoperitoneum vs Intestinal Perforation – do ABDOMINAL PARACENTESIS ● IP has (+) organism in gram stain Batch Clingy ▪ Risk Factors: mechanical ventilation and central venous lines, abnormal neurologic conditions predisposing to aspiration pneumonia, poor nutrition, severe underlying disease, and prolonged hospitalization ▪ Asymptomatic pneumothorax usually unilateral, 1-2 % of infants ▪ Symptomatic pneumothorax less common ▪ Risk Factors: MAS, RDS, Assisted ventilation (HFV), urinary tract anomalies, oligohydramnios ▪ Most common cause: overdistention → alveolar rupture 23 ▪ ↑ Mediastinal pressure → compressed pulmonary veins at hilum interfering with pulmonary venous return to the heart and cardiac output → pulmonary air embolism, cutaneous blanching, air in IVC, air-filled heart and vessels on chest radiograph → death CONGENITAL DIAPHRAGMA-TIC HERNIA ▪ Major limiting factor for survival: Pulmonary Hypoplasia – ↓ pulmonary mass and the number of bronchial divisions, respiratory bronchioles, and alveoli ▪ Pathology: Abnormal septa in terminal saccules, thickened alveoli, thickened pulmonary arterioles ▪ Biochemical abnormalities: surfactant deficiencies, ↑ glycogen in the alveoli, ↓ phosphatidylcholine, total DNA and total lung protein all of which contribute to limited gas exchange ▪ Associated anomalies: (30%) CNS lesions, esophageal atresia, omphalocele, CV lesions ▪ Chromosomal syndromes: trisomy 21, 13, 18. Fryns, Brachmann-deLange, Pallister-Killian, Turner Syndrome ▪ Respiratory distress – immediately after birth or may have honeymoon period of up to 48 hrs which the baby is stable ▪ Early RD within 6hrs after birth – poor prognostic sign ▪ Scaphoid abdomen, ↑ chest wall diameter, bowel sound heard in the chest, ↓ BS bilateral, point of max cardiac impulse may be displaced ▪ Delayed presentation: vomiting (intestinal obstruction or mild respi sx), incarceration → ischemia, sepsis shock, GBS sepsis ▪ Prenatal UTZ between 16-24wks AOG – polyhydramnios, chest mass, mediastinal shift, gastric bubble or liver in thoracic cavity, fetal hydrops Predictors of outcome ▪ Liver position in the chest, observed to expected total lung volume, and observed to expected lung to head ratio ▪ After delivery, do CXR to confirm the diagnosis ▪ Passage of nasal gastric tube DDX: eventration or cystic lung lesion ▪ Delivery at tertiary center ▪ Endotracheal intubation, NGT/OGT for decompression ▪ Arterial and central venous lines ▪ Preductal arterial SpO2 ≥ 85% ▪ Gentle ventilation with permissive hypercapnia ▪ Avoid Pulmonary HTN ▪ Inotropes – if (+) LV dysfunction ▪ Sedation and paralysis – severe respiratory failure and hypoxemia Ventilation strategy ▪ Conventional MV – PIP<25, PEEP 3-5cm, PaCO2 <65-70 mmHg ▪ HFOV – if PIP>25 is required to maintain ventilation ▪ ECMO – if CV and HFOV fails, used before repair of defect, lasts up to 2-4 wks; goal: oxygenation, CO2 Elimination without inducing volutrauma iNO – reduces ductal shunting and pulmonary pressures and improved ventilation, used as bridge to ECMO Surgical repair – 48 hours after stabilization and resolution of pulmonary HTN ESOPHAGEAL ATRESIA (EA) and TRACHEOESOPHAGEAL FISTULA (TEF) EA ▪ Most common anomaly of the esophagus ▪ 1.7 per 10,000 live births (of these 90% have TEF) ▪ Most common form: Type C – upper esophagus ends in a blind pouch and TEF is connected in distal esophagus EA ▪ Frothing and bubbling at the mouth and nose after birth ▪ Episodes of coughing, cyanosis, and respiratory distress. ▪ Exact cause: unknown ▪ Risk Factors: advanced maternal age, European ethnicity, obesity, low socioeconomic status, and tobacco smoking ▪ 50% nonsyndromic without other anomalies, and the rest have associated Isolated TEF in the absence of EA ▪ “H-type” fistula ▪ Chronic respiratory problems, including refractory bronchospasm and recurrent PPS Oral Exam Reviewer 2020 ▪ Inability to pass an OGT/NGT ▪ Prenatal imaging: absence of the fetal stomach bubble and maternal polyhydramnios ▪ Plain radiography: EA w/ TEF: coiled feeding tube in the esophageal pouch and/or an air-distended stomach Pure EA: airless scaphoid abdomen ▪ Patent airway, pre-operative proximal pouch decompression – prevent aspiration of secretions ▪ Antibiotics – prevent consequent pneumonia ▪ Prone position – minimizes movement of gastric secretions into a distal fistula ▪ Esophageal suctioning - minimizes aspiration from blind pouch ▪ Surgical ligation of the sided thoracotomy ▪ Postop – monitor for worsening pulmonary hypertension, bleeding, chylothorax, bowel obstruction ▪ Overall survival: 71% POOR PROGNOSIS ▪ Major anomaly ▪ Symptoms before 24hrs ▪ Severe pulmonary hypoplasia ▪ Herniation to the contralateral lung ▪ Need for ECMO; size of defect MORBIDITIES Pulmonary problems ▪ Decreased forced expiratory flow at 50% of vital capacity and decreased peak expiratory flow, highest risk: ECMO requiring and patch repair GERD (>50%) ▪ More common in CDH that involves esophageal hiatus Intestinal Obstruction (20%) ▪ Result from midgut volvulus, adhesions, or recurrent hernia that became incarcerated Recurrent diaphragmatic hernia (5-20% ▪ Patch repairs highest risk Delayed growth in the 1st 2y of life ▪ Factors: poor intake, GERD, higher caloric requirement d/t energy required to breathe Neurocognitive defects (67% in ECMO, 24% no ECMO) ▪ Transient and permanent developmental delay, abnormal hearing/vision, seizures ▪ Pectus excavatum ▪ Scoliosis ▪ <1500g at birth and those with severe associated cardiac anomalies – highest risk for mortality COMPLICATIONS OF SURGERY ▪ Anastomotic leak, refistulization, and anastomotic stricture ▪ GERD – from intrinsic abnormalities of esophageal function + delayed gastric emptying → respiratory disease (reactive Batch Clingy Tension pneumothorax ▪ Accumulation of air w/in the pleural space sufficient to elevate intrapleural pressure above atmospheric pressure. Compression of vena cava and torsion of great vessels may interfere with venous return ▪ Communication between the abdominal and thoracic cavities ± abdominal contents ▪ 1:2000 males, 1:5000 females ▪ 90% Bochdalek – posterolateral, 80-90% at left side ▪ 2-6% Morgagni – retrosternal ▪ Others: Hiatal (esophageal), Paraesophageal (adjacent to hiatus) 24 anomalies, most often associated with (VACTERL) syndrome. ▪ Genetic factors have a role as suggested by discrete mutations in syndromic cases: Feingold syndrome (N-MYC), CHARGE syndrome (CHD7), and anophthalmia-esophageal-genital syndrome (SOX2). pneumonias Isolated TEF: esophagogram w/ contrast medium injected under pressure ▪ Bronchoscopy/endoscopy TEF and primary end-to-end anastomosis of the esophagus via R-sided thoracotomy ▪ Preterm: primary closure may be delayed by temporizing with fistula ligation and gastrostomy tube placement airway disease) that complicates EA and TEF and worsens the frequent anastomotic strictures after repair of EA ▪ Tracheomalacia that improves as the child grows Complex DISEASE MECONIUM ILEUS ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Impaction of inspissated meconium in the distal small bowel CLINICAL MANIFESTATIONS ▪ Abdominal distention ▪ Vomiting often bilious ▪ 30% of neonatal intestinal obstruction ▪ Can present as early as in utero with volvulus or perforation, peritoneal ascites, meconium peritonitis, hydrops, fetal loss ▪ Common in cystic fibrosis (CF) PATHOGENESIS ▪ CF → lack of fetal pancreatic enzymes inhibits digestive mechanisms → meconium becomes viscid and mucilaginous MECONIUM PLUG SYNDROME ▪ Doughy or cordlike masses of intestines thru the abdominal wall DIAGNOSIS ▪ (+) NBS for CF → do sweat testing when >2kg, at least 36 wks corrected AOG ▪ Genetic testing confirms CF ▪ Prenatal UTZ: Enlarged bowel loops/mass with distention of the proximal small bowel ▪ Plain radiograph: Small bowel obstruction ▪ At points of heaviest meconium concentration: infiltrated gas may create bubbly granular appearance TREATMENT ▪ High osmolarity gastrografin enema COMPLICATIONS / PROGNOSIS ▪ Most survive neonatal period ▪ If unsuccessful/perforation is suspected: Laparotomy – ileum is opened at largest diameter of impaction, irrigation ▪ If associated with CF: long term prognosis depends on severity of underlying disease ▪ 50% have associated intestinal atresia, stenosis/ volvulus which requires surgery ▪ Some requires bowel resection w/ temporary double barrel enterostomy ff by serial irrigations and distal refeeding / primary anastomosis at initial operation ▪ Intestinal obstruction in the distal colon, rectum, anal canal caused by disproportionately low amount of water in the intestinal lumen ▪ Initial treatment ● Glycerin suppository or ● Rectal irrigation with isotonic saline ▪ Associated w/ small left colon syndrome in IDM, CF, Hirschsprung Disease, maternal opiate use, magnesium sulfate therapy ▪ Gastrografin enema ● Hyperosmolar water soluble, radiopaque solution ● Diagnostic & therapeutic ● Draws fluid rapidly into the intestinal lumen and loosens the inspissated material ▪ Can cause intrauterine intestinal obstruction, meconium peritonitis unrelated to cystic fibrosis ▪ 30% spontaneously resolves ▪ Anorectal plugs → mucosal ulceration from bowel wall erosion and intestinal perforation *see Constipation* Batch Clingy HIRSCHSPRUNG DISEASE PPS Oral Exam Reviewer 2020 25 Complex DISEASE ETIOLOGY / INCIDENCY / PATHOGENESIS Most common life-threatening emergency of the GIT ▪ 5-10% in <1500g ▪ Mortality rate: 20-30% ▪ Increase with decrease BW and AOG CLINICAL MANIFESTATIONS ▪ Onset: 2nd or 3rd wk of life but can be late as 3mos in VLBW DIAGNOSIS ▪ Neutropenia, anemia, thrombocytopenia, coagulopathy, metabolic disease ▪ 1st signs: nonspecific, lethargy, temperature instability, abdominal distention, feeding intolerance, bloody stools ▪ Plain abdominal radiograph Pneumatosis intestinalis ● Air in the bowel wall ● Diagnostic Portal venous gas – severe Pneumoperitoneum—perforation ▪ Cause: multifactorial ▪ Hypotension, respiratory failure common ▪ Major Risk Factors: ● Prematurity ● Bacterial colonization of the gut ● Formula Feeding ▪ NEC in term: secondary disease (history of birth asphyxia, down syndrome, congenital heart disease, rotavirus infection, gastroschisis, Hirschsprung disease) NECROTIZING ENTEROCOLITIS PATHOGENESIS ▪ Mucosal ischemia and subsequent necrosis, gas accumulation in the submucosa of the bowel wall (pneumatosis intestinalis) → perforation, peritonitis, sepsis, death ▪ Most commonly involved: distal part of the ileum and proximal segment of colon ▪ Fatal cases: may extend from stomach to rectum (NEC totalis) ▪ Exposure to metabolic substrate in the context of immature intestinal immunity, microbial dysbiosis and mucosal ischemia ▪ Progression is rapid but unusual to progress from mild to severe after 72 hrs ▪ UTZ with Doppler: evaluate free fluid, abscess, bowel wall thickness, peristalsis, perfusion ▪ DDx: infections, GI obstruction, volvulus, isolated intestinal perforation TREATMENT ▪ Supportive care and prevention of further injury w/ cessation of feeding, nasogastric decompression and IV fluids ▪ Attention to respiratory status, coagulation profile, acid base and electrolyte balances ▪ Do Blood CS then start systemic antibiotics (Gram positive, Gram negative, anaerobes) ▪ Remove umbilical catheters ▪ Assisted ventilation in apnea or abdominal distention contributes to hypoxia and hypercapnia ▪ Crystalloid/ blood products, CV support with fluid boluses and or inotropes and correction of hematologic, metabolic, electrolyte abnormalities ▪ Perforation on radiograph – absolute indication for surgery ▪ Relative indications: Progressive clinical deterioration despite maximum medical mgt Single fixed bowel loop Abdominal wall erythema ▪ Surgery should be performed after intestinal necrosis develops but before perforation and peritonitis. ▪ Options: Primary Peritoneal Drainage or Exploratory Laparotomy with resection of the necrotic intestine and usually stoma creation COMPLICATIONS / PROGNOSIS ▪ Medical management fails in 20-40% of patients with pneumatosis intestinalis at diagnosis, of these 20-50% die. EARLY POSTOP COMPLICATIONS ▪ Wound infection ▪ Dehiscence ▪ Stomal problems – prolapse, necrosis LATE POSTOP COMPLICATIONS ▪ Intestinal strictures (10%) ▪ Short bowel syndrome (malabsorption, growth failure, malnutrition) ▪ Complications related to central venous catheters – sepsis, thrombosis ▪ Cholestatic jaundice ▪ Adverse growth and neurodevelopmental outcomes PREVENTION ▪ Use of human milk, fortification is essential for preterm infants ▪ No clear consensus on use of probiotics, prebiotics and synbiotics ▪ Inhibitors of gastric acid secretion (H2 blockers, PPI) or prolonged empirical antibiotics in early neonatal period increases risk of NEC and should be avoided. Batch Clingy ▪ Genetic predisposition: variants in genes regulating immunomodulation and inflammation (TLR4, IL6), apoptosis and cellular repair (plt activating factor), oxidant stress (VEGF, arginine, nitric oxide) PPS Oral Exam Reviewer 2020 26 Chronic DISEASE DEFINITION PATHOGENESIS ▪ Structural defect arising from a localized error in morphogenesis that results in the abnormal formation of a tissue or organ Caused by: ▪ Gene mutations ● Genes are often transcription factors, part of evolutionarily conserved signal transduction pathways, or regulatory proteins required for key developmental events ▪ Chromosome aberrations and copy number variants ▪ Environmental factors ▪ Interactions between genetic and environmental factors MALFORMATION EXAMPLES Trisomy 21, Teratogens Spondylocostal dysostosis (SCD) ▪ Vertebral segmentation defects assoc w/ other malformations, such as NTDs ▪ Etiologically heterogeneous, mutations in the gene coding for delta-like 3 (DLL3), a ligand of the Notch receptors. APPROACH TO DIAGNOSIS / LABORATORY TESTS Medical History ▪ Pedigree/ family history (3 generation) ▪ Perinatal history ▪ Maternal exposures to teratogenic drugs or chemicals ▪ Natural history of phenotype Smith-Lemli-Opitz syndrome (SLOS) ▪ Mutations in the sterol delta-7-dehydrocholesterol reductase (DHCR7) gene, (enzyme for cholesterol biosynthesis) ▪ Syndactyly in 2nd and 3rd toes; postaxial polydactyly, anteverted nose; ptosis; cryptorchidism; and holoprosencephaly Physical Exam ▪ Evaluate size and formation of body structures ▪ Size and shape of head ▪ Eye position and shape ▪ Categorize defects to major/minor ● Major: significant dysfunction or require surgical correction Rubinstein-Taybi syndrome ▪ Heterozygous, loss-of-function deleterious sequence variants in a gene coding for a transcriptional coactivator called CREB-binding protein (CBP) and from deleterious sequence variants in the EP300 gene ▪ Developmental delays and intellectual disability, broad and angulated thumbs and halluces, and congenital heart disease. Imaging Studies ▪ Full Skeletal Survey – short/disproportionate stature ▪ Brain Imaging – microcephaly, hypotonia, neurologic symptoms ▪ Echocardiography ▪ Renal ultrasonography X-linked lissencephaly ▪ Severe neuronal migration defect, smooth brain with reduction or absence of gyri and sulci in males and intellectual disability and seizures in females ▪ Deleterious sequence variants in DCX protein (regulates the activity of dynein motors that contribute to movement of the cell nucleus during neuronal migration) Diagnosis ▪ Assessment based on specificity ▪ Prioritize rarer selection or pathognomonic findings ▪ Feature index of textbook or computerized database (OMIM) Down syndrome ▪ Extra copy of an entire chromosome 21 or, less frequently, extra copy of the DS critical region on chromosome 21. Chromosome 21 contains 250 genes, and thus DS have an (↑) dosage of the numerous genes encoded by this chromosome that causes their physical differences TREATMENT Anticipatory guidance & medical monitoring for syndrome specific medical risks Genetic counseling, include recurrence risk provision Lab Studies & Genetic Test ▪ Cytogenetic studies w/ Giemsa banded chromosome analysis or karyotyping - GOLD STD ▪ Array CGH and SNP arrays enable copy number variant detection and, in the case of SNP arrays, evaluation of loss of heterozygosity. ▪ Abnormal organization of cells into tissues DYSPLASIA PPS Oral Exam Reviewer 2020 Ethanol ▪ Causes fetal alcohol syndrome (FAS), fetal alcohol spectrum disorder (FASD), or fetal alcohol effects (FAE) ▪ Microcephaly, developmental delays, hyperactivity, and facial dysmorphic features ▪ Toxic to the developing central nervous system (CNS), causes cell death in developing neurons Neurocutaneous melanosis sequence with atypical migration of melanocyte precursor cells from the neural crest to the periphery, manifesting as melanocytic hamartomas of skin and meninges Batch Clingy Neural tube defects ▪ Multifactorial, defects in folate sensitive enzymes or folic acid uptake 27 ▪ Alteration in shape or structure of a structure or organ that has developed, or differentiated, normally Major Intrinsic Causes: ▪ Primary neuromuscular disorders ▪ Oligohydramnios caused by renal defects DEFORMATION Defect resulting from the destruction of a structure that had formed normally before the insult DISRUPTION MULTIPLE ANOMALIES Fetal movement is required for the proper development of the musculoskeletal system, and restriction of fetal movement – can cause MSK deformations Syndrome Pattern of multiple abnormalities that are related by pathophysiology, resulting from a single, defined etiology Sequences Multiple malformations that are caused by a single event, although the sequence itself can have different etiologies Major Extrinsic Causes: ▪ Fetal crowding and restriction of fetal movement ▪ Oligohydramnios from chronic leakage of AF, abnormal shape of amniotic cavity d/t uterine shape, volume of AF and size & shape of fetus, breech Entanglement ▪ Followed by tearing apart or amputation of normally developed structure usually a digit/limb Interruption to the blood supply ▪ Infarction, necrosis, and resorption of structures distal to the insult ▪ If early AOG, usually involves atresia/ absence of body part ▪ Oligohydramnios produces deformations by in utero compression of limbs ● Dislocated hips, equinovarus foot deformity), crumpled ears, or small thorax ▪ Chromosome deletion syndromes may also be identified with specific and sensitive FISH analysis ▪ Molecular testing ▪ Most children with deformations from extrinsic causes are otherwise completely normal, and their prognosis is usually excellent. Correction typically occurs spontaneously. ▪ Deformations caused by intrinsic factors, such as multiple joint contractures resulting from CNS or peripheral nervous system defects, have a different prognosis and may be much more significant for the child ▪ Amnionic membrane rupture sequence → to amputation of fingers/ toes, tissue fibrosis, and tissue bands ▪ The prognosis for a disruptive defect is determined entirely by the extent and location of the tissue loss. ▪ Sanger sequencing – for single nucleotide variants, exons, genes ▪ Panel testing – multiple relevant genes can be interrogated for single nucleotide variants, gene deletions and duplications ▪ Whole Exome Sequencing – examines approx 200,000 exons or 1-2% of DNA that comprises the coding regions of the genome. Performed w/ a trio approach (px and both parents tested simultaneously) so that inheritance pattern or segregation of deleterious sequence variants can be determined thus simplifying analysis ▪ Whole genome sequencing - examines all the DNA content including noncoding regions and includes analysis for cytogenetic rearrangements in addition to copy number loss or gain. Pierre-Robin Sequence ▪ Small jaw (mandibular hypoplasia) → glossoptosis (tongue relatively larger for oral cavity hence drops back) → blocking closure of posterior palatal shelves → U shaped cleft palate DiGeorge sequence of primary 4th brachial arch and 3rd and 4th pharyngeal pouch defects, leading to aplasia or hypoplasia of the thymus and parathyroid glands, aortic arch anomalies, and micrognathia Batch Clingy Association Non random grouping of malformations in which there is an unclear, or unknown, relationship among the malformations, such that they do not fit the criteria for a syndrome or sequence PPS Oral Exam Reviewer 2020 28 Chronic MANAGEMENT ▪ Programs aimed at cognitive training, stimulation, development, education ● Can perform activities of daily living; guardian for complex financial, legal, medical decisions ▪ Anticipatory guidance: screening, evaluation, and care for chronic disorders ▪ Special Olympics: recommend sports participation ● X-ray (full extension & flexion views) of neck → sports w/ hyperextension, radical flexion, pressure on neck/upper spine (swimming, butterfly stroke, diving, pentathlon, high jump, equestrian sports, gymnastics, football, soccer, alpine skinning, warm-up exercise) ● (+) atlantoaxial instability → participation if guardians request; written certification from MD & acknowledgment of risks by guardian Batch Clingy TRISOMY ▪ Most common form of aneuploidy; Can occur in all cells or mosaic ▪ Exhibit consistent and specific phenotype depending on chromosome involved ▪ ↑ with advanced maternal age: women who are ≥35yr at delivery → offer genetic counseling & prenatal diagnosis (serum screening, UTZ, cell-free fetal DNA detection, amniocentesis, chorionic villus sampling) DISEASE EPIDEMIOLOGY/PATHOGENESIS CLINICAL MANIFESTATIONS DIAGNOSIS ▪ Most common type of trisomy in liveborn infants Cognitive impairment + Congenital anomalies + Dysmorphic features ▪ Women in 2nd trim → quad screen: 4 maternal ▪ Most common genetic etiology of moderate intellectual ▪ Developmental delay: universal serum tests (free β-hCG, unconjugated estriol, disability ● Social dev’t relatively spared (but ASD may occur) inhibin, and α-fetoprotein) ▪ Incidence in live births ~1 in 733 → twice at conception ● Difficulty in expressive language ● 80% of pregnancies (5% false positive) (early pregnancy losses) ▪ CHD (50%): AVSD, VSD, isolated secundum ASD, PDA, TOF ▪ 1st trim: fetal nuchal translucency (NT) thickness ▪ ~95%: 3 copies of chromosome 21 ▪ Pulmonary complications: recurrent respiratory infection, sleep-disordered ● Alone: ≤70% detection rate ▪ 97% supernumerary chromosome maternal origin, errors breathing, laryngo- and tracheobronchochomalacia, tracheal bronchus, ● W/ maternal serum β-hCG and pregnancyin meiosis pulmonary HTN, asthma associated plasma protein-A (PAPP-A): 87% ▪ 4%: Robertsonian translocation ▪ Congenital and acquired GI anomalies (celiac disease) ● Integrated screen w/ 2nd trim screening: 95% ▪ 1%: mosaic ▪ Hypothyroidism ▪ Detection of cell-free fetal DNA in maternal plasma ▪ Partial trisomy: only a part of long arm of chromosome 21 ▪ Megakaryoblastic leukemia, immune dysfunction, DM, seizures, alopecia, JIA, ● 98% detection rate; noninvasive; replace TRISOMY 21 in triplicate hearing & vision problems conventional 1st- and 2nd-trimester screens (Down syndrome) ▪ Least common: no visible chromosome abnormality ▪ Alzheimer disease–like dementia: 4th decade (incidence 2-3x higher than ▪ Chromosome analysis indicated in every person sporadic Alzheimer) suspected of having Down syndrome ▪ Most males sterile; some females able to reproduce, 50% chance of having ● Translocation identified → parental chromosome trisomy 21 studies → determine translocation carrier → high ▪ 15%: misalignment of the 1st cervical vertebra (C1) → risk for spinal cord injury recurrence risk for another affected child w/ neck hyperextension or extreme flexion (translocation (21;21) carriers: 100% recurrence risk ▪ ~18-38%: psychiatric comorbidity: inattentiveness, stubbornness, need for for chromosomally abnormal child) routine; aggression & self-injurious behavior less common (respond to educational, behavioral, or pharmacologic interventions) ▪ Life expectancy: ~50-55yo ▪ Not possible to distinguish the phenotypes → mosaic tend to be milder PPS Oral Exam Reviewer 2020 29 TRISOMY 13 (Patau syndrome) ▪ Relatively less common ▪ Characteristic set of congenital anomalies and severe intellectual disability ▪ Prenatal UTZ: IUGR w/ polyhydramnios, abnormal hand positioning ("clenched hands"), choroid plexus cysts (Uptodate) ▪ Intensive tx: resuscitation & surgical procedures → may prolong survival ▪ "Noninterventional paradigm" supportive not intensive tx → high mortality, severe ID in survivors >1yr, lack of cure (not universally accepted) (Uptodate) ▪ Tx depend on type of major/life-threatening abnormalities ▪ Multidisciplinary team ▪ "Noninterventional paradigm" → not universally accepted (Uptodate) Batch Clingy TRISOMY 18 (Edwards syndrome) ▪ Classic triad: micro/anophthalmia, cleft lip and/or palate, and postaxial polydactyly ▪ Prenatal UTZ: characteristic CNS defects ▪ Survive >1yr: severe ID, seizures, failure to thrive (Uptodate) PPS Oral Exam Reviewer 2020 30 ▪ Complete or partial monosomy of X chromosome ▪ Single X maternal origin: 80% ▪ ½ 45,X chromosome complement; ~15% mosaics ▪ ~1 in 1,500-2,500 liveborn female ● assoc w/ spontaneous abortion (~95–99% conceptions miscarried) TURNER SYNDROME ▪ Mechanism of chromosome loss unknown; risk does not increase w/ maternal age ▪ Phenotype → X-linked gene that escape inactivation ▪ SHOX: major locus w/in pseudoautosomal region of the X chromosome (PAR1) ● important for growth in Turner syndrome, Leri-Weill syndrome, idiopathic short stature (rare) ▪ Normal ovarian function: Xp & 2 supergenes on Xq Recombinant human growth hormone ▪ ↑Ht velocity& ultimate stature ▪ Initiated early childhood and/or evidence of ht velocity attenuation (specific Turner syndrome growth curves) ▪ Monitor IGF-1 → significantly ↑, dose of GH ↓ ▪ Can cause excessive growth of the hands and feet ▪ Not significantly aggravate carb tolerance/cause adverse events Oxandrolone ▪ Alone/in combination w/ GH ▪ Synthetic anabolic steroid → weak androgenic effects → monitor for sx of pubarche, hepatotoxicity (rare) Estrogen therapy ▪ Improve verbal and nonverbal memory ▪ Psychological preparedness ● 12yr: normal pace of puberty w/out interfering w/ GH effect on ht ▪ Delay → deleterious to bone health ▪ Glucose tolerance worsens, but fat-free mass, BP & general physical fitness improve ▪ Neurocognitive profile unaffected by estrogen status ▪ Oral estrogen: conjugated estrogen (Premarin) 0.15-0.625mg QD; micronized estradiol (Estrace) 0.5mg QD, 3-6mo ▪ Transdermal patches (bypass 1st hepatic metabolism, ↓ dose): 6.25µg QD gradual ↑ over 2yr to adult dose 100-200µg QD ▪ May be cycled (days 1-23), progestin (Provera) 5-10mg QD on day 10-23 → withdrawal bleeding 1wk after ▪ Combination OCP Psychosocial support ▪ Psych eval: time of dx, behavior/cognition sx, before school entry Batch Clingy sexual infantilism, webbed neck, and cubitus valgus in adult females ▪ Newborn: SGA/LBW, webbing of the neck, protruding ears, lymphedema of the hands and feet, or phenotypically normal ▪ Short stature cardinal finding → chromosomal analysis: considered routinely in short females ● UTZ of heart, kidneys and ovaries after diagnosis ▪ CHD (40%): bicuspid aortic valves, coarctation of the aorta, aortic stenosis, MVP, anomalous pulmonary venous drainage ● Complete cardiac eval (webbed neck ↑ chance of CoA) ▪ Renal anomalies: pelvic kidney, horseshoe kidney, double collecting system, complete absence of 1 kidney, UPJO ● ↑ HTN & UTI ▪ Gonadal dysgenesis ● Process of oocyte loss accelerated (absence of 1 X chromosome; nearly all oocytes gone by 2yr) → gonads streaks of fibrous tissue - early onset of adrenarche (↑DHEAS); but delayed pubarche - lack of secondary sex characteristics; but 10-20% spontaneous breast devt - primary amenorrhea; small % (+)menstrual period & pregnancy - risk of miscarriage, offspring ↑ risk of trisomy 21, premature menopause ● Gonadotropins (particularly FSH): ↑during infancy → progressive ↓ 2-3yr → nadir 6-8yr → ↑(adult castrate level)10-11yr ▪ Autoimmune thyroid disease, w/ or w/out goiter (10–30%) ● Thyroid peroxidase and/or thyroglobulin antibodies → checked periodically; if (+) → thyroxine & TSH ▪ Age-dependent abnormality in carbohydrate metabolism: abnormal glucose tolerance & insulin resistance ▪ Cholesterol levels ↑ ▪ IBD (Crohn disease & ulcerative colitis), GI bleeding (abnormal mesenteric vasculature), delayed gastric emptying ▪ Celiac disease (4–6%): tissue transglutaminase immunoglobulin A antibodies → around age 4yr then q2-5yr ▪ Sternal malformations (lateral chest radiography); congenital hip dysplasia ▪ Most common skeletal abnormalities: shortening of the 4th metatarsal and metacarpal bones, epiphyseal dysgenesis in the joints of the knees & elbows, inadequate osseous mineralization, Madelung deformity, scoliosi s (10%), inadequate osseous mineralization ▪ Eye findings: anterior segment dysgenesis & keratoconus ▪ Pigmented nevi, melanocytic nevi ▪ Recurrent bilateral otitis media (~75%) ▪ Sensorineural hearing deficits common → ↑freq w/ age ▪ Gross and fine motor sensory integration problem → fail to walk before 15mo ▪ Normal intelligence in most; early language dysfunction → raise question about developmental delay ▪ ↑ risk for social isolation, immaturity, anxiety ▪ Dyslexia, nonverbal learning disability, ADHD, deficits in perceptual spatial skills ▪ Gonadoblastoma: 7-10% of Y-positive patients ● Prophylactic gonadectomy (timing being reevaluated) PPS Oral Exam Reviewer 2020 31 MANAGEMENT ▪ Inhibitors of mGluR (overexpressed in fragile X): clinical trials ▪ Minocycline (lowers MMP-9): preliminary trials → short-term improvements in anxiety, mood, and the clinical Global Impression Scale Batch Clingy FRAGILE CHROMOSOME SITES ▪ Regions of chromosomes → tendency for separation, breakage, attenuation under particular growth conditions ▪ At least 120 chromosomal loci (many heritable) identified EPIDEMIOLOGY / PATHOGENESIS CLINICAL MANIFESTATIONS / DIAGNOSIS DISEASE ▪ Distal long arm of chromosome Xq27.3 ▪ Male: intellectual disability, autistic behavior, postpubertal macroorchidism, hyperextensible finger joints, characteristic facial features ▪ 3% of males w/ ID (long face, large ears, prominent square jaw) ▪ FRAXE on Xq28 → mild intellectual disability ▪ Females: varying degrees of intellectual disability and/or learning disabilities ▪ CGG repeat expansion → silence gene producing fragile ▪ DNA testing: expansion of a triplet DNA repeat inside the FMR1 gene on X chromosome >200 repeats FRAGILE X X mental retardation protein (FMRP) → FMRP deficiency ● Involves area of gene that contains number of trinucleotide (CGG) repeats (typically <50 in unaffected) SYNDROME ● upregulates metabotropic glutamate receptor ● Larger triplet repeat expansion = more significant is the intellectual disability (mGluR) 5 pathway & alters the expression of matrix ● Fragile X–associated tremor/ataxia syndrome: males w/ premutation triple repeat expansions (55-200 repeats) → adult, late-onset, metalloproteinase (MMP) 9 progressive neurodegenerative disorder ● Females with premutation triple repeat expansions → ↑risk for premature ovarian failure (POF) PPS Oral Exam Reviewer 2020 32 Complex AMINO ACID DISORDERS ETIOLOGY / INCIDENCE / PATHOGENESIS Classic Homocystinuria ▪ Cystathionine β-synthetase deficiency ▪ Most common IE of methionine metabolism ▪ 1 in 200,000 to 1 in 350,000 live births ▪ Autosomal recessive trait ▪ Gene for cystathionine β-synthase (CBS) is on chromosome 21q22.3. ▪ Most affected patients are compound heterozygotes for 2 different alleles ▪ Heterozygous carriers are asymptomatic HOMOCYSTINURIA Homocysteine -Intermediate compound of methionine -Remethylated to methionine by methionine synthase w/c requires metabolite of folic acid (5-methyltetrahydrofolate) as a methyl donor and a metabolite of vitamin B12 (methylcobalamin) as a cofactor MAPLE SYRUP URINE DISEASE (MSUD) Defects in Methylcobalamin Formation ▪ 7 distinct defects in the intracellular metabolism of cobalamin may interfere with the formation of methylcobalamin → cblC, cblD (including cblD variant 1), cblE (methionine synthase reductase), cblG (methionine synthase), cblF, cblJ, and cblX. ▪ Patients w/ cblC, cblD, cblF, cblJ, and cblX → methylmalonic acidemia + homocystinuria – formation of both adenosyl-cobalamin and methylcobalamin is impaired ▪ Patients with cblE, cblG, and cblD variant 1 defects are unable to form methylcobalamin → homocystinuria only Deficiency of Methylenetetrahydrofolate Reductase (MTHFR) ▪ This enzyme reduces 5,10-methylene- tetrahydrofolate to form 5-methyltetra- hydrofolate, which provides the methyl group needed for remethylation of homocysteine to methionine ▪ Autosomal recessive trait Branched-chain α-keto acid dehydrogenase deficiency ▪ Used in decarboxylation of leucine, isoleucine & valine using thiamine pyrophosphate Vit B1 as coenzyme ▪ This enzyme consists of 4subunits: E1α, E1β, E2, and E3 ▪ E3 subunit is shared with 2 other dehydrogenases, PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS ▪ Birth: asymptomatic ▪ Infancy: nonspecific, FTT & developmental delay ▪ >3yo: ● Subluxation of ocular lens (ectopa lentis) → astigmatism, glaucoma, staphyloma, cataracts, retinal detachment, and optic atrophy ● Progressive intellectual disability, psychiatric and behavioral disorders, seizures ● Skeletal deformities resembling Marfan (tall with elongated limbs and arachnodactyly), scoliosis, pectus excavatum or pectus carinatum, genu valgum, pes cavus, high-arched palate, crowding of the teeth, fair complexion, blue eyes, peculiar malar flush, generalized osteoporosis esp. spine ● Thromboembolic episodes (d/t elevated homocysteine levels → abn angiogenesis and inhibition of fibrinolytic activity) of large & small vessels → optic atrophy, paralysis, cor pulmonale, severe HTN from renal infarcts ● Spontaneous pneumothorax, acute pancreatitis – rare complications ▪ 1st few months of life Nonspecific, vomiting, poor feeding, failure to thrive, lethargy, hypotonia, seizures, developmental delay ▪ Late-onset forms Neurocognitive defects, psychosis, peripheral neuropathy DIAGNOSIS ▪ ↑ Methionine & homocysteine ● Confirmed by molecular analysis of cystathionine B-synthase (CBS) or by assay of the enzyme in cultured fibroblasts, phytohemagglutininstimulated lymphocytes, or liver biopsy ▪ Prenatal Diagnosis DNA analysis or enzyme assay of cultured amniotic cells ▪ Megaloblastic anemia, hyperhomo- cysteinemia, homocystinuria, hypome- thioninemia ▪ DNA testing or by complementation studies in cultured fibroblasts TREATMENT ▪ Dietary protein restriction ▪ Methionine restriction with cysteine supplementation ▪ Vitamin B6 supplementation – high doses 100-500mg/day ▪ Betaine (trimethylglycine, 6g/day for adults or 200-250mkday for children) lowers homocysteine levels in body fluids by remethylating homocysteine to methionine which may result in elevation of plasma methionine levels ▪ Folate (1-5mg/day) and vitamin B12 for vitamin B6–nonresponsive COMPLICATIONS / PROGNOSIS ▪ Vitamin B6 – responsive: fewer complications and later age of onset of complications than w/ vitamin B6– nonresponsive form ▪ Cerebral edema in vit B6-nonresponsive ▪ Vitamin B12 in the form of high dose hydroxycobalamin ▪ Improves w/ treatment ▪ Prenatal Diagnosis Studies in amniotic cell cultures ▪ Renal artery thrombosis, HUS, pulmonary HTN, and optic nerve atrophy have been reported ▪ Apnea, seizure, microcephaly, coma, death ▪ Developmental delay, ataxia, motor abnormalities, peripheral neuropathy, psychiatric manifestations ▪ Thromboembolism also observed ▪ Exposure to nitrous oxide (inhibits methionine synthase) → neurologic deterioration → death ▪ Might present before NBS results are available ▪ Difficulty feeding, vomiting, lethargy → coma, hypertonicity, muscular rigidity, opisthotonic posturing, convulsions → death ▪ Periods of hypertonicity may alternate w/ bouts of ▪ Moderate homocystinemia and homocystinuria ▪ Methionine concentration is low or low-normal (vs classic) ▪ Confirmed by molecular analysis of MTHFR ▪ Cultured fibroblasts or leukocytes. Prenatal Diagnosis Molecular analysis of MTHFR or measuring MTHFR enzyme activity in chorionic villus cells/amniocytes ▪ Ketoacidosis, hypoglycemia ▪ Peculiar odor of maple syrup in urine, sweat, and cerumen ▪ Amino acid analysis – markedly ↑ leucine, ▪ Folic acid + vitamin B6 + vitamin B12 + methionine + betaine ▪ Hydration and hemodialysis ● Emergency treatment for symptomatic neonates ● Rapid removal of the BCAAs & their metabolites from tissues & body fluids ▪ Improved intellectual outcome expected if treatment is initiated before first crisis ▪ Developmental delay in severe cases ▪ Recurrent episodes of ketoacidosis, cerebral edema and death especially when Batch Clingy DISEASE 33 Phenylalanine hydroxylase deficiency or Tetrahydrobiopterin (BH4) (cofactor of PAH) biosynthesis or recycling defects PHENYLKETONURIA (PKU) ▪ Excess phenylalanine metabolized to phenylketones (phenylpyruvate & phenylacetate) → excreted in urine ▪ PAH converts PA to tyrosine (for production of the NTs epinephrine, NE, Dopa) ▪ High levels of PA saturate transport system across BBB → inhibited cerebral uptake of other large neutral amino acids (branched chain AAs, tyrosine, and tryptophan) → impaired brain protein synthesis TYPE I: Fumarylacetoacetate hydrolase deficiency ▪ Liver, kidney & nerve damage caused by metabolites of tyrosine degradation esp. fumarylacetoacetate & succinylacetone TYROSINEMIA ▪ Autosomal recessive trait ▪ FAH gene maps to chromosome 15q25.1 ▪ Common in French Canadians in Quebec PPS Oral Exam Reviewer 2020 flaccidity → boxing & bicycling repetitive movements of extremities ▪ ↑ Leucine competitively inhibits the uptake of other amino acids by the large neutral amino acid (LNAA) transporter. Once taken by the brain tissue, leucine is metabolized by BCAA amino transferase to ⍺ketoisocaproic acid → disrupted metabolism of neurotransmitters and amino acids glutamate, GABA, glutamine, alanine and aspartate ▪ Can reversibly inhibit oxidative phosphorylation → cerebral lactic acidosis → detrimental to neurons & glia → acute encephalopathy & brain edema (Leucinosis) isoleucine, valine & alloisoleucine, ↓ alanine ▪ At birth: Asymptomatic ▪ After a few months: Microcephaly, seizures, pale pigmentation, seborrheic, eczematoid rash ▪ Later years: Abnormal posturing, purposeless hand movements, rhythmic rocking, athetosis, mental retardation, behavioral or psychiatric disturbances, low bone mineral density, osteopenia ▪ Musty/Mousy Odor Severe PAH deficiency/Classic PKU: ▪ Plasma phenylalanine >20mg/dL Patients with BH4 defects ▪ Additional neurologic problems 2o to dopamine and serotonin deficiency ▪ Extrapyramidal signs, hypersalivation, swallowing difficulties ▪ Sx progressive & marked diurnal fluctuation ▪ Hyperprolactinemia d/t hypothalamic dopamine deficiency ▪ Might present before NBS results are available. Most at 2-6 mos of age. ▪ Earlier presentation, poorer prognosis BH4 defects ▪ Measure Neopterin & Biopterin levels ▪ ↓ Dopamine & serotonin metabolites in CSF analysis ▪ BH4 loading test with oral BH4 20mg/kg – normalize PA & PA: tyrosine ratio within 4-12hrs ▪ Molecular testing & enzyme assay ▪ Severe liver failure, jaundice, ascites, and bleeding diathesis – herald onset of disease, precipitated by intercurrent illness that produces catabolic state ▪ Boiled cabbage odor – due to ↑ methionine metabolites ▪ Peripheral neuropathy resembling acute porphyria, triggered by minor infection, severe pain in the legs → extensor hypertonia of neck & trunk, paralytic ileus, marked weakness → respiratory failure, seizures ● Crisis lasts 1-7 days but recuperation from paralytic crises require weeks to months ▪ Renal Fanconi syndrome (hyperphospha- turia, hypophosphatemia, normal AG metab acidosis) → vitD resistant rickets, nephromegaly, nephrocalcinosis ▪ Urine – ↑ Leucine, isoleucine, valine and their ketoacids (few drops of 2,4-dinitrophenylhydrazine reagent (0.1% in 0.1N HCl) to urine → yellow ppt of 2,4-dinitrophenylhydrazone) ▪ Neuroimaging: cerebral edema (cerebellum, dorsal brainstem, cerebral peduncle & internal capsule) → after recovery, hypomyelination & cerebral atrophy Mild hyperphenylalaninemia: ▪ Between 2-10mg/dL PAH deficiency ▪ Tandem mass spectrometry ▪ Blood PA level rise as early as 4hrs after birth ▪ NBS obtained in 1st 24-48hrs of life after feeding protein ▪ ↑ Succinylacetone in serum and urine ▪ ↑ AFP ▪ ↑ Liver transaminases ▪ ↓ Liver synthesized coagulation factors ▪ Hypoglycemia hyperphosphaturia, hypophosphatemia, generalized aminoaciduria Prenatal screening Measurement of succinylacetone in AF ▪ Sufficient calories & nutrients – reverse catabolic state ▪ Mannitol, diuretics, hypertonic saline – if with Cerebral edema ▪ Supplementation w/ isoleucine & valine – compete with leucine for the LNAA transporter at the BBB → ↓ leucine entry into CNS and help in the prevention and tx of leucine encephalopathy ▪ Dietary protein restriction ▪ Leucine, isoleucine, valine restriction ▪ Thiamine supplementation (10-100 mg/day) for thiamine-responsive px ▪ Liver transplantation ▪ Dietary protein restriction ● Start at PA level >10mg/dL or if persistent >6mg/dL ▪ Phenylalanine restriction – maintain level at 2-6mg/dL ▪ Sapropterin dihydrochloride – a synthetic BH4 analogue; for responsive patients ▪ Administration of LNAAs ▪ Some may require sapropterin dihydrochloride plus dopa/carbidopa, 5hydroxytryptophan, and/or other medications “e” Mgt for Symptomatic Neonates: ▪ Dietary protein restriction ▪ Phenylalanine and tyrosine restriction ▪ NTBC (nitisinone) – selective enzyme inhibitor of the tyrosine degradative pathway ▪ Liver transplantation when HCC is suspected ill, surgery or fasting ▪ Acrodermatitis enteropathica –in infants with very low plasma isoleucine/valine ▪ Normal development if diet is instituted early although a mild decrease in IQ and behavioral difficulties relative to unaffected sibs ▪ Patients with biopterin defects: ↑ risk for neurologic problems (seizures, dystonia) ▪ Liver disease could progress despite dietary treatment ▪ NTBC treatment improves liver, kidney, neurologic function, and reduces risk for hepatocellular carcinoma ▪ Liver transplantation might still be required Prenatal diagnosis DNA analysis of amniocytes or of chorionic villi if familial pathogenic variants are known Batch Clingy pyruvate dehydrogenase and α-ketoglutarate dehydrogenase ▪ Deficiency of any of these subunits causes MSUD 34 ▪ Rare autosomal recessive disorder ▪ Caused by tyrosine aminotransferase (TAT) gene pathogenic variants, causing deficiency of cytosolic TAT activity in liver ▪ TAT maps to chromosome 16q22 TYPE III: 4-Hydroxy-phenylpyruvate deficiency ▪ Age of presentation: 1-17 mos ▪ Autosomal recessive dioxygenase GENERALITIES ▪ Catabolism of amino acids → ammonia production, which in high concentration is toxic to the CNS ▪ Mammals detoxify ammonia to urea through a series of reactions known as the urea cycle which is composed of 5 enzymes: carbamoyl phosphate synthetase 1 (CPS1), ornithine transcarbamylase (OTC), argininosuccinate synthetase (ASS), argininosuccinate lyase (ASL), and arginase 1 ▪ A 6th enzyme, N-acetylglutamate (NAG) synthetase (NAGS), catalyzes synthesis of NAG, which is an obligatory activator (effector) of the CPS1 enzyme ▪ 1 in 35,000 live births ▪ Most common genetic causes of hyperammonemia in infants Goals of Treatment in Acute Hyperammonemia ▪ Removal of ammonia from the body in a form other than urea ▪ Minimize endogenous protein breakdown and favor endogenous protein synthesis by providing adequate calories & essential amino acids DISEASE ARGININEMIA ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Arginase deficiency ▪ Autosomal recessive ▪ 2 genetically distinct arginases in humans: Cytosolic (ARG1) in liver & erythrocytes, (ARG 2) in renal & brain mitochondria PPS Oral Exam Reviewer 2020 ▪ Developmental delay, seizures, intermittent ataxia, selfinjurious behavior ▪ Liver and renal abnormalities absent ▪ Assay of plasma tyrosine concentration: marked hypertyrosinemia (>500μmol/L and may reach 1,100-2,750μmol/L) ▪ 4-hydroxyphenylpyruvic acid and its metabolites are ↑ in urine ▪ Phenylalanine and tyrosine restriction ▪ Eye and skin lesions resolve with treatment and intellectual outcome improves ▪ Sustained moderate ↑ in plasma tyrosine (350-700 mmol/L on a N diet) ▪ (+) 4-hydroxyphenylpyruvic acid and its metabolites in urine ▪ Low-phenylalanine, low-tyrosine diet ▪ Trial of Vitamin C – cofactor of 4-HPPD ▪ Improved intellectual outcome UREA CYCLE DISORDERS ACUTE TREATMENT OF HYPERAMMONEMIA ▪ IVF + electrolytes + glucose (10–15%) + lipids (1-2 g/kg/day) + minimal protein (0.25g/kg/day) ▪ Kidneys clear ammonia poorly, its removal from the body must be expedited by formation of compounds with a high renal clearance ▪ Acylation therapy – uses an exogenous organic acid that is acylated endogenously with nonessential amino acids to form a nontoxic compound with high renal clearance. Eg. sodium salts of benzoic acid and phenylacetic acid. ▪ Benzoate forms hippurate with endogenous glycine in the liver ▪ Phenyl acetate conjugates with glutamine to form phenylacetylglutamine, which is readily excreted in the urine. ▪ IV infusion of arginine, effective in all patients (except with arginase deficiency). It supplies the urea cycle with ornithine which leads to formation of citrulline & arginosuccinate which are less toxic than ammonia and more readily excreted by the kidneys ▪ Hemodialysis – if initial ammonia level is <500 μmol/L & if ongoing treatment fails within 4-6hrs ▪ Extracorporeal Detoxification – if >500 μmol/L ▪ Exchange Transfusion –if dialysis cannot be employed promptly or when the patient is a newborn with hyperbilirubinemia ▪ Oral neomycin limits growth of intestinal bacteria that can produce ammonia. Limited use in patients (e.g., neonates) in whom reduction of hyperammonemia is an urgent priority. ▪ Oral lactulose acidifies the intestinal lumen → ↓ diffusion of ammonia across the intestinal epithelium. Limited applicability in newborns, who have a high risk of acidemia and dehydration. ▪ Cooling as therapeutic adjunct in NB with metabolic encephalopathy caused by hyperammonemia CLINICAL MANIFESTATIONS DIAGNOSIS ▪ Rarely symptomatic in neonatal period ▪ Marked ↑ arginine in plasma & CSF ▪ Insidious onset ▪ Urinary orotic acid can be ↑ ▪ Progressive spastic di-/tetraplegia, scissoring of the lower ▪ Determination of amino acids in plasma extremities, choreoathetotis, loss of developmental ▪ Guanidino compounds (α-keto-guanidino- valeric acid and milestones, failure to thrive, opisthotonus, seizures, cerebral α-keto-argininic acid) are markedly ↑ in urine LONG TERM THERAPY ▪ Protein restriction, sodium benzoate (250 mg/kg/24 hr), sodium phenylbutyrate (250-500 mg/kg/24hr), and arginine (200-400 mg/kg/24hr) or citrulline (in patients with OTC deficiency, 200-400 mg/kg/24 hr) is effective in maintaining blood deficiency ▪ Arginine and citrulline are contraindicated in patients with argininemia ▪ Patients w/ difficulty taking sodium phenylbutyrate: trial of glycerol phenylbutyrate (not for <2 months of age) ▪ Growth parameters, especially head circumference, and nutritional indices (blood albumin, prealbumin, pH, electrolytes, amino acids, zinc, selenium) should be followed closely ▪ Catabolic states (infections, fasting) that may trigger hyperammonemia should be avoided. Must be treated vigorously if they occur ▪ Avoid valproic acid (can elevate blood ammonia) TREATMENT ▪ Dietary protein restriction ▪ Alternative pathway drugs for removing ammonia (sodium benzoate and phenylbutyrate) ▪ Liver transplantation COMPLICATIONS / PROGNOSIS ▪ Improved neurologic outcome but some develop intellectual disability Batch Clingy TYPE II: Tyrosine aminotransferase deficiency ▪ Survivors develop chronic liver disease with ↑ risk of HCC and cirrhosis Ocular manifestations ▪ As early as 6 mos ▪ Excessive tearing, redness, pain, photophobia, corneal lesions caused by tyrosine deposition Skin lesions ▪ Later in life ▪ Painful, nonpruritic hyperkeratotic plaques on soles, palms, fingertips Intellectual disability ▪ Mild to moderate 35 ▪ Argininosuccinate acid lyase deficiency ▪ Autosomal recessive ▪ 1 in 220, 000 live births ▪ ASL gene on chromosome 7q11.21 ARGININOSUCCINIC ACIDEMIA edema, death ▪ Low risk of symptomatic hyperammonemia – hepatomegaly may be present ▪ Might present before NBS results available ▪ Anorexia, vomiting, lethargy → coma, seizures, intellectual disability, failure to thrive, hypertension, gallstones, liver fibrosis, hepatomegaly ▪ If untreated, trichorrhexis nodosa – dry and brittle hair ▪ Confirmed by assaying arginase activity in erythrocytes or by the identification of the mutant gene. ▪ Hyperammonemia, moderate ↑ in liver enzymes ▪ Nonspecific ↑ in plasma glutamine and alanine ▪ Moderate ↑ in plasma citrulline (less than in citrullinemia) ▪ Marked ↑ in the concentration of argininosuccinic acid in plasma, urine, and CSF Prenatal diagnosis ▪ Enzyme assay, identification of variants in ASL gene; argininosuccinic acid elevated in AF ▪ Argininosuccinate synthetase deficiency ▪ Autosomal recessive ▪ 1:250,000 live births ▪ Gene ASS1 on chromosome 9q34.11 CITRULLINEMIA TYPE I CITRULLINEMIA TYPE II ▪ Citrin deficiency ▪ Citrin ● Aspartate-glutamate carrier protein ● Mitochondrial transporter encoded by a gene (SLC25A13) located on chromosome 7q21.3 ● Transports aspartate from mitochondria into cytoplasm and replenishes the cytosolic aspartate pool required for converting citrulline to argininosuccinic acid ▪ If aspartate is unavailable to the cytoplasmic component of the urea cycle, urea will not be formed at a normal rate, and citrulline will accumulate ▪ ASS activity is diminished in the liver of these patients, but no pathogenic variant in the ASS1 gene has been found. It is postulated that citrin deficiency interferes with translation of messenger RNA for ASS enzyme in the liver. Severe/Neonatal Form ▪ Most common, 1st few days of life ▪ Refusal to eat, vomiting, tachypnea, lethargy → deep coma, seizures, hepatomegaly, increased ICP, bulging fontanelle, dilated pupils Subacute/Mild Form ▪ After 1yr of life ▪ FTT, frequent vomiting, developmental delay, dry brittle hair ▪ Anorexia, vomiting, lethargy, seizures, coma, possibly leading to death Neonatal-onset form ▪ <1yo ▪ FTT, hepato(spleno)megaly, cholestatic jaundice Early childhood-onset form ▪ Dietary preference for protein-rich and/or lipid-rich foods ▪ Aversion to carbohydrate-rich foods ▪ Failure to thrive, pancreatitis Adult-onset form ▪ Neuropsychiatric problems – disorientation, delirium, delusion, aberrant behavior, tremors, frank psychosis Neonatal-Onset ▪ Hypoglycemia, abnormal AA profile ▪ Mild to moderately ↑ plasma ammonia ▪ Mild-mod conjugated hyperbilirubinemia, marked hypoproteinemia ▪ Clotting dysfunction (prolonged PT, PTT) ▪ ↑ serum glutamyltransferase and alkaline phosphatase activities ▪ Liver transaminases are usually normal. ▪ Ammonia & citrulline are usually normal, some w/ mod elevations. ▪ ↑ Methionine, tyrosine, alanine, threonine, galactose, AFP ▪ Liver biopsy: fatty infiltration, cholestasis with dilated canaliculi, and a moderate degree of fibrosis Adult-onset ▪ Moderate hyperammonemia and hypercitrullinemia ▪ Fatty liver ETIOLOGY / INCIDENCE / PATHOGENESIS GLUTARIC ACIDEMIA TYPE I (GAI) ▪ Glutaryl-CoA dehydrogenase deficiency ▪ Glutaric acid is an intermediate in the degradation of PPS Oral Exam Reviewer 2020 ▪ Improved intellectual outcome if treatment is initiated early ▪ Developmental delay if severe ▪ Recurrent hyperammonemic episodes Common Sequelae ▪ Intellectual disability ▪ Persistent hepatomegaly with mild increases in liver enzymes ▪ Bleeding tendencies as a result of abnormal clotting factors ▪ Improved intellectual outcome if treatment is initiated early ▪ Developmental delay if severe ▪ Recurrent hyperammonemic episodes Neonatal-onset ▪ Lactose-free diet ▪ MCT and fat-soluble vitamin supplementation Neonatal-onset ▪ Transient neonatal cholestasis and variable hepatic dysfunction ▪ Some develop cirrhosis and have a poor prognosis ▪ Hyperammonemia w/ marked ↑ plasma glutamine and alanine with ↓ arginine ▪ Blood level of urea is usually low ▪ Greatly ↑ plasma citrulline (50-100x) ▪ Moderately ↑ urinary excretion of orotic acid → crystalluria (orotate precipitation) ▪ Confirmed by DNA analysis/enzyme assay Early childhood-onset ▪ Dyslipidemia ▪ Normal–slightly ↑ plasma ammonia DISEASE “e” Mgt for Symptomatic Neonates: ▪ Dietary protein restriction ▪ Essential amino acid supplementation ▪ Arginine supplementation ▪ Alternative pathway drugs for removing ammonia (sodium benzoate and phenylbutyrate) ▪ Liver transplantation ORGANIC ACIDEMIAS CLINICAL MANIFESTATIONS / DIAGNOSIS PHYSICAL EXAM ▪ Rarely symptomatic in neonatal period, although ▪ Mild to moderate metabolic acidosis, ketosis, macrocephaly may be present (at birth/1styr) hypoglycemia, hyperammonemia, ↑ serum Early childhood-onset Same diet as above ▪ MCT and fat-soluble vitamin supplementation ▪ Sodium pyruvate supplementation Adult-onset ▪ Low calorie, low carbs, high protein diet – diet enriched with protein and lipids helps restore cytosolic aspartate pool and stimulate ureagenesis ▪ MCT, arginine, and sodium pyruvate supplementation ▪ Liver transplantation TREATMENT “e” Mgt for Symptomatic Neonates: ▪ Cessation of protein intake for 24 hr Early childhood-onset ▪ Variable Adult-onset ▪ Poor prognosis, improved with liver transplantation ▪ Pancreatitis, hyperlipidemia, hepatoma - major complications COMPLICATIONS / PROGNOSIS ▪ Improved intellectual treatment is initiated early outcome if Batch Clingy ▪ ARG1 in chromosome 6q23.2 36 lysine, hydroxylysine, tryptophan ▪ GCDH located on chromosome 19p13.2 ▪ Delayed onset of motor milestones, irritability, feeding problems ▪ Onset of the condition is usually heralded by acute encephalopathic findings – loss of normal developmental milestones (head control, rolling over, or sitting), seizures, generalized rigidity, opisthotonos, choreoathetosis, and dystonia caused by acute striatal injury. These symptoms may occur suddenly in an apparently normal infant after a minor infection. ▪ Insidious form ● Hypotonia & choreoathetosis → rigidity & dystonia ● Acute episodes of metabolic decompensation with vomiting, ketosis, seizures, and coma also occurs. GLUTARIC ACIDEMIA TYPE II (GAII) ▪ Electron transfer flavoprotein (ETF) deficiency; ETF dehydrogenase deficiency ▪ ETF and ETF-DH transfer electrons into the mitochondrial ETC from dehydrogenation reactions catalyzed by VLCAD, MCAD, and SCAD, as well as by glutaryl-CoA dehydrogenase and 4 enzymes involved in BCAA oxidation ▪ Deficiencies of ETF or ETF-DH → illness w/ combined features of impaired fatty acid oxidation and impaired oxidation of amino acids. ▪ Commonly manifests in the neonatal period: hypotonia, hepatomegaly, congenital anomalies – facial dysmorphism and cystic kidney disease, coma ▪ Sweaty Feet Odor (d/t isovaleryl-CoA dehydrogenase inhibition ▪ Generally lethal ▪ Late-onset forms variable, rarely have structural birth defects ▪ Partial deficiency may mimic MCAD deficiency or other milder fatty acid oxidation defects; have attacks of fasting hypoketotic coma. ▪ 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency ▪ This enzyme catalyzes the conversion of 3-HMG CoA to acetoacetate and is a rate-limiting enzyme for ketogenesis ▪ Autosomal recessive ▪ 3-HMG-CoA lyase is encoded by gene HMGCL 3-HYDROXY-3METHYLGLUTARIC ACIDURIA ▪ 30% 1st few days of life ▪ >60% bet 3-11 mos old ▪ Similar to 3-HMG-CoA synthase deficiency – acute hypoketotic hypoglycemia, vomiting, severe hypoglycemia, hypotonia, acidosis with mild or no ketosis, dehydration, lethargy, ataxia, coma, hepatomegaly ▪ Episodic hypoglycemia may contribute to developmental delay transaminases ▪ High concentrations of glutaric acid in urine, blood, and CSF ▪ ↑ Glutarylcarnitine (C5-DC) in blood & urine ▪ Replacement of lost calories using carbohydrates or lipids, IV L-carnitine, IV dextrose ▪ Prompt treatment of infection ▪ Control of fever ▪ NBS: glutarylcarnitine levels ▪ Brain Imaging: ↑ Extraaxial (particularly frontal) fluid with stretched bridging veins, striatal lesions, dilated lateral ventricles, cortical atrophy (mainly in frontotemporal region), and fibrosis Prenatal diagnosis ▪ ↑ Glutaric acid in amniotic fluid ▪ Enzyme activity in amniocytes or chorionic villus samples ▪ Identification of the known pathogenic variants in GCDH ▪ Metabolic acidosis ▪ Hypoglycemia, hyperammonemia ▪ Acylcarnitine profile and urinary organic acids: abnormalities corresponding to blocks in the oxidation of fatty acids (ethylmalonate and C6-C10 dicarboxylic acids), lysine (glutarate), BCAAs (isovaleryl-, isobutyryl-, ⍺-methylbutyryl-glycine) ▪ Confirmed by genetic testing for ETF (2 genes, A and B) and ETF dehydrogenase Partial deficiency ▪ Urinary organic acid profile: primarily elevations of dicarboxylic acids and ethylmalonate, derived from short-chain fatty acid intermediates ▪ Secondary carnitine deficiency is present ▪ Hypoglycemia, moderate to severe hyperammonemia, acidosis ▪ Mild or no ketosis ▪ Cat Urine Odor – urinary excretion of 3-hydroxy-3methylglutaric acid and other proximal intermediate metabolites of leucine catabolism (3-methylglutaric acid, 3-methylglutaconic acid, and 3hydroxyisovaleric acid) ▪ Dietary protein restriction ▪ Lysine & tryptophan restriction ▪ Arginine supplementation ▪ Riboflavin and carnitine supplementation ▪ Poor neurologic outcome if treatment is started after acute neurologic injury occurs ▪ Treatment might slow neurologic deterioration ▪ Prone to subdural hematoma and retinal hemorrhage following minor falls and head traumas ▪ If with movement disorder: Baclofen, diazepam, trihexy-phenidyl, and injectable botulinum toxin A “e” Mgt for Symptomatic Neonates: ▪ Dietary low-fat/low-protein diet ▪ Carnitine supplementation ▪ Riboflavin supplementation for responsive cases (mild forms) ▪ Treatment for neonatal-onset forms is of limited benefit but might be helpful for patients with late-onset disease ▪ Most severely affected infants do not survive neonatal period ▪ Frequent feedings ▪ Low-fat / low-protein (leucine-restricted) diet ▪ Carnitine supplementation ▪ Glucose infusion for hypoglycemia ▪ Bicarbonate to correct acidosis ▪ RRT for severe recalcitrant hyperammonemia ▪ Improved intellectual outcome if treatment is initiated early ▪ Developmental delay if severe ▪ Recurrent hypoglycemic episodes decrease in frequency and severity with age Long term complications Dilated cardiomyopathy, hepatic steatosis, and pancreatitis ▪ Isobutyryl-CoA dehydrogenase deficiency ISOBUTYRIC ACIDEMIA PPS Oral Exam Reviewer 2020 ▪ Uncertain because number of cases is small ▪ May be benign ▪ Case reports of cardiomyopathy associated with carnitine deficiency ▪ Carnitine supplementation if deficiency present Batch Clingy ▪ Glutaric and dicarboxylic acids may also be ↑ in urine during acute attacks ▪ Secondary carnitine deficiency ▪ Confirmed by molecular analysis of HMGCL or by enzyme assay in cultured fibroblasts, leukocytes, or liver specimens 37 ▪ Might present before NBS results available ▪ Vomiting, lethargy, convulsions, and coma, possibly death ▪ Sweaty Feet / Rancid Cheese Odor ISOVALERIC ACIDEMIA (IVA) ▪ Thrombocytopenia, leukopenia, occ pancytopenia, BM suppression, hypocalcemia, anemia, ketoacidosis, moderate to severe hyperammonemia ▪ Accumulating derivatives of isovaleric acid, isovalerylcarnitine (C5-carnitine w/c is found in NBS), isovalerylglycine, and 3-hydroxyisovaleric acid ▪ Confirmed by molecular analysis of IVD gene Prenatal diagnosis ▪ Enzyme assay in cultured amniocytes ▪ IVD gene analysis 3-KETOTHIOLASE DEFICIENCY 2-METHYL- BUTYRYLGLYCINURIA 3METHYLCROTONYLGLYCI NURIA 2-METHYL-3HYDROXYBUTYRIC ACIDEMIA ▪ Mitochondrial acetoacetyl CoA thiolase deficiency ▪ This reversible mitochondrial enzyme is involved in the final steps of isoleucine catabolism and in ketolysis ▪ In the isoleucine catabolic pathway, the enzyme cleaves 2-methylacetoacetyl-CoA into propionyl-CoA and acetyl coA ▪ In the fatty acid oxidation pathway, the enzyme generates 2 moles of acetyl-CoA from 1 mole of acetoacetyl-CoA. The same enzyme synthesizes 2methyacetoacetate-CoA and acetoacetyl-CoA in the reverse direction ▪ Autosomal recessive ▪ ACAT1 gene located on chromosome 11q22.3 ▪ Might present before NBS results are available ▪ Hallmark: ketoacidosis – triggered by infections, prolonged fasting and large protein load ▪ Cognitive impairment, vomiting, lethargy and coma, possibly death ▪ Abnormal odor ▪ 2-Methylbutyryl-CoA dehydrogenase deficiency (aka: short/ branched chain acyl-CoA dehydrogenase) ▪ Most patients ascertained by NBS are clinically asymptomatic and remain so, although several patients have been described with a variety of problems ▪ Onset: between 3 weeks-3yo ▪ Highly variable phenotype ▪ Many patients asymptomatic ▪ Some w/ developmental delay w/o episodes of metabolic decompensation, seizures, hyperammonemia, and metabolic acidosis ▪ Severe 3-MCC deficiency: Acute episode of vomiting, hypotonia, lethargy, and convulsions after a minor infection, w/c may progress to Reye syndrome, coma ▪ 3-Methylcrotonyl-CoA carboxylase deficiency (3-MCC) ▪ Heteromeric enzyme consisting of ⍺(biotin containing) and β subunits, encoded by genes MCCC1 and MCCC2, respectively ▪ 1:2400 - 1:68, 000 live births ▪ Autosomal recessive ▪ Gene for ⍺-subunit (MCC1) located on chromosome 3q27.1; β subunit (MCC2) on chromosome 5q13.2 Maternal 3-MCC deficiency ▪ Mothers have variable neurologic phenotype ▪ 2-Methyl-3-hydroxybutyryl CoA dehydroge- nase deficiency (aka:17-beta-hydroxysteroid dehydrogenase X deficiency) Mild: some males may have normal neurologic development. Affected females have less severe disease. Severe: Neurodegenerative disease, associated with mental retardation, rigidity, choreoathetosis, seizures, and cerebral atrophy ▪ Clinical variability has been observed in this X-linked disorder PPS Oral Exam Reviewer 2020 ▪ Thrombocytopenia, leukopenia, anemia ▪ Possible basal ganglia damage ▪ Ketoacidosis, hypo/ hyperglycemia ▪ Mild-mod hyperammonemia ▪ Urine: large amounts of 2-methyl- acetoacetate and its decarboxylated products butanone, 2-methyl-3hydroxybutyrate, and tiglylglycine ▪ Plasma acylcarnitine profile: ↑ C5:1 and C5-OH carnitines ▪ Confirmed by molecular analysis of ACAT1 gene or enzyme assay of leukocytes or cultured fibroblasts ▪ Mild to mod metabolic acidosis, ketosis, hypoglycemia, hyperammonemia, ↑ serum transaminase ▪ Urine: Large amounts of 3-hydroxyisovaleric acid and 3-methylcrotonylglycine ▪ ↑ 3-hydroxyisovalerylcarnitine (C5-OH) in NBS ▪ Severe secondary carnitine deficiency ▪ Confirmed by molecular analysis or measurement of enzyme activity “e” Mgt for Symptomatic Neonates: ▪ Hydration, reversal of catabolic state, correction of met acidosis, facilitate isovaleric acid excretion ▪ Dietary protein restriction <24hr ▪ Leucine restriction ▪ L-Carnitine supplementation (100 mg/kg/24hr oral) – ↑ removal of isovaleric acid by forming isovalerylcarnitine (excreted in urine) ▪ Glycine supplementation (250 mg/kg/ 24hr) – enhance formation of isovalerylglycine (high urinary clearance) ▪ Renal replacement therapy if all else fails “e” Mgt for Symptomatic Neonates: ▪ IVF and bicarbonate treatment ▪ 10% glucose w/ electrolytes to suppress protein catabolism, lipolysis & ketogenesis ▪ Dietary protein restriction ▪ Isoleucine restriction ▪ Avoidance of fasting ▪ L-Carnitine 100mg/kg/day) supplementation ▪ Improved intellectual outcome if diagnosed and treated early ▪ If treated appropriately, most have normal development ▪ Recurrent metabolic episodes ▪ Highly variable clinical course ▪ Improved intellectual outcome if diagnosed and treated early ▪ If recognized and treated appropriately, some patients have normal development ▪ Recurrent metabolic episodes (50- ▪ The need for treatment with a low-protein diet has not been established ▪ Generally good ▪ Hydration ▪ Glucose & sodium bicarbonate – correct hypoglycemia and metabolic acidosis ▪ Dietary protein restriction ▪ Selected amino acid (leucine) restriction ▪ L- Carnitine supplementation ▪ Generally, good ▪ Mother might benefit from carnitine supplementation if she has carnitine insufficiency ▪ Dietary protein restriction improves the metabolic markers but does not alter clinical outcome ▪ Mother treatment generally improves with ▪ Poor for severe affected patients but better for mildly affected patients Batch Clingy ▪ Isovaleryl-CoA dehydrogenase deficiency → impaired leucine degradation 38 ▪ Deficiency of either mutase or its coenzyme → accumulation of methylmalonic acid and its precursors METHYLMALONIC ACIDEMIA (MMA) Group of metabolic disorders of diverse etiology characterized by impaired conversion of methylmalonyl CoA into succinyl-CoA ▪ Two biochemical forms of deficiency: mut0 – no detectable enzyme activity, not responsive to hydroxocobalamin therapy mut− – indicates residual, although insufficient, mutase activity ▪ 1:50,000 to 1:100,000 live births ▪ Gene for mutase (MUT) on the short arm of chromosome 6p12.3 “e” Mgt for Symptomatic Neonates: ▪ Ketosis, metabolic acidosis, hyperglycinemia, hypoglycemia, anemia, neutropenia, thrombocytopenia ▪ Hyperammonemia (inhibition of proximal urea cycle in the mitochondrial matrix) ▪ Large quantities of methylmalonic acid in body fluid ▪ Metabolites of propionic acid (3-hydroxypropionate and methylcitrate) in the urine ▪ ↑ Propionylcarnitine (C3) “e” Mgt for Symptomatic Neonates: ▪ Dietary protein restriction ▪ Isoleucine, methionine, threonine, valine restriction ▪ Daily hydroxycobalamin IM injection if px responsive ▪ Oral carnitine supplementation ▪ Antibiotic suppression of gut flora (metronidazole or neomycin) ▪ Liver &/or kidney transplantation might be required ▪ Confirmed by identifying pathogenic variants in the causal gene, by measuring propionate incorporation with complementation analysis in cultured fibroblasts, and by measuring the specific activity of the mutase enzyme in biopsies or cell extracts. vitamin B12 (adenosylcobalamin) ▪ Propionyl-CoA carboxylase deficiency ▪ Propionic acid is an intermediate metabolite of isoleucine, valine, threonine, methionine, odd-chain fatty acids, and side chains of cholesterol ▪ Normally, propionic acid in the form of propionyl-CoA undergoes carboxylation to D-methylmalonyl-CoA, catalyzed by the mitochondrial enzyme propionyl-CoA carboxylase which requires biotin as a cofactor. ▪ Autosomal recessive ▪ 1:105,000 to 1:250,000 live births ▪ Gene for ⍺ subunit (PCCA) is located on chromosome 13q32.3 and that of the β-subunit (PCCB) is mapped to chromosome 3q22.3 ▪ Variable, depending on specific disorder Mild: ▪ Episodes of metabolic decompensation less frequent, but still at risk for intellectual diability seizures, long QTc interval, severe cardiomyopathy Severe: ▪ 1st few days of life ▪ Might present before NBS results are available ▪ Poor feeding, vomiting, seizures, hypotonia, lethargy, dehydration, a sepsis like picture, and clinical signs of severe ketoacidosis progress rapidly to coma and death. ▪ Moderate to severe intellectual disability and neurologic manifestations reflective of extrapyramidal (dystonia, choreoathethosis, tremor) & pyramidal (paraplegia) ▪ Damage to basal ganglia esp. to globus pallidus (metabolic stroke) d/t metabolic decompensation ▪ Thrombocytopenia, leukopenia, anemia, neutropenia ▪ Ketoacidosis, hypoglycemia ▪ Moderate to severe Hyperammonemia (perturbed biochemical and pH environment of the mitochondrial matrix) ▪ Mild to moderate ↑ in blood lactate and lysine; N to ↓ plasma glutamine ▪ Concentrations of propionylcarnitine, 3hydroxypropionic acid, and methylcitric acid ↑ in plasma & urine ▪ ↑ Propionylglycine & other intermediate metabolites of branched chain amino acid ▪ Secondary carnitine deficiency ▪ NBS: elevated propionylcarnitine (C3) ▪ Brain Imaging: cerebral atrophy, delayed myelination, and abnormalities in the globus pallidus and other parts of the basal ganglia ▪ Confirmed by molecular analysis of PCCA and PCCB or by measuring the enzyme activity in leukocytes or cultured fibroblasts Prenatal diagnosis PPS Oral Exam Reviewer 2020 ▪ Improved outcome if diagnosed and treated early ▪ Recurrent metabolic episodes and longterm sequelae may still occur ▪ Renal failure often develops despite appropriate therapy Long-term Sequelae Psychomotor retardation, seizures, pyramidal and extrapyramidal movement disorder, basal ganglia strokes, interstitial renal disease, cardiomyopathy, pancreatitis, BM suppression, osteopenia, optic nerve atrophy ▪ Close monitoring of blood pH, essential amino acid levels, blood and urinary concentrations of methylmalonate, and growth parameters as well as kidney function, vision, hearing, and bone mineral density NBS: measuring propionylcarnitine (C3) Defect affecting metabolism PROPIONIC ACIDEMIA (PA) Mild: ▪ Later in life ▪ Hypotonia, FTT, developmental delay Severe: ▪ 1st few days of life ▪ Lethargy, feeding problems, vomiting, sepsis-like picture, tachypnea (from metabolic ketoacidosis), hypotonia ▪ May progress to hyperammonemic encephalopathy, coma, and death if left untreated ▪ Injury to basal ganglia, metabolic stroke → movement disorder Same as above, plus: ▪ Daily intramuscular injection of hydroxocobalamin ▪ Daily oral betaine supplementation, if methylcobalamin also involved ▪ Dietary protein restriction <24hr ▪ If enteral feedings cannot be tolerated after 48hrs of protein restriction, give parenteral nutrition ▪ Hydration w/solutions containing glucose; correction of acidosis ▪ Isoleucine, methionine, threonine, valine restriction ▪ Carnitine supplementation ▪ Liver transplantation (recurrent hyperammonemia, frequent metabolic decompensations, poor growth) ▪ Suppression of propionogenic gut microflora (metronidazole or neomycin) – prolonged use of metronidazole avoided d/t reversible peripheral neuropathy and ↑ QTc interval hence baseline & interval ECG before & after metro ▪ Hemodialysis to remove ammonia & other toxic compounds ▪ N-carbamoylglutamate (carglumic acid) and nitrogen scavengers ▪ mut0 and severe forms of cblB deficiency – least favorable prognosis ▪ mut- and cblA defects – better outcome ▪ Improved outcome if diagnosed and treated early ▪ Recurrent metabolic episodes Long-term Sequelae Cardiomyopathy and pancreatitis, FTT, optic nerve atrophy, pancreatitis, cardiomyopathy, osteopenia, permanent neurodevelopmental deficit such as tremor, dystonia, chorea, and spasticity, osteoporosis leading to fractures ▪ Over restriction of methionine esp in 1styrs of life may contribute to reduced brain growth & microcephaly ▪ Bicarbonate substitution (citric acid/sodium citrate) to correct chronic acidosis ▪ Close monitoring of plasma ammonia, plasma amino acids obtained 3-4hrs after the last typical meal (especially isoleucine, leucine, valine, threonine) and growth parameters7 Batch Clingy ▪ Methylmalonyl-CoA mutase deficiency ▪ Converts L-methylmalonyl-CoA to succinyl-CoA w/c requires adenosylcobalamin, (metabolite of B12) as coenzyme 39 ▪ Identification of variants in PCCA or PCCB /measuring enzyme activity in cultured amniotic cells MULTIPLE CARBOXYLASE DEFICIENCY (Defects of Biotin Cycle) ▪ Holocarboxylase synthetase deficiency ▪ Free biotin (water soluble vitamin that is cofactor for all 4 carboxylase enzymes in humans: pyruvate carboxylase, acetyl-CoA carboxylase, propionyl-CoA carboxylase, and 3methylcrotonyl-CoA carboxylase. The latter 2 are involved in the catabolic pathways of leucine, isoleucine, and valine) must form a covalent bond with the apocarboxylases to produce the activated enzyme (holocarboxylase) ▪ Binding catalyzed by holocarboxylase synthetase ▪ Autosomal recessive ▪ May present in the newborn period (few hrs after birth to as late as 8yo) After birth: Breathing difficulties, tachypnea, problems, vomiting, hypotonia apnea, feeding If untreated: Generalized erythematous rash w/exfoliation & alopecia, FTT, irritability, seizures, lethargy, coma, impaired T-cell immunity hence susceptible to infection, seizures, and developmental delay ▪ Tomcat Urine – peculiar odor in urine ▪ Metabolic ketoacidosis ▪ Hyperammonemia ▪ Presence of a variety of organic acids (lactic acid,3methylcrotonic acid, 3-methylcrotonylglycine, tiglylglycine, 3-OH propionicacid, methylcitric acid, and 3hydroxyisovaleric acid) in body fluids (sodium benzoate, sodium phenylacetate, sodium phenylbutyrate) can aid in the treatment of acute hyperammenomia ▪ Biotin supplementation (10-20 mg/day PO) ▪ Most patients supplementation respond to biotin ▪ Poor or no response to biotin: may have significant residual neurologic impairment ▪ NBS: elevated C5-OH -carnitine on tandem mass spectrometry ▪ Confirmed by identification of pathogenic variants in HLCS or by enzyme assay in lymphocytes or cultured fibroblasts Prenatal diagnosis Molecular analysis of the known pathogenic variants in HLCS or by assaying enzyme activity in cultured amniotic cells. BIOTINIDASE DEFICIENCY DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Biotinidase deficiency ▪ Delay in onset of symptoms may be d/t presence of free biotin derived from mother or the diet ▪ Autosomal recessive ▪ 1 in 60, 000 live births ▪ Gene for biotinidase (BTD) is located on chromosome 3p25.1 ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Carnitine uptake defect ● Only genetic defect in which carnitine deficiency is the cause, rather than the consequence ▪ Involves the plasma membrane sodium gradientdependent carnitine transporter present in heart, muscle, & kidney CARNITINE UPTAKE DEFECT OTHER CLINICAL MANIFESTATIONS / DIAGNOSIS PHYSICAL EXAM ▪ Generally, does not manifest in neonatal period, ▪ Lab findings and the pattern of organic acids in body fluids same several months or years old w/ holocarboxylase synthetase deficiency (see above) ▪ Symptoms similar to holocarboxylase synthetase ▪ Measurement of the enzyme activity in the serum or by the deficiency identification of the mutant gene ▪ Atopic or seborrheic dermatitis, candidiasis, alopecia, ataxia, seizures (usually myoclonic), hypotonia, Prenatal diagnosis developmental delay, optic nerve atrophy, sensorineural Identification of the known pathogenic variants in BTD, or less hearing loss, & immunodeficiency from impaired T-cell frequently by the measurement of the enzyme activity in the function amniotic cells FATTY ACID OXIDATION CLINICAL MANIFESTATIONS / DIAGNOSIS PHYSICAL EXAM ▪ Does not generally manifest in neonatal period (1-4yo) ▪ Hypoketotic hypoglycemia ▪ Extremely ↓ carnitine levels in plasma and muscle ▪ Progressive Cardiomyopathy (1-2% of normal) ▪ Skeletal myopathy ▪ Fasting ketogenesis may be normal because liver ▪ Inability to tolerate prolonged fasting carnitine transport is normal, but it may become impaired if dietary carnitine intake is interrupted Fasting urinary organic acid profile ▪ Hypoketotic dicarboxylic aciduria pattern if hepatic fatty acid oxidation is impaired but is otherwise unremarkable ▪ Severe reduction in renal carnitine threshold or by in vitro assay of carnitine uptake using cultured fibroblasts or lymphoblasts PPS Oral Exam Reviewer 2020 TREATMENT ▪ Free biotin (5-20 mg/day) TREATMENT ▪ Carnitine supplementation ● High-dose, high-frequency ● Oral carnitine (100-200 mkday) COMPLICATIONS / PROGNOSIS ▪ Good COMPLICATIONS / PROGNOSIS ▪ Good response to treatment, prevents and/or reverses cardiomyopathy, skeletal myopathy, and impaired ketogenesis Batch Clingy DISEASE 40 ▪ CPT II deficiency CARNITINE PALMITOYLTRANSFERASE TYPE II (CPT II) DEFICIENCY Partial Translocase deficiency ▪ Milder disease ▪ Without cardiac involvement Severe form (neonatal period) ▪ Lethargy → coma, cardiomyopathy with ventricular arrhythmias, hepatic dysfunction, skeletal myopathy, congenital malformation (brain and cystic renal disease), hypoketotic hypoglycemia, mild facial anomalies Intermediate form (infancy) ▪ Similar (but milder) features, w/o congenital brain and kidney malformations ▪ Fasting induced hepatic failure, cardiomyopathy, and skeletal myopathy w/ hypoketotic hypoglycemia, but not assoc. w/ severe developmental changes ▪ LCHAD deficiency/TFP deficiency ▪ LCHAD enzyme is part of the MTP, which also contains 2 other steps in β-oxidation: long-chain 2,3-enoyl CoA hydratase and long-chain β-ketothiolase LONG-CHAIN 3HYDROXYACYL-COA DEHYDROGENASE (LCHAD) DEFICIENCY or TRIFUNCTIONAL PROTEIN (TFP) DEFICIENCY MEDIUM-CHAIN ACYLCOA DEHYDROGENASE (MCAD) DEFICIENCY ▪ Commonly manifests in neonatal period ▪ Lethargy → coma, hepatomegaly/hepatic dysfunction, Cardiomyopathy with ventricular arrhythmia, skeletal myopathy ▪ MCAD deficiency ▪ Most common fatty acid oxidation disorder ▪ MCAD missense mutation, an A-G transition at cDNA position 985 (c.985A>G) that changes a lysine to glutamic acid at residue 329 (p.K329E) Mild, late-onset form (childhood/adult) ▪ Weakness and exercise-induced rhabdomyolysis ▪ Aching muscle pain & myoglobinuria that may cause renal failure Severe form (neonatal period) ▪ Cardiomyopathy with arrhythmia, hypotonia, hepatic dysfunction, hypoketotic hypoglycemia, sudden death ▪ Adult: mutation, c.338C>T, p.S113L; produces a heat-labile protein that is unstable to (↑) muscle temperature during exercise → myopathic presentation Diagnosis Molecular genetic analysis and demonstrating deficient enzyme activity in muscle or other tissues and in cultured fibroblasts ▪ Acute hypoketotic hypoglycemia similar to MCAD deficiency ▪ ↑ Blood spot or plasma 3-hydroxy acylcarnitines of chain lengths C16-C18 ▪ ↑ Levels of 3-hydroxydicarboxylic acids of chain lengths C6-C14. ▪ Secondary carnitine deficiency ▪ Mutation in the ⍺-subunit, E474Q ▪ Severe neonatal cases: poor outcome and early death ▪ Patient with later onset might respond to treatment, but they often succumb to chronic skeletal-muscle weakness or cardiac arrhythmias Severe ▪ Avoid fasting, may require continuous enteral feeding ▪ High-carbohydrate, low-fat diet, including MCT oil ▪ Carnitine supplementation Severe ▪ Have poor outcome and early death Intermediate ▪ Treated with an attenuated regimen of the above Late-onset ▪ Generally do well Intermediate ▪ Milder problems than those with neonatal-onset form Late-onset ▪ Treated with an attenuated regimen of the above Severe ▪ Avoid fasting ▪ High-carbohydrate, low-fat diet with MCT oil supplementation (to bypass the defect in long-chain fatty acid oxidation and docosahexaenoic acid (for protection against the retinal changes) ▪ Carnitine supplementation ▪ Liver transplantation ▪ Prognosis for severe form is guarded despite therapy ▪ Early diagnosis and treatment generally improve outcome for patients with infantile/childhood form but risk of peripheral neuropathy and visual impairment persists Infantile/childhood form ▪ Milder form of “severe” disease and later develop rhabdomyolysis, peripheral neuropathy, and pigmentary retinopathy (d/t toxic effects of fatty acid metabolites) Infantile/childhood ▪ Treat with an attenuated regimen of the above, plus supplementation with docosahexanoic acid Maternal disease ▪ Heterozygous pregnant women are at risk for acute fatty liver of pregnancy, HELLP syndrome, if they are carrying an affected (homozygous or compound heterozygous) fetus ▪ Generally, does not manifest in neonatal period ▪ 1st 3mos to 5yrs of life ▪ Triggered by prolonged fasting (>12-16hrs) ▪ Breastfed are at higher risk because of early poor caloric intake Maternal treatment ▪ Early delivery ▪ Carnitine supplementation if deficient ▪ Prognosis guarded for pregnant woman ▪ Avoid fasting – limit overnight fasting periods to <10-12hrs ▪ Fluid containing 10% dextrose to correct & prevent hypoglycemia and to suppress lipolysis as rapidly as possible ▪ Heart healthy diet (≤30%) ▪ Carnitine supplementation ▪ Excellent intellectual and physical outcome generally seen if treatment is initiated before irreversible neurologic damage occurs, but some may sustain permanent brain injury ▪ Recurrent episodes of lethargy, vomiting, coma, seizures, and possibly sudden death PPS Oral Exam Reviewer 2020 ▪ Creatine kinase ↑ to 5,000-100,000 units/L ▪ Muscle biopsy: increased deposition of neutral fat ▪ Avoid fasting ▪ Continuous enteral feeding in severe cases ▪ High-carbohydrate, low-fat diet ▪ Carnitine supplementation ▪ Hypoketotic hypoglycemia, ↑ liver enzymes, ↑ blood ammonia, prolonged PT, PTT ▪ Liver biopsy: micro- or macrovesicular steatosis from triglyceride accumulation ▪ Low concentrations of ketones and ↑ levels of medium-chain dicarboxylic acids (adipic, suberic, and sebacic acids) ▪ Fasting tolerance improves with age; Batch Clingy CARNITINE/ ACYLCARNITINE TRANSLOCASE (CACT) DEFICIENCY ▪ Carnitine/acylcarnitine translocase deficiency ▪ This defect of the inner mitochondrial membrane carrier protein for long-chain acylcarnitines blocks the entry of long-chain fatty acids into the mitochondria for oxidation ▪ Characterized by a severe and generalized impairment of fatty acid oxidation. ▪ Mutations in the organic cation/carnitine transporter (OCTN2) underlie this disorder ▪ Hypoketotic hypoglycemia ± hyperammonemia ▪ ↑ Plasma long-chain acylcarnitines of chain lengths C16-C18 are reported ▪ Confirmed using genetic analysis ▪ Functional carnitine: acylcarnitine translocase activity measured in cultured fibroblasts or lymphoblasts 41 ▪ Cardiomyopathy not generally seen ▪ Liver may be slightly enlarged with fat deposition ▪ SCAD deficiency ▪ Harmless ▪ Autosomal recessive ▪ Most px detected by NBS are asymptomatic ▪ Some with severe manifestations – dysmorphic facial features, feeding difficulties/failure to thrive, metabolic acidosis, ketotic hypoglycemia, lethargy, developmental delay, seizures, hypotonia, dystonia, myopathy ▪ VLCAD deficiency ▪ No ability to oxidize physiologic long-chain fatty acids ▪ Usually more severely affected than those with MCAD Severe (neonatal) ▪ Lethargy progressing to coma ▪ Cardiomyopathy & ventricular arrhythmias, hepatic dysfunction, skeletal myopathy, hypoketotic hypoglycemia VERY LONG-CHAIN ACYLCOA DEHYDROGENASE (VLCAD) DEFICIENCY Intermediate (infancy) ▪ Similar (but milder) features than the severe form ▪ Nightly (optional) ▪ 2DED: LV may be hypertrophic/dilated with poor contractility ▪ Lab features similar to those of MCAD ▪ Nonketotic dicarboxylic aciduria with (↑) levels of C6-12 dicarboxylic acids. Severe ▪ Avoid fasting, consider continuous enteral feeding ▪ High-carbohydrate, low-fat diet supplemented with MCT oil ▪ Carnitine supplementation Diagnosis ▪ Suggested by an abnormal acyl dicarboxylic acids or blood spot C14:0, 14:1, 14:2 acylcarnitine Late-onset (childhood/adulthood) ▪ Weakness and exercise-induced rhabdomyolysis DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Galactose-1-phosphate uridyltransferase deficiency ▪ Autosomal recessive ▪ 1 in 47,000 live births GALACTOSE-1PHOSPHATE URIDYLTRANSFE-RASE (GALT) DEFICIENCY ▪ 2 FORMS: Complete/Near Complete deficiency (Classic Galactosemia) ● Serious disease with onset by 2nd half of 1stwk of life ● 1 in 60,000 live births ● Without the transferase enzyme, infant unable to metabolize galactose-1-phosphate, accumulation of which → injury to kidney, liver, brain Partial Transferase Deficiency GALACTOSEMIAS CLINICAL MANIFESTATIONS DIAGNOSIS ▪ May present in the neonatal period ▪ (+) Reducing substance in urine while on diet ▪ Lethargy, poor feeding, jaundice with hepatic containing human/cow milk /formula containing dysfunction, hepatomegaly, vomiting, hypoglycemia, lactose via chromatography/ enzymatic test specific seizure, irritability, poor weight gain, failure to gain for galactose weight ▪ Amino acids may be detected in urine since they are excreted together with glucose because of a If untreated proximal renal tubular syndrome ▪ Nuclear cataracts, vitreous hemorrhage, hepatic ▪ Confirmed by Direct enzyme assay using failure, cirrhosis, ascites, splenomegaly, or intellectual erythrocytes disability, ↑ risk for E. coli neonatal sepsis, pseudotumor cerebri causing bulging fontanel ▪ Death from liver, kidney failure, sepsis Chronic problems ▪ Growth failure, cirrhosis, cataracts, intellectual disabilities, ovarian failure supplementation ▪ Normal diet ▪ Carnitine supplementation demonstrates deficiency if testing risk of illness also decreases ▪ The need for and efficacy of treatment is unknown Severe: Poor outcome and early death Intermediate & Late: Generally, do well Intermediate ▪ Treated with an attenuated regimen of the above Late-onset ▪ Treated with an attenuated regimen of the above TREATMENT ▪ Strict dietary galactose restriction with adequate calcium supplementation – reverses growth failure and renal and hepatic dysfunction ▪ Use non–lactose-containing milk substitutes (casein hydrolysates, soybeanbased formula) COMPLICATIONS / PROGNOSIS ▪ Improved intellectual outcome, but may have milder problems such as speech delay, if diagnosed and treated early ▪ Cataracts regress, and most patients have no impairment of vision ▪ Ovarian failure with 1o/2o amenorrhea, decreased bone mineral density, developmental delay & learning disabilities, that ↑ in severity with age may still develop ▪ Hypergonadotropic hypogonadism (80-90% of women) ▪ Recurrent metabolic episodes may occur seizures, Partial transferase deficiency ▪ Generally asymptomatic ▪ More common than classic, dx in NBS because of moderately ↑ blood galactose and/or low transferase activity PPS Oral Exam Reviewer 2020 cornstarch Batch Clingy SHORT-CHAIN ACYL-COA DEHYDROGENASE (SCAD) DEFICIENCY ▪ Secondary carnitine deficiency: reflects competition between ↑ acylcarnitine levels and free carnitine for transport at the renal tubular plasma membrane ▪ ↑ Plasma C6:0, C8:0, C10:0, and C10:1 acylcarnitine species and ↑ urinary acylglycines, (hexanoylglycine, suberyl gly- cine, 3-phenylpropionylglycine) ▪ Confirmed by A985G mutation or sequencing the MCAD gene. ▪ Neonatal hypoglycemia w/ normal ketone ▪ NBS: ↑ butyrylcarnitine (C4-carnitine) ▪ ↑ Excretion of urinary ethylmalonic acid and butyrylglycine. 42 UDP-GALACTOSE-4EPIMERASE (GALE) DEFICIENCY (Epimerase Deficiency Galactosemia) DISEASE Generalized epimerase deficiency ▪ Poor feeding, vomiting, weight loss, jaundice, liver dysfunction, hepatomegaly, hypotonia, cataracts, generalized aminoaciduria, nerve deafness Peripheral or intermediate form ▪ Benign; detected in NBS ▪ Generally, remain clinically well since enzyme deficiency limited to leukocytes & erythrocytes ▪ May tolerate a normal diet ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ ⍺-L-Iduronidase deficiency HURLER SYNDROME (MPS I) MPS I – mutations of the IUA gene on chromosome 4p16.3 encoding ⍺-Liduronidase Homozygous nonsense mutations: severe Missense mutations: milder; preserve some residual enzyme activity POMPE DISEASE (GSD II) (Acid Maltase Deficiency) ▪ Cataracts, with no other medical problems ▪ Pseudotumor Cerebri – rare complication ▪ Heterozygous carriers: risk for presenile cataracts ▪ -Glucosidase deficiency – enzyme responsible for the degradation of glycogen in lysosomes ▪ Enzyme defect → lysosomal glycogen accumulation in multiple tissues and cell types, predominantly cardiac, skeletal, & smooth muscle cells ▪ As disease progresses: lysosomal rupture & leakage → presence of cytoplasmic glycogen as well ▪ Autosomal recessive PPS Oral Exam Reviewer 2020 ▪ ↑ Concentration of blood galactose levels, provided the infant has been fed a lactose containing formula ▪ Absence of galactokinase activity in erythrocytes or fibroblasts ▪ Transferase activity is normal ▪ Abnormally accumulated metabolites similar to transferase deficiency ▪ ↑ in cellular uridine diphosphate (UDP) galactose ▪ Confirmed by assay of epimerase in erythrocytes Prenatal diagnosis ▪ Enzyme assay of cultured amniotic fluid cells LYSOSOMAL STORAGE DISEASES DIAGNOSIS Severe ▪ Failed neonatal hearing tests ▪ Onset w/in 1st year of life; progressive ▪ Radiograph of chest, spine, pelvis, hands: skeletal ▪ Short stature, coarse facial features, hirsutism, dysplasia (Dysostosis multiplex); earliest large tongue symptoms: thick ribs & ovoid vertebral bodies ▪ Corneal clouding, glaucoma, retinal degeneration, ▪ Other skeletal abnormalities: and hearing loss ● Enlarged, coarsely trabeculated diaphyses of ▪ Cardiomyopathy/ valvular disease (incompetence the long bones w/ irregular metaphyses and of mitral & aortic valves, narrowing of coronary epiphyses arteries) ● With disease progression, macrocephaly ▪ Hepatosplenomegaly, inguinal hernias, stiff joints develops w/ thickened calvarium, premature ▪ Hydrocephalus → progressive ventricular closure of lambdoid and sagittal sutures, shallow enlargement & increased ICP & spinal cord orbits, enlarged J-shaped sella, and abnormal compression, intellectual disabilities spacing of teeth with dentigerous cysts ▪ Recurrent URTI, ear infection, noisy breathing, ▪ Assay the urinary excretion of GAGs; persistent copious nasal discharge, obstructive semiquantitative spot tests for ↑ urinary GAG airway disease excretion ▪ Limited language skills d/t intellectual disability, ▪ Quantitative analysis of single GAG and/or combined conductive & neurosensory hearing loss oligosaccharides by mass spectrometry detection and enlarged tongue tests: type-specific profiles in urine, serum, plasma, ▪ Death by cardiorespiratory failure by 10yo and dried blot spots ▪ Confirmed by enzyme assay on serum, leukocytes, Mild cultured fibroblasts ▪ Normal intellectual development and moderate ▪ Gene panels somatic manifestations at age 2yo, with variable disease progression Early infantile form ▪↑ CK activity, AST, ALT, LDH ▪ Within the first 6mos of life; progressive -Hypotonia, macroglossia, cardiomegaly or ▪ Urine glucose tetrasaccharide – glycogen hypertrophic cardiomyopathy, bulbar weakness, breakdown metabolite, biomarker for disease hepatomegaly, progressive weakness (floppy infant severity & treatment response appearance) ▪ Death in the first 2y of life (Fanaroff), 1 year ▪ CXR – Massive cardiomegaly (Nelsons) if untreated w/ ERT ▪ ECG – High-voltage QRS complex, WPW syndrome, shortened PR interval Later-onset form (Juvenile, Childhood, Adult) ▪ 2DED: Thickening of both ventricles and/or CLINICAL MANIFESTATIONS ▪ Milk restriction only ▪ Rapid initiation of milk restriction may resolve cataracts that were already present ▪ Good ▪ Lactose/ required ▪ Similar to GALT deficiency, with no evidence of ovarian failure if maintained on proper diet galactose-restricted diet is ▪ Galactose-restricted diet is not generally required for peripheral form TREATMENT Severe ▪ Initiate enzyme replacement therapy following diagnosis: appropriate for mild CNS involvement or to stabilize extraneural manifestations in young patients before stem cell transplantation. ▪ Hematopoietic stem cell transplant (HSCT) if <2yo with baseline mental developmental index >70; careful consideration of risk vs. benefit advised if >2yo Mild ▪ HSCT not recommended for mild form because of treatment complications ▪ Symptomatic therapy ● Focuses on respiratory and CV complications, hearing loss, carpal tunnel syndrome, spinal cord compression, hydrocephalus ▪ Enzyme replacement therapy w/ recombinant human -glucosidase ASAP ▪ CRIM-negative patients should receive immune tolerance induction before initiating therapy ▪ MTX, rituximab, IVIG – immunomodulation therapy for patients who develop enzyme neutralizing antibodies after starting ERT Late-onset form: ▪ Enzyme replacement therapy Good COMPLICATIONS / PROGNOSIS Severe ▪ Early initiation of enzyme replacement therapy followed by HSCT improves outcome, except for skeletal changes ▪ Comprehensive symptomatic treatment HSCT ▪ ↑ Life expectancy with resolution or improvement of growth, hepatosplenomegaly, joint stiffness, facial appearance, skin changes, OSA, heart disease, communicating hydrocephalus, hearing loss. Enzyme activity in serum & urinary GAG excretion normalize. ERT w/ -L-iduronidase ▪ Reduces organomegaly and ameliorates rate of growth, improves joint mobility, reduces the number of episodes of sleep apnea and urinary GAG excretion. ▪ Rapid initiation of enzyme replacement therapy (ERT) improves outcome Late-onset ▪ Variable depending on age of onset and age of initiation of therapy Batch Clingy GALACTOKINASE (GALK) DEFICIENCY ▪ Galactokinase (GALK) deficiency ▪ This enzyme catalyzes the phosphorylation of galactose ▪ Primary metabolites that accumulate are galactose & galactitol ▪ 2 genes that encode galactokinase: GK1 on chromosome 17q24 and GK2 on chromosome 15. ▪ UDP-galactose-4- epimerase deficiency ▪ GALE located on chromosome 1 at 1p36 43 ▪ 1 in 40,000 live births in Caucasians ▪ 1 in 18,000 in Han Chinese ▪ Gene for acid ⍺-glucosidase (GAA) is on chromosome 17q25.2. ▪ Generally, >12 months ▪ Slowly progressive, proximal limb girdle muscle weakness (within 1yr to 6th decade of life, lower>upper limb) – mostly pelvic girdle, paraspinal muscles, diaphragm ▪ Respiratory insufficiency: somnolence, morning headache, orthopnea, exertional dyspnea → sleep disordered breathing & RF ▪ Cardiac involvement – rhythm disturbances, less severe cardiomyopathy ▪ Lingual weakness, ptosis & dilation of blood vessels (basilar artery, ascending aorta) → basilar artery aneurysm w/rupture ▪ GI: postprandial bloating, dysphagia, early satiety, diarrhea, chronic constipation, IBD ▪ GU: bladder & bowel incontinence, weak urine stream, dribbling intraventricular septum and/or LVOT obstruction ▪ Muscle Biopsy: Vacuoles that stain (+) glycogen; acid phosphatase increased (compensatory increase if lysosomal enzymes) ▪ EM: Glycogen accumulation within a membranous sac and in the cytoplasm ▪ EMG: Myopathic features with excessive electrical irritability of muscle fibers and pseudomyotonic discharges ▪ Peripheral Nerve Biopsy: Glycogen accumulation in neurons and Schwann cells ▪ Enzyme assay: Dried blood spots, leukocytes, blood mononuclear cells, muscle, or cultured skin fibroblasts – deficient acid α-glucosidase activity. ▪ High protein diet ▪ Respiratory muscle strength training Approaches under development ▪ Chaperone molecules to enhance rhGAA delivery ▪ neoGAA, which is a second-generation ERT with a high number of mannose-6-phosphate (M6P) tags that enhances M6P receptor targeting and enzyme uptake ▪ Gene therapy to correct endogenous enzyme production pathways ▪ Confirmed by Gene sequencing: 2 pathogenic variants in the GAA gene Prenatal diagnosis Amniocytes/chorionic villi X-LINKED ADRENOLEUKODYSTROPHY Most common peroxisomal disorder ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Impaired peroxisomal metabolism of very longchain fatty acids ▪ Accumulation of saturated VLCFAs (carbon chain length of ≥24) & progressive dysfunction of adrenal cortex & CNS d/t genetically deficient peroxisomal degradation of FA ▪ Defective gene (ABCD1) codes for peroxisomal membrane protein (ALD protein) → disruption of transport of saturated fatty acids into the peroxisome → continued elongation of progressively longer FA ▪ Excess hexacosanoic acid (C26:0) ● Most characteristic feature ● Increases the viscosity of the plasma membrane, which may interfere with receptor & other cellular function ▪ Males 1 in 21,000 ▪ Combined males & heterozygous females 1 in 17,000 ▪ Lamellar cytoplasmic inclusions (w/c probably consists of cholesterol esterified with VLCFA) in adrenocortical cells, testicular Leydig cells, and nervous system macrophages; most prominent in zona fasciculata of the adrenal cortex (cells distended w/ abnormal lipids) ▪ Severity of illness & rate of progression correlate with intensity of the inflammatory response which may be partially cytokine mediated and may involve an autoimmune response triggered in an unknown way by the excess of VLCFAs. Mitochondrial PPS Oral Exam Reviewer 2020 PEROXISOMAL DISORDERS CLINICAL MANIFESTATIONS / DIAGNOSIS PHYSICAL EXAM Childhood-onset (4-8 yo) ▪ High levels of VLCFAs in plasma, RBCs, or ▪ M > F, X-linked recessive cultured skin fibroblast ▪ Intracerebral inflammatory response → demyelination → ▪ Mutation analysis – most reliable method for accumulation of perivascular lymphocytes the identification of carriers ▪ Adrenal insufficiency, impaired cortisol response to ACTH ▪ Neuroimaging MRI: stimulation, mild hyperpigmentation ● White matter lesions, symmetric, involve ▪ Regression of cognition, behavior, vision (involvement of splenium of the corpus callosum and parietooccipital cortex) periventricular white matter in the posterior ▪ Hyperactivity, inattention, and worsening school performance parietal and occipital lobes ▪ Loss of hearing (Impaired Auditory discrimination) and motor ● Garland of contrast enhancement adjacent function follow the initial symptoms → total disability & anterior to the posterior hypodense lesions ▪ Spatial orientation impaired, ataxia, poor handwriting, w/c corresponds to zone of intense perivascular seizures, strabismus. lymphocytic infiltration where the BBB breaks ▪ Progress rapidly with ↑ spasticity and paralysis, visual and down hearing loss, and loss of ability to speak or swallow. ▪ ↑ ACTH in plasma & subnormal rise of cortisol ▪ Death/vegetative state generally by 1.9 years levels in plasma after IV injection of 250μg of ▪ May continue veg. state for >10yrs ACTH Adolescent ALD (10-21 yo) ▪ Like childhood form but progression slower ▪ 10%: acute status epilepticus, adrenal encephalopathy, coma crisis, acute Later-onset (late adolescence/a dulthood) ▪ M>F, with females more mildly affected ▪ Adrenomyeloneuropathy beginning in late 20s (paraparesis, sphincter and sexual dysfunction, and adrenal insufficiency) – distal axonopathy that affects long tracts of the spinal cord TREATMENT COMPLICATIONS / PROGNOSIS Childhood-onset ▪ Immediate HSCT – if neurologic involvement is not yet present ▪ Bone marrow – derived cells do express ALDP, the protein that is deficient in ALD. Favorable effect cause by modification of the brain inflammatory response. Does not arrest the course in those who already had severe brain involvement and may accelerate disease progression. Not recommended in patients with performance IQ significantly <80. Childhood-onset ▪ May improve neurologic outcome (further follow-up studies are underway) Later-onset ▪ HSCT ▪ Corticosteroid replacement therapy ▪ Lorenzo’s oil ● 4:1 mixture of glyceryl trioleate and glyceryl trierucate ● Lower plasma levels of VLCFAs, but not to alter disease progression in males with cerebral disease Supportive Treatment ▪ Parent support groups, low-level resource services within a regular school program to home- and hospital – based teaching programs ▪ As leukodystrophy progresses, modulation of Later-onset ▪ Limited benefits of HSCT for neurologic problems after onset of symptoms ▪ Adrenal insufficiency – avoidable complication ▪ The most difficult neurologic problems are those related to bed rest, contracture, coma, and swallowing disturbances Other complications ▪ Behavioral disturbances and injuries associated with defects of spatial orientation, impaired vision, hearing, seizures ▪ Brain MRI every 6mos in neurologically asymptomatic boys and adolescents aged 3-15 yr. If the MRI is normal, BMT not indicated. Genetic Counselling and Prevention ▪ Identification of asymptomatic males permits institution of steroid replacement therapy when appropriate and prevents adrenal crisis ▪ Plasma VLCFA assay is recommended in all male patients with Addison disease ▪ DNA analysis permits accurate identification of carriers Batch Clingy DISEASE 44 damage and oxidative stress also appear to contribute. ▪ Inflammatory response mild/absent ▪ Adrenal insufficiency precedes and dominates neurologic abnormalities Addison only (male w/ addison, 25% have defect in ALD) ▪ Intact CNS/subtle neuro symptoms ▪ Many acquire adrenomyeloneuropathy in adulthood muscle tone & support of bulbar muscular function are major concerns. Baclofen in gradually increasing doses (5mg BID to 25mg QID) for treatment of acute episodic painful muscle spasms. Changing the diet to soft and pureed or eventually use of gastrostomy tube ▪ Anticonvulsants for seizures Batch Clingy Asymptomatic ALD ▪ With biochemical defect but free or CNS/Endo disturbances PPS Oral Exam Reviewer 2020 45 Batch Clingy PPS Oral Exam Reviewer 2020 46 Batch Clingy PPS Oral Exam Reviewer 2020 47 Batch Clingy PPS Oral Exam Reviewer 2020 48 Chronic ETIOLOGY / INCIDENCE / PATHOGENESIS DUCHENNE MUSCULAR DYSTROPHY (DMD) ▪ Most common hereditary neuromuscular disease ▪ X-lined recessive trait ▪ 1 in 3600 infant boys ▪ Abnormal gene at Xp21 locus (one of the largest genes) CLINICAL MANIFESTATIONS Characteristic clinical features: progressive weakness, intellectual impairment, hypertrophy of the calves, with proliferation of connective tissue and fibrosis in muscle ▪ Rarely symptomatic at birth or early infancy, some are mildly hypotonic ▪ Early gross motor skills (rolling over, sitting, standing) – achieved or mildly delayed ▪ Distinctive facies – not an early feature; facial muscle weakness is late event (in later childhood, a “transverse” or horizontal smile may be seen) ▪ Gower’s sign – may be seen at 3yo; always evident at 5 or 6yo ▪ Trendelenburg (waddling) gait appears at this time as well Common presentation in toddlers: delayed walking, frequent falling, toe walking, trouble running or going upstairs, developmental delay, malignant hyperthermia after anesthesia CONGE NITAL NEUROMUSCULAR DISORDERS HYPOXIC-ISCHEMIC ENCEPHALOPATHY BECKER MUSCULAR DYSTROPHY ▪ Similar to DMD ▪ Onset after 5 or 7yo ▪ Genetic defect at same locus of DMD CONGENITAL MYOTONIC DYSTROPHY ▪ Second most common muscular dystrophy ▪ Autosomal dominant ▪ Genetic defect causing dysfunction in multiple organ systems (striated muscle, smooth muscle, endocrinopathies, immunologic deficiencies, cataracts, dysmorphic facies, risk for malignancies, intellectual impairment, neuro abnormalities) TYPES Classic myotonic dystrophy (type 1) ▪ DM1 or Steinert disease ▪ Caused by CTG trinucleotide expansion on chromosome 19q13.3 in the 3’ untranslated region of DMPK (gene that encodes serine-threonine protein kinsase) Type 2 (DM2) – unstable CCTG tetranucleotide repeat expansion on chromosome 3q21 of an intron of the zinc finger 9 protein gene Late form (DM3) – identified at locus 15q21-q24 Hypoxemia – ↓ arterial concentration of oxygen resulting in Hypoxia – ↓ oxygenation to cells or organs Ischemia – blood flow to cells or organs that is inadequate to maintain physiologic function Major neonatal encephalopathies or seizure – due to PERINATAL EVENTS PPS Oral Exam Reviewer 2020 ▪ Age of complete loss of ambulation: 10-14yo ▪ Contractures most often involve ankles, knees, hips and elbows, neck lateral rotation, shoulders, elbows ▪ Scoliosis is common ▪ Cardiomyopathy – occurs after loss of independent ambulation ▪ Intellectual impairment occurs in majority although 20-30% have IQ <70 ▪ Milder and more protracted course than DMD ▪ Remain ambulatory until late adolescence or adulthood ▪ Given the increased level of activity, cardiac manifestations may be evident earlier ▪ Learning disabilities are less common ▪ DM1 symptomatic at any age ▪ DM2 rarely expressed in infancy or early childhood Facial appearance is characteristic: inverted V-shaped upper lip, thin cheeks, scalloped concave temporalis muscle, head narrow, palate high and arched, tongue thin and atrophic, long thin cylindrical contour neck ▪ Weakness is often mild in first few years (childhood onset form) or may not even become evident until adulthood (classic/adult-onset form) ▪ Progressive wasting of DISTAL muscles – exception to general rule of myopathies having proximal and neuropathies having distal distribution patterns ▪ Proximal muscles eventually undergo atrophy, scapular winging appears ▪ Gower’s sign is progressive ▪ Tendon stretch reflex usually preserved ▪ Myotonia evident until about 5yo → not a painful muscle spasm ▪ Poorly articulated speech IUGR with ↑vascular resistance ▪ 1st indication of chronic fetal hypoxia before peripartum period ▪ Variable or late deceleration pattern At delivery – meconium stained amniotic fluid → fetal distress occurred ▪ Depressed, fail to breathe, remain hypotonic or change to hypertonic state ▪ Pallor, cyanosis, periodic breathing with apnea, slow heart rate and unresponsiveness to stimulation – signs of HIE ▪ Cerebral edema develops next 24 hours – profound brainstem depression → during this DIAGNOSIS Serum CK level ▪ Consistently greatly elevated (even in pre symptomatic stages) ▪ 15,000-35,000 IU/L (Normal is <160 IU/L) 2DED and ECG ▪ Done every other year ▪ Yearly if have symptoms or reaches 10 yo) EMG ▪ Shows characteristic myopathic features but is not specific – no denervation found Genetic evaluation Muscle biopsy ▪ Primary dx test: DNA analysis of blood ▪ Classic EMG myogram is not found in infants but evident in early school years ▪ Serum CK and other enzymes may be normal or slightly elevated in hundreds (never thousands) ▪ ECG should be done yearly ▪ UTZ of abdomen and Xray of chest and abdomen w/ GI motility or swallowing studies ▪ Endocrine assessment (coz hypothyroidism and adrenocortical insufficieny is common), and glucose tolerance test ▪ Immunologic studies Differential Diagnosis for seizure ▪ IVH, hypocalcemia, hypoglycemia, CNS infection Diffusion weighted MRI – imaging of choice, most sensitive for detecting hypoxic brain injury CT scan – limited UTZ – limited in term TREATMENT / COMPLICATIONS ▪ No medical cure at this time ▪ Mainstay: Supportive and preventive care ▪ Glucocorticoids – slow decline in muscle strength and increase time of independent ambulation (initiate at 46yo) ▪ Cardiac care: ACE inhibitors, ARBS or B blockers ▪ Promptly treat pulmonary infections ▪ Preserve good nutritional state ▪ Physiotherapy COMPLICATIONS DMD ▪ Death occurs in late teens to 20s ▪ Causes: ● Respiratory failure during sleep ● Intractable heart failure ● Pneumonia ● Aspiration or airway obstruction BMD ▪ Lifespan is into 40-50s ▪ Cause of death: cardiac and pulmonary complications ▪ No specific medical treatment, but the cardiac, endocrine, GI, and ocular complications can be treated ▪ Anesthesia precautions ▪ 20-30% die in neonatal period and 33-50% of survivors are left with permanent neurodevelopmental abnormalities (CP, ↓ IQ, learning/cognitive impairment) ▪ Cerebral or body systemic therapeutic hypothermia reduces mortality and major neurodev impairment at 18mo of age (33.5C within 1st 6 hours after birth and maintained for 72 hr) Batch Clingy DISEASE 49 Fetal hypoxia may be caused by: ▪ Inadequate oxygenation of maternal blood from hypoventilation during anesthesia ▪ Low maternal BP from acute blood loss, spinal anesthesia, or compression of vena cava by gravid uterus ▪ Uterine tetany – inadequate relaxation of uterus for placental filling ▪ Abruption placenta ▪ Compression or knotting of cord ▪ Placental insufficiency from toxemia After birth hypoxia may be caused by: ▪ CHD or severe pulmo disease ▪ Severe anemia (hemorrhagic/hemolytic disease) ▪ Shock (sepsis, blood loss, IVH) ▪ Failure to breathe after birth because of in utero CNS injury or drug-induced suppression PATHOGENESIS Hypoxia and ischemia → anaerobic metabolism → ↑ lactate and inorganic phosphates Glutamate accumulates in damaged tissue → overactivation of NMDA, AMPA and kainate receptors → ↑ cellular permeability to Na and Ca ions → intracellular accumulation → cytotoxic edema and neuronal death Initial response: shunt blood through the ductus venosus, ductus arteriosus, foramen ovale with maintenance of perfusion of the brain, heart, adrenals Effects ▪ Congestion and petechiae in pericardium, pleura, thymus, heart, adrenals and meninges time seizures may occur (although most often sec to HIE, seizures may also be caused by vascular events, metab derangements, CNS infection and cerebral dysgenesis or genetic disorders ▪ Heart failure and cardiogenic shock, PPHN< RDS, GI perforation, hematuria → associated with asphyxia due to inadequate perfusion CNS Multiorgan Systemic Effects of Asphyxia EFFECTS HIE, infarction, intracranial hemorrhage, seizures, cerebral edema, hypotonia, hypertonia CV Myocardial ischemia, poor contractility, cardiac stunning, tricuspid insufficiency Pulmo Pulmo HTN, pulmo hemorrhage, RDS Renal Acute tubular or cortical necrosis Adrenal Adrenal hemorrhage GI Perforation, ulceration with hemorrhage, necrosis SYSTEM Skin Inappropriate secretion of ADH, hyponat, hypogly, hypocal, myoglobinuria Subcutaneous fat necrosis Hema DIC Metabolic Poor predictive variables for death/disability after HIE: ▪ Low (0-3) 10 min Apgar score ▪ Need for CPR in delivery room ▪ Delayed onset (≥20 min) of spontaneous breathing ▪ Severe neuro signs (coma, hypotonia, hypertonia) ▪ Seizures onset ≤12hrs or difficult to treat ▪ Severe, prolonged (~7 days) EEG findings including burst suppression pattern ▪ Oliguria/anuria >24hrs ▪ Abnormal neuro exam ≥14 days aEEG (amplitude integrated EEG) – who is at risk for long term brain injury ▪ Hypothermia decreases rate of apoptosis, suppresses neurotoxic mediators like glutamate, free radicals, nitric oxide, lactate Phenobarbital (DOC) ▪ Loading dose 20mg/kg ▪ Additional 5-10mg/kg ▪ Maintenance 3-5mg/kg For refractory seizures → Phenytoin or Lorazepam (first line), Levetiracetam (first or second line) ▪ Monitor vital signs, prevent hypotension, acid base balance COMPLICATIONS ▪ Greatest risk of adverse outcome → severe fetal acidosis (pH <6.7) and a base deficit >25mmol/L ▪ Brain death after HIE is diagnosed from clinical findings of: ● Coma unresponsive to pain, auditory or visual stimulation ● Apnea with PCO2 rising from 40 to >60mmHg ● Absence of brainstem reflexes ▪ Because of inconsistencies, there is no universal agreement reached regarding the definition of neonatal brain death Stages of HIE in Term infants PPS Oral Exam Reviewer 2020 STAGE 1 STAGE 2 STAGE 3 Hyperalert Lethargic Stupor, coma Muscle tone Normal Hypotonic Flaccid Posture Normal Flexion Decerebrate Reflexes/clonus Hyperactive Hyperactive Absent Myoclonus Present Present Absent Moro reflex Strong Weak Absent Pupils Mydriasis Miosis Unequal, poor light reflex Seizures None Common Decerebration Batch Clingy SIGNS Consciousness 50 EEG findings Normal Low voltage changing to seizure activity Duration <24hr if progresses; otherwise may remain normal 24hrs to 14 days Days to weeks Outcome Good Variable Death, deficits Burst suppression to isoelectric severe INTRAVENTRICULAR HEMORRHAGE STATIC ENCEPHALOPATHY Nonprogressive Brain Disorders like Cerebral Palsy and Mental Retardation PERIVENTRICULAR LEUKOMALACIA (PVL) ▪ Involves intrauterine and postnatal events ▪ Hypoxia, venous obstruction from IVH, or undetected fetal stress may result in decreased perfusion to the brain, leading to periventricular hemorrhage and necrosis ▪ Risk for PVL increases in infants with IVH or ventriculomegaly CEREBRAL PALSY ▪ Group of permanent disorders of movement and posture → activity limitation attributed to nonprogressive disturbance in the fetal or infant brain ▪ Considered as a static encephalopathy but some features like movement disorders and orthopedic complications can progress over time ▪ 80% of cases point to antenatal factors, few had congenital anomalies external to CNS, 10% have evidence of intrapartum asphyxia. ▪ Intrauterine exposure to maternal infection was associated with significant ↑ in CP in normal BW infants ▪ Intracerebral hemorrhage and periventricular leukomalacia are the major lesions that contribute to CP in preterm infants PPS Oral Exam Reviewer 2020 ▪ Preterms <32 weeks evaluated with routine cranial UTZ to check ▪ Infants <1000gms highest risk, UTZ within 3-7th day of life ▪ Ff up at 36-40 weeks AOG to check for PVL MRI – more sensitive for evaluation of extensive periventricular injury, more predictive of longterm outcome GRADING Grade 1 – bleeding isolated to subependymal area Grade 2 – bleeding within ventricle without ventricular dilatation Grade 3 – IVH with ventricular dilatation Grade 4 – IVH with Parenchymal hemorrhage ▪ Usually clinically asymptomatic until neuro sequelae of white matter damage become apparent in later infancy – as spastic motor deficits ▪ May be present at birth but occurs later ▪ Divided into major motor syndromes ▪ Commonly associated with a spectrum of developmental disabilities (intellectual impairment, epilepsy, visual, hearing, speech, cognitive and behavioral abnormalities) Classification of Cerebral Palsy and Major Causes MOTOR NEUROPATHO/MRI MAJOR CAUSES SYNDROME Spastic diplegia (35%) PVL, periventricular cysts or scars in white matter, enlarged ventricles, squared off posterior ventricles Prematurity Ischemia Infection Endocrine/metabolic Spastic quadriplegia (20%) PVL, multicystic encephalomalacia, cortical malformations Endocrine/ metabolic/ genetic/ developmental ▪ Antenatal steroids – 24-34 weeks at risk for preterm delivery → decrease risk IVH ▪ Post hemorrhagic hydrocephalus – VPS insertion ▪ Symptomatic Seizures – anticonvulsant drugs Anemia and coagulopathy – PRBC or FFP Insert VP shunt Serial LP and ventricular taps – temporizing but not therapeutic COMPLICATIONS ▪ Post hemorrhagic hydrocephalus ▪ Hydrocephalus – 2-4 weeks after PROGNOSIS ▪ Neuro development (CP, mental devt index if <1000gm) MRI (check table from previous column) Multidisciplinary team ▪ Neurodev pediatrician ▪ Child neuro ▪ Rehab med ▪ OT, PT, speech patho ▪ Social workers ▪ Educators ▪ Dev psychologists ▪ MgSO4 given to mothers in premature labor with birth imminent <32wks AOG showed significant reduction of CP at 2yo ▪ Cooling term infants with HIE to 33.3C for 3 days starting within 6hr birth decreases dyskinetic or spastic quadriplegia form of CP ▪ Spastic diplegia → assistance of adaptive equipment (orthoses, walkers, poles, standing frames) ▪ Quadriplegia → motorized wheelchair, feeding devices modified typewriters, customized seating arrangements Batch Clingy ▪ Develops spontaneously, or due to trauma and asphyxia, often with preterm infants (IVH) ▪ Can be associated with fetal alloimmune thrombocytopenia, cerebral hemorrhage, or porencephalic cyst ▪ VLBW infants – IVH and PVL IVH ▪ Deterioration on 2nd or 3rd day of life ▪ Preterm ▪ Hypotension, apnea, pallor or cyanosis, poor suck, abnormal eye signs, high pitched shrill ▪ Occur in the gelatinous subependymal germinal matrix cry, convulsions, decreased muscle tone ▪ Immature blood vessels plus poor tissue support ▪ Metabolic acidosis, shock ▪ Decreased hematocrit or failure to rise after transfusion 51 ▪ Primarily in infants born <32wks AOG ▪ Alveolar hypoplasia with concomitant small airway dysfunction and impaired pulmonary vascular growth ▪ Result of lung injury in infants requiring mech vent and supplemental O2 ▪ Interference with lung anatomic maturation CAUSES ▪ Early gestational age ▪ Low birth weight ▪ Lung barotrauma ▪ Exposure to hyperoxia ▪ Lung inflammation ▪ Pre and postnatal infections ▪ Potential modifier genes ▪ Epigenetic factors BRONCHOPULMONARY DYSPLASIA / CHRONIC LUNG DISEASE New BPD ▪ BW <1000 grams ▪ <28 weeks AOG Features in new BPD ▪ Alveolar hypoplasia ▪ Variable saccular wall fibrosis ▪ Minimal airway disease Hemiplegia (25%) Stroke: in utero or neonatal Focal infarct or cortical, subcortical damage Cortical malformations Thrombophilic disorders, infection, genetic/ developmental, periventricular hemorrhagic infarction Extrapyramidal (athetoid, dyskinetic) (15%) Asphyxia: symmetric scars in putamen and thalamus Kernicterus Mitochondrial Asphyxia, kernicterus, mitochondrial, genetic/ metabolic ▪ Tachypnea, head bobbing, retractions when ill or at baseline depending on severity ▪ Breath sounds may be clear but many have baseline wheeze or coarse crackles ▪ Persistent fixed wheeze or stridor suggests subglottic stenosis or large airway malacia ▪ Fine crackles present in those prone to fluid overload ▪ No improvement on 3rd or 4th day of ventilation → increased need for O2 and ventilator support ▪ RD worsens → hypoxia, hypercapnia, O2 dependence, R sided heart failure ▪ Alveolar collapse and volutrauma from mech vent → lung injury ▪ Free radicals from O2, immaturity, infection, PDA, malnutrition → contribute to BPD COMPLICATIONS Quality of life Trauma Infection CXR: air trapping, focal atelectasis, interstitial changes and/or peribronchial thickening Chronic respiratory insufficiency ▪ ↑ Serum bicarbonate ▪ ↑ CO2 ▪ Hypoxemia ▪ Polycythemia ▪ After discharge, high risk of rehospitalization ▪ Nutritional support – calories ▪ Fluid restriction Drugs ▪ Furosemide + KCl supplementation – for fluid overload, decreases PIE and PVR ▪ Inhaled bronchodilators (albuterol, CV or DV) Maintenance of oxygenation ▪ Severe may require chronic O2 support ▪ Target O2 sat is ≥ 92% FOUR PATHOLOGIC STAGES ▪ Acute lung injury ▪ Exudative bronchiolitis ▪ Proliferative bronchiolitis ▪ Obliterative fibroproliferative bronchiolitis Chronic respi insufficiency ▪ Tracheostomy, mech ventilation FEATURES MILD MODERATE SEVERE <32wk AOG at birth O2 reqt for at least 28 days Breathing RA at 36wk PMA or at discharge, whichever comes first <30% supplemental O2 at 36wks PMA or at discharge, whichever comes first >30% supplemental O2 and/or PPV at 36wk PMA or at discharge whichever comes first >32wk AOG at birth O2 reqt for at least 28 days Breathing RA at 56 DOL or at discharge, whichever comes first <30% supplemental O2 at 56 DOL or at discharge, whichever comes first >30% supplemental O2 at 56 DOL or at discharge, whichever comes first ▪ Occurrence indirectly proportional to gestational age ▪ Vit A supplementation in VLBW infants ↓ risk of BPD ▪ Rhizotomy ▪ Serial botulinum toxin injection ▪ Prevention of respiratory viral illness ▪ Monitor for development of pulmonary HTN PROGNOSIS ▪ Prognosis better for neonates >1500gms ▪ Prognosis is generally good for infants with BPD and most are weaned off by 1yr of life (those requiring mech vent at home are often weaned during toddlerhood) ▪ Mortality – highest in those who are ventilator dependent >6mos COMPLICATIONS ▪ Growth failure, psychomotor retardation, parental stress, nephrolithiasis, osteopenia, electrolyte imbalance ▪ Airway problems – subglottic stenosis, vocal cord paralysis, ▪ Cardiac – pulmonary HTN, systemic HTN ▪ Aspiration from dysphagia and/or GER ▪ Pulmonary exacerbation is triggered during viral respiratory infections Batch Clingy ▪ Severe BPD – prolonged mech vent; gradual weaning PPS Oral Exam Reviewer 2020 52 Acute DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Imbalance of caloric intake and expenditure ▪ Affected by environmental factors – food, physical activity, changes in health behavior and sleep, genetics Sensitive periods (increased risk for obesity): infancy, adiposity rebound (when body fat is lowest approx. 5.5 yo), adolescence OBESITY Neuroendocrine Feedback loops – appetite and weight negative feedback system ▪ Links adipose tissue, GI tract and CNS ▪ GI hormones – CCK, glucagon like peptide 1, peptide YY and vagal neuronal feedback- promote satiety ▪ Ghrelin – stimulates appetite ▪ Adipose tissue – feedback regulates energy levels to brain through release of leptin (low leptin stimulates food intake, high leptin inhibits hunger) and adiponectin (reduced levels if obese, increased if fasting ▪ Peptide YY – reduces food intake via the vagal-brainstemhypothalamic pathway (lower levels in obese children) ▪ Result of abnormally high blood cortisol levels of other glucocorticoids ▪ Can be iatrogenic or result of endogenous cortisol secretion (adrenal gland tumor or hypersecretion of ACTH by pituitary) ▪ Most common cause – exogenous administration of glucocorticoid hormones ▪ Endogenous Cushing (infants) – by functioning adrenocortical tumor (signs of cortisolism along with hypersecretion of steroids, androgens, estrogen and aldosterone) ▪ Endogenous Cushing (children >7 yrs) – Cushing disease, excessive ACTH secreted by pituitary adenoma (microadenoma) → bilateral adrenal hyperplasia CUSHING SYNDROME ▪ Ectopic ACTH secretion (ACTH dependent - uncommon in children) – islet cell carcinoma of pancreas, NB, Wilms, thymic carcinoid ▪ ACTH independent CS – nodular hyperplasia and adenoma formation occurs in McCune Albright syndrome ▪ Mutation of G protein where ACTH receptor normally signals, if mutation present, cortisol and cell division stimulated independently CLINICAL MANIFESTATIONS Endocrine causes – may show SLOW linear growth Genetic causes – may manifest with extreme hyperphagia, coexisting dysmorphic features, cognitive impairment, vision and hearing abnormalities and short stature DIAGNOSIS Overweight – BMI in 85–95th percentile Obese – BMI ≥95th percentile Check comorbidities Developmental delay, hearing impairment, difficulty snoring, sleeping or daytime sleepiness → sleep apnea Family history – parental obesity (increased risk) ▪ Check BP, acanthosis nigricans (insulin resistance), hirsutism (PCOS), Tanner staging ▪ Labs – FBS, Triglycerides, LDL, HDL, liver function tests ▪ 24-hour food recall ▪ Seen in infants <1yo ▪ More severe in infants ▪ Rounded face with prominent cheeks and flushed appearance (moon facies) ▪ Generalized obesity common Children with adrenal tumors ▪ Signs of abnormal masculinization occur ▪ Hirsutism of face and trunk ▪ Pubic hair, acne deepening of voice ▪ Enlargement of clitoris (girls) ▪ Impaired growth (length below 3 rd percentile ▪ Hypertension → may lead to heart failure ▪ Increased susceptibility to infection Older children ▪ Obesity and short stature ▪ Severe obesity of face and trunk compared with extremities ▪ Striae on hips, abdomen and thighs ▪ Puberty delayed, amenorrhea ▪ Weakness, headache and emotional lability COMPLICATIONS ▪Increased risk for morbidity -CVD, type 2 DM, HTN, hyperlipidemia, nonalcoholic fatty liver disease (cirrhosis) ▪ Mechanical – OSA, orthopedic complications ▪ Low self-esteem, depression, eating disorders Cortisol levels – diurnal (normally elevated 8am, decrease to <50% by midnight) ▪ Cushing – midnight cortisol levels high (>4.4 mg/dl) ▪ Can be measured saliva samples ↑ Urinary excretion of free cortisol – measure 24hour urine sample Single dose Dexamethasone suppression test ▪ 25-30mg/kg (max 2mg) given at 11pm ▪ In normal people – plasma cortisol at 8am <5mcg/dL Glucose tolerance test – abnormal Electrolytes – may be decreased If Cushing syndrome established – determine if caused by: ▪ Pituitary adenoma – ACTH may be normal ▪ ACTH secreting tumor – high ACTH ▪ Cortisol secreting adrenal tumor – ACTH suppressed Bolus of CRH ▪ Patients with ACTH dependent CS – exaggerated ACTH and cortisol response ▪ Adrenal tumors – no increase in ACTH and cortisol TREATMENT / COMPLICATIONS / PROGNOSIS Nutrition + exercise + cognitive behavioral approaches ▪ Recommendations about caloric intake ▪ Eating patterns ▪ Traffic light diet (groups food into green, yellow, red) ▪ Limit screen time and more physical activity Adolescents – Sibutramine (NE or serotonin reuptake inhibitor) and Orlistat (intestinal lipase inhibitor) Bariatric surgery – considered only in kids with: ▪ Complete or near complete skeletal maturity ▪ BMI >40 ▪ Medical complication of obesity after they have failed 6mos of a multidisciplinary weight management program Transsphenoidal pituitary microsurgery ▪ TOC ▪ Relapses tx with re-operation or pituitary irradiation Cyproheptadine ▪ Centrally acting serotonin antagonist that blocks ACTH release ▪ Used in adults, rare in children Adrenalectomy ▪ If pit adenoma no response to tx or if ACTH secreted by ectopic metastatic tumor ▪ May lead to ↑ ACTH secretion by unresected pit adenoma → Nelson syndrome ▪ s/p Adrenalectomy – preop and post op replacement tx with corticosteroid COMPLICATIONS Post op: sepsis, pancreatitis, thrombosis, poor wound healing, sudden collapse CT scan – detects adrenal tumors >1.5cm MRI – may detect ACTH secreting pituitary adenomas Bilateral inferior petrosal blood sampling – measure ACTH before and after CRH, required to localize tumor DIFFERENTIAL DIAGNOSIS Cushing – Obesity + striae + HTN Simple Obesity ▪ Tall (CS short or decelerating growth) ▪ Salivary nighttime levels of cortisol are normal PPS Oral Exam Reviewer 2020 Batch Clingy ▪ HTN, hyperglycemia, osteoporosis 53 ▪ Cortisol secretion suppressed by oral dexamethasone Generalized glucocorticoid resistance ▪ ↑ Cortisol and ACTH HTN, hypokalemia, precocious puberty Complex DISEASE PRADER WILLI SYNDROME ETIOLOGY / INCIDENCE / PATHOGENESIS Uniparental disomy – both chromosomes of a pair or areas of 1 chromosome inherited from single parent UPD chromosome 15 – Prader willi (maternal UPD, paternal contribution missing) and Angelman syndrome ▪ Paternal deficiency of HB11-85 snoRNAs (small nucleolar RNAs) ▪ Early in life: hypotonic that they cannot consume enough calories to maintain weight ▪ NGT feeding, FTT common ▪ 1st 4 yrs of life – muscle tone improves ▪ Voracious appetite DIAGNOSIS TREATMENT / COMPLICATIONS / PROGNOSIS ▪ Initial management of hypotonia and feeding ▪ Evaluate for hypogonadism or hypopituitarism ▪ Management of obesity ▪ Monitoring for scoliosis ▪ Therapy for behavioral issues ▪ Surgery – cryptorchidism, scoliosis, tonsillectomy (if with OSA) Batch Clingy ▪ Associated with advanced maternal age ▪ Embryo starts off as trisomy 15 then loss of paternal chromosome CLINICAL MANIFESTATIONS ▪ Hypotonia of prenatal onset ▪ Postnatal growth delay ▪ Almond shaped eyes, small hands and feet, developmental disability ▪ Hypogonadotrophic hypogonadism and obesity after infancy PPS Oral Exam Reviewer 2020 54 Complex PREDIABETES DIABETES MELLITUS TYPE 1 ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Individuals with abnormalities in blood glucose homeostasis who are at ↑ risk of diabetes ▪ Not a clinical entity but a risk factor for future diabetes and CV disease ▪ Often associated with metabolic syndrome – insulin resistance, compensatory hyperinsulinemia, obesity, dyslipidemia, HTN ▪ Most common type in childhood (median age 7-15yo) ▪ Autoimmune destruction of pancreatic islet B cells → permanent insulin deficiency ▪ Genetic predisposition: HLA and other genes ▪ Unknown environmental insult – triggers autoimmune process ▪ Factors: ● Cow’s milk feeding at early age ● Viral infections (Coxsackie, CMV, enteroviruses, mumps) – direct infection of B cells → lysis and release of self-antigens; clearest evidence is in congenital rubella syndrome (70% associated with B cell autoimmunity, development of type 1 DM in 40, time lag may be as high as 20yrs, no association if rubella appears after birth or when vaccinated) ● Vit D deficiency and environmental ▪ T cell mediated, Ab to islet cell antigens months to years before onset of B cell dysfunction ▪ Less common types – genetic defects of insulin receptor or inherited abnormalities in sensing of glucose concentration by pancreatic B cells FINDINGS / CLINICAL MANIFESTATIONS Defined by: ▪ Impaired fasting glucose (FBS 100-125mg/dL) ▪ Impaired glucose tolerance (2h postprandial glucose 140-199mg/dL) ▪ HbA1c values of 5.7-6.4% ▪ FBS of 99mg/dL upper limit of normal ▪ Associated features: Lean or weight loss at diagnosis, thyroid autoimmunity, celiac disease ▪ DKA at presentation (25%) Based on 4 glucose abnormalities ▪ FBS >126mg/dl ▪ Random venous plasma glucose >200mg/dL with symptoms of hyperglycemia ▪ Abnormal OGTT with 2hr post prandial >200mg/dL ▪ HbA1C >6.5% Classic Presentation: POLYURIA + POLYDIPSIA + POLYPHAGIA AND WEIGHT LOSS (reflecting hyperglycemic and catabolic physiologic state) ▪ Other symptoms: fatigue, weakness, general feeling of malaise ▪ Hyperglycemia – insulin secretory capacity inadequate to enhance peripheral glucose uptake and suppress hepatic and renal glucose production ▪ Insulin deficiency – causes postprandial hyperglycemia then fasting hyperglycemia ▪ Ketogenesis – sign of more complete insulin deficiency ▪ Lack of suppression of gluconeogenesis and glycogenolysis exacerbates hyperglycemia ▪ Fatty acid oxidation generates ketones → B hydroxybutyrate, acetoacetate, acetone ● Protein stores in muscle and fat stores in adipose tissue are metabolized to provide substrates for gluconeogenesis and fatty acid oxidation DIABETES MELLITUS TYPE 2 DIABETIC KETOACIDOSIS ▪ Insulin resistance and progressive non-autoimmune B cell failure ▪ Common in adults but with ↑ incidence in childhood and adolescence due to rise in obesity (rarely younger than 10yo) ▪ No insulin → persistent partial hepatic oxidation of fatty acids to ketone bodies ▪ 2/3 ketone bodies (organic acids) → elevated anion gap ▪ Lactic acidosis – severe dehydration, ↓ tissue perfusion ▪ Osmotic diuresis from hyperglycemia → dehydration ▪ Vomiting from acidosis and tachypnea → ↑ dehydration PPS Oral Exam Reviewer 2020 ▪ Glycosuria –serum glucose exceeds renal threshold for glucose reabsorption (160-190mg/dl) → polyuria/nocturia begins ● Osmotic diuresis (lose Na, K, electrolytes) → dehydration ● Female patients develop vulvovaginal candidiasis from chronic glycosuria ▪ Polydipsia and polyphagia – triggered by daily losses of as high as 5L water and 250g glucose (representing 1000 calories or 50% average daily caloric intake ▪ Weight loss – persistent catabolic state and loss of calories through glycosuria and ketonuria When disease progress, ketoacids accumulate → abdominal pain, nausea, emesis → inability to replace urinary water losses → dehydration accelerates manifested by weakness, orthostasis and further weight loss → DKA ▪ Presentation is more insidious: excessive weight gain and fatigue, or incidental glycosuria on routine PE ▪ DKA at presentation (5-20%) ▪ Acanthosis nigricans (dark pigmentation of skin creases in nape of neck) – present in majority w/ accompanying hyperinsulinemia ▪ Associated features: PCOS, HTN, hyperlipidemia, fatty liver disease, family history ▪ Dehydration, N/V, lethargy, altered mental status, in extreme cases – coma Advanced ketoacidosis symptoms ▪ Abdominal pain – mimics acute abdomen (2 vomiting or paralytic ileus) *presence of polyuria (despite dehydration) – differentiates DKA from GI disorders ▪ Kussmaul respiration – deep, heavy, non-labored rapid breathing, respiratory compensation for acidosis ▪ Fruity breath odor (from acetone) – detected patient’s breath TREATMENT ▪ Lifestyle modification Goal: maintain blood glucose as close to normal and delay onset of complications (retinopathy, nephropathy, neuropathy) ▪ <5yo – maintain between 80-180 ▪ School age – 80-150 ▪ Adolescents – 70-130 Insulin ▪ Fast acting insulin during meals ▪ Long acting insulin at bedtime Nutrition ▪ CHO 50-65% ▪ Protein 12-20% ▪ Fat 30% ▪ 3 meals and 3 snacks Blood glucose testing ▪ Before each meal and bedtime ▪ If sick, measure urine ketones also Long term control: HgbA1C – reflect blood glucose for the past 3 months ▪ <6yo – target 7.5%-8.5% ▪ 6-13yo – <8% ▪ 13-18yo – <7.5% COMPLICATIONS ▪ Annual ophthalmologic exam for retinopathy ▪ Check urine for microalbuminuria annually ▪ Chronic autoimmune lymphocytic thyroiditis hypothyroidism common → ▪ Lifestyle modification ▪ Metformin ▪ Insulin ▪ NPO ▪ Monitor I&O and neurologic status ▪ Have Mannitol at bedside (1g/kg IV push for cerebral edema) TREATMENT PROTOCOL 1st hour (Quick Volume Expansion) ▪ Initial IV bolus 10-20cc/kg PNSS or LRS Batch Clingy DISEASE 55 ▪ Electrolyte abnormalities – lost in urine ▪ Hydrogen ions accumulate – IC K exchanged for H ions ● Serum K ↑, then ↓ when excreted by kidney ● ↓ Serum K ominous sign of body K depletion ▪ Phosphate depletion – ↑ renal phosphate excretion ▪ Na depletion – renal loss Na and GI losses (vomiting) Classification Of DKA NORMAL CO2 20-28 7.35pH 7.45 No change MILD 16-20 7.257.35 Oriented Alert but fatigued Clinical MODERATE 10-15 ▪ Prolonged corrected Q-T interval ▪ Diminished neurocognitive function ▪ Coma ▪ Fever- uncommon, search for infectious sources SEVERE <10 7.15-7.25 <7.15 Kussmaul respirations Kussmaul or depressed respirations Oriented but sleepy Arousable Sleepy to depressed sensorium Coma Honeymoon Phase ▪ If new onset, beta cell mass not yet completely destroyed ▪ Period of stable glucose control usually starts first few weeks of therapy, can last for 2 years Cardinal biochemical abnormalities ▪ ↑ Blood and urine ketones ▪ ↑ Anion gap ▪ ↓ Serum bicarbonate and pH ▪ ↑ Effective serum abnormality ▪ Hyponatremia is commonly present with hyperglycemia – 2o to osmotic dilution as water shifts to ECF ▪ Potassium and phosphate depletion is common after prolonged polyuria but may be masked by acidosis which leads to extracellular shifting of these ions COMPLICATIONS ▪ Cerebral edema (can lead to herniation) – most serious complication ▪ Osmolar shift → fluid accumulation in IC compartment and cell swelling ▪ ↓ Sensorium, sudden severe HA, vomiting, change in vital signs (bradycardia, HTN, apnea), dilated pupil, ophthalmoplegia, or seizure ▪ ↑ Risk for cerebral edema ● Higher initial BUN concentration, lower initial PCO2, failure of serum Na to increase, treat wit HCO3 ● Signs – obtundation, papilledema, pupillary dilation or inequality, HTN, bradycardia, apnea ● Tx – rapid use of IV Mannitol, endotracheal intubation, ventilation ▪ Thrombosis or infarction ▪ ATN with ARF by severe dehydration ▪ Pancreatitis ▪ Arrhythmias 2o to electrolyte abnormalities ▪ Pulmonary edema, bowel ischemia ▪ Peripheral edema – elevations in ADH and aldosterone ▪ Mauriac syndrome – growth failure and hepatomegaly 2 o to excess glycogen accumulated in liver, related to chronic underinsulinization LONG TERM COMPLICATIONS ▪ Diabetic retinopathy ▪ Diabetic nephropathy ▪ Diabetic neuropathy ▪ Skeletal complications (ex. fractures) ● To avoid rapid shifts osmolality ● Use NSS for replacement 1st 4-6hrs then 0.45 NaCl ▪ Insulin drip at 0.05 to 0.10u/kg/hr ● Serum glucose should ↓ at a rate of 100mg/dL/hr 2nd hour until DKA resolution ▪ 0.45% NaCl plus continue insulin drip ▪ 20meq/L Kphos and 20meq/L KAc ● If K<3meq/L, give 0.5-1.0meq/kg as oral K solution or ↑ IV K to 80meq/L ▪ 5% glucose if blood sugar <250mg/dL ● If serum glucose <250-300 – glucose infusion in IV should be done ● Insulin infusion should not be discontinued before resolution of acidosis Variable ▪ Oral intake w/ subcutaneous insulin ▪ No emesis, CO2 ≥16meq/L, normal electrolytes NOTE: Initial IV bolus is considered part of total fluid allowed in the first 24 hours and subtracted before calculating IV rate Acidosis ▪ Insulin decreases production of FFA and protein catabolism, enhances glucose usage in tissues ▪ Avoid HCO3 – ↑ in CNS acidosis (↑ diffusion of CO2 in BBB) ▪ Acidosis corrected – urine ketones may rise Electrolyte imbalance ▪ Serum K can ↓rapidly as insulin and glucose therapy improves acidosis → potassium exchanged for IC hydrogen ion ▪ If with adequate UO, add K to IV fluids ▪ Give 50% KCl and 50% K phosphate (20-40meq/L) ▪ If serum K >6, don’t add K! Monitoring ▪ Initial labs – serum glucose, Na, K, Cl, HCO3, BUN, Crea, Calcium, Phosphate, and Mg, ABGs for arterial and venous pH, and urinalysis ▪ Serum glucose monitored every hour ▪ Electrolytes every 2-3 hours ▪ Ca, Ph, Mg measured every 4-6hrs ▪ Assess neuro and mental status ▪ If with HA or deterioration of mental status, check for cerebral edema Batch Clingy Transition to outpatient ▪ If acidosis corrected, patient tolerates oral feedings → insulin d/c ▪ SC insulin given 30 mins before d/c IV insulin ▪ Insulin starting 0.5-0.7u/kg.24hrs (prepubertal) ▪ 0.7-1 u/kg/24hr for adolescents ▪ Serum glucose assessed before each meal, at bedtime, and periodically at 2 or 3 am PPS Oral Exam Reviewer 2020 56 Complex DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS CLINICAL MANIFESTATIONS Adrenal cortex ▪ Outer: Zona glomerulosa – mineralocorticoids (aldosterone) ▪ Middle: Zona fasciculata – glucocorticoids (cortisol) ▪ Inner: Zona reticularis – adrenal androgens Adrenal medulla ▪ Neuroendocrine (chromaffin) cells – catecholamines (dopamine, norepinephrine, epinephrine) ▪ Glial (sustentacular) cells LABORATORY FINDINGS / DIAGNOSIS TREATMENT ▪ Hypothalamus (CRH) → Pituitary (ACTH, Corticotropin) → ACTH → synthesis and release of cortisol and adrenal androgens ▪ Primary Adrenal insufficiency or cortisol deficiency (defect in adrenal gland) → over secretion of ACTH ▪ Cortisol deficiency (from ACTH or CRH deficiency) → ↓ serum ACTH and cortisol ▪ Endogenous glucocorticoids feedback – inhibit ACTH and CRH ▪ RAA and K – regulate aldosterone secretion ▪ Fetal CRH-ACTH adrenal axis operational in utero, deficiencies in cortisol synthesis lead to EXCESSIVE ACTH ex. 21 hydroxylase def – fetal adrenal gland secretes excess androgens, virilizing the fetus ADDISON DISEASE ▪ Acquired primary adrenal insufficiency ▪ Autoimmune destruction of adrenal cortex (marked lymphocytic infiltration) ▪ Most patients have antiadrenal cytoplasmic antibodies in their plasma ▪ 21-hydroxylase (CYP21) is the most commonly occurring biochemically defined autoantigen ▪ Form of 1o adrenal insufficiency with absence of glucocorticoid and mineralocorticoid ▪ Hyperpigmentation ▪ Salt craving ▪ Orthostatic hypotension ▪ Fasting hypoglycemia ▪ Anorexia ▪ Weakness ▪ Episodes of shock during severe illness ▪ Most definitive test: measurement of serum cortisol before and after administration of ACTH ▪ ↓ Cortisol ▪ ↑ ACTH ▪ ↓ Na (90%) ▪ ↑ K (occur later) and Ca ▪ ↑ Plasma renin ▪ Hypoglycemia and ketosis ▪ Eosinophilia, lymphocytosis, anemia ▪ ↑ Urinary excretion of Na and Cl, ↓ urinary K ▪ Glucocorticoid replacement (Hydrocortisone) and mineralocorticoid replacement (Fludrocortisone) if salt wasting + NaCl supplementation ● Double or triple doses during stressful states (ex. Infection or surgery) ▪ Surgical correction ambiguous genitalia (ex. Vaginoplasty and clitoral recession for females) ▪ Elevated serum androgens – suppressed with glucocorticoids ▪ If diagnosed prenatally, can start prenatal treatment with Dexamethasone 20ug/kg prepregnancy maternal weight daily in 2 or 3 divided doses COMPLICATIONS ▪ Overtreatment – growth stunting and excessive weight gain (Cushingoid features) ▪ Undertreatment – excessive height gain, skeletal advance and early appearance of puberty, testicular adrenal tumors for males Treatment must be immediate and vigorous: ▪ D5PNSS – correct hypoglycemia, hypovolemia, and hyponatremia ▪ Avoid hypotonic fluids – can precipitate or exacerbate hyponatremia ▪ If severe hyperK, give IV calcium or bicarbonate, intrarectal K binding resin, or IV glucose and insulin ▪ Water soluble form of IV HAA: Hydrocortisone sodium succinate *caution if w/ concomitant hypothyroidism as thyroxine can increase cortisol clearance Once acute manifestations are under control: ▪ Replacement treatment with hydrocortisone ▪ Mineralocorticoid replacement w/ fludrocortisone (monitored PPS Oral Exam Reviewer 2020 Batch Clingy ADRENAL DISORDERS ▪ Normal variation serum cortisol and ACTH levels – diurnal – ↑ in morning (highest at 8am) and ↓ at night (reduced by 50%) → lost in Cushing syndrome – ↑ midnight cortisol levels CONGENITAL ADRENAL HYPERPLASIA ▪ Female – virilization of external genitalia (uterus, ovaries and FT ▪ ↓ Cortisol ▪ Autosomal recessive remains unaffected) – mild clitoromegaly to complete fusion of ▪ ↑ ACTH ▪ Most common cause of adrenocortical insufficiency in infancy labioscrotal folds, to severe clitoromegaly making a phallus ▪ Very high baseline and ACTH stimulated 17-OHP (hundred to ▪ 21 hydroxylase deficiency (>90%) ▪ Male – appears normal at birth thousand-fold) ● Glucocorticoid deficiency only (simple virilizing disease) ● May present with mineralocorticoid deficiency (salt wasting Late onset CAH ▪ If with salt wasting crisis: hyponatremia, hyperkalemia, ↑ crisis). These 2 forms are collectively termed classic 21▪ Years after birth plasma renin hydroxylase deficiency. (Nonclassic has mildly elevated androgens ▪ Milder manifestations without ambiguous genitalia and may be asymptomatic) ▪ Acne, hirsutism, irregular menstrual cycles or amenorrhea (may Late onset CAH – ACTH stimulation test shows abnormally high ● Blocks conversion of 17OHP to 11-deoxycortisol → ↓ cortisol be confused with PCOS) response → ↑↑ ACTH → synthesis of steroids, shunting to overproduction of androgens ▪ Next most common cause of CAH is 11B hydroxylase deficiency 57 PHEOCHROMOCYTOMA ▪ Catecholamine secreting tumors arising from chromaffin cells ▪ Most common site of origin is adrenal medulla, R>L ▪ May be associated with genetic syndromes such as von Hippel Lindau disease ▪ Component of multiple endocrine neoplasia (MEN2A, MEN2B) ▪ Abnormally ↑ cortisol or other glucocorticoids ▪ Progressive central or generalized obesity ▪ Moon facies ▪ Short stature ▪ Hirsutism ▪ Weakness ▪ Nuchal fat pad ▪ Acne, striae ▪ HTN, hyperglycemia ▪ Delayed puberty ▪ Amenorrhea ▪ Emotional lability ▪ Osteoporosis ▪ HTN ▪ HA, palpitations, abdominal pain, dizziness, pallor, vomiting, sweating, seizures ▪ If severe, precordial pains radiating to arms, pulmonary edema, cardiac and hepatic enlargement, growth failure ▪ Internal and external genitalia formed at 6-13wks AOG ▪ Fetal gonad and external genitalia – bipotential, can develop into male and female phenotype ▪ Presence of SRY → differentiates into a testis → Leydig cells secrete testosterone → converted to DHT → enlargement, rogation, and fusion of the labioscrotal folds into scrotum → penis ▪ Female phenotype develops unless specific male influences alter development ▪ Total absence of androgens → female HYPOFUNCTION OF THE TESTES ▪ From puberty onwards → testosterone deficiency, infertility, or ▪ During fetal life, can be a component of disorders of sex both development and lead to degrees of ambiguous genitalia ▪ No evidence for prepubertal children since they do not yet produce significant amount of testosterone and are not yet producing sperm DISORDERS OF GONADS (Sexual Development) ▪ ↑ 24-hour urinary cortisol secretion ▪ Single dose dexamethasone suppression test (25-30ug/kg) given at 11pm → ↓ cortisol level at 8am in normal individuals ▪ ↑ Nighttime salivary cortisol levels ▪ 2 step dexamethasone (30 and 120ug/kg/day in 4 divided doses) ● In pituitary Cushing syndrome, the larger dose suppresses cortisol levels ● In ACTH independent Cushing syndrome, cortisol levels are not suppressed CT scan detects adrenal tumors >1.5cm MRI may detect ACTH secreting pituitary adenomas ▪ Proteinuria, glucosuria ▪ Polycythemia ▪ ↑ Blood and 24hr urinary metanephrine ▪ Urinary excretion of VMA is increased but vanilla containing foods and fruits can give falsely elevated results hence no longer routinely measured CT scan or MRI to localize tumors APPROACH to infant with GENITAL AMBIGUITY ▪ PE – noted where urethral opening lies and check fusion of anterior portion of labioscrotal folds ▪ If vaginal opening open but clitoris enlarged – late exposure to androgens ▪ Fully formed scrotum, normal and small penis (microphallus) – normal exposure to androgen during 9-13wks AOG ▪ GOAL: identify life threatening disorders (CAH) ▪ 46XY – small phallus that does not increase in size after androgen therapy → usually raised as female, some revert to male gender Primary hypogonadism (testes): ▪ 47XXY Klinefelter syndrome, males with >1 X chromosome, and XX males ▪ Testes and penis are abnormally small at birth ▪ 2o sex characteristics will not develop ▪ Long extremities (eunuchoid), fat accumulation in hips, buttocks, breasts, and abdomen ▪ ↑ Gonadotropins (hypergonadotropic) ▪ ↑ FSH and LH ▪ ↓ Testosterone at all ages ▪ Continuously ↑ AMH at puberty ▪ Noonan syndrome ▪ Short stature, webbing of neck, pectus carinatum or excavatum, cubitus valgus, R sided CHD (pulmo valvular stenosis, hypertrophic cardiomyopathy or ASD), characteristic facies, cryptorchidism, small testes ▪ High frequency sensorineural hearing loss ▪ Hepatosplenomegaly, hematologic diseases ▪ Serum inhibin-B – marker of Sertoli cell function for Noonan syndrome Secondary hypogonadism (pituitary gonadotropic hormone deficiency): tumors, infiltrative disease, autoimmune disease, trauma, stroke ▪ Kallman syndrome – most common form of hypogonadotropic hypogonadism ▪ Anosmia or hyposmia, synkinesia, hearing loss, midfacial defects, renal agenesis ▪ Low or absent levels (hypogonadotropic) ▪ Chromosomal analysis ▪ MRI to look for tumors and other anomalies in the hypothalamic-pituitary region PPS Oral Exam Reviewer 2020 ▪ Surgical removal Replace deficient hormones ▪ Cortisol in CAH ▪ Testosterone in child with androgen biosynthetic defects ▪ Human growth hormone therapy in Noonan and Turner ▪ Oxandrolone ± growth hormone for Turner ▪ Replacement therapy w/ estrogen is indicated for Turner but no consensus as to when to initiate Psychological support Batch Clingy CUSHING SYNDROME ▪ Most common cause – exogenous administration (long term steroids) ▪ Endogenous – adrenal adenoma, carcinoma, nodular adrenal hyperplasia, ACTH secreting pituitary microadenoma (→ bilateral adrenal hyperplasia → Cushing disease → most common endogenous etiology in children >7yo) by plasma renin activity and Na and K) ▪ Replacement of DHEA is still controversial ▪ Transsphenoidal pituitary microsurgery – treatment of choice in pituitary Cushing disease ▪ Parenteral glucocorticoids – given during and after surgery to avoid acute adrenal insufficiency 58 HYPOFUNCTION OF THE OVARIES Primary (hypergonadotropic): Premature ovarian failure – arrest of normal ovarian function before 40yo ▪ 45X Turner syndrome ▪ Sexual infantilism, webbed neck, cubitus valgus, broad chest, widely spaced nipples, short stature (cardinal finding), cardiac defects (bicuspid aortic valve, CoA, TAPVR), renal malformations, IBD, autoimmune thyroid disease, recurrent bilateral otitis media ▪ UTZ of heart, kidneys and ovaries + thyroid peroxidase antibodies and thyroglobulin antibodies + transglutaminase immunoglobulin A antibodies to screen for celiac disease for Turner’s syndrome ▪ Females with Noonan syndrome (46XX chromosomes) and ▪ Show similar anomalies with Turner normal sexual maturation usually occurs but delayed on 2yr average ▪ Central (hypogonadotropic): Multiple pituitary hormone deficiencies, chronic diseases ▪ Hypopituitarism ▪ Isolated deficiency of gonadotropins Hypothalamus secretes GnRH → pituitary releases LH and FSH → gonads secrete sex steroids (testosterone from testes, estradiol and progesterone from ovaries) Serum gonadotropin levels obtained – determine if HYPO or HYPER gonadotropic Females: FSH – ovarian production of estrogen, in puberty causes formation and support of corpus luteum Males: LH – production of testosterone from Leydig cells, later FSH stimulates development and support of seminiferous tubules Sex steroids and inhibin (produced by gonads) –suppress the secretion of gonadotropins Ultrasound of pelvic structures Permanent condition – replacement with sex steroids ▪ Transdermal estradiol or conjugated estrogens breakthrough bleeding happens ▪ Combined progesterone and estrogen used after until ▪ Males – testosterone enanthate and cypionate given IM once every 4 weeks GnRH released in episodic pulses vary during development and menstrual period ▪ Onset of puberty, ↑ at night then during the day ▪ Adrenarche then Gonadarche ▪ Constitutional delay- may be given low dose steroids for 3-6 months Females – thelarche (due to gonadarche), pubarche (due to adrenarche) then menarche (2-3 yrs later) ▪ Adequate calcium intake (risk for osteoporosis) Males – Scrotal thinning → enlargement of testes and appearance of pubic hair ▪ Turner – promote growth with exogenous human GH supplementation and induction of secondary sexual characteristics DELAYED PUBERTY – girls 13yrs old, boys 14yrs old DISORDERS OF PUBERTY Constitutional delay in Growth and Adolescence ▪ With family hx delayed puberty ▪ Delayed onset of pubertal development ▪ Significant bone age delay (2 SD below mean, 1.5-2y delay) Hypogonadotrophic Hypogonadism ▪ Difficult to distinguish from constitutional delay ▪ Normal dev’t in childhood and early puberty ▪ In adulthood, may have eunuchoid proportions Isolated Gonadotropin Deficiency ▪ Inability to release gonadotropin but no other pituitary abnormality ▪ Kallman syndrome – isolated gonadotropin deficiency with disorders of olfaction Syndromes of Hypogonadotropic Hypogonadism ▪ Anorexia nervosa – 1o or 2o amenorrhea, pubertal development PPS Oral Exam Reviewer 2020 ▪ ↓ Gonadotropin function when dieting, malnutrition, or if with chronic disease resulting in weight loss (<80% ideal weight) Batch Clingy Abnormalities of the CNS ▪ CNS tumors (pituitary adenoma, glioma, prolactinoma, craniopharyngoma) ▪ Idiopathic hypopituitarism – congenital absence of various combinations of pituitary hormones 59 absent or minimal ▪ Athletic amenorrhea – female athlete triad: anorexia, amenorrhea, osteoporosis ▪ Chronic or systemic illness (cystic fibrosis, DM, IBD) → pubertal delay or amenorrhea ▪ Hypothyroidism – inhibits onset of puberty and delays menstrual periods Hypergonadotropic Hypogonadism ▪ Ovarian failure – Turner syndrome (45XO) (common cause of ovarian failure and short stature) Klinefelter syndrome (47XXY) – most common cause of testicular failure Primary Amenorrhea ▪ Mayer Rokitansky Kauser Huster syndrome – congenital absence of uterus ▪ Imperforate hymen or vaginal septum ▪ Androgen insensitivity – normal feminization, absence of pubic and axillary hair, 1o amenorrhea ▪ All mullerian structures (ovaries, FT, vagina) lacking SEXUAL PRECOCITY (precocious puberty) – 2o sexual development <9yo boys, <8yo girls ▪ Determine which part of normal puberty present and whether estrogen effect of androgen effects or both are present Central Precocious Puberty (GnRH dependent Precocious Puberty) ▪ Premature activation of hypothalamic-pituitary-gonadal axis ▪ Endocrine and physical aspect is normal but too early ▪ Diff r/o – CNS diseases (hydrocephalus, meningitis, encephalitis, suprasellar cysts, head trauma, epilepsy, mental retardation, irradiation) ▪ Hamartomas, germinomas, optic or hypothalamic gliomas, astrocytomas, ependymomas ▪ Tall stature ▪ Advanced bone age ▪ ↑Sex steroid and pulsatile gonadotropin secretion ▪ ↑ Response of LH to GnRH Peripheral Precocious Puberty (GnRH Independent Precocious Puberty) ▪ Does not involve HPG axis ▪ Mc Cune Albright – most common cause, girls > boys ▪ Precocious gonadarche ▪ Bone disorder with polyostotic fibrous dyplasia ▪ Hyperpigmented macules (café au lait) ▪ Adrenal carcinomas and adenomas Males –familial GnRH independent sexual precocity with premature Leydig cell maturation ▪ HCG secreting tumors VARIATIONS in PUBERTAL DEVELOPMENT Isolated premature Thelarche ▪ ↑ Gonadotropins and ↓ sex steroid levels resulting from primary gonadal failure Labs ▪ Sex steroid levels (testosterone, androstenedione) ▪ LH (pubertal) and FSH (prepubertal) ▪ Thyroid hormone estradiol, DHEAS, ▪ Long acting analogs of GnRH (Leuprolide) – suppress gonadotropin secretion by downregulating GnRH receptors MRI of CNS ▪ Isolated appearance of uni- or bilateral breast tissue in girls (6mos to 3yrs) ▪ Inhibitor of testosterone synthesis (ketoconazole), antiandrogen (spironolactone) and aromatase inhibitor (testolactone) ▪ Surgical removal if with tumor ▪ If no other signs, evaluate every 6-12mos to monitor if with precocious puberty Gynecomastia ▪ 45-75% boys ▪ Prepubertal – suggests unusual source of estrogen, exogenous sources or from endogenous sources PPS Oral Exam Reviewer 2020 ▪ Ultrasound to check hyperplastic adrenals or ovarian tumor Batch Clingy Isolated Premature Adrenarche (Pubarche) ▪ Girls <6-7yo, boys <9yo ▪ If w/ other signs investigate for virilization, r/o CAH 60 Calcium and phosphate – regulated by 3 hormones ▪ Total serum Calcium measured, but ionized calcium preferable because it more nearly reflect physiologic adequacy Parathyroid hormone ▪ Secreted in response to a decrease in serum ionized calcium level (Ca ↓, PTH ↑) ▪ Stimulates 1a-hydroxylase in the kidney → enhanced production of 1-25dihydroxycholecalciferol (1,25(OH2)D3) → induced calcium binding protein (calbindin 3) in intestinal mucosa → Ca absorption ▪ Mobilizes calcium from bone to serum and enhances reabsorption of Ca by kidney while inducing PO4 excretion → ↑ Ca, ↓ PO4 Vitamin D ▪ 1,25 dihydroxyvitamin D – enhances Ca absorption from the GIT, increases serum Ca levels and bone mineralization Calcitonin ▪ Secreted by parafollicular (C cells) of thyroid gland ▪ Inhibits bone resorption by decreasing the number and activity disease) ▪ Greater effect on fetus rather than child/adult HYPOCALCEMIA ▪ Common in neonates between 12 and 72 hr of life (especially in premature, asphyxiated, born to diabetic moms) ▪ Albumin major reservoir of protein bound calcium DISORDERS OF PARATHYROID ▪ ↓ Serum Ca is low, ↑ phosphorus ▪ N or ↓ alkaline phosphatase ▪ ↓ 1,25(OH2)D3 (high in severe hypocalcemia) ▪ N magnesium (but should always be checked in hypoCa) ▪ ↓ PTH ▪ Severe tetany or seizures – 5-10mL or 1-3mg/kg of IV 10% calcium gluconate at 0.5-1ml/min, monitor cardiac status for bradycardia ▪ Hypoparathyroidism – administer Vit D (calcitriol) and calcium; monitor levels to avoid nephrocalcinosis and renal impairment Bone radiographs: ↑ density in metaphyses or lamina dura Xray or CT scan of skull: calcifications in basal ganglia of bone-resorbing osteoclasts (hence used in treatment of Paget ECG: prolonged QT interval ▪ Sxs result from ↑ neuromuscular activity – muscle cramps, carpopedal spasm (tetany), weakness, paresthesia, laryngospasm, seizure like activity ▪ Teeth erupt late, enamel formation is irregular → teeth may appear soft ▪ Dry scaly skin, nails have horizontal lines, cataracts ▪ Tetany (Chovstek sign) – tapping facial nerve in front of ear ▪ Trousseau sign – carpal spasms exhibited when arterial blood flow to hand occluded for 3-5 mins (BP inflated to 15mmHg above SBP) ▪ ↑ Serum calcium ▪ ↓ Serum phosphorus and magnesium ▪ N calcitonin ▪ Surgical exploration – all cases ▪ Others have been treated successfully with bisphosphonates and calcimimetics Radiology: resorption of subperiosteal bone best seen on phalanges of hands (most consistent finding) Batch Clingy PRIMARY HYPOPARATHYROIDISM ▪ Causes hypocalcemia but does not cause rickets Etiologies: ▪ Congenital malformation: DiGeorge syndrome - del 22q11.2; Hypocalcemia occurs in 60%, transitory in majority (assoc abnormalities: conotruncal heart defects, velopharyngeal insufficiency, cleft palate, renal anomalies, thymic aplasia) ▪ Surgical procedures: thyroidectomy or parathyroidectomy ▪ Autoimmune – can destroy thyroid gland, often associated with Addison disease and chronic mucocutaneous ca ndidiasis PSEUDOHYPOPARATHYROIDISM (hypocalcemia and hyperphosphatemia) ▪ Autosomal dominant; may present at birth or later ▪ Associated with Albright hereditary osteodystrophy ▪ Short stature, stocky body, round facies, short 4th & 5th MCPs, subcutaneous calcification, dev delay ▪ Hypomagnesemia – may cause 2o hypocalcemia (giving calcium will be ineffective, but giving magnesium promptly corrects both hypoMg and hypoCa) HYPERPARATHYROIDISM ▪ Muscle weakness, HA, fatigue, anorexia, abdominal pain, n/v, ▪ Excessive production from primary defect of the parathyroid constipation, polydipsia, polyuria, weight loss, fever glands like adenoma or hyperplasia (primary ▪ Nephrocalcinosis, osseous changes hyperparathyroidism) ▪ More often compensatory, aimed at correcting hypocalcemic states (2o hypoparathyroidism) PPS Oral Exam Reviewer 2020 61 RICKETS ▪ Disease of growing bone caused by unmineralized matrix at the growth plates in children only before fusion of the epiphyses Causes: Nutritional Vit D deficiency ▪ Calcium not absorbed from intestine ▪ Poor vitamin D intake and avoidance of sunlight → rickets ▪ Fat malabsorption from hepatobiliary disease (biliary atresia, neonatal heap) → Vit D deficiency (because it’s a fat-soluble vitamin) ▪ Defects in vit D metabolism by liver and kidney can also cause rickets Phosphorus deficiency ▪ Familial hypophosphatemic rickets ● Defect in mineral metabolism – failure of kidney to adequately reabsorb filtered phosphate ● ↓ Serum pH, ↑ urine pH ● X linked, more in males ▪ Growth plate thickens; proportion of osteoid (extensive) → bone becomes soft and metaphyses of long bones widen (→ classic symptoms of widening of wrist and ankles, bone deformities, craniotabes) ▪ Older infants – poor linear growth, bowing of the legs on weight bearing (windswept deformity), thickening at wrists and knees, prominence of costochondral junctions (rachitic rosary) Assess mineral and Vit D status: ▪ Calcium ▪ Phosphate ▪ PTH ▪ 25OHD ▪ Rickets – treat with 1,25 dihydroxyvitamin D and supplemental calcium Radiology: rachitic changes easily visualized on PA radiographs of the wrist, fraying and cupping ▪ Nutritional rickets – given Vit D as one large does or weekly doses ▪ Hypophosphatemic rickets – PO4 supplementation with vitamin D therapy Obtain: ▪ Dietary history ▪ Cutaneous synthesis ▪ Maternal risk factors ▪ Medication use ▪ History of renal disease Calcium deficiency ▪ Rickets of prematurity Batch Clingy Renal losses PPS Oral Exam Reviewer 2020 62 Complex Idiopathic GH deficiency – most common cause of BOTH congenital and acquired GH deficiency Less often by anatomic defects of the pituitary gland (pituitary aplasia) and other midline defects Hereditary forms of GH deficiency – heterogenous defects of gene for GH, GRF or GH receptor Classic GH deficiency – ↓ to absent secretions of GH GROWTH HORMONE DEFICIENCY Acquired GH deficiency ▪ Late onset growth failure suggests tumor of hypothalamus or pituitary ▪ Causes: radiotherapy for malignancy, meningitis, histiocytosis, trauma Congenital GH deficiency ▪ Normal or near normal BL and BW at term ▪ Growth rate slows after birth (2-3yo) – become shorter for age, tends to have elevated weight to height ratio, appears chubby and short ▪ If severe, GH production or action fall >4 SD below the mean by 1yo Classic GH deficiency ▪ Cherubian facies (chubby immature appearance) ▪ High pitched voice from immature larynx ▪ Normal intellect, growth, and age appropriate speech (unless hypogly or midline defects occur) Males with isolated GH deficiency ▪ Micropenis – stretched penile length <2cm, N 3-5cm) ▪ Fasting hypoglycemia GH resistance/GH insensitivity (rare) ▪ Abnormal number or function of GH receptors (ex. Laron syndrome) PE: ▪ Round head, short and broad face, prominent frontal bone, depressed nose bridge and saddle shaped, small nose, underdeveloped mandible and chin, teeth erupt late and is overcrowded ▪ Short neck, small larynx, high pitched voice until puberty, small hands, and feet ▪ Genitals small for age, sexual maturation delayed or absent ▪ Intelligence normal for age FAILURE TO THRIVE (FTT) ▪ Children who are not growing as expected (related to environmental and psychosocial causes) ▪ Studies advocated use of term MALNUTRITION ▪ Malnutrition = undernutrition → imbalance between nutrient requirements and intake or delivery → deficits of energy, protein or micronutrients that negatively affect growth and development → wasting, stunting (months after) ▪ Weight that falls or remains <3rd percentile for age ▪ <80% median height of the child ▪ Weight that decreases 2 major percentiles on the chart ▪ Acute malnutrition – duration of <3 mos PPS Oral Exam Reviewer 2020 ▪ WFH is usually normal, but an excess of body fat and a deficiency of muscle mass contribute to a pudgy appearance ▪ Inadequate weight-for-corrected age, failure to gain adequate weight over a period of time (weight gain velocity), height velocity, weight-for-height, BMI (review WHO growth charts) ▪ Include prenatal history, family, and travel history ▪ List symptoms – vomiting, diarrhea, fever, respiratory symptoms ▪ Feeding history (quantity, quality, frequency of meals), feeding behavior ▪ Psychosocial assessment of the family ▪ Complete PE and dev screening, neuro exam PE findings: ▪ ↓ Subcutaneous fat LABORATORY FINDINGS / DIAGNOSIS Screening tests ▪ Metabolic panel for kidney and liver function ▪ CBC r/o anemia ▪ Celiac panel r/o celiac disease ▪ Carotene and folic acid levels to r/o malabsorption Definitive: demonstration of absent or ↓ levels of GH in response to stimulation 2 GH tests ▪ Target hormone measurement – IGF-1, IGFBP3 ▪ Provocative tests: Arginine, levodopa, Insulin, clonidine, or glucagon administration (to rapidly ↑ GH level) ▪ Thyroid hormone is a prerequisite for normal GH synthesis hence must always be assessed first before provocative testing. CT scan – suprasellar calcifications associated with craniopharyngioma, or bony changes accompanying histiocytosis MRI – small anterior pituitary gland, missing or attenuated pituitary stalk, ectopic posterior pituitary Plain film of hand – bone age DIFFERENTIAL DIAGNOSIS ▪ Hormonal: Primary hypothyroidism, Cushing disease ▪ Systemic ● IBD, Celiac disease, occult renal disease, anemia ● Patients with systemic conditions have a greater deficit of weight than length ▪ Severe psychosocial deprivation ▪ Genetic: Turner syndrome, SHOX gene defects ▪ Chronic illness ▪ Undernutrition ▪ Nonsyndromic family trait ▪ Constitutional delay of growth and development ▪ CBC w/ PC (IDA) ▪ Screen BLL ▪ Urinalysis ▪ Urine CS ▪ Serum electrolytes (renal function) ▪ TSH (check for hypothyroidism) ▪ Liver function tests ▪ Screen for TB ▪ Secure patient’s newborn screening results TREATMENT Recombinant hGH (rhGH) – given subcutaneously once daily, no long-acting form available yet Approved indications for rhGH use accdg to US FDA: 1. GH deficiency 2. Turner syndrome 3. Chronic renal failure before transplant 4. Idiopathic short stature 5. SGA short stature 6. Prader-Willi syndrome 7. SHOX gene abnormality 8. Noonan syndrome Recommended initial dose: 0.16-0.24mg/kg/wk (22-25ug/kg/day) ▪ GH therapy is started as soon as possible and continued until near-final height is achieved Criteria to stop: ▪ Decision by parent that he/she is tall enough ▪ Growth rate <1inch/yr ▪ Bone age >14y/o in girls and >16y/o in boys COMPLICATIONS ▪ Some patients develop hypothyroidism or adrenal insufficiency during GH treatment hence periodic evaluation of thyroid and adrenal function is indicated ▪ GH treatment influences glucose homeostasis hence ↑ risk for type 2 diabetes ▪ Address the child’s nutritional requirements and social issues of family ▪ Caloric supplementation Nutritional management ▪ 1.5x expected calorie and protein intake for their age ▪ Initiation of catch up growth may take 2 weeks ▪ Consider a speech therapist for suck-and-swallow evaluation ▪ If no response after 2-3mos of outpatient therapy, admit with NGT feed Batch Clingy ▪ Presence of height 3.5 SD below mean, height velocity below 5th percentile for age, or height below target height for midparental height DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS CLINICAL MANIFESTATIONS ▪ May be accompanied by hypoadrenalism, hypothyroidism, and Infants: apnea, cyanosis, severe hypoglycemia, w/ or w/o seizures, gonadotropin deficiency prolonged neonatal cholestatic jaundice, nystagmus 63 ▪ Causes: Environmental, GIT, Congenital, Infection, Metabolic, Neurologic, Renal and Hematologic Illness-related causes mechanisms: ▪ Failure to ingest sufficient calories / starvation (ex. Cardiac failure or fluid restriction) ▪ ↑ Nutrient losses (ex. enteropathy / diarrhea) ▪ ↑ Metabolic demands (ex. Burns) ▪ Altered nutrient absorption or utilization (ex. Cystic fibrosis or short bowel syndrome) CONGENITAL HYPOTHYROIDISM ▪ Caused by dysgenesis (aplasia, agenesis, hypoplasia, or ectopia) or dyshormogenesis (rare) ▪ Bamforth-Lazarus syndrome: thyroid dysgenesis, spiky or curly hair, cleft palate, choanal atresia, bifid epiglottis ▪ Goiter – reflects IEM in pathway of iodide incorporation, or reflects transplacental passage of antithyroid drugs of mom (thyroid glands respond normally to ↑ TSH stimulation, a goiter is almost always present) ▪ ↓ Free T4, ↑ TSH ▪ NBS (measures TSH) – results after 1 week ▪ If positive, do immediate confirmatory test ▪ Pendred syndrome: sensorineural deafness and goiter HYPOTHYROIDISM Primary – defect of thyroid dysgenesis Central or 2o – isolated TSH deficiency MALNUTRITION *check failure to CENTRAL (SECONDARY) HYPOTHYROIDISM ▪ Associated with developmental defects of the pituitary or hypothalamus ▪ Not detected by neonatal screening ▪ Suspected if with ↓ growth velocity, especially if NOT associated with weight loss Hashimoto thyroiditis (chronic lymphocytic autoimmune thyroiditis) ● Most common cause of acquired hypothyroidism ● Autoimmune process targeted against thyroid gland with lymphocytic infiltration and lymphoid follicle and germinal center formation preceding fibrosis and atrophy ▪ May have associated autoimmune diseases (DM type I, adrenal insufficiency, hypoparathyroidism) ▪ Iodine deficiency – endemic cretinism ▪ Drugs with iodides can cause hypothyroidism through WolffChaikoff effect: Expectorants, nutritional supplements ▪ Other drugs that can cause hypothyroidism: Amiodarone, phenytoin, phenobarbital, valproate, lithium, rifampin, thalidomide, etc. ▪ Not a cause of permanent developmental delay ▪ Critical window for maternal and child undernutrition: between a woman’s pregnancy and her child’s second birthday, later coined “the first 1,000 days” PPS Oral Exam Reviewer 2020 ▪ ↓ Muscle mass ▪ Dermatitis ▪ Hepatomegaly ▪ Cheilosis or edema ▪ Delayed puberty (check tanner stage) COMPLICATIONS ▪ Malnutrition infection cycle ▪ Refeeding syndrome (ph, Ca, Mg and K) → life threatening cardiac, pulmonary or neurologic problems MUAC – useful when weight may be distorted by use of steroids or fluid status Hand-grip strength – for ≥6yo, more accurate than MUAC because muscle function reacts earlier to changes in nutritional status than muscle mass ▪ Initially asymptomatic at birth – due to transplacental passage of maternal T4, although despite this, infants with 1o hypothyroidism have ↑ TSH and ↓ T4 levels which is detected during screening, hence, symptoms more evident weeks or months later ▪ After birth, nonspecific symptoms: prolonged jaundice, large fontanels (clue to early recognition), ↑ HC (due to myxedema of brain), cry little, sleep much, poor appetite, sluggish, respiratory difficulties, constipation, abdominal distention, umbilical hernia, hypothermia of extremities, mottled skin, edema of genitals, slow pulse, murmurs, cardiomegaly, asymptomatic pericardial effusion ▪ Thyroid hormones crucial for maturation and differentiation of tissues (bone and brain) ▪ Slowing of growth – first clinical manifestation ▪ Goiter – common presenting feature Hashimoto thyroiditis ▪ Firm, nontender euthyroid, hypothyroid, or rarely hyperthyroid (hashitoxicosis) diffuse goiter with pebble like surface ▪ Onset: >6yo ▪ Peak incidence: adolescence ▪ F>M ▪ Ectodermal – poor growth, dull facies, thick lips, periorbital edema, dry scaly skin, brittle hair, diminished sweating, carotenemia, vitiligo ▪ Circulatory – sinus bradycardia, cold extremities, cold intolerance, pallor, ECG low voltage QRS complex ▪ Neuromuscular – weakness, hypotonia, constipation, umbilical hernia, myxedema, myalgia, dev delay, physical and mental lethargy, delayed reflexes, paresthesia, cerebellar ataxia ▪ Skeletal – delayed bone age ▪ Metabolic – serous effusions, weight gain, menstrual irregularity, arthralgia, macrocytosis, hypercholesterolemia, hyperprolactinemia, precocious puberty Criteria for identifying children with severe acute malnutrition for treatment: ▪ Measure MUAC and assess WFL/WFH status of infants and Newborn screening ▪ TSH, T4 (T3 are often normal and not helpful in diagnosis) ▪ Delay of osseous development (distal femoral and proximal tibial epiphyses are absent) Ultrasound – presence or absence of anatomically normal gland (but can miss ectopic glands) Scintigraphy – demonstrate ectopic thyroid gland ▪ Demonstration of ectopic thyroid tissue is diagnostic for thyroid dysgenesis and establishes need for lifelong treatment ▪ Serum TSH and fT4 ▪ Thyroglobulin or TPO – diagnostic for autoimmune thyroiditis Ultrasonography – not indicated unless PE raises suspicion of thyroid nodule Biopsy or thyroid scan – not indicated but thyroid scan with ↓ uptake can differentiate hashitoxicosis from Graves Levothyroxine ▪ 10-15ug/kg/day (37.5-50ug/day for term infants) ▪ Initiate at < 1 month after birth, prognosis for normal intellect is excellent ▪ If after 6 months, likelihood for normal intellect decreased ▪ Should not be mixed with soy protein formulas, concentrated iron, or calcium → can ↓ absorption Monitoring: TSH and fT4 ▪ Every 1-2mos in the 1st 6mos, then ▪ Every 2-4mos between 6mos to 3yo L-T4 ▪ 1-3yo: 4-6ug/kg ▪ 3-10yo: 3-5ug/kg ▪ 10-16yo: 2-4ug/kg Monitoring: TSH ▪ Every 4-6mos, and ▪ 4-6wks after any change in dosage Bone age x-ray may suggest the duration and severity of hypothyroidism based on degree of bone age delay 10 Steps to Recovery: 1. Treat/Prevent hypoglycemia 2. Treat/Prevent hypothermia Batch Clingy ▪ Symmetric FTT (weight, height, HC) – long standing malnutrition, chromosomal abnormalities, congenital infection, or teratogenic exposures 64 thrive for malnutrition based on nelson’s *the following in this table are from pps and who guidelines on malnutrition ▪ ↑ Nutritional needs to support rapid growth and development ▪ At most risk of malnutrition and infection ▪ Chronic exposure to inadequate nutrition = STUNTING ▪ Outcome of brain development by age 2 years determines person’s mental capacity for the rest of his/her life ▪ Children who get right nutrition during first 1,000 days are 10x more likely to overcome life threatening conditions, 4.6x more grades in school, earn 21% more wages as adults children 6-59mos of age ▪ Examine for B pitting edema ▪ Those who have MUAC <115cm, WFH/WFL below -3 z score or who have any degree of B edema should be referred for full assessment and admitted to a program for management of severe acute malnutrition 3. 4. 5. 6. 7. 8. 9. 10. Treat/Prevent dehydration Correct electrolyte imbalance Treat the infection Correct the micronutrient deficiencies Start cautious feeding Rebuild wasted tissue (Catch-up growth) Provide stimulation, play and loving care Prepare for follow-up after discharge Proven services, practices, and policy interventions to prevent malnutrition based on PPS: Adolescence/Pregnancy ▪ Improved use of locally available food ▪ Food fortification including salt iodization ▪ Micronutrient supplementation ▪ Deworming ▪ Fortified food supplements for undernourished moms ▪ Antenatal care including HIV testing Birth ▪ Delayed cord clamping ▪ Neonatal Vit K administration ▪ Early initiation of breastfeeding ▪ Appropriate infant practices ▪ Immunization ▪ Newborn Screening 0-5 months ▪ Exclusive breastfeeding ▪ Appropriate infant feeding practice ▪ Immunization ▪ Vit A supplementation in first 8 weeks after delivery ▪ Multi-micronutrient supplementation ▪ Improved use of locally available foods, fortified foods Batch Clingy 6-23 months ▪ Complementary feeding ▪ Continued breastfeeding ▪ Micronutrient supplementation: Vit A, Zinc ▪ Deworming ▪ Food fortification ▪ Handwashing ▪ Improved water and sanitation practice ▪ Immunization PPS Oral Exam Reviewer 2020 65 Acute ETIOLOGY / INCIDENCE / PATHOGENESIS Rotavirus – most common cause Bacteria: Nontyphoidal Salmonella, Shigella, Campylobacter, Yersinia Parasites: Gardia intestinalis, Cryptosporidium spp, Cyclospora cayetenensis, E. histolytica ▪ Viral AGE → cytolytic infection of the small intestinal villus tips → ↓ absorption of water, disaccharide malabsorption, inflammation and cytokine activation ▪ Rotavirus NSP4 (enterotoxin) produces secretory diarrhea → activation of CAMP and CGMP → secretion of water and electrolytes activates the enteric nervous system → ↓ gastric emptying and ↑ intestinal mobility ▪ Bacterial AGE possesses an invasive phenotype → elicitation of inflammatory cytokines with or without toxin production ▪ Shigella → ulcers and microabscesses ACUTE GASTROENTERITIS TREATMENT COMPLICATIONS / PROGNOSIS CLINICAL MANIFESTATIONS LABORATORY FINDINGS / DIAGNOSIS Diarrhea – passage of ≥3 abnormally loose or liquid stools per day Medical History ▪ Duration of diarrhea ▪ Description of stools – frequency, amount, presence of blood or mucus ▪ Fever – duration, magnitude ▪ Vomiting – onset, amount, frequency ▪ Amount and type of solid or liquid oral intake Evaluate clinical signs of dehydration ▪ Urine output ▪ Sunken eyes ▪ Mental status ▪ Thirst ▪ Recent weight measurement ▪ Skin turgor, capillary refill time Viral Diarrhea ▪ Vomiting → passage of watery non bloody stools with fever ▪ Contains mucus but lacks fecal leukocytes Bacterial Diarrhea ▪ Fever >40C ▪ Overt fecal blood ▪ Abdominal pain ▪ No vomiting before diarrhea onset ▪ High stool frequency (>10 per day) Bacterial agents can present with 1 of 5 syndromes: ▪ Acute diarrhea ▪ Bloody diarrhea ▪ Invasive, nonfocal disease (enteric fever) ▪ Extraintestinal invasive infections ▪ Vertical transmission STEC ▪ Crampy abdominal pain, nonbloody diarrhea ▪ Large volume stools, rarely with fever (STEC hemorrhagic colitis) ETEC – traveler’s diarrhea C. difficile – pseudomembranous colitis (diarrhea, abdominal cramps, fever with 2-5mm raised yellowish plaques on colonic mucosa) LABS Stool specimens can be examined for: ▪ Mucus ▪ Blood ▪ Neutrophils ▪ Fecal lactoferrin *>5 leukocytes/hpf or (+) lactoferrin enteropathogen Hydration ▪ Correct dehydration in 4-6 hours ▪ Oral rehydration – preferred method, ORS 75 ▪ IVF resuscitation necessary: ● Shock ● Decreased level of consciousness ● Ileus ● Intussusception ● Carbohydrate intolerance ● Severe emesis ● High stool output (>10ml/kg/hr) Enteral feeding and Diet selection ▪ Continued breastfeeding and refeeding with age-appropriate unrestricted diet ▪ Withdrawal of milk and replacement with specialized lactose-free formulations are unnecessary Zinc supplementation ▪ Reduces duration and severity, preventS recurrences ▪ Oral zinc 20mg/day for 10-14 days → bacterial Stool cultures should be restricted to the ff: ▪ Clinical features suggestive of bacterial AGE ▪ Moderate or severe disease ▪ Immunocompromised ▪ Outbreaks with suspected HUS ▪ Highly suggestive epidemiologic history Rectal swab – done if the child has not passed stool and antibiotics will be started Measure serum electrolytes: ▪ Severe dehydration ▪ IVF are administered ▪ Skin feels doughy without delayed recoil ▪ Inappropriate rehydration fluids have been administered Blood culture – if with systemic bacterial infection Probiotics – does not support a recommendation for use in all settings Ondansetron – reduces incidence of emesis ▪ Sublingual dose of 4mg (4-11yo), 8mg (>11yo) Antibiotic therapy ▪ Treat severe infections ▪ Prevent complications in high risk hosts ▪ Limit spread of infection PREVENTION ▪ Exclusive breastfeeding for 6 months ▪ Vitamin A supplementation ▪ Rotavirus immunization COMPLICATIONS ▪ Dehydration ▪ Electrolyte and Acid-Base Derangements ▪ Poor growth and nutrition ▪ Secondary infection ▪ Micronutrient deficiencies ▪ Intussusception (viral AGE) ▪ Bacteremia Shigella & C. difficile are assoc. with: ▪ Toxic megacolon ▪ Intestinal perforation ▪ Rectal Prolapse PPS Oral Exam Reviewer 2020 Batch Clingy DISEASE 66 ▪ Stool antigen testing or PCR ▪ Indirect hemagglutination – most sensitive Trophozoites invade mucosa of large intestine, secrete substances that cause intestinal secretion and damage → inflammatory type of diarrhea Tissue invasion → flask shaped ulcers → if spreads to the liver → abscess ▪ Giardia duodenalis ▪ 1-5 years old common PARASITIC INFECTION (Giardia lamblia) Amebic colitis ▪ Colicky abdominal pain ▪ Frequent bowel movements (6-8/day) ▪ Tenesmus ▪ Common in 1-5 years old ▪ Persistent diarrhea ▪ Malabsorption with fatty stools ▪ Abdominal pain ▪ Bloating ▪ Trophozoites colonize the lumen of duodenum and proximal jejunum ▪ Flattening of intestinal epithelium ▪ Cysts and trophozoites of G. duodenalis seen in stool specimen (3 specimens increase sensitivity to 90%) ▪ Stool Enzyme Immunoassay ▪ Direct fluorescent antibody test ALTERNATIVE ▪ Albendazole >6yo: 400mg QD for 5 days ▪ Quinacrine 6mkday ÷ 3 doses for 5 days ▪ Foodborne, waterborne ▪ Spread by fecal-oral route ▪ Caused by excessive intake of sweetened liquids (sorbitol), overwhelming the disaccharide absorptive capacity of the intestine ▪ <4 yrs old TODDLER’S DIARRHEA ▪ Frequent watery stools in the setting of normal growth and weight gain ▪ Clinical diagnosis ▪ Distinguish secretory from osmotic: put on NPO, if diarrhea stops → osmotic ▪ Reduce or change beverage intake ▪ CDI – from mild, self-limited diarrhea to explosive watery diarrhea with occult blood or mucus ▪ Pseudomembranous colitis – bloody diarrhea, fever, abdominal pain, cramps, nausea, vomiting ▪ C. difficile toxin in stool of a symptomatic patient – tissue culture ▪ Discontinue antibiotics ▪ Metronidazole 7.5mkdose TID or QID x 10 days PO or ▪ Vancomycin 10mkdose QID x 10 days PO ▪ Type of osmotic diarrhea – malabsorbed substance pulls water into bowel lumen ▪ Fermentation of malabsorbed substances (lactose) often occurs, resulting in gas, cramps and acidic stools ▪ Clostridium difficile ▪ Antibiotic associated diarrhea ▪ Produce toxin A and B PSEUDOMEMBRANOUS COLITIS COMPLICATIONS ▪ Acute necrotizing colitis ▪ Ameboma ▪ Toxic megacolon ▪ Extraintestinal extension ▪ Local perforation and peritonitis ▪ Tinidazole >3yo: 50 mg/kg once or ▪ Nitazoxanide 1-3yo: 100mg (5mL) BID for 3 days 4-11yo: 200mg (10mL) BID for 3 days >12yo: 500mg BID for 3 days, or ▪ Metronidazole 15mkday ÷ 3 doses for 5-7 days Toxin producing strain → impaired normal GIT response, induce an inflammatory response → diarrhea and pseudomembrane formation PPS Oral Exam Reviewer 2020 ▪ Small gut involvement, bacteremia, abscess formation, toxic megacolon and death may occur ▪ Symptoms begin a week after colonization and develop during or weeks after antibiotic exposure, more severe in immunocompromised ▪ EIA ▪ NAAT PROGNOSIS ▪ 95% response rate to initial treatment ▪ Recurrence rate 5-20% within 4 weeks of treatment Batch Clingy PROTOZOAL INFECTION (Amoebiasis) ▪ 4 species: E. dispar – most prevalent; does not cause symptomatic disease E. moshkovskii – can cause diarrhea in infatns and children E. histolytica – main pathogenic species E. Bangladeshi Hemolytic Uremic Syndrome (HUS) ▪ Leading cause of acquired renal failure in children ▪ 5-10% of patients infected with STEC ▪ Diagnosed 2-14 days after the onset of diarrhea ▪ Unlikely to occur once diarrhea has remained resolved for 2-3 days with no evidence of hemolysis ▪ Risk Factors: 6mos to 4yo, bloody diarrhea, fever, ↑ leukocyte count, treatment with antibiotics and antimotility agents ▪ Metronidazole 35-50mkday ÷ 3 doses for 7-10 days or ▪ Tinidazole 50mkday QD for 3 days Followed by ▪ Paromomycin 25-35mkday ÷ 3 doses for 7 days or ▪ Iodoquinol 30-40mkday ÷ 3 doses for 20 days or ▪ Diloxonide furoate for >2yo 20mkday ÷ 3 doses for 7 days 67 Complex DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Immune-mediated systemic disorder elici ted by gluten ▪ Affects genetically susceptible individuals ▪ Strongest association with HLA DQ2.5 gene MALABSORPTION (Celiac Disease) CLINICAL MANIFESTATIONS ▪ Malabsorption and malnutrition ▪ 1st 2yrs of life – FTT, chronic diarrhea, vomiting, abdominal distention, muscle wasting, anorexia, irritability ▪ Extraintestinal – IDA, unresponsive to iron therapy, short stature, endocrinopathies, arthritis, arthralgia, epilepsy, cardiomyopathy LABORATORY FINDINGS / DIAGNOSIS ▪ Small bowel biopsy – definitive ● Villous atrophy with hyperplasia of the crypts ● Abnormal surface epithelium TREATMENT / COMPLICATIONS / PROGNOSIS ▪ Gluten free diet ▪ Wheat, barley and rye free diet ▪ Anti TG2A antibodies ▪ Serum total IgA to exclude IgA deficiency ▪ T cell mediated chronic inflammatory disorder ▪ Altered processing by intraluminal enzymes, changes in intestinal permeability and activation of innate immunity mechanisms precede the activation of the adaptive immune response ▪ Idiopathic chronic intestinal inflammation ▪ Unpredictable exacerbations and remissions ▪ Most common onset: Preadolescent/ adolescent era and young adulthood ▪ Genetic and environmental influences ULCERATIVE COLITIS (UC) ▪ Localized to the colon ▪ Remission and relapse Associated with type 1 DM, autoimmune thyroid disease, Addison disease, Sjögren syndrome, rheumatoid arthritis, autoimmune cholangitis, autoimmune hepatitis, primary biliary cholangitis, selective IgA deficiency and Down, Turner, and Williams syndromes ▪ Blood, mucus, pus in the stool ▪ Diarrhea ▪ Fever, diarrhea, hypoalbuminemia, leukocytosis and >5 stools/day for 5d → FULMINANT COLITIS ▪ CBC: anemia, leukocytosis if severe ▪ Hypoalbuminemia ▪ Elevated ESR and CRP ▪ Flexible sigmoidoscopy – confirm the presence of UC ▪ Colonoscopy – extent of disease ▪ Upper endoscopy – more sensitive than contrast studies in detecting proximal CD involvement ▪ Video capsule endoscopy – may be done except if there is stricturing disease Endoscopic findings: ▪ Diffuse carpeting of the distal or entire colon with tiny ulcers and loss of haustral folds ▪ Extraintestinal manifestations: pyoderma gangrenosum, sclerosing cholangitis, chronic active hepatitis, ankylosing spondylitis INFLAMMATORY BOWEL DISEASE ▪ Colitis, diarrhea, blood, and mucus in stool ▪ Small bowel involvement – loss of appetite, crampy, postprandial pain, poor growth, delayed puberty, fever, anemia, lethargy ▪ Severe CD with fibrosis→ partial or complete small bowel obstruction Perianal disease – skin tags and fistulas Extraintestinal manifestations more common– arthritis, erythema nodosum, uveitis or iritis, renal or gallstones Endoscopic findings: ▪ Larger ulcerations with linear branching, skip areas usually present (cobblestone appearance) Aminosalicylates ▪ 5-ASA (Mesalamine 50-100mkday) – packaged in capsules to be taken as suppository ▪ Sulfasalazine 30-100mkday – least expensive ▪ If cannot be controlled by ASA, use steroids (Prednisone 1-2mkday) ▪ Immunosuppressive – 6 mercaptopurine & azathioprine → if failed treatment, use cyclosporine or anti-tumor necrosis factor ▪ Probiotics (for adults) – prevent pouchitis ▪ Surgical colectomy with ileoanal anastomosis – option for unresponsive disease, reduce risk of colon cancer Step up: steroids and less immunomodulating agents Top down: start with strong DMARDs ▪ Responds less to ASA ▪ Metronidazole – first line for perianal disease and if with infectious complications ▪ Steroids – induces remission; Prednisone or Budesonide ▪ Immunomodulators – 6-MP, azathioprine, MTX ▪ Biologics – Infliximab, Adalimumab, Vedolizumab ▪ Enteral Nutrition Therapy – effective than steroids for mucosal healing ▪ Surgery – but must be limited and length of bowel resection must be minimized; >50% recurrence rate Batch Clingy CROHN DISEASE (CD) ▪ Any region of the alimentary tract from mouth to anus ▪ Eccentric and segmental often with skip areas ▪ Transmural involvement COMPLICATIONS ▪ Toxic megacolon – fever, abdominal distention and pain, massively dilated colon, anemia, and low serum albumin because of fecal protein losses PPS Oral Exam Reviewer 2020 68 Acute DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS CLINICAL MANIFESTATIONS DIAGNOSIS ▪ Delay or difficulty in defecation present for >1month and causes significant distress to the patient ▪ Contraction of gluteal muscles → stiffening legs while lying down, holding on to furniture while standing, squatting quietly in corners ▪ Acute stressor precedes the chronic course Encopresis – voluntary or involuntary passage of feces into inappropriate places at least 1x a month for 3 consecutive months once a chronologic or developmental age of 4 has been reached ROME IV CRITERIA Neonates and Toddlers: Must include 1 month of ≥2 of the following in infants to 4 mos old: • ≤2 defections weekly • History of excessive stool retention • History of hard/painful bowel movements In toilet trained children, the following additional criteria may be used: • History of large-diameter stools • Presence of a large fecal mass in the rectum • At least 1 weekly episode of incontinence after being toilet trained • History of large-diameter stools that may clog the toilet Children and Adolescents (Developmental age of ≥4yo): Must include ≥2 of the following ≥1/wk for ≥1mo with insufficient criteria to diagnose irritable bowel syndrome • ≤ 2 defecations in the toilet per week • ≥1 episode of fecal incontinence per week • History of retentive posturing or excessive volitional stool retention • History of painful or hard bowel movements • Presence of a large fecal mass in the rectum • History of large diameter stools that can obstruct the toilet • After appropriate evaluation, symptoms cannot be fully explained by another medical condition ▪ Large volume of stools palpated in the suprapubic area ▪ Dilated rectal vault with guaiac negative stool ▪ Underwear soiling FUNCTIONAL CONSTIPATION TREATMENT / COMPLICATIONS / PROGNOSIS Rapid Rectal Disimpaction ▪ Glycerin suppositories ▪ Phosphate enema Slow oral disimpaction for children Over 2-3 days ▪ PEG with electrolytes Over 5-7 days ▪ PEG without electrolytes ▪ Milk of magnesia ▪ Mineral oil ▪ Lactulose or Sorbitol Maintenance Therapy Long Term ▪ Milk of magnesia ▪ Mineral oil ▪ Lactulose or sorbitol ▪ PEG 3350 Short Term ▪ Senna ▪ Glycerin enemas ▪ Bisacodyl suppository Nonretentive Fecal Incontinence ≥1mo history of the following symptoms: • Defecation into places inappropriate to the sociocultural context • No evidence of fecal retention • After appropriate evaluation, symptoms cannot be fully explained by another medical condition Complex HIRSCHSPRUNG DISEASE Congenital Aganglionic Megacolon ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Absence of ganglion cells in the submucosal and myenteric plexus ▪ Most common cause of lower intestinal obstruction in neonates CLINICAL MANIFESTATIONS ▪ Usually diagnosed in the neonatal period ▪ Failure to pass meconium in 48 hours ▪ M:F ratio: 4:1 for short-segment disease, 2:1 for total aganglionosis ▪ May be associated with other congenital defects ▪ Arrest of neuronal migration from proximal to distal bowel → absence of neuronal innervation ▪ Absence of submucosal and myenteric plexus → inadequate relaxation of the bowel wall and bowel wall hypertonicity → intestinal obstruction ▪ Signs of obstruction: distended abdomen, bilious emesis/aspirate with feeding intolerance ▪ Constipation starting in infancy that has not responded to medical management ▪ DRE: explosive discharge of foul-smelling feces and gas ▪ Small pellet stools, ribbon-like with fluid consistency ▪ 80% – limited to the rectosigmoid ▪ 10-15% – long segment (proximal to the sigmoid colon) ▪ 5% – total colonic aganglionosis PPS Oral Exam Reviewer 2020 Enterocolitis → diarrhea, abdominal tenderness, sepsis, signs of bowel obstruction Currarino triad (older patients) – anorectal, sacral and presacral anomalies DIAGNOSIS Rectal suction biopsy – gold standard ▪ Demonstrate hypertrophied nerve bundles that stain positively for acethylcholinesterase Anorectal Manometry ▪ Shows failure of the internal anal sphincter to relax to rectal distention and absence of rectoanal inhibitory reflux Contrast enema ▪ Abrupt narrow transition zone (normal dilated proximal colon and obstructed distal aganglionic segment) ▪ If no transition zone, compare rectal vs sigmoid diameter ▪ Check 24 hour delayed contrast films for delayed contrasts TREATMENT / COMPLICATIONS / PROGNOSIS ▪ Placement of temporary ostomy ▪ Endorectal pull through ▪ Anal botulism injection – for ultrashort-segment Hirschsprung disease ▪ If total colonic aganglionosis → ileal-anal anastomosis PROGNOSIS Satisfactory prognosis post-surgery COMPLICATION Hirschsprung associated enterocolitis – leading cause of death Batch Clingy DISEASE 69 CONGENITAL HYPOTHYROIDISM SPINAL CORD ABNORMALITIES ▪ Usually true constipation in neonatal period due to Hirschsprung, intestinal pseudoobstruction and hypothyroidism ▪ Constipation that does not respond to treatment ▪ Large abdomen with umbilical hernia ▪ Defective rectal filling because of defective colonic peristalsis ▪ Leads to excessive drying of stool—distention of colon and rectum lessens defecation reflex and effectiveness of peristalsis Tethered cord – spinal cord ends below L2 ▪ Other features: prolonged jaundice, large fontanels, abdominal distention, lethargy and poor feeding, edema, umbilical hernia, large tongue, dry skin, hoarse cry ▪ Presence of an open myelomeningocele or by cutaneous manifestations of dysraphism MRI ▪ Diagnostic of choice for anatomy of tethering lesion ▪ May present with 1 of 4 manifestations: neurologic, orthopedic, bowel/bladder and/or pain symptoms UTZ of the bladder and urodynamic studies ▪ Assessment of bladder function Constipation progressing to incontinence – lax sphincter due to impaired innervation GI symptoms: anorexia, abdominal pain, vomiting and constipation – seen in kids with BLL >20mcg/dl Blood lead levels – gold standard ▪ ≥5ug/dl means exposure and requires second testing ▪ Spinal cord is fixed at any point → movement is restricted → spinal cord and nerve roots are stretched ▪ Fixed spinal cord regardless of cause (i.e. lipomyelomeningocoele, myelocystocele and diastematomyelia) + pain, neurologic dysfunction, bowel/bladder dysfunction → TETHERED CORD SYNDROME Paint chips – most common source ▪ Most common pathway – hand to mouth activity of children ▪ Absorbed → disseminated via blood → bound to erythrocytes ▪ Retained lead → accumulates in bone ▪ Affects heme pathway → lead inhibitory effects → accumulation of excess amounts of heme precursors → increases erythrocyte protoporphyrin levels (>35ug/dl) → toxicity ▪ Also competes with calcium – prevents normal intracellular and intercellular signaling ▪ Prevents development of normal tertiary brain structure → ADHD Levothyroxine – 10-15ug/kg/day PO PROGNOSIS Delay in diagnosis and treatment → severe delay in growth and development, irreversible brain damage ▪ BLL of 45ug/dl → chelation therapy ▪ KUB radiograph → radiopaque flecks ▪Prophylactic surgery before deterioration occurs ▪ Microsurgical dissection with release of SC attachment to the overlying dura PROGNOSIS Good outcome however, patients with symptomatic tethered cord who undergo repair of a myelomeningocele or a lipomyelomeningocele → recurrent tethering and recurrent symptoms Chelation with 2,3 DMSA acid: 350mg/m2BSA/dose (not 10 mg/kg) q8h PO for 5 days → q12h for 14 days Dimercaprol: 300-500mg/m2BSA/day; IM only divided q4h for 3-5 days. Only for blood lead level ≥70 μg/dL Penicillamine 10 mkday for 2 wk increasing to 25-40 mg/kg/day; oral, divided q12h. For 12-20 wk 2 CaNa2EDTA: 1,000-1,500 mg/m body surface area/day; IV infusion— continuous or intermittent; IM divided q6h or q12h for 5 days Successful intervention → greatest fall in BLL occurs 1st 2 months after therapy is initiated Batch Clingy LEAD POISONING CNS symptoms: Lead Encephalopathy, headache and changes in mentation, lethargy, papilledema, seizures and coma (due to increasing cerebral edema and inc ICP); BLL of 70 to above 100mcg/dl Newborn screening – measure levels of T4, then if low measure TSH PPS Oral Exam Reviewer 2020 70 Acute DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Most common surgical emergency in childhood ▪ Peak in the second decade (10-18yo) ▪ Uncommon in < 5 years old ▪ Luminal obstruction, inspissated fecal material, lymphoid hyperplasia, ingested foreign body, parasites, tumors → increasing intraluminal pressure, lymphatic and venous congestion and edema → impaired arterial perfusion → ischemia of the appendiceal wall → bacterial proliferation and invasion of the wall and necrosis ACUTE APPENDICITIS CLINICAL MANIFESTATIONS ▪ Visceral inflammation → periumbilical pain ▪ Somatic pain → migration to the RLQ ▪ Nausea and vomiting follow the onset of abdominal pain by several hours ▪ Anorexia – classic and consistent finding ▪ Diarrhea and urinary symptoms – 2o to inflamed peritoneum ▪ Fever ▪ Localized abdominal tenderness ▪ Rebound tenderness ▪ Guarding ▪ Rovsing, Dunphy, Psoas, and Obturator signs LABORATORY FINDINGS / DIAGNOSIS ▪ Mildly elevated WBC (11,000-16,000) with left shift ▪ Plain Film of the abdomen – for complicated cases i.e. small bowel obstruction ▪ Ultrasound – first choice ● Wall thickness>/= 6mm ● Luminal distention ● Lack of compressibility ● Complex mass in the RLQ ● Appendicolith ● 25-60% cannot visualize the appendix ▪ CT scan – gold standard; fat stranding TREATMENT / COMPLICATIONS / PROGNOSIS ▪ Broad spectrum antibiotic (Zosyn or equivalent) – directed for anaerobes and gram-negative organisms ▪ Surgical – (Laparoscopic) Appendectomy ▪ If with perforation, after IV antibiotics, discharge with 7-10 days of Ciprofloxacin and Metronidazole Pediatric Appendicitis Score: <4 less likely, >8 highly likely Alvarado MANTRELS rule scores 1 point 2 points Pain migration to RLQ RLQ tenderness Anorexia WBC >10,000 Nausea/vomiting Rebound pain Temp at least 37oC WBC shift >75% neut *score of 7 or greater increase likelihood MALROTATION WITH MIDGUT VOLVULUS ▪ Sudden onset of severe paroxysmal colicky pain that recurs at frequent intervals ▪ Straining efforts with knees flexed and loud cries ▪ Lethargy disproportionate to the abdominal pain ▪ Bilious vomiting ▪ Currant jelly stool ▪ GIT infection or introduction of new food proteins → swollen peyer patches and lymphoid hyperplasia → lead points (more common >2yo) ▪ Most commonly ileocolic ▪ Upper portion of bowel (intussusceptum), invaginates into the lower (intussuscipiens) → obstruction in venous return → engorgement, edema, bleeding Predictive value: Pain + palpable sausage-shaped abdominal mass + currant jelly stool: <30% Pain + palpable mass + vomiting: > 90 % If with rectal bleeding = 100% ▪ Incomplete rotation of the intestine during fetal development ▪ Intestinal nonrotation or incomplete rotation around the SMA ▪ Nonrotation → jejunum and ileum occupy the right side, colon on the left and cecum move to the RLQ ▪ Mesentery including SMA → tethered by a narrow stalk → twist and cause a midgut volvulus ▪ Ladd bands (abnormal mesenteric attachments) extend from cecum to duodenum → partial obstruction ▪ 75-85% present in the 1st yr of life ▪ Bilious emesis, acute bowel obstruction ▪ Malabsorption, protein losing enteropathy ▪ Volvulus → life threatening → septic PPS Oral Exam Reviewer 2020 Abdominal Ultrasound ▪ Tubular mass in longitudinal views and doughnut or target appearance ▪ Coiled spring sign if with contrast ▪ Manual operative reduction if reduction by air or barium (w/o perforation) ▪ If bowel not viable – resection of intussusception PROGNOSIS ▪ Fatal if untreated ▪ 10% recurrence rate, 2-5% after surgical reduction Plain Abdominal Film ▪ Gasless abdomen, double bubble sign Upper GI series – gold standard ▪ Checks for position of ligament of Treitz, corkscrew appearance of bowel, bird’s beak appearance Abdominal Ultrasound ▪ Duodenal obstruction, thickened bowel loops to the right of the spine, free peritoneal fluid ▪ Surgical – bowel untwisted, and Ladd bands divided (Ladd’s Procedure) ▪ Mesentery flattened out and cecum moved to left side of abdomen COMPLICATION Extensive intestinal ischemia → short bowel syndrome Batch Clingy INTUSSUSCEPTION ▪ A portion of the alimentary tract is telescoped to an adjacent segment ▪ Most common cause of intestinal bowel obstruction between 5 months and 3 years of age ▪ Most common abdominal emergency in children <2yo ▪ 90% are idiopathic 71 ▪ Nonreducible mass in the inguinal canal ▪ Can include small bowel, appendix, omentum, colon or rarely Meckel’s diverticulum ▪ Rarely ovary or FT in girls ▪ Irritability, pain in groin and abdomen, feeding intolerance ▪ Abdominal distention, vomiting ▪ Onset of ischemic changes → bilious vomiting or feculent ▪ Bloody stools ▪ Greatest risk: <6 months old ▪ Tense non fluctuant mass in THE inguinal region and can extend to scrotum or labia majora ▪ Mass is well defined, tender, and does not reduce ▪ With edema and erythema of overlying skin, fever, signs of obstruction ▪ Testes may be normal or swollen because of venous congestion ▪ Clinical diagnosis ▪ Xray – features of partial or complete intestinal obstruction ▪ Gas within the incarcerated segments INCARCERATED HERNIA *also see Genital Disorders* Strangulated hernia – contents have become necrotic and gangrenous ▪ In children, non-reducible hernias rapidly progress to strangulation with potential infarction ▪ Pressure on herniated viscera → impaired lymphatic and venous drainage → compression, arterial occlusion → gangrene or perforation ▪ Most common <1yo ▪ Beyond 1-2yo → female preponderance (1:10) ▪ E. coli- most common cause URINARY TRACT INFECTION *also see Renal Disorders* If incarcerated – NGT, IV fluids, correct electrolyte imbalance Surgery – reduce contents, separate from spermatic cord vessels, and high ligation of sac at the internal ring (patent processus vaginalis) PROGNOSIS ▪ Wound infection increases to 5-7% if incarcerated Acute Pyelonephritis ▪ Abdominal, back or flank pain ▪ Fever (>39C for >24h in males and >48h in females) ▪ Malaise ▪ Nausea, vomiting ▪ Diarrhea Urine Culture – gold standard ▪ Suprapubic tap – gram (-) bacteria (any count) ▪ Transurethral catheterization – >50,000cfu/ml of single organism ▪ Midstream urine sample (3 specimens: 95% probability) ▪ If count is <105 but patient is symptomatic, infection likely Ultrasound – first line type of imaging Cystitis ▪ Dysuria, urgency, frequency, suprapubic pain, incontinence ▪ Malodorous urine Bottom up approach: UTZ + VCUG Top down approach: DMSA → VCUG COMPLICATIONS ▪ Recurrent hernia – 50% within 1yr of repair, 75% by 2yrs ▪ Iatrogenic cryptorchidism (trapped testicle) ▪ Injury to the vas deferens and infertility Acute Pyelonephritis ▪ 7 to 14 days oral/parenteral antibiotics ▪ IV: Ceftriaxone (50mday, not to exceed 2 g) or cefepime (100mkday q12h) or cefotaxime (100-150mkday in 3-4 divided doses) Cystitis ▪ 3 to 5 days of TMP-SMX COMPLICATIONS ▪ Renal scarring ▪ ESRD *see Diarrhea* *see Constipation* Batch Clingy ACUTE GASTROENTERITIS CONSTIPATION ▪ Ascending infection ▪ Attachment of type II P fimbriae (mannose resistant) to umbrella cells of the bladder → form intracellular bacterial communities → become filamentous → evades attack by WBCs in the urine → form quiescent intracellular reservoirs → source of recurrent infections and antiBiotic resistance Surgery ▪ Timing of operative repair includes age, general condition, and comorbid conditions ▪ If <1yo – prompt repair, high chance of incarceration ▪ If >1yo, less chance of incarceration PPS Oral Exam Reviewer 2020 72 Complex DISEASE BILIARY ATRESIA Noncystic obliterative cholangiopathy ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Most frequent identifiable cause of obstructive jaundice in the first 3 months of life ▪ Most common form is the 3rd type or non-patency of the entire extrahepatic biliary system and intrahepatic bile ducts at the hilum (85%) ▪ 85-90% are normal at birth and have a postnatal progressive obliteration of bile ducts ▪ May be associated with other congenital anomalies (BASM syndrome) or an immune or infection mediated process CLINICAL MANIFESTATIONS ▪ Generally, acholic stools with onset at about 2 weeks old ▪ Average birth weight ▪ Hepatomegaly with firm to hard consistency DIAGNOSIS ▪ Ultrasound: Triangular cord sign ▪ Normal hepatic uptake of dye on radionucleotide scan (HIDA, DISIDA) with absent duodenal excretion ▪ Biopsy: Bile duct proliferation, bile plugs, portal or perilobular fibrosis and edema, and intact lobular structure BA Jaundice at birth Jaundice presentation ▪ Impedance of bile flow through the intrahepatic and extrahepatic biliary trees after it is formed ▪ Bile plugging of the interlobular bile ducts, portal expansion and bile duct proliferation in association with centrilobular cholate injury Dark yellow urine Hepatomegaly Splenomegaly Acholic stools Alpha-fetoprotein CHOLEDOCHAL CYST ▪ Junction of the CBD and pancreatic duct before their entry into the Sphincter of Oddi → reflux of pancreatic enzymes → inflammation, localized weakness and dilatation of the duct ▪ Uncorrectable lesions – obstruction at or above the porta hepatis → Kasai Portoenterostomy ▪ 90% success rate if performed before the 8 th week of life Differential Diagnosis: BA vs Idiopathic Neonatal Hepatitis ▪ Female predominance ▪ Congenital dilatations of the common bile duct ▪ Most common variant is Saccular or Fusiform dilation of the CBD (Type 1) ▪ 75% appear in childhood TREATMENT / COMPLICATIONS / PROGNOSIS ▪ Correctable lesions – patent distal portion of extrahepatic duct, distal atresia → surgical resection ▪ Cholestatic Jaundice ▪ Severe liver dysfunction – ascites and coagulopathy ▪ Older Child: abdominal pain, jaundice, mass Features of acute cholangitis ▪ Fever ▪ RUQ tenderness ▪ Jaundice ▪ Leukocytosis INH Never Rarely 2-4 weeks 2-4 weeks yes yes yes Sometimes (cirrhosis) yes May be absent PROGNOSIS Life expectancy with untreated EHBA > 2 yrs ▪ 75% with SX <2months ▪ 10% with SX >3 months yes More common Transient/ incomplete Frequently (+) ▪ Ultrasound ▪ MRI – for preoperative assessment of choledochal cyst anatomy ▪ Primary excision of choledochojejunostomy the cyst and a Roux-en-Y COMPLICATIONS ▪ Cholangiocarcinoma ▪ Recurrent cholangitis ▪ Distal congenital stenotic segment of the biliary tree → ↑ intraluminal pressure and proximal biliary dilatation Batch Clingy ▪ Part of the spectrum of an infectious disease PPS Oral Exam Reviewer 2020 73 Complex ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Mechanisms of TBI: motor vehicle crashes, falls, assaults, and abusive head trauma ▪ Most TBIs in children are from closed-head injuries Primary injury – produces irreversible tissue disruption 2 types of secondary injury ● Ultimate damage seen in the injured brain evolves over hours or days ● Injured brain is vulnerable to additional insults because injury disrupts normal autoregulatory defense mechanisms ▪ Epidural, subdural and parenchymal intracranial hemorrhages can result ▪ Injury to gray or white matter is also commonly seen (e.g. focal cerebral contusions, diffuse cerebral swelling, axonal injury, and injury to the cerebellum or brainstem) CLINICAL MANIFESTATIONS ▪ Classified as mild, moderate, or severe MILD (from 2018 CDC Guideline on the Diagnosis and Mgt. of mTBI among Children) ▪ Acute brain injury resulting from mechanical energy to the head from external physical forces including: ● ≥1 of the ff: confusion or disorientation, loss of consciousness for ≤30 mins, post-traumatic amnesia for <24hrs, and/or transient neurological abnormalities (e.g. focal signs, symptoms, or seizure) ● GCS score of 13-15 30 mins. post-injury or later upon presentation for healthcare MODERATE ▪ GCS 9-12 ▪ Has the potential for deterioration especially in those with significant contusion due to progressive swelling SEVERE ▪ GCS 3-8 ▪ Coma is seen immediately after injury and is sustained Development of increased ICP with impending herniation may be heralded by: new-onset or worsening HA, depressed LOC, VS changes (hypertension, bradycardia, irregular respirations), and signs of 6 th or 3rd CN palsy ▪ In cases of epidural hematoma, child may be alert on presentation but may deteriorate after a period of hours ▪ “Talk-and-die” scenario in children with diffuse swelling DIAGNOSIS ▪ Diagnosis is generally obvious from the hx and PE ▪ Cranial CT should be obtained immediately after resuscitation and stabilization For mild TBI, the 2018 CDC Guidelines recommends that: ▪ HCP should not routinely obtain head CT for diagnostic purposes in children with mTBI ▪ HCP should use validated clinical decision rules to identify children with mTBI at risk for intracranial injury. Risk factors include: ● Age <2yo ● Vomiting ● Loss of consciousness ● Severe mechanism of injury ● Severe or worsening headache ● Amnesia ● Non frontal scalp hematoma ● GCS less than 15 ● Clinical suspicion for skull fracture ▪ Skull radiographs should not be used in the diagnosis of pediatric mTBI TREATMENT ▪ Infants and children with severe or moderate TBI receive ICU monitoring ▪ Initial stabilization: rapid sequence intubation with spine precautions, maintenance of normal extracerebral hemodynamics, MAP, and temperature ▪ Euvolemia is the target; plain saline is the fluid of choice ▪ Vasopressors may be needed as guided by monitoring of CVP ▪ Trauma survey should be performed COMPLICATIONS / PROGNOSIS ▪ Mortality rates range between 10-30% ▪ Ability to control ICP is related to patient survival ▪ Motor and cognitive sequelae resulting from severe TBI generally benefit from rehabilitation Cerebral herniation – pupillary dilation, systemic hypertension, bradycardia, extensor posturing, should be treated as a medical emergency ● Hyperventilation with FiO2 of 100% ● IV thiopental or pentobarbital ● Mannitol (0.2-1g/kg IV) or hypertonic saline (3% solution, 510 mL/kg) ▪ ICP should be maintained at <20mmHg ▪ First-tier therapy: ● Elevation of head of bed ● Midline positioning of the head ● Controlled mechanical ventilation ● Analgesia and sedation ● EEG monitoring if under neuromuscular blockade to check for status epilepticus ● Use of osmolar agents (e.g. hypertonic saline and mannitol) ▪ Second-tier therapy: ● Decompressive craniectomy for refractory traumatic intracranial hypertension ● Pentobarbital infusion ● Mild hypothermia (32-34OC) ● Hyperthermia must be avoided Batch Clingy (see Nelson’s 21st ed., Fig. 85.13 for Approach to Management of a child with severe TBI) PPS Oral Exam Reviewer 2020 74 Acute MIGRAINE without AURA ▪ Most common form of migraine in both children and adults ▪ Status migrainosus – migraine beyond 72hrs Triggers: skipping meals, inadequate or irregular sleep, dehydration and weather changes, menstrual periods, RED FLAGS FOR HA d/t ↑ ICP ▪ Daily or near daily morning vomiting ▪ HA waking the child up from sleep ▪ Remits with maintenance of upright posture ▪ Diagnostic Criteria (ICHD-3 beta) – recurrent (at least 5 attacks) lasting 4-72hrs in children and adolescents (may be as short as 2 hours) Has 2 of the ff. 4 characteristics: ▪ Unilateral/focal (more commonly bilateral in younger children) ▪ Pulsating quality (difficult to elicit in young children; drawing or demonstration might help) ▪ Moderate or severe pain intensity ▪ Aggravation by or causing avoidance of routine physical activity (e.g. reduction in play or physical activity; limitation or restriction of sports activity or exercise during attack) During HA, at least 1 of the ff: ▪ Nausea and/or vomiting (sporadic episodes that are not worsening is more likely to be migraine; getting up and going about normal, upright activities make it worse) ▪ Photophobia and phonophobia (more apparent as the child matures) MIGRAINE MIGRAINE with AURA ▪ Aura: neurologic warning that a migraine is going to occur Importance of aura duration: ▪ Lasting >5mins – differentiates migraine aura from a seizure with postictal headache) ▪ 60-min maximum – to separate migraine aura from a more prolonged neurologic event like TIA HEMIPLEGIC MIGRAINE MIGRAINE WITH BRAINSTEM AURA ▪ Basilar-type migraine ▪ Formerly considered a disease of the basilar artery PPS Oral Exam Reviewer 2020 In younger children, n/v may outweigh HA itself → overlap with GI periodic diseases (e.g. cyclic vomiting, abdominal migraine) → ↑ propensity for the later development of migraine TYPICAL AURA ▪ Visual, sensory, or dysphasic, lasts >5 mins and <60 mins with the HA starting w/in 60mins ▪ Visual aura: photopsia (flashes of light or light bulbs going off everywhere) is the most common; less likely types are fortification spectra or shimmering scotoma ▪ Sensory aura: less common, unilateral, insects crawling from hand up to face with numbness ▪ Dysphasic aura: least common, difficulty to respond verbally ATYPICAL AURA – hemiplegia (weakness), vertigo, distortion (Alice in Wonderland syndrome) Transient unilateral weakness lasting a few hours, may persist for days Vertigo, tinnitus, diplopia, blurred vision, scotoma, ataxia, occipital HA ANCILLARIES / DIAGNOSIS ▪ Thorough history and PE including a neurologic examination Family history ▪ Headaches and any other neurologic, psychiatric and general health conditions ▪ For identification of migraine within the family as well as possible secondary headache disorders ▪ Familial penetrance of migraine is so robust that the absence of a family history of migraine or its equivalent phenomena should raise the concern that the diagnosis may not be migraine and warrants further history taking, referral to a headache specialist, or investigation Neuroimaging: warranted when neuro exam is abnormal or unusual neurologic features occur during the migraine ▪ HA that awakens the child from sleep ▪ HA present on first awakening and remit with upright posture ▪ Brief HA that only occur when coughing or bending over ▪ HA mostly in the occipital area ▪ Migrainous headache with absolutely (-) family hx of migraine or its equivalent *see Nelson’s 21st ed., Table 613.7 for more indications MRI: Imaging of choice If child has HA that instantaneously at its worst at onset (thunderclap headache) – CT scan looking for blood is the best initial test. If negative, do LP to look for xanthochromia of the CSF ▪ Other labs or EEG is not beneficial in a typical migraine without aura or migraine with aura TREATMENT Goals of treatment (American Academy of Neurology) ▪ ↓ HA frequency, severity, duration, and disability ▪ ↓ Reliance on poorly tolerated, ineffective, or unwanted acute pharmacotherapies ▪ Improvement in quality of life ▪ Avoidance of acute HA medication escalation ▪ Education and enabling of patients to manage their disease to enhance personal control of their migraine ▪ ↓ HA-related distress and psychological symptoms ACUTE TREATMENT Goal: headache relief within 1hr with return to function 10 of 10 headaches *2019 AAN Clinical Practice Guidelines ▪ Initial tx: Ibuprofen 10 mg/kg ▪ For adolescents: triptans can be given ▪ If with contraindication to NSAIDs: Paracetamol 15 mg/kg ▪ If ibuprofen is not effective: Naproxen in similar doses ▪ Limit NSAIDs to ≤2-3 times/week ▪ Prevent medication-overuse headaches ▪ If weekly allowance of analgesics has been maximized, next step is to only use hydrating fluids for the rest of the week ▪ If migraine is especially severe, NSAIDs alone may not be sufficient; a triptan may be considered (e.g. almotriptan (12-17 y/o); rizatriptan (as young as 6 y/o); intranasal Zolmitriptan (12 y/o and older)) ▪ If 1 triptan fails to provide pain relief, offer an alternative triptan ▪ SE: tightness in the jaw, chest, and fingers, feeling of grogginess and fatigue ▪ For an acute attack, NSAIDs can be repeated once in 34hrs; triptans can be repeated once in 2hrs ▪ Other therapies: antiemetics (e.g. prochlorperazine and metoclopramide) PREVENTIVE THERAPY Indications: HAs are frequent (>1HA/wk) or disabling (missing school, home or social activities or having a PedMIDAS score >20) Goal: ↓ Frequency (to ≤1-2HA /mo); ↓ Level of disability ▪ Given 4-6 mos then tapered off ▪ Prophylactic agents: Flunarizine – only medication which demonstrated a substantial level of effectiveness Batch Clingy PRIMARY HEADACHE: recurrent, episodic; often sporadic; most common forms: migraine & tension-type headaches; neurologic exam should be normal SECONDARY HEADACHE: symptom of an underlying illness; common causes: head trauma, viral illness, sinusitis; serious causes: increased ICP DISEASE ETIOLOGY / INCIDENCE CLINICAL MANIFESTATIONS ▪ Most frequent type of recurrent headache ▪ Episodic HA moderate to severe intensity, focal in location, throbbing ▪ Occur in up to 10.6% of children between 5-15 years old and up to 28% quality, associated with nausea, vomiting, light sensitivity and sound of older adolescents sensitivity ▪ May be associated with typical (visual, sensory or dysphasic) or atypical aura (hemiplegia, Alice in wonderland) 75 CHILDHOOD PERIODIC SYNDROMES ▪ Group of potentially related symptoms that occur in increased frequency in children with migraine Hallmark: recurrent episodic nature of the events Cyclic Vomiting ▪ Many have a FHx of migraine and have a higher than average likelihood of developing migraine as they grow older ▪ Diagnosis of exclusion ▪ Must be differentiated from GI disorders, abnormal GIT motility and pelviureteric junction, metabolic causes (e.g. disorders of amino acid metabolism, organic acidosis, CHO metabolism), acute porphyria and CNS lesions ▪ Uncontrollable vomiting → dehydration ▪ Occurs in a predictable time schedule Abdominal Migraine ▪ To meet the criteria, child must complain at the time of the abdominal pain at least 2 of the ff: anorexia, nausea, vomiting and pallor ▪ Migraine without the headache ▪ Episodic ▪ Mid abdominal pain with pain free periods between attacks ▪ Dull pain, may be moderate to severe, persisting for 1-72hrs, may be periumbilical or poorly localized Diagnostic Criteria (ICHD-3 beta) ▪ At least 10 eps of HA occurring on <1d/mo on average (<12d/year) ▪ Lasting 30 mins to 7 days ▪ At least 2 of the ff. 4: ● Bilateral location ● Pressing or tightening (nonpulsating) quality ● Mild or moderate intensity ● Not aggravated by routine physical activity ▪ Both of the ff: ● No nausea/vomiting ● No more than 1 photophobia or phonohobia ▪ Common in children and adolescents (prevalence may be as high as 48%) ▪ An underlying psychological stressor is often suspected but difficult to identify/confirm TENSION HEADACHE ACUTE SINUSITIS Subtypes ▪ Infrequent (<12 times per year) ▪ Frequent (1-15 times per month) ▪ Chronic (>25 times per month) Amitriptyline – SE: sleepiness, wt. gain; may cause prolonged QT syndrome so avoid in patients w/ arrhythmia Antiepileptics (e.g. topiramate, valproic acid, levetiracetam) – effective for migraine prophylaxis in adults but limited studies in children w/ migraine Propanolol ▪ B-blocker best studied in pediatric migraine prevention ▪ CI: asthma or allergic disorders, diabetes Cyproheptadine – may be effective in very young children ▪ GI: colic, motion sickness, recurrent abdominal pain, recurrent vomiting including cyclic vomiting and abdominal migraine ▪ Sleep disorders: sleep walking sleep talking, night terrors, sometimes seizures BEHAVIORAL THERAPY ▪ Adherence to treatment plan is important ▪ Avoid triggers (e.g. skipping meals, dehydration, decreased or altered sleep), adequate fluid intake without caffeine, regular exercise, adequate sleep ▪ Thorough history and PE including a neurologic examination ensure exclusion of secondary etiologies Same general principles and medications used in migraine can be applied to children with TTH ▪ Simple analgesics (e.g. ibuprofen or acetaminophen) can be effective for acute treatment ▪ Amitriptyline has the most evidence of effective prevention of TTH ▪ Biobehavioral intervention can be useful as well *see Fever* Complex DISEASE CLINICAL MANIFESTATIONS Can present with clinical features observed in primary headache disorders (i.e. tension-type, migraine, and cervicogenic headaches) ANCILLARIES / DIAGNOSIS TREATMENT ▪ In the absence of purulent nasal discharge, fever or chronic cough, dx of sinus headache should not be made ▪ HA present for >15days/mo for >3mos and ▪ Intake of a simple analgesic >15 days/mo and/or ▪ Prescription medications >10 days/mo Batch Clingy SECONDARY CAUSES OF HEADACHE ETIOLOGY / INCIDENCE POST TRAUMATIC HEADACHE ICHD-3b classification ▪ Acute (last less than 3 mos) ▪ Persistent (last > 3 mos) ▪ Begin within 7 days after injury to the head or after regaining consciousness (may be arbitrary) SINUS HEADACHE ▪ Most over diagnosed form of recurrent headaches ▪ When HA are recurrent and respond within hours to analgesics, migraine should be considered first MEDICATION OVERUSE HEADACHE ▪ Suspect if with increasing use of analgesics with decreased effectiveness or frequently wearing off PPS Oral Exam Reviewer 2020 76 IDIOPATHIC INTRACRANIAL HYPERTENSION Pseudotumor Cerebri BRAIN TUMORS DIFFERENTIAL DIAGNOSIS Serious causes – due to increased ICP ▪ Can be due to mass (tumor, vascular malformation, cystic structure) → mass effect and local pressure on dura, or ▪ Intrinsic ↑ in pressure (i.e. idiopathic intracranial hypertension) → diffuse pressure on the dura ● Etiology may be d/t intake of excessive amounts of soluble compounds, hormonal changes, or blockage in venous drainage ▪ If ↑ ICP is suspected, MRI with MRA and MRV should be performed followed by LP (if no mass or vascular anomaly noted) Other causes not due to ↑ ICP: AVMs, collagen vascular diseases affecting CNS, hypertensive encephalopathy, acute subarachnoid hemorrhage, stroke ▪ Frequently considered a potential cause of HA with papilledema in ▪ Most frequent symptom: chronic (weeks-to-months), progressive, children with normal brain MRI findings frontal HA that may worsen with postural changes or a Valsalva ▪ A large proportion of children with possible/probable IIH will have a maneuver secondary IH identified after thorough Hx/PE. ▪ Vomiting may occur, rarely persistent & insidious ▪ Transient visual obscuration (transient graying out or vision loss often CAUSES associated with postural changes or Valsalva maneuvers) ▪ Metabolic Disorders: galactosemia, hypoparathyroidism, ▪ Diplopia secondary to abducens nerve dysfunction ▪ Pulsatile tinnitus hypophospatasia, prolonged corticosteroid therapy or rapid withdrawal, ▪ Children are alert and lack constitutional symptoms refeeding syndrome, hypervitaminosis A, severe vit A deficiency, Addison disease, obesity, menarche, OCP use, pregnancy ▪ Papilledema with an enlarged blind spot is the most consistent sign ▪ Infection: chronic OM, mastoiditis, GBS ▪ Presence of focal neurologic signs – investigate other causes ▪ Drugs: doxycycline, minocycline, tetracycline, nitrofurantoin, isotretinoin, sodium valproate ▪ Hematologic Disorders: polycythemia, various anemias, WAS ▪ Obstruction of intracranial drainage by cerebral venous thrombosis ▪ Second most common malignancy overall ▪ Depends on tumor location, type, and age of child ▪ Most common solid organ malignancy in childhood and adolescence ▪ Some Ssx are related to obstruction of CSF drainage paths by the tumor → ↑ ICP or focal brain dysfunction ▪ Familial syndromes account for approx. 5% of cases SUPRATENTORIAL LESIONS ▪ Cranial exposure to ionizing radiation is associated with a higher ▪ Subtle changes in personality, mentation, speech incidence ▪ More frequently associated with focal disorders – motor weaknesses, ▪ Incidence is highest in infants and children <5yrs old sensory changes, speech disorders, seizures, reflex asymmetry ▪ Premature hand preference in infants ▪ Age related differences in primary location: 0-1 y/o: supratentorial tumors predominate – choroid plexus tumors and INFRATENTORIAL LESIONS teratoma ▪ Classic triad: HA, n/v, and papilledema 1-10 y/o: infratentorial tumors predominate – medulloblastoma, juvenile ▪ Blurred vision, diplopia, and nystagmus are also associated PA After 10 y/o: supratentorial tumors again predominate – diffuse BRAINSTEM LESIONS astrocytoma most common ▪ Gaze palsy, multiple CN palsy, upper motor neuron deficits (hemiparesis, hyperreflexia, clonus) Cranial MRI: papilledema or enlargement of the optic never sheaths/pituitary fossa MR venography: to exclude a venous thrombosis and identify tapering of the lateral sinuses (commonly seen in intracranial hypertension) Measurement of CSF pressure: ideally, using an electronic transducer (minimize falsely elevated readings) ▪ When LP opening pressure is measured under GA, it is important to record a normal end-tidal partial pressure of CO2 MRI – neuroimaging standard Serum and CSF measurements of β-HCG and alpha fetoprotein to check germ cell tumors LP with CSF analysis ▪ For tumors that spread to meninges ▪ Contraindicated in those with hydrocephalus 2o to CSF obstruction ▪ Any cause of secondary IH should be treated ▪ Obese children with IIH need a weight-loss regimen ▪ Acetazolamide (10-30 mkday) probably is an effective regimen ▪ Serial monitoring of visual function and serial optic nerve examination is essential ▪LP may be diagnostic and therapeutic COMPLICATIONS ▪ Optic atrophy ▪ Visual impairment COMPLICATIONS 50% with chronic problems ▪ Chronic neurologic deficit – focal and sensory abnormalities ▪ Seizure disorders ▪ Neurocognitive deficits - developmental delays, learning disabilities ▪ Neuroendocrine deficiencies – hyperthyroidism, growth failure, delay or absence of puberty SUPRASELLAR TUMORS ▪ Neuroendocrine deficits (DI, galactorrhea, precocious puberty, delayed puberty and hypothyroidism) ▪ Diencephalic syndrome: FTT, emaciation despite normal caloric intake, inappropriately normal or happy affect ▪ Parinaud syndrome: paresis of upward gaze, pupillary dilation reactive to accommodation but not to light, nystagmus to convergence or retraction, eyelid retraction SPINAL CORD TUMORS PPS Oral Exam Reviewer 2020 Batch Clingy OPTIC PATHWAY TUMORS ▪ Visual disturbances, ↓ visual acuity, Marcus Gunn pupil, nystagmus, visual field defects 77 ▪ Long nerve tract motor and/or sensory deficits, bowel and bladder deficits, back or radicular pain TYPES OF BRAIN TUMOR 1) ASTROCYTOMA (40%) Low grade astrocytoma (LGA): predominant in children; indolent clinical course ▪ Surgery + chemoRT ▪ Pilocytic astrocytoma (PA) ● Most common astrocytoma in children ● Classic sites cerebellum & optic pathway region ▪ Seen as contrast enhancing nodule within the wall of a cystic mass ▪ Presence of Rosenthal fibers helps establish dx ▪ Diffuse astrocytoma (DA) ● 2nd most common astrocytoma ● Occur anywhere in the CNS with a predilection to supratentorial locations ▪ MRI: lack of enhancement after contrast infusion Medulloblastoma ▪ Cerebellar tumor ▪ Occur predominantly in males ▪ Median age: 5-7yrs old 5) PINEAL PARENCHYMAL TUMORS ▪ 2nd most common malignancy in the pineal region after germ cell tumors Pineoblastoma – subgroup of childhood PNETs 6) CRANIOPHARYNGIOMA 7) GERM CELL TUMORS ▪ Arise in midline structure of pineal and suprasellar regions ▪ Peak incidence: 10-12 y/o 8) TUMORS OF THE BRAINSTEM 9) METASTATIC TUMORS ▪ Uncommon ▪ Rarely can occur from lymphoma, neuroblastoma, rhabdomyosarcoma, Ewing sarcoma, osteosarcoma and clear cell sarcoma of the kidney ▪ Childhood ALL & non-Hodgkin lymphoma can spread to leptomeninges causes sx of communicating hydrocephalus PPS Oral Exam Reviewer 2020 ▪ Noninvasive but extends into the ventricular lumen; can lead to hydrocephalus ▪ Signs of ↑ ICP, macrocephaly, focal neuro deficits ▪ Ssx of ↑ICP and cerebellar dysfunction ▪ Irradiation with surgery ▪ Surgery ▪ Histology: small, blue round cell tumor; presence of Homer Wright rosettes ▪ Surgery + chemoRT ▪ Chemotherapy ▪ Endocrine abnormalities – growth failure, delayed sex maturation) ▪ Visual field changes – decreased visual acuity, decreased visual field deficits ▪ Surgery – main treatment ▪ No role for chemotherapy ▪ Alpha fetoprotein and β-hCG useful in diagnosis and response to treatment ▪ Motor weakness, CN deficits, cerebellar deficits, signs of increased ICP ▪ Depends if germinoma or non-germinomatous ▪ Surgery ± radiation and chemo ▪ Surgical resection for focal and dorsally exophytic tumors ▪ Radiation therapy for the others Batch Clingy Malignant astrocytoma: less common in children, expressed p53 prognostic 2) EPENDYMAL TUMORS (10%) ▪ From ependymal lining of ventricular system; approx. 70% occur in posterior fossa ▪ Mean age: 6yrs old 3) CHOROID PLEXUS TUMORS ▪ Most common CNS tumor in children <1yo ▪ Account for 10-20% of tumors in infants 4) EMBRYONAL TUMORS or PRIMITIVE NEUROECTODERMAL TUMORS (PNETs) ▪ Most common group of malignant CNS tumors of childhood ▪ Have the potential to metastasize to the neuraxis and beyond 78 Acute PPS Oral Exam Reviewer 2020 TREATMENT Parent education ▪ If seizure lasts >5mins, acute treatment with lorazepam, midazolam or diazepam is needed ▪ Antiepileptic therapy, continuous or intermittent, is not recommended for children with ≥1 simple FS – risk of SE and lack of demonstrated long-term benefits ▪ Antipyretics can decrease the discomfort of the child but do not reduce the risk of having a recurrent FS COMPLICATIONS / PROGNOSIS ▪ SFS do not have an increased risk of mortality ▪ CFS may have an approx. 2-fold longterm increase in mortality rates, as compared to the general population, over the subsequent 2 yr, prob. 2O to a coexisting pathology 2018 DOH National Antibiotic Guidelines ▪ Subdural effusions (10-30%) ▪ SIADH → hyponatremia and ↓ serum osmolality → exacerbate cerebral edema or result in seizures ▪ Risk for nosocomial infections >2mos – 5yo ▪ S. pneumoniae, Hib, N. meningitides ▪ Ceftriaxone 100mkday q12-24h OR ▪ Chloramphenicol 100 mkday q8h ▪ If penicillin- or cephalopsporin-resistant S. pneumoniae, add: Vancomycin 15-20 mg/kg q8-12h ▪ If Hib is suspected, add Dexamethasone 0.15mkday q6h x 4 days and Rifampin prophylaxis to eradicate carrier state >5 – 18 yo ▪ S. pneumoniae, N. meningitides ▪ Ceftriaxone 100mkday q12-24h OR ▪ Chloramphenicol 100mkday q8h 2015 PIDSP/CNSP CPG on Acute Bacterial Meningitis Drug of choice for specific etiology ▪ Hib: Ceftriaxone for 7-10 days (alt: chloramphenicol) ▪ S. pneumoniae: Penicillin for 10-14 days (alt: chloramphenicol or ceftriaxone) ▪ N. meningitides: Penicillin for 7 days (alt: ampicillin, ceftriaxone, chloramphenicol, and cefotaxime) ▪ Highest mortality rates are observed with pneumococcal meningitis ▪ Seizures that persist after the D4 of illness suggest poor prognosis and can be refractory to treatment ▪ Severe neurodevelopmental sequelae may occur in 10-20% of patients: ● Sensorineural hearing loss (most common) ● Cognitive impairment ● Recurrent seizures ● Language delay ● Visual impairment ● Behavioral problems ▪ Vaccination (PCV, MCV and Hib) and antibiotic prophylaxis (all close contacts of patients with Hib & meningococcal Batch Clingy SEIZURE – a transient occurrence of signs and/or symptoms resulting from abnormal excessive or synchronous neuronal activity in the brain DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS CLINICAL MANIFESTATIONS DIAGNOSIS ▪ Occur between the ages of 6 and 60mos (peak 12SIMPLE FEBRILE SEIZURE Lumbar Puncture 18mos) ▪ Generalized, tonic-clonic ▪ Meningitis should be considered ▪ Temperature ≥38oC ▪ Maximum of 15mins ▪ Should be performed in: ▪ Not the result of CNS infection or any metabolic ▪ Not recurrent within a 24h-period ● All infants younger than 6mos of age who present with fever imbalance and seizure ▪ Occur in the absence of a history of prior afebrile COMPLEX FEBRILE SEIZURE ● Child is ill-appearing seizures ▪ More prolonged (>15mins) and/or ● At any age if there are clinical signs or symptoms of concern ▪ Focal and/or ▪ An option in: ▪ Between 2% and 5% of neurologically healthy ▪ Recurs within 24hrs ● A child 6-12mos of age who is deficient in Hib and S. infants and children experience at least one, usually pneumoniae immunizations or for whom the immunization status is simple, febrile seizure (see Nelson’s 21st Ed., Table 611.6 for Risk unknown Factors for Occurrence of Subsequent ● Children pretreated with antibiotics FEBRILE SEIZURE (see Nelson’s 21st Ed., Table 611.5 for Risk Factors for Epilepsy After a FS) Recurrence of Febrile Seizures) EEG ▪ Need not be performed in a child presenting with the first FS and ▪ Pathophysiology remains unclear is otherwise neurologically healthy ▪ HHV-6 and HHV-7 infections – high reported rate of ▪ Restricted to special cases in which epilepsy is highly suspected association with FS (done after 2 weeks have passed) ▪ The genetic contribution to the incidence of FS is manifested by a positive FHx for FS in many patients Neuroimaging ▪ In some families, it is inherited as an autosomal ▪ Not recommended in evaluating a first simple FS dominant trait ▪ Workup of children with complex FS needs to be individualized and can include EEG and neuroimaging if the child is neurologically abnormal ▪ CNS infection primarily affecting the meninges Common symptoms Lumbar Puncture ▪ Typically, due to bacterial colonization of the ▪ HA, N/V, anorexia, photophobia, ▪ Most important step in the diagnosis nasopharynx with subsequent invasion into the restlessness, altered state of consciousness, ▪ Obtain CSF for cytology, Gram stain and culture bloodstream → breach BBB and enter the CNS to irritability ● Neutrophilic pleocytosis (CSF leukocyte count >1,000/mm3 cause infection and inflammation with 75-95% PMN), ↑ protein and ↓ glucose concentrations could Common signs be suggestive of a diagnosis of bacterial meningitis; findings usually ETIOLOGIES ▪ Fever, neck pain and rigidity, focal persist for several days after administration of appropriate Nelson’s 21st Ed. (in order of incidence) neurologic deficits, seizures, obtundation, antibiotics ▪ S. pneumoniae coma ● Gram stain is positive in >70% of patients with untreated ▪ N. meningitides bacterial meningitis; can be negative as early as 2-4hrs after ▪ H. influenza Type b ▪ Rash of meningococcemia – initial petechial administration of antibiotics ACUTE BACTERIAL rash → ecchymotic and purpuric lesions ● CSF culture is the gold standard for the diagnosis MENINGITIS 2015 PIDSP/CNSP CPG on Acute Bacterial Meningitis ▪ Kernig sign and Brudzinski sign (both not ▪ Contraindications: 3mos to <5yo consistently present in those <12-18mos) ● Evidence of increased ICP such as 3rd or 6th CN palsy with a ↓ ▪ H. influenza Type b ▪ Signs of increased ICP LOC, or the Cushing reflex ▪ S. pneumoniae ● Severe CP compromise requiring prompt resuscitation ≥5yo ● Infection of the skin overlying the site of the LP ▪ S. pneumoniae – most common ▪ If LP is delayed, empiric antibiotic therapy should be initiated ▪ N. meningitides – occur in epidemics or sporadically Neuroimaging ▪ Routine scans prior to LP are not recommended unless the patient is at risk for ↑ ICP Blood cultures ▪ Performed in all patients with suspected meningitis 79 ▪ An acute inflammatory process involving the meninges and/or brain parenchymal tissue VIRAL MENINGO ENCEPHALITIS BRAIN ABSCESS TOXIC INGESTION METABOLIC & ELECTROLYTE DISTURBANCES ▪ Neurologic damage is caused by direct invasion and destruction of neural tissues by actively multiplying viruses or by a host reaction to viral antigens ● In HSV-1 encephalitis, the temporal lobes are severely affected ● Arbovirus - affects the entire brain ● Rabies – predilection for the basal structures ▪ Contiguous spread from an assoc. infection – meningitis, OM, mastoiditis, sinusitis, SSTI of the face or scalp, orbital cellulitis, or dental infections ● Direct compromise of the BBB due to penetrating head injuries or surgical procedures ● Embolic phenomena (endocarditis) ● CHD with R to L shunts ● Immunodeficiency ● Infection of foreign material inserted to the CNS EEG ▪ Typically shows diffuse slow-wave activity ▪ Exanthems can precede or accompany CNS signs, especially with enteroviruses, VZV, measles, rubella and WNV ▪ Nuchal rigidity without significant localizing neuro changes MRI ▪ May demonstrate focal brain lesions that correlate with clinical disease ▪ Asymptomatic early in the course ▪ Non-specific symptoms – low grade fever, headache, lethargy ▪ Vomiting, severe HA, seizures, papilledema, focal neuro signs, coma Brain MRI with contrast – diagnostic test of choice Cranial CT scan – can be an alternative to provide more rapid imaging results but cannot provide the fine tissue detail offered by MRI Lumbar Puncture – not routinely recommended (could cause herniation) (see Nelson’s 21st Ed., Table 621.4 for Antibiotics Used for the Treatment of Bacterial Meningitis) ▪ For most causes, no effective antiviral agents exist ● Members of the herpesvirus family can be treated with acyclovir, ganciclovir, cidofovir and foscarnet meningitis) ▪ Recovery depends on the severity of the clinical illness, the specific causative agent, and the age of the child ▪ Supportive and rehabilitative efforts are very important after recovery ▪ Treatment is primarily supportive care ● IV fluids for poor oral intake ● NSAIDs for symptomatic relief of HA ● Monitor for seizures, cerebral edema, fluid and electrolyte imbalance, aspiration, respiratory failure, cardiac arrest ▪ Empiric therapy combination (3rd gen cephalosporin and metronidazole) often with vancomycin to provide MRSA coverage – duration depends on the organism and response to treatment but is most typically 6wks ▪ Aspiration of the abscess – recommended for diagnostic cultures and decompression unless contraindicated ▪ Mortality rate: 5-10% Long-term sequelae ▪ Hemiparesis ▪ Seizures ▪ Hydrocephalus ▪ CN abnormalities ▪ Behavioral and learning difficulties ETIOLOGIES ▪ Streptococci – most common ▪ S. aureus – 2nd most common ▪ Gram negative and anaerobes ▪ Seizures can occur in the setting of alcohol or BDZ withdrawal ▪ Can occur acutely in the setting of alcohol intoxication or illicit drug use (amphetamines, cocaine, narcotics) ▪ Other drugs: INH, aminophylline/theophylline, TCA, oral hypoglycemic agents ▪ Hypernatremia → brain hemorrhage due to movement of water out of the cell ▪ Hyponatremia → cerebral edema ▪ Hypomagnesemia → secondary hypocalcemia ▪ Hypocalcemia ▪ Hypophosphatemia ▪ Hypoglycemia *see Traumatic Brain Injury* Batch Clingy TRAUMA Etiologies ▪ Most common: enteroviruses, parechoviruses ▪ Most common arboviral cause: West Nile virus, Japanese encephalitis virus ▪ HSV type 1 – important cause of severe, sporadic encephalitis ▪ VZV may cause CNS infection in a close temporal relationship with chickenpox ▪ CMV – can occur with congenital infection or disseminated disease in immunocompromised hosts ▪ CNS Ssx are generally preceded by a nonspecific febrile illness of a few days’ duration ▪ Presenting manifestations in older children: HA (often frontal or generalized) and hyperesthesia; in infants: irritability, lethargy ▪ Fever, N/V, photophobia, pain the neck, back and legs ▪ Focal neurologic signs may be persistent, fluctuating, or migratory CBC, CRP, ESR and procalcitonin should not be used to routinely determine which patients should receive antibiotics CSF Analysis ▪ Pleocytosis of leukocytes with counts typically <1,000/mm3 ▪ Very early in the disease, cells are often PMN whereas mononuclear cells predominate for the remainder of the duration ▪ Protein tends to be elevated ▪ Glucose level is typically normal ▪ Primary means of detecting nonbacterial pathogens is nucleic acid amplification PPS Oral Exam Reviewer 2020 80 Complex ETIOLOGY / INCIDENCE / PATHOGENESIS The ILAE has refined the definition of SE to reflect ▪ The time at which treatment should be initiated (t1), and ▪ Time at which continuous seizure activity leads to long-term sequelae (t2) ▪ Continuous convulsive activity or recurrent generalized convulsive seizure activity without regaining of consciousness (t1 = 5 min; t2 ≥ 30 min); focal seizures with impaired awareness (t1 = 10 min; t2 = 30 min) and absence SE (t1 = 10-15 min; t2 = unknown) STATUS EPILEPTICUS PATHOGENESIS ▪ Failure of desensitization of AMPA glutamate receptors → persistent ↑ excitability, and ▪ ↓ GABA-mediated inhibition 2o to intra- cellular internalization of GABAA receptors ETIOLOGY ▪ New-onset epilepsy of any type ▪ Drug intoxication, drug & alcohol abuse ▪ Drug withdrawal or overdose in patients taking AEDs ▪ Hypoglycemia, hypocalcemia, hyponatremia, hypomagnesemia ▪ Acute head trauma ▪ Meningoencephalitis ▪ Autoimmune encephalitis ▪ Stroke (ischemic or hemorrhagic) ▪ IEM, MELAS ▪ Brain tumors ▪ Disorder of the brain characterized by an enduring predisposition to generate seizures ● At least 2 unprovoked (or reflex) seizures occurring >24hrs apart ● Enough EEG and clinical information to convincingly demonstrate an enduring predisposition to develop recurrences EPILEPSY Epileptic syndrome ▪ ≥1 specific seizure type, has a specific age of onset and a specific prognosis 4 distinct, often sequential, mechanistic processes Underlying etiology ▪ Any pathology or pathologic process that can disrupt neuronal function and connectivity (e.g. disorder in ion channel function and/or structure, gene mutations affecting neurotransmitter function, PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS CONVULSIVE SE ▪ GTC, tonic, or clonic ▪ Most common type of SE FEBRILE SE ▪ Most common type of SE in children NONCONVULSIVE SE ▪ Confusional state, dementia, hyperactivity with behavioral problems, fluctuating impairment of consciousness, fluctuating mental status, hallucinations, paranoia, aggressiveness, catatonia, and/or psychotic symptoms REFRACTORY SE ▪ Failed to respond to therapy, usually with at least 2 medications SUPERREFRACTORY SE ▪ Failed to resolve, or recurs within 24hrs or more despite therapy that includes a continuous infusion such as midazolam and/or pentobarbital GENERALIZED MOTOR SEIZURES ▪ Starts with LOC, upward rolling of the eyes, and a generalized tonic contraction → clonic phase that shows slowing of the rhythmic contractions until the seizure stops ABSENCE SEIZURES ▪ Usually starts at 5-8yo ▪ Do not have an aura, usually lasts for only a few seconds ▪ Eyelid flutter or upward rolling of the eyes ▪ No postictal period ▪ Immediate resumption of what the patient was doing before the seizure ▪ EEG: 3-Hz spike and slow wave discharges DIAGNOSIS Labs ▪ Glucose, Na, Ca, Mg, CBC, basic metabolic panel ▪ AED levels in patients known already to be taking these drugs ▪ Toxic drug screens and screens for IEM, if warranted EEG ▪ Helpful in ruling out pseudo-SE or other movement disorders ▪ Helpful in identifying the type of SE Neuroimaging ▪ Considered once the child is stabilized, especially if indicated by the clinical manifestations, by an asymmetric or focal nature of the EEG abnormalities or by lack of knowledge of the underlying etiology TREATMENT ▪ SE is a medical emergency ▪ Secure airway, breathing and circulation ▪ Admit to PICU for completion of therapy and monitoring American Epilepsy Society SE Guidelines ▪ BZD – started for convulsive seizures lasting >5mins (IV lorazepam, IV diazepam or IM midazolam) ▪ If seizures persist 5 min after the initial BZD dose, give a 2nd dose ▪ If still unsuccessful, fosphenytoin, valproate or levetiracetam is the recommended option ● IV phenobarbital is an alternative option if the above are not available ▪ If seizure persists, decision must be made regarding re-dosing with another second-line agent or proceeding to a continuous infusion COMPLICATIONS / PROGNOSIS ▪ Mortality rate: 4-5%, mostly 2o to the underlying etiology rather than to the seizures ▪ SE carries an approximately 14% risk of new neurologic deficits, most of them 2o to the underlying pathology (see Nelson’s 21st Ed., Fig 611.7 for the Proposed Algorithm for SE) Diagnosis of select generalized epilepsy syndromes DRAVET SYNDROME ▪ Myoclonic epilepsy of infancy ▪ Starts as focal febrile SE or focal FS and later manifests as myoclonic and other seizure types WEST SYNDROME ▪ Starts between 2-12mos ▪ Triad: infantile epileptic spams that occurs in clusters, developmental regression, and typical EEG (hypsarrhythmia) LENNOX-GASTAUT SYNDROME ▪ Starts between 2-10yo ▪ Triad: developmental delay, multiple seizure types that include atypical absences, and myoclonic, astatic, and tonic seizures and specific EEG findings ▪ Educate the family and the child ▪ Drug therapy should be based on the type of seizure and the epilepsy syndrome ● Oxcarbazepine and levetiracetam –first choice for focal seizures and epilepsies ● Levetiracetam – first drug to use in other primary generalized seizures ▪ Monotherapy should be the goal ● Dose is increased until complete control is achieved or until SE prohibit further increases ● Only then may another drug be added, and the initial drug be subsequently tapered ▪ Epilepsy carry a risk of increased mortality rates (two or more times the standardized mortality rates of the general population) mostly related to conditions assoc. with or underlying the epilepsy, poor seizure control and poor compliance with prescribed therapies (see Nelson’s 21st Ed., Table 611.7 for Sports and Special Considerations for the Child with Epilepsy) ▪ Dravet syndrome – BZD; keto diet can be useful ▪ West syndrome – hormonal therapy (either ACTH or oral steroids) Batch Clingy DISEASE 81 autoAb) Epileptogenesis ▪ Mechanism through which the brain, or part of it, turns epileptic ▪ Repeat seizures → rewiring of the brain and longterm epilepsy Epileptic state of increased excitability ▪ Dysregulation of glutamatergic excitation vs. GABAergic inhibition occurs Seizure-related neuronal injury FOCAL SEIZURES WITH PRESERVED AWARENESS ▪ Can be sensory seizures (aura) or brief motor seizures (most common) ▪ Brief motor seizures include focal tonic, clonic, or atonic szs with a Jacksonian march, adversive head and eye movements to the contralateral side or postictal Todd paralysis FOCAL SEIZURES WITH IMPAIRED AWARENESS ▪ Last 1-2 min and preceded by an aura ▪ ↓ Responsiveness, staring, looking around seemingly purposelessly and automatisms (automatic semipurposeful movements of the mouth, or the extremities) ▪ LGS – treatment varies according to predominant seizure type DOOSE SYNDROME ▪ Myoclonic astatic epilepsy ▪ Similar but milder than LGS ▪ Usually do not have tonic seizures or polyspike bursts in sleep Diagnosis of select epilepsy syndromes with focal seizures ▪ Absence seizures – treat initially with ethosuximide; alternatives are lamotrigine and valproate BENIGN CHILDHOOD EPILEPSY WITH CENTROTEMPORAL SPIKES ▪ Most common ▪ Starts at 3-10yo; outgrown by adolescence ▪ Child wakes up at night due to a focal seizure with preserved awareness causing buccal and throat tingling and tonic or clonic contractions of one side of the face, with drooling and inability to speak but with preserved consciousness and comprehension ▪ EEG: wide-based centrotemporal spikes that are markedly increased in freq. during drowsiness and sleep BENIGN EPILEPSY WITH OCCIPITAL SPIKES ▪ Panayiotopolous type: Occur in early childhood; manifests with focal seizures with impaired awareness and ictal vomiting ▪ Gastaut type: Appear later in childhood as focal seizures with impaired awareness, visual auras, and migraine HAs that occur independently or postictally ▪ Both are typically outgrown in a few years *see Headache* Batch Clingy BRAIN TUMOR ▪ EEG: 1- to 2-Hz spike and slow waves, polyspike bursts in sleep and slow background in wakefulness PPS Oral Exam Reviewer 2020 82 Complex DISEASE GENERALITIES / PATHOGENESIS ▪ A diverse group of conditions that result from impaired circulation and/or absorption of CSF Normal physiology: CSF is produced in the choroid plexus → lateral ventricles → foramen of Monro → 3rd ventricle → aqueduct of Sylvius → 4th ventricle → foramen of Luschka and Magendie → basal cisterns → convexities of the cerebral hemispheres → absorbed by arachnoid villi Normal rate of production: 20 mL/hr Normal value: 50mL in infants; 150mL in adults Obstructive/Non-communicating ▪ Obstruction within ventricular system ▪ Most commonly because of an abnormality of the aqueduct of Sylvius (e.g. aqueductal stenosis, aqueductal gliosis 2O neonatal meningitis or SAH in a premature infant) or a lesion in the fourth ventricle (e.g. posterior fossa brain tumors, Chiari malformation and the DandyWalker syndrome) HYDROCEPHALUS Non-obstructive/communicating ▪ Obliteration of subarachnoid cisterns or malfunction of arachnoid villi ▪ Most commonly follows a SAH 2O to IVH in a premature infant ▪ Pneumococcal and TB meningitis have a propensity to produce a thick, tenacious exudate that obstructs the basal cisterns ▪ Leukemic infiltrates can seed the subarachnoid space and produce communicating hydrocephalus CLINICAL MANIFESTATIONS Infants ▪ Accelerated rate of head enlargement ▪ Wide bulging anterior fontanel ▪ Dilated scalp veins ▪ Broad forehead ▪ Eyes deviate downward (setting sun eye sign) ▪ Long tract signs (brisk tendon reflexes, spasticity, clonus, and Babinski sign) ▪ Macewen sign: cracked pot sound on percussion of skull (indicates separation of sutures) Foreshortened occiput: Chiari malformation Prominent occiput: Dandy-Walker Older children ▪ Signs are subtler ▪ Irritability, lethargy, poor appetite, vomiting ▪ Headache is a prominent symptom ▪ Gradual change in personality and deterioration in academic productivity suggest a slowly progressive form of hydrocephalus Chiari Malformation Type I ▪ Produces s/sx during adolescence or adult life; not assoc. with hydrocephalus ▪ Recurrent HA, neck pain, urinary frequency and progressive LE spasticity Type II ▪ Progressive hydrocephalus w/ myelomeningocoele ▪ 10% produce symptoms during infancy (stridor, weak cry and apnea) ▪ Relieved by shunting or decompression of posterior fossa Neuroimaging: ▪ Plain skull films: separation of sutures, erosion of posterior clinoids (in older children), increase in convolutional markings on the inside of the skull (d/t long-standing increased ICP) ▪ CT/MRI and UTZ – most important studies to identify cause and severity of hydrocephalus DIFFERENTIALS FOR INCREASED HEAD SIZE ▪ Chronic anemia (thalassemia), rickets, osteogenesis imperfect, epiphyseal dysplasia Megalencephaly ▪ Anatomic disorder of brain growth ▪ Brain weight:volume ratio of >98th percentile for age (≥ 2 SD above the mean) ▪ Causes: storage and degenerative diseases; anatomic and genetic causes ▪ Benign familial megalencephaly: most common anatomic cause ▪ Sotos syndrome (cerebral gigantism): most common megalencephalic syndrome ▪ High forehead with frontal bossing, sparse hair in frontoparietal syndrome, downslanting palpebral fissures, apparent hypertelorism, long narrow face, prominent mandible, malar flushing ▪ Hypotonia, poor coordination and speech delay TREATMENT / PROGNOSIS Depends on cause ▪ Acetazolamide and furosemide: temporary relief by reducing rate of CSF production ▪ VP shunt in most cases COMPLICATIONS OF SHUNTING ▪ Headache ▪ Papilledema ▪ Emesis ▪ Mental status changes ▪ Bacterial infection PROGNOSIS ▪ Depends on the cause of dilated ventricles ▪ Increased risk for dev disabilities ● Mean IQ is reduced ● Abnormalities in memory function ● Vision problems (strabismus, visuospatial abnormalities, visual field defects, optic atrophy) ▪ Some show aggressive and delinquent behavior ▪ Accelerated pubertal development in pts. with shunted hydrocephalus because of ↑ gonadotropin secretion in response to increased ICP Hydranencephaly ▪ Cerebral hemispheres absent or represented by membranous sacs ▪ Midbrain and brainstem intact ▪ Transillumination shows absence of cerebral hemispheres ▪ Irritable, feeds poorly, develops seizures and spastic quadriparesis, little or no cognitive dev’t Batch Clingy Dandy walker formation ▪ Cystic expansion of 4th ventricle in posterior fossa and midline cerebellar hypoplasia (dev failure of roof of 4 th ventricle during embryogenesis) ▪ 90% have hydrocephalus with assoc. anomalies (agenesis of posterior cerebellar vermis and corpus callosum) ▪ Rapid ↑ in head size and prominent occiput ANCILLARIES / DIAGNOSIS ▪ Inspect, palpate, auscultate skull and spine ▪ Inspect back for midline lesions ▪ Prominent forehead or abnormalities in the shape of the occiput can suggest hydrocephalus ▪ Cranial bruit: vein of Galen AVM ▪ (+) Transillumination: massive dilation of the ventricular system or DandyWalker syndrome ▪ (+) Chorioretinitis: suggestive of intrauterine infection (e.g. toxoplasmosis) as cause of hydrocephalus PPS Oral Exam Reviewer 2020 83 ▪ Nontraumatic intracranial hemorrhage: intraparenchymal bleeds or intraventricular hemorrhage ▪ Bleeding outside the brain: SAH, subdural and epidural hematomas HEMORRHAGIC STROKE Intracranial Hemorrhage ▪ Cause and risk factors for HS include vascular malformations and systemic disorders (see Nelson’s 21 st ed., Table 619.4) ● Arteriovenous malformations: most common cause of childhood SAH and intraparenchymal HS ● Other vascular malformations: cavernomas, dural AV fistulas, vein of Galen malformations ● Cerebral aneurysms: less common cause of SAH; suggest underlying disorder ● Bleeding from a preexisting brain tumor is another common cause ● Additional causes: hypertensive hemorrhage, hematologic disorders (e.g. ITP, hemophilia, DIC), anticoagulant therapy, or illicit drug use ● Ischemic infarcts may undergo hemorrhagic transformation ▪ Vary according to location, cause and rate of bleeding ▪ CT scan is highly sensitive ▪ LP may be required to exclude SAH ▪ Angiography – to exclude vascular abnormalities ▪ Cranial UTZ in neonatal HS – detect neonatal parenchymal bleeds, especially in the preterm, where bleeds are located centrally within the cranium (germinal matrix bleeding and intraventricular hemorrhage) and in the cerebellum Acute hemorrhages ▪ Instantaneous (thunderclap headache) ▪ Loss of consciousness ▪ Nuchal rigidity ▪ Focal neurologic deficits ▪ Seizures PROGNOSIS ▪ Likely depends on lesion size, location, and etiology ▪ Compared with arterial ischemic stroke, HS mortality rate is higher but long-term deficits are less common ▪ Recurrence risk for those with structural lesions is significant Vascular malformations ▪ Pulsatile tinnitus ▪ Cranial bruit ▪ Macrocephaly ▪ High output heart failure Abusive head trauma ▪ Subtle scalp, suborbital, or ear bruising ▪ Retinal hemorrhages in ▪ Chronic failure to thrive ▪ Emergent neurosurgical intervention ▪ Neuroprotection ▪ Reversal of anticoagulant therapy (e.g. vit K or FFP) but other medical interventions like FVII are unstudied in children ▪ Definitive repair or removal of the vascular malformation multiple layers Abusive head trauma: may present as primary subdural or parenchymal hemorrhage ● Epidural hematoma: nearly always caused by trauma (e.g. middle meningeal artery injury assoc. w/ skull fracture) ● Subdural hematoma: can occur spontaneously or with trivial trauma *see Headache* *see Seizures* Batch Clingy BRAIN TUMOR CNS INFECTIONS PPS Oral Exam Reviewer 2020 84 Chronic DEFINITION / EPIDEMIOLOGY ETIOPATHOGENESIS DIAGNOSTIC CRITERIA / SYMPTOMS DIFFERENTIALS SCREENING AND ASSESSMENT MANAGEMENT ▪ Neurobiologic disorder with onset in early childhood ▪ Result from disrupted neural connectivity ▪ Primarily impacted by genetic variations affecting early brain development ● Animal models and studies of individuals with ASD indicate changes in brain volume and neural cell density in the limbic system, cerebellum, and frontotemporal regions ● Numerous genes involved in brain development and synaptic function have been associated with ASD DSM-V criteria focus on symptoms in 2 primary domains: social communication and social interaction and restrictive and repetitive behavior ● Symptoms should be present since the early developmental period ● Significantly impact functioning ● Not better explained by other diagnosis (see Nelson’s 21st ed., Table 54.1 for full DSM-V criteria) Language disorder ▪ Impaired social communication and play but social and play skills are at par ▪ No associated restricted/ repetitive behavior Universal screening at age 18mos and 24mos ▪ Screening should also occur when there is increased risk for ASD (child with an older sibling who has ASD or concern for possible ASD) ▪ MCHAT-R/FU: most common screening tool ▪ Thorough history and detailed physical examination including direct behavioral observations of communication and play Developmental and educational programming: primary treatment for ASD ▪ Earlier age at initiation of tx and higher intensity leads to better outcomes Key features: ▪ Impaired social communication and social interaction and ▪ Restricted and repetitive behaviors ▪ 4:1 male predominance ▪ Prevalence increased in siblings (up to 10%) particularly in identical twins ▪ No racial or ethnic differences in prevalence ▪ There is also evidence for environmental contributions ● Older maternal or paternal age may increase the risk ● Factors influencing the intrauterine environment (maternal obesity or overweight, short interval from prior pregnancy, premature birth, certain perinatal infections) AUTISM SPECTRUM DISORDER ▪ Multiple research studies and metaanalyses have failed to show an association of vaccines with ASD Social Communication and Social Interaction ▪ Difficulty understanding and engaging in social relationships ▪ Pervasive and impact 3 major areas: Social-emotional reciprocity ▪ Young child with ASD may not respond to name calling, exhibit limited showing and sharing behaviors, prefers solitary play ▪ Older child with ASD may have an interest in peers but does not know how to initiate or join in play Nonverbal communication ▪ Reduced eye contact and gestures ▪ Limited range of facial expression Understanding of social relationships ▪ Limited insight regarding social relationships Restrictive and Repetitive Behavior (At least 2 of 4 symptoms) ▪ Stereotyped motor movements or speech (e.g. hand flapping, spinning, echolalia, lining up objects) ▪ Insistence on sameness (i.e. insist certain routines or schedules) ▪ Restricted interests (i.e. intense interest that seem out of the norm in comparison to same-age peers) ▪ Hypo- or Hyperreactivity to Sensory Input ▪ Most common neurobehavioral disorder of childhood ATTENTIONDEFICIT/ HYPERACTIVITY DISORDER Characterized by: ▪ Inattention ▪ Poor impulse control ▪ ↓ Self-inhibitory capacity and motor overactivity ▪ Motor restlessness ▪ 5-10% of school-age children are affected ▪ Mothers of children with ADHD are PPS Oral Exam Reviewer 2020 Dopamine hypothesis ▪ Disturbances in the dopamine system may be related to the onset of ADHD ▪ Brain MRI studies indicated a reduction or even loss of the normal hemispheric asymmetry in the brain as well as smaller brain volumes in the prefrontal cortex and basal ganglia (both rich in dopamine receptors) ▪ Dopaminergic MOA of medication treatment for ADHD ▪ Genetic studies have primarily implicated 2 candidate genes: Dopamine transporter gene (DAT1) and Dopamine 4 Intellectual disability/GDD ▪ Delays in social and communication skills as well as stereotyped behavior but social and communication skills are commensurate with their cognitive and adaptive functioning Hearing loss ▪ Poor response to name but typically develop nonverbal communication and play skills ▪ No stereotyped or restricted behavior patterns ADHD ▪ May present with reduced eye contact and response to name caused by poor attention rather than lack of social awareness ▪ No impairments in shared enjoyment and social reciprocity or repetitive behaviors ▪ Examination should include measurement of HC, evaluation for dysmorphic features, screening for tuberous sclerosis with Wood lamp exam, genetic testing, audiology examination and a lead test (in children with pica) Additional medical or behavioral health treatment is often required for mgt of co-occurring conditions in ASD There are currently no medications that treat the core symptoms of ASD Social anxiety ▪ Shy children may have reduced eye contact and social initiation; Anxious children can be resistant to change ▪ Have preserved social interest and insight and will not exhibit high levels of stereotyped behavior Reactive attachment disorder ▪ Difficult to distinguish from ASD particularly in younger children with hx of trauma but social behaviors improve with positive caretaking ▪ Symptoms vary from motor restlessness and aggressive and disruptive behavior (common in preschool children) to disorganized, distractible and inattentive symptoms (typical in older adolescents and adults) ▪ Difficult to diagnose in preschoolers because distractibility and inattention are often considered developmental norms during this period Chronic illness ▪ E.g. migraine, absence seizures, asthma/allergies, hema d/o, diabetes, childhood CA ▪ Can impair children’s attention and school performance because of either the disease itself or the medications used to treat or control the underlying illness ▪ DSM-V criteria state that the behavior must be developmentally inappropriate (substantially different from that of other children of the same age and developmental level) beginning before 12yo, present for at least 6 mos. in 2 or more settings as reported Substance abuse ▪ Can result in declining school performance and inattentive behavior Sleep disorders (e.g. OSA) ▪ The clinical interview allows a comprehensive understanding of whether the symptoms meet the diagnostic criteria for ADHD ▪ Behavior rating scales are useful in establishing the magnitude and pervasiveness of symptoms but not sufficient alone to make a diagnosis Behaviorally Oriented treatments ▪ Goal: identify target behaviors that cause impairment in the child’s life and for the child to work on improving his/her skill in these areas ▪ Modestly successful at improving core ADHD symptoms ▪ First-line treatment in preschool age children ▪ No laboratory tests are available to identify ADHD ● Presence of HTN, ataxia, or symptoms of sleep or thyroid d/o Medications ▪ Most widely used medications are the presynaptic dopaminergic agonists called psychostimulants Batch Clingy DISEASE 85 receptor gene (DRD4) by independent observers and not secondary to another disorder ▪ Three different presentations of ADHD ● Inattentive: more common in females; assoc. w/ high rates of anxiety and low mood ● Hyperactive-impulsive: more common in males together w/ combined type ● Combined (see Nelson’s 21st ed., Table 49.1 for full DSM-V criteria) ▪ Group of permanent disorders of movement and posture causing activity limitation ▪ Most common and costly form of chronic motor disability that begins in childhood CEREBRAL PALSY Attributed to nonprogressive disturbances in the developing fetal or infant brain SPASTIC HEMIPLEGIA ▪ 34% had injury to the white matter in utero ▪ 27% had a focal lesion resulted from a stroke ▪ Other children with hemiplegic CP had malformations from multiple causes: infections, disorders of neuronal migration SPASTIC DIPLEGIA ▪ Strongly associated with damage to immature WM during the vulnerable period of immature oligodendroglia (20-34 wks AOG) ▪ 15% of cases result from in utero lesions in infants who go on to delivery at term Periventricular leukomalacia ▪ Most common neuropathologic finding ▪ Visualized in MRI SPASTIC QUADRIPLEGIA ▪ Most severe form of CP ▪ Most common lesions: severe PVL and multicystic cortical encephalomalacia PPS Oral Exam Reviewer 2020 (see Nelson’s 21st Ed., Table 616.1 for Classification of Cerebral Palsy and Major Causes) ▪ ↓ Spontaneous movements on the affected side ▪ Hand preference at a very early age ▪ Arm often more involved than the leg ▪ Walking is delayed until 18-24mos with a circumductive gait ▪ Ankle clonus and Babinski sign may be present ▪ Approx. 25% have cognitive abnormalities including ID ▪ 1/3 have a seizure disorder that usually develops in the first 2yrs ▪ Bilateral spasticity of the legs > arms ▪ Infants uses arms normally when crawling but drags the legs behind (commando crawl) instead of 4limbed crawl ▪ Spastic legs with brisk reflexes, ankle clonus and bilateral Babinski ▪ Scissoring posture of the LE when suspended by the axillae ▪ Likelihood of seizures is minimal ▪ Normal intellectual development ▪ Some may have learning disabilities or vision abnormalities ▪ Marked motor impairment of all extremities ▪ High assoc. with ID and seizures ▪ Swallowing difficulties → aspiration pneumonia and growth failure ▪ ↑ Tone and spasticity in all extremities, ↓ ▪ Can result in behavioral and emotional symptoms that can resemble or exacerbate ADHD should prompt work-up ▪ Identify any possible vision or hearing problems Depression and anxiety disorders ▪ Can mimic sx of ADHD but can also be a comorbid condition (e.g. methylphenidate, dexmethylphenidate, amphetamine) ▪ Children with a positive personal or FHx of cardiomyopathy, arrhythmias, or syncope require an ECG and possible cardio consult before starting stimulants OCD ▪ Can mimic ADHD esp. when recurrent and persistent thoughts, impulses or images are intrusive and interfere with normal activities Adjustment disorders ▪ Can result in symptoms similar to ADHD ▪ Hereditary spastic diplegias ▪ Monoamine transmitter disorders IEM ▪ History and PE should r/o a progressive disorder of the CNS ▪ MRI – to determine the location and extent of structural lesions or assoc. congenital malformations ▪ Test of hearing and visual function ▪ IV MgSO4 – for mothers in premature labor with birth imminent <32wks showed significant ↓ in the risk of CP at 2yo ▪ Cooling term infants with HIE to 33.3OC x 3 days within 6h of birth – ↓ the risk of dyskinetic or spastic quadriplegia ▪ Multidisciplinary mgt. is needed ▪ Drugs for spasticity: Baclofen, Botox ▪ Movement of the good side is constrained with casts ▪ The impaired extremities perform exercises that induce improved hand and arm functioning ▪ Treated initially with assistive equipment – orthoses, walkers, poles, standing frames ▪ Rhizotomy procedure – produces considerable improvement in selected patients with severe spastic diplegia and little or no basal ganglia involvement ▪ Motorized wheelchairs ▪ Special feeding devices ▪ Modified typewriters ▪ Customized seating arrangements Batch Clingy more likely to experience birth complica-tions (e.g. toxemia, lengthy labor, complicated delivery) ● Maternal drug use ● Maternal smoking, alcohol use during pregnancy, and prenatal or postnatal exposure to lead 86 ATHETOID (extrapyramidal) CP ▪ Most likely to be associated to birth asphyxia ● Assoc. with bilaterally symmetric lesions in the posterior putamen and ventrolateral thalamus ▪ Can also be caused by kernicterus ▪ MRI: lesions in the globus pallidus bilaterally ▪ Group of disorders that have in common significant impairment in general intellectual function, social skills, and adaptive behavior ▪ >2/3 will not have a readily identifiable underlying diagnosis ▪ 2 overlapping populations of children with ID with differing corresponding etiologies ▪ Mild ID: assoc. more with environmental influences; highest risk among children of low socioeconomic status ▪ Most common biologic causes of mild ID: genetic or chromosomal syndromes with multiple, major, or minor congenital anomalies, IUGR, prematurity, perinatal insults, intrauterine exposure to drugs and sex chromosomal abnormalities ▪ Severe ID: more freq. linked to biologic and genetic causes (e.g. chromosomal and other geneic and epigenetic disorders, abnormalities of brain development, IEM, neurodegenerative d/o) ▪ Nonsyndromic severe ID may be a result of inherited or de novo gene mutations, as well as microdeletions or microduplications not detected on standard chromosomal analysis Global Developmental Delay ▪ Diagnosis given to children <5yo who display significant delay (> 2SD) in acquiring early childhood devt’l. milestones in ≥2 domains (receptive and expressive language, gross and fine motor function, cognition, social and personal development, ADLs) ▪ Not all children who meet criteria for GDD go on to meet criteria for ID after 5 yo INTELLECTUAL DISABILITY spontaneous movements, brisk reflexes, plantar extensor responses ▪ Speech and visual abnormalities ▪ Infants are characteristically hypotonic with poor head control and marked head lag ▪ Develop variably increased tone with rigidity and dystonia ▪ UE are more affected than LE ▪ Tongue thrust and drooling are prominent ▪ Speech is affected d/t oropharyngeal muscle involvement ▪ UMN signs are not present, seizures are uncommon, intellect is preserved Significant impairment in general intellectual function ▪ Performance on an individually administered test of intelligence that is approx. 2 SD below the mean Significant impairment in adaptive behavior ▪ The degree that the cognitive dysfunction impairs daily function ▪ Adaptive behavior: skills required for people to function in their everyday lives ▪ 3 broad set skills: conceptual, social and practical ● Conceptual skill: language, reading, writing, time, number concepts, and self-direction ● Social skills: interpersonal skills, personal and social responsibility, self-esteem, gullibility, naiveté, and ability to follow rules, obey laws, and avoid victimization ● Practical skills: ADLs (dressing, feeding, toileting/bathing, mobility), instrumental ADLs (housework, managing money, taking medication, shopping, preparing meals, using phone), occupational skills, and maintenance of a safe environment ▪ For a deficit to be present, a significant delay in at least 1 of the 3 skill areas must be present Onset before age 18 y/o or adulthood ▪ Distinguishes dysfunctions that originate during the developmental period ▪ Diagnosis of ID may be made after 18yo but the cognitive and adaptive dysfunction must have been manifested before age 18 ▪ Sensory deficits – severe hearing and vision loss ▪ Communication disorders ▪ Refractory seizure disorders ▪ Poorly controlled mood disorders ▪ Unmanaged severe attention deficits can mimic ID ▪ Many children with CP or ASD also have ID (see Nelson’s 21st ed., Fig. 53.1 for algorithm for the evaluation of the child with unexplained GDD or ID) Microarray analysis: replaced karyotyping as first-tier testing ▪ A child with a progressive neurologic disorder, developmental regression or acute behavioral changes needs metabolic investigation ▪ A child with seizure-like episodes should have an EEG ▪ MRI of the brain may be useful in some instances ▪ Interdisciplinary mgt ▪ Periodic reevaluation ▪ Family counseling ▪ Transition to adulthood (see Nelson’s 21st ed., Table 53.5 for Suggested Evaluation of the Child with ID or GDD) Bayley Scales of Infant and Toddler Development ▪ Most commonly used infant intelligence test ▪ Used in children between 1-42 mos. ▪ Differentiates infants with severe ID from typically developing infants Wechsler Scales ▪ Most commonly used test for children older than 3yo Batch Clingy (see Nelson’s 21st ed., Table 53.4 for Common Presentations of Intellectual Disability by Age) PPS Oral Exam Reviewer 2020 87 SLD with an impairment in math / Math learning disability (MLD) ▪ Difficulties mastering number sense, number facts, or fluent calculation and difficulties with math reasoning (DSM-V) SPECIFIC LEARNING DISORDER (SLD) SLD with impairment in written expression (IWE) Oral language ▪ Complex process that typically develops in the absence of formal instruction Written language ▪ Requires instruction in acquisition (word reading), understanding (reading comprehension), and expression (spelling and composition) Broad Cognitive Processes ▪ Children with MLD have significantly poorer math achievement than children with low IQ and have severe deficits in math not accounted for by their cognitive functioning ▪ Children with MLD have shown deficits in working memory and are often slower to complete math problems than their typically developing peers Math Specific Processes ▪ Procedural Errors ▪ The type of errors made by children with MLD are typical for any child but with a 23year lag in understanding the concept ▪ Memory for Math Facts ▪ Committing math facts to or retrieving facts from memory have consistently been found to be problematic for children with MLD Children with IEW have skill deficits in: Transcription ▪ Tend to write slowly, or when writing at reasonable speed, the legibility degrades Oral Language ▪ Writing difficulties are assoc. with deficits in both expression and comprehension of oral language ▪ Symptoms in the DSM-V criteria must be present for a minimum of 6 months and persist despite interventions to address the learning challenges ▪ Risk Factors for MLD: ● Child is at or below the 20 th percentile in any math area, as reflected by standardized testing or ongoing measures of progress monitoring ● Teacher expresses concern about the child’s ability to “take the next step” in math ● Positive FHx for MLD ▪ Parents think they have to “reteach” math concepts to their child ▪ Explicit instruction on solving specific types of problems: most effective intervention (see Nelson’s 21st ed., Table 51.3 for full DSM-V criteria) ▪ Weak graphomotor skills may not necessarily require intervention from an OT but may help ▪ Explicit instruction of writing strategies combined with implementation and coaching in selfregulation Dysgraphia ▪ Often used synonymously with IWE but the two are distinct conditions ▪ Primarily a deficit in motor output (paper/pencil skills) and IWE is a conceptual weakness in developing, organizing, and elaborating on ideas in writing Executive Functions ▪ Poor writers seldom engage in the necessary planning and struggle to selfmonitor and revise effectively Batch Clingy Working Memory ▪ Weak WM limits the space available to devote to other tasks PPS Oral Exam Reviewer 2020 88 IRON DEFICIENCY ANEMIA G6PD DEFICIENCY THALASSEMIA ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ 2% of adolescent girls – secondary to adolescent growth spurt and menstrual blood loss ▪ Teenagers who are or have been pregnant – highest risk of IDA (>30%) ▪ Breastfed infants absorb iron 2-3x more efficiently than infants fed cow’s milk but are at risk of developing IDA if without regular intake of iron-fortified food by 6mos old ▪ In neonates, iron is in circulating Hgb → Hgb levels fall during the first 2-3 mos of life → iron is recycled and sufficient for blood formation in the first 6-9 mos of life (if term) ▪ In infants and toddlers, due to excessive consumption of cow’s milk (low iron content, blood loss from milk protein colitis) in a child who is often overweight or bottle feeding beyond 12mos old ▪ May be due to blood loss – menstrual losses, recurrent nosebleeds, intravascular hemolysis with hemoglobinuria (e.g. in malaria), peptic ulcer, Meckel diverticulum, polyp, hemangioma, IBD, chronic diarrhea, chronic intestinal blood loss induced by exposure to whole cow’s milk protein ▪ Hookworm infections (developing countries) – Trichuris trichiura, Plasmodium ▪ Gastric bypass procedures / H. pylori infection – interferes with iron absorption since iron is absorbed in the proximal duodenum by the assistance of gastric acid Acute CLINICAL MANIFESTATIONS LABORATORY FINDINGS/DIAGNOSIS ▪ Most are asymptomatic ▪ ↓ serum ferritin (depleted tissue iron stores) → ↓ serum iron ▪ Pallor – most recognized clinical sign (Hgb 7-8mg/dL) → ↑ serum transferrin → ↓ transferrin saturation ▪ Depleted iron stores → iron is unavailable to form with Mild to Moderate IDA (Hgb 6-10g/dL) protoporphyrin to form heme → impaired Hgb synthesis → no ▪ Mild irritability only because of effective compensatory available Hgb in each cell → ↓and varied RBC size (↑ RDW), ↓ mechanisms (↑2,3 DPG; shift of O2 dissociation curve) MCV and MCH, ↓ RBC count ▪ ↑ TIBC Severe (Hgb <5g/dL) ▪ N or mod ↑ reticulocyte count ▪ Irritability, anorexia, lethargy, systolic flow murmurs ▪ Elliptocytic or cigar shaped cells are often seen ▪ Tachycardia and high output cardiac failure can occur ▪ Bone marrow iron staining – most accurate method of diagnosing IDA; invasive, expensive, unnecessary Non-hematologic systemic effects: ▪ N WBC count, often ↑ platelet ▪ Impaired neurocognitive function in infancy → may ▪ Occult blood test (stool) – to exclude blood loss as case of IDA lead to irreversible cognitive defects ▪ Pica (nonnutritive substances) → ingestion of leadSerum ferritin containing substances → plumbism ▪ Iron-storage protein, provides an estimate of body iron stores in ▪ Pagophagia (ice) the absence of inflammatory disease ▪ A low SF value is diagnostic of IDA in a patient with anemia Serum transferrin ▪ Iron binding capacity of the serum Increase in Hgb ≥1g/dL after 1 mo of iron therapy – most practical means to establish the diagnosis ▪ Most frequent disease involving enzymes of the HMP ▪ Asymptomatic ▪ Precipitous fall in Hgb and Hct ▪ X-linked deficiency; inheritance of any of a large number of abnormal ▪ Neonatal jaundice and hemolytic anemia if triggered by ▪ If severe, haptoglobulin (Hgb binding protein) are saturated, and alleles of the gene responsible for the synthesis of the G6PD protein infection, drugs (ASA, sulfonamides, rasburicase, free Hgb may appear in plasma and urine ▪ 4.9% global prevalence, M>F antimalarials), or ingestion of fava beans ▪ Heinz bodies (precipitated Hgb) – seen only within the first 3-4 ▪ 2 clinical syndromes ▪ Hemolysis after 24-48hrs of ingestion of substances days of illness in unstained RBCs, rapidly cleared from the blood ● Episodic hemolytic anemia with oxidant properties → hemoglobinuria and jaundice ▪ Bite cells ● Chronic, nonspherocytic hemolytic anemia if severe → ↓ Hgb level ▪ Polychromasia – bluish, larger RBCs; represent reticulocytosis ▪ Glucose 6 phosphate → G6PD → phosphogluconic acid → NADPH → ▪ The dx is suspected when G6PD is within the low-normal range GSH (reduced, functional form of glutathione) ▪ Favism – acute severe hemolytic syndrome (more of in the presence of a high reticulocyte count ▪ GSH – provides protection against oxidant threats from certain drugs G6PD B-variant); fava beans contain divicine, isouramil, ▪ Newborn screening – confirmatory test to check enzyme activity and infection that can cause precipitation of Hgb (Heinz bodies) or and convicine → production of H2O2 and other reactive damage RBC membrane O2 products ▪ Disorders of globin chain production – imbalance between the α and β globin chains ▪ Primary pathology stems from the quantity of globin produced ▪ Inadequate β-globin gene production leading to ↓HbA + unbalanced α- and β-globin chain production leading to ineffective erythropoiesis α – THALASSEMIA α-thalassemia trait – microcytic anemia that can be α-thalassemia trait – N Hgb analysis except in the NB period; ▪ Absence or ↓ α-globin production due to deletions of α-globin genes mistaken as IDA ↓MCV and MCH α0-thalassemia – no α-chains produced from that chromosome (--/) α+-thalassemia – ↓ amount of α-globin chain (-alpha/) Silent carrier – deletion of 1 α-globin gene; common in African Americans α-thalassemia trait – deletion of 2 α-globin genes HbH Disease – deletion of 3 α-globin genes Hydrops fetalis – complete absence of the α-globin chain (4) ▪ ↓ α-globin chains → excess of β- and γ-globin chains → Bart Hgb in PPS Oral Exam Reviewer 2020 HbH Disease – mild splenomegaly, scleral icterus, cholelithiasis Hydrops fetalis ● In utero anemia, fetal loss d/t insufficient α-chains needed to produce HbF ● Intrauterine infusions may rescue the fetus but congenital abnormalities and neurodevelopmental delay often result HbH Disease – diagnosed in the NB period: >25% HbH, (+) Bart Hgb; marked microcytosis, anemia, Hgb range 7-11g/dL, MCV 5173fL Silent Carrier – not identified hematologically; diagnosed after the birth of a child with a 2-gene deletion or HbH DIFFERENTIALS Alpha/Beta thalassemia ▪ N/↑ RBC ▪ N serum ferritin, TIBC, and transferrin saturation Anemia of Inflammation ▪ Usually normocytic but can also be microcytic ▪ ↑ Serum ferritin ▪ ↓ TIBC TREATMENT ▪ 3-6mkday elemental iron QD or BID (max 150-200 mg/day) for 2-3 months ▪ Dietary counselling – limit cow’s milk, increase dietary iron ▪ If mild anemia – repeat CBC after 4 weeks. Hgb must in↑ by at least 1-2g/dL and has often normalized ▪ If severe, reticulocytosis within 2-4d. Hgb will then start to ↑ by 0.1-0.4 g/dL/day ▪ Blood transfusion – if with heart failure or with ongoing blood loss Lead poisoning ▪ Microcytic PREVENTION ▪ Breastfeeding ▪ At 4mos, add supplemental iron ▪ If not BF, use iron-fortified formula (12mg Fe/L) for the 1st year then limit cow’s milk to <20-24oz/d thereafter – encourages the ingestion of foods richer in iron, prevents blood loss as a result of cow’s milk-induced enteropathy ▪ Routine screening (H&H) at 9-12mos, earlier if at 4 mos assessed to be high risk for iron deficiency ▪ Prevention of hemolysis is the most important therapeutic measure ▪ The usual doses of ASA and TMP-SMX do not cause clinically relevant hemolysis in the A- variety ▪ ASA used for acute rheumatic fever (60-100mkday) may produce a severe hemolytic episode ▪ Infants with severe neonatal jaundice require G6PD deficiency testing ▪ If with severe hemolysis, may require blood transfusions although recovery is the rule when the oxidant agent is discontinued ▪ Parental and Family Testing ▪ Detailed family/genetic counseling ▪ Lifelong transfusion dependence if severe HbH Disease ▪ On going monitoring of growth and organ dysfunction ▪ Dietary supplement with folate ▪ MVTs without iron – because of risk of iron overload ▪ Vit D supplementation if level is low ▪ Adequate dietary calcium intake ▪ Intermittent transfusion requirements during intercurrent infection ▪ Splenectomy in some patients → ASA or other anticoagulant therapy postop due to high risk of thrombosis ▪ Avoid oxidative medications Batch Clingy DISEASE 89 β0-thalassemia – absence of production of β-globin; homozygous for the β0 gene; 15-20% may have a clinical course similar to β-thalassemia intermedia β+-thalassemia – ↓ amount of normal β-globin (HbA); 25% may have transfusion-dependent thalassemia β-thalassemia major – transfusion-dependent β-thalassemia intermedia – non-transfusion dependent Carrier – has a single β-globin mutation ▪ ↓ β-globin chains → excess in α-globin chains → formation of αglobin tetramers (α4) w/c appear as RBC inclusions → precipitate in RBC precursors → damaged RBC membrane → shortened RBC survival → anemia + ↑ erythroid production → early erythroid precursor death in the BM ▪ Chronic anemia + ↑ erythroid drive → ↑ iron absorption from the GIT → 2o hemosiderosis-induced organ injury ▪ No/↓ β-globin chains → α-chains combine with γ-chains → HbF (α 2γ2) Signs of Ineffective Erythropoiesis ● Growth failure ● Bone deformities 2o to BM expansion ● Hepatosplenomegaly Classic presentation of a child with severe disease – 1o seen in countries w/o access to chronic transfusion therapy; may develop in patients with mod anemia d/t severe compensatory ineffective erythropoiesis ● Thalassemic Facies – maxillary hyperplasia, flat nasal bridge, frontal bossing ● Pathologic bone fractures ● Marked hepatosplenomegaly ● Cachexia Carrier – generally asymptomatic, microcytosis and mild anemia except for β0-thalassemia – progressive anemia, profound weakness, cardiac decompensation during the 2 nd 6mos of life β-thalassemia intermedia ▪ May not be chronically transfused after infancy but may be sporadically transfused throughout their lifetime ▪ Microcytic anemia w/ Hgb ~7g/dL (range 6-10g/dL) ▪ Many develop hemosiderosis 2 o to increased GI absorption of iron-requiring chelation β-thalassemia major – symptomatic only after birth when HbA predominates Thalassemia Trait ▪ Often misdiagnosed as IDA; use a short course of iron to identify if further evaluation is needed ▪ Persistently N RDW, ↓ MCV ▪ On Hb analysis, ↑ HbA2 and variably ↑ HbF ▪ Microcytosis, hypochromia, target cells, nucleated RBCs, marked anisopoikilocytosis, relative reticulocytopenia ▪ Progressive fall in Hgb to <6g/dL unless given transfusions ▪ Inappropriately low reticulocyte count (<8%) compared to the degree of anemia caused by ineffective erythropoiesis ▪ Elevated unconjugated serum bilirubin ▪ Newborn Screening ● Not definitive ● Detection of only HbF on Hgb electrophoresis in those with βthalassemia major ● Detection of Hgb FE pattern in Hgb E β0-thalassemia ▪ DNA Diagnosis + testing for common genetic modifiers of the clinical phenotype is recommended COMPLICATIONS ▪ Transfusion-induced hemosiderosis ● Major clinical complication of transfusion-dependent thalassemia ● Physiologically, the body cannot eliminate excess iron ● Fe is deposited in the liver → endocrine organs (hypothyroidism, hypogonadotropic gonadism, GH deficiency, hypoparathyroidism, DM) → heart (heart failure, arrhythmias) ● Prevented by iron chelation therapy ▪ Heart Disease – leading cause of death in inadequately chelated patients ▪ Even if the child does not receive transfusions, iron eventually accumulates with ↑ serum ferritin and transferrin saturation PREVENTIVE MONITORING ▪ Cardiac Disease ● Serial echocardiograms – evaluate cardiac function & PAP ● Cardiac T2* MRI – after ~8y of chronic transfusion therapy (when cardiac hemosiderosis may occur) ● Periodic ECG – after 10yo due to risk of arrhythmia from cardiac iron overload ▪ Endocrine Disease ● Monitoring for dysfunction starting at 5yo or after at least 3y of chronic transfusions ● Monitor height, weight, pubertal assessment, and sitting height semiannually ● Bone density starting in the 2nd decade of life d/t risk of osteopenia ● Nutritional assessments ● Vit C, D, and zinc replacement ● Routine assessment of fertility ▪ Psychosocial Support ● Culturally sensitive anticipatory counselling ● Early social service consultation to address financial and social issues ▪ At-risk couples for hydrops fetalis should be offered molecular diagnosis on fetal tissue obtained early in pregnancy TRANSFUSION THERAPY ▪ Improves the quality of life and reduces the complications of severe disease ▪ 1mL pRBC contains ~1mg of iron ▪ Transfuse every 3-4 weeks ▪ Goal: maintain a pretransfusion Hgb of 9.5-10.5g/dL ▪ Give RBCs depleted of leukocytes and matched for D, C, c, E, e, and Kell antigens ▪ If stem cell transplant candidate, transfuse CMV-safe units IRON OVERLOAD MONITORING ▪ Serial serum ferritin levels – screen for iron balance trends but may not accurately predict quantitative iron stores ▪ Quantitative Liver Iron approved by R2 or R2* MRI – best indicator of total body iron stores; should be obtained after starting chronic transfusion therapy ▪ Quantitative cardiac iron by T2* MRI – obtained at 10yo, earlier if w/ severe iron overload or if transfusion & chelation history is unknown CHELATION THERAPY ▪ To prevent hemosiderosis-induced tissue injury ▪ Start as soon as the patient becomes significantly iron-overloaded: ● Generally, occurs after 1yr of transfusion therapy ● Serum ferritin >1000ng/dL and/or ● Liver iron concentration >5000mcg/g dry weight IRON CHELATORS ▪ Deferoxamine (Desferal) – SC/IV due to poor oral bioavailability and short half-life (<30mins) ● 25 to 60mg/kg (in heavily iron-overloaded patients) ● May be given as continuous infusion over at least 8h/day, 5-7days/wk ● AE: local skin reaction, ototoxicity, retinal changes, bone dysplasia with truncal shortening ▪ Deferasirox – PO QD, half-life >16h ● Dispersible tablet – 20 to 40mkday depending on the iron burden; dissolved in water or juice ● Film-coated tablet and granule – dose is 30% lower, 14-28mkday ● AE: GI symptoms, hepatic transaminitis (>5x the UL of N), potential kidney damage (occur commonly in the setting of dehydration) ▪ Deferiprone – 75-99mkday PO TID, half-life 3h ● Effectively enters cardiac tissue thus, more effective in reducing cardiac hemosiderosis ● AE: transient agranulocytosis (1%) ▪ Deferoxamine + Deferiprone for those with increased cardiac iron HYDROXYUREA ▪ DNA antimetabolite; increases HbF production; 10-20mkday ▪ Used in β-thalassemia intermedia patients ▪ Mean increase in Hgb of 1g/dL (range 0.1-2.5g/dL) ▪ Reduces risk of leg ulcers, pulmonary HTN, and extramedullary hematopoiesis ▪ AE: cytopenias HEMATOPOIETIC STEM CELL TRANSPLANTATION PPS Oral Exam Reviewer 2020 Batch Clingy fetal life (γ4) → HbH (β4) after birth → ▪ Most frequent in SE Asia (5-10% carry alleles) β – THALASSEMIA ▪ Usually requires a β-thalassemia mutation in both β-globin genes ▪ Ineffective erythropoiesis + massive marrow expansion 90 ▪ All children who have an HLA-matched sibling should be offered BM transplant ▪ At least 90% survival, 80% event-free survival ▪ Clinically, lead is purely a toxicant ▪ May occur in utero thru redistribution from endogenous stores i.e. the mother’s skeleton or newly acquired from ongoing environmental exposure (lead readily crosses the placenta from maternal blood) ▪ Batteries, paint, cable sheathing, cosmetics, mineral supplements, plastics, toys, traditional medicines LEAD POISONING ▪ Non-nutritive hand-to-mouth activity of young children – most common pathway for lead to enter the body, some through the skin ▪ Lead is more absorbed in an empty stomach; calcium and iron decrease lead absorption by direct competition for binding sites ▪ Lead circulates in the body bound to erythrocytes (97%) ▪ Most retained lead accumulates in bone (resides for years) but it has multiple effects in all cells Mechanisms of Lead Toxicity ▪ Binds to enzymes with sulfhydryl groups → changed contour → decreased function; accumulation of excess amounts of heme precursors are toxic ▪ Competes with calcium → abnormal intra- and intercellular signaling (e.g. neurotransmitter release) ▪ Affects the normal neuronal pruning process → elimination of multiple intercellular brain connections → prevention of the development of the normal tertiary brain structure GI Symptoms (BLL >20mcg/dL) ▪ Anorexia, abdominal pain, vomiting, and constipation, occurring and recurring over a period of weeks CNS Symptoms (BLL >100mcg/dL; may be seen as low as 70mcg/dL) ▪ Related to worsening cerebral edema and increased ICP ▪ Headache, change in mentation, lethargy, papilledema, seizures, coma → death ▪ Blood Lead Level (BLL) – gold standard ● ≥5mcg/dL – (+) exposure; requires second round of testing ● ≥45mcg/dL – requires prompt chelation therapy ▪ Erythrocyte Protoporphyrin (EP) Level ● >35mcg/dL – (+) lead poisoning, iron deficiency, or recent inflammatory disease ● Useful for monitoring biochemical lead toxicity ● If elevated, can be an indicator of lead effect and a useful means of assessing the success of treatment ● Will begin to fall a few weeks after successful interventions ▪ X-ray fluorescence (XRF) – assess bone lead stores; not available for clinical use in children ▪ Urine lead level – spontaneous lead excretion is low even with high BLLs; excretion may be stimulated by treatment with chelating agents ▪ KUB Radiograph – radiopaque flecks in the intestinal tract consistent with recent ingestion of lead-containing plaster or paint chips; absence of flecks does not rule out lead poisoning; used in children with acute symptoms and whose BLL result is not immediately available Other organs ▪ Renal tubular dysfunction (BLL >100) ▪ Reversible fanconi syndrome ▪ ↓ RBC survival → hemolytic anemia ▪ Iron deficiency and hemoglobinopathies → anemia ▪ Peripheral neuropathy → wrist drop, foot drop, HTN Physiologic Anemia of Prematurity ▪ Decline is more extreme and rapid ▪ At 3-6 wks old, Hgb 7-9 g/dL; may be lower in very small premature infants ▪ Same mechanism as term infants but exaggerated; intensified by blood loss from repeated phlebotomies ▪ Shortened RBC lifespan (40-60 days), accelerated RBC mass expansion ▪ In utero, liver produces EPO. If born preterm, still relies on the liver (no switch to kidney yet) → diminished responsiveness to anemia Drug treatment ▪ Given if BLL ≥45mcg/dL ▪ 2,3-DMSA and Penicillamine – given PO ▪ CaNa2EDTA and BAL dimercaprol – given IV ▪ BLL 44-70 mcg/dL – monotherapy (preferably DMSA) ▪ BLL >70mcg/dL – two-drug treatment ● CaNa2EDTA + DMSA or BAL – if (-) encephalopathy ● CaNa2EDTA + BAL – if (+) encephalopathy ▪ No drug removes ALL lead from the body ▪ Within days to weeks after completion of treatment, BLL rises, due to presence of lead in bone. If BLL rebounds to ≥45mcg/dL, repeat chelation. Drug-related toxicities ▪ GI distress, transient elevation in transaminases, active urinary sediment, neutropenia PREVENTION ▪ Handwashing immediately before nutritive hand-to-mouth activity Term ▪ No treatment ▪ Ensure proper diet Preterm ▪ Transfusions may be needed (pRBC 10-15mL/kg) ▪ If feeding well and growing normally with no iatrogenic blood loss, no treatment ▪ Iron therapy (1-2mkday of elemental iron) starting at 1 month of age until 1yo Batch Clingy PHYSIOLOGIC ANEMIA OF INFANCY ▪ Physiologic adaptation to extrauterine life ▪ At birth, term infants have higher Hgb levels and larger RBCs than older children and adults ▪ 1st week of life, decline in levels and persists for 6-8 weeks ▪ Onset of respiration, more oxygen for Hgb binding → switch from HbF (high oxygen affinity) to adult Hgb (low oxygen affinity) synthesis → increase in blood oxygen content and delivery → downregulation of EPO production → suppression of erythropoiesis → removed aged RBCs are not replaced → decreased Hgb levels ▪ Hgb level declines until tissue oxygen needs > O2 delivery – reached between 8-12 wks old (Hgb 11g/dL) → increased EPO production → erythropoiesis resumes ▪ May be aggravated by folic acid deficiency ▪ Shorter height – for every 10mcg increase in BLL, the child is 1cm shorter ▪ Decreased cognitive test scores ▪ Delayed puberty – chronic lead exposure ▪ Hyperactivity in young school aged children ▪ Aggression and behaviors predictive of later juvenile delinquency in older children with higher bone lead content SPLENECTOMY ▪ For those who develop hypersplenism – falling steady-state Hgb level and/or a rising transfusion requirement Main goals: Prevent cognitive/behavioral effects from occurring and prevent further ingestion ▪ Identify and eliminate environmental sources of lead exposure ▪ Reduce nonnutritive hand-to-mouth activity ▪ Dietary counseling – ensure sufficient intake of calcium (1g/day) and iron (6mg/day for infants, 12mg/day for adolescents) PPS Oral Exam Reviewer 2020 91 Complex DISEASE ANEMIA OF CHRONIC DISEASE (Anemia of Inflammation) ETIOLOGY/INCIDENCE/ PATHOGENESIS ▪ Found in conditions where there is ongoing immune activation – infections, malignancies, chronic renal disease, autoimmunity, GVHD CLINICAL MANIFESTATIONS ▪ Mild to moderate anemia ▪ The important signs and symptoms are those of the underlying disease ▪ ↓ RBC life span ▪ Inflammation-associated excess synthesis of hepcidin (key regulatory protein that controls intestinal iron absorption and tissue distribution) → alterations in Fe metabolism ▪ Low levels of serum Fe → accumulation of Fe in RE macrophages → functional Fe deficiency → impaired heme synthesis and Fe-restricted erythropoiesis → anemia DIAGNOSIS ▪ Hgb is generally at 6-9 g/dL ▪ Often normocytic, normochromic anemia but some may have modest hypochromia and microcytosis if there is concomitant Fe deficiency ▪ N/↓ absolute reticulocyte count ▪ Leukocytosis ▪ ↓ serum iron, low to normal serum transferrin ▪ Serum ferritin may be elevated secondary to inflammation ▪ BM has normal cellularity; adequate/↓ RBC precursors ▪ ↑ Marrow hemosiderin; granulocytic hyperplasia may be present ▪ Soluble transferring receptor (sTfR) – diagnostic test to distinguish ACD from IDA; N in ACD, ↑ in IDA TREATMENT ▪ Treat the underlying disorder ▪ If the associated systemic disease can be controlled, the anemia will improve or resolve ▪ Transfusions raise the Hgb concentration temporarily but are rarely indicated ▪ Recombinant human EPO increase the Hgb level and improve activity and the sense of well-being. Give iron to produce optimal effect. ▪ ACD does not respond to iron alone unless there is concomitant deficiency Chronic LEUKEMIA ETIOLOGY / INCIDENCE / PATHOGENESIS CLINCAL MANIFESTATIONS LABORATORY FINDINGS / DIAGNOSIS ▪ Most common malignant neoplasms in childhood (31% of all malignancies in <15yo) ▪ Group of malignant diseases where genetic abnormalities in a hematopoietic cell give rise to an unregulated clonal proliferation of cells ▪ Cells have ↑ proliferation and ↓ spontaneous apoptosis → disrupted normal marrow function → marrow failure ▪ See table 522.1 for the factors predisposing to childhood leukemia ACUTE LYMPHOBLASTIC LEUKEMIA ▪ Initial presentation is nonspecific and brief ▪ Polymerase Chain Reaction (PCR) and Fluorescence in situ ▪ 77% of childhood leukemia ▪ Anorexia, fatigue, malaise, irritability, intermittent lowhybridization (FISH) – pinpoint molecular genetic abnormalities, ▪ Peak incidence at 2-3yo and more common in boys grade fever, bone/joint pain (lower extremities) can be used to detect small numbers of malignant cells at ▪ More common in children with chromosomal ▪ Severe pain that can wake the patient at night diagnosis as well as during follow-up (MRD, minimal residual abnormalities – Down syndrome, Blood syndrome, ataxia▪ Sometimes, symptoms may be of several months’ disease) telangiectasia, Fanconi anemia duration, may be localized predominantly to the ▪ Morphology alone is often adequate to establish a diagnosis ▪ Unknown etiology, but most are thought to be d/t somatic bones/joints, and may include joint swelling ▪ Diagnosis is strongly suggested by PBS findings indicating BM mutations in lymphoid cells ▪ Exquisite tenderness over the bone, joint swelling, and failure effusion ▪ Anemia, thrombocytopenia, most present with total leukocyte B-lymphoblastic Leukemia (85%) ▪ Eventually, SSx of BM failure – pallor, fatigue, exercise counts <10,000 ▪ Onset between 1-10yo intolerance, bruising, oral mucosal bleeding/ epistaxis, ▪ ± Leukemic cells – often reported as “atypical lymphocytes” ▪ Median leukocyte count at presentation: 33,000 listlessness, purpura and petechiae, mucous membrane ▪ Elevated LDH T-lymphoblastic Leukemia (15%) hemorrhage ▪ When PBS suggests leukemia, examine the BM – BMA biopsy, ▪ Has a higher leukocyte count ▪ With marrow involvement, (+) deep bone pain, (-) flow cytometry, cytogenetics, molecular studies Burkitt Leukemia tenderness ▪ >25% Lymphoblasts in the BM – diagnostic of ALL ▪ Rare leukemia of mature B cells ▪ Signs of organ infiltration – lymphadenopathy, ▪ CSF Examination ▪ One of the most rapidly growing cancers in humans hepatosplenomegaly, testicular enlargement, CNS ● Included in the initial evaluation (if traumatic initial LP, risk involvement (HA, seizures, cranial neuropathies) of CNS relapse) ▪ Signs of ↑ ICP (papilledema, retinal hemorrhage, CN ● If (+) lymphoblasts and ↑ leukocyte count, (+) overt CNS or palsies) – CNS leukemia meningeal leukemia o ▪ Respiratory distress – 2 to severe anemia or mediastinal node compression of the airways (e.g. in thymus/nodes; DIFFERENTIAL DIAGNOSIS frequently seen in adolescent boys with T-cell ALL) ▪ Pancytopenia – aplastic anemia, myelofibrosis, familial HLH ▪ Failure of a single cell line – ITP, congenital/ acquired neutropenia, transient erythroblastopenia of childhood ▪ Acute onset of fever & lymphadenopathy – infectious mononucleosis ▪ Fever, bone pain, (-) tenderness, joint swelling – JIA PPS Oral Exam Reviewer 2020 TREATMENT ▪ Treatment is the single most important prognostic factor in ALL ▪ Risk-directed therapy ● Accounts for age at diagnosis, initial WBC count, immunophenotypic and cytogenetic characteristics of blast populations, rapidity of early treatment response (how quickly the leukemic cells can be cleared from the marrow/peripheral blood), and MRD assessment at the end of induction therapy ● Standard Risk: age 1-10y, WBC <50,000 ● High Risk: age <1 or >10, WBC >50,000, T-cell immunophenotype, slow response to initial therapy ▪ Imatinib – inhibits the BCR-ABL kinase resulting from the translocation (t (9;22), Philadelphia chromosome) Remission Induction Phase ▪ Initial therapy; to eradicate the leukemic cells from the BM ▪ Given for 4 weeks – Vincristine weekly + CS (dexamethasone/ prednisone) + 1 dose asparaginase ▪ If high risk, add daunomycin weekly ▪ Give intrathecal (IT) chemotherapy at the start of treatment and at least once more during induction ▪ Remission – <5% blasts in the marrow + return of neutrophil & platelet counts to near-normal after 4-5 weeks of treatment Consolidation Phase ▪ Intensive CNS therapy + continued intensive systemic therapy ▪ To prevent later CNS relapses (↓ to <5%) ▪ Give repeated IT chemotherapy by LP Intensification Phase ▪ To eradicate residual disease Batch Clingy DISEASE 92 SUPPORTIVE CARE ▪ Allopurinol/urate oxidase ● Prevent or treat kidney failure associated with ↑ serum uric acid ▪ Erythrocyte and Platelet Transfusion ● For severe myelosuppression produced by chemotherapy – requires a high index of suspicion ▪ Aggressive empirical antimicrobial therapy ● For sepsis in children with febrile neutropenia ▪ Antibiotic prophylaxis ● For Pneumocystiis jiroveci pneumonia during chemotherapy and for several months after treatment completion ▪ Watch out for tumor lysis syndrome as therapy is initiated in patients with high WBC count PROGNOSIS Overall 5yr survival rate: 90% ▪ 14-28 weeks of multiagent chemotherapy – cytarabine, MTX, asparaginase, vincristine ▪ Delayed intensification – aggressive treatment ▪ Interim maintenance – nontoxic phase Maintenance Phase ▪ Lasts for 2-3 years ▪ Daily mercaptopurine, weekly oral MTX, with intermittent doses of vincristine and a corticosteroid ▪ After chemotherapy for 2-3 years, most achieve remission at the end of induction phase ▪ Minimal Residual Disease ● Provide an estimate of the burden of leukemic cells present in the marrow ● If high at the end of induction phase, poorer prognosis and higher risk of relapse ● MRD >0.01% on D29 of induction – significant risk factor for shorter eventfree survival for all risk categories, compared with patients with (-) MRD ▪ Adolescents and young adults have an inferior prognosis compared to children <15yo Acute Promyelocytic Leukemia (APL) ▪ Subtype that is more common in certain parts of the world ▪ Gene rearrangement involving the retinoic acid receptor ▪ Responsive to all-trans-retinoic acid (ATRA, tretinoin) + anthracyclines + cytarabine PPS Oral Exam Reviewer 2020 ▪ BM failure → replacement of BM by malignant cells → SSx of AML ▪ May present with any or all the findings associated with marrow failure in ALL ▪ Blueberry muffin lesions – subcutaneous nodules; seen especially in infants ▪ Infiltration of the gingiva ▪ Signs & lab findings of DIC (especially in APL) ▪ Chloromas/granulocytic sarcomas – discrete masses, can occur in the absence of apparent BM involvement; associated with t(8;21) translocation ▪ BMA biopsy – hypercellular marrow consisting of a monotonous pattern of cells, >20% blast cells with features similar to those that characterize early differentiation states of the myeloidmonocyte-megakaryocyte series of blood cells ▪ Flow cytometry + stains – assist in identifying myeloperoxidasecontaining cells, confirming both the myelogenous origin of the leukemia and the diagnosis SUPPORTIVE CARE ▪ Prolonged hospitalization, filgrastim (GCSF), prophylactic antimicrobials ● Due to prolonged BM suppression with a very high incidence of serious infections especially viridans streptococcal sepsis and fungal infection PROGNOSIS ▪ Remission in 85-90% of patients ▪ Up to 5% die of either infection or bleeding before a remission can be achieved ▪ 60-70% survival rate Batch Clingy ACUTE MYELOGENOUS LEUKEMIA ▪ 11% of childhood leukemia ▪ Frequency increases in adolescence – 36% of leukemia in 15-19-year-olds RELAPSE ▪ Outcomes remain poor among those who relapse ▪ Most important prognostic indicators – time of diagnosis and site of relapsed disease Bone Marrow Relapse (15-20% of patients) ● Carries the most serious implications especially if it occurs during or shortly after completion of therapy ● Treatment: Intensive chemo with agents not previously used followed by allogeneic stem cell transplantation CNS Relapse (<5%) ● May be discovered at a routine LP in an asymptomatic patient ● SSx of ↑ ICP and isolated CN palsies ● Diagnosis: (+) Leukemic cells in the CSF ● Treatment: IT chemotherapy, cranial/craniospinal irradiation, must include systemic chemotherapy (because of high risk for subsequent BM relapse) Testicular Relapse (<2% of boys) ● Painless swelling of 1 or both testes ● Diagnosis: biopsy of the affected testis ● Treatment: Systemic chemotherapy ± local irradiation ▪ Induction Phase ● Aggressive multiagent chemotherapy – anthracycline + high dose cytarabine ▪ Post remission therapy – chosen based on the 1) cytogenetic and molecular markers of the leukemia and 2) response to induction chemotherapy (MRD assessment) ▪ Stem cell transplantation ● Recommended only after a relapse for selected patients with favorable prognostic features and improved with chemotherapy alone ● May be beneficial in 1st remission to those who have inferior outcome with chemotherapy 93 2) Transient Myeloproliferative Disorder ▪ 10% of neonates with Down syndrome ▪ Hepatosplenomegaly ▪ High leukocyte count, blast cells in the peripheral blood, anemia, thrombocytopenia CHRONIC MYELOGENOUS LEUKEMIA ▪ 2-3% of childhood leukemia ▪ 99% are characterized by a specific translocation, t(9;22)(q34;q11) – Philadelphia chromosome ▪ Fever, fatigue, weight loss, anorexia ▪ Splenomegaly may be present → LUQ pain ▪ Diagnosis is suggested by a high WBC count with myeloid cells at all stages of differentiation in the PBS and BM ▪ Diagnosis is confirmed by cytogenetic and molecular studies that show (+) Philadelphia chromosome and the BCR-ABL gene rearrangement – characteristic of CML but seen in a small percentage of ALL JUVENILE MYELOMONOCYTIC LEUKEMIA ▪ <1% of childhood leukemia ▪ Affects children <2yo ▪ Patients with NF1 and Noonan syndrome have a predilection for this type of leukemia INFANT LEUKEMIA ▪ 2% of leukemia during childhood occurs before 1 year old ▪ ALL: AML = 2:1 ▪ Leukemic clones in cord blood at birth before symptoms appear ▪ Chromosome translocation can occur in utero LYMPHOMA ▪ 3rd most common cancer in adolescents (>25% of newly diagnosed cancers in children 15-19 yo) HODGKIN LYMPHOMA (HL) ▪ 6% of childhood cancers ▪ Most common malignancy in adolescents (15%) ▪ Bimodal age distribution – 15-35yo and >50yo ▪ In developing countries, the early peak occurs before adolescence ▪ Male predominance but decreases with age ▪ EBV Infection – 4-fold higher risk of developing HL ▪ Arise in lymphoid tissue and spread to adjacent LN areas ▪ Hematogenous spread → involvement of the liver, spleen, bone, BM, brain ▪ Reed-Sternberg (RS) Cell ● Pathognomonic of HL ● Large cell with multiple/multilobulated nuclei ● Hallmark of HL but can be found in infectious PPS Oral Exam Reviewer 2020 Chronic Phase ▪ ↑ Leukocyte count with a predominance of mature forms but with ↑ numbers of immature granulocytes ▪ Mild anemia, thrombocytosis ▪ Terminates 3-4y after onset Accelerated Phase (Blast crisis) ▪ Blood counts rise dramatically ▪ Clinical picture is indistinguishable from acute leukemia ▪ Hyperleukocytosis → ↑ blood viscosity → ↓ CNS infection → neurologic symptoms ▪ Rash, lymphadenopathy, splenomegaly, hemorrhagic manifestations ▪ ↑ Leukocyte count ▪ ↑ Monocytes, thrombocytopenia, anemia erythroblasts ▪ BM: myelodysplastic pattern with <20% blasts with (+) ▪ Sensitive to MTX and other antimetabolites – results in substantial toxicity when standard doses are administered ▪ ALL: outcome of treatment is slightly inferior to that of other children d/t lack of good prognostic characteristics – ETV6-RUNX1, trisomies, IKZF1 ▪ AML: better outcome than non-Down syndrome children; >80% long-term survival rate ▪ Can require temporary transfusion support ▪ Do not require chemotherapy unless with life-threatening complications ▪ Abnormal PE and blood count resolve within the 1 st 3mos of life ▪ Close follow-up – 20-30% will develop typical leukemia (often acute megakaryocytic leukemia) by 3yo (mean onset: 16mos) ▪ Imatinib ● Standard treatment for pediatric CML ● Inhibit the BCR-ABL tyrosine kinase ● Produce major cytogenetic responses in >70% of patients ▪ Hydroxyurea – controls the disabling or threatening signs and symptoms during the chronic phase; gradually returns the leukocyte count to normal ▪ Stem cell transplantation offers the best opportunity for cure but outcomes are still poor ▪ Extensive tissue infiltration – organomegaly, including CNS disease ▪ Diffuse pulmonary infiltration by leukemic cells → leukemia cutis (subcutaneous nodules) and tachypnea ▪ >80% have rearrangements of the KMT2A (MLL) gene – found at the site of 11q23 band translocation, majority are t(4;11) ▪ Hyperleukocytosis ▪ Large, irregular lymphoblasts, with a phenotype (-) CD10 marker ▪ Poor prognosis ▪ Painless, nontender, firm, rubbery, cervical, or supraclavicular lymphadenopathy with some mediastinal involvement ▪ Hepatosplenomegaly ▪ SSx of airway obstruction – dyspnea, hypoxia, cough ▪ Pleural or pericardial effusion ▪ Hepatocellular dysfunction ▪ Bone marrow infiltration – anemia, neutropenia, thrombocytopenia ▪ B symptoms ● Unexplained fever >38oC ● Weight loss >10% total body weight over 6mos ● Drenching night sweats ● Others (not of prognostic significance): pruritus, lethargy, anorexia, pain ▪ Chest Radiography ● Any patient with persistent, unexplained lymphadenopathy unassociated with inflammation or infection ● To identify the presence of a large mediastinal mass before undergoing lymph node biopsy ● Determines “bulk” disease ▪ Formal Excisional Biopsy ● Preferred over needle biopsy to ensure that adequate tissue is obtained ▪ Once the diagnosis is established, determine the extent of disease ● CT scan of the chest – defines the extent of the mass, hilar nodes, and pulmonary parenchymal involvement ● CT scans of the neck, abdomen, and pelvis ● BMA biopsy – to rule out advanced disease ● Bone scan – for patients with bone pain and/or ↑ alkaline ▪ Risk adapted – chemotherapy ± low-dose involved-field radiation therapy ▪ Determined largely by disease stage, ± B symptoms, and presence of bulky nodal disease Batch Clingy DOWN SYNDROME and 1) Acute Leukemia ▪ Leukemia is 15-20x more frequently in children with Down syndrome than in the general population ▪ In the 1st 3yrs of life, AML > ALL 94 mononucleosis, NHL, etc. ● Arises from the germinal center B cells but has lost most B-cell gene expression and function ▪ HLA-A – asymptomatic patients ▪ HLA-B – patients with any B symptoms ▪ HLA-E – extralymphatic disease 2o to direct extension of an involved LN region phosphatase ● Fluorodeoxyglucose (PET) scan – prognostic in HL ▪ CBC to identify abnormalities that might suggest BM involvement; ESR; Serum ferritin – if abnormal at diagnosis, used to evaluate treatment effects PROGNOSIS ▪ 85-90% event-free survival rate (EFS) – for those with favorable prognostic factors and early-stage disease ▪ >95% overall survival rate (OSR) at 5yrs ▪ 80-85% EFS for those with advanced-stage disease ▪ After relapse, those who relapse >12mo after treatment have the best prognosis and a long-term survival rate of 40-50% POOR PROGNOSTIC FEATURES ▪ Reactive infiltration of eosinophils and CD68 + macrophages ▪ ↑ IL-1, IL-6, and TNF ▪ Tumor bulk ▪ Presence of B symptoms ▪ Decreased/slow response to therapy NONHODGKIN LYMPHOMA (NHL) ▪ 60% of lymphomas in children ▪ 90-95% survival rate for localized disease ▪ 80-95% survival rate for advanced disease Lymphoblastic Lymphoma (LBL) PPS Oral Exam Reviewer 2020 ▪ Depend on the pathologic subtype and sites of involvement ▪ 70% present with advanced disease (stage III or IV), including extranodal disease with bone marrow and CNS involvement ▪ Painless, rapid, LN involvement ▪ For initial diagnosis: CBC, electrolytes, LDH, BUA, Ca, phosphorus, BUN, creatinine, bilirubin, alanine & aspartate aminotransferase, BMA, LP, CXR, Abdominal UTZ ▪ For staging: CT of the neck, chest, abdomen, pelvis ▪ For suspected CNS disease: MRI of the brain and spine ▪ PET Scan – for functional imaging and for judging treatment RELAPSE ▪ Most occur within the 1st 3years after diagnosis, but may occur as late as 10 yrs after ▪ Myeloablative Therapy and Autologous Stem Cell Transplant ± RT – for patients who achieve an initial chemosensitive response but relapse or progress before 12mos from diagnosis ▪ Multiagent systemic chemotherapy and/or immunotherapy with intrathecal chemotherapy ▪ Surgery – used mainly for diagnosis ▪ Radiation Therapy for special circumstances – CNS involvement in LBL or the presence of acute superior mediastinal syndrome or paraplegias ▪ Newly diagnosed patients (esp. BL/LBL) are at high risk for TLS Batch Clingy COMPLETE RESPONSE ▪ Complete resolution of disease on clinical examination and imaging studies, or at least 70-80% reduction of disease, and ▪ A change from initial positivity to negativity on PET scanning – reflects residual fibrosis 95 ▪ Arises from precursor T lymphocytes, less often from B lymphocytes Mature B-cell Lymphoma ▪ Burkitt Lymphoma (BL) – express the CD20 antigen ▪ Diffuse large B-cell Lymphoma (DLBCL) – express the CD20 antigen ● Germinal center B-cell-like – majority of DLBCL cases; favorable prognosis ● Activated B-cell-like – poorer prognosis ● Mediastinal B-cell subtype – shares molecular signature akin to Hodgkin Lymphoma Anaplastic Large Cell Lymphoma (ALCL) ▪ Most are of mature T-cell origin; express the CD30 antigen ▪ Cough/dyspnea with thoracic involvement ▪ Superior mediastinal syndrome – 2o to a large mediastinal mass → obstruction of blood flow or respiratory airways ▪ Ascites, ↑ abdominal girth ▪ Intestinal obstruction with an abdominal mass ▪ Nasal congestion, ear pain, hearing loss, tonsil enlargement with Waldeyer ring involvement ▪ Localized bone pain ▪ Tumor Lysis Syndrome (TLS) ● 2o to rapid cell turnover; common in BL ● ↑ BUA, phosphate, potassium, ↓ calcium ▪ Can present as a life-threatening oncologic emergency COMPLICATIONS ▪ Multiagent Chemo: Serious mucositis, infections, and cytopenias that require RBC and platelet transfusions, electrolyte imbalance, poor nutrition ▪ Long-term complications: Risk of growth retardation cardiac toxicity, gonadal toxicity with infertility, secondary malignancies PROGNOSIS ▪ Excellent for most forms ▪ Localized disease: 90-100% survival rate ▪ Advanced disease: 80-95% ● Vigorous hydration + frequent electrolyte monitoring + xanthine oxidase inhibitor (allopurinol 10mkday PO TID) or urate oxidase (rasburicase 0.2mkday IV QD x 5d) ▪ Rituximab – mAb directed at CD20’ when combined with standard chemotherapy, improves outcome Lymphoblastic Lymphoma ▪ Requires 12-24mos of therapy – chemo, IT, cranial radiation (if CNS +) ▪ Best results are obtained using therapeutic approaches mirroring those of childhood acute leukemia – induction. Consolidation, interim maintenance, reinduction (if advanced disease), year-long maintenance phase with 6-MP & MTX ▪ If relapse disease, poor outcome (10% OS) BL and DLBCL ▪ Give the same mature B-cell NHL chemoimmunotherapy regimen ▪ If with localized disease, chemo for 6 weeks; good prognosis – 4yr overall survival rate of 90% ▪ If with advanced disease, chemo for 4-6 months – overall survival rate of 79-90% Primary Mediastinal B-cell Lymphoma (PMBCL) ▪ 2% of mature B-NHLs ▪ Prolonged infusional chemotherapy, rituximab, chimeric antigen receptor cells expressing anti-CD19 mAbs Anaplastic Large Cell Lymphoma ▪ If localized disease, surgical resection alone ▪ If advanced, multiagent chemotherapy Relapsed Non-Hodgkin Lymphoma ▪ Reinduction chemotherapy + allogeneic/autologous stem cell transplantation except ALCL (prolonged low-dose vinblastine) ▪ MRD – prognostic marker, aids in risk stratification Batch Clingy LBL ▪ Symptomatic mediastinal mass ▪ Has a predilection for spreading to the BM, CNS, testes BL ▪ Diffuse leukemia presentation or massive abdominal (sporadic type) or head & neck (endemic type) tumor ▪ Can metastasize to the BM or CNS ALCL ▪ B symptoms can be seen but are not prognostic ▪ Manifests either as a 1o cutaneous manifestation (10%) or a systemic disease (90%) with dissemination to liver, spleen, lung, or mediastinum; rare BM/CNS involvement response ▪ Tumor tissue should be tested by flow cytometry for immunophenotypic origin (T, B, or null) and cytogenetics (karyotype) ▪ FISH/RT-PCR – for specific gene translocations, T- and B-cell gene rearrangement studies, molecular profiling PPS Oral Exam Reviewer 2020 96 Batch Clingy PPS Oral Exam Reviewer 2020 97 Acute DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Most result from blunt trauma – fall, athletic activities, motor vehicle crash Risk Factors ▪ Child – greater risk of blunt renal injury than adults because the kidneys are not located behind the ribs ▪ Pre-existing renal anomaly – hydronephrosis 2o to UPJO, horseshoe kidney, renal ectopia ▪ Fall → deceleration injury → injury to the renal pedicle → interrupted blood flow to the kidney ▪ Blunt lower abdominal trauma in a full bladder → bladder rupture ▪ Straddle injury → trauma to the bulbous urethra TRAUMA CLINICAL MANIFESTATIONS ▪ Gross or microscopic hematuria ▪ Bleeding from the urethral meatus ▪ Abdominal or flank pain ▪ Flank mass ▪ Fractured lower ribs or lumbar transverse processes ▪ Perineal or scrotal hematoma ▪ In >50% of cases, major injuries to the brain, spinal cord, skeleton, lungs, or abdominal organs Penile Injury ▪ Uncommon ▪ Can be d/t partial or complete glans amputation during NB circumcision → immediate surgical repair ▪ Can be during toilet training → injury to the glans if the toilet lid falls while urinating → ● Hematoma covering the distal half of the glans, no difficulty urinating → no extensive evaluation ● Inadvertent hair coil injury or strangulation injury → very narrow constriction → severe distal penile swelling and pain LABORATORY FINDINGS / DIAGNOSIS See Figure 561.1 ▪ If with significant abdominal injury, gross hematuria or >50RBCs/HPF, or suspicion of renal injury (deceleration injury, flank pain or bruise), do renal imaging ▪ Catheterize the bladder unless blood is dripping from the urethral meatus – potential urethral injury ● Passing a catheter in the presence of urethral injury → ↑ extent of the damage, convert a partial membranous urethral tear into total disruption ● Do a retrograde urethrogram ▪ 3-phase spiral CT scan ● To evaluate the kidneys, uterus, bladder ● Delayed images – to detect renal extravasation of blood or urine ▪ If with a pelvic fracture, suspect a urethral transection injury TREATMENT Kidney Injury (See Table 561.1) ▪ If minor, bed rest and VS monitoring until abdominal or flank discomfort and gross hematuria have resolved ▪ If major, ● Admit to ICU ● IV antibiotics ● Manage nonoperatively – because Gerota’s fascia often causes tamponade of bleeding from the kidney and dramatic healing of the parenchyma can occur even with significant urinary extravasation ● 10% undergo surgical exploration d/t associated abdominal injuries ▪ The kidney can be salvaged by emergency renal revascularization only if the kidney is explored within 2-3hrs of the injury ▪ Long-term Complications: loss of renal function, renin mediated HTN Bladder Rupture ▪ If intraperitoneal, bladder repair Testicular Injury ▪ Uncommon in children because of the small size of the testes and their mobility within the scrotum ▪ Often result from blunt trauma during athletic activity ▪ Significant scrotal swelling, testicular pain and tenderness ▪ Ultrasound: rupture of the tunica albuginea (capsule of the testis) + surrounding hemorrhage Membranous Urethral Injury ▪ Temporary suprapubic cystostomy with continuous bladder drainage for 3-6mos → open or endoscopic urethroplasty ▪ Late complications: erectile dysfunction, urethral stricture, urinary incontinence Penile Injury ▪ If strangulation injury, identify and incise the hair → prompt resolution of the edema ▪ If penetrating penile injury, emergency debridement & repair Zipper Injury ▪ Affects either the scrotum or foreskin ▪ Occurs generally in males who do not wear underwear Testicular Injury ▪ Prompt surgical treatment increases the salvage rate SEXUALLY TRANSMITTED DISEASE / CHILD ABUSE PPS Oral Exam Reviewer 2020 ▪ Painful genital ulceration + inguinal lymphadenopathy (usually unilateral) ▪ Incubation period: 4-7 days ▪ Small, inflammatory papule on the preputial orifice or frenulum in men and on the labia, fourchette, or perineal region in women → pustular, eroded, and ulcerative in 2-3 days ▪ Ulcer edge is ragged and undermined ▪ Painful, tender inguinal lymphadenitis (>50% of cases, mostly men) → can become fluctuant → buboes (can spontaneously rupture) ▪ Without treatment, persists for weeks to months ▪ Diagnosis in infants and children is strong evidence of sexual abuse ▪ Diagnosis is established by the clinical presentation and the exclusion of both syphilis and HSV infections ▪ Gram stain of ulcer secretions: gram-negative coccobacilli in parallel clusters (school of fish) ▪ Culture: expensive, requires special media, only 80% sensitive ▪ Evaluate for other STIs (syphilis, HBV, HIV, chlamydia, gonorrhea) – 10% have a concomitant syphilis or genital herpes DIFFERENTIAL DIAGNOSIS ▪ Lymphogranuloma venereum ▪ Genital herpes – vesicular lesions with a history of recurrence Alternative regimens: ▪ Erythromycin 500mg PO TID x 7 days ▪ Ciprofloxacin 500mg PO BID x 3 days if ≥18yo ▪ Fluctuant nodes may require drainage ▪ Symptoms often resolve within 3-7 days ▪ Relapses can be treated successfully with original treatment regimen Batch Clingy CHANCROID ▪ Haemophilus ducreyi – fastidious, gram (-) bacillus ▪ Risk factor for HIV transmission ▪ Male circumcision lowers the risk for chancroid Zipper Injury ▪ Cut the zipper with bone or metal cutters ▪ Most are resistant to penicillin and ampicillin because of plasmid-mediated β-lactamase production ▪ Easy to treat if recognized early ▪ Azithromycin 1g PO x 1 dose OR Ceftriaxone 250mg/IM x 1 dose 98 ▪ Suspect resistance if with persistence of the ulcer and the organism after therapy ▪ All sexual contacts should be evaluated and treated ▪ If initial HIV/syphilis testing is negative, re-test in 3mos because of the high rate of co-infections COMPLICATIONS ▪ Phimosis ▪ Secondary bacterial infection ▪ Bubo formation ▪ Increased risk for HIV transmission UTI VULVOVAGINI-TIS GONORRHEA, SYPHILIS, CHLAMYDIA HIV, HBV TRICHOMONIASIS *see Renal Disorders* *see Genital Disorders* *see Common Bacterial Infections* *see Common Viral Illnesses* *see Parasitic Infections* Complex DISEASE CLINICAL MANIFESTATIONS LABORATORY FINDINGS / DIAGNOSIS TREATMENT *see Renal Disorders* Batch Clingy UROLITHIASIS ETIOLOGY / INCIDENCE / PATHOGENESIS PPS Oral Exam Reviewer 2020 99 EPIDEMIOLOGY / PATHOGENESIS ▪ Most common <1yo ▪ 1st year of life – M:F 2.8:5.4 ● 20% in febrile uncircumcised males <1yo ▪ Beyond 1-2yr — M:F 1:10 ● Female 1st UTI often by 5yo; peak during infancy, toilet training & onset of sexual activity ▪ Colonic bacteria: E. coli (54–67%), Klebsiella spp, Proteus spp, Enterococcus, Pseudomonas; S saprophyticus, GBS; less common, S aureus, Candida spp, & Salmonella spp ▪ 2-24mo risk factor ● Female: white race, <12 mo, >39°C, fever >2days, absence of another source ● Male: nonblack race, >39°C, fever >24hr, absence of another source URINARY TRACT INFECTION ▪ Ascending infections; rarely, hematogenous ▪ Simple & compound papillae in kidney: antireflux mech prevent urine in renal pelvis to enter collecting tubules → some compound papillae (upper & lower) allow intrarenal reflux → infected urine stimulate immunologic & inflammatory response → renal injury and scarring (highest risk <2yo) ▪ Grade III, IV, or V VUR & febrile UTI: 90% (+) acute pyelonephritis on renal scintigraphy ▪ Bowel-bladder dysfunction ● Toilet training → retain urine to stay dry → uninhibited contractions of bladder forcing urine out → ↑pressure, turbulent urine flow, incomplete bladder emptying → bacteriuria ● School-age children who refuse to use school bathroom → urinary retention ● Constipation w/ fecal impaction ▪ Obstructive uropathy → hydronephrosis → urinary stasis ▪ Neurogenic bladder → clean intermittent catheterization → organisms w/ more antibiotic resistance ▪ Bacterial pili or fimbriae: ● Type I → mannose sensitive (D mannose block attachment to target cells) → no role in pyelonephritis ● Type II → mannose resistant, receptor is glycosphingolipid in uroepithelial membrane & RBC: Gal 1-4Gal w/c agglutinate by P blood group erythrocyte → “P fimbriae” ● E coli: 76-94% (+) P fimbriae, 19-23% cystitis strain ● Attachment to mannose receptor on umbrella cells → take E. coli into cell → replicate in nutrient-rich environment → intracellular bacterial communities (IBCs); E coli become filamentous → bladder shed infected cells → IBCs break out & repopulate the urine → filamentous E coli evade WBC → taken up in bladder wall → quiescent intracellular reservoirs (QIRs) → protected from antibiotics → recurrent infections PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS / LABORATORY FINDINGS UTI = symptoms + positive culture (urine culture necessary for confirmation & appropriate therapy) PYELONEPHRITIS: parenchymal involvement ▪ Most common serious bacterial infection in <24mo w/ fever without focus ▪ Fever: may be only sx; consider >39C w/out another source for >24hr in males and > 48hr in females ▪ Abdominal, back or flank pain, malaise, N/V, occ diarrhea ▪ Newborns: nonspecific, poor feeding, irritability, jaundice, weight loss Acute pyelitis: no parenchymal involvement Acute lobar nephronia: localized renal parenchymal mass by acute focal infection w/out liquefaction; more in older children; may be early stage renal abscess Renal abscess → hematogenous spread w/ S aureus or occur ffg pyelonephritis by uropathogens; most unilateral & R-sided → ↑risk for renal scarring Perinephric abscess → sec to contiguous infection in perirenal area (vertebral osteomyelitis, psoas abscess) or pyelonephritis that dissects to renal capsule → diffuse throughout the capsule, not walled of; if no communication w/ collecting system, normal UA/culture Xanthogranulomatous pyelonephritis: rare, granulomatous inflammation w/ giant cells & foamy histiocytes; manifest as renal mass, acute or chronic infection; risk factor: renal calculi, obstruction, Proteus ssp, E coli; may require total/partial nephrectomy CYSTITIS: bladder involvement ▪ Dysuria, urgency, frequency, suprapubic pain, incontinence, malodorous urine ▪ Does not cause high fever & renal injury Acute hemorrhagic cystitis: E coli, adenovirus types 11 and 21 (adenovirus cystitis: M>F, self-limiting, hematuria ~4days); immunocompromised (adenovirus & polyomavirus) Eosinophilic cystitis (hematuria) or interstitial cystitis (irritative voiding sx): (-) culture UA & culture ▪ Toilet-trained children: midstream urine; introitus cleaned; uncircumcised males, prepuce retracted; prepuce not retractable → may be unreliable, contaminated with skin flora ▪ Not toilet trained: catheterized/suprapubic aspirate ● Application of adhesive, sealed, sterile collection bag after disinfection of skin: useful if negative → NPV from “bag” specimen: 99%; ↑rate of contamination, mixed organisms → not used if tx planned immediately after collection ▪ Nitrites & leukocyte esterase: bacterial metabolism of nitrate to nitrite: 4hrs → not detected if organism does not convert (notably COMPLICATIONS / PROGNOSIS Goal of imaging studies: identify anatomic abnormalities, determine active renal involvement, assess whether renal function is normal or at risk “Bottom-up” ▪ Renal sonogram + VCUG → identify upper and lower UT abnormalities: VUR, bladder–bowel dysfunction, bladder abnormalities (paraureteral diverticulum) “Top-down” ▪ Reduce VCUG exam ▪ DMSA renal scan → identify areas of acute pyelonephritis: photopenic, enlarged kidney → ~50% develop renal scarring ▪ (+) DMSA → VCUG → (+) scan in 90% of children w/ dilating reflux COMPLICATIONS ▪ Chronic renal damage → arterial HTN & end-stage renal insufficiency ▪ Renal scarring: ↑between days 2-3 of fever; ↑w/ eps of pyelonephritis; grade of reflux → parental education, prompt tx ▪ Bacteremia in pyelonephritis: 3–20% of cases; most common <90 days old & obstructive uropathy → blood culture before antibiotic ▪ Atypical features: failure to respond w/in 48hr of appropriate antibiotics, poor urine flow, abdominal flank or suprapubic mass, non–E coli pathogen, urosepsis, ↑crea TREATMENT / PREVENTION ▪ Acute cystitis: tx promptly to prevent pyelonephritis; severe sx → presumptive tx pending culture; mild sx → tx delayed til culture ● TMP-SMX 6-12mg TMP/kg/day ÷ 2doses, 3-5days ● Nitrofurantoin 5-7mkday ÷ 3-4doses (advantage: active against Klebsiella and Enterobacter) ● Amoxicillin 50mkday ÷ 2doses (effective initial tx, ↑ resistance) ▪ Pyelonephritis: 7-14days, agent capable of reaching significant tissue levels; oral & IV equally efficacious → IV: dehydrated, vomiting, unable to drink, urosepsis ● Ceftriaxone 50mkday (not to exceed 2g) ● Cefepime 100mkday q12 ● Cefotaxime 100-150mkday ÷ 3-4doses ● Oral 3rd-gen ceph (cefixime): choice for oral tx ● Cephalexin: considered given ↑resistance of Gram (-) to amoxicillin ● Nitrofurantoin: not routine w/ febrile UTI; does not achieve significant renal tissue levels ● Ciprofloxacin: alternative agent for resistant, particularly P aeruginosa, >17yo, weigh risk vs benefit (short course therapy for younger w/ P aeruginosa); levofloxacin, alternative quinolone w/ good safety profile ● IM loading dose ceftri ffd by oral 3rd gen ceph ▪ Culture: negative w/in 24hr of tx → repeat culture after tx not routine ▪ Acute lobar nephronia: same antibiotics as pyelonephritis; 14-21 days ▪ Renal/perirenal abscess or with infection in obstructed urinary tract: IV antibiotics (10-14days IV ffd by 2-4wk oral) + percutaneous drainage typically attempted prior to surgery ● Immediate percutaneous drainage: abscess >3-5 cm; <3cm may initially be treated w/ antibiotics alone ● 48-hr trial of IV antibiotics before sx in otherwise stable children ● Kidney loss: 10–20% of cases of renal abscess ● ID of causative organism: advantage of percutaneous drainage of perinephric abscess (infection isolated from collecting system based on the location) ▪ PNSP: antibiotic switch → poor response after 28-72hr, culture has diff sensitivity ● Probiotic & cranberry for UTI prevention inconclusive ● Breastfeeding & circumcision may ↓ risk ▪ Identify & manage predisposing factor ● Behavioral modification for constipation ● Bowel-bladder dysfunction: hx of cystitis, wetting, Vincent’s curtsy (female squat on heels in response to uninhibited bladder contraction), recurrent upper tract infection; VCUG: tightening the pelvic floor during urination seen as a spinning-top urethra, UTZ: residual urine, thick bladder wall, urodynamics: intermittent stream w/ ↑activity in pelvic floor muscles ▪ Prophylaxis ● ↓ recurrence; rate of renal scarring same; ↑rate of UTI by resistant organism; need for daily meds ● AAP: does not recommend routine use w/ 1st ep of pyelonephritis in anatomically normal urinary tract ● PNSP: prophylaxis reduce risk of recurrences, considered in Batch Clingy Acute DISEASE 100 Enterococcus) or (+) urinary frequency ▪ Febrile infants <60 days old: pyuria, nitrite or leukocyte esterase, ↑Sn&Sp ▪ Microscopic hematuria: alone does not suggest UTI ▪ WBC casts (rare) suggest renal involvement ▪ Pyuria: >3-6WBC/hpf, indicative in symptomatic; asymptomatic bacteriuria can have pyuria ● sterile pyuria: (+) leukocytes (-) culture → partially treated bacterial UTIs, viral, urolithiasis, renal TB, renal abscess, UTI in urinary obstruction, urethritis due to STI, inflammation near ureter/bladder (appendicitis, Crohn dx), Kawasaki dx, schistosomiasis, neoplasm, renal transplant rejection, interstitial nephritis (eosinophils) ▪ Urine at room temp >60min → overgrowth of minor contaminant → refrigeration (storing urine til culture) ▪ Culture >50,000 CFU/mL of single pathogen (suprapubic/catheter) and UA (+) pyuria or bacteriuria in symptomatic child = UTI ▪ Bag sample UA (+) in symptomatic child → obtain catheter sample for culture susceptible patients wherein benefit outweigh risk ● Urologic conditions that might benefit: neuropathic bladder, urinary tract stasis and obstruction, severe VUR, urinary calculi AAP UTI and VUR in Infants and Children 2010 ▪ Prophylactic dose: ¼ to ½ of therapeutic dose for acute infection ● TMP-SMX: TMP 2mg/kg as a single dose or 5mg/kg of TMP twice per week ● Nitrofurantoin: 1-2mg/kg as a single daily dose ● Cephalexin: 10mg/kg as a single daily dose ● Ampicillin: 20mg/kg as a single daily dose ● Amoxicillin: 10mg/kg as a single daily dose NOTE: ▪ ↑ Resistance of E coli: ampi & amox are less effective; not used for beyond the first 2 postnatal months ▪ Neonatal period: avoid TMP-SMZ ▪ Toilet-trained children: at bedtime (not evidence-based) CBC: leukocytosis & neutrophilia ↑ESR, procalcitonin, CRP KUB UTZ: 1st line imaging (enlarged kidney w/ possible mass if w/ acute lobar nephronia/renal abscess); 2-24mo 1st ep UTI VCUG → hydronephrosis, scarring, suggestive of reflux, obstructive uropathy, atypical complex features, recurrent febrile UTI CT scan: more Sn&Sp for lobar nephronia (wedge-shaped, lowerdensity area after contrast) Acute Uncomplicated UTI Preferred regimen Infants < 2 months Cefotaxime Age Weight <7days >7days <1200g 50mg/kg/dose q 12hr >7days >1200g 50mg/kg/dose q 8 hr PLUS Amikacin Age E. coli, Klebsiella, Enterobacter, Dose 50mg/kg/dose q 12hr >1month 100-200 mg/kg/d divided q 6hr Weight Dose 0-4 week <1200g 7.5mg/kg OD <7days 1200-2000g 7.5mg/kg OD <7days >2000g 7.5-10mg/kg OD >7days >1200-2000g 7.5 mg/kg OD >7days >2000g 10g/kg OD Duration of Treatment: 10-14 days >2 months to 18 years Oral options PPS Oral Exam Reviewer 2020 Comments If there are signs of sepsis, treat as neonatal sepsis. Adjust therapy based on culture. Early onset is usually due to maternal transmission. Use ceftriaxone if cefotaxime is not available and the neonate is not jaundiced. UTI, recurrent, catheter related or with other co-morbidities. These patients require a referral to a pediatric infectious disease specialist, pediatric nephrologist and pediatric urologist. Etiology Enterobacteria ceae, Pseudomonas aeruginosa, Enterococcus Preferred regimen Ceftriaxone Age <7days Dose 50mg/kg/dose q 24hr >7days <2000g 50mg/kg/dose q 24hr >7days >200g 50-75 mg/kg/dose q 24hr If Pseudomonas is suspected, use Ceftazidime instead if Cefotaxime. Infants & children: 50-100 mg/kg/dose q24 Adjust antibiotics depending on results of culture. AND/OR Amikacin Age Oral therapy is equally effective to IV therapy. IV therapy is preferred for Weight Comments Use Cefotaxime instead of Ceftriaxone in jaundiced patients. Weight Dose 0-4 week <1200g 7.5mg/kg OD <7days 1200-2000g 7.5mg/kg OD <7days >2000g 7.5-10mg/kg OD >7days >1200-2000g 7.5 mg/kg OD >7days >2000g 10g/kg OD Infants and children: 15-22.5 mg/kg/d as single daily dose Cephalosporins are not active against Enterococcus. Batch Clingy Etiology E. coli, Klebsiella, Enterobacter, Enterococcus, Group B Strep 101 Amoxicillin-clavulanate: <40 kg: 20-40 mg (amoxicillin)/kg/d q8h or 25-45 mg/kg/d q12h using the 20 mg/5mL or 400 mg/5mL >40 kg: 500-875 mg q8h; maximum dose: 2g/d seriously ill children and for those who cannot take oral therapy. or q8h; max dose: 24 g/d Treat for 7-14 days depending on response. Switch to oral therapy once patient has been afebrile for 24h and able to take oral medications. OR Cephalosporins are not useful if Enterococcus is suspected. Nitrofurantoin should NOT be used for pyelonephritis and renal sepsis due to poor serum concentrations. Cefuroxime >3 mos - 12 yrs: 20 - 30 mg/kg/d q12h PO Adolescents: Cefuroxime 250-500 mg q12h PO OR Nitrofurantoin (only for cystitis) 5-7 mg/kg/d q6h, maximum dose: 400 mg/d IV options Ampicillin-Sulbactam100-200 mg/kg/d of ampicillin q6h IM or IV infusion over 10-15 min OR Clinical response is expected in 24-48 hours. Antibiotic coverage should be reassessed if still unwell in 24-48h Cefuroxime 75-150 mg/kg/d q8h; max dose: 6 g/d. For those >40 kg, use adult dose. Duration of therapy (IV/PO): 7-14 days Test Leukocyte esterase Nitrite Leukocyte esterase or nitrite positive Microscopy (WBC) Microscopy (bacteria) Leukocyte esterase test, nitrite, or microscopy positive WBC >5/HPF in centrifuged WBC >10/uL in uncentrifuged Bacteriuria (+) Bacteriuria (-) Nitrite (+) Nitrite (-) Collection Method Suprapubic aspiration Catheterization Clean catch, midstream urine ACUTE GLOMERULONEPHRITIS ▪ Tea/cola colored urine (painless hematuria but assoc w/ flank pain when acute/severe) ACUTE POSTSTREPTOCOCCAL GLOMERULONEPHRITIS ▪ Nephritogenic strain of GABHS ▪ 1-2wk after pharyngitis; 3-6wk after pyoderma ▪ Glomeruli: enlarged & relatively bloodless, diffuse mesangial cell proliferation, polymorphonuclear leukocyte infiltration (early), crescents & interstitial inflammation (severe); IF: “lumpy-bumpy” deposits of Ig & complement; EM: electrondense deposits/“humps” on epithelial side of glomerular BM ▪ Mechanisms of immunologic injury → molecular mimicry; in situ immune complex formation; complement activation ▪ Most common: 5-12yr; uncommon <3yr PPS Oral Exam Reviewer 2020 ▪ UA: RBC casts, proteinuria, polymorphonuclear leukocytes; CBC: normochromic anemia (hemodilution, ↓grade hemolysis); ↓C3 in > 90% in acute phase ▪ Confirm: clear evidence of prior streptococcal infection → (+) throat culture (support dx or carrier state), ASO titer (↑pharyngeal infection, rare in skin infections), antideoxyribonuclease B level (best single Ab titer document cutaneous infection), streptozyme screen (measure multiple Ab to different streptococcal antigens) ▪ MRI: severe neurologic sx, PRES in parietooccipital areas (T2weighted images) ▪ CXR → sx of heart failure or pulmonary edema Sensitivity (Range) % 83 (67–94) 53 (15–82) 93 (90–100) 73 (32–100) 81 (16–99) Specificity (Range) % 78 (64–92) 98 (90-100) 72 (58–91) 81 (45–98) 83 (11–100) 99.8 (99–100) 70 (60–92) Pyuria (+) Pyuria (-) Send for urine CS Treat as UTI Start antibiotics Send for urine CS Start antibiotics if with symptoms Leukocyte (+) Send for urine CS Treat as UTI Start antibiotics Send for urine CS Start antibiotics if with symptoms Colony Count (cfu/ml) Any growth >105 104 - 105 >104 (boy) >105 (girl) (3 specimens) >105 (girl) (2 specimens) >105 (girl) (1 specimen) ▪ Nephrotic syndrome: <5% ▪ HTN 60% → HTN encephalopathy 10% → neurologic sequelae often reversible → severe prolonged HTN → intracranial bleeding ▪ HyperK, hyperphosphatemia, hypoCa, acidosis, seizures, uremia ▪ Acute renal failure → dialysis ▪ Hypervolemia → pulmonary edema & heart failure ▪ Complete recovery: >95% ● Recurrence extremely rare ▪ Severe acute phase → glomerulosclerosis and chronic renal Send for urine CS Treat as UTI Start antibiotics Not UTI Leukocyte (-) Send for urine CS Treat as UTI Start antibiotics Not UTI Probability of infection (%) >99% 95% Infection likely Infection likely 95% 90% 85% ▪ Acute phase: generally, resolves w/in 6-8wk ● Urinary protein excretion; HTN: normalize 4-6wk after onset ● C3: 6-8wk ● Persistent microscopic hematuria: persist for 1-2yr ▪ Management → renal dysfunction and HTN ▪ 10-day antibiotic therapy w/ penicillin: limit spread of nephritogenic organisms, not affect natural history ▪ Sodium and fluid restriction, diuretics, and pharmacotherapy w/ Cachannel antagonists, vasodilators, or ACE inhibitors → hypertension ▪ Prevention: early systemic antibiotic therapy for streptococcal infections does not eliminate risk of GN (family of patients w/ acute GN, Batch Clingy Citrobacter 102 ▪ Other causes of postinfectious GN: coagulase-positive/negative staphylococci, S pneumoniae, Gram(-) bacteria, influenza, parvovirus Diseases commonly manifesting as AGN ▪ Postinfectious GN ▪ IgA Nephropathy ▪ MPGN ▪ HSP Nephritis ▪ SLE Nephritis ▪ Granulomatosis with polyangiitis (formerly Wegener granulomatosis) ▪ Microscopic PAN (please see NOTE below) ▪ Goodpasture syndrome ▪ HUS (page 2718) IgA NEPHROPATHY (Berger nephropathy) ▪ Most common chronic glomerular disease in children ▪ Immune complex disease → excessive amounts of poorly galactosylated IgA1 in the serum → production of IgG and IgA autoantibodies ▪ Predominance of IgA (often accompanied by C3) w/in mesangial glomerular deposits in absence of systemic disease → mesangial proliferation w/ epithelial cell crescent formation & sclerosis ▪ Findings similar to HSP → hypothesis → disease of same spectrum ▪ Familial clustering: linkage to 6q22-23; high predisposition in Southeast Asia, ↓prevalence Africa ▪ M>F MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS (MPGN, Mesangiocapillary glomerulonephritis) ▪ Primary (idiopathic) or secondary (subacute & chronic infection: hep B & C, syphilis, subacute bacterial endocarditis, infected shunts, especially ventriculoatrial shunts → shunt nephritis), lupus nephritis ▪ Recent URI, skin, GI infection → postinfectious GN, HUS, HSP nephritis ▪ Rash & joint complaints → HSP or SLE Nephritis Please see Fig 536.1 and Fig 537.6 NOTE: On page2307, Microscopic Polyangiitis (MPA) was previously the microscopic variant of polyarteritis Primary MPGN ▪ Type I: common; accentuated lobular pattern from diffuse mesangial expansion, ↑mesangial cells & matrix; thickened glomerular capillary walls; crescents → poor prognosis; IF: C3, lesser amt of Ig in mesangium & peripheral capillary walls (lobular pattern); EM: deposits in mesangial and subendothelial regions ▪ Type II (dense deposit disease): similar LM findings; IF: C3 prominent w/out concomitant Ig; EM: lamina densa undergoes dense transformation w/out immune complex-type deposits (C3GN related but separate category) Primary & secondary type I indistinguishable; immune complexes trapped in glomerular subendothelial space → injury → proliferative response & mesangial expansion (complement activation through classic pathway in 50%) ▪ Type II → not mediated by immune complexes; pathogenesis not known → deranged complement regulation ● ↓C3, other complement normal; C3 nephritic factor (anti– C3 convertase antibody) → activates alternative complement pathway HSP NEPHRITIS ▪ ~50% develop renal manifestations (>8 yr 3-fold greater risk) ▪ Severity of systemic manifestations not correlated w/ severity of nephritis ▪ Similar pathogenesis, nearly identical renal histology w/ IgA nephropathy PPS Oral Exam Reviewer 2020 ▪ Biopsy: ARF, nephrotic syndrome, no evidence of strep infection, normal complement level; hematuria & proteinuria, ↓renal fx, ↓C3 level >2mo after onset ● Persistent hypocomplementemia: chronic form of postinfectious GN, MPGN ● Purpuric rash: can’t diff from HSP w/out biopsy ▪ Gross hematuria: w/in 1-2days of onset of URI or GI infection, may be assoc w/ loin pain ▪ Proteinuria: <1,000mg/24hr in patient w/ asymptomatic microscopic hematuria ▪ Mild to moderate HTN if w/ nephritic or nephrotic syndrome (rarely severe to cause HTN emergency) ▪ Normal C3; IgA levels no diagnostic value (↑ in only 15% of pedia) disease: <2% especially young children → at risk → culture for group A β-hemolytic streptococci → treat; family pets, particularly dogs, reported as carriers) ▪ Most: does not lead to significant kidney damage ▪ Progressive disease develops in 20–30% of adult patients 15-20yr after onset (need for careful long-term ffup) ▪ Poor prognostic indicators: persistent HTN; ↓renal function; significant, increasing, or prolonged proteinuria; histology: diffuse mesangial proliferation, extensive glomerular crescents, glomerulosclerosis, & diffuse tubulointerstitial changes, including inflammation & fibrosis ▪ BP control, mgt of proteinuria: ACEIs, ARBs → ↓proteinuria, ↓rate of progression (individual/ combination) ▪ Fish oil (antiinflam omega-3 PUFA): ↓rate of progression in adults ▪ RAS blockade ineffective, proteinuria persists → steroids → improve renal function (patients w/ eGFR >60mL/ min/m2) ▪ Cyclophosphamide/azathioprine → not appeared effective, RCTs in progress ▪ Tonsillectomy w/out significant tonsillitis in assoc w/ IgA nephropathy → not recommended ▪ Targeted-release oral budesonide combined with RAS blockade (pilot study to reduce proteinuria) ▪ Kidney transplant → recurrent disease frequent ▪ Most common in 2nd decade of life; M=F; more common in Caucasian ▪ Equal proportions w/ nephrotic syndrome, acute nephritic syndrome, or persistent asymptomatic microscopic hematuria and proteinuria ▪ ↓C3 ▪ Diagnosis by renal biopsy: nephrotic syndrome in older child, significant proteinuria w/ microscopic hematuria, hypocomplementemia >2mo in child w/ acute nephritis ● C3 but no Ig deposition: genetic testing & functional assays (defects of complement cascade regulation) ▪ Untreated, idiopathic MPGN, regardless of type: poor prognosis ● 10yr: 50% ESRD ● 20yr: 90% lost renal fx ▪ Nephrotic syndrome & HTN at presentation → renal failure more rapidly ▪ Determine if idiopathic or secondary (lupus or chronic infection) → tx causative disease ▪ No definitive therapy → extended course of alternate-day prednisone (years) → preserve renal function ▪ C3GN: interruption of complement activation pathways → complement factor H replacement, eculizumab (anti–C5 antibody) → prevent progression of renal disease ▪ Classic tetrad: palpable purpura, arthritis/arthralgia, abdominal pain, evidence for renal dx Mild: isolated microscopic hematuria w/out significant proteinuria Severe: combined acute nephritic & nephrotic syndrome (hematuria, HTN, renal insufficiency, significant proteinuria) ▪ Most urinary abnormalities by 1mo, nearly all by 3-6mo after onset ▪ Mild: majority → spontaneous & complete resolution of nephritis; occ progress despite resolution of other HSP features ▪ Untreated → CKD ▪ Moderate/severe HSP nephritis → likely to progress to CKD ▪ Approach in severe clinical renal involvement (nephrotic range proteinuria, HTN, ↑crea) ● Prednisone (1mkday for 3mo) ● ACE inhibitors ● persists: Azathioprine, MMF Batch Clingy ▪ Facial/body edema ▪ Hypertension ▪ Oliguria (renal dysfunction) 103 Although GN not typical (page 1322), PAN part of differential diagnostic algorithm (Fig 537.6) presenting with hematuria. ▪ Mucosal infection/food antigen trigger ↑production of pathogenic IgA1 → defective glycosylation of hinge region of IgA1 + autoantibodies → immune complexes deposited in glomerular mesangium ▪ IF: pathognomonic IgA deposits in glomerular mesangium of HSP ▪ UA 1x/wk during active clinical disease → 1x/mo up to 6mos ▪ All UA normal during ffup interval: nephritis unlikely to develop ▪ Kidney biopsy: significant proteinuria (urine protein >1g/day or UPCr > 1.0), significant HTN, ↑ crea SLE NEPHRITIS ▪ More common & more severe in children ▪ 80% of pediatric px w/ SLE; common w/in 1styr of dx ▪ Most impt cause of morbidity & mortality in SLE ▪ Result of deposition of circulating immune complexes & direct binding of autoAb to glomerular components w/ resultant complement stimulation ▪ Renal histopathology: gold standard ▪ IF: full-house immune staining; granular deposition of Ig isotypes (IgG, IgM, IgA), complements (C3, C4, C1q) in glomerular mesangium and capillary walls WHO classification (1980) ▪ Class I nephritis (minimal mesangial LN) ● LM: no histologic abnormalities; IM/EM: mesangial immune deposits ▪ Class II nephritis (mesangial proliferative nephritis) ● Mesangial hypercellularity, ↑matrix, mesangial deposits containing Ig & complement ▪ Class III nephritis ● <50% glomeruli w/ involvement ▪ Class IV nephritis ● >50% glomeruli w/ involvement ● Poorer outcomes; successfully tx w/ aggressive immunosuppressive therapy ▪ Class V nephritis (membranous LN) ● Isolated lesion, resembles idiopathic membranous nephropathy w/ subepithelial immune deposits *WHO class III & class IV: interrelated, characterized by mesangial and endocapillary lesions; subclassification scheme: severity of proliferative lesion → segmental vs global *Chronic injury → glomerular sclerosis *Active disease: capillary walls thickened sec to subendothelial deposits (wire-loop lesion), necrosis, crescent formation GRANULOMATOSIS WITH POLYANGIITIS (GPA) ▪ Wegener granulomatosis ▪ Necrotizing granulomatous small & medium vessel vasculitis, targets upper & lower respiratory tract and kidneys ▪ Occurs at all ages; female predominance, 3-4:1 ● Pediatric GPA prevalent in Caucasian ▪ EULAR/PReS Classification Criteria for Pediatric-Onset GPA (Dx: 3 out of 6) ● Histopath showing granulomatous inflam ● Upper airway involvement ● Laryngeal, tracheal or bronchial involvement ● ANCA positivity ● Renal involvement ● Proteinuria, hematuria, RBC casts, necrotizing pauci- PPS Oral Exam Reviewer 2020 International Society of Nephrology and the Renal Pathology Society (2004) [Widely preferred but most RCTs based on WHO] Class I (Minimal mesangial LN) – No renal findings Class II (Mesangial proliferative LN) ● Mild clinical renal dx; minimally active urinary sediment; mild to moderate proteinuria (never nephrotic) but may have active serology Class III (Focal proliferative LN, <50% glomeruli) [A. Active; A/C. Active and chronic; C. Chronic] ● More active sediment changes; often active serology; ↑proteinuria (>25% nephrotic); HTN may be present; some evolve into class IV pattern; active lesions require tx; chronic do not Class IV (Diffuse proliferative LN, >50% glomeruli); all may be w/ segmental or global involvement (S or G) [A. Active; A/C. Active and chronic; C. Chronic] ● Most severe renal involvement w/ active sediment, HTN, heavy proteinuria (freq nephrotic syndrome) often ↓GFR; serology very active. Active lesion require tx Class V (Membranous LN glomerulonephritis) ● Significant proteinuria (often nephrotic) w/ less active lupus serology Class VI (Advanced sclerosing LN) ● >90% glomerulosclerosis; no tx prevent renal failure GPA ▪ Early disease: nonspecific constitutional sx (fever, malaise, wt loss, myalgias, arthralgias) ▪ Upper airway involvement: sinusitis, nasal ulceration, epistaxis, otitis media, hearing loss ▪ Lower respiratory tract: cough, wheezing, dyspnea, hemoptysis ▪ Childhood GPA: more frequently complicated by subglottic stenosis ▪ Inflammation-induced damage to the nasal cartilage → saddle nose deformity ▪ Pulmonary hemorrhage → rapid respiratory failure ▪ Ophthalmic involvement: conjunctivitis, scleritis, uveitis, optic neuritis, invasive orbital pseudotumor (causing proptosis) ▪ Perineural vasculitis or direct compression on nerves by granulomatous lesions → cranial & peripheral neuropathies ▪ Renal survival (defined as CKD w/out need for ESRD therapy): 80% 10yr post-diagnosis of SLE nephritis ▪ Highest risk for progression to ESRD: diffuse proliferative WHO class IV lupus nephritis, poor renal function at presentation, persistent nephrotic-range proteinuria ▪ ↑ Risk of malignancy/infertility: receiving cumulative dose of >20g of cyclophosphamide or other immunosuppressant therapies ▪ Upper respiratory tract lesions can invade orbit → threaten optic nerve ▪ Lesions in the ear → permanent hearing loss ▪ Respiratory complications: life-threatening pulmonary hemorrhage & upper airway obstruction (subglottic stenosis) ▪ Chronic lung dx sec to granulomatous inflammation, cavitary lesions, scarring → infectious complications ▪ Chronic GN → ESRD (advanced/undertreated disease) ▪ Course variable ▪ Relapse: 60% of patients ▪ Mortality ↓ w/ introduction of cyclophosphamide & other immunosuppressive agents ▪ Compared w/ adults, children more likely to develop multiorgan involvement, renal involvement & subglottic ▪ Severe histologic manifestations (>50% glomerular crescents): IV methylprednisolone pulses (3days); ffd by prednisone (3mo) + azathioprine or MMF (extended course) ▪ Most severe histology (>75% glomerular crescents), progressive renal failure: IV steroids + plasmapheresis ▪ ESRD: renal transplant ▪ Immunosuppression: cornerstone of therapy → clinical remission (normalization of renal function & proteinuria); serologic remission (normalization of anti-DNA antibody, C3, C4) ▪ Prednisone 1-2mkday in divided doses → slow steroid taper over 4-6mo beginning 4-6wk after achieving serologic remission ▪ Severe forms (WHO classes III and IV): steroid therapy alone insufficient to induce remission ● Induction phase: 6 monthly IV infusions of cyclophosphamide 5001,000mg/m2; pulse IV methylprednisolone 1,000mg/m2 + oral steroids; alternative: MMF 600mg/m2/dose BID (SE: diarrhea, leucopenia, teratogenicity) ● Maintenance phase: cyclophosphamide (Cytoxan) infusions q3mo for 18mo (SE: infections, hair loss, hemorrhagic cystitis, gonadal failure); MMF; azathioprine 1.5-2mkdose QD (steroid-sparing agent in WHO class I or II) ▪ Rituximab, chimeric monoclonal antibody specific for human CD20: resistance to conventional tx ▪ Plasmapheresis: ineffective unless w/ accompanying TTP or antineutrophilic cytoplasmic antibody–associated dx ▪ Belimumab, fully humanized monoclonal antibody against type II transmembrane protein: requires further study ▪ Hydrochloroquine: for extrarenal SLE manifestations; effect in maintaining remission in lupus nephritis ▪ Antihypertensive, ACE inhibitors, ARBs ▪ Lower respiratory tract/kidneys significantly involved: initial induction tx: prednisone (2mg/kg/day) or IV methylprednisolone (30mg/kg/day, 3days) + daily oral or monthly IV cyclophosphamide ▪ Rituximab, mono-clonal antibody to CD20 on activated B cells, option for induction (study primarily in adults) ▪ Plasmapheresis + methylprednisolone: severe dx (pulmonary hemorrhage or ESRD: potential for ↓dialysis dependency) ▪ Transition to less toxic maintenance medication: methotrexate, azathioprine, MMF w/in 3-6mo once in remission ▪ TMP-SMX (one 180mg/800mg tab 3days/wk): prophylaxis (Pneumocystis jiroveci); ↓upper respiratory bacterial colonization w/ S aureus (trigger dx activity) ▪ Disease limited to upper respiratory tract: steroids (1-2 mg/kg/day), methotrexate (0.5-1.0mg/kg/wk) Batch Clingy nodosa. 104 MICROSCOPIC POLYANGIITIS (MPA) ▪ Small vessel necrotizing vasculitis; clinically similar to GPA w/out granulomas & upper airway involvement ▪ Rare in children ▪ Cardinal histologic feature (GPA/MPA): necrotizing vasculitis ● Kidney biopsy: crescentic GN w/ little or no immune complex deposition (“pauci-immune”) ● Granulomatous inflammation common in GPA & Churg Strauss Syndrome (CSS); not present in MPA ● Perivascular eosinophilic infiltrates distinguish CSS from MPA & GPA ▪ Etiology of ANCA-associated vasculitis unknown ● Neutrophils, monocytes & endothelial cells involved in pathogenesis ● Neutrophils & monocytes activated by ANCAs, specifically ANCA-associated antigens proteinase-3 (PR3) & myeloperoxidase (MPO) → release proinflammatory cytokines (TNF-α, IL-8) ● Localization of inflammatory cells to endothelium → vascular damage POLYARTERITIS NODOSA ▪ Systemic necrotizing vasculitis → small & medium-size arteries; aneurysms & stenoses form at irregular intervals throughout affected arteries ▪ Cutaneous PAN limited to the skin ▪ PAN rare in childhood ● Boys & girls equally affected; mean age 9yr ▪ Cause unknown ▪ Devt ffg infections (GAS, chronic hep B) → may represent postinfectious autoimmune response ● Other: EBV, MTB, CMV, parvo B19, hep C virus ▪ Assoc w/ familial Mediterranean fever ▪ Biopsy: necrotizing vasculitis w/ granulocytes & monocytes infiltrating walls of small & medium-size arteries → usually segmental, at vessel bifurcations ▪ Granulomatous inflammation not present ▪ Deposition of complement & immune complex rare ▪ Different stages of inflammation found: mild inflammatory changes to panmural fibrinoid necrosis assoc w/ aneurysm formation, thrombosis, vascular occlusion ▪ Immune complexes → pathogenic → poorly understood ▪ Inflamed vessel wall thickened & narrowed → impeding blood flow → contributing to end-organ damage ▪ No clear genetic association ● Deficiency in adenosine deaminase 2 (DADA2) (caused by mutation in CECR1 gene) → familial form of vasculitis in Georgian Jewish patients w/ autosomal recessive inheritance Proposed Classification Criteria: Pediatric-Onset PAN (5 criteria: Sn 89.6%, Sp 99.6%) ● Histopath: necrotizing vasculitis in medium/small art PPS Oral Exam Reviewer 2020 ▪ Hematuria, proteinuria, HTN → signal renal disease ▪ Cutaneous lesions: palpable purpura & ulcers ▪ Venous thromboembolism: rare but potentially fatal complication MPA ▪ closely resemble GPA, sinus disease less common ▪ systemic features dominant: fever, malaise, wt loss, myalgia, arthralgia ▪ predominantly affects kidney & lungs ▪ distinguished from PAN: (+) ANCAs, tendency for small vessel involvement ▪ no clear correlation between ANCA titers & disease activity/relapse ▪ DDx: ANCAs absent in other granulomatous diseases (sarcoidosis, TB); Goodpasture syndrome (Ab to GBM); meds (PTU, hydralazine, minocycline) associated w/ drug-induced ANCA (perinuclear ANCA) vasculitis; SLE and HSP (present as pulmonary hemorrhage, nephritis) ▪ Presentation variable, reflect distribution of inflamed vessels ▪ Constitutional sx present in children at disease onset ▪ Wt loss, severe abdominal pain → mesenteric arterial inflammation & ischemia ▪ Renovascular arteritis → HTN, hematuria, proteinuria; GN not typical ▪ Cutaneous: purpura, livedo reticularis, ulcerations, digital ischemia, painful nodules ▪ CNS: CVA, TIA, psychosis, ischemic motor/sensory peripheral neuropathy (mononeuritis multiplex) ▪ Myocarditis, coronary arteritis → heart failure, MI, pericarditis, arrhythmias ▪ Arthralgias, arthritis, myalgias ▪ Pulmonary vasculature usually spared ▪ Biopsy or angiography: demonstration of vessel involvement ● Biopsy of cutaneous lesions: small/medium vessel vasculitis ● Kidney biopsy: necrotizing arteritis ● Sural nerve biopsy: vasculitis ▪ EMG: peripheral neuropathy identifies affected nerves ▪ Conventional arteriography: gold standard diagnostic, reveals areas of aneurysmal dilation & segmental stenosis, classic “beads on a string” ▪ MRA & CTA: less invasive, gaining acceptance; not effective identifying small vessel dx or in younger children ▪ Nonspecific lab: ↑ESR, CRP; anemia, leukocytosis, hypergammaglob; abnormal urine sediment, proteinuria, hematuria → renal disease; ↑hepatic enzyme→ hep B/C; serologic tests for hepatitis (hep B surface antigen, hep C Ab) ▪ DDx: early skin lesions → resemble HSP (finding of nodular lesions & presence of systemic features distinguish PAN); pulmonary vascular involvement very rare in PAN, lesions suggest ANCA-associated stenosis *Renal involvement uncommon in CSS (inflammation of upper & lower respiratory tracts: recurrent rhinitis/sinusitis, nasal polyposis, nonfixed pulmonary lesions, difficult-to-treat asthma; eosinophilia (>10%) w/ pulmonary infiltrates before vasculitic phase; mepolizumab, anti-IL5 monoclonal Ab, may have role in tx) Feature S/sx of small vessel vasculitis IgA-dominant immune deposits Circulating antineutrophil cytoplasmic antibodies Necrotizing vasculitis Granulomatous inflammation Asthma and eosinophilia ▪ Cutaneous nodules → ulcerate, become infected ▪ Renovascular involvement → HTN, chronic renal disease ▪ Cardiac involvement → ↓cardiac function, CAD ▪ Mesenteric vasculitis → predispose to bowel infarction, rupture, malabsorption ▪ Stroke and rupture of hepatic arterial aneurysm: uncommon ▪ Course varies from mild disease w/ few complications to severe, multiorgan disease ↑morbidity & mortality ▪ Poor prognostic factors ● ↑ Crea, proteinuria, severe GI involvement, cardiomyopathy, CNS involvement ▪ Early and aggressive immunosuppressive tx: ↑clinical remission, minimize potential long term vascular complications ▪ Compared w/ adults, childhood PAN less mortality ▪ Cutaneous PAN → unlikely to transition to systemic disease HSP + + GPA + - CSS + - MPA + - - +(PR3) +(MPO > PR3) +(MPO) - + + - + + + + - ▪ Mainstay: prednisone (1-2mg/kg/day) or IV pulse methylprednisolone (30 mg/kg/day) ▪ Oral/IV cyclophosphamide: used as adjunctive tx ▪ Plasma exchange: life-threatening ▪ Hepatitis B identified → antiviral therapy ▪ Cutaneous PAN: less intense → steroids alone, NSAIDs, methotrexate ▪ Azathioprine, MMF, IVIG, thalidomide, cyclosporine, anti-TNF agents (infliximab) → refractory cutaneous or systemic PAN (clinical trials lacking) ▪ Infectious trigger for PAN → antibiotic prophylaxis considered Batch Clingy immune GN 105 HEMOLYTIC UREMIC SYNDROME ▪ Most common form of thrombotic microangiopathy (TMA) in children ▪ Common cause of community-acquired AKI in young children ▪ Most common form: 90% diarrhea-associated HUS (STEC-HUS → E coli O157:H7 most common; Asia & South Africa, Shigella dysenteriae type 1) ● Undercooked meat, unpasteurized milk and apple cider; cross-contaminated cuttingboards; water supply; petting farms ● Enteropathic organism → produce toxin → absorbed from colonic mucosa to systemic circulation → bind to endothelial cells in glomerulus → endothelial cell damage; Shiga toxin directly activate platelets → aggregation → mechanical injury to RBCs passing through thrombotic microvasculature → severe nonimmune anemia w/ (-)direct Coombs test ▪ Pneumococcal-associated HUS → neuraminidase cleaves sialic acid on membranes of endothelial cells, RBC, platelets → expose ThomsenFriedenreich (T) antigen → IgM react w/ T antigen → hemolysis → anemia w/ (+)direct Coombs test ▪ Genetic form (atypical, nondiarrheal): 2nd major category → probably predispose but not cause dx → absence of preceding diarrheal prodrome → insidious onset/relapsing (trigger; mild nonspecific infection) ▪ In each form of HUS PPS Oral Exam Reviewer 2020 vasculitis/Goodpasture disease; other: SLE (characteristic targetorgan involvement, autoantibodies); prolonged fever & weight loss → IBD/malignancy ▪ Combination of pulmonary hemorrhage with acute AGN ▪ Hemoptysis from pulmonary hemorrhage → can be life-threatening ▪ AGN: hematuria, nephritic urinary sediment w/ cellular casts, proteinuria, HTN →rapidly progressive course → renal failure w/in days to weeks of clinical ● Less common: anti-GBM nephritis → isolated, rapidly progressive GN w/out pulmonary hemorrhage ▪ Fever may be present; other systemic complaints (malaise, arthralgia) usually absent (presence raise suspicion for systemic vasculitis) ▪ (+) anti-GBM Ab in serum and/or kidney; C3 normal ▪ ↑ANCA: 10–40% (along with the anti-GBM Ab; patients doubly positive → more severe disease) ▪ Anti-GBM Ab titer correlated w/ severity of renal involvement → accuracy of serology variable → biopsy (unless contraindicated) → histology guide therapy ▪ DDx: pulmonary-renal syndrome → SLE, HSP, nephrotic syndrome– associated pulmonary embolism, GPA, MPA Triad: microangiopathic hemolytic anemia, thrombocytopenia, renal insufficiency ▪ Microangiopathic hemolytic anemia w/ schistocytes; mild at presentation, rapidly progresses ▪ Thrombocytopenia usually 20,000-100,000/mm3; petechiae, severe bleeding rare ▪ Normal partial thromboplastin & prothrombin times ▪ Leukocytosis ▪ UA: microscopic hematuria, low-grade proteinuria ▪ Renal failure + severe hemolysis → life-threatening hyperkalemia ▪ Volume overload, HTN, severe anemia → heart failure ▪ ≤20%: severe CNS involvement: seizures, significant encephalopathy → HTN & focal ischemia sec to microvascular CNS thrombosis ▪ Intestinal complications: severe inflammatory colitis, ischemic enteritis, bowel perforation, intussusception, pancreatitis Diarrhea form: most common in preschool & school-age ▪ Onset of HUS: 5-7days after onset of gastroenteritis (fever, vomiting, abdominal pain, diarrhea w/ or w/out blood) ▪ Sudden onset of pallor, weakness, lethargy → onset of HUS: dev of microangiopathic hemolytic anemia ▪ Oliguria in early stage may be masked by diarrhea ▪ Significant dehydration or volume overload depend on whether ▪ Untreated, poor prognosis ▪ Tx early, good renal outcome in majority ▪ Oligoanuria, ↑proportion of glomerular crescents, kidney failure requiring dialysis → worse renal & patient survival rate ▪ Tx initiated emergently: plasmapheresis, ↑dose IV methylprednisolone, cyclophosphamide → induce remission, improve survival times ▪ Plasmapheresis: remove circulating anti-GBM Ab ▪ Initial immunosuppression w/ steroids & cyclophosphamide: inhibit ongoing Ab production ▪ Rituximab: substitute → cyclophosphamide toxicity ▪ Tx guided by clinical response & serial anti-GBM titers ▪ Maintenance therapy: lower dose of prednisone and azathioprine (or MMF) continued for 6-9mo ▪ Survive acute pulmonary hemorrhage & rapidly progressive GN → still progress to ESRD despite immunosuppressive therapy → kidney transplant → relapse & recurrent dx after transplant uncommon ▪ Mortality rate: diarrhea-associated HUS is <5% w/ early tx; 50%: severe AKI requiring dialysis usually about 2wk → most recover renal function completely, 5% remain dependent on dialysis, 30% some degree of chronic renal insufficiency ▪ Annual exam: no residual urinary abnormalities after 1yr → long-term sequelae unlikely ▪ Recovery of platelet count → renal recovery ~5days later → resolution of anemia ▪ Not associated w/ diarrhea: more severe ▪ Supportive care: F&E balance; control of HTN, early institution of dialysis (oliguric, anuric, hyperkalemia) ▪ RBC transfusion: pneumococci-associated HUS → washed → remove residual plasma → endogenous IgM → accelerate disease ▪ Platelets: generally, not administered → consumed by active coagulation → worsen ▪ Antibiotic to clear STEC → ↑toxin release, not recommended ▪ Tx of causative pneumococcal infection → important ▪ Plasma infusion, plasmapheresis: serious CNS involvement BUT contraindicated in pneumococcal associated HUS ▪ Eculizumab, anti-C5 antibody: atypical familial HUS; give meningococcal vaccine prior Pneumococci-associated HUS: >80% require dialysis, mortality rate 20% Genetic form: insidiously progressive/ relapsing, poor prognosis → ID specific factor deficiencies for directed therapy Kidney biopsy ▪ Risks during active phase; dx by clinical criteria ▪ Early changes: thickening of capillary walls (swelling of endothelial cells) & accumulation of fibrillar material between endothelial cells and GBM → narrowing capillary lumens; platelet–fibrin thrombi in glomerular capillaries, thrombi in afferent arterioles & small arteries w/ fibrinoid necrosis of arterial wall → renal cortical necrosis ▪ Late findings: glomerular sclerosis & obsolescence sec to severe direct glomerular involvement or glomerular ischemia from arteriolar involvement Batch Clingy ● Angiographic abnormalities: aneurysm, stenosis, or occlusion of medium/small artery not from noninflammatory cause ● Cutaneous findings: livedo reticularis, tender subcutaneous nodules, superficial skin ulcers, deep skin ulcers, digital necrosis, nail bed infarctions, or splinter hemorrhages ● Muscle involvement: myalgia/muscle tenderness ● Hypertension: SBP/DBP >95th percentile for height ● Peripheral neuropathy: sensory peripheral neuropathy, motor mononeuritis multiplex ● Renal involvement: proteinuria (>300mg/24hr equiv), hematuria/RBC casts, impaired renal function (GFR <50% normal) GOODPASTURE DISEASE ▪ Autoimmune disease: pulmonary hemorrhage, rapidly progressive GN, ↑anti–GBM antibody titer ▪ Rare in childhood ▪ Ab against epitopes of type IV collagen; located w/in alveolar basement membrane & GBM ▪ Acquired conformational change in noncollagenous 1 domain of alpha 3-chain of type IV collagen → production of pathologic autoAb → high affinity to GBM → rapidly progressive kidney disease ▪ Kidney biopsy: crescentic GN; IF: pathognomonic continuous linear deposition of IgG along GBM 106 ▪ Nephrotic Range Proteinuria (>3.5g/24hr OR UpCr Ratio >2) ▪ Hypoalbumine mia (≤2.5g/dL) ▪ Edema ▪ Hyperlipidemia (chol >200 mg/dL) 1o/Idiopathic (90%) ▪ Minimal Change Disease (most common) ▪ FSGS ▪ MPGN (GN Associated) ▪ C3 Glomerulopathy ▪ Membranous Nephropathy 2o to Systemic Disease ▪ SLE ▪ HSP ▪ Malignancy (Lymphoma and Leukemia ▪ Infections (Hepatitis, HIV, Malaria) ▪ Hereditary Proteinuria Syndrome Please see Table 545.1 Please see Table 545.2 MINIMAL CHANGE NEPHROTIC SYNDROME ▪ ~85%; glomeruli appear normal or minimal ↑mesangial cells & matrix, IF: (-), EM: effacement of epithelial cell foot processes; >95% respond to steroid ▪ M>F (2:1); 85-90% <6yr (most commonly 2-6yr); only 20-30% of adol ▪ absence of HTN and gross hematuria (nephritic features) MESANGIAL PROLIFERATION ▪ LM: diffuse ↑mesangial cells & matrix on light microscopy; IF: trace to 1+ mesangial IgM and/or IgA; EM: mesangial cells & matrix; effacement of epithelial cell foot processes; ~50% respond to steroid FOCAL SEGMENTAL GLOMERULOSCLEROSIS (FSGS) ▪ LM: mesangial cell proliferation & segmental scarring; IF: (+)IgM & C3 in segmental sclerosis; EM: segmental scarring of glomerular tuft w/ obliteration of glomerular capillary lumen; 20% respond to steroid; often progressive to ESRD ▪ Most common cause of ESRD in adol PPS Oral Exam Reviewer 2020 enteritis or renal insufficiency from HUS predominates ▪ Stool culture often negative Pneumococci-associated HUS: pneumonia, empyema, bacteremia Genetic form: risk for recurrence, severe prognosis, benefit from different therapy Edema: most common presenting sx ▪ Underfill hypothesis: proteinuria → ↓plasma protein level → ↓intravascular oncotic pressure → edema & secretion of vasopressin and atrial natriuretic factor → ↑Na and water retention ▪ Overfill hypothesis: primary Na retention → volume expansion → edema ▪ Misdiagnosed as allergic disorder: periorbital swelling that ↓at end of day ▪ DDx of marked edema: protein-losing enteropathy, hepatic/heart failure, acute/chronic GN, protein malnutrition ▪ Goal: gradual reduction of edema w/ diuretics, Na restriction, cautious use of IV albumin Hyperlipidemia ▪ ↑Synthesis & ↓catabolism of lipids; ↑cholesterol, triglycerides, LDL, VLDL; HDL unchanged or low; ↑CV in adults, in children not as serious ↑ Susceptibility to infection: cellulitis, spontaneous bacterial peritonitis, bacteremia ▪ Hypoglobulinemia: urinary loss of IgG, complement factors (predominantly C3 & C5), alternative pathway factors B & D → impaired opsonization ▪ Encapsulated bacteria ▪ Spontaneous bacterial peritonitis: fever, abdominal pain, peritoneal signs → pneumococcus (most frequent), Gram (-) bacteria → peritoneal leukocyte counts >250 cells/µL ▪ Appropriate cultures, treated promptly & empirically Hypercoagulability ▪ Vascular stasis (hemoconcentration, intravascular volume depletion), ↑Plt number & aggregability, changes in coagulation factor levels; ↑ fibrinogen + urinary losses of antithrombotic factors (antithrombin III & protein S) ▪ DVT → cerebral venous sinus, renal vein, pulmonary veins ▪ Low in children (2–5%) compared with adults; potential for serious consequences ▪ Initial ep/relapse: follow infections, rxn to insect bites ▪ Rule out secondary forms: children ≥10yo, C3 level, ANA, anti dsDNA; hepatitis B & C, HIV in high-risk ▪ Kidney biopsy: ≥ 12yr, does not fit picture of MCNS ▪ 3+ or 4+ proteinuria; microscopic hematuria ▪ Spot UPCr > 2.0 ▪ Creatinine normal; ↑ if diminished renal perfusion from contraction of intravascular volume ▪ Albumin < 2.5 g/dL; ↑serum cholesterol & triglyceride levels ▪ Serum complement: normal ▪ Steroid-resistant nephrotic syndrome (most often FSGS) → poorer prognosis → ESRD requiring dialysis/kidney transplant Response: attainment of remission w/in initial 4wk of steroid Remission: UPCr ratio <0.2 or <1+ protein on urine dipstick for 3 consecutive days ▪ MCD respond to daily prednisone w/in the first 23wk Relapse: ↑ First morning UPCr >0.2 or 2+ and higher for 3 consecutive days Frequently relapsing: 2 or more relapses w/in 6mo after initial therapy or 4 relapses in a 12-mo period Steroid dependent: relapse during steroid tapering or a relapse w/in 2wk of discontinuation Steroid resistance: inability to induce remission w/in 4wk of daily steroid – Trace + ++ +++ ++++ PROTEINURIA Negative 10-20 mg/dL 30mg/dL 100mg/dL 300mg/dL 1000-2000 mg/dL ▪ Prednisone/prednisolone single daily dose 60mg/m2/day or 2mkday (max 60mg daily) for 4-6wk; ffd by alternate-day prednisone (starting at 40mg/m2 QD or 1.5mg/kg QD) ranging 8wk to 5mo w/ tapering ● Duration controversial; prolonged tx w/ tapering 2-5mo ↓incidence of relapse; KDIGO: at least 12wk ▪ Severe symptomatic edema (large pleural effusions, ascites, severe genital edema) → hospitalized ● HypoNa+: fluid & Na restriction (<1,500 mg daily) ● Diuresis: loop diuretics (furosemide) extreme caution → aggressive diuresis → intravascular volume depletion, ↑risk for ARF & intravascular thrombosis ● Edema w/ intravascular volume depletion (hemoconc, hypotension, tachycardia), IV 25% albumin (0.5-1.0g/kg) slow infusion ffd by IV furosemide 1-2mkdose → monitor volume status, BP, elec balance, renal function → complication (esp rapid infusion): volume overload (HTN, heart failure, pulmonary edema) ▪ Dyslipidemia: low-fat diet (limit to <30% of calories w/ saturated fat intake of <10% calories); dietary cholesterol <300mg/day ▪ Parent educ: sx of cellulitis, peritonitis, bacteremia ● Blood culture; broad coverage antibiotics: include Pneumococcus, Gram(-) bacteria; 3rd-gen ceph common choice ● Spontaneous bacterial peritonitis: peritoneal fluid cell count, GS, culture ▪ Thromboembolism: imaging confirm presence of clot ● Heparin, LMW heparin, warfarin ▪ Relapses → triggered by URI or GI infections ● Tx similar to initial ep except daily prednisone course shortened ● Duration of alternate-day therapy depend on freq of relapses ▪ Steroid resistance → failure to achieve remission after 8wk of steroid → kidney biopsy, eval kidney function, quantitation of urine protein excretion → reduced life expectancy ▪ Steroid-dependent, frequent relapsers, steroid-resistant → alternative (particularly steroid toxicity: cushingoid appearance, HTN, cataracts, and/or growth failure) ▪ Cyclophosphamide (2mg/kg single PO dose, 8-12wk) prolong remission, ↓number of relapse in children w/ frequently relapsing & steroiddependent NS; SE: neutropenia, disseminated varicella, hemorrhagic cystitis, alopecia, sterility, ↑risk future malignancy ● Alternate-day prednisone often continued ● WBC monitored weekly; withheld if <5,000/mm3 ● Cumulative threshold dose >250mg/kg: oligospermia/ azoospermia ▪ Calcineurin inhibitors (cyclosporine/tacrolimus): initial tx for steroidresistant; SE: HTN, nephrotoxicity, hirsutism, gingival hyperplasia ▪ Mycophenolate: maintain remission in steroid-dependent, frequently relapsing NS ▪ Levamisole, antihelminthic w/ immunomodulating effect: ↓risk of relapse in comparison to prednisone Batch Clingy NEPHROTIC SYNDROME ● Capillary & arteriolar endothelial injury in kidney → localized thrombosis → ↓glomerular filtration ● Microvascular injury → progressive platelet aggregation → consumptive thrombocytopenia ● Mechanical damage to RBC passing through damaged/thrombotic microvasculature → microangiopathic hemolytic anemia ▪ 1-3 per 100,000 children <16yr ▪ W/out tx: ↑risk of death, most commonly from infections ▪ 80%: respond to steroids ▪ Podocyte, GBM and fenestrated glomerular endothelium → glomerular filtration barrier ▪ Podocyte: structural support of the capillary loop; major filtration barrier to proteins; involved in synthesis & repair of GBM ▪ Slit diaphragm: major impediments to protein permeability; not simple passive filters, consist of numerous proteins → contribute to complex signaling pathways, play important role in podocyte function ▪ Nephrin, podocin, CD2AP, α-actinin 4: important component protein of slit diaphragm ▪ Immune & nonimmune insults to podocyte → foot process effacement, ↓functional podocytes, altered slit diaphragm → ↑permeability of the glomerular capillary wall → massive proteinuria & hypoalbuminemia ▪ Pathogenesis of idiopathic nephrotic syndrome (hypothesis): either immune mediated (systemic podocyte-derived circulating factor) or genetic (mutations assoc w/ steroid-resistant nephrotic syndrome) ▪ Genetic screening cohort: mutations in NPHS1, WT1, and NPHS2 → steroid-resistant nephrotic syndrome and congenital nephrotic syndrome 107 SECONDARY NEPHROTIC SYNDROME ▪ > 8yr w/ HTN, hematuria, renal dysfunction, extrarenal sx (rash, arthralgias, fever), ↓serum complement levels ▪ Present at birth: edema caused by massive proteinuria ▪ Delivered w/ enlarged placenta (>25% of infant’s wt) ▪ Severe hypoalbuminemia, hyperlipidemia, hypogammaglobulinemia: from loss of filtering selectivity at glomerular filtration barrier ▪ Prenatal dx: ↑maternal and amniotic AFP levels ▪ Denys-Drash syndrome (mutations in the WT1 gene): abnormal podocyte function; early-onset nephrotic syndrome, progressive renal insufficiency, ambiguous genitalia, Wilms tumors ▪ Pierson syndrome: mutations in LAMB2 gene → abnormalities of β2-laminin, critical component of the glomerular and ocular basement membranes → bilateral microcoria (fixed narrowing of pupil) ▪ Galloway-Mowat syndrome: microcephaly w/ hiatal hernia & congenital nephrotic syndrome; kidney biopsy: loss of or poor basement membrane formation or permeation of their basement membranes with fibrils ▪ Clinical dx: severe generalized edema, poor growth & nutrition w/ hypoalbuminemia, ↑susceptibility to infections, hypothyroidism (urinary loss of thyroxin-binding globulin), ↑risk of thrombotic events Most: progressive renal insufficiency Batch Clingy CONGENITAL NEPHROTIC SYNDROME ▪ Nephrotic syndrome manifesting at birth or w/in the first 3mos of life ▪ Primary (syndromes inherited as autosomal recessive disorders) or secondary to utero infections (CMV, toxoplasmosis, syphilis, hep B and C, HIV), infantile SLE, mercury exposure ▪ Structural and functional abnormalities of glomerular filtration ▪ European cohort of children: 85% mutations in four genes: NPHS1, NPHS2, WT1 (components of glomerular filtration barrier) and LAMB2 ● Finnish type: mutations in NPHS1 or NPHS2 gene → encodes nephrin and podocin ▪ Rituximab: prolong remissions steroid-dependent and/or steroidresistant NS; less effective in patients treated w/ calcineurin inhibitors & steroids and w/ multidrug-resistant NS ▪ ACE inhibitor/ARBs: ↓proteinuria in steroid-resistant ▪ PCV13 & PPSV23, influenza vaccine to child & contacts ● Defer live vaccines til on prednisone 1mkday or 2mg/kg on alternate days; contraindicated receiving cyclophosphamide/ cyclosporine ● Close contact w/ varicella infection, give VZIG ● Immunize household contacts w/ live vaccines → minimize risk of transfer; avoid direct exposure of child to GI or respiratory secretions of vaccinated contacts for 3-6wk after vaccination ▪ Intensive supportive care; IV γ-globulin, albumin & diuretic infusions; high amounts of protein (3-4g/kg), lipids, high caloric intake to maintain nutrition, vitamin and thyroid hormone replacement; unilateral nephrectomy; ACE inhibitor, angiotensin II receptor inhibitors, and prostaglandin synthesis inhibitors ● Conservative fails, suffer from persistent anasarca/repeated severe infections → bilateral nephrectomy, chronic dialysis ● Renal transplant: definitive treatment (recurrence reported after transplant) ● Some centers: more aggressive, bilateral nephrectomy at 1-2yr of age, weight >7kg, initiation of peritoneal dialysis w/ subsequent kidney transplantation ▪ Secondary congenital nephrotic syndrome resolves w/ tx of underlying cause PPS Oral Exam Reviewer 2020 108 Complex URINARY LITHIASIS EPIDEMIOLOGY / PATHOGENESIS ▪ Genetic, climatic, dietary, and socioeconomic factors ▪ Adolescents: 10x more likely to have symptomatic calculus than children 0-3yr ▪ ↑stone disease (in US): obesity, changes in dietary habits (↑Na and fructose intake, ↓Ca and water intake) ▪ Southeast Asia: urinary calculi endemic, related to diet ▪ Contamination of infant formula w/ the organic base and illegally added nitrogen-containing food additive melamine: China (2008) ▪ ~90%: contain Ca; 60% composed of Ca oxalate ▪ Renal calculi: crystals that form on calyx ▪ Bladder calculi: formed in the kidney → traveled down ureter; form primarily in bladder ▪ ↓Urine volume, ↓urine pH, Ca, sodium, oxalate, urate: promote stone formation ▪ Many inorganic (citrate, magnesium) and organic (glycosaminoglycans, osteopontin) substance: inhibit stone formation ▪ Stone formation depends on four factors ● Matrix: mixture of protein, nonamino sugars, glucosamine, water & organic ash that make up 2–9% of dry wt of stones; arranged in organized concentric laminations ● Precipitation-crystallization: supersaturation of urine w/ specific ions → crystals aggregate by chemical & electrical forces; ↑saturation of urine w/ ions → ↑rate of nucleation, crystal growth, aggregation → ↑stone formation & growth ● Epitaxy: aggregation of crystals of diff composition but similar lattice structure → forming stones of heterogeneous nature; lattice structures of Ca oxalate & monosodium urate have similar structure → Ca oxalate aggregate on nucleus of monosodium urate crystals ● Absence of inhibitors of stone formation (citrate, diphosphonate, magnesium ion) in the urine CLINICAL MANIFESTATIONS / LABORATORY FINDINGS ▪ Gross or microscopic hematuria ▪ Calculus: ureteral or renal pelvic obstruction → severe flank (renal colic) or abdominal pain ● Areas of narrowing: ureteropelvic junction, where ureter crosses iliac vessels, ureterovesical junction (most narrow segment); ureter progressively narrows distally ● Pain radiates anteriorly to scrotum or labia; intermittent (periods of obstruction of urine flow ↑pressure in collecting system) ● Distal ureter: irritative sx, dysuria, urgency, frequency; stone pass into bladder→ asymptomatic ● In the urethra: dysuria & diff voiding (particularly in males) ● Some children: pass small amounts of gravel-like material ▪ ~90%: calcified to some degree → radiopaque on plain abdominal film ● Only a few mm in diameter → difficult to see (particularly in the ureter) ● Struvite (magnesium ammonium phosphate) stones: radiopaque ● Cystine, xanthine, uric acid calculi: may be radiolucent but often slightly opacified ▪ Suspected renal colic → most accurate: unenhanced spiral CT of abdomen & pelvis; Sn & Sp 96% in delineating number and location of calculi, demonstrates if involved kidney is hydronephrotic ● Pediatric imaging centers use low-dose CT (similar to 3 CXR) ● Alternative: plain radiograph of abdomen & pelvis plus renal UTZ → hydronephrosis, possibly calculus on radiograph; calculus not visualized on UTZ unless adjacent to bladder/renal pelvis; renal calculi <3mm typically not seen ● Balance risks of CT imaging against ↓Sn of plain abdominal film plus sonography ● Difficult to determine whether hydronephrosis is sec to obstructing stone, UPJO or both Please refer to Table 562.3 Urine Chemistry: Normal Values Metabolic Evaluation of Children with Hypercalciuria RestricFasting Type Serum Ca ted Ca Ca (Urine) (Urine) Absorptive N N or ↑ N Ca Load (Urine) PTH (serum) ↑ ↑ Renal N ↑ ↑ ↑ N Resorptive ↑ ↑ ↑ ↑ ↑ ▪ Metabolic evaluation (note: structural, infectious, metabolic factors often coexist) ● Eval not undertaken in child in the process of passing stone → altered diet and PPS Oral Exam Reviewer 2020 COMPLICATIONS / PROGNOSIS ▪ May be asymptomatic (but uncommon to pass a ureteral calculus w/out sx) ▪ Child diagnosed w/ calculus: serial plain xrays or renal UTZ → follow status of calculus, ↑/↓ size, has moved; renal pelvic calculus → suspect UPJO ▪ Decision to remove: location, size, composition (if known), presence of obstruction and/or infection TREATMENT / PREVENTION ▪ Pain: NSAIDs, opiates ▪ Small ureteral calculi pass spontaneously (child might experience severe renal colic) ▪ Narrowest segment (ureterovesical junction): calculi <5mm will pass 80–90% of the time ▪ α-adrenergic blocker tamsulosin, 0.4mg at bedtime → ↓ureteral pressure below stone, ↓frequency of peristaltic contractions → medical expulsive therapy ▪ Ureteral stent: relieve pain, dilate ureter (allow to pass) ▪ Uric acid calculus or furosemide-associated calculus → dissolution alkaline therapy ▪ Calculus does not pass, assoc UTI → removal: lithotripsy; ESWL; percutaneous nephrostolithotomy; laparoscopic removal (Please see Table 562.5 for comparison) ▪ Maintaining continuous ↑UO: ↑fluid intake effective method of preventing further stones → continued at night → necessary for child to get up at least once a night to urinate and drink more water ▪ Daily fluid intake of 2-2.5 L in adolescent stone formers, ↑intake during summer ▪ ↓sodium, ↓protein diet: ↓urine Ca & oxalate excretion ▪ Avoid excess Ca intake, however, children require Ca for bone development → Ca restriction avoided ▪ Thiazide diuretics: ↓renal Ca excretion ▪ Potassium citrate (inhibitor of Ca stones) 1-2mEq/kg/24hr ● Source of citrate: lemonade (4oz lemon juice = 84mEq citric acid); daily mixture of 4oz of reconstituted lemon juice in 2L of water ↑urinary citrate level ▪ Neutral orthophosphate (difficult cases, poorly tolerated) ▪ Uric acid: allopurinol (xanthine oxidase inhibitor), ↓production uric acid and 2,8-dihydroxyadenine; urine alkalinization (≥6.5) w/ Na bicarbonate/Na citrate ▪ Cystine: more soluble when urinary pH >7.5 ● D-penicillamine, chelating agent binds to cysteine or homocysteine; poorly tolerated, effective in dissolving & prevent recurrence when hydration & alkalinization fail ▪ N-Acetylcysteine: low toxicity, control cystinuria (lack long-term experience) ▪ Type 1 RTA: correct metabolic acidosis, replace lost Na and K+ (sodium or potassium citrate therapy) ▪ Primary hyperoxaluria: liver transplant (defective hepatic enzymes); ideally, performed before renal failure ▪ Severe cases: kidney transplant Batch Clingy DISEASE 109 hydration status, effect of obstruction on the kidney → alter result ● Hypercalciuria → studies of Ca excretion w/ dietary Ca restriction & Ca loading are necessary Calcium Oxalate and Calcium Phosphate Calculi ▪ Most common metabolic abnormality: normocalcemic hypercalciuria; 30-60% of children w/ Ca stones ▪ Hypercalciuria: absorptive, renal or resorptive ● Primary disturbance in absorptive hypercalciuria: intestinal hyperabsorption of Ca ● Renal hypercalciuria: impaired reabsorption of Ca → mild hypocalcemia → ↑PTH → ↑intestinal absorption of Ca & ↑mobilization of Ca stores ● Resorptive hypercalciuria: uncommon; primary hyperparathyroidism → ↑PTH secretion ▪ Hyperoxaluria: ↑Ca oxalate precipitation ● Primary hyperoxaluria: rare autosomal recessive disorder ● Secondary hyperoxaluria: more common, ↑intake of oxalate (tea, coffee, spinach, rhubarb), precursors (vit C), pyridoxine deficiency, intestinal malabsorption ▪ Enteric hyperoxaluria: IBD, pancreatic insufficiency, biliary disease → GI malabsorption of fatty acids w/c bind intraluminal Ca → form salts excreted in feces ● Normally, Ca forms complex w/ oxalate → ↓oxalate absorption ▪ Hypocitraturia: ↓excretion of citrate (inhibitor of Ca stone: form complex w/ Ca, ↑solubility, inhibit aggregation of Ca phosphate & Ca oxalate crystal) ● Idiopathic; chronic diarrhea, intestinal malabsorption & RTA ▪ Type 1 RTA: urine pH never <5.8, hyperchloremic hypokalemic acidosis → nephrolithiasis, nephrocalcinosis, muscle weakness, osteomalacia ▪ Cystic fibrosis ▪ Hyperuricosuria: epitactic growth of Ca oxalate around nucleus of uric acid crystals; action of uric acid as counterinhibitor of urinary mucopolysaccharides ▪ Heterozygous cystinuria: mechanism unknown; may be similar to uric acid ▪ Sarcoidosis: ↑sensitivity to vit D3 → ↑absorption of Ca from GI ▪ Lesch-Nyhan syndrome: excessive uric acid synthesis ▪ Immobility: hypercalciuria by mobilization of Ca stores ▪ High dose steroids: hypercalciuria & Ca oxalate precipitation ▪ Furosemide: NICU → severe hypercalciuria, urolithiasis, nephrocalcinosis ▪ Idiopathic: complete metabolic evaluation performed prior to this dx RENAL TB (Nelson’s + PPS TB Guidelines) ▪ Rare in children, incubation several years (up to 1520yr after primary infection): ● Local study on extrapulmonary TB, constituted 3% ● GU tract TB mostly seen in >7yo ▪ Tubercle bacilli reach kidney during lymphohematogenous dissemination → tubercles in glomeruli → caseating sloughing lesions discharge TB bacilli into tubules PPS Oral Exam Reviewer 2020 ▪ Infection can be unilateral or bilateral, can spread caudad to bladder ▪ Clinically silent in early stages → sterile pyuria and microscopic hematuria ▪ Dysuria, flank or abdominal pain, and gross hematuria develop as disease progresses ▪ (PPS: insidious in onset; 75% sx related to urinary tract inflammation) ▪ Suspected in presence of destructive pulmonary TB w/ persistent, painless, sterile pyuria, assoc albuminuria & hematuria ▪ Urine cultures for M tuberculosis: positive 80–90% of cases (PPS: repeated culture advisable; complete urologic investigation mandatory for those w/ persistent pyuria) Cystine Calculi ▪ Cystinuria: rare autosomal recessive disorder of epithelial cells of renal tubule → prevent absorption of 4 dibasic amino acids (cystine, ornithine, arginine, lysine) → excessive urinary excretion → ↓solubility of cystine ▪ Acidic urine → ↑rate of precipitation Struvite Calculi ▪ UTI (urea-splitting organisms: Proteus, occ Klebsiella, E coli, Pseudomonas) → urinary alkalinization, ↑production of ammonia → precipitation of magnesium ammonium phosphate (struvite) & Ca phosphate ▪ calculi → staghorn configuration → obstruction, perpetuate infection, gradual kidney damage ▪ metabolic abnormalities, neuropathic bladder (particularly undergone urinary tract reconstructive procedure) Uric Acid Calculi ▪ Hyperuricosuria w/ or w/out hyperuricemia ▪ Stones: radiolucent ▪ Inborn errors of purine metabolism, Lesch-Nyhan syndrome, G6PD, short-bowel syndrome, rapid turnover of purine w/ tumors & myeloproliferative diseases ▪ Uric acid calculi/“sludge” fill entire upper collecting system → renal failure, anuria ▪ Present w/in Ca containing stones → more than one predisposing factor for stone formation Indinavir Calculi ▪ Indinavir sulfate: protease inhibitor for HIV infection → 4% acquire symptomatic nephrolithiasis ● 12% excreted unchanged in urine → rectangles, fan-shaped or starburst crystals ● Soluble at pH <5.5; urinary acidification w/ ammonium chloride or ascorbic acid ▪ Mostly radiolucent Nephrocalcinosis ▪ Ca deposition w/in the renal tissue ▪ Furosemide (premature neonates → hypercalciuria), distal RTA, hyperparathyroidism, medullary sponge kidney, hypophosphatemic rickets, sarcoidosis, cortical necrosis, hyperoxaluria, prolonged immobilization, Cushing syndrome, hyperuricosuria, monogenetic causes of HTN, renal candidiasis ▪ Superinfection by other bacteria common → PPS delay recognition of TB 2HRZE/4HR ▪ Hydronephrosis or ureteral strictures ▪ Category I: Pulmonary or Extra-pulmonary TB, new (whether ▪ PPS: children whose urine reveal presence of bacteriologically-confirmed or clinically-diagnosed) except CNS/bones or tubercle bacilli are considered to be highly joints infectious & should be isolated until urine 2HRZES/1HRZE/5HRE sterile ▪ Category II: Pulmonary or Extra-pulmonary TB, previous treated drugsusceptible TB (whether bacteriologically-confirmed or clinically- Batch Clingy Please see Table 562.1 110 ▪ Organism recovered from urine in cases of miliary TB & some w/ PTB in absence of renal parenchymal dx ▪ True renal TB: small caseous in renal parenchyma ▪ Infection spreads locally to ureters, prostrate or epididymis ▪ Genital tract TB: uncommon in prepubescent boys & girls ● Lymphohematogenous spread, direct spread from intestinal/urinary tract or bone ● PPS: frequently, other forms of TB w/ initial infection (pleural effusion) ● Adolescent girls: can develop genital tract TB during primary infection → fallopian tubes 90–100% of cases; endometrium 50%; ovaries 25%; cervix 5% ▪ Non–anion gap (hyperchloremic) metabolic acidosis in normal/near-normal GFR Genital TB: systemic manifestation usually absent, CXR normal in majority, TST usually reactive ▪ Adolescent female: lower abdominal pain, dysmenorrhea or amenorrhea, lower abdominal mass, free peritoneal fluid; PPS: considered in mothers when managing newborns suspected of infection (congenital TB) in perinatal period; TB of external genitalia → consider child abuse ▪ Adolescent male: epididymitis or orchitis; present as painless, unilateral nodular swelling of the scrotum; involvement of glans penis extremely rare; PPS: may develop primary TB of penis after circumcision presenting as massive inguinal lymphadenopathy ▪ Genital abnormalities, (+) TST, adolescent → genital tract TB 4 main types: ▪ Proximal (type II) (impaired HCO3 reabsorp) ▪ Classic distal (type I) (failure to secrete acid) ▪ Hyperkalemic (type IV) ▪ Combined proximal and distal (type III) ▪ Hx: growth & devt, diarrhea, family hx of mental retardation, failure to thrive, ESRD, infant deaths, or miscarriages ▪ PE: growth parameters and volume status, dysmorphic features ▪ Patient with suspected RTA → confirm normal anion gap metabolic acidosis, elec imbalance, assess renal fx, r/o other causes of bicarb loss ● Metabolic acidosis in diarrheal dehydration: common, improve w/ fluid correction; depletes total-body bicarb stores, persistent acidosis despite volume restoration → allow several days for reconstitution of total-body bicarb stores before full eval for RTA ● Acidosis persists → serum elecs, BUN, Ca+, phosphorus, crea, pH; traumatic blood draws, small vol of blood in adult-size tubes, prolonged specimen transport time at room temp → falsely low bicarb levels, ↑serum K+ ● Blood anion gap: [Na+] − [Cl− + HCO3−] - <12: absence of anion gap; >20: presence of anion gap - anion gap: lactic acidosis, DKA, IEM, ingested toxins - tachypnea → ABG → possibility of mixed acid–base disorder ▪ Urine pH → distal (pH <5.5) vs proximal (pH >6.0) ● Urine anion gap ([urine Na+ + urine K+] − urine Cl−) confirm diagnosis of distal RTA ● (+) gap: deficiency of ammoniagenesis → possibility of distal RTA ● (-) gap: proximal tubule bicarb wasting (or GI wasting) diagnosed) ▪ Prognosis: depend on underlying disease ▪ Treated isolated proximal or distal RTA → improvement in growth, provided serum bicarb in normal range ▪ Systemic illness & Fanconi syndrome → growth failure, rickets, sx related to underlying dx ▪ Rickets: bone pain, growth retardation, osteopenia, pathologic fractures Finding Plasma [K+] Urine pH with acidosis Urine net charge Fanconi lesion Fractional bicarbonate excretion U−BPco2 Response to therapy Associated features ▪ UA: glycosuria, proteinuria, hematuria → global tubular damage or dysfunction ▪ Random or 24hr urine Ca and crea → identify hypercalciuria ▪ Renal UTZ: identify underlying structural abnormalities, obstructive uropathies, nephrocalcinosis ▪ Mainstay of therapy in all forms: Bicarbonate replacement Proximal Classic Distal Low <5.5 Negative Present w/ acquired pRTA >10–15% during alkali therapy Normal Least responsive Low >5.5 Positive Absent Fanconi syndrome 2–5% Low Responsive Nephrocalcinosis/ hyperglobulinemia Generalized Distal Dysfunction High <5.5 or >5.5 Positive Absent 5–10% Low Less responsive Renal insufficiency Batch Clingy RENAL TUBULAR ACIDOSIS ▪ Acid–base balance by kidneys: reabsorption of filtered HCO3− & excretion of H+ ● H+ secretion from tubule cells into lumen → key in reabsorption of HCO3− and formation of titratable acid (H+ bound to buffers, HPO42−) & NH4+ ions ● Loss of filtered HCO3− equivalent to addition of H+ to body; all filtered HCO3− absorbed before dietary H+ can be excreted ● ~90% of filtered bicarbonate absorbed in proximal tubule; 10% in distal segments, mostly thick ascending limb and outer medullary collecting tubule ● In proximal tubule & TAL of loop of Henle, H+ from water → secreted by the Na+-H+ exchanger on luminal membrane; H+ combines w/ filtered HCO3− → formation of H2CO3 → splits into water and CO2 in presence of carbonic anhydrase IV ● CO2 diffuse freely back into cell → combine with OH− (from H2O) to form HCO3− in presence of carbonic anhydrase II → returns to systemic circulation via Na+HCO3 − cotransporter at basolateral membrane of cell ● In collecting tubule, H+ secreted into lumen by H+ATPase (adenosine triphosphatase) and HCO3− returned to systemic circulation by HCO3–-Cl− exchanger on basolateral membrane ● H+ secreted proximally and distally in excess of filtered HCO3− → excreted in the urine as titratable acid (H2PO4−) or as NH4+ ▪ AFB of large volumes of urine sediment: positive 50–70% of cases ▪ TST nonreactive: up to 20% of patients ▪ Pyelogram or CT scan: mass lesions, dilation of the proximal ureters, multiple small filling defects, and hydronephrosis if ureteral stricture present PPS Oral Exam Reviewer 2020 111 DISTAL (TYPE I) RTA ▪ Result from damaged/impaired functioning of one or more transporters or proteins involved in the acidification process (including H+/ATPase, HCO3−/Cl− anion exchangers, or components of the aldosterone pathway) ▪ Shared features w/ pRTA: non–anion gap metabolic acidosis, growth failure ▪ Distinguishing features: nephrocalcinosis, hypercalciuria; phosphate & massive bicarb wasting generally absent ACUTE KIDNEY INJURY Abrupt loss of kidney function → ↓GFR, accumulation of waste products HYPERKALEMIC (TYPE IV) RTA ▪ Impaired aldosterone production (hypoaldosteronism) or impaired renal responsiveness to aldosterone (pseudohypoaldosteronism) ▪ Aldosterone → direct effect on H+/ATPase (responsible for hydrogen secretion); potent stimulant for K+ secretion in collecting tubule ● no aldosterone → hyperkalemia; further affects acid–base status → inhibiting ammoniagenesis and H+ excretion ▪ Prerenal (prerenal azotemia): ↓effective circulating arterial volume (dehydration, sepsis, hemorrhage, severe hypoalbuminemia, cardiac failure) → inadequate renal perfusion, ↓GFR; absent structural kidney damage ▪ Intrinsic renal: renal parenchymal damage (sustained hypoperfusion, ischemic/hypoxic injury, nephrotoxic insults; assoc w/ cardiac, oncologic, urologic, renal, and genetic disorders or prematurity) ● severe and prolonged ischemic/hypoxic injury & PPS Oral Exam Reviewer 2020 ▪ Growth failure in the 1st year of life; polyuria, dehydration (from sodium loss), anorexia, vomiting, constipation, hypotonia ▪ Fanconi syndrome: rickets sec to phosphate wasting ▪ UA: isolated pRTA generally unremarkable (urine pH acidic (<5.5) because distal acidification intact); Fanconi syndrome: phosphaturia, aminoaciduria, glycosuria, uricosuria, ↑urinary sodium or potassium ▪ Isolated autosomal recessive pRTA: mutation in gene encoding Na bicarbonate cotransporter NBC1; ocular abnormalities (band keratopathy, cataracts, glaucoma, often leading to blindness), short stature, enamel defects of teeth, intellectual impairment, occ basal ganglia calcification ● Autosomal dominant pattern: identified in a single pedigree w/ 9 members: hyperchloremic metabolic acidosis; normal ability to acidify urine &renal function, growth retardation. ▪ Cystinosis: mutations in CTNS gene; defect of cysteine metabolism → accumulation of cystine crystals in major organs of body (kidney, liver, eye, brain) ● Infantile or nephropathic cystinosis: most severe form; present in 1st or 2nd year of life w/ severe tubular dysfunction, growth failure → not treated, ESRD by end of 1st decade ▪ Lowe syndrome (oculocerebrorenal syndrome of Lowe): rare X-linked disorder; mutations in the OCRL1 gene; congenital cataracts, mental retardation, Fanconi syndrome ▪ Impaired H+ ion excretion → urine pH cannot be reduced to <5.5 despite severe metabolic acidosis ▪ Loss of Na bicarbonate distally (lack of H+ to bind to in tubular lumen) → ↑Clabsorption → hyperchloremia ▪ Inability to secrete H+ → compensated for by ↑K+ secretion distally → hypoK+ ▪ Hypercalciuria → nephrocalcinosis/nephrolithiasis ▪ Chronic metabolic acidosis → impaired urinary citrate excretion → hypocitraturia → ↑risk of Ca deposition in tubules ▪ Mobilization of organic components from bone to serve as buffers to chronic acidosis → bone dx ▪ Medullary sponge kidney: relatively rare sporadic disorder ● cystic dilation of terminal portions of collecting ducts as they enter renal pyramids; UTZ: medullary nephrocalcinosis; normal renal function through adulthood → complications: nephrolithiasis, pyelonephritis, hyposthenuria, distal RTA; assoc w/ Beckwith-Wiedemann syndrome or hemihypertrophy ▪ Aldosterone deficiency → adrenal gland disorders (Addison disease, some forms of CAH) ▪ Aldosterone unresponsiveness: more common cause in children → can occur transiently (acute pyelonephritis, acute urinary obstruction) or chronically (obstructive uropathy) ▪ Growth failure in 1st few years of life; polyuria & dehydration (from salt wasting); rarely (esp pseudohypoaldosteronism type 1) life-threatening hyperK+ ▪ Obstructive uropathy → acute pyelonephritis ▪ Hyperkalemic non–anion gap metabolic acidosis ▪ Alkaline/acidic urine ▪ ↑Urinary Na w/ inappropriately ↓urinary K+ (lack aldosterone effect) ▪ Defined as: ↑serum creatinine by ≥0.3 mg/dL from baseline within 48h or ↑serum creatinine to ≥1.5times baseline within the prior 7 days or urine volume ≤ 0.5 mL/kg/hr for 6 hr ▪ Carefully taken hx → define cause of AKI ● Infant, 3-day hx of vomiting & diarrhea → prerenal (volume depletion) or consider HUS ● 6yo, recent pharyngitis, periorbital edema, HTN, gross hematuria → intrinsic (postinfectious GN) ● Critically ill, hx of protracted hypotension/exposure to nephrotoxic meds → ATN Rickets: hypophosphatemia & phosphaturia from generalized proximal tubular dysfunction ▪ Bicarb & oral phosphate → heal rickets ▪ VitD – offset 2o hyperparathyroidism complicating oral phosphate therapy ▪ Require large quantities of bicarbonate → up to 20 mEq/kg/24hr ▪ Na bicarbonate or Na citrate (Bicitra or Shohl solution) ▪ Fanconi syndrome: phosphate supplementation Bone demineralization w/out overt rickets ▪ Dissolution of bone → Ca carbonate in bone → serves as buffer against metabolic acidosis ▪ Administration of sufficient bicarb → reverse acidosis → reverse bone dissolution & hypercalciuria ▪ Base rqmt: 2-4 mEq/kg/24hr (individualize) ▪ Monitor hypercalciuria ● Symptomatic (recurrent eps of gross hematuria), nephrocalcinosis, nephrolithiasis → thiazide diuretic → ↓urine Ca excretion ▪ Chronic tx for hyperK+ → sodium–potassium exchange resin (sodium polystyrene sulfonate) Please see Table 550.5 ▪ Functional AKI induced by dehydration → usually reversible w/ early fluid therapy ▪ Mortality rate: depends on underlying dx process rather than on renal failure itself ● AKI caused by renal-limited condition: ↓mortality rate (<1%) ● AKI related to multiorgan failure: ▪ Volume status → no overload/cardiac failure → isotonic saline, 20mL/kg over 30min → hypovolemic patient void w/in 2 hr → failure suggests intrinsic/postrenal AKI ▪ Diuretic: after adequate BV established → furosemide 2-4mg/kg single IV dose (alternative: bumetanide 0.1mg/kg) → UO not improved → continuous diuretic infusion ▪ ↑renal cortical blood flow → dopamine 2-3µg/kg/min (no controlled data but clinicians use) ▪ mannitol: prevent pigment (myoglobin, hemoglobin)-induced renal Batch Clingy PROXIMAL (TYPE II) RTA ▪ Inherited and persistent from birth or occur as transient phenomenon during infancy ▪ Occurs as component of global proximal tubular dysfunction or Fanconi syndrome (low-molecular-weight proteinuria, glycosuria, phosphaturia, aminoaciduria, pRTA) ● ifosfamide (Wilms tumor & other malignancies) → secondary Fanconi syndrome 112 nephrotoxic insult → ATN → most often critically ill infants & children → pathology: tubular cell necrosis; mechanism of injury: alteration in intrarenal hemodynamics, tubular obstruction, passive backleak of glomerular filtrate across injured tubular cells into peritubular capillaries ● tumor lysis syndrome: specific form related to spontaneous or chemotherapy-induced cell lysis in lymphoproliferative malignancies → primarily caused by obstruction of tubules by uric acid crystals ● acute interstitial nephritis: result of hypersensitivity rxn to therapeutic/infectious agent ▪ Postrenal: obstruction of urinary tract ● 2 functioning kidneys: obstruction must be bilateral to result in AKI → relief of obstruction → recovery, except w/ renal dysplasia or prolonged obstruction ● Neonate, hx of hydronephrosis on prenatal UTZ, palpable bladder → congenital urinary tract obstruction (probably related to posterior urethral valves) ▪ PE: careful attention to volume status ● Tachycardia, dry mucous membrane, poor peripheral perfusion → inadequate circulating volume → prerenal AKI ● HTN, peripheral edema, rales, cardiac gallop → volume overload → intrinsic AKI from GN or ATN ● Rash, arthritis → SLE or HSP nephritis ● Palpable flank masses → renal vein thrombosis, tumors, cystic disease, urinary tract obstruction ▪ Anemia: dilutional or hemolytic (SLE, renal vein thrombosis, HUS) ▪ Leukopenia (SLE, sepsis) ▪ Thrombocytopenia (SLE, renal vein thrombosis, sepsis, HUS) ▪ Hyponatremia (dilutional) ▪ Metabolic acidosis ▪ ↑Serum BUN, crea, uric acid, potassium, phosphate (diminished renal function) ● Serum creatinine: measure kidney function but insensitive & delayed measure following AKI ▪ Hypocalcemia (hyperphosphatemia) ▪ ↓ Serum C3 level (postinfectious GN, SLE, MPGN) ▪ Antibodies detected: streptococcal (PSGN), nuclear (SLE), neutrophil cytoplasmic (granulomatosis with polyangiitis, microscopic polyarteritis), glomerular basement membrane (Goodpasture disease) ▪ Hematuria, proteinuria, RBC or granular urinary casts: intrinsic AKI (glomerular dx, ATN) ▪ WBC and WBC casts w/ low-grade hematuria & proteinuria: tubulointerstitial dx ▪ Urinary eosinophils: drug-induced tubulointerstitial nephritis ▪ Urinary indices: prerenal vs intrinsic ● Prerenal AKI: ↑specific gravity (>1.020), ↑urine osm (UOsm >500mOsm/kg), ↓urine sodium (UNa <20mEq/L), fractional excretion of sodium <1% (<2.5% in neonates) ● Intrinsic AKI: specific gravity of <1.010, ↓urine osm (UOsm <350 mOsm/kg), ↑urine sodium (UNa >40mEq/L), fractional excretion of sodium >2% (>10% in neonates) ▪ CXR: cardiomegaly, pulmonary congestion (fluid overload), pleural effusions ▪ renal UTZ: hydronephrosis/hydroureter (urinary tract obstruction), nephromegaly (consistent w/ intrinsic renal disease) ▪ renal biopsy: determine precise cause of AKI (patient not have clearly defined pre/postrenal AKI) ↑mortality rate (>50%) ▪ Recovery of renal function: depend on disorder that precipitated AKI ▪ Sequelae of AKI → chronic renal insufficiency, HTN, RTA, urinary concentrating defect INDICATIONS FOR DIALYSIS 1. Anuria/oliguria 2. Volume overload with HTN/ pulmonary edema refractory to diuretic therapy 3. Persistent hyperK+ 4. Severe metabolic acidosis unresponsive to mgt 5. Uremia (encephalopathy, pericarditis, neuropathy) 6. Ca:phosphorus imbalance w/ uncontrolled hypoCa tetany 7. Additional: inability to provide adequate nutrition due to severe fluid restriction Intermittent hemodialysis Relatively stable hemodynamic status; 3-4hr sessions using pump-driven extracorporeal circuit & large central venous catheter; 37times/wk Peritoneal dialysis Neonates & infants; hyperosmolar dialysate dwell for 45-60min, drained by gravity, repeated for 8-24hr/day (based on F&E), anticoagulation not necessary; surgically/percutaneously placed peritoneal dialysis catheter; contraindicated w/ significant abdominal pathology Please see Table 550.4 Urinalysis, Urine Chemistries and Osmolality in AKI PPS Oral Exam Reviewer 2020 CRRT Unstable, concomitant sepsis, multiorgan failure in ICU; continuous (24hr/day) using specialized pump-driven machine; doublelumen catheter (IJV/femoral vein) failure ▪ no response to diuretic challenge → discontinue; fluid restriction ● Relatively normal intravascular volume: 400mL/m2/24hr (insensible losses) plus fluid equal to UO for that day ● Extrarenal (blood, GI tract) fluid losses: replaced, mL for mL, w/ appropriate fluids ● Markedly hypervolemic patients: further fluid restriction → omit replacement of insensible fluid losses, UO, extrarenal losses ● Fluid intake, urine/stool output, body weight, serum chem monitored daily ▪ Hyperkalemia (>6 mEq/L) ● Earliest ECG change: peaked T waves → ffd by widened QRS intervals, ST segment depression, ventricular arrhythmias, cardiac arrest ● Exogenous sources (dietary, IV, TPN) eliminated ● Sodium polystyrene sulfonate resin (Kayexalate) 1g/kg, orally or by retention enema ● More severe elevation (>7 mEq/L) esp w/ ECG change → emergency measures Calcium gluc 10% sol, 100mg/kg/dose (max 3000mg/dose) Sodium bicarbonate, 1-2mEq/kg IV, over 5-10min Regular insulin, 0.1 units/kg (with glucose 50% solution, 1 mL/kg, over 1hr) ▪ Mild metabolic acidosis: retention of H+ ions, phosphate, sulfate; rarely requires treatment ● Severe (arterial pH <7.15; serum bicarb <8mEq/L) or contributes to hyperK+ → correct partially: IV bicarbonate to raise arterial pH to 7.20 (approximates serum bicarb 12 mEq/L); remainder of correction: oral Na bicarbonate after normalization of serum Ca and phosphorus ● IV bicarb → ↓ionized Ca conc → precipitate tetany ▪ Hypocalcemia: primary tx by ↓phosphorus level (↓diet phosphorus, phosphate binders); IV Ca not given except tetany ▪ Hyponatremia: dilutional → fluid restriction; hypertonic (3%) saline for symptomatic hypoNa+ or serum Na <120mEq/L acute correction of serum Na to 125 mEq/L mEq sodium required = 0.6 x wt in Kg x (125 – serum Na) ▪ GI bleeding: uremic platelet dysfunction, ↑stress, heparin exposure (hemodialysis or CRRT) → H2 blockers (ranitidine) ▪ HTN: hyperreninemia, expansion of extracellular fluid volume ● Salt & water restriction, diuretic administration ● Rapid: isradipine (0.05-0.15mg/kg/dose, max 5mg QID) ● Longer-acting oral agents for maintenance: - CCB (amlodipine 0.1-0.6mg/kg/24hr QD or BID) - β blockers (labetalol 4-40mg/kg/24hr BID or TID) ● Severe symptomatic HTN (urgency or emergency) - continuous infusions of nicardipine (0.5-5.0 µg/kg/min), Na nitroprusside (0.5-10.0µg/kg/min), labetalol (0.25-3.0mg/kg/hr), esmolol (150-300 µg/kg/min) ▪ seizure: benzodiazepines (most effective for acute control) → tx precipitating cause ▪ mild dilutional anemia (hgb 9-10g/dL) ● HUS, SLE, active bleeding, prolonged AKI → pRBC transfusion hgb falls below 7g/dL ● Hypervolemic → slow BT (4-6hr; pRBC 10 mL/kg) ● Fresh, washed RBC: ↓hyperkalemia & risk of sensitization Batch Clingy (BUN, Crea), dysregulation of extracellular volume & electrolyte homeostasis 113 ▪ Clinical presentation depends on underlying etiology and CKD stage ▪ CAKUT & some genetic forms of renal disease (familial nephronophthisis): growth failure, vomiting, polyuria, polydipsia ▪ UTI: common w/ urologic abnormalities ▪ Glomerular forms of CKD: edema, HTN, hematuria, proteinuria; severe GN, malnutrition ▪ Develop uremic symptoms (worsening fatigue, weakness, nausea, vomiting, anorexia, poor sleep patterns), edema, HTN, fluid overload, regardless of cause ▪ PE: focus on overall growth & devt, eval of BP, skin (pallor), extremities (edema; bony abnormalities of rickets seen in untreated renal osteodystrophy) Please see Fig 550.2 ▪ ↑BUN & serum crea; hyperK+, hypoNa+ (sec to either renal salt wasting vs volume overload), hyperNa+ (loss of free water), acidosis, hypocalcemia, hyperphosphatemia, ↑uric acid ▪ Heavy proteinuria → hypoalbuminemia ▪ CBC: normochromic, normocytic anemia; dyslipidemia ▪ GN: hematuria & proteinuria; congenital lesions such as renal dysplasia, ↓sp gravity w/ minimal other abnormalities ▪ Renal function: estimated by GFR; inulin clearance → gold standard (no longer readily available); other methods: iohexol or various radioisotopes (99mTc-DTPA, 51Cr-EDTA, 125Iothalamate) ▪ Estimating GFR by endogenous markers (creatinine and/or cystatin C) most utilized eGFR (mL/min/1.73m2) = 0.43 × height (cm)/serum creatinine (mg/dL) ▪ CKD Mineral & Bone Disease (CKD-MBD): systemic disorders of Ca, phosphorus, PTH, & vit D metabolism → bone disorders (renal osteodystrophy), vascular and soft tissue calcification ● High-turnover bone disease (osteitis fibrosa cystica): most common; hypoCa, hyperphosphatemia, ↑ALP & PTH values, Xray: subperiosteal bone resorption, metaphyseal widening; bone pain, fractures w/ minor trauma; rachitic changes, varus & valgus deformities of long bones, SCFE ● Low-turnover bone disease (adynamic renal osteodystrophy): PTH oversuppression, hyperCa, ↓ALP; pediatric dialysis px receiving tx for secondary hyperparathyroidism ● Vascular calcification ▪ Plans for RRT initiated at stage 4 CKD (GFR <30 mL/min/1.73m2) ▪ Dialysis initiation considered as GFR approaches 10-15mL/min/BSA ▪ 85% of children from birth-5yr: peritoneal dialysis → excess body water removed by osmotic gradient created by relatively ↑dextrose conc in dialysis fluid; wastes removed by diffusion from peritoneal capillaries into dialysis fluid ● Continuous ambulatory PD or automated therapy using cycler ● Cycler-driven PD: allow uninterrupted day of activities (↓school interruption), ↓number of dialysis catheter connections & disconnections (↓risk of peritonitis), less strict fluid & dietary restriction, ↓time required to perform dialysis (↓caregiver fatigue) ● PD not as efficient as hemodialysis, done at least 6x/wk; CI: anatomic abnormalities (surgical adhesions, omphalocele, gastroschisis, bladder exstrophy), peritoneal injury (sec to previous severe peritoneal infections), lack of appropriate caregiver ▪ 50% of children ≥13yr: hemodialysis ● Performed in hospital or OPD; IJV preferred catheter site ● Intensified HD programs (short daily HD, intermittent nocturnal HD, daily nocturnal HD): improved control of BP, fluid overload, phosphorus, anemia, improved growth; CI: PPS Oral Exam Reviewer 2020 ▪ Variable timing of CKD progression (minimal renal injury to onset of ESRD) ▪ Nonmodifiable risk factors assoc w/ more rapid progression: older age, glomerular etiology, CKD severity, onset of puberty ▪ Potential modifiable risk factors: ↑BP, persistent nephrotic range proteinuria, anemia, dyslipidemia, no ACE/ARB use ▪ Prompt tx of infection & dehydration → minimize additional loss of renal parenchyma ▪ Potentially beneficial: avoid tobacco, prevention of obesity, avoid potential nephrotoxic meds (NSAIDs, illegal street drugs, herbal and/or homeopathic meds) ▪ Dialysis-associated infection (peritonitis, hemodialysis-related bloodstream infections): leading cause of hospitalization, 2nd-leading cause of death in pediatric dialysis patients ▪ Ultimate goal: successful kidney transplant → most normal lifestyle, improved mortality & morbidity rates Batch Clingy CHRONIC KIDNEY FAILURE ▪ CKD determined by presence of kidney damage and level (or severity) of kidney function (GFR) ▪ ESRD: patients treated w/ dialysis/kidney transplantation; subset of patients with stage 5 CKD ▪ pediatric CKD prevalence ~18 per 1M ▪ Etiology: congenital, acquired, inherited, metabolic renal dx; subdivided into nonglomerular or glomerular in origin ● <5yr: congenital abnormalities of kidney & urinary tract (renal hypoplasia, dysplasia, obstructive uropathy); prenatal UTZ ● >5yr: acquired or inherited forms of GN ▪ Hyperfiltration injury: important final common pathway of glomerular destruction, independent of underlying cause ● Nephrons lost → remaining nephrons undergo structural & functional hypertrophy (↑glomerular blood flow) → compensatory hyperfiltration (temporarily preserves renal function) → ↑hydrostatic pressure, toxic effect of ↑protein traffic → ↑excretory burden ▪ Proteinuria: direct toxic effect of protein on tubular cells & recruit monocytes and macrophages → glomerular sclerosis & tubulointerstitial fibrosis ▪ Uncontrolled HTN → arteriolar nephrosclerosis; ↑hyperfiltration injury ▪ Hyperphosphatemia → Ca phosphate deposition in renal interstitium & blood vessels ▪ Hyperlipidemia: oxidant-mediated injury ▪ Progression of tubulointerstitial fibrosis → primary determinant of CKD progression ● (+) Hypervolemia or hyperkalemia, BT during dialysis ▪ Nutrition: Na, K & phosphorus restricted; protein moderately restricted, maximize caloric intake (minimize accumulation of nitrogenous wastes) ▪ Supportive: screen for and tx metabolic complications ▪ Close monitoring of blood/urine studies (quantitative proteinuria: spot/24hr UPCr ratio), clinical symptomatology ▪ Gold std for BP eval: ambulatory BP monitoring ● Masked HTN: normal office BP, abnormal ABPM; 35% of pediatric pre dialysis CKD patient; 4-fold ↑risk of LVH ▪ Nutrition: calories balanced → carbs, unsaturated fat in physiologic ranges, protein restriction not suggested → adverse effect on growth & dev; CKD st 2-5 100% DRI of vit & trace elements; water-soluble vit supplement for dialysis ▪ CKD-MBD: ↓phosphorus intake; low-phosphorus formula (Similac PM 60/40); phosphate binders (given w/ meals) at onset of hyperphosphatemia ● ergocalciferol, cholecalciferol → correct 25OH vit D insufficiency → delay onset of sec hyperparathyroidism in predialysis CKD patients, improve bone mineralization (25OH vitamin D sufficiency ≥30ng/mL) ● active vit D sterols (calcitriol, paricalcitol, doxercalciferol) → ↑Ca & phosphorus absorption from GI ↓PTH values ▪ ↑BP or edema: Na restriction, diuretic therapy, fluid restriction ▪ HyperK+: ↓K+ intake, oral alkalinizing agents, Kayexalate ▪ Metabolic acidosis: Bicitra (1mEq Na citrate/mL) or Na bicarb tab (650mg = 8mEq of base) → maintain serum bicarb ≥22mEq/L ▪ Short stature: recombinant human growth hormone (rHuGH) SQ daily → reach 50th %tile for midparental height, achieve final adult height, or undergoes kidney transplantation ▪ Anemia (inadequate erythropoietin production, renal function falls below 40 mL/min/BSA): - hgb <5% for age and gender; alternatively: - <11g/dL: 0.5-5yr - <11.5g/dL: 5-12yr - <12g/dL: F >12yr, M 12-15yr - <13g/dL: M >15yr ● iron supplement (oral/IV): transferrin saturation (TSAT) ≤20%, ferritin ≤100ng/mL ● erythropoiesis-stimulating agents (ESAs) (erythropoietin & darbepoetin alfa) ↓need for transfusion ▪ HTN: SBP or DBP >90% for age, gender, height → titrate meds to achieve SBP or DBP <50% (esp w/ proteinuria) ● Na restriction (<2g/24hr), lifestyle modifications → healthy weight ● ACE inhibitors (enalapril/lisinopril), ARB (losartan): anti-HTN irrespective of level of proteinuric renal dx, potential ability to slow CKD ● thiazide (hydrochlorothiazide, chlorothiazide), loop diuretic (furosemide): related to salt and fluid retention; thiazide become ineffective when eGFR falls below 30mL/min/BSA ● Ca channel blockers (amlodipine), β-blockers (propranolol, atenolol), centrally acting agents (clonidine) → adjunctive agent, BP cannot be controlled ▪ Immunization: receive all standard immunizations; withhold live vaccines → receiving immunosuppressive meds (make every attempt to administer live vaccine before kidney transplant); yearly influenza 114 (studies suggest CKD child might respond suboptimally to immunizations) ▪ Dose adjustment for drugs excreted by kidney → maximize effectiveness, minimize risk of toxicity → lengthening interval bet doses, ↓absolute dose or both Batch Clingy inadequate vascular access PPS Oral Exam Reviewer 2020 115 Acute UNDESCENDED TESTES (Cryptorchidism) RETRACTILE TESTES HYDROCELE INGUINAL HERNIA EPIDEMIOLOGY / PATHOGENESIS ▪ Most common disorder of sexual differentiation in males ▪ At birth, ~4.5% of males ▪ Testicular descent: 7-8mo AOG → 30% of preterm male; 3.4% at term ▪ 50% descend spontaneously 1st 3mo of life (incidence ↓1.5% at 6mo) → temporary testosterone surge (minipuberty during 1st 2mo + significant penile growth) ▪ Not descended by 4mo → remain undescended ▪ 10% bilateral ▪ Some older males: scrotal testis “ascends” (low inguinal position) → requires orchiopexy ▪ 1–2% post-hernia repair → secondary cryptorchidism: scar tissue along spermatic cord ▪ Process of descent: genetic, hormonal, mechanical factors ● Testosterone, dihydrotestosterone, MIF, gubernaculum, intraabdominal pressure & genitofemoral nerve ▪ 7-8 weeks AOG: testis develops in abdomen (ILF-3 controls transabdominal phase) ▪ 10-11 weeks AOG: Leydig cells produce testosterone: differentiation of Wolffian (mesonephric) duct → epididymis, vas deferens, seminal vesicle, ejaculatory duct ▪ 32-36 weeks AOG: begins descent → gubernaculum guides testis into scrotum → patent processus vaginalis (hernia sac) normally involutes ▪ Normally descended testes (neonatal period) → pulled into suprascrotal position by cremasteric reflex (Uptodate) ▪Accumulation of fluid in tunica vaginalis ▪1-2% of neonates ▪noncommunicating (processus vaginalis obliterated) → disappears by 1yo ▪Patent processus vaginalis → persists; small in morning, larger during day ▪Abdominoscrotal hydrocele → large, tense, extending into lower abdominal cavity (rare) ▪Older male: inflammatory condition → testicular torsion, torsion of appendix testis, epididymitis, testicular tumor, trauma ▪ Overall incidence 0.8–4.5% term; ~30% premature and LBW ▪ 99%: congenital indirect hernias (patent processus vaginalis) PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS / DIAGNOSIS ▪ Classified as ● Abdominal: nonpalpable → will not descend after 3mo of age ● Peeping: abdominal but can be pushed into the upper part of the inguinal canal ● Inguinal ● Gliding: can be pushed into scrotum but retracts immediately to the pubic tubercle ● Ectopic: superficial inguinal pouch or, rarely, perineal ▪ PE: undress child → nondominant hand: over pubic tubercle, pushed inferiorly toward scrotum → dominant hand: examine scrotum & inguinal canal, palpate testis → most are palpable just distal to inguinal canal over pubic tubercle ▪ Nonpalpable → soap test → soap applied to inguinal canal and examiner’s hand → reduces friction ▪ Pulling on scrotum pull a high inguinal testis ▪ Contralateral testicular hypertrophy (not 100% diagnostic) ▪nonpalpable testes → 50% viable testes in abdomen/high inguinal canal, 50% atrophic (w/in scrotum, sec to spermatic cord torsion in utero/vanishing testis) ▪ Sonography: identify if testis present; abdominal testis & atrophic testis not identified; inguinal/scrotal UTZ: obese males w/ nonpalpable testis ▪ CT scan: relatively accurate, radiation exposure significant ▪ MRI: more accurate; disadvantage: sedation in young ▪ Routine imaging discouraged: not 100% accurate, not add to decision making >1yo: brisk cremasteric reflex, child anxious/ticklish → testis difficult to manipulate into scrotum → examine w/ legs in relaxed frogleg position: testis manipulated into scrotum comfortably → retractile ▪ Smooth & nontender ▪ Transillumination: confirms fluid-filled nature ▪ Palpate testis → testis tumor ▪ Testis nonpalpable → UTZ: testis present and normal ▪ Compression of fluid-filled mass → completely reduces hydrocele → inguinal hernia ▪ ~50% present in 1st yr: asymptomatic/minimal sx ▪ Classic hx: intermittent groin, labial (usually upper portion), or scrotal swelling/bulge, spontaneously reduces → gradually enlarge, more persistent, difficult to reduce DIFFERENTIAL DIAGNOSIS ▪ Disorder of sex development ● Newborn phenotypic male w/ bilateral nonpalpable testes → virilized female w/ CAH ▪ Retractile testes ● misdiagnosed as undescended COMPLICATIONS / PROGNOSIS ▪ Poor testicular growth, infertility, testicular malignancy, assoc hernia, torsion of cryptorchid testis, psychological effect of empty scrotum ▪ Risk of a germ cell malignancy: 4x higher than gen population ● Peak age: 15-45yo ● Most common: seminoma (65%) (↓ by orchiopexy; after orchiopexy, nonseminomatous tumors (65%) (selftesticular exam) ▪ Acquired/ascending undescended testis: usually 4-10yo, hx of retractile testis, incomplete involution of processus vaginalis, restricted spermatic cord growth during male’s somatic growth TREATMENT ▪ Surgery at 6mo appropriate; should be tx surgically by 9-15mo (no spontaneous descent after 4mos) ▪ Orchiopexy: mobilization of testis and spermatic cord + correction of indirect inguinal hernia; OPD procedure: 98% success rate ▪ Two-stage orchiopexy: high abdominal testis ▪ Nonpalpable testis: diagnostic laparoscopy → assessment if intraabdominal ▪ Orchiectomy: difficult cases, testis atrophic ▪ Testicular prostheses: older children & adol; psychological effect ● Saline testicular implant ● Solid silicone “carving block” implants ● Placement early in childhood for anorchia (absence of both testes) *Please see Table 560.1 American Urological Association for Evaluation and Treatment and Fig 560.1 for Management Algorithm Monitor q6-12mo → 1/3 develop acquired undescended testis → orchiopexy (<7yo greatest risk) (Data not available) generally thought not ↑risk for infertility/malignancy Difficulty differentiating between undescended, retrac- tile, ectopic testes (any age) → refer to urologist (Uptodate) ▪Inguinal Hernia ▪ Varicocele: painless/dull ache; “bag of worms” ▪ Spermatocele: painless, cystic ▪ Testicular CA: painless, hard, (-) transillumination ▪ HSP: bilateral, painful, (+) purpura ▪ Epididymitis: pain, erythema ▪ Testicular torsion ▪ Scrotal hematoma ▪Meconium peritonitis ▪ Long-term risk of communicating hydrocele: inguinal hernia ▪ Congenital hydrocele → resolve by 12mo (reabsorption of hydrocele fluid) ▪ Large and tense → consider early surgical correction → verify presence of hernia; spontaneous disappearance rare ▪ Persist beyond 12-18mo → often communicating → repair ▪ Surgical correction similar to herniorrhaphy ▪ Older male: diagnostic laparoscopy → determine presence of patent processus vaginalis Incarcerated inguinal hernia ▪ 2/3 in 1st yr of life; greatest in <6mo ▪ Prematurity ↑risk ▪ Content: small bowel, appendix, omentum, ▪ Prompt repair: prevent incarceration & complications ▪ Full-term, healthy infants (<1yr): w/in 2-3wk ffg dx *based on Table 560.3/560.4 Inguinal–scrotal mass ▪ Acute hydrocele: consolable, tolerates feeding, flat inguinal region (transillumination misleading thin wall of infant intestine “+”) Batch Clingy DISEASE 116 INDIRECT INGUINAL HERNIA → 12wk AOG: processus vaginalis present → 28wk AOG: testes descend from retroperitoneum (urogenital ridge) to internal ring → 2836wk AOG: final descent → last few weeks of gestation/shortly after birth: obliteration of processus vaginalis ▪ Patency rate of PV at birth ≈80%; 1styr of life ≈40%; persistent at 2yr ≈20% of males ▪ Females: obliterates earlier ~7mo AOG (could explain ↓incidence) ▪ Involution left PV precedes right → 60% R-sided indirect inguinal hernias (30% L-sided, 10% bilateral) Ectopic testes → cordlike structures of gubernaculum pass to perineum/femoral region ▪ Reason for failure of PV closure unknown → common in cryptorchidism & prematurity COMPLETE INGUINAL HERNIA: protrusion of contents extending into scrotum ▪ Males: hernia sac contains intestines ▪ Females: often have ovary and fallopian tube DIRECT (ACQUIRED) INGUINAL HERNIA ▪ Weakness in transversus abdominis (floor of inguinal canal) ▪ Originate medial to deep inferior epigastric vessels & external to cremasteric fascia; protrudes directly through posterior wall of inguinal canal not through external ring ▪ 1/3: hx of prior indirect hernia repair (possible missed dx direct hernia/injury to floor muscles); (+) connective tissue dx: Ehlers-Danlos syndrome, Marfan syndrome, mucopolysaccharidosis (Hunter-Hurler syndrome) IMPERFORATE HYMEN FEMORAL HERNIA: medial to femoral vein, descends inferior to inguinal ligament along femoral canal → protrudes on medial aspect of thigh, below inguinal region, does not enter scrotum/labia ▪ Most common obstructive anomaly ▪ Familial occurrence reported ▪ ~1 in 1000 PPS Oral Exam Reviewer 2020 ▪ Most visible: crying, irritable, straining, coughing; after bathing or urination ▪ Hallmark sx: smooth, firm mass; emerge through external inguinal ring lateral to pubic tubercle; enlarges w/ ↑intraabdominal pressure → inspect protruding mass/examining finger invaginating the scrotum to palpate at the external ring → reduce spontaneously as child relaxes/by gentle pressure ▪ Bimanual exam: palpate internal ring per rectum, other hand gentle pressure on inguinal region over internal ring → intraabdominal viscera palpated extending through internal ring ● Quiet infant: stretch the infant out supine w/ legs extended, arms held straight above the head → infants struggle to get free → ↑intraabdominal pressure ● Older children: ask to perform the Valsalva maneuver by blowing a balloon/coughing; examine while standing, after voiding ▪ Torsion of undescended testis: painful erythematous mass; absence of gonad ipsilateral side ▪ Epididymitis/orchitis: swelling & tenderness of testis, confined to the scrotum; assoc urinary sx ▪ Suppurative inguinal lymphadenitis: superficial infected, crusted skin lesion; more inferior and lateral location Ambiguous genitalia ▪ Disorders of sexual devt → inguinal hernia often containing a gonad ▪ Female patient w/ inguinal hernia + bilateral inguinal mass, consider testicular feminization syndrome ▪ Silk glove sign: rolling spermatic cord beneath index finger at pubic tubercle → feeling of layers of hernia sac as they slide over spermatic cord colon, bladder, rarely, Meckel diverticulum; females: ovary, fallopian tube or both, rarely, uterus in infants ▪ Pressure on herniated viscera → impaired lymphatic & venous drainage → swelling → ↑compression → total occlusion of arterial supply → progressive ischemic changes → gangrene (strangulated hernia) and/or perforation ▪ Irritable, feeding intolerance, abdominal distention, pain ▪ Inconsolable, no flatus/stool → complete obstruction → bilious/feculent vomiting; bloody stool ▪ Nonreducible, tense, tender mass, erythema (extending from inguinal area to scrotum/labia) ▪ Abdominal x-ray: partial/ complete obstruction, gas w/in incarcerated bowel segments seen below the inguinal ligament or w/in scrotum. SURGICAL COMPLICATIONS ▪ Wound infection: fever & irritability 3-5days post-sx; wound warm, erythema, fluctuant; mgt: draining, antibiotics, daily wound dressing; most common: Gram(+) (Stap & Strep spp), consider MRSA; heal in 1-2wk ▪ Recurrent hernia ▪ Iatrogenic cryptorchidism: malposition of testis ▪ Injury to vas deferens & male fertility: injured from incarcerated hernia or during OR; underreported (unlikely recognized until adulthood & possibly only if bilateral) ▪ ‘E’ repair ↑risk for testicular atrophy, bowel ischemia, wound infection & recurrence than elective ▪ Sliding hernias: content adherent w/in sac, not reducible → in females: fallopian tube/ovary palpated as firm, slightly mobile, nontender mas ▪ Clinical diagnosis → reduce spontaneously in young → unequivocal PE → UTZ (distinguish hernia, hydrocele, lymphadenopathy) or refer to surgeon (Older child: ↓risk of incarceration, parents reassured & educated → plan for period of observation (take picture at home) ▪ Diagnostic laparoscopy: increasingly used for suspected hernia (particularly infants: ↑risk incarceration) ▪ 30%: testicular atrophy after incarceration <3mo ▪ 70% incarcerated requiring ‘E’ repair: <11mo ▪ Children >1yo: ↓risk of incarceration, less urgency → elective, OPD → full recovery in 48hrs ▪ Prophylactic antibiotics – used if w/ co-morbids (CHD, VPS, BPD) ▪ Preterm & LBW: controversy in timing → before NICU discharge: ↑risk incarceration but also ↑risk complications, hemodynamic instability, wound infection, recurrent hernia; open repair preferred: can be done under gen anes or regional (avoid intubation in BPD, chronic lung disease) ▪ Incarcerated, irreducible hernia without strangulation/ obstruction, peritonitis, hemodynamic instability: nonopera-tive, manual reduction under sedation → observe feeding tolerance → herniorrhaphy after 1-4days (less edema, easier handling of sac, ↓ complications) ▪ Irreducible, prolonged incarceration, peritoneal irritation, obstruction → NGT, IVF, broad-spectrum antibiotics, correct F&E imbalance → surgery ▪ Female: sliding hernias, irreducible, (-) strangulation → repair w/in 24-48hrs ▪ Necrotic ovaries & testes at OR → not predict future functionality (ischemic appearing testes: survive ~50%, testicular atrophy 2.5–15%, avoid testicular resection unless frank necrosis present) ▪ 85%: unilateral inguinal hernia → controversy in contralateral groin exploration → pro: avoid parental anxiety, 2nd surg & anes, contralateral incarceration; con: injury, ↑OR & anes time, often unnecessary OPEN INGUINAL REPAIR: standard of care; ↓rate of recurrence, vas deferens injury, testicular atrophy LAPARASCOPIC REPAIR: better cosmesis, faster recovery, greater ability to visualize & repair contralateral hernia; ↑risk assoc w/ gen anes, hemodynamic effect of abdominal insufflation (acidosis), technical challenge Post op pain: acetaminophen 24-48hr; older child: brief period of post op narcotic ▪ Newborn period & early infancy: Mucocolpos ● Bulging membrane ● From maternal estrogen stimulation of the vaginal ▪ Variants: hymenal membranes do not undergo complete resorption → microperforate, cribriform, septate-shaped ▪ Can obstruct urinary outflow ▪ Resection: prevent/relieve outflow tract obstruction ● Symptomatic, done at time of dx ● Elective excision: after age 1-2yr to puberty, Batch Clingy ● Full-term newborn infants: 3.5–5.0% ● Preterm & LBW: 9-11% ● Preterm (<28wk AOG) & VLBW: 30% ● (+) Family hx: 11.5% ▪ M>F → 8:1 ratio but ● bilateral inguinal hernias: ↑females (~25%) vs males (~12%) ▪ Rare in children ● Direct/acquired hernia 0.5–1.0% ● Femoral hernia <0.5%: more females 2:1 ratio ▪ ~50% manifest 1st yr of life (↑1st 6mo) 117 OVARIAN TORSION VULVOVAGINITIS Vulvitis External genital pruritus, burning, redness, rash ▪ Adnexal torsion (ovary and/or fallopian tube): 5th most common gynecologic emergency ▪ Children and adolescents > adults ▪ Can occur w/ normal adnexa ▪ More often: adnexa enlarged by cystic (follicular, tubal) changes or ovarian (teratoma, cystadenoma) neoplasms ▪ Torsion → venous outflow obstruction → fallopian tube/ovary swells, hemorrhagic → arterial flow interrupted → necrosis ▪ Most common gynecologic-based problem for prepubertal children: incidence 17–50% ▪ Peak age: 4-8yo ▪ Caused by inadequate OR excessive hygiene (chemical irritants) ▪ ~75% nonspecific: lack of vaginal estrogenization → atrophy, alkalinic pH; poor perianal hygiene; proximity of anus to vagina w/out geographic barriers (flattened labia, no pubic hair ▪ Infectious vulvovaginitis: fecal/respiratory pathogens (E coli, S pyogenes, S aureus, H influenzae, E vermicularis; rarely Candida spp) → transmitted by improper toilet hygiene & manually from the nasopharynx to the vagina ● STI in prepubertal children: cooperation w/ CPU → acquired after neonatal period → gonorrhea, syphilis, chlamydia virtually 100% sexual contact; HPV, HSV unclear Vaginitis Inflammation of vagina; discharge w/ or w/out odor or bleeding mucosa ● Reabsorbed if not too large/symptomatic ▪ More often diagnosed at menarche: Hematocolpos ● Menstrual fluid accumulates ● Bulging blue-black membrane ● Cyclic abdominal pain/pelvic mass ● Primary amenorrhea ● Normal secondary sex characteristics ▪ Acute lower abdominal pain: episodic or constant ▪ Nausea, vomiting, bowel/bladder sx, peritonitis ▪ Pelvic UTZ: unilateral enlargement of adnexa, with or without Doppler flow, free pelvic fluid, “whirlpool sign,” “beak sign” ▪ Hx: hygiene (wiping from front to back); exposure to chemical irritants (bath soap, bubble bath, detergents, pools/hot tubs); recent diarrhea, perianal itching, nighttime itching; possibility of foreign objects into the vagina (young child unlikely to recall) ▪ Infectious vulvovaginitis ● Perianal redness, introital inflammation, yellowgreen/mildly bloody discharge ● Child grabbing genital area or “digging” in underwear (stained yellow-brown discharge) ● Failed attempt to tx w/ antifungal med (leads to more irritation) ▪ Diaper dermatitis ● Most common dermatologic problem in infancy (1/2 of all diaper-wearing children) → moisture, contact w/ urine and feces irritates skin, colonization with Candida spp ↑severity ▪ Physiologic leukorrhea ● Neonates & peripubertal girls → white/clear/mucus discharge (physiologic effect of estrogen) ● Complaints of moisture and mucus → hygiene measures may help; reassure the px and mother hymen → interfere w/ tampon use → resection elective (patient preference) ▪ Acute pelvic pain + adnexal mass ▪ Ectopic pregnancy: (+) hcg, UTZ, (+) vaginal bleeding ▪ Ruptured ovarian cyst: UTZ (also show hemoperitoneum/free pelvic fluid), classic hx: sudden onset pelvic pain midcycle often ffg sexual intercourse ▪ Tubo-ovarian abscess: indolent course, (+) fever, UTZ (complex multilocular mass) (Uptodate) ▪ Culture: cotton swabs or urethral (Calgiswab) swabs moistened with nonbacteriostatic saline ▪ NAAT: gonorrhea, chlamydia; special media/collection procedure: Shigella, H influenzae ▪ Shigella: blood-tinged purulent discharge; Candida: common cause diaper rash, uncommon cause of vaginitis prepubertal (alkaline pH) ▪ Pinworms: transparent adhesive tape/anal swab ▪ Foreign body: serosanguineous vaginal discharge, foul odor, fails to respond to hygiene measures → vaginal irrigation, vaginoscopy ideally prior to menses; discouraged in very young (under anes) ▪ Prepubertal child: heal best if topical estrogen placed at site for a few days ▪ Not known how long adnexa remain viable ▪ Observation for viability: necrotic-appearing recover function (Doppler flow and follicular devt 6wk postop) → long-term preservation of fertility ▪ Untreated: pelvic thrombophlebitis, hemorrhage, infection, peritonitis, and autoamputation of the adnexa ▪ Prompt detorsion ▪ Cystectomy completed if possible: ↓risk of recurrence ▪ Removal of fallopian tube/ovary: reserved for grossly necrotic tissue, malignant tumors (FS) ▪ Oophoropexy (plication) of affected & contralateral adnexa: controversial ▪ Diabetic, immunocompromised, taking prolonged antibiotics: ↑risk for fungal vaginitis ▪ Untreated lichen sclerosis: labial resorption, obliteration of clitoris, narrowing of introitus, painful fissures, infection ▪ Condition improved by hygiene ▪ Specific vulvovaginitis: directed at the organism causing symptoms ▪ Nonspecific vulvovaginitis: Sitz baths, avoid harsh soaps/chemicals, tight clothing that abrades the perineum, fabric softeners; use cotton underwear, parent counseling ▪ Diaper dermatitis: hygiene measures (↑freq of diaper change, allowing infant to be diaper free, freq bathing, application of water-repellant barriers: zinc oxide); persists or classic satellite lesions of Candida → tx w/ topical antifungal ▪ Genital ulcer: pain mgt, urinary diversion w/ catheter; topical Xylocaine 2% jelly, sitz baths, good hygiene, acetaminophen (NSAIDSs avoided, possible causative link); topical steroids (clobetasol 0.05% ointment), oral for recurrent outbreak/extensive dx ▪ Lichen sclerosis: eval q6-12 for recurrence *Please see Table 564.1 Specific Vulvar Disorders in Children and Table 564.2 Antibiotic Recommendations for Specific Vulvovaginal Infections ▪ Acute genital ulcers (Please see Fig 564.3 Algorithm) ● Young adolescents not sexually active ● 1/3 associated w/ oral aphthous ulcers; painful red/white lesion evolve into sharply demarcated red- PPS Oral Exam Reviewer 2020 Batch Clingy ▪ Labial agglutination (adhesion) ● Secondary to inflammatory response + hypoestrogenic state ● Asymptomatic: seen on routine exam ● Sx: difficulty voiding, persistent infection, pain → tx 118 rimmed ulcers w/ necrotic or eschar-like base ● 10-14 days until remission ● Flu-like prodrome (fever, nausea, abdominal pain); dysuria and vulvar pain → urinary diversion ● Biopsy nondiagnostic (acute/chronic inflammatory changes) Batch Clingy ▪ Dermatoses (check skin elsewhere on body) ● Lichen sclerosis ● Vitiligo acquired skin depigmentation (autoimmune against epidermal melanocytes); test for hypothyroidism, Graves disease, Addison disease, pernicious anemia, insulin-dependent DM) ● Vulvar psoriasis PPS Oral Exam Reviewer 2020 119 Complex DISEASE EPIDEMIOLOGY PATHOGENESIS Primary HTN ▪ Older school-age, adolescents ▪ ↑ Prevalence in parallel w/ obesity epidemic PRIMARY HYPERTENSION ▪ ≥6 years, (+) family hx in a parent and/or grandparent, overweight and/or obesity (AAP) ▪ Older school age & adolescent ▪ BP values at/or only slightly above 95th %tile for age ▪ Isolated systolic HTN more consistent w/ primary HTN (diastolic HTN suggest a secondary cause) ▪ Multifactorial; obesity, genetic alterations in Ca and Na transport, vascular smooth muscle reactivity, RAAS, sympathetic nervous system overactivity, insulin resistance, ↑uric acid levels ▪ Salt-sensitive hypertension: some children/adol, ameliorated w/ wt loss and Na restriction Children w/ BP >90th %tile: 2.4-fold greater risk of HTN as adults Adolescent HTN: independent predictor of ESRD and LV dysfunction in middle-aged men Infants and young children: prevalence <1% → 2o HTN *Please see Table 472.2/472.3/472.4 Risk factors for CV disease: ▪ Obesity ▪ ↑serum cholesterol ▪ ↑Na intake ▪ Sedentary lifestyle ▪ Alcohol and tobacco use – ↑arterial wall rigidity & blood viscosity assoc w/ exposure to tobacco) RENAL/RENOVASCULAR DISEASE ▪ Renal dx (chronic GN, reflux or obstructive nephropathy, HUS, polycystic kidney dx, congenital anomalies of kidney & urinary tract) + renovascular HTN = ~90% of secondary HTN ▪ Renal parenchymal dx & renal artery stenosis → water & sodium retention sec to ↑renin secretion ▪ ↑index of suspicion: HTN in <6yo CLINICAL MANIFESTATIONS / DIAGNOSIS Primary hypertension: usually asymptomatic ▪ BP elevation mild, detected during routine exam ▪ May be obese Secondary hypertension: mild to severe BP elevation ▪ Unless sustained or rising rapidly, does not usually produce symptoms ▪ Sx reflect underlying dx – growth failure: CKD Acute severe hypertension ▪ BP elevation >st 2 HTN ▪ Severe sx represent acute target-organ injury Subclinical hypertensive target-organ injury: common in children w/ 1o HTN *Please see Table 472.5/472.6 and Fig 472.5 MANAGEMENT / PREVENTION ▪ Prevention of ↑BP → prevent of CV disease & stroke ▪ Asymptomatic mild HTN w/out target-organ damage: lifestyle modification w/ dietary changes & regular exercise ● Weight loss: obesity related HTN ● DASH diet beneficial ↓BP in adolescents – Dietary Approaches to Stop Hypertension: ↓Na intake (adult: standard diet 2300mg, low sodium diet 1500mg); ↑K+, Ca, Mg-containing food; 6-8servings whole grains, 4-5 servings fruits, 4-5servings veg per day, ↓fat dairy ● Regular aerobic physical activity: at least 30-60min most days; sedentary activities to <2hr/day Indications for Pharmacologic Therapy: Symptomatic HTN, stage 2 HTN without modifiable risk factor, HTN in DM or CKD, persistent HTN despite nonpharmacologic measures ▪ Initiate as single agent at low dose → ↑ dose until goal BP achieved, max dose reached, SE develop ▪ Add 2nd drug from diff class, complementary MOA ▪ BP control not achieved → add 3rd drug or refer Initial agents: ACEIs, ARBs, CCBs, thiazide diuretics → tailored to etiology Recommended goal: BP <90th percentile for age or <130/80 mmHg (whichever is lower) → lower for child w/ CKD ▪ ACEIs/ARBs: children with diabetes and microalbuminuria or proteinuric renal disease *Please see Table 472.8/472.9 HYPERTENSION (Nelson’s + AAP) ACUTE SEVERE HTN (accelerated HTN or hypertensive crisis) CARDIAC/VASCULAR ▪ Coarctation of aorta: discrete narrowing of aortic arch (generally level of aortic isthmus) ● Right arm BP ≥20mmHg than lower extremity BP ● Repair: infants o surgical, adol w/ angioplasty/stenting ▪ Long-segment narrowing of abdominal aorta: refractory HTN, upper extremity SBP exceeds lower extremity SBP by 20mmHg ▪ Abdominal aortic obstruction may have neurofibromatosis, Williams syndrome, Alagille syndrome, Takayasu arteritis Patients PPS Oral Exam Reviewer 2020 Ambulatory BP monitoring ▪ Records BP q20-30min for 24hrs; compute mean daytime/sleep BP, mean BP over 24hr; cuff placed in nondominant arm; journal of sleep & awake times, meds, events; feasible in ≥6-7yo ▪ Determine proportion of BP in HTN range (BP load), appropriate ↓during sleep ▪ ICU admission, arterial line for continuous BP monitoring, IV drug infusion: labetalol, nicardipine, Na nitroprusside ▪ Rapid ↓BP: interfere w/ adequate organ perfusion ▪ Stepwise reduction in pressure: not >25% of planned reduction over the 1st 8hr → gradual normalization over next 24-48hr ▪ Less severe symptoms (headache, N&V): clonidine, isradipine (if oral meds tolerated), short-acting IV hydralazine, labetalol Secondary HTN: tx underlying disease (chronic renal dx, Batch Clingy ▪ Headache, dizziness, nausea/vomiting – HTN urgency ▪ More severe cases, retinopathy, encephalopathy, cardiac failure, renal injury, seizures – HTN emergency ▪ No absolute distinction (nomenclature lead to confusion); treatment depend on clinical judgment ▪ HTN encephalopathy (generalized or PRES) – headache, vomiting, ↑ temp, visual disturbances, ataxia, ↓ LOC, imaging abnormalities, seizures ▪ ↓vision (cortical blindness), papilledema, congestive heart failure, accelerated deterioration of renal function 120 with coarctation ▪ Recoarctation in repaired patients: 4 extremity BP & echo; ambulatory BP monitoring - gold std for HTN after coarctation repair ENDOCRINE ▪ Thyroid, parathyroid, adrenal glands ▪ Hyperthyroidism: systolic HTN & tachycardia common; DBP not usually↑ ▪ Hypercalcemia (sec to hyperparathyroidism or other): ↑vascular tone, mild ↑BP ▪ Adrenocortical disorders – aldosterone-secreting tumors, Naretaining CAH, Cushing syndrome: ↑mineralocorticoid secretion ▪ Real/apparent mineralocorticoid excess → ↓renin level (± hypokalemia) ▪ Pheochromocytoma: catecholamine-secreting tumors → cardiac & peripheral vascular effects of epi & norepi; sustained rather than intermittent/exercise-induced HTN ▪ Pseudohyperaldosteronism: ↓renin ▪ Liddle syndrome: mineralocorticoid excess, glucocorticoidremediable aldosteronism DRUGS ▪ Cocaine: rapid ↑BP → seizures/intracranial hemorrhage ▪ Phencyclidine: transient become persistent in chronic abusers ▪ Tobacco ▪ Sympathomimetic agents – nasal decongestants, appetite suppressants, stimulants for ADHD: peripheral vasoconstriction, varying degrees of cardiac stimulation ▪ OCP ▪ Immunosuppressant agents: cyclosporine & tacrolimus in organ transplant recipients, effect is exacerbated by co-administration of steroids ▪ Heavy metal (lead, cadmium, mercury) (nocturnal dip, normal decrease >10% from awake values) ▪ Diagnose whitecoat HTN (↑BP in office, normal ABPM), masked hypertension (normal in office, ↑ABPM) ▪ BP pattern in high risk: CKD, solid organ transplant, DM, severe obesity ▪ Correlated w/ target organ damage than office readings ▪ AAP: performed for confirmation of HTN w/ office BP in elevated BP category for 1 year or more or with stage 1 HTN over 3 clinic visits (limited to children ≥5yr who can tolerate procedure, for whom reference data available) hyperthyroidism, pheochromocytoma, CoA, renovascular HTN) Renovascular stenosis: anti-HTN, angioplasty, surgery; bilateral renovascular HTN or renovascular dx in solitary kidney suspected → drugs acting on RAAS contraindicated (↓GFR, AKI) ▪ Recognition of ↑BP → evaluate underlying causes of HTN, comorbidities, screening for target-organ damage Birth hx: prematurity, perinatal events (UA catheterization, renal artery thrombosis, BPD) Family hx: metabolic & renal dx, early CV events, secondary HTN Growth parameters: detect chronic disease BP of 4 extremities: coarctation (thoracic/abdominal) of aorta Detect renal disease: UA, electrolytes, BUN, creatinine, CBC; renal UTZ ↑suspicion of secondary HTN: assess for discrepancies in renal size, structural abnormalities Hypokalemia: renovascular HTN, many monogenic forms (Liddle syndrome, glucocorticoid remedial aldosteronism, and apparent mineralocorticoid excess) Hyperkalemia: Gordon syndrome Renovascular HTN: assoc w/ other dx or isolated ▪ MR angiography: Sn 80%, Sp 63% ▪ CT angiography: Sn 88%, Sp 81% ▪ Intraarterial angiography needed to detect intrarenal vascular stenoses & in infants/young children (noninvasive imaging not helpful because of small vessel size) ▪ Doppler renal UTZ: poor cooperation, imaging difficulties related to obesity, operator inexperience; Sn ~60–65%, Sp 95% (AAP: normal-wt, ≥8yrs, cooperative) Screen for comorbidities ↑CV risk: dyslipidemia, glucose intolerance ▪ Non fasting lipid panel → do fasting panel if abnormal Screen for sleep-disordered breathing → assoc w/ ↑BP, particularly overweight Most common extracranial solid tumor in children NEUROBLASTOMA Most commonly diagnosed malignancy in infants >15% of mortality from cancer ▪ Median age at dx: 22 mo ▪ 90% diagnosed by 5yr ▪ Incidence: slightly ↑ in boys PPS Oral Exam Reviewer 2020 Embryonal Ca of peripheral sympathetic nervous system Derived from primordial neural crest cells, variable degrees of neural differentiation: tumors w/ primarily undifferentiated small round cells (neuroblastoma) to consisting of mature and maturing schwannian stroma with ganglion cells (ganglioneuroblastoma or ganglioneuroma) ▪ Resemble other small round blue cell tumors: rhabdomyosarcoma, Ewing sarcoma, non-Hodgkin lymphoma LVH: most common manifestation of target-organ damage ▪ LV mass measurements: indexed to height (account for effect of body size & BSA) ▪ LV mass >51g/m2.7 or LV mass >115g/BSA for boys, >95g/BSA for girls ▪ AAP: echocardiography obtained when tx w/ anti-HTN considered Localized disease: asymptomatic mass or sx from mass effect – spinal cord compression, bowel obstruction, superior vena cava syndrome Metastatic disease: fever, irritability, failure to thrive, bone pain, cytopenias, bluish SQ nodules, orbital proptosis, periorbital ecchymoses ▪ Neurologic sx: originating in superior cervical ganglion → Horner syndrome ▪ Paraspinal neuroblastoma tumors → invade neural foramina → SC and nerve root compression ▪ Paraneoplastic syndrome of autoimmune origin (opsoclonus-myoclonus–ataxia syndrome) rapid, uncontrollable jerking eye & body movements, poor coordination, LOW RISK: surgery for stage L1 & L2; observation for asymptomatic stage MS: cure rate >90% ▪ Chemotherapy/radiation for rare child w/ local recurrence → curative ▪ SC compression at dx: urgent chemo, surgery, or RT → avoid neurologic damage ▪ Stage MS: favorable prognosis ▪ Many regress spontaneously w/out therapy ▪ Chemotherapy/resection of primary tumor → not improve survival rates → infants w/ massive liver involvement & respiratory Batch Clingy HTN retinopathy: ↑carotid intima-to-media thickness, ↑vascular stiffness 121 ▪ May develop at any site of sympathetic nervous system tissue ▪ ½ arise in adrenal glands; remainder originate in paraspinal sympathetic ganglia ▪ Metastatic spread (more common in children >1yo at dx): local invasion, distant hematogenous, lymphatic routes; most common sites: regional/distant lymph nodes, long bones and skull, bone marrow, liver, and skin (lung & brain metastases rare, <3%) cognitive dysfunction ▪ Some tumors: produce catecholamines (↑sweating and HTN), vasoactive intestinal peptide (profound secretory diarrhea) ▪ Extensive tumors: tumor lysis syndrome, DIC ▪ Infants <18mo: stage MS → widespread SQ tumor nodules, massive liver involvement, limited BM disease, small 1o tumor w/out bone involvement or other metastases; can spontaneously regress Plain radiography, CT, MRI: mass or multiple masses; often contains calcification, hemorrhage Prenatal dx of perinatal neuroblastoma (sometimes possible) Tumor markers: catecholamine metabolites homovanillic acid (HVA) & vanillylmandelic acid (VMA) ↑in urine ~95% ▪ W/out biopsy: dx w/ small round blue tumor cells in BM samples + ↑HVA & VMA Metastatic evaluation: CT/MRI of chest & abdomen, bone scans (detect cortical bone involvement); at least 2 independent BMA & biopsies to evaluate marrow dx ▪ Iodine-123 metaiodobenzylguanidine (123 I-MIBG): define extent of dx ▪ MRI of spine: suspected or potential spinal cord compression; imaging of brain not routine (unless dictated by presentation) International Neuroblastoma Risk Group (INRG) Staging System (INSS): stage based on extent of disease as determined by imaging at diagnosis ▪ Extent of locoregional disease is based on specific local image-defined risk factors (IDRFs) ▪ L1 (previously INSS st 1): localized, confined to 1 body compartment w/out any IDRFs ▪ L2 (INSS st 2 & 3): localized tumors w/ presence of IDRFs ▪ M (INSS st 4): disseminated tumors w/ metastases to bones, BM, liver, distant lymph nodes, and other organs ▪ MS (previously st 4S): neuroblastoma in children <18mo, w/ dissemination to liver, skin, or bone marrow w/out bone involvement & w/ primary tumor that would otherwise be staged as L1/L2 10% occur in children (6-14yo) PHEOCHROMOCYTOMA Most common site of origin (~90%): adrenal medulla ▪ May develop anywhere along abdominal sympathetic chain, located near aorta at the level of inferior mesenteric artery or at its bifurcation ▪ Also appear in the periadrenal area, urinary bladder or ureteral walls, thoracic cavity, cervical region ▪ Vary from 1-10cm diameter ▪ R side > L side ▪ >20% bilateral PPS Oral Exam Reviewer 2020 ▪ Catecholamine-secreting tumors arising from chromaffin cells ▪ Associated w/ genetic syndromes: von Hippel-Lindau dx, as component of MEN2A and MEN2B, rarely w/ neurofibromatosis (type 1) or tuberous sclerosis ▪ Classic features of von Hippel-Landau syndrome (1 in 36,000): retinal & CNS hemangioblastomas, renal clear cell Ca, pheochromocytomas ● Germline mutations in VHL tumor-suppressor gene on chromosome 3p25-26 ▪ Mutations of RET protooncogene on chromosome 10q11.2 → MEN2A & MEN2B ● Medullary thyroid Ca & parathyroid tumors ● ~50%: pheochromocytoma (mutations at codon 634 of RET gene: ↑ risk) ▪ NF 1: mutations present in NF1 gene on chromosome 17q11.2 PROGNOSIS Varies w/ histologic features of tumor as dictated by presence & amount of schwannian stroma, degree of tumor cell differentiation, and mitosis-karyorrhexis index ▪ Detected by surveillance of known carriers (asymptomatic) ▪ HTN ← excessive secretion of metanephrines, epinephrine & norepinephrine ▪ Paroxysmal HTN (sustained rather than paroxysmal in children) ● Paroxysms: usually infrequent at first → frequent → continuous ● Between attacks, free of symptoms; during attacks, HA, palpitations, abdominal pain, dizziness, pallor, vomiting, sweating ▪ Seizures & other manifestation of HTN encephalopathy ▪ Severe: precordial pains radiate into arms, pulmonary edema, cardiac & hepatic enlargement ▪ Exacerbated by exercise, nonprescription meds containing stimulants (pseudoephedrine) ▪ Good appetite; hypermetabolic state may not gain weight → severe cachexia ▪ Polyuria, polydipsia: suggest diabetes insipidus ▪ Growth failure ▪ BP range: SBP 180-260mmHg, DBP 120-210mmHg diastolic compromise ▪ Chemotherapy/radiation alleviate sx; child requiring tx for sx: survival rate 81% INTERMEDIATE RISK: surgery, chemo, RT ▪ Chemotherapy: moderate doses of cisplatin or carboplatin, cyclophosphamide, etoposide and doxorubicin given over several mos ▪ RT: tumors w/ incomplete response to chemo ▪ L2 & M disease (both w/ favorable characteristics): >90% survival ▪ Determination of underlying biologic features: Shimada pathologic classification and MYCN gene amplification → unfavorable characteristics → more aggressive tx HIGH RISK: intensive chemotherapy, high-dose chemotherapy with autologous stem cell rescue, surgery, RT, and 13-cis -retinoic acid (isotretinoin, Accutane) ▪ Induction chemotx: cyclophosphamide, topotecan, doxorubicin, vincristine, cisplatin, and etoposide ↓ resection of residual primary tumor ↓ high-dose chemotherapy w/ autologous stem cell rescue & focal RT to tumor sites ▪ Long-term survival rates 25-35%; frequent relapses → recurrent neuroblastoma: <50% response rate to alternative regimens ▪ Remove surgically ▪ Careful preoperative, intraoperative, postoperative management → manipulation & excision → ↑ catecholamine secretion that ↑BP ↑HR ▪ Preoperative α- and β-adrenergic blockade required ▪ Tumors often multiple in children → thorough transabdominal exploration of all usual sites ▪ Appropriate choice of anes & expansion of blood volume with appropriate fluids before and during surgery critical → avoid precipitous ↓BP during OR or w/in 48hr postop ▪ Surveillance continues postop ▪ May appear malignant histologically but accurate indicators of malignancy → presence of metastatic dx or local invasiveness that precludes complete resection, or both ▪ ~10% of all adrenal pheochromocytomas are malignant → rare in childhood Batch Clingy Etiology remains unknown ▪ Familial neuroblastoma: 1–2%, younger age at dx, linked to mutations in PHOX2B & ALK genes ▪ BARD1 gene: major genetic contributor to neuroblastoma risk ▪ Assoc w/ other neural crest disorders: Hirschsprung disease, central hypoventilation syndrome, NF type 1, potentially congenital cardiovascular malformations ▪ ↑incidence in children w/ Beckwith-Wiedemann syndrome and hemihypertrophy ▪ Assoc w/ some maternal and paternal occupational chemical exposures, farming, work related to electronics; no single environmental exposure shown to directly cause 122 ▪ 30–40% found in both adrenal & extraadrenal areas or only in an extraadrenal area Pheochromocytoma: may occur in kindreds along w/ paragangliomas, particularly in head & neck ▪ Mutations in SDHB, SDHD, rarely SDHC genes (encoding subunits of mitochondrial enzyme succinate dehydrogenase) ▪ ~50% w/ SDHB → malignant Assoc w gastrointestinal stromal tumors (GISTs; association termed CarneyStratakis dyad) and/or pulmonary chondromas (CarneyStratakis triad) & adrenocortical tumors; heterogenous genetic etiologies, often involve mutations in SDH gene ▪ Ophthalmologic exam: papilledema, hemorrhages, exudate, arterial constriction ▪ Urine: protein, few casts, occ glucose; gross hematuria suggest tumor is in bladder wall ▪ Occ polycythemia ▪ Normally, plasma norepinephrine derived from both adrenal gland & adrenergic nerve endings; epinephrine derived primarily from adrenal gland ● In contrast to adults (↑norepinephrine & epinephrine), children predominantly excrete norepinephrine in urine ● Urinary excretion of metanephrines (particularly normetanephrine) ↑ ● Daily urinary excretion by unaffected children ↑ w/ age ▪ ↑Urinary VMA (3-methoxy-4-hydroxymandelic acid), major metabolite of epinephrine and norepinephrine (vanilla-containing foods & fruits: falsely ↑ no longer routinely measured) ▪ ↑ Plasma free catecholamines and metanephrines ▪ Best Sn & Sp: plasma normetanephrine using gender-specific pedia reference; next best: plasma norepinephrine (plasma metanephrine & epinephrine not reliably ↑ in children) ● Avoid caffeinated drinks, acetaminophen (interfere w/ plasma normetanephrine assays); sample obtained from indwelling IV catheter, avoid acute stress assoc w/ venipuncture ▪ Pediatric malignant pheochromocytomas more frequently in extraadrenal sites; often assoc w/ mutations in the SDHB gene ▪ Prolonged ff-up: functioning tumors at other sites manifest many years after initial OR ▪ Exam relatives of affected patient → reveal asymptomatic individuals w/ unsuspected tumors ▪ CT/MRI: localize most tumors in adrenal gland; extraadrenal tumors difficult to detect ▪ 123 MIBG: taken up by chromaffin tissue anywhere in body; useful for localizing small tumors ▪ PET-CT/PET-MRI w/ MIBG, DOPA, succinate, or FDG: highly Sn; for difficult to localize tumors ▪ Venous catheterization w/ sampling of blood at different levels for catecholamine determinations: only rarely necessary for localizing tumor Batch Clingy DIFFERENTIAL DIAGNOSIS Renal or renovascular disease; CoA; hyperthyroidism; cerebral disorders, DI, DM; Cushing syndrome; deficiencies of 11β-hydroxylase, 17α-hydroxylase, or 11βhydroxysteroid dehydrogenase (type 2 isozyme); primary aldosteronism; adrenocortical tumors; and, rarely, essential hypertension ● Nonfunctioning kidney: result from compression of ureter/renal artery by pheochromocytoma ● Paroxysmal HTN: assoc w/ porphyria or familial dysautonomia ● HTN in NF: renal vascular involvement or concurrent pheochromocytoma ● Neuroblastoma, ganglioneuroblastoma, ganglioneuroma freq produce catecholamines - urinary catecholamines higher w/ pheochromocytoma - dopamine & homovanillic acid usually higher in neuroblastoma - secreting neurogenic tumors: HTN, excessive sweating, flushing, pallor, rash, polyuria, polydipsia chronic diarrhea assoc w/ these tumors, particularly ganglioneuroma (sufficiently persistent to suggest celiac disease) PPS Oral Exam Reviewer 2020 123 Complex Batch Clingy ▪ Chest pain is a common presenting complaint among children and adolescents. ▪ Many causative factors: ● Idiopathic (12-85%) ● Musculoskeletal (15-76%) ● Pulmonary (12-21%) ● Psychogenic (4-17%) ● GI (4-8%) ● Miscellaneous (4-25%) ▪ Children younger than 12 years old are more likely to have cardiopulmonary cause, adolescents older than 12 years old more likely to have idiopathic or psychogenic causes PPS Oral Exam Reviewer 2020 124 DISEASE MUSCLE STRAIN ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Most identifiable cause of chest pain due to its association with localized tenderness elicited by specific manipulation of the thorax ▪ Involves the ribs, costochondral junctions, costal cartilages, intercostal muscles, sternum, clavicle or spine ▪ Occurs in young children with URTI – frequent coughing → straining of extrinsic muscles; among teenagers active in gymnastics and most other sports ▪ Self-limited with intermittent exacerbations during adolescence ▪ Same as above ▪ Unilateral sharp, stabbing pain along the upper 2 or more contiguous costochondral joints ▪ Pain exacerbated by deep breathing and lasts from a few seconds to a few minutes LABORATORY FINDINGS / DIAGNOSIS ▪ Clinically diagnosed based on history and physical examination ▪ Diagnosis of exclusion TREATMENT / COMPLICATIONS / PREVENTION ▪ Reassurance, analgesia, rest or any combination of these 3 measures ▪ NSAIDs for 1 week – decrease inflammation and pain ▪ In most circumstances, allaying the fears of the patient and parents by counselling them about the benign nature of condition helps to relieve concern and reduce the degree of chest pain Batch Clingy COSTOCHONDRITIS Costosternal Syndrome CLINICAL MANIFESTATIONS ▪ Localized or pinpoint pain ▪ Pain is worse with movement, certain body positions, coughing, inspiration ▪ Some are described in relations to specific patterns of muscular pain (i.e. Pectoral syndrome, coracoid syndrome, xiphoid process syndrome) PPS Oral Exam Reviewer 2020 125 PLEURAL EFFUSION 3 STAGES Exudative/Stage of uncomplicated effusion ▪ Airway and parenchymal infection → aspiration of the microorganisms into subpleural alveoli → causes migration and adherence of PMNs ▪ Oxygen metabolites and products of activated PMNs → endothelial injury of subpleural and pleural vessels, and increase capillary permeability ▪ The parapneumonic fluid initially tends to be small volume of sterile, PMN-dominant exudate PPS Oral Exam Reviewer 2020 ▪ Dyspnea ▪ Chest pain ▪ Respiratory symptoms ▪ Older children: sharp pleuritic pain ● Pain on inspiration with subsequent shallow breaths or cough ● Due to stretching of parietal pleura ● As the effusion increases and separates the pleural membranes, pleuritic pain becomes a dull ache and disappears ▪ Dullness and decreased breath sound over the affected area ▪ Decreased vocal fremitus ▪ Fullness of intercostal spaces ▪ Pleural rub ● Due to roughened pleural surfaces ● Can be present in the early phase ● Disappears as fluid accumulates CXR: identifies severity of lung contusion – correlates with impairment of oxygenation, CO2 exchange, and duration of mechanical ventilation ▪ Minor – supportive care in the form of supplemental oxygen, pulmonary toilet, and fluid restriction ▪ Severe – require mechanical ventilation in up to 30% of children CT scan: more sensitive, for better imaging of chest Long-term outcomes ▪ Spontaneous resolution ▪ Normal lung function CXR (PA view) ▪ Primary tool for diagnosing pleural effusion ▪ Obliteration of costophrenic sinus is the earliest diagnostic sign of the effusion ▪ Lateral decubitus film – quality and quantity of underlying parenchyma ▪ IV antibiotics directed at the underlying infection ● Continued until the child is afebrile for at least 7-10 days ● Oral antibiotics administered for 1-3 weeks Ultrasound ▪ Can differentiate pleural thickening from effusion, amount of fluid ▪ Helps identify best site for thoracentesis or insertion of thoracostomy tube ▪ Detects loculations and determine quality of effusion ▪ Drainage of infected fluid by thoracentesis or CTT ● Less effective at the 2nd stage of disease ● When CTT reaches 30-50ml/day and symptoms improve, chest tube may be removed ▪ VATS or early lung decortication ● Considered in selected children who have parapneumonic effusion or empyema whose clinical course does not improve, severe lung trapping, multiple loculi or an extensive pleural peel, or empyema caused by infectious bacteria other than S. aureus Pleural fluid examination (see table below) ▪ Most patients with uncomplicated parapneumonic effusion respond well to appropriate antibiotic therapy and do not require thoracostomy, with no residual lung damage ▪ Patients with empyema have more prolonged and complicated Batch Clingy CONTUSION Pulmonary Contusion ▪ Range in severity from being an isolated asymptomatic radiographic finding, to significant respiratory failure requiring mechanical ventilation ▪ Direct lung injury from thoracic trauma → transmission of force through the relatively thin and compliant chest wall to the underlying organs → pulmonary contusions, lacerations, traumatic pneumatoceles ▪ 34 – 100% of children with thoracic trauma ▪ Results in damage at the level of the alveoli → hemorrhage and edema → poor gas exchange ▪ Fluid in the pleural space Causes: ▪ Infection (50-70% parapneumonic effusion) ▪ CHF (5-15%) ▪ Malignancy (rare) Bacterial causes: S. aureus (<2yo), S. pneumoniae (25%), H. influenzae, Group A streptococci 126 Fibrinopurulent/Second or Bacterial invasive stage ▪ Endothelial injury more pronounced → pleural fluid formation increases – characterized by ↑ PMNs, ↓ glucose (increased glycolysis by PMNs and bacterial metabolism) ▪ ↓ pH, ↑ LDH ▪ Pleural fluid is clottable because procoagulants may move in the space and fibrinolytic activity may be lost due to mesothelial injury hospital courses Stage of organization (third stage of empyema) ▪ Thick, purulent coagulum ▪ The resultant inelastic peel impairs pleural fluid drainage and inhibits lung expansion Type of Effusion STARLING PRINCIPLE ▪ Fluid accumulates in the pleural cavity whenever filtration > removal mechanism, and may be the result of ↑ filtration associated with impaired absorption or of normal filtration associated with inadequate removal Pleural/Serum Concentration Ratio CHON LDH CHON LDH pH level Transudate < 3 g/dL < 2/3 < 0.5 < 0.6 > 7.45 Exudate ≥ 3 g/dL > 2/3 ≥ 0.5 ≥ 0.6 < 7.3 *Pleural LDH should be <2x the upper level of serum LDH ▪ Infants and toddlers: vomiting, feeding problems, poor weight gain ▪ Peripheral eosinophilia ▪ Elevated IgE ▪ Older children and adolescents: solid food dysphagia, occasional food impactions or strictures, heartburn, chest or epigastric pain ▪ Search for food (aerodigestive) and environmental allergies via skin prick (IgE mediated) and patch (non-IgE mediated) tests to guide decisions regarding dietary elimination and future food challenges ▪ Incidence: 5 per 100,000 ▪ Prevalence: 29.5 per 100,000 ▪ Mostly male; mean age at diagnosis: 7 years old ▪ Duration of symptoms: 3 years ESOPHAGITIS ▪ Pathogenesis involves mainly T helper type 2 cytokine-mediated (IL-5 & 13) pathways leading to production of a potent eosinophil chemoattractant, eotaxin-3, by esophageal epithelium INFECTIVE ESOPHAGITIS ▪ Often affects immunocompromised children ▪ EA: fungi – Candida albicans, Torulopsis glabrata ▪ Candida – leading cause immunocompromised children in immunocompetent PPS Oral Exam Reviewer 2020 Topically acting swallowed corticosteroids (ie fluticasone without spacer, viscous budesonide suspension) – for those who refuse, fail to adhere, or have a poor response to restricted diets ▪ Histologic remission is observed in 65-77% children and adults treated with fluticasone for 3 months ▪ Odynophagia ▪ Dysphagia ▪ Retrosternal or chest pain ▪ Fever, nausea, vomiting and PILL ESOPHAGITIS ▪ Produced by contact with a damaging agent ▪ Common meds: tetracycline, doxycycline, KCl, FeSO4, NSAIDs, cloxacillin and alendronate Dietary restrictions ▪ Elimination diets guided by circumstantial evidence and food allergy test results ▪ “6 food elimination diet” removing the major food allergens ▪ Elemental diet composed exclusively of an amino acid-based formula ▪ Elimination diets are generally successful, with highest histologic response observed in nearly 91% on elemental diet and in 72% to empiric dietary elimination Candida – concurrent oropharyngeal infection ▪ Acute discomfort ▪ Progressive retrosternal pain ▪ Odynophagia ▪ Dysphagia Diagnosis is made by endoscopy ▪ White plaques in Candida ▪ Multiple superficial ulcers or volcano ulcers in HSV ▪ Single deep ulcer in CMV Histologic exam ▪ Yeast and pseudohyphae in Candida ▪ Tissue invasion distinguishes esophagitis from mere colonization Endoscopy – focal lesion often localized to 1 of the anatomic narrowed regions of the esophagus or to an unsuspected pathologic narrowing ▪ Appropriate antimicrobial agents ▪ Supportive ▪ Sucralfate, antacids, topical anesthetics and bland or liquid diets – often used, lacks evidence ▪ Offending pill may be restarted after complete resolution of Batch Clingy ▪ ↑ Capillary permeability – infection, SLE, toxin, tumors ▪ ↑ Capillary hydrostatic pressure – CHF, pericarditis ▪ ↓ Hydrostatic or interstitial space pressure – post thoracentesis or trapped lung ▪ ↓ Plasma oncotic pressure – hypoalbuminemic state, nephrosis, hepatic cirrhosis ▪ ↑ Oncotic pressure of interstitial space – pulmonary infarction EOSINOPHILIC ESOPHAGITIS ▪ Esophageal dysfunction and infiltration of esophageal epithelium by >15 eosinophils/hpf ▪ Many have other atopic diseases and associated with food allergies ▪ EREFS (endoscopic reference score – Edema, Rings, Exudates, Furrows, Strictures) Pleural Liquid Concentration 127 ▪ Most often, offending tablet is ingested at bedtime with inadequate water ▪ Imbalance between myocardial oxygen supply and demand ▪ If left untreated → angina pectoris, myocardial stunning, myocardial hibernation ▪ Under the most severe instances, acute coronary syndromes like myocardial infarctions ▪ Increased myocardial oxygen demand in the presence of a severe fixed stenosis ▪ Coronary spasm due to local release of vasoactive mediators ▪ Transient thrombus formation ISCHEMIA ▪ Main mechanisms by which MI can occur ● Reduction in myocardial supply of oxygen ● Increase in myocardial oxygen demand Chest pain is more likely ischemic in nature when associated with: ▪ Exertion more than at rest ▪ Dyspnea ▪ Diaphoresis ▪ Syncope and characterized as: ▪ Substernal pressure or burning rather than pain ▪ Pressure that radiates to neck or arm ▪ Fairly reproducible with similar activity ▪ Short lived (2-10 mins) ECG ▪ Whether the ischemia is subendocardial or transmural will affect the ST changes and the interpretation of the ECG ▪ Acute transmural ischemia – tall peaked T waves with ST elevation are produced from epicardial injury representing the ischemic zone at risk of myocardial injury ▪ The location that represents the ischemic zone is represented by the placement of the surface ECG symptoms, if deemed necessary, though with clear emphasis on ingestion with adequate volume of water, usually at least 4 oz -Immediate therapeutic interventions involves reducing myocardial oxygen demand with betablocker therapy, antiplatelet agents, diagnostic testing to delineate the cause, and finally definitive treatment Cardiac troponins T and I and creatine kinase MB isoenzyme (CK-MB) ▪ Biomarkers of myocardial injury ▪ When elevated, signify myocardial damage with good sensitivity and specificity ▪ After injury occurs, within 2 hours, these enzymes will rise and may continue to rise for several hours ▪ By 12 hours, the CK-MB will begin to decrease and by 24 hours the sensitivity of the troponins remains high whereas the CK-MB sensitivity diminishes Cardiac MRI (CMR) and CT ▪ Tests of choice for diagnosing anomalous coronary arteries, coronary fistulae, coronary aneurysm, and ostial coronary artery disease following coronary reimplantation ▪ More common in adolescents ▪ Children and adolescents may have a history of anxiety and/or stressful life events that are not easily apparent but can have a large impact on their perception of pain ANXIETY/ STRESS ▪ Within anxiety disorders, chest pain and other symptoms are temporally related to stressful situations. Anxiety-related psychogenic chest pain is 4x as likely to occur in a patient with a family history of chest pain (often an adult with a history of cardiac ischemia), and the pain is often fleeting and vague in its description *see Chronic Cough* *see Arrhythmia* ▪ Hardest to treat, often requiring consultation with behavioral pediatricians, counselors, and/or psychologists ▪ Rapid breathing ▪ Dyspnea and anxiety to the degree that systemic symptoms result – paresthesia, dizziness, lightheadedness, palpitations, and confusion ▪ Sharp, non-radiating pain over the left precordium – easiest to diagnose when these symptoms arise without exertion ▪ Rest ▪ Detect underlying cause ▪ Referral to psychiatrist as needed Batch Clingy GERD RHYTHM DISTURBANCE ▪ Most common etiologies ● Hyperventilation ● Underlying psychiatric illness – anxiety, depression, or somatoform disorders (conversion or somatization) HYPERVENTILATION SYNDROME ▪ Most often in the setting of a panic attack ▪ Can occur as an acute one-time episode or can represent an underlying panic disorder if recurrent ▪ Somatic symptoms – breathlessness, fatigue, nervousness, near-syncope, palpitations ▪ Demonstrate more bodily worries, more limitation of general activity, and more school absences ▪ Psychologic forms of chest pain is typically recurrent with stressors. PPS Oral Exam Reviewer 2020 128 Complex ATRIAL SEPTAL DEFECT ASD ▪ Despite the large pulmonary blood flow, PAP is initially normal – absence of a high-pressure communication between the pulmonary and systemic circulations ATRIOVENTRICULAR SEPTAL DEFECTS ▪ Ostium primum and Atrioventricular canal/Endocardial cushion defects ▪ Deficiency of AV septum ▪ Severity of the AV valve abnormality varies ▪ Complete AV septal defect common in children with Down syndrome Basic abnormality ▪ Combination of L-to-R shunt across atrial defect and ▪ Mitral (or occ tricuspid) insufficiency ▪ Complete AV septal defects: L-to-R shunting occurs at atrial and ventricular level ▪ Often asymptomatic ▪ PE same as secundum ASD, but with apical holosystolic murmur ▪ Hyperdynamic precordium ▪ Normal or accentuated S1 ▪ Wide, fixed splitting of S2 ▪ Pulmonary SEM preceded by click ▪ Low-pitched, mid-diastolic rumbling murmur at the lower left sternal edge or apex Complete AV septal defects ▪ Infancy: CHF and intercurrent pulmonary infection ▪ Mod to marked cardiac enlargement ▪ Systolic thrill at lower sternal border ▪ If small with minimal L-to-R shunts, no RV enlargement: closure not required ▪ Infants with small to moderate-sized ASDs: watch closely – defects often grow smaller in the 1 st year of life Transcatheter device or surgical closure for: ● All symptomatic patients ● Asymptomatic patients with Qp:Qs ratio ≥2:1 ● RV enlargement ▪ Done after the 1st YOL and before school entry ▪ Repair is preferred during early childhood because surgical mortality and morbidity are significantly greater in adulthood Percutaneous catheter device closure ▪ Procedure of choice for most patients ▪ Uses an atrial septal occlusion device, implanted transvenously in the cardiac catheterization laboratory PROGNOSIS ▪ Results after surgical or device closure in children with moderate to large shunts are excellent ▪ Secundum ASDs are well tolerated during childhood ▪ Significant symptoms do not usually appear until the 3rd decade or later CXR (complete AV septal defects): ▪ Moderate to severe cardiac enlargement ▪ Large PA ▪ ↑ Pulmonary vascularity Surgical treatment of complete AVSD: ▪ More complex ▪ Highly successful ▪ Must be performed during infancy ECG (complete AV septal defects) – distinctive and diagnostic: ▪ Superior orientation of the mean frontal QRS axis with axis deviation to the RUQ (QRS negative in both lead I and lead aVF) ▪ Counterclockwise inscription of QRS vector loop (Q wave in leads I and aVL) ▪ Signs of biventricular hypertrophy or isolated RV hypertrophy ▪ RV conduction delay (rSR’ pattern in leads V3R and V1) ▪ N or tall P waves ▪ Occasional prolongation of the PR interval Pulmonary arterial banding – reserved for patients with lesions that make early corrective surgery too risky 2DED: “Gooseneck” deformity of the LVOT PPS Oral Exam Reviewer 2020 TREATMENT / COMPLICATIONS / PREVENTION PROGNOSIS If unrepaired complete AVSD, depends on: ▪ Magnitude of L-to-R shunt ▪ Degree of PVR elevation ▪ Severity of AV valve insufficiency COMPLICATIONS ▪ Pulmonary HPN ▪ Early tendency to increase PVR Batch Clingy DISEASE ACYANOTIC HEART DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS CLINICAL MANIFESTATIONS LABORATORY FINDINGS /DIAGNOSIS ▪ May occur in any portion of the atrial septum – secundum, primum, or sinus venosus, depending on which embryonic septal stru cture has failed to develop normally ▪ Majority are sporadic OSTIUM SECUNDUM DEFECT ▪ Often asymptomatic CXR ▪ Most common form of ASD ▪ Younger children: subtle failure to thrive ▪ RA and RV enlargement depending on size of the shunt best ▪ Normal AV valves ▪ Older children: varying degrees of exercise intolerance appreciated on lateral view (RV protrudes anteriorly as its ▪ May be single or multiple (fenestrated atrial septum) volume increases) ▪ Openings >2cm in diameter are common in symptomatic older ▪ Widely split and fixed S2 during all phases of respiration ▪ Enlarged PA children – constantly ↑ RV diastolic volume, prolonged ejection ▪ ↑ Pulmonary vascularity ▪ May be associated with MVP and partial anomalous pulmonary venous time in all phases of respiration return (PAPVR) ECG ▪ Systolic ejection murmur (SEM) ▪ RV volume overload ▪ Degree of L-to-R shunting depends on the size of the defect, the ● Not > grade 3/6, medium-pitched, without harsh ▪ RAD or N QRS axis relative compliance of the RV and LV, and the relative vascular qualities ▪ Minor RV conduction delay (rSR’ pattern in the R precordial resistance in the pulmonary and systemic circulations ● Seldom accompanied by a thrill leads) ● Best heard at the left middle and upper sternal border ▪ In large defects, a considerable shunt of oxygenated blood flows from ● Produced by the ↑ flow across the RV outflow tract 2DED LA to RA. This blood is added to the usual venous return to the RA and is into the PA ▪ RV volume overload pumped by the RV to the lungs (ratio of pulmonary-to-systemic blood ▪ ↑ RV end-diastolic dimension flow (Qp:Qs) is between 2:1 and 4:1) ▪ Short rumbling mid-diastolic murmur ▪ Flattening and abnormal motion of the ventricular septum ● d/t the ↑ volume of blood flow across the TV (septal motion either flattened or reversed) ▪ Paucity of symptoms in infants with ASDs is related to the structure of ● Audible at the left lower sternal border the RV in early life – thick and less compliant muscular wall → limited Lto-R shunt ▪ As the infant grows and PVR drops, the RV wall becomes thinner → ↑ L-to-R shunt across the ASD → ↑ blood flow through the R side of the heart → RA and RV enlargement, PA dilation ▪ ↑ Pulmonary blood flow returns to the LA → LA enlargement 129 VENTRICULAR SEPTAL DEFECT (VSD) Small VSDs with trivial L-to-R shunts and normal PAP – most common and asymptomatic CXR: cardiomegaly with prominent ventricles, LA and PA ▪ Physical size of the VSD – major determinant of the size of the L-to-R shunt ▪ Large defects: level of PVR to SVR – major determinant of the shunt’s magnitude ▪ When the PVR/SVR ratio approaches 1:1, the shunt becomes bidirectional → Eisenmenger syndrome → signs of CHF ▪ Wall of ductus deficient in both the mucoid endothelial layer and the muscular media ▪ F>M ▪ Associated with maternal rubella and prematurity PATENT DUCTUS ARTERIOSUS DISEASE ▪ High aortic pressure → blood flows through the aorta → PDA: blood shunts L-to-R from aorta to pulmonary artery ▪ Extent of the shunt depends on 1) size of the ductus, 2) PVR/SVR ratio ▪ Large PDA: PAP elevated to systemic levels during systole and diastole ETIOLOGY / INCIDENCE / PATHOGENESIS 4 COMPONENTS ▪ Obstruction to RV outflow (pulmonary stenosis) ▪ Malalignment type of VSD ▪ Overriding aorta ▪ RVH TETRALOGY OF FALLOT PPS Oral Exam Reviewer 2020 ECG normal but may suggest LVH Large VSDs – signs of CHF: ▪ Dyspnea ▪ Feeding difficulties ▪ Poor growth ▪ Profuse perspiration ▪ Recurrent pulmonary infections in early infancy ▪ Loud, harsh or blowing holosystolic murmur at the L left sternal border with a thrill ▪ Prominence of the left precordium ▪ Palpable parasternal lift ▪ Laterally displaced apical impulse ▪ Apical thrust Small PDA: asymptomatic Large PDA: ▪ Growth retardation ▪ Bounding peripheral arterial pulses ▪ Wide pulse pressure ▪ Classic continuous murmur (machinery-like) begins after S1, reaches max intensity at end of systole, and wanes in late diastole ▪ Thrill, maximal in the 2nd left ICS, radiating toward the left clavicle 2DED ▪ Estimates shunts size ▪ Calculates pressure gradient across the defect PROGNOSIS Results of primary surgical repair are excellent; long-term prognosis excellent ECG ▪ Normal if small ▪ Large: LVH or biventricular hypertrophy CXR (large PDA) ▪ Prominent PA ▪ ↑ Pulmonary vascular markings CYANOTIC HEART DISEASE CLINICAL MANIFESTATIONS LABORATORY FINDINGS / DIAGNOSIS “tet” or “blue” spells – paroxysmal hypercyanotic attacks CXR: Coeur en sabot (boot shaped heart) ▪ 1st year of life ▪ Frequently in the morning or after episodes of crying ECG ▪ Relieved with squatting ▪ RAD, RVH ▪ Dominant R wave in right precordial chest leads (V1, V2), or an Older children with unrepaired tetralogy: RSR’ pattern ▪ Dyspnea on exertion ▪ Dusky blue skin, gray sclerae with engorged blood vessels 2DED ▪ Marked clubbing of fingers and toes ▪ Extent or aortic override of the septum ▪ Location and degree of RVOT obstruction ▪ Left anterior hemithorax may bulge anteriorly (long-standing ▪ Size of the pulmonary valve annulum and main and proximal RVH) branch pulmonary arteries and side of the aortic arch ▪ Substernal RV impulse ▪ Systolic thrill along the left sternal border in the 3rd and 4th Cardiac Catheterization parasternal spaces ▪ Systolic pressure in RV = systemic pressure ▪ Loud and harsh systolic ejection murmur most intense at the left upper sternal border COMPLICATIONS Cerebral thromboses ▪ In cerebral veins or dural sinuses; <2yo ▪ Sequelae of extreme polycythemia and dehydration ▪ Tx: adequate hydration, supportive measures; if extremely polycythemic, phlebotomy and volume replacement with albumin Small defects ▪ Close spontaneously ▪ 1st 2 years of life ▪ Small muscular VSDs more likely to close Goals in infants with large VSD: ▪ Control symptoms of CHF ▪ Prevent pulmonary vascular disease Indications for surgical closure ▪ Any age with large defect and clinical symptoms and failure to thrive cannot be controlled medically ▪ Infants 6-12 mos old with moderate to large defects assoc with pulmonary HPN ▪ Older than 24 mos old with Qp:Qs ratio greater than 2:1 Antibiotic prophylaxis is no longer recommended for dental visits or surgical procedures Irrespective of age, requires either catheter or surgical closure good prognosis Small PDA: ▪ Closure to prevent bacterial endarteritis or other late complications ▪ Closed with coils Moderate to large PDA: ▪ Closure to treat heart failure and prevent the pulmonary vascular disease ▪ Closed with umbrella-like device TREATMENT / COMPLICATIONS / PREVENTION “Tet” spells: ▪ Place infant on the abdomen in knee-chest position (make sure infant’s clothing not restrictive) ▪ Oxygen ▪ Morphine/SC in a dose not >0.2 mg/kg ▪ If tet spell is unusually severe, metabolic acidosis may develop → rapid correction with IV Na bicarbonate ▪ If still resistant, intubation and anesthetic sedation IV phenylephrine ▪ ↑ SVR ▪ Improve RV outflow ▪ Decrease the R-to-L shunt B-adrenergic blocker ▪ IV propranolol (0.1-0.2mg/kg) Corrective open-heart surgery ▪ Early infancy and critically ill newborn ▪ Early physiologic correction allows improved growth of the branch pulmonary arteries. ▪ Infants with less severe cyanosis and can be maintained with good growth and absence of hypercyanotic spells, primary repair is performed electively Batch Clingy ▪ Most common cardiac malformation ▪ 25% of CHDs ▪ Most common are the membranous type 130 or saline Brain abscess ▪ >2yo ▪ Low-grade fever, gradual change in behavior, HA, n/v, seizure ▪ Do CT or MRI to confirm ▪ Tx: surgical drainage + antibiotics Bacterial endocarditis: right infundibulum or pulmonic, aortic, or tricuspid valve Heart failure TRANSPOSITION OF THE GREAT ARTERIES (TGA) ▪ A risk factor is GDM L-TGA “Physiologically corrected TGA” ▪ L – looped TGA ▪ AV relationships are discordant (atrioventricular discordance/ventricular inversion): RA → LV; LA → RV ▪ Desaturated systemic venous blood returns via the vena cava to a normal RA → bicuspid atrioventricular (mitral) valve → R-sided ventricle (with architecture and smooth wall morphologic features of the normal LV) → transposed PA → lungs → oxygenated pulmonary venous blood returns to a normal LA → L-sided ventricle (has trabeculated morphologic features of a normal RV) → transposed aorta ▪ The double inversion of the atrioventricular and ventriculoarterial relationships result in desaturated RA blood appropriately flowing to the lungs, and oxygenated pulmonary venous blood appropriately flowing to the aorta (physiologically corrected circulation) ▪ Total mixing of systemic venous and pulmonary venous blood flow within the heart TOTAL ANOMALOUS PULMONARY VENOUS RETURN (TAPVR) PPS Oral Exam Reviewer 2020 ▪ Cyanosis and tachypnea within the few hours or days of life → survival in immediate NB period provided by foramen ovale and ductus arteriosus ECG: normal ▪ S2 usually single and loud ▪ Soft systolic ejection murmur 2DED – diagnostic; confirms: ▪ Transposed ventricular-arterial connections size of the interatrial communication ▪ Ductus arteriosus ▪ Degree of mixing Cardiac catheterization ▪ Done if unusual coronary artery anomaly is suspected, or require emergency balloon atrial septostomy (Rashkind procedure) Hypoxemia depends on ▪ Degree of atrial level shunting ▪ Whether the ductus is partially open or totally closed CXR: classic egg-shaped heart ECG ▪ Atrioventricular conduction disturbances ▪ Abnormal P waves ▪ Absent Q waves in V6 ▪ Abnormal Q waves in leads III, aVR, aVF, V1 ▪ Upright T waves across the precordium Palliative systemic-in-pulmonary artery shunt (Blalock-Taussig shunt) ▪ Indication: co-morbidities such as other major congenital anomalies or prematurity ▪ Augment pulmonary artery blood flow to decrease the amt of hypoxia and improve linear growth ▪ Augment growth of the branch pulmonary arteries ▪ After successful total correction, patients are asymptomatic and can lead unrestricted lives Prostaglandin E1 (PGE1, 0.01-0.02 ug/kg/min); maintains patency of the ductus arteriosus Rashkind balloon atrial septostomy ▪ In infants who remain severely hypoxic or acidotic despite PGE1 infusion ▪ If significant delay in surgery is necessary Arterial switch (Jatene) procedure ▪ Surgical treatment of choice for neonates (d-TGA with intact VSD) ithin the first 2 weeks of life ▪ >95% Survival rate for uncomplicated d-TGA Atrial switch procedure (Mustard or Senning operation) ▪ Excellent early survival (85-90%) ▪ Significant long-term morbidities ▪ Reserved for those whose anatomy are not candidates for the arterial switch procedure Double switch procedure: ▪ Atrial switch – reroute the systemic and pulmonary venous returns ▪ Arterial switch – reroute ventricular outflows CXR: suggest abnormal position of the great arteries ▪ Ascending aorta occupies the upper left border of the cardiac silhouette 2DED: atrioventricular discordance ▪ Manifestations depend on the presence or absence of obstruction of the venous channels ▪ If obstructed pulmonary venous return → severe pulmonary congestion and pulmonary hypertension ▪ Neonates with severe obstruction to pulmonary venous return: severe cyanosis, respiratory distress Mild or no obstruction to pulmonary venous return: ▪ Heart failure as the pulmonary vascular resistance falls ▪ Mild to moderate degrees of desaturation ECG: RVH – usually qR pattern in V3R and V1 and P waves are frequently tall and spiked Obstructed TAPVR – surgical emergency because prostaglandin therapy is usually not effective CXR ▪ Neonates with marked pulmonary venous obstruction – very dramatic perihilar pattern of pulmonary edema ▪ In older children: snowman appearance of heart ▪ Surgical correction: infancy ▪If surgery cannot be performed urgently, ECMO may be required to maintain oxygenation 2DED ▪ Any vein with doppler flow away from the heart is pathognomonic ▪ Large RA and RV PROGNOSIS ▪ Early post-op results are generally good, even for critically ill neonates Batch Clingy d-TGA ▪ 5% of all CHDs ▪ Aorta is anterior and to the right of the pulmonary artery (d – dextropositioned aorta) ▪ Systemic and pulmonary circulations exist as 2 parallel circuits: ● Aorta arises from the anterior RV and systemic veins return to the LA ▪ AV concordance (RA → RV; LA → LV) o at 4-6 months of age 131 TRUNCUS ARTERIOSUS ▪ Single arterial trunk arises from the heart and supplies the systemic, pulmonary and coronary circulations ▪ Total mixing lesion with complete admixture of pulmonary and systemic venous return ▪ VSD always present, TA overrides VSD ▪ Ventricles are at systemic pressure and eject blood into the truncus ▪ When PVR is relatively high immediately after birth, pulmonary blood flow may be normal. But as PVR drops in the 1st month of life, blood flow to the lungs is greatly increased and heart failure ensues. ▪ If lesion is left untreated, PVR eventually increases, pulmonary blood flow decreases, and cyanosis becomes more prominent (Eisenmenger physiology) Immediate NB period ▪ Signs of heart failure absent ▪ Murmur & minimal cyanosis may be only finding ▪ No outlet from the RA to RV ▪ Entire systemic venous return leaves the RA and enters the L side of the heart through the foramen ovale or atrial septal defect ▪ Physiology of the circulation and the clinical presentation will depend on the presence of other congenital heart defects ▪ Some degrees of cyanosis at birth, extent depending on the degree of limitation to pulmonary blood flow ▪ Increased LV impulse ▪ Single S2 ▪ Pulses in lower extremities may be weak, or absent if with CoA 1-2 mos old ▪ Heart failure with mild cyanosis ▪ Wide pulse pressure and bounding pulses ▪ Hyperdynamic precordium ▪ S2 is loud and single ▪ Systolic ejection murmur, accompanied by a thrill audible along the left sternal border and frequently preceded by an early systolic ejection click ▪ Apical mid-diastolic rumbling murmur audible as heart failure develops TRICUSPID ATRESIA ▪ Enlarged PA ▪ N or small LA and LV Cardiac catheterization ▪ Done if there is question about the drainage of 1 or more pulmonary veins ▪ Shows that O2 sat of blood in both atria, ventricles and aorta is similar ECG: RVH, LVH or combined ventricular hypertrophy CXR ▪ Cardiac enlargement, with prominence of both ventricles ▪ Prominent shadow that follows the normal course of the ascending aorta and aortic knob 2DED: diagnostic ▪ Large truncal artery overriding the VSD ▪ Pattern of origin of the branch pulmonary arteries ▪ Evaluate truncal valve regurgitation ECG: ▪ LAD and LV hypertrophy – unique features ▪ Right precordial leads: prominent R wave replaced by an rS complex ▪ Left precordial leads: ● qR complex, followed by a normal, flat, biphasic, or inverted T wave ● RV6 is normal or tall ● V1 is deep ● P waves usually biphasic, with initial component tall and spiked in lead II ● Combination of cyanosis and left axis deviation is highly suggestive Cardiac catheterization: Indicated only if questions remain after echo, shows normal or slightly elevated right atrial pressure with a prominent a wave COARCTATION OF THE AORTA ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ 98% are juxtaductal coarctation – just below the origin of the left subclavian artery, at the origin of the ductus arteriosus ▪ Discrete juxtaductal coarctation: ascending aortic blood flows through the narrowed segment to reach the descending aorta ▪ More severe juxtaductal coarctation: RV blood is ejected through the ductus to supply the descending aorta (perfusion of lower part of the body is RV output dependent) PPS Oral Exam Reviewer 2020 ▪ First few weeks of life, infants can be managed with anti-congestive medications ▪ As PVR falls in infants, heart failure symptoms worsen and surgery is indicated, within the 1st few months ● VSD is closed ● PA separated from truncus ● Continuity is established between RV and pulmonary arteries ▪ Delay in surgery → increase the likelihood of pulmonary vascular disease ▪ Surgical results have been excellent Cardiac catheterization: L-to-R shunt at the ventricular level, with Rto-L shunting into the truncus 2DED: fibromuscular membrane in place of a tricuspid valve, a variably small RV, VSD and large LV DISEASE COMPLICATIONS ▪ Patients where diagnosis was delayed, or with severe obstruction, recurrent stenosis and development of pulmonary veno-occlusive disease may occur ▪ Aggressive veno-occlusive disease: heart-lung transplantation is only option CLINICAL MANIFESTATIONS ▪ Classic sign: disparity in pulsation and BP in the arms and legs – femoral, popliteal, posterior tibial and dorsalis pedis pulses are weak ▪ Radial-femoral delays Blood flow to the descending aorta is dependent on collaterals Femoral pulse is felt after the radial pulse Severely cyanotic neonates: ▪ IV infusion of PGE1 (0.01-0.02 ug/kg/min) until a surgical aortopulmonary shunt procedure can be performed ▪ Blalock-Taussig procedure: preferred anastomosis Next stage surgery: bidirectional Glenn shunt ▪ Creation of an anastomosis between SVC and pulmonary arteries ▪ 2-6 mos old ▪ Reduces volume load on the LV ▪ Lessens the chance of LV dysfunction Modified Fontan operations ▪ Preferred approach to larger surgical management ▪ Between 2 and 3 years old, after patient is ambulatory ▪ Anastomose RA or atrial appendage directly to PA Cavopulmonary isolation procedure ▪ Modified modified Fontan ▪ IVC anastomose to pulmonary arteries ▪ Decrease risk of right atrial dilation LABORATORY FINDINGS / DIAGNOSIS CXR ▪ Mildly or moderately enlarged heart – d/t LV prominence ▪ Prominent shadow in left superior mediastinum – d/t enlarged L subclavian artery ▪ Notching of the inferior border of the ribs (inverted 3 sign) – d/t pressure erosion by enlarged collateral vessels TREATMENT / COMPLICATIONS / PREVENTION PGE1 ▪ Neonates with severe coarctation ▪ Reopen the ductus, re-establish adequate LE blood flow Anti-congestive meds ▪ Older infants with heart failure but good perfusion: ▪ Improve clinical status before surgical intervention ECG ▪ Delay is unwarranted, especially after the 2nd decade of life Batch Clingy ▪ Systolic murmurs at left sternal border ▪ Gallop rhythm 132 ▪ Unless surgically corrected in infancy, coA results in development of an extensive collateral circulation to by-pass coA ▪ Occurs twice as often in males vs females ▪ Short systolic murmur at the 3rd and 4th ICS, L sternal border, transmitted to the L infrascapular area and occ to the neck ▪ Normal in young children ▪ LV hypertrophy in older infants 2DED: demonstrate specific site of obstruction PULMONARY STENOSIS ▪ Frequently associated with other types of CHDs ▪ High incidence in infants with congenital rubella syndrome ▪ 5% of cardiac malformations ▪ One of the causes of sudden cardiac death AORTIC STENOSIS ▪ Most common form: valvular aortic stenoses – leaflets are thickened, commissures are fused to varying degrees ▪ Subvalvular (subaortic) stenosis – discrete fibromuscular shelf below the AV → an important form of LVOT obstruction ▪ Supravalvular aortic stenosis – least common ▪ More frequent in males (3:1) DISEASE ▪ Valvular lesions begin as small verrucae composed of fibrin and blood cells along the borders of ≥1 heart valves → repeated attacks of RF → new verrucae form near the previous ones → mural endocardium and chordae tendinae become involved CLINICAL MANIFESTATIONS ▪ ARF + echo findings of MV/AV involvement ▪ Subclinical carditis – (+) 2DED evidence of mitral or aortic valvulitis, (-) auscultatory findings ECG: in severe stenosis, RVH and RAE CXR: cardiomegaly, prominence of MPA 2DED: acceleration of blood flow through stenoses CXR: prominent ascending aorta, N heart size 2DED ▪ Site and severity of obstruction ▪ Number of valve leaflets and morphology, and presence of a subaortic membrane or supravalvar stenosis ▪ Catheter balloon dilation, sometimes with placement of an intravascular stent ▪ Balloon valvuloplasty: procedure of choice for moderate to severe valvular AS ▪ Surgical treatment reserved for 1) extremely dysplastic aortic valves, not amenable to balloon therapy, 2) subvalvar or valvar stenosis ▪ Konno procedure: AS in association with severe tunnel-like sub-aortic obstruction PROGNOSIS In older infants and children with mild to moderate aortic stenosis, prognosis is reasonably good, although disease progression over 5-10 years is common LABORATORY FINDINGS / DIAGNOSIS TREATMENT / COMPLICATIONS / PREVENTION ▪ Do 2DED for all cases of confirmed or suspected ARF Batch Clingy RHEUMATIC HEART DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Most important sequela of acute rheumatic fever (ARF) ▪ Mitral valve most often affected → aortic valve ▪ Severe coarctation: cardiac enlargement, pulmonary congestion ▪ Continuous SEM in widespread locations ▪ Immediate NB period: soft SEM over either or both lung fields and disappears by 1-2mos old ▪ Less severe: asymptomatic, normal growth and development, murmur is discovered during routine PE ▪ Louder, harsher, and longer murmur with greater degree of obstruction ▪ Murmur is audible maximally at the R upper sternal border and radiates to the neck and left midsternal border accompanied by a thrill in the suprasternal notch ▪ Normal splitting of S2 in mild to moderate obstruction Severe AS: ▪ Heart failure ▪ Cardiomegaly ▪ Pulmonary edema ▪ Weak pulses in all extremities ▪ Pale or grayish skin ▪ ↓ S1 d/t ↓ compliance of the thickened LV ▪ Significant number of infants operated before 1yo require revision later in childhood; must be monitored for recoarctation and aortic anastomotic aneurysm (if recoarctation occur, balloon angioplasty is procedure of choice) PPS Oral Exam Reviewer 2020 133 MITRAL INSUFFICIENCY Structural changes: ▪ Loss of valvular substance ▪ Changes to the subvalvular apparatus ▪ Elongation of chordae Structural changes in the valve → ↑ volume load → LV dilates → LA also dilates to accommodate the regurgitant volume → ↑ LA pressure → pulmonary congestion and symptoms of L-sided heart failure MITRAL STENOSIS ▪ Fibrosis of the mitral ring, commissural adhesions and contracture of the valve leaflets, chordae and papillary muscles over time ▪ Chronic, takes >10yrs for lesion to be fully established Significant MS → ↑ LA pressure → enlargement and hypertrophy of LA, pulmonary venous HPN, ↑PVR → overt pulmonary HPN → RV hypertrophy and RA dilation → RV dilation, TR, R-sided heart failure AORTIC INSUFFICIENCY ▪ Poor coaptation of the leaflets or leaflet prolapse Chronic AI → valve sclerosis → distortion and retraction of the cusps → regurgitation of blood → LV volume overload → dilation and hypertrophy of LV Mild ▪ No signs of heart failure ▪ High-pitched holosystolic murmur at the apex, radiating to the axilla Severe ▪ Signs of acute or chronic heart failure ▪ Heart enlarged ▪ Apical systolic thrill ▪ S2 may be accentuated if pulmonary HPN is present ▪ 3rd heart sound or gallop prominent Mild: asymptomatic Severe: exercise intolerance and dyspnea ▪ Orthopnea, PND, overt pulmonary edema, atrial arrhythmias ▪ Pulmonary HPN: functional tricuspid insufficiency, hepatomegaly, ascites, edema, increased JVP, accentuated S2 ▪ Parasternal RV lift ▪ Loud first heart sound ▪ Opening snap of mitral valve ▪ Long, low-pitched, rumbling mitral diastolic murmur with presystolic accentuation at the apex ▪ Palpitations d/t large SV ▪ Sweating and heat intolerance d/t excessive vasodilation ▪ Wide pulse pressure, with bounding peripheral pulse (water-hammer or Corrigan pulse) Severe AI ▪ Enlarged heart ▪ LV apical heave, diastolic thrill ▪ Murmur begins with S2 and continues until late in diastole, heard over the upper L and mid-L sternal border with radiation to the apex and upper R sternal border, with high-pitched blowing quality ECG ▪ Severe: longer duration and often bifid P waves, LVH, RVH CXR ▪ Severe: Prominent LA and LV, congestion of perihilar vessels if with pulmonary venous HPN 2DED Acute MI: ▪ LAE and LVH ▪ Mitral annular dilation ▪ Chordal elongation ▪ Chordal rupture resulting in flail leaflet ▪ Nodular leaflet tips ▪ Prolapse of anterior MV leaflet tip Chronic MI: ▪ Leaflet and chordal thickening ▪ Chordal fusion ▪ Restricted leaflet ECG ▪ Severe – prominent and notched P waves, RVH ▪ Late manifestations: AFib, atrial arrhythmia CXR (Severe) ▪ LAE; prominent PA, RA, RV ▪ Kerley B lines in lower lung periphery 2DED ▪ Thickened MV and chordal apparatus ▪ Restricted motion of the valve ▪ Elbow or dog leg appearance of anterior leaflet of MV ▪ LA dilation ▪ Narrow jet with flow acceleration ▪ Variable degrees of tricuspid insufficiency ECG: LVH, prominent P waves CXR: Enlargement of LV and aorta ▪ Mild: prophylaxis against recurrences of RF ▪ Significant MI: Prophylaxis + corticosteroids Afterload-reducing agents (ie. ACEI or ARBs) ▪ Reduce regurgitant volume ▪ Attenuate pathologic compensatory mechanisms ▪ Preserve LV function Diuretics Surgery for: ▪ Despite adequate medical therapy, have persistent heart failure ▪ Dyspnea with moderate activity ▪ Progressive cardiomegaly, often with pulmonary HPN In patients with prosthetic MV replacement, prophylaxis against bacterial endocarditis is warranted for dental procedures. ▪ Diuretics and B-blockers – for symptom control ▪ Surgical valvotomy or balloon catheter mitral valvuloplasty ▪ ACEIs or ARBs ▪ Prophylaxis against ARF recurrence ▪ Surgical intervention (aortic valve replacement) – done in advance of heart failure, pulmonary edema and angina 2DED ▪ Dilated LV ▪ Diastolic mitral valve flutter or oscillations ▪ Aortic valve may show irregular or focal thickening ▪ Decreased systolic excursion ▪ Coaptation defect and leaflet prolapse Batch Clingy Austin Flint murmur: apical presystolic murmur (resembles MS) PPS Oral Exam Reviewer 2020 134 Complex DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS CLINICAL MANIFESTATIONS LABORATORY FINDINGS / DIAGNOSIS TREATMENT / COMPLICATIONS / PREVENTION ▪ Sinus rhythm with heart rate greater than the upper limit of normal for age ▪ Usually precipitated by fever or hyperthermia, pain, anxiety, and severe infection ▪ May also be compensatory to loss of intravascular volume (e.g. diarrhea, blood loss), or compromised cardiac output SUPRAVENTRICULAR TACHYCARDIA ▪ Includes all forms of paroxysmal or incessant tachycardia except ventricular tachycardia ▪ May occur when the patient is at rest or exercising ▪ In infants, may be precipitated by acute infection ▪ May be exacerbated by exposure to caffeine, nonprescription decongestants, or bronchodilators ▪ Abrupt onset and termination ▪ 1 in 250,000 to 1 in 250 ▪ Peak incidence in the 1st 2mos of life Older children ▪ Heart rate >180 bpm ▪ Palpitations, chest pain, abdominal pain, syncope during exertion, rarely, sudden cardiac death Three Mechanisms: AV re-entry tachycardia (AVRT) ▪ Most common ▪ Ues an accessory pathway as the retrograde limb and the AV node as the antegrade limb of the tachycardia ▪ Vagal stimulation – place the face in ice water (older children) or place an ice bag over the face (infants) (diving reflex) ▪ Vagal maneuvers – valsalva maneuver, straining, breath holding, or standing on their head ▪ If vagal stimulation failed, give Adenosine – drug of choice, 0.1mg/kg (max 6mg), by rapid bolus followed by saline flush using central line or antecubital vein. If SVT fails to terminate, give a 2nd dose at 0.2mg/kg (max 12mg) 2DED to determine presence of heart disease and intracardiac thrombus ▪ If with symptoms of severe heart failure, do synchronized cardioversion (0.5-2 J/kg) ▪ As maintenance therapy, for patients without antegrade accessory pathway, β-adrenergic blockers are mainstay of drug therapy. Contraindicated in WPW. AV node re-entry tachycardia (AVNRT) ▪ Rare in infancy ▪ Involves the slow antegrade limb and the fast pathway as the retrograde limb of the tachycardia or “slow-fast” pathway most of the time Atrial tachycardia ▪ Has a focus in the atrium which beats faster than the sinus node; ectopic or automatic tachycardias VENTRICULAR TACHYCARDIA ▪ May be associated with myocarditis, anomalous origin of a coronary artery, arrhythmogenic cardiomyopathy, MVP, primary cardiac tumors, dilated or hypertrophic cardiomyopathy, drug use (cocaine, amphetamine) ▪ May develop years after intraventricular surgery (especially TOF and related defects) or occur without obvious organic heart disease ▪ In neonates, may be associated with an anomalous left coronary artery or a myocardial tumor VENTRICULAR FIBRILLATION ▪ Chaotic rhythm that results in death unless an effective ventricular beat is rapidly established ▪ Lethal event ▪ May be seen in the setting of hypoxemia, anesthesia, hyper- or hypokalemia, hereditary arrhythmias PPS Oral Exam Reviewer 2020 ECG or rhythm strip ▪ Wide QRS tachycardia ▪ HR >120 bpm or >25% of the sinus rate ▪ At least 3 PVCs at >120bpm ▪ Clear capture and fusion beats – confirm the diagnosis 2DED to document presence of heart disease ▪ Syncope or sudden death ECG: Rapid, irregular and polymorphic complexes ▪ Must be promptly treated because may result to hypotension and degeneration into Vfib ▪ IV amiodarone, lidocaine or procainamide – initial drugs of choice if hemodynamically stable. If successful, correct underlying abnormalities after. ▪ Amiodarone – treatment of choice during cardiac arrest ▪ Immediate direct current (DC) cardioversion (VT with pulse: synchronized cardioversion; pulseless VT: defibrillation) if hemodynamically unstable ▪ Chronic tx: amiodarone, B-blocker, sotalol, phenytoin ▪ Defibrillation at 2 J/kg as initial dose + PALS algorithm ▪ If defibrillation is ineffective or VF recurs, give amiodarone or lidocaine per IV and repeat defibrillation ▪ Electrophysiology study (EPS) – indicated for patients who survived VF unless a clearly reversible cause is identified ▪ For patients in whom no correctable abnormality can be found, an Batch Clingy SINUS TACHYCARDIA Infants ▪ Heart rate >220 bpm ▪ Signs and symptoms of congestive heart failure, rapid heartbeat, irritability, poor feeding, hypothermia, poor perfusion ECG ▪ Narrow QRS complex tachycardia ▪ No visible P wave ▪ Fixed R-R interval 135 ATRIAL FLUTTER ▪ HR 250-300bpm in children ▪ HR 400-600bpm in neonates ▪ Diagnosis is confirmed by ECG: rapid, regular atrial sawtoothed flutter waves Intraatrial Reentrant Tachycardia ▪ In older children, usually occurs in the setting of CHD; neonates usually have normal hearts ▪ May occur during acute infectious illnesses but is most often seen in patients with large stretched atria (ie. Long-standing mitral or tricuspid insufficiency, tricuspid atresia, Ebstein anomaly or rheumatic mitral stenosis) ▪ Can occur after palliative or corrective intraatrial surgery ▪ HR 400-700bpm FIRST DEGREE ▪ May be found in normal children, patients with acute rheumatic fever, myocarditis, congenital heart disease (ASD, AVSD), Duchenne muscular dystrophy, myotonic dystrophy, trichinosis ▪ Can be due to anti-arrhythmic drugs HEART BLOCK ECG ▪ No distinct P waves ▪ Undulating baseline with narrow QRS complexes ▪ Changing R-R intervals ▪ Irregularly irregular ventricular rates – due to variability of conduction from the atria to the AV node to the ventricles COMPLICATIONS ▪ If uncontrolled, may precipitate heart failure ▪ Chronic atrial flutter in the setting of CHD may be at ↑ risk for thromboembolism and stroke, and should undergo anticoagulation before elective cardioversion ▪ Rate control is the best initial treatment, most effectively with calcium channel blockers to limit the ventricular rate during atrial fibrillation ▪ Do immediate DC cardioversion if hemodynamically unstable PROGNOSIS ▪ ↑ Risk for thromboembolism and stroke → anti-coagulation with warfarin ECG ▪ Sinus rhythm ▪ Rate is below the normal for age ▪ If asymptomatic, no intervention needed ECG ▪ Has a P wave preceding each QRS complex ▪ PR interval is prolonged but constant – all atrial impulses are conducted to the ventricle ▪ No active intervention necessary ▪ Address the underlying cause SECOND DEGREE ▪ Not every atrial impulse is conducted to the ventricle Mobitz type 1 ▪ Wenckebach Phenomenon ▪ Possible causes: myocarditis, digitalis toxicity, and CHDs ▪ Asymptomatic ECG: Progressive prolongation of the PR interval until a P wave is not conducted (drop beat) ▪ In the absence of heart failure or hemodynamic compromise, no treatment is needed Mobitz type 2 ▪ Regular non-conducted P waves such that atrial rate exceeds the ventricular rate ▪ Less common conduction defect but has more potential to cause syncope, and may be progressive ▪May be asymptomatic ▪ When there is progression of the bradycardia, syncope can occur ECG: ▪ Identical P waves ▪ PR interval for conducted P waves is constant ▪ No progressive conduction delay or subsequent shortening ▪ No need for acute intervention unless associated with symptomatic bradycardia, hemodynamic compromise, or heart failure ▪ Treatment is indicated for post-operative cases with advanced second-degree AV block that is not expected to resolve and for cases PPS Oral Exam Reviewer 2020 Batch Clingy SINUS BRADYCARDIA ▪ Mechanism of common atrial flutter: reentrant rhythm originating in the RA circling the tricuspid valve annulus ▪ Because the AV node cannot transmit such rapid impulses, some degree of AV block is virtually present ATRIAL FIBRILLATION ▪ Atrial excitation is chaotic and more rapid and produces irregularly irregular ventricular response and pulse ▪ Often associated with atrial enlargement or disease ▪ May be seen in older children with rheumatic MV stenosis ▪ May be due to thyrotoxicosis, pulmonary embolism, pericarditis, or cardiomyopathy, in a previously normal older child ▪Complication of dilated atria from congenital or acquired heart disease, particularly RHD among Filipino adolescents ▪ Seen in normal children during sleep or in any condition with increased vagal tone, in athletes, in patients with sinus node dysfunction, in children with increased intracranial, intrathoracic or intraabdominal pressure implantable cardioverter defibrillator (ICD) is almost always indicated because of the high risk of sudden death ▪ Synchronized DC cardioversion – treatment of choice; usually converts it immediately to sinus rhythm ▪ β-blockers or calcium channel blockers – may be used to slow the ventricular response in atrial flutter by prolonging AV node refractory period ▪ Class I agents (procainamide or propaferone) or class III agents (amiodarone or sotalol) – may be used to maintain sinus rhythm ▪ Catheter ablation has been used in patients with normal hearts and those with CHD with moderate success 136 ▪ Possible causes: myocarditis, digoxin toxicity, and CHDs of the PR interval after a blocked beat which persist at least 7 days after cardiac surgery ▪ When symptomatic, do implantation of a permanent pacemaker Holter monitoring may be indicated in some cases to document whether symptoms occur together with this rhythm. THIRD DEGREE ▪ No impulse from the atria reach the ventricles ▪ Absence of meaningful association between atrial (P wave) and ventricular (QRS complex) depolarization, despite regular and constant P-P (distance between P waves) and R-R (distance between QRS complex) intervals ▪ Atria and ventricles fire electrical impulses independently from each other ▪ Can be congenital or acquired (myocarditis, diphtheria) and cardiac surgery involving area around AV node like VSD closure or repair of complex lesions ▪ Known complication of myocardial abscess secondary to endocarditis ▪ 1 in 22,000 ▪ Some are asymptomatic ▪ Most will present with signs of ↓ cardiac output (from easy fatigabil ity to syncope) ▪ In older children with normal hearts, often asymptomatic, although syncope and sudden death may occur ▪ Infants and toddlers may have night terrors, tiredness with frequent naps and irritability ▪ Prominent peripheral pulse – d/t the compensatory large ventricular stroke volume and peripheral vasodilation ▪ JVP occur irregularly and may be large when the atrium contracts against a closed tricuspid valve (cannon wave) ▪ Systolic murmurs along the left sternal border and apical mid-diastolic murmurs are unusual ▪ Variable first heart sound – d/t variable ventricular filling with AV dissociation ▪ AV block results in enlargement of the heart based on ↑ diastolic ventricular filling ECG ▪ Wide QRS complex ▪ Varied PR intervals ▪ Atrial rate > ventricular rate ▪ Confirms the diagnosis Holter monitoring – to diagnose variation in heart rate, pauses, ventricular tachycardia and other arrhythmias ▪ Permanent pacemaker implantation for symptomatic children and for infants with a HR < 55 bpm ▪ Cardiac pacing – for neonates with HR <55bpm, evidence of heart failure, wide complex rhythms, or CHD (with ventricular rates <70 bpm) ▪ Isoproterenol, atropine, epinephrine – may be tried to temporarily ↑ the heart rate until pacemaker placement can be arranged PROGNOSIS: Usually favorable Batch Clingy CONGENITAL AV BLOCK ▪ Infants born to mothers with SLE (develops within 1 st 3-6 months after birth) or Sjogren’s syndrome ▪ Autoimmune injury of the fetal conduction system by maternally derived IgG antibodies (anti-SSA/Ro, anti-SSB/La) in a mother with overt or asymptomatic SLE or Sjogren syndrome ▪ Ventricular rate lies within the range of 15-75bpm PPS Oral Exam Reviewer 2020 137 Complex ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Clinical and pathological syndrome that results from either ventricular dysfunction or volume or pressure overload ▪ Heart is unable to pump enough blood in a quantity commensurate with body requirements, to dispose of venous return adequately, or a combination of the 2. ▪ Overall incidence and prevalence are unknown CONGESTIVE HEART FAILURE ▪ May be due to CHD or acquired heart diseases ▪ CHD with volume or pressure overload ● Most common cause of CHF in the pediatric age group (90% manifests before 1 yo) ● VSD, PDA, and AVSD – most common causes of CHF in the 1 st 6 months of life ▪ In acquired heart disease, the most common causes are RHD and myocarditis CO = HR x SV SV: determined by preload (volume work), contractility (intrinsic myocardial function), and afterload (pressure work) ▪ ↑ Preload may stretch the myocardial fibers to a degree that CO can no longer be increased ▪ Salt and water retention (preload) would only contribute to pulmonary and systemic congestion and increased diastolic stress for the myocardium ▪ Tachycardia and enhanced contractility may initially augment CO but impose greater workload on the heart, as would vasoconstriction (increased afterload) ▪ LV remodeling with progressive structural deterioration induced by catecholamines, angiotensin II and aldosterone, can lead to more severe heart failure and increase the risk of ventricular arrhythmias ▪ Inflammation of the heart muscle arising from the interplay of a causative agent and the host’s immune response ▪ EA: Coxsackievirus (particularly Coxsackie B), adenovirus, parvovirus, EBV, parechovirus, influenza virus and CMV ▪ Virus – most common causative agents in children MYOCARDITIS ▪ True incidence is underestimated because the disease can be subclinical or mild ▪ Bimodal distribution – prominent peaks in infants and mid-teenage years ▪ Myocardial inflammation aggravated by the immunological response of the host → myocyte injury → ↓ cardiac contractility or ↓ LV systolic function → compromised CO, inadequate oxygen delivery to the body, heart failure ▪ Myocardial insult arises from both direct damage by the pathogen and action of inflammatory cells and mediators PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS ▪ Poor growth, feeding difficulties, failure to thrive, respiratory distress, exercise intolerance, fatigue ▪ Older children: puffy eyelids, swollen feet, tachypnea, dyspnea on exertion, orthopnea ▪ Tachycardia, gallop rhythm, weak and thready pulses – compensatory responses to impaired cardiac function ▪ Increased perspiration and cold wet skin – signs of ↑ sympathetic response; frequently seen in infants LABORATORY FINDINGS / DIAGNOSIS CXR: cardiomegaly ECG: ▪ Nonspecific changes ST-segment or T-wave changes ▪ For diagnosis of heart defect and rhythm disorders 2DED ▪ Reveals ventricular chamber size and systolic/diastolic function ▪ Determines the cause of CHF ▪ Useful in the serial evaluation of efficacy of therapy Inotropes ▪ Augment cardiac contractility ▪ Digitalis, catecholamines, PDE inhibitors (milrinone) Diuretics ▪ Loop diuretics – most potent, diuretics of choice in heart failure ▪ Aldosterone antagonist – anti-aldosterone and potassium sparing effect, good drug to combine with loop diuretics Vasodilators ▪ ↓ Either preload, afterload or both ▪ ↓ Afterload reduction → ↓ wall tension, ↑ CO → ventricular emptying ▪ ACE inhibitors – vasodilators of choice d/t its angiotensin II suppressing effects Carvedilol ▪ β- and α-blocker ▪ Improve cardiac function and symptoms in children with cardiomyopathy PROGNOSIS ▪ In contrast to HF secondary to CHD, the outcome of children with cardiomyopathy remains poor, with a 5-year risk for death or cardiac transplantation of around 50% for patients with dilated cardiomyopathy (DCM) SUPPORTIVE MEASURES ▪ Rest – semi-fowler position; oxygen ▪ Diet: ↑ Na, give potassium supplementation ▪ Improve physical and metabolic requirements: temperature, pH, glucose, calcium, hgb ▪ Control heart rate ▪ Ensure electrolyte and metabolic balance Cardiac MRI ▪ Standard imaging modality for diagnosis ▪ Presence and extent of edema, hyperemic capillary leak, myocyte necrosis, LV dysfunction Troponin – more sensitive and specific in the diagnosis Endomyocardial biopsy ▪ For identifying inflammatory cell infiltrates or myocyte damage ▪ For performing molecular viral analysis ▪ Primary therapy is supportive and centered on management of symptoms and complications – beta blockers and ACE inhibitors ▪ Diuretics ▪ Non-pharmacologic therapy: oxygen, bed rest, high back rest, correction of anemia and metabolic disturbances ▪ Cardiomegaly ▪ Signs of pulmonary venous congestion – d/t L-sided heart failure ▪ Wheezing and rales ▪ Hepatomegaly – sign of systemic venous congestion ▪ Distended neck veins ▪ Ankle edema ▪ Infants and young children ● Fulminant presentation ● Fever, respiratory distress, tachycardia, hypotension ● Gallop rhythm ● Cardiac murmur (apical systolic murmur of mitral insufficiency) ▪ Chest discomfort, palpitation, easy fatigability, syncope TREATMENT / COMPLICATIONS / PREVENTION GOALS ▪ Relief of symptoms ▪ Correction of the underlying causes ▪ Elimination of precipitating factors ▪ ↓ Morbidity and ↑ survival PROGNOSIS ▪ Better in children and adolescents ▪ Poor in newborns (75% mortality) 2DED ▪ ↓ Ventricular systolic function ▪ Cardiac chamber enlargement ▪ Mitral insufficiency ▪ Occ pericardial effusion Batch Clingy DISEASE 138 PERICARDITIS ▪ In the Philippines, the common etiologies are: ● Tuberculosis ● Rheumatic fever ● Bacterial infections Chest pain ▪ Main presenting manifestation ▪ Relieved by sitting up and leaning forward ▪ Aggravated by lying supine ▪ True incidence is difficult to ascertain because asymptomatic or minimally symptomatic episodes are likely undetected Pericardial friction rub ▪ Pathognomonic of a small amount of pericardial fluid ▪ Best heard along the left sternal border with the patient leaning forward ▪ Amount of fluid in the non-distensible pericardial space becomes excessive → ↑ pressure within the pericardium → pressure is transmitted to the heart → compressed chambers → impaired filling ▪ Small to moderate amounts of pericardial effusions can be welltolerated and clinically silent 2DED ▪ Most sensitive for identifying the size and location of a pericardial effusion ▪ Treatment is aimed at the underlying pathology ▪ Salicylates or NSAIDs – for chest pain ▪ IV antibiotics – for 4-6 weeks ▪ Corticosteroids – for severe rheumatic carditis, post-pericardiotomy syndrome, and TB pericarditis ▪ Pericardiocentesis – for detecting etiology, relieving symptoms in large effusions, and preventing cardiac tamponade ▪ Tachycardia, cough, dyspnea, abdominal pain, vomiting, fever ▪ Muffled heart sounds when fluid accumulation is large CARDIAC TAMPONADE ▪ Complication of pericarditis ▪ Pulsus paradoxus ▪ Neck vein engorgement ▪ ↑ central venous pressure, narrow pulse pressure ▪ 2DED: Compression and collapse of the RA and/or RV Bacterial pericarditis: purulent effusion Tuberculous fluid: fibrinous or hemorrhagic ▪ Tuberculous and bacterial effusions are usually moderate to large amount, while effusions secondary to rheumatic fever are small and serous CARDIOMYOPATHY ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Most common form of cardiomyopathy ▪ LV dilation + ↓ LV systolic function ▪ Common indication for cardiac transplantation ▪ Most common etiology: idiopathic ▪ Up to 50% are genetic ▪ In 20-50% of cases, DCM is familial with autosomal dominant inheritance DILATED CARDIOMYOPATHY (DCM) ▪ Annual incidence in children <18yo: 0.57 cases per 100,000 per year ▪ Higher in males, blacks, and infants <1yo ▪ Systolic dysfunction and myocyte injury ▪ The resulting myocardial damage, ventricular enlargement, and poor function likely occur by final common pathways HYPERTROPHIC CARDIO-MYOPATHY ▪ ↑ LV wall thickness in the absence of structural heart disease or hypertension ▪ ↑ Ventricular myocardial wall thickness, N or ↑ systolic function, and diastolic (relaxation) abnormalities ▪ Heterogenous, relatively common, and potentially life-threatening form of cardiomyopathy ▪ Genetic disorder frequently occurs because of mutations in sarcomere or cytoskeletal components of the cardiomyocte ▪ Hypertrophic and fibrotic cardiac muscle demonstrates relaxation abnormalities (diminished compliance) and LV filling may be impaired PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS ▪ Similar to CHF ▪ Include palpitations, syncope, sudden death ▪ Irritability or lethargy, failure to thrive, nausea, vomiting, abdominal pain ▪ Respiratory symptoms – tachypnea, wheezing, cough or dyspnea on exertion) ▪ Infrequently, may present acutely with pallor, altered mentation, hypotension, shock ▪ Tachycardia with narrow pulse pressure ▪ Hepatic enlargement ▪ Rales or wheezing ▪ ↑ Precordial cardiac impulse ▪ Heart may be enlarged to palpation or percussion ▪ Gallop rhythm ▪ Sudden death during physical exertion ▪ Palpitations, chest pain, easy fatigability, dyspnea, dizziness, syncope ▪ Overactive precordial impulse with a lift or heave ▪ Systolic ejection murmur in the aortic region not associated with an ejection click ▪ Apical blowing murmur of mitral insufficiency LABORATORY FINDINGS / DIFFERENTIAL DIAGNOSIS TREATMENT / COMPLICATIONS / PREVENTION ECG ▪ Atrial or ventricular hypertrophy ▪ Nonspecific T-wave abnormalities ▪ Occasionally, atrial or ventricular arrhythmias ACE inhibitors / ARBs + β-blockers (carvedilol/metop) ▪ Reverse remodeling → improve ventricular size and function Furosemide ▪ ↓ Symptoms of pulmonary or systemic venous congestion CXR ▪ Cardiomegaly ▪ Pulmonary vascular prominence ▪ Pleural effusion Implantable cardiac defibrillators – considered for patients with a high risk of sudden cardiac arrest Pacemakers – Improve patients with certain underlying electrical derangements 2DED ▪ Diagnostic ▪ LV enlargement ▪ ↓ Ventricular contractility ▪ Occ, globular (remodeled) LV contour ▪ Evidence of pulmonary hypertension, MR, or other structural cardiac or coronary abnormalities PROGNOSIS ▪ 1- and 5-years freedom from death or transplantation in patients diagnosed with DCM is 60-70% and 50-60% ▪ Independent factors at DCM diagnosis for subsequent death or transplantation: ● Older age ● Heart failure ● Lower LV fractional shortening z score ● Underlying etiology B-blockers (propranolol, atenolol, metoprolol) or calcium channel blockers (verapamil) ▪ ↓ LVOT obstruction, modify LV hypertrophy, improve ventricular filling ▪ Has an anti-arrhythmic benefit ▪ Reduce symptoms ECG ▪ LV hypertrophy ▪ ST segment and T-wave abnormalities (T-wave inversion in the left precordial leads) CXR ▪ Normal or mildly ↑ heart size ▪ Prominent LV 2DED ▪ Diagnostic in identifying, localizing and quantifying the Anti-arrhythmic therapy – if with atrial or ventricular arrhythmias Indications for implantable cardioverter-defibrillator ▪ Documented, previously aborted sudden cardiac arrest Batch Clingy DISEASE 139 (diastolic dysfunction) ▪ Systolic function is preserved or even hyperdynamic but systolic dysfunction may occur late and is a predictor for death or need for cardiac transplant degree of myocardial hypertrophy ▪ Defines, localizes, and quantifies the degree of LVOT obstruction ▪ Demonstrates and quantifies the degree of mitral insufficiency and diastolic dysfunction ▪ Interventricular septum is disproportionately involved → asymmetric septal hypertrophy ▪ Left ventricle is predominantly affected ▪ Cardiac myofibrils and myofilaments demonstrate disarray ▪ Near-normal ventricular chamber size and wall thickness with preserved systolic function ▪ Dramatically impaired diastolic function → ↑ filling pressures and atrial enlargement ▪ <5% of cardiomyopathy cases ▪ F>M RESTRICTIVE CARDIOMYOPATHY ▪ Abnormal ventricular myocardial compliance + high ventricular diastolic pressure → dramatic atrial dilation 2DED ▪ Diagnostic ▪ Normal-sized ventricles with preserved systolic function ▪ Dramatic enlargement of the atria ▪ Abnormal filling parameters ▪ Fibrofatty infiltration and replacement of the normal RV myocardium and occasionally the LV → RV (and LV) systolic and diastolic dysfunction and arrhythmias ▪ Most patients have no symptoms at birth ▪ Rare ▪ Prevalence: 1:5000 in general population Major clinical findings ▪ Global and regional RV and LV dysfunction ▪ Ventricular tachyarrhythmias ▪ Mutations create problems with intercellular adhesion, intracellular calcium, intercellular signaling, and combinations thereof ▪ As many of these myocardial structural and functional changes develop over time, it is important to understand their pathophysiology to remain attuned to earlier pediatric manifestations MRI ▪ Thickened or calcified pericardium ▪ Criteria for diagnosis based on the repolarizing and depolarizing abnormalities ▪ Restrict from competitive sports and strenuous physical activity ▪ Recreational exercise activities should be tailored based on overall clinical status ▪ Screen first-degree relatives of patients using ECG and 2DED Cardiac transplantation ▪ Treatment of choice ▪ Results are excellent in those without pulmonary hypertension or severe CHF COMPLICATIONS Atrial tachyarrhythmias, complete heart block, thromboemboli PROGNOSIS Generally poor with progressive clinical deterioration ▪ Heart transplantation ▪ Myocardial changes manifest during the pediatric years ▪ In the early phase, no detectable structural abnormalities in the myocardium – patients are still at ↑ risk of ventricular arrhythmias and sudden cardiac death (SCD) ▪ Later phase: symptomatic arrhythmias, clinical evidence of RV or biventricular failure Batch Clingy ARRHYTHMOGE-NIC RIGHT VENTRICULAR CARDIO-MYOPATHY (ARVC/D) ▪ Edema, hepatomegaly, ascites – d/t abnormal ventricular filling (diastolic heart failure) ▪ Cough, dyspnea, or pulmonary edema – d/t elevation of L-sided filling pressures ▪ Chest pain, shortness of breath, syncope/ near-syncope, or sudden death during activity ▪ Pulmonary hypertension and pulmonary vascular disease ▪ Prominent gallop rhythm PROGNOSIS ▪ <1yo or those with inborn errors of metabolism, malformation syndromes, or a mixed HCM/DCM have a significantly poorer prognosis ▪ The risk of sudden death in older patients is greater in those with a personal or family history of cardiac arrest, Vtach, exercise hypotension, syncope, or excessive ventricular wall thickness ECG ▪ Prominent P waves ▪ Normal QRS voltage ▪ Nonspecific ST and T-wave changes CXR ▪ Normal or ▪ Prominent atrial shadow ▪ Pulmonary vascular redistribution ▪ Strong family history of sudden death ▪ Ventricular wall end-diastolic dimensions of >3cm ▪ Unexplained syncope, non-sustained ventricular tachycardia, or blunted or hypotensive blood pressure response to exercise PPS Oral Exam Reviewer 2020 140 Acute ETIOLOGY / INCIDENCE / PATHOGENESIS Etiologic Agents ▪ Viruses (predominant): Adenovirus, coronavirus, CMV, EBV, Enteroviruses, HSV, HIV, human metapneumovirus, influenza, measles, parainfluenza, RSV, rhinovirus ▪ Bacteria: GABHS (most important), Acranobacterium haemolyticum, F. necrophorum, C. diphtheriae, N. gonorrhea, Group C Strep, Group G Strep, mycoplasma, F. tularensis, C. pneumonia, C. trachomatis Environmental Exposures ▪ Smoke, air pollutants, allergens PATHOGENESIS ▪ Colonization in the pharynx ▪ GAS (M Protein) ACUTE PHARYNGITIS PERITONSILLAR ABSCESS ▪ Commonly seen in adolescents with recurrent ATP ▪ Group A Strep + mixed oropharyngeal anaerobes ▪ Bacterial invasion through the tonsillar capsule → cellulitis/ abscess in surrounding tissues PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS Viral (General) – conjunctivitis, coryza, cough, diarrhea, hoarseness, discrete ulcerative stomatitis, viral exanthema HSV – gingivostomatitis, ulcerating vesicles, high fever, difficulty taking in fluids Herpangina – discrete papulovesicular lesions or ulcerations (posterior pharynx), severe throat pain HFMD (Coxsakie A16, A6; Enterovirus 71) ▪ Same with herpangina but also involves palms and soles Adenovirus (Pharyngoconjuctival fever) ▪ Pharyngitis resolves in 7days, conjunctivitis may persist up to 14 days ▪ Swimming pool exposures Measles ▪ Intense, diffuse pharyngeal erythema, koplik spots HIV (Acute Retroviral Syndrome) ▪ Non-exudative pharyngitis, fever, arthralgia, myalgia, adenopathy, maculopapular Rash GABHS ▪ Sudden onset sore throat ▪ 5-15 years old ▪ Fever, HA, N/V, abdominal pain, tonsillopharyngeal exudates, palatal petechiae, anterior cervical adenitis (tender nodes) ▪ Winter and early spring presentation ▪ History of exposure to strep pharyngitis ▪ Scarlatiniform rash Fusobacterium necrophorum ▪ 15-30 years old; toxic looking ▪ Lemierre Syndrome (etiologic agent in 80% of cases) ▪ Internal jugular vein septic thrombophlebitis Gonococcal ▪ Asymptomatic ▪ Acute ulcerative or exudative pharyngitis, fever, cervical lymphadenitis Diphtheria ▪ Bull neck, gray pharyngeal pseudomembrane F. tularensis ▪ Ingestion of water, milk or undercooked meat ▪ Severe throat pain, tonsillitis, cervical adenitis, oral ulcerations, pseudomembrane formation ▪ Sore Throat, fever, trismus, muffled or garbled voice, dysphagia ▪ Asymmetric tonsillar bulge with displacement of uvula (poorly visualized because of trismus) LABORATORY FINDINGS / DIAGNOSIS ▪ Throat Culture – gold standard ▪ Rapid Antigen Detection Tests ● Very high specificity ≥95%; if positive, diagnosis is accurate ● Less sensitive than culture; if negative, must confirm with culture ▪ Testing should only be done if signs and symptoms point to bacterial rather than viral etiology ▪ GAS Molecular Tests ● Amplification of specific GAS gene in <1hr ● Sens & Spec ≥98% when compared with standard throat culture ● May not exhibit spectrum bias ● Very sensitive that it may cause unnecessary treatment to carriers ● Prone to contamination with exogenous GAS DNA from other swabs ●More expensive than culture ▪ Special culture media ● Done if suspected organism is not GAS ● A. haemolyticum ▪ CBC – atypical lymphocytes and a positive mononucleosis slide agglutination test → EBV RECURRENT PHARYNGITIS ● ≥7 episodes in the previous year, or ● ≥5 in each of the preceding 2yrs, Or ● ≥3 in each of the previous 3yrs ▪ CT Scan ▪ Ultrasound – differentiates cellulitis vs. abscess; avoids radiation TREATMENT / COMPLICATIONS / PROGNOSIS ▪ Amoxicillin 50mkday (PO) QD x 10 days ▪ Penicillin V (PO) ● <27 kg: 250mg BID x 10 days ● > 27 kg: 500mg BID x 10 days ▪ Benzathine Penicillin G (IM) once ● < 27 kg: 600,000 IU ● > 27 kg: 1,200,000 IU ▪ Benzathine Penicillin G (900,000 IU) + Procaine Penicillin G (300,000 IU) per IM for one dose For Penicillin Allergic Patients: ▪ 1st generation cephalosporins (Varies) x 10 days ● Do not use if with history of anaphylaxis to beta-lactam ▪ Erythromycin Ethylsuccinate 40mkday (Max 1,000 mg/day) BID x 10 days ▪ Erythromycin Estolate 20-40mkday (Max 1,000mg/day) BID x 10 days ▪ Clarithromycin 15mkday (Max 500mg/day) BID x 10 days ▪ Azithromycin ● D1: 12mkday (Max 500mg/day) QD ● D2-5: 6mkday (Max 250mg/day) QD ▪ Clindamycin 20mkday (Max 1800mg/day) TID x 10 days ▪ Tetracyclines, fluoroquinolones, or sulfonamides should not be used to treat GAS pharyngitis Supportive Management ▪ Paracetamol or Ibuprofen ▪ Anesthetic spray/lozenges (benzocaine, phenol, menthol) COMPLICATIONS ▪ AOM, sinusitis, parapharyngeal Abscess ▪ Acute renal failure, PSGN ▪ Poststreptococcal reactive arthritis ▪ Pediatric autoimmune neuropsychiatric disorders associated with streptococci (PANDAS) or childhood acute neuropsychiatric symptoms (CANS) or pediatric acute-onset neuropsychiatric syndrome (PANS) ▪ Chronic GAS Carriage ▪ Directed antibiotic therapy ▪ Surgical Drainage – needle aspiration; incision and drainage ▪ Tonsillectomy COMPLICATIONS ▪ Aspiration Pneumonitis (if abscess ruptures) ▪ Overall, 10% recurrence risk ▪ Recurrence in 5% of s/p needle aspiration → requires I&D Batch Clingy DISEASE 141 ▪ Epstein-Barr Virus (EBV) – 90% Type 1/A – more prevalent; induces in vitro growth transformation of B lymphocytes more efficiently ▪ 5-10% of cases: CMV, T. gondii, adenovirus, hepatitis virus, HIV ▪ Socioeconomically disadvantaged societies ▪ Incidence: 20-70 per 100,000 person-years ▪ Young adults: 100 in 100,000 person-years INFECTIOUS MONONUCLEOSIS (IM) TRANSMISSION ▪ Via oral secretions – viral shedding consistently for > 6mos after acute infection then intermittently for life ▪ Sexual Contact ▪ Transmission → oral epithelial cells and tonsillar B lymphocytes → viremia → blood stream → lymphoreticular system (liver and spleen) ▪ Malaise, fatigue, acute or prolonged fever (> 1 week), headache, sore throat (moderate to severe), nausea, abdominal pain, myalgia ▪ Marked tonsillar enlargement ▪ Palatal petechiae ▪ Lymphadenopathy ● Anterior and posterior cervical nodes – most common ● Epitrochlear lymphadenopathy – suggestive of Mononucleosis ● Axillary, inguinal ▪ Splenomegaly (50%) ▪ Hepatomegaly (10%) ▪ Ampicillin Rash (3-15%) Infectious Gianotti-Crosti syndrome ▪ Symmetric rash on the cheeks with multiple erythematous papules or plaques ▪ 15-50 days ▪ Appears like atopic dermatitis at the extremities or buttocks ▪ Clinical diagnosis ● Consider IM if failure to improve within 48-72 hours of treatment for strep. ▪ Leukocytosis (10,000-20,000) (90%) ● 2/3 lymphocytes; 20-40% are atypical ▪ Mild thrombocytopenia (50,000 - 200,000) (50%) ▪ Mild elevation of liver enzymes ▪ Monospot (Heterophile antibodies) ● Cross-reactive immunoglobulin M (IgM) antibodies that agglutinate mammalian erythrocytes but are not EBV-specific ● Very useful if supported by strong clinical manifestations ▪ Detection of Viral DNA ▪ Rest, adequate fluid and nutrition ▪ Symptomatic treatment ▪ Avoidance of contact sports and strenuous athletic activities if with splenomegaly ▪ Antiviral therapy is not recommended ▪Short course steroids if with airway obstruction COMPLICATIONS ▪ Splenic Rupture ▪ Airway Obstruction ▪ Seizures ▪ Alice in Wonderland Syndrome ● Metamorphosia ● Perceptual distortions of sizes, shapes and spatial relationships ▪ Hemophagocytic lymphohistiocytosis (HLH) ▪ Burkitt Lyphoma ▪ Hodgkin Lymphoma ▪ Aggressive NK Cell Leukemia ▪ Nasopharyngeal Carcinoma ▪ Gastric Carcinoma Batch Clingy ▪ Incubation period: 6 weeks ▪ Classic Triad: fatigue, pharyngitis, generalized lymphadenopathy (90%) ▪ Incubation period: 30-50 days ▪ Shorter in children ▪ Clinical presentation may be silent in children and fast in adults PPS Oral Exam Reviewer 2020 142 Acute Cough (<2 Weeks) | Acute COMMON COLDS ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Rhinovirus (50%), RSV, Human metapneumovirus, Parainfluenza viruses, Adenoviruses, Influenza virus, Enteroviruses, Human Coronavirus ▪ Seasonal prevalence in other countries ▪ Average of 6-8 colds per year ▪ 10-15% of children have at least 12 infections per year → decreases to 2-3/year by adulthood ▪ Direct hand contact (self-inoculation) ▪ Inhalation of small particles ▪ Deposition of large particles on nasal or conjunctival mucosa PATHOGENESIS ▪ HRV – large particles ▪ Influenza Virus – genetic drift and shift ▪ Coronavirus – multiple distinct strains can induce short-term protective immunity PATHOLOGY ▪ Viral infection of nasal epithelium → destruction → acute inflammatory response → cytokine release and infiltration of mucosa by inflammatory cells → symptoms ▪ Peak of viral shedding: 3-5 days after inoculation ▪ Viral shedding may persist for up to 2 weeks ETIOLOGY ▪ Genetics ▪ Environment – virus, allergen, irritant exposure (cold, dry, air, exercise) ASTHMA WITHOUT EXACERBATION EPIDEMIOLOGY (MORE COMMON) ▪ Males, poor ▪ 80% of all asthmatic patients report disease onset before 6 years old EARLY CHILDHOOD RF FOR PERSISTENT ASTHMA Major ▪ Parent asthma ▪ Eczema ▪ Inhalant Allergen Sensitization Minor ▪ Allergic rhinitis ▪ Wheezing apart from colds ▪ ≥4% Peripheral blood eosinophils ▪ Food allergen sensitization ▪ Allergy in young children with recurrent cough and/or wheeze is the strongest identifiable factor for the persistence of childhood asthma. PATHOGENESIS ▪ Airflow obstruction in asthma is the result of numerous pathologic processes. ▪ Bronchoconstriction of muscle layer + inflammatory infiltration of PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS Infants: fever, nasal discharge Older children: ▪ Colds (2-3 days after infection) ▪ Sore or scratchy throat – resolves quickly (2-3 days) then nasal symptoms predominate ▪ Nasal obstruction, rhinorrhea ▪ Hoarseness ▪ Cough (2/3 of cases) – may persist for 1-2 weeks DIAGNOSIS ▪ Clinical ▪ Routine laboratory studies are not helpful for the diagnosis and management of the common cold ▪ Hansel stain – nasal smear for eosinophils to rule out allergic rhinitis ▪ Cultures and PCR – may be useful if GABHS and Bordetella pertussis is suspected Occasional Symptoms: ▪ Headache ▪ Difficulty sleeping ▪ Irritability ▪ Decreased appetite ▪ Vomiting, diarrhea ▪ Increased nasal secretion ▪ Change in color or consistency of mucus ▪ Swollen erythematous nasal turbinates ▪ Anterior cervical lymphadenopathy ▪ Conjunctival injection ▪ Intermittent dry cough and expiratory wheezing – most common chronic symptoms ▪ Shortness of breath ▪ Chest congestion ▪ Chest tightness ▪ Non focal chest pain – common in younger children ▪ Worse symptoms at night, triggered by physical activities ▪ Usually triggered by environmental changes ▪ Decreased breath sounds in some lung fields (right lower posterior lung field is common) ▪ Rhonchi and crackles (or rales) TREATMENT / COMPLICATIONS Supportive care ▪ Maintain adequate oral hydration (warm liquids) ▪ No use of non-prescription cold and cough medications < 6 years old ▪ Zinc – reduces duration if given within 24 hours of symptoms ▪ NSAIDs ▪ Decongestants, saline drops ▪ Antihistamines ▪ Honey Antibacterial therapy ▪ Of no benefit ▪ Should be avoided to minimize possible adverse effects and antibiotic resistance ▪ Ribavirin – severe RSV infections ▪ Oseltamivir or Zanamivir – influenza COMPLICATIONS ▪ Acute Otitis Media – most common, 5-30% ▪ Sinusitis ▪ Asthma Exacerbation Not effective: ▪ Vitamin C, guaifenesin, inhalation of warm, humidified air, echinacea ▪ Risk factors + clinical presentation ▪ Improvement within 10mins from SABA administration ▪ Pulmonary Function Testing ▪ Forced expiratory airflow ▪ Spirometry – can be done as young as 6yo Key components of optimal asthma management ▪ Assessment and monitoring of disease activity ▪ Education to enhance patient and family knowledge and skills for self-management ▪ Identification and management of precipitating factors and comorbid conditions that worsen asthma ▪ Appropriate selection of medications ▪ Long term goal: asthma control For Classification See Table 169.7 of Nelson’s 21st ADOLESCENTS Preferred reliever: low dose ICS-formoterol or SABA Preferred controller: ▪ Step 1: ICS-formoterol PRN or low dose ICS with SABA ▪ Step 2: Daily low-dose ICS or low dose ICS-formoterol PRN or daily LRTA or low dose ICS with SABA ▪ Step 3: Low dose ICS-LABA or medium-dose ICS or low dose ICS+LTRA ▪ Step 4: Medium dose ICS-LABA; High-dose ICS, add on tiotropium or LRTA ▪ Step 5: High dose ICS-LABA or low dose OCS CHILDREN 6-11 YEARS OLD Preferred reliever: SABA PRN Preferred controller: ▪ Step 1: No preferred; May give low dose ICS with SABA or daily low dose ICS ▪ Step 2: Daily low-dose ICS or daily LRTA or low dose ICS with SABA Batch Clingy DISEASE 143 different cell types → epithelial damage and desquamation ▪ Step 3: Low dose ICS-LABA or medium-dose ICS or low dose ICS+LTRA ▪ Step 4: Medium dose ICS-LABA and refer for expert advice; Highdose ICS, add on tiotropium or LRTA ▪ Step 5: Refer for Phenotypic assessment, High dose ICS-LABA or low dose OCS Regular clinic visits every 2-6 weeks for reassessment of asthma control. PEF Monitoring Stepwise Approach *See GINA* See Table 169.11-13 (Nelsons) COMPLICATION Status Asthmaticus PNEUMONIA MINIMUM/ LOW RISK ETIOLOGY Non-infectious: aspiration (food or gastric acid, foreign body, hydrocarbons, lipoid substances), hypersensitivity reactions, drugor radiation-induced pneumonitis Infectious causes: ▪ S. pneumoniae (pneumococcus) – most common in children 3wks to 4yrs of age ▪ Mycoplasma pneumoniae ▪ C. pneumoniae – most frequent in children age ≥5yo ▪ Group A streptococcus (Streptococcus pyogenes) ▪ Staphylococcus aureus ▪ Viral pathogens – most common cause of LRTIs in 1mo – 5yo PATHOGENESIS Viral pneumonia – direct injury of the respiratory epithelium → swelling and airway obstruction → atelectasis, interstitial edema, hypoxemia from VQ mismatch Bacterial pneumonia – colonization of trachea → seeding to the PPS Oral Exam Reviewer 2020 ▪ Preceded by several days of URTI (rhinitis, cough) ▪ Tachypnea – most consistent ▪ Viral – low grade fever, retractions (subcostal, intercostal, suprasternal), alar flaring, fever, crackles, wheeze ▪ Bacterial ● Fever, cough, chest pain, drowsiness, intermittent periods of restlessness, rapid respirations, anxiety, delirium ● Early: grunting, diminished breath sounds, scattered crackles, rhonchi are commonly heard over the affected lung field. A lag in respiratory excursion often occurs on the affected side. ● Abdominal distention, abdominal pain ▪ Chest Radiograph (PA-Lat) ● Viral – hyperinflation with bilateral interstitial infiltrates and peribronchial cuffing ● Pneumococcal – confluent lobar consolidation ▪ CBC – high WBC count ▪ ESR, procalcitonin, CRP Definitive diagnosis: viral genome or antigen detection (for viruses) and sputum culture (Bacteria) ▪ Blood CS only 10% have yield ▪ Gram Stain ▪ Culture and Sensitivity Other exams: ▪ Cold agglutinin – Mycoplasma ▪ ASO – Streptococcus ▪ Ultrasound ▪ Amoxicillin 90 mkday q12 (Nelson’s) ▪ Amoxicillin 40-50mkday (Max 1500) divided to 3 doses in low resistance areas (PAPP) Alternatives ▪ Cefuroxime, co-amoxiclav ▪ Azithromycin 10mkday QD for 3days, or 10mkday at day 1 then 5mkday for D2-5 (max dose 500 mg) Suspected Mycoplasma or Chlamydia – macrolide ▪ Azithromycin, Clarithromycin 15mkday in 2 doses x 7 days (Max 1000 mg) or Doxycycline See Table 428.7 for antibiotic choices in Nelson’s Antibiotic duration: ▪ Until 72 hours afebrile but not less than 10 days all-in-all ▪ 5-7 days is acceptable for OPD Adjuncts: ▪ Oral zinc (10mg/day for <12mos, 20mg/day for ≥ 12mos given for 7days) ▪ Bubble CPAP – improves mortality from pneumonia with hypoxemia compared with standard oxygen therapy in settings Batch Clingy ▪ Inflammation of the lung parenchyma ▪ Leading cause of death globally ▪ Risk Factors: Poverty 144 lung tissue → infection to the lung parenchyma → properties of invading organism determines presentation ▪ Vitamin D3 ▪ Probiotics Recurrent pneumonia – ≥2 episodes in a single year or ≥3 episodes ever with radiographic clearing between occurrences COMPLICATIONS ▪ Pleural effusion, empyema ▪ Asthma after 5 years in 45% of cases ▪ Pericarditis ▪ Acute Respiratory Failure ▪ Pneumothorax ▪ Lung Abscess Rarely, ▪ Meningitis, endocarditis ▪ Suppurative arthritis, osteomyelitis Acute Cough in Distress | Acute ETIOLOGY / INCIDENCE / PATHOGENESIS Etiology – RSV (50%), Parainfluenza, Human metapneumovirus, Rhinovirus, Bocavirus, Adenovirus ▪ Most common diagnosis ▪ More common in males ▪ Only 5-8% of wheezing illness prior to school age develop asthma in adulthood BRONCHIOLITIS RISK FACTORS ▪ Second-hand tobacco smoke ▪ Formula-fed ▪ Crowded conditions ▪ Infants born from mothers who smoked during pregnancy ▪ History of asthma and allergies PATHOGENESIS ▪ Bronchiolar obstruction with edema, mucus and cellular debris ▪ Increased resistance in smaller airways → expiratory wheezing, air trapping, and lung hyperinflation ▪ Complete obstruction → atelectasis CLINICAL MANIFESTATIONS ▪ Sneezing and clear rhinorrhea initially ▪ Diminished appetite ▪ Fever ▪ Respiratory distress ensues, with paroxysmal cough, dyspnea, irritability ▪ Apnea may precede lower respiratory signs early in the disease, particularly with very young infants. ▪ Term infants at a postconceptual age of <44wks and preterm infants at postconceptual age <48wks are at highest risk for apneic events DIAGNOSIS ▪ Clinical ▪ CXR and labs not routinely indicated ▪ Viral testing (PCR, rapid immunofluorescence) – not routinely recommended but may be helpful if such testing prevents more invasive evaluations ▪ If with failure to thrive, consider chronic diseases such as cystic fibrosis or immunodeficiency DIFFERENTIAL DIAGNOSIS Asthma ▪ Tachypnea, wheezing, crackles, rhonchi – 2o to inflammation of the small airways ▪ Expiratory time may be prolonged ▪ Work of breathing may be markedly increased, with nasal flaring and retractions ▪ Complete obstruction to airflow can eliminate the turbulence that causes wheezing thus, the lack of audible wheezing is not reassuring if the infant shows other signs of respiratory distress ▪ Poorly audible breath sounds – suggest severe disease with nearly complete bronchiolar obstruction COMPLICATIONS ▪ Otitis Media ▪ Respiratory Arrest or Failure ▪ Severe dehydration and electrolyte imbalance ▪ A majority of deaths due to bronchiolitis occur in children with complex medical conditions or comorbidities such as bronchopulmonary dysplasia, congenital heart disease, or immunodeficiency ▪ Wheezing with an onset after the first 18-36mos of life is one of the strongest predictors of eventual asthma in both cohorts THREE PATTERNS OF INFANT WHEEZE ▪ Transient early wheezing – 20%, lower lung function at birth which improves with growth resulting in resolution of wheezing by age 3 ▪ Persistent wheezing – 14%, declining lung function and wheezing before and after age 3 ▪ Late-onset wheezing –15%, relatively stable lung function and wheezing that does not begin until after age 3 PNEUMONIA, MODERATE RISK Respiratory Rate ▪ 3-12mos: >60 to <70/min ▪ 1-5yrs: >50/min ▪ >5yrs: >35/min ▪ Intercostal or subcostal retractions ▪ Head bobbing, cyanosis ▪ Irritability PPS Oral Exam Reviewer 2020 TREATMENT / COMPLICATIONS ▪ Supportive management o sPO2 > 90% o Fluids via IV or NGT o Suction secretions ▪ Admit if with respiratory distress (hypoxia, inability to feed, apnea, extreme tachypnea) ▪ Chest physiotherapy – no benefit to children with bronchiolitis ▪ Pharmacologic agents – largely proven ineffective in the management of bronchiolitis Same as mild but with focus on assessment of gas exchange: ▪ O2 saturation, ABG ▪ CRP, procalcitonin ▪ CXR PA-Lat ▪ CBC (WBC) ▪ Chest UTZ (Effusion) ▪ Gram Stain, Culture and Sensitivity May manage as outpatient if the following are NOT present: ▪ Age less than 2 years old ▪ Convulsions ▪ CXR with effusion, lung abscess, air leak, or multilobar consolidation ▪ O2 Sat <95% ▪ Otherwise, admit to ward Batch Clingy DISEASE 145 EPIDEMIOLOGY ▪ Older infants and toddlers ▪ M>F ▪ Most common object: food items (nuts, seeds, hot dogs, hard candy, gum, bones, raw fruits and vegetables) FOREIGN BODY LARYNGOTRACHEITIS (Croup) PATHOGENESIS ▪ Young children are more at risk to aspirate a foreign body largely because of their developmental vulnerabilities and their underdeveloped ability to swallow food. ▪ An infant is able to suck and swallow and is equipped with involuntary reflexes (gag, cough, and glottis closure) that help to protect against aspiration during swallowing. ▪ Despite a strong gag reflex, a child's airway is more vulnerable to obstruction than an adult's airway ▪ Young children are more likely to experience significant blockage by small foreign bodies due to their smaller airway diameter. ▪ Mucus and secretions may form a seal around the foreign body, making it more difficult to dislodge by forced air ▪ In addition, the force of air generated by an infant or young child's cough is less effective in dislodging an airway obstruction. ETIOLOGY AND EPIDEMIOLOGY ▪ Parainfluenza virus types 1, 2, and 3 (75%) ▪ Other viruses: influenza A and B, adenovirus, respiratory syncytial virus, and measles. ▪ 3 months to 5 years old ▪ Peak: 2nd year of life ▪ Males PATHOGENESIS ▪ Viral infection (invasion) at the glottic and subglottic region PPS Oral Exam Reviewer 2020 Additional: Serum Na, K ▪ May range from asymptomatic to severe respiratory distress ▪ History taking – most important ▪ Comprehensive PE of the orifices ▪ Radiograph – look for 2o findings (air trapping, asymmetric hyperinflation, obstructive emphysema, atelectasis, mediastinal shift, consolidation) ▪ Bronchoscopy – done if clinically suggestive despite negative imaging Areas involved: ▪ Larynx – hoarseness, cough, stridor, dyspnea ▪ Trachea – dysphonia, dysphagia, dry cough, biphasic stridor PRESENTATION IN 3 STAGES Initial event: Paroxysms of coughing, choking, gagging, and airway obstruction immediately after aspiration of the FB; the child is sometimes able to expel the foreign body during this stage. Asymptomatic interval: The foreign body becomes lodged, reflexes fatigue, and the immediate irritating symptoms subside. ▪ If with immunization: Penicillin G 100,000 units/kg/day in 4 divided doses ▪ If incomplete or unsure immunization: ampicillin 100mkday divided to 4 doses. ▪ Oral medications may be given if not O2 requiring ▪ MRSA: Vancomycin + Referral to ID If clinically suspected or confirmed influenza virus infection: ▪ Oseltamivir (start within 48 hours) ● 3-8mos: 3mkdose BID x 5days ● 9-11mos: 3.5mkdose BID x 5days ● >12mos: BW <15 kg at 30 mg BID x 5days >15-23 kg at 45 mg BID x 5days >23-40 kg at 60 mg BID x 5days >40 kg at 75 mg BID x 5days ▪ Zanamivir >7yo: 10mg (two 5-mg inhalations) BID x 5days within 36 hours of onset of influenza-like symptoms ▪ Heimlich Maneuver – If laryngeal foreign body is suspected ▪ Endoscopic removal COMPLICATIONS ▪ Complete obstruction of airway – most serious complication ▪ Air trapping ▪ Atelectasis For bronchoscopy ▪ Bronchospasm ▪ Desaturation ▪ Bleeding ▪ Airway edema Complications: Obstruction, erosion, or infection develops. Patient may have include fever, cough, hemoptysis, pneumonia, atelectasis ▪ Acute or chronic complications have been reported in almost 15% of cases of foreign bodies of the airway. ▪ Rhinorrhea, pharyngitis, mild cough, and low-grade fever for 1-3 days before apparent upper airway obstruction ▪ Barking cough, hoarseness, and inspiratory stridor ▪ Symptoms are worse at night ▪ Prefer to sit up in bed or be held upright ▪ Hoarseness, coryza, inflamed pharynx, slight tachypnea ▪ Nasal flaring, retractions, persistent stridor ▪ Clinical Diagnosis ▪ Radiograph: subglottic narrowing, or steeple sign but not diagnostic COMPLICATIONS ▪ Bacterial tracheitis ▪ Otitis media ▪ Pneumonia ▪ Airway management Nebulized racemic epinephrine – established treatment for moderate or severe croup ▪ 0.25-0.5 ml of 2.25% racemic epi in 3 mL of normal saline every 20 mins. ▪ Duration of activity < 2 hours ▪ No rebound effect. Oral dexamethasone – single dose of 0.6mg/kg, a dose as low as 0.15mg/kg may be just as effective Batch Clingy ▪ Choking is a leading cause of morbidity and mortality among children, especially those <4yo ▪ Convulsions ▪ CRT >3secs ▪ Pallor ▪ Moderately malnourished, unable to drink ▪ CXR: effusion, abscess, air leak or multilobar consolidation ▪ O2 Sat 91-94% 146 EPIGLOTTITIS BACTERIAL TRACHEITIS ASTHMA IN EXACERBATION ▪ Staphylococcus aureus – most common ▪ S. pneumoniae, S. pyogenes, M. catarrhalis, non-typeable H. influenzae ▪ Mean Age: 5-7 years old ▪ Males ▪ Bacterial infection preceded by a viral respiratory infection (LTB) ▪ More common than epiglottitis in vaccinated populations ▪ Brassy cough, high fever, toxic-looking ▪ Lies flat, no drooling, no dysphagia ▪ Mucosal swelling at the level of the cricoid cartilage, complicated by copious, thick, purulent secretions, sometimes causing pseudomembranes ▪ Progressive ↑ in symptoms of shortness of breath, cough, wheezing or chest tightness and progressive ↓ in lung function ▪ Usual Triggers: ● Viral infections ● Allergen, food allergen, outdoor air pollution ● Seasonal changes ● Poor adherence to ICS ▪ Worsen during sleep (midnight – 8 AM) Mild Breathless-ness Can lie down Sentences May be agitated Increased Increased Severe While at rest (stops feeding) Sits upright Words Drowsy or confused Often > 30 cpm Usually not Common Usual Pulse Rate (bpm) Moderate; Often endexpiratory <100 Pulsus Paradoxus Absent < 10 mmHg Loud; throughout exhalation 100-120 May be present > 10-25 mmHg Usually loud; throughout inhalation and exhalation >120 Often present > 20-40 mmHg (child) Wheeze FUNCTIONAL ASSESSMENT Peak Expiratory Flow PaO2 PCO2 SaO2 at sea level PPS Oral Exam Reviewer 2020 > 70% Normal < 42 mmHg > 95% Hypercapnea ▪ Laryngoscopy under OR-ICU set-up. ▪ Lateral Radiograph – thumb sign ▪ Cultures: blood, epiglottic surface, CSF 10-40% are resistant to ampicillin ▪ Clinical diagnosis – upper airway disease with high fever, purulent secretions, and absence of epiglottitis features ▪ Incentive Spirometry ▪ Peak Expiratory Flow (PEF) ▪ Forced Expiratory Volume in 1 Second (FEV1) Moderate While at rest (difficulty feeding) Prefers sitting Phrases Usually agitated While walking Talks in.. Alertness SIGNS RR Use of accessory muscles ▪ High fever, sore throat, dyspnea, and rapidly progressing respiratory obstruction. ▪ Previously healthy child suddenly develops a sore throat and fever → toxic-looking within a matter of hours: difficulty in swallowing, labored breathing, drooling, hyperextended neck (tripod position) ▪ Air hunger with restlessness → cyanosis, coma ▪ Stridor is a late finding and suggests near-complete airway obstruction ▪ The barking cough typical of croup is rare. ~ 40-69% or response lasts <2 hours > 60 mmHg < 42 mmHg 90-95% < 40% < 60 mmHg; possible cyanosis > 42 mmHg <90% ▪ Secure airway ▪ Prepare for possible intubation in an OR-ICU set-up ▪ O2 supplementation (unless patient is agitated with mask) ▪ Start on ceftriaxone, cefepime, or meropenem ▪ Complete antibiotics for 10d ▪ Racemic epinephrine and corticosteroids are ineffective Rifampicin Prophylaxis ▪ 20mkday x 4 days (max dose: 600 mg) ▪ <4yo, incompletely immunized ▪ <12mos old, incomplete primary vaccination ▪ Immunocompromised COMPLICATIONS Respiratory arrest or failure ▪ Supplemental oxygen ▪ Antibiotics: Vancomycin or clindamycin and a 3rdgeneration cephalosporin (e.g. ceftriaxone or cefepime) COMPLICATIONS ▪ Cardiorespiratory Arrest ▪ Toxic Shock Syndrome ▪ Written action plan: medication instructions and when to come back ▪ Increase controller medications from previous following the recommended stepwise approach, if: ● Asthma symptoms are interfering with normal activities ● PEF has fallen by >20% for >2days ▪ The need for repeated doses of SABA over more than 1–2 days signals the need to review, and possibly increase, controller treatment if this has not already been done. ▪ Combination low dose ICS (budesonide or beclometasone) with formoterol maintenance and reliever regimen ● Effective in improving asthma symptom control ● Reduces exacerbations requiring OCS and hospitalizations ● Formoterol max dose: 72 mcg ● Not approved in 4-11 years old ● Do not use with other ICS or ICS-LABA ▪ Leukotriene Receptors Antagonists ▪ Oral Corticosteroids – 6-11yo 1-2mkday x 3-5d (max 40/day), with indications: ● Failure to improve with controller in 2-3d ● Rapid deterioration ● PEF or FEV1 <60% ● With history of severe exacerbations ▪ If with 1-2 exacerbations/year despite Step 4-5 therapy, refer to specialist COMPLICATION ▪ Status Asthmaticus Batch Clingy ▪ H. influenzae type b – most common (pre-vaccination) ▪ Other etiologic agents: S. pyogenes, S. pneumoniae, nontypeable H. influenzae, S. aureus ▪ Usually seen in 2-4 years old ▪ Some cases in 1st year of life and in 7-year-old children 147 Acute Cough in Distress | Complex ETIOLOGY / INCIDENCE / PATHOGENESIS PNEUMONIA HIGH RISK CLINICAL MANIFESTATIONS ▪ Respiratory Rate ● 3-12mos: >70/min ● 1-5years: >50/min ● >5years: >35/min ▪ Intercostal, subcostal, or suprasternal retractions ▪ Head bobbing, cyanosis ▪ Grunting ▪ Apnea ▪ Lethargic/stupor/coma ▪ Convulsions ▪ Shock ▪ Pallor ▪ Severely malnourished ▪ Unable to drink DIAGNOSIS Similar with severe but with focus on assessment of gas exchange: ▪ O2 Saturation, ABG ▪ CRP, procalcitonin ▪ CXR PA-Lat ▪ CBC (WBC) ▪ Chest UTZ (Effusion) ▪ Gram Stain, Culture and Sensitivity Additional labs to investigate metabolic derangements ▪ ABG ▪ Serum Na, K ▪ CXR: effusion, abscess, air leak or multilobar consolidation ▪ O2 Sat ≤90% ARDS ▪ Respiratory failure - oxygenation and ventilation are insufficient to meet the metabolic demands of the body. ● PaO2 <60 mm Hg when breathing room air ● PaCO2 >50 mm Hg resulting in acidosis ▪ ALI → ARDS ● Onset: within 1 week of insult ● Origin: Not fully explained by cardiac failure or fluid overload ● Chest imaging: new infiltrate(s) consistent with acute pulmonary parenchymal disease ▪ Non-invasive MV ● PF Ratio < 300 ● SF Ratio < 264 ▪ Invasive MV Hypoxic respiratory failure – intrapulmonary shunting and venous admixture or insufficient diffusion of O2 from alveoli into pulmonary capillaries Hypercarbic respiratory failure – decreased minute ventilation (i.e. TV x RR) ▪ Asthma exacerbation which does not improve with standard therapy STATUS ASTHMATICUS PPS Oral Exam Reviewer 2020 ▪ Depends on the site involved ● Alar flaring, tachypnea, retractions, grunting, dyspnea ● Wheezing ● Altered sensorium, unresponsiveness, lethargy ● Poor cry, rapid shallow respirations ▪ Pulse oximetry monitoring – keep between 94-96% ▪ Capnography – determine the effectiveness of ventilation and pulmonary circulation ▪ Clinical diagnosis + Blood Gas ● PaO2 <60 mm Hg when breathing room air ● PaCO2 >50 mm Hg resulting in acidosis ▪ Extreme dyspnea, prolonged exhalation ▪ Anxiety ▪ Upright, leaning forward ▪ Unable to talk, drowsy or confused ▪ Paradoxical breathing ▪ Absent breath sounds ▪ Bradycardia ▪ PEF < 25% ▪ Hypoxia despite O2 supplementation, hypercapnea ▪ Signs of severe exacerbation with no response within 1-2 hours from administering SABA ▪ Recurrence of exacerbation within 48 hours of administering oral corticosteroids TREATMENT / COMPLICATIONS ▪ Refer to specialist ▪ May manage as outpatient if the following are NOT PRESENT: ● Age less than 2 years old ● Convulsions ● CXR with effusion, lung abscess, air leak or multilobar consolidation ● O2 Sat < 95% ▪ Otherwise, admit to ward ▪ If with immunization: Penicillin G 100,000 units/kg/day in 4 divided doses ▪ If incomplete or unsure immunization: ampicillin 100mkday divided to 4 doses ▪ Oral medications may be given if not O2 requiring ▪ MRSA: Vancomycin + Referral to ID If clinically suspected or confirmed influenza virus infection ▪ Oseltamivir: (start within 48 hours) ● 3-8mos: 3mkdose BID x 5 days ● 9-11mos: 3.5mkdose BID x 5 days ● >12mos: BW <15 kg at 30 mg BID x 5 days >15-23 kg at 45 mg BID x 5 days >23-40 kg at 60 mg BID x 5 days >40 kg at 75 mg BID x 5 days ▪ Zanamivir: >7yo: 10 mg (two 5-mg inhalations) BID x 5 days, within 36 hours of onset of influenza-like symptoms. Goals: ▪ Ensure a patent airway ▪ Adequate oxygenation ▪ Removal of Carbon Dioxide ▪ Oxygen administration ▪ Airway adjuncts (oropharyngeal and NP airway) ▪ Inhaled gases – Helium-oxygen mixtures, nitric oxide ▪ Positive Pressure Respiratory Support – high-flow nasal cannula, CPAP, NIPPV ▪ Intubation – sedated or rapid sequence intubation ● PaO2 <60 mmHg while breathing >60% oxygen ● PaCO2 >60 mmHg ● pH <7.25 ▪ ICU Admission ▪ O2 via FM to maintain 94-98% O2 Sat ▪ SABA: 2-6 puffs or 2.5mg diluted in 3 mL NSS; 2-3 puffs per hour may be given. ▪ Systemic Steroids: ● 1-2yo: 1-2mkday (max 20) ● 2-5yo: 30 mg ● IV Methylpred 1mkday q6 ▪ Ipratropium Bromide: 2 puffs or 250 mcg/neb q 20 x 1 hour ▪ Magnesium sulfate Batch Clingy DISEASE 148 Subacute (2-4 weeks) | Acute ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Nonspecific bronchial inflammation often following a viral infection ▪ Usually viral in origin: influenza ▪ Common bacterial etiology: pertussis, diphtheria, S. aureus, S. pneumoniae (not necessarily) BRONCHITIS CLINICAL MANIFESTATIONS ▪ Cough is prominent followed by constitutional symptoms (fever and malaise). Protracted cough may last 1-3 weeks ▪ Nonspecific URTI (rhinitis) → frequent, dry, hacking cough which may or may not be productive 3-4 days after ▪ Sputum becomes purulent due to leukocyte migration ▪ Emesis of sputum that was swallowed ▪ Chest pain Early findings: no or low-grade fever, upper respiratory signs – nasopharyngitis, conjunctivitis, rhinitis, clear BS Later findings: coarse and fine crackles, scattered high-pitched wheezing ▪ M. tuberculosis complex: M. tuberculosis, M. bovis, M. africanum, M. microti, M. canetti CLINICAL STAGES ▪ Exposure – recent contact, TST or IGRA (-), CXR and PE is normal ▪ Tuberculosis infection (TBI) – TST positive (hallmark) or IGRA positive, asymptomatic with normal PE, CXR may show granuloma or calcifications. ▪ Disease – symptomatic and apparent radiographic evidence. EPIDEMIOLOGY ▪ Ethnic minorities (Asian, non-Hispanic Black and Hispanic Children) ▪ 3 cases per 100,000 persons TUBERCULOSIS PATHOGENESIS ▪ Multiplication of tubercle bacilli in the alveoli → survives in non-activated macrophages and carried to LN → intensifies over 2-12 weeks → tissue hypersensitivity → fibrosis and encapsulation → enlarging LN → may cause obstruction ▪ Seeding to different organs (RES, brain, kidneys, bones) or dissemination especially in immunocompromised patients Primary Pulmonary Disease ▪ Hallmark: relatively large size of the regional lymphadenitis compared with the relatively small size of the initial lung focus ▪ Usual sequence: hilar lymphadenopathy → focal hyperinflation → atelectasis ▪ Nonproductive cough and mild dyspnea (most common), fever, night sweats, anorexia, ↓ activity ▪ Infants: failure-to-thrive, poor weight gain, wheezing, ↓ breath sounds Progressive Primary Pulmonary Disease ▪ High fever, severe cough with sputum production, weight loss, night sweats ▪ Diminished breath sounds, rales, dullness or egophony Reactivation Tuberculosis ▪ CXR: extensive infiltrates, thick-walled cavities in the upper lobes ▪ Older child and adolescents: fever, anorexia, malaise, weight loss, night sweats, productive cough, hemoptysis, chest pain Pleural Effusion ▪ Commonly asymptomatic ▪ Tuberculous pleural effusion is uncommon in children <6yo and rare in children <2yo ▪ Sudden onset, low to high fever, shortness of breath, chest pain on deep inspiration, and diminished breath sounds Pericardial Disease (Pericarditis) ▪ Low-grade fever, malaise, weight loss ▪ Pericardial friction rub, distant heart sounds, pulsus paradoxus PPS Oral Exam Reviewer 2020 DIAGNOSIS ▪ CXR: Normal or with ↑ bronchial markings ▪ Absence of abnormality of VS (tachycardia, tachypnea, fever) and a normal physical examination of the chest reduce the likelihood of pneumonia TREATMENT / COMPLICATIONS ▪ No specific therapy ▪ No need for antibiotics ▪ Cough suppressants may be used judiciously Nonprescription cough and cold medicines ▪ Should not be used in children <4yo ▪ Use with caution if 4-11yo Positive TST or IGRA + abnormal chest radiograph consistent with TB + history of recent exposure to an adult with infectious TB – highly suggestive of the clinical diagnosis of TB disease ▪ Isoniazid (H) 10-15mkday (max 300) ▪ Rifampin (R)15-20mkday (max 600) ▪ Pyrazinamide (Z) 30-40mkday (max 2000) ▪ Ethambutol (E) 20mkday (max 2500) Chest X-ray and other imaging Tuberculin Skin Testing (TST) ▪ Tuberculin sensitivity develops 3wks to 3mos (often in 4-8wks) after inhalation of organisms ▪ False negatives: Very young age, malnutrition, immunosuppression by disease or drugs, viral infections (measles, mumps, varicella, influenza), vaccination with live-virus vaccines, overwhelming TB, corticosteroid therapy Interferon-γ Release Assay (IGRA) ▪ Cannot differentiate between TBI and TB disease ▪ Advantages: only one patient encounter (vs two with TST) and the lack of cross-reaction with BCG vaccination Mycobacterial Sampling, Susceptibility and Culture ▪ Induced sputum with a jet nebulizer, inhaled saline, and chest percussion followed by NP suctioning is effective in children as young as 1yo ▪ Early morning gastric acid as specimen ▪ Negative cultures never exclude the diagnosis of tuberculosis in a child. Nucleic Acid Amplification Tests ▪ Sensitivity 25–83%, and specificity has varied from 80–100%. Gene Xpert MTB/RIF ▪ Real-time PCR assay ▪ Simultaneously detects rifampin resistance – often used as a proxy for MDR TB CSF Analysis (CNS TB) CT Scan ▪ CNS infection ▪ Usual regimen: 6 months: 2 HRZE 4 HR ▪ Bone and Joint: 12 months: 2 HRZE 10 HR ▪ Surgical debridement in bone and joint disease ▪ VP Shunt in CNS disease ▪ HIV-infected children: 6-9mos or 6mos after culture of sputum becomes sterile; treatment should be daily ▪ Treatment of drug-resistant tuberculosis is successful only when at least 2 bactericidal drugs are given to which the infecting strain of M. tuberculosis is susceptible. Drug-Resistant Tuberculosis Primary resistance – infected with M. tuberculosis that is already resistant to a particular drug Secondary resistance - drug- resistant organisms emerge as the dominant population during treatment; usually due to poor adherence ▪ Treatment duration of 9mo with RZE is usually adequate for isoniazid-resistant tuberculosis ▪ For IR resistance, total duration of therapy must be extended to 12-24mos, intermittent regimens should not be used 2nd Line Treatment ▪ Bedaquiline – children ≥12yo and >33 kg, with the same indications specified for delamanid ▪ Delamanid – children ≥6yo and ≥20 kg in whom a 4-drug regimen plus pyrazinamide cannot be used because of drug resistance ▪ Have baseline ECG and QTc monitoring for both Bedaquiline and Delaminid. ▪ Linezolid, clofazimine Corticosteroids ▪ Indicated in TB meningitis, endobronchial TB, and pericardial Batch Clingy DISEASE 149 Lymphohematogenous (Disseminated) Disease ▪ Depends on the burden of organisms released from the primary focus to distant sites and the adequacy of the host's immune response ▪ Indolent and prolonged ▪ Spiking fever TB ▪ Prednisone 1-2mkday in 1-2 divided doses for 4-6 weeks Latent Tuberculosis Infection ▪ 6-9 months of isoniazid ▪ 3 months of rifampicin and isoniazid ▪ 4-6 months of rifampicin (daily) ▪ Isoniazid and rifapentine (once weekly) x 12 wks Miliary tuberculosis ▪ Occurs 2-6 months after initial infection ▪ Most common in infants and young children, malnourished and immunosuppressed patients ▪ Insidious with early systemic signs – anorexia, weight loss, low-grade fever ▪ Generalized lymphadenopathy & hepatosplenomegaly within several weeks ▪ Dyspnea, cough, rales, wheezing Window Prophylaxis ▪ Isoniazid should be given to children <5yo who have a (-) TST or IGRA result but who have a known recent exposure to an adult with potentially contagious TB disease. ▪ TST or IGRA done at 8-10 weeks after exposure ● If positive, complete LTBI treatment ● If negative, stop LTBI treatment Upper Respiratory Tract Disease ▪ Laryngeal TB: croup-like cough, sore throat, hoarseness, dysphagia ▪ TB of the middle ear: painless unilateral otorrhea, tinnitus, ↓ hearing, facial paralysis, perforated TM Lymph Node Disease (Scrofula) ▪ Most common form of extrapulmonary tuberculosis in children ▪ Acute, with rapid enlargement, tenderness, and fluctuance of LN, high fever ▪ Lesion is discrete, nontender, and firm but not hard, usually unilateral but may be bilateral, involving multiple nodes and cause matted nodes COMPLICATIONS ▪ Respiratory distress ▪ Pneumothorax ▪ TB effusion ▪ Pneumomediastinum Nontuberculous mycobacteria (NTM) ▪ Cat-scratch disease (Bartonella henselae), tularemia, brucellosis, toxoplasmosis, pyogenic infection Central Nervous System Disease ▪ Most serious complication; fatal without prompt and appropriate treatment ▪ SIADH ▪ Stage 1: nonspecific symptoms – fever, HA, irritability, drowsiness, malaise. No focal neurologic signs but infants can experience a stagnation or loss of developmental milestones. ▪ Stage 2: lethargy, nuchal rigidity, seizures, (+) Kernig and Brudzinski, hypertonia, vomiting, CN palsies, and other focal neurologic signs – encephalitis, disorientation, movement D/O, speech impairment ▪ Stage 3: coma, hemi- or paraplegia, HTN, decerebrate posturing, deterioration of vital signs, death Bone and Joint Disease (Pott disease) ▪ Vertebrae – most common involvement ▪ May present with gibbus formation (vertebrae) ▪ Sterile polyarticular (large joints) CHLAMYDIA PNEUMONIA ▪ TB peritonitis – most often in young ▪ TB enteritis – commonly the jejunum and ileum near Peyer patches and the appendix; shallow ulcers that cause pain, diarrhea or constipation, weight loss, low-grade fever ▪ Renal TB – sterile pyuria, microscopic/gross hematuria, dysuria, flank, or abdominal pain ▪ Genital tract TB – fallopian tubes (90–100%), endometrium (50%), ovaries (25%), cervix (5%); lower abdominal pain, dysmenorrhea, amenorrhea ▪ Congenital TB – most common route of infection for the neonate is postnatal airborne transmission from an adult with infectious pulmonary TB ▪ Perinatal Disease – may be present at birth but usually begins by the 2nd or 3rd week of life; family history of TB; respiratory distress, fever, hepatic or splenic enlargement, poor feeding, lethargy or irritability, lymphadenopathy, abdominal distention, failure to thrive, ear drainage, skin lesions ▪ C. pneumoniae, C. trachomatis ▪ Mild to moderate: constitutional symptoms – fever, malaise, headache, cough, pharyngitis PPS Oral Exam Reviewer 2020 ▪ Isolation of organism in tissue culture (at posterior pharynx) CXR: diffuse lobar infiltrates with small pleural effusion (appears ▪ Tetracycline ▪ Macrolides Batch Clingy Abdominal and Gastrointestinal Disease ▪ Most common lesion is a painless ulcer on the mucosa, palate, or tonsil with enlargement of the regional LNs 150 PATHOGENESIS ▪ Person to person transmission ▪ Elementary body (EB) → attach to host cells via electrostatic binding → endocytosis → differentiate to Reticulate body (RB) in 36 hours that undergo binary fission → after 48 hours, cytolysis or exocytosis ▪ Mycoplasma pneumoniae ▪ Complete absence of a cell wall → dependence to host cells for obtaining essential nutrients; intrinsic resistance to β-lactam agents MYCOPLASMA PNEUMONIA EPIDEMIOLOGY ▪ ~20% of all CAP in middle and high school children ▪ 5% in < 5 yr ▪ 16% in 5-9 yr ▪ 23% in 10-17 yr ▪ Transmission rates of 40 to > 80% for household adult and children contacts, respectively. ▪ Incubation period: 2-3 weeks ▪ Severe: pneumonia with pleural effusion and empyema ▪ Symptoms of asthma and acute chest syndrome in sickle cell disease patients ▪ Rales, wheeze worse than the clinical status) CBC: may be elevated with left shift Microimmunofluorescence (MIF): for acute infection, 4-fold increase in IgG titer or an IgG titer of ≥16; use of a single elevated IgG titer was discouraged ▪Tracheobronchitis and atypical pneumonia – most common ▪ Abrupt onset but with gradual development of headache, malaise, fever, and sore throat ▪ Hoarseness and non-productive cough ▪ Clinical hallmark: worsens during the 1st wk of illness, symptoms generally resolve within 2 wk; cough up to 4 weeks, may be accompanied by prolonged wheezing ▪ Auscultation may be normal or dry rales ▪ PCR throat swab ▪ Culture on special media (SP4 Agar media) – mulberry colonies after 2-3 weeks ▪ ELISA to detect IgM and IgM against M. pneumonia CXR: interstitial or bronchopneumonic, most commonly involving lower lobes; bilateral diffuse infiltrates, lobar pneumonia or hilar lymphadenopathy in up to 30%; may have pleural effusion CBC: normal ESR, CRP: elevated PATHOGENESIS ▪ Attaches to sialic acid receptors in the cilia of respiratory epithelial cells of the respiratory tract → ciliostasis and sloughing off of cells and hydrogen peroxide production → biofilm production → hinders antibiotic penetration and immune system recognition ▪ Community-Acquired Respiratory Distress Syndrome (CARDS) – exotoxin involved in severe or even fatal disease ▪ At Lower Respiratory Tract: polyclonal activation of B & T lymphocytes + CD4+ cells → cytokine production + cold agglutinins (IgM Ab) RHINOSINUSITIS ▪ Viral from common colds ▪ S. pneumoniae (~30), nontypeable H. influenzae (~30%), M. catarrhalis (~10%). ▪ MRSA, α- and β-hemolytic streptococci, coagulase-negative staphylococci ▪ Nasal congestion, purulent nasal discharge (unilateral or bilateral), fever, cough ▪ Halitosis, hyposmia, periorbital edema ▪ Headache, facial pain, maxillary tooth discomfort, pain or pressure exacerbated by bending forward Predisposing conditions: viral URTI, AR, tobacco smoke exposure, immune deficiencies, cystic fibrosis, ciliary dysfunction, abnormalities of phagocyte function, GER, anatomic defects (cleft palate), nasal polyps, cocaine abuse, nasal foreign bodies ▪ Erythema and swelling of the nasal mucosa with purulent nasal discharge, sinus tenderness ▪ Transillumination: opaque sinus ▪ Acute <30 days ▪ Subacute 1-3mos ▪ Chronic >3mos PATHOGENESIS ▪ Viral URTI → viral rhinosinusitis (MRI: mucosal thickening, PPS Oral Exam Reviewer 2020 Suggestive of sinusitis ▪ Persistence of nasal congestion, rhinorrhea (of any quality), and daytime cough ≥10d without improvement ▪ Severe symptoms of temperature ≥39°C (102°F) with purulent nasal discharge for ≥ 3 days ▪ Worsening symptoms: recurrence of symptoms after an initial improvement or new symptoms of fever, nasal discharge, daytime ▪ Clinical Diagnosis: 2 Major or 1 major with 2 or more minor ▪ Sinus aspirate culture – only accurate method of diagnosing ▪ CT Scan – opacification, mucosal thickening, or presence of an airfluid level ● Erythromycin 40mkday PO BID x 10 days ● Clarithromycin 15mkday PO BID x 10 days ● Azithromycin 10mkday PO on day 1, and then 5mkday PO on days 2-5 ▪ Quinolones COMPLICATIONS ▪ Acute chest syndrome in Sickle Cell Disease ▪ Acute otitis media ▪ Persistent cough for several weeks ▪ Treatment may be more effective when started within 3-4 days of illness onset ▪ Clarithromycin 15mkday BID x 10 days ▪ Azithromycin 10mkday on D1, 5mkday on D2-5 ▪ Doxycycline 100mg BID x 7-14 days – child >8yo ▪ Levofloxacin 750mg QD x 7-14days – not recommended as a first-line therapy in children Macrolide-Resistant: ▪ Doxycycline 2-4mkday QD or BID x 10 days (max 200 mg) – >8yo ▪ Levofloxacin 10mkdose q12 children <5yrs or QD in older children COMPLICATIONS ▪ Large pleural effusions with lobar infiltrates and necrotizing pneumonia in sickle cell disease and immunodeficiencies, Down Syndrome, and Chronic Cardiopulmonary Disease ▪ Encephalitis, ADEM, transverse myelitis, cerebellar ataxia, aseptic meningitis, Guillain-Barré syndrome, Bell palsy, peripheral neuropathy ▪ Maculopapular rash, urticaria, SJS, Gianotti-Crosti syndrome, erythema nodosum ▪ Thrombocytopenia, aplastic anemia, coagulation defects ▪ Arthritis, cardiac complications (pericarditis, myocarditis, and rheumatic fever-like syndrome) ▪ Uncomplicated mild to moderate acute bacterial sinusitis: Amoxicillin 45mkday BID ▪ Penicillin-allergic: cefdinir, cefuroxime axetil, cefpodoxime, or cefixime, levofloxacin ▪ Children with risk factors (antibiotic treatment in the preceding 1-3mos, daycare attendance, or age <2yo) for the presence of resistant bacterial species, and for children who fail to respond to initial therapy with amoxicillin within 72hrs, or with severe sinusitis: ● Co-Amoxiclav 80-90mkday of amoxicillin ● Ceftriaxone 50mkday IV or IM ▪ Refer to ENT COMPLICATIONS Orbital complications: periorbital cellulitis, orbital cellulitis ▪ CT Scan ▪ Refer to Ophtha and ENT Batch Clingy EPIDEMIOLOGY ▪ Affects all ages ▪ 2-19% of CAP ▪ Droplet transmission 151 edema, inflammation) ▪ Nose blowing → propels nasal secretions to sinuses → bacteria from nasopharynx enters sinuses → inflammation and edema → blocked sinus drainage, impaired mucociliary clearance of bacteria cough ▪ Surgical drainage ▪ IV antibiotics Intracranial complications: epidural abscess, meningitis, cavernous sinus thrombosis, subdural empyema and brain abscess ▪ Cranial CT Scan ▪ IV antibiotics ▪ Surgical drainage (50%) Pott puffy tumor – osteomyelitis of frontal bone characterized by edema and swelling of the forehead Mucoceles Major Symptoms: purulent anterior nasal discharge, purulent or discolored posterior nasal discharge, nasal congestion or obstruction, facial congestion or fullness, facial pain or pressure, hyposmia or anosmia, fever (For acute sinusitis only) Minor Symptoms: headache, ear pain, pressure or fullness, halitosis, dental pain, cough, fever (for subacute or chronic sinusitis), fatigue Chronic Cough (>4 weeks) | Complex DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ B. pertussis (causes pandemic), B. parapertussis – exclusive pathogens of humans and some primates which causes the epidemic ▪ B. holmesii – in immunocompromised hosts without cough illness; causes pertussis-like cough illness in small outbreaks in healthy persons ▪ B. bronchiseptica – common animal pathogen CLINICAL MANIFESTATIONS Catarrhal Stage ▪ 1-2 weeks ▪ Nasal congestion, rhinorrhea, low-grade fever, sneezing, lacrimation, conjunctival suffusion ▪ Pertussis transmitted via droplet → hemagglutinin, agglutinogens, perctactin allows attachment to ciliated respiratory epithelial cells ▪ Pertussis toxin (major virulence protein) – increases histamine sensitivity, insulin secretion, leukocyte dysfunction ▪ Tracheal cytotoxin + PT – inhibits the clearance of bacterial Paroxysmal Stage ▪ 2-6 weeks ▪ Onset of cough – dry, intermittent, irritative hack, → inexorable paroxysms (hallmark) ▪ Machine-gun burst of uninterrupted cough on a single exhalation, chin and chest held forward, tongue protruding maximally, eyes bulging and watering, face purple, until coughing ceases and a loud whoop ▪ Posttussive emesis, exhaustion ▪ Extremely contagious (100% attack rate) via droplet with high rates of subclinical infection (80%) in households ▪ Incubation period: 3-12 days Convalescent Stage ▪ ≥2 weeks ▪ Number, severity, and duration of episodes diminishes DIAGNOSIS ▪ Clinical: predominant complain of cough with the absence of fever, malaise/myalgia, exanthem or enanthem, sore throat, hoarseness, tachypnea, wheezes, rales Clinical case definition: Cough of ≥14 days duration with at least 1 associated symptom of paroxysms, whoop, or post tussive vomiting has sensitivity of 81% and specificity of 58%. CBC: Leukocytosis (15,000-100,000 cells/μL), absolute lymphocytosis (catarrhal stage) CXR: perihilar infiltrate or edema and variable atelectasis PCR via nasopharyngeal wash: test of choice Culture: nasopharyngeal TREATMENT / COMPLICATIONS Hospitalization ▪ <3mos old ▪ 3-6mos old unless paroxysms are not severe or if with comorbidities Azithromycin – drug of choice in all age groups for both treatment and prophylaxis ▪ < 6mos: 10mkday QD ▪ > 6mos: D1 10mkday QD then D2-5 5mkday QD Erythromycin ▪ May cause infantile hypertrophic pyloric stenosis in neonates < 14days old ▪ 40-50mkday in 4 doses x 14 days Serologic tests: most sensitive tests in immunized individuals PERTUSSIS In infants <3mos old: ▪ Shorter catarrhal phase (often unnoticed) ▪ Chokes, gasps, gags, and flail extremities ▪ Reddened face, apnea, cyanosis ▪ Lengthy paroxysmal and convalescent stages in young infants Clarithromycin ▪ >1mos: 15mkday in 2doses x 7days Trimethoprim-sulfamethoxazole (TMP-SMX) ▪ Alternative for infants >2mos old and children unable to receive azithromycin ▪ TMP 8mkday in 2doses x 14days Place on droplet precaution and isolation until 5 days after initiation of azithromycin therapy. Batch Clingy COMPLICATIONS ▪ Apnea ▪ Bradycardia ▪ Otitis Media ▪ Progressive Pulmonary Hypertension ▪ Pneumonia ▪ Seizures, encephalopathy ▪ Death PPS Oral Exam Reviewer 2020 152 Physiologic GER – retrograde movement of gastric contents across the LES into the esophagus Pathologic GERD – reflux produces bothersome symptoms or impairs nutrition ▪ Associated with OM, sinusitis, lymphoid hyperplasia, hoarseness, vocal cord nodules, laryngeal edema ▪ Worsens laryngomalacia or BPD if present ▪ Genetic predispositions: family clustering of GERD symptoms, endoscopic esophagitis, hiatal hernia, Barrett esophagus, adenocarcinoma GASTROESOPHAGEAL REFLUX DISEASE (GERD) PATHOPHYSIOLOGY ▪ Factors determining reflux symptoms: duration and frequency of exposure, causticity of gastric regurgitant, susceptibility to damage ▪ Frequency of episodes is ↑ by insufficient LES tone, abnormal frequency of LES relaxations, and hiatal herniation that prevents the LES pressure from being proportionately augmented by the crura during abdominal straining ▪ Duration of episodes is ↑ by lack of swallowing (e.g., during sleep) and by defective esophageal peristalsis ▪ Transient LES relaxation – 1o mechanism allowing reflux VASCULAR RING ▪ Vascular anomalies that result from abnormal development of the aortic arch complex ▪ Double aortic arch – most common complete vascular ring, encircling both the trachea and esophagus, compressing both ▪ Pulmonary artery sling ▪ Left aortic arch with aberrant right subclavian artery – most common open (incomplete) vascular ring ▪ May have congenital heart disease TRACHEOMALACIA ▪ Chondromalacia of a central airway → insufficient cartilage to maintain airway patency throughout the respiratory cycle ▪ Recurrent regurgitation (± vomiting) ▪ Weight loss or poor weight gain ▪ Irritability ▪ Ruminative behavior ▪ Heartburn or chest pain ▪ Hematemesis ▪ Dysphagia or odynophagia ▪ Wheezing, stridor ▪ Cough ▪ Hoarseness, laryngeal/pharyngeal Inflammation ▪ Recurrent Pneumonia ▪ Anemia ▪ Dental Erosion, feeding refusal ▪ Dystonic neck posturing (Sandifer Syndrome) Infants: ▪ Regurgitation (postprandial) ▪ Signs of esophagitis – irritability, arching, choking, gagging, feeding aversion ▪ Respiratory symptoms – obstructive apnea, stridor ▪ Failure to thrive ▪ Symptoms resolve spontaneously in most infants by 12-24mos ▪ Thorough history and PE (with use of standardized questionnaires) – infant GER questionnaire ▪ Extended esophageal pH monitoring –used to diagnose acidic reflux ▪ Empirical antireflux therapy – trial of PPI treatment, mostly established in adults ▪ Barium Studies – to rule out other diseases ▪ Endoscopy – usually for complications ▪ Intraluminal impedance – diagnoses GERD and assesses esophageal function ▪ Laryngotracheobronchoscopy – to establish GERD as cause of respiratory symptoms DIFFERENTIAL DIAGNOSIS ▪ Milk and other food allergies ▪ Eosinophilic esophagitis ▪ Pyloric stenosis, intestinal obstruction (especially malrotation with intermittent volvulus) ▪ Non-esophageal inflammatory diseases ▪ Infections, IEM, hydronephrosis, ↑ ICP, rumination, bulimia Older children: ▪ Regurgitation ▪ Abdominal or chest pain ▪ Airway manifestations (laryngitis, sinusitis) Waxing and Waning ▪ Respiratory symptoms ▪ Dysphagia ▪ Barium esophagogram ▪ CT or MRI with angiography ▪ Symptoms appear within the 1st 2wks, increase in severity for up to 6mos ▪ Gradual improvement can begin at any time ▪ Flexible or rigid bronchoscopy ▪ Fluoroscopy – poorly sensitive ▪ MRI or CT Scan – if secondary to vascular ring AIRWAY ANOMALY Primary: congenital absence of tracheal-supporting cartilages Secondary: EA, TEF, vascular rings (double aortic arch), tracheal compression from an aberrant innominate artery, tracheal compression from mediastinal mases, abnormally soft tracheal cartilages associated with connective tissue disease, prolonged mechanical ventilation, chronic lung disease ▪ M:F = 2:1 ▪ 45% to 75% of congenital causes of stridor LARYNGEAL CLEFT ▪ Deficiency in the midline of the posterior larynx PPS Oral Exam Reviewer 2020 ▪ Stridor especially with crying, agitation, or feeding ▪ Low-pitched monophonic wheezing predominantly during expiration – most prominent over the central airways ▪ Wheezing – loudest at trachea Conservative therapy and lifestyle modification ▪ Normalization of abnormal feeding techniques, volumes, frequencies ● Thickening feeds ● Hypoallergenic diet ● Avoidance of smoke exposure ● Avoid acidic or reflux-inducing food and beverages: tomatoes, chocolate, mint, juices, carbonated and caffeinated drinks, alcohol ● Weight reduction ● Positioning measures ▪ Antacids ▪ H2RA: cimetidine, famotidine, nizatidine, and ranitidine ▪ PPI: omeprazole, lansoprazole, pantoprazole, rabeprazole, esomeprazole ▪ Prokinetic agents: metoclopramide (dopamine-2 and 5-HT3 antagonist), bethanechol (cholinergic agonist), erythromycin (motilin receptor agonist) ▪ Surgery: fundoplication COMPLICATIONS ▪ Esophagitis, esophageal stricture, barrett adenocarcinoma of the esophagus ▪ Failure to thrive ▪ Aspiration ▪ Laryngeal penetration ▪ Apnea in infants ▪ Reflux Laryngitis ▪ Dental erosions ▪ Surgery esophagus, ▪ Postural drainage ▪ Nebulized ipratropium bromide ▪ CPAP via tracheostomy (if severe) ▪ Surgery (aortopexy and bronchopexy) if symptomatic or cyanotic COMPLICATIONS ▪ Prolonged recovery from common respiratory infections ▪ Prolonged bacterial bronchitis ▪ Prognosis for secondary causes varies with the comorbidity ▪ Aspiration (frequent) ▪ Recurrent respiratory infections ▪ Larygoscopy and bronchoscopy with palpation of posterior larynx – gold standard ▪ Stabilize airway ▪ Control GERD Batch Clingy ▪ Most common esophageal disorder in children of all ages 153 ▪ Associated with tracheal agenesis, TEF, and multiple congenital anomalies (G syndrome, Opitz-Frias syndrome, Pallister-Hall syndrome) 4 Types: Type I – mild; interarytenoid notch extends to the true vocal cords; 60% asymptomatic with no surgery needed Type II – extends beyond the vocal cords to, but not through, the cricoid cartilage Type III – extends through cricoid into cervical trachea Type IV – extends inferiorly into the cervical or thoracic trachea; no separation between the trachea and esophagus → laryngotracheoesophageal cleft BRONCHIECTASIS ▪ Cough and copious purulent sputum – most common ▪ Etiology, as complications of: ▪ Hemoptysis ● Cystic Fibrosis, primary ciliary dyskinesia ▪ Fever ● Foreign body aspiration ▪ Anorexia ● Aspiration of Gastric Contents ▪ Crackles and wheezing ● Immune Deficiency Syndromes ▪ Digital Clubbing ● Infections ▪ Severe: Hypoxemia ● Williams-Campbell Syndrome (absence of annular bronchial cartilage) Prebronchiectasis – chronic or recurrent endobronchial infection with ● Marnier-Kuhn syndrome (congenital nonspecific HRCT changes; may be reversible tracheobronchomegaly) HRCT bronchiectasis – clinical symptoms with HRCT evidence of bronchial dilation; may persist, progress, or improve and resolve ▪ Lower lobes – most commonly affected Established bronchiectasis – like the previous but with no resolution ▪ Irreversible abnormal dilation and anatomic distortion of within 2yrs the bronchial tree ▪ Common end stage of many nonspecific and unrelated Exacerbations antecedent events ▪ 1 Major + 1 Lab Criteria; 2 Major; 1 major + 2 minor ▪ Major Criteria: wet cough >72hrs, increased cough frequency > 72hrs Incidence ▪ Laboratory Criteria: CRP > mg/L, serum IL-6 >2 ng/L, serum amyloid A ▪ Low- and middle-income countries >5mg/L, ↑ neutrophil percentage ▪ Females > Males ▪ Minor Criteria: change in sputum color, breathlessness, chest pain, crackles/crepitations, wheeze PATHOGENESIS Three Mechanisms 1. Obstruction 2. Infections 3. Inflammation: inflammatory factors activate neutrophils → destruction of cells and induction of goblet cell hyperplasia ▪ Early stages: bronchiolar wall thickening and destruction of elastin → bronchial dilatation ▪ Later stages: bronchial walls develop cartilage destruction with associated pulmonary artery/ arteriole vascular remodeling → pulmonary HTN ▪ May have microorganisms in the isolate: S. pneumoniae, H. influenzae non–type b, M. catarrhalis, M. pneumoniae Psychogenic Cough ▪ Abrupt & loud cough – harsh, honking, barking quality ▪ Tic Cough/Somatic Cough Disorder/Habit Cough ▪ Absent if the physician listens outside the examination room and ▪ May be preceded by an URTI reappears upon direct attention to child. ▪ Esophagogram ▪ FEES ▪ Type I – endoscopic surgery ▪ Type II and up – open procedure ▪ Pulmonary function studies: obstructive, restrictive, or mixed ▪ Investigate possible cause (sweat test, immunologic work-up ▪ Thin-section HRCT scanning – gold standard ▪ “tram lines”, “signet ring appearance”, varicose (bronchi with beaded contour), cystic (cysts in strings and clusters) ▪ Airway clearance techniques (e.g., gravity-assisted drainage, active cycle of breathing, positive expiratory pressure [PEP], acapella, high-frequency chest wall oscillation) ▪ Antibiotics for 2-4wks depending on isolate: Amoxicillin/clavulanic acid (22.5mkdose BID) ▪ Bronchodilators ▪ Long-term prophylactic macrolide antibiotics or nebulized antibiotics ▪ Inhaled hypertonic saline ▪ Inhaled corticosteroids ▪ Lung transplantation Forms: ▪ Cylindrical – regular, diffuse dilation of the bronchial unit ▪ Varicose – Irregular, greater dilatation with local constrictions ▪ Saccular (cystic) bronchial dilation progresses; ballooning of bronchi; most severe COMPLICATIONS ▪ Recurrent pulmonary illness ▪ Stunted growth ▪ Osteopenia ▪ Osteoporosis ▪ Cough lasting weeks-months ▪ Refractory to treatment ▪ Disappears with sleep or distraction ▪ Assurance that a pathologic lung condition is absent, and child may resume full activity ▪ Speech therapy techniques ▪ Self-hypnosis Batch Clingy TRACHEOESOPHAGEAL FISTULA PPS Oral Exam Reviewer 2020 154 Acute DISEASE COMMON COLDS ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Rhinoviruses – 50% ▪ RSV ▪ Human Metapneumovirus ▪ Parainfluenza virus ▪ Adenovirus ▪ Influenza virus ▪ Nonpolio enterovirus ▪ Human coronaviruses ▪ Direct hand contact ▪ Inhalation of small particle aerosol ▪ Deposition of large particle aerosols ▪ Viral infection of the nasal epithelium → destruction of epithelial lining → acute inflammatory response 2 factors for expression of AR: ▪ Sensitivity to allergen ▪ Presence of allergen in the environment Early Phase: degranulation of mast cells, release of preformed and newly generated inflammatory mediators Late Phase: 4-8hrs after allergen exposure, inflammatory cells infiltrate the nasal mucosa LABORATORY FINDINGS / DIAGNOSIS ▪ Clinical ▪ Routine lab tests – not helpful ▪ Sore or scratchy throat ▪ Nasal obstruction ▪ Rhinorrhea ▪ Cough ▪ Swollen, erythematous nasal turbinates ▪ Abnormal middle ear pressure ▪ Anterior CLAD ▪ Conjunctival injection ▪ Change in the color and consistency of nasal secretion → accumulation of PMNs ▪ Allergic salute ▪ Nasal crease ▪ Nasal congestion ▪ Itching, sneezing ▪ Clear rhinorrhea ▪ Conjunctival irritation ▪ Allergic gape – open mouth breathing ▪ Allergic shiners ▪ Skin test ▪ Prep: Withhold montelukast for 1 day, most sedating antihistamine for 3-4 days, and nonsedating antihistamines for 5-7 days ▪ Serum IgE DIFFERENTIAL DIAGNOSIS ▪ Nonallergic rhinitides – no sIgE elevation ▪ Vasomotor rhinitis ▪ Rhinitis medicamentosa ▪ Structural problems – nasal polyps, septal deviation TREATMENT ▪ Mainly supportive ▪ Antiviral treatment if indicated (Oseltamivir, Zanamivir) – should be started within 48hrs of symptom onset ▪ Nonprescription cough and cold products → not used for infants and <6yo ▪ Zinc – reduces symptom duration if given within 24hrs ▪ Topical adrenergic agents ● Oxymetazoline, phenylephrine ● Avoid prolonged use to prevent Rhinitis Medicamentosa ● Not used for <6yo ▪ 2nd gen antihistamine – anticholinergic effect ▪ Honey (5-10ml) – for children >1yo to relieve nocturnal cough ▪ Antihistamines (second-generation) ▪ +/- pseudoephedrine ▪ Intranasal decongestants ▪ Intranasal corticosteroids – most effective therapy ▪ Immunotherapy COMPLICATIONS / PROGNOSIS ▪ AOM – most common complication ▪ Sinusitis – rhinorrhea or daytime cough without improvement for 10-14 days ▪ Exacerbation of asthma ▪ Antibiotic resistance ▪ Chronic sinusitis ▪ Eustachian tube obstruction ▪ Middle ear effusion ▪ OSA ▪ Up to 78% of patients with asthma have AR, and 38% of patients with AR have asthma Batch Clingy ALLERGIC RHINITIS CLINICAL MANIFESTATIONS Infants: fever and nasal discharge predominate PPS Oral Exam Reviewer 2020 155 Acute DISEASE ETIOLOGY / PATHOLOGY / PATHOGENESIS ▪ Spongiosis – marked intercellular edema CLINICAL MANIFESTATIONS ▪ Severely dry skin – hallmark LABORATORY FINDINGS / DIAGNOSIS Labs: Nonspecific ▪ Chronic lichenified AD: hyperplastic epidermis with hyperkeratosis and minimal spongiosis Cardinal features: ▪ Intense pruritus ▪ Cutaneous reactivity DIFFERENTIAL DIAGNOSIS ▪ Seborrheic Dermatitis ▪ Nummular Dermatitis ▪ Irritant Contact Dermatitis ▪ Allergic Contact Dermatitis ▪ Lichen Simplex Chronicus ▪ Asteatotic Eczema ▪ Dermatophyte infection ▪ Scabies, Impetigo, HIV ▪ Icthyosis vulgaris ▪ Acrodermatitis enterohepatica (zinc) ▪ Biotin deficiency, Pellagra (niacin) ▪ Kwashiorkor, PKU ▪ Cutaneous T-cell lymphoma ▪ Langerhans cell histiocytosis ▪ Atopic eczema (IgE mediated) ▪ Nonatopic eczema ▪ Increased expression of IL4 & IL13 ▪ Genetic mutations in Filaggrin gene ▪ Lichenification & fibrotic papules – chronic AD ATOPIC DERMATITIS Atopic Eczema TREATMENT / COMPLICATIONS / PROGNOSIS ▪ Moisturizers – first line of therapy ▪ Topical corticosteroids – cornerstone of therapy ▪ Topical Calcineurin Inhibitors (>2yo) ▪ Topical PDE-4 Inhibitor (Crisaborole) ▪ Antihistamine ▪ Systemic Corticosteroids ▪ Cyclosporine ▪ Dupilumab ▪ Antimetabolites (MMF, MTX, Azathioprine) ▪ Phototherapy COMPLICATIONS Eczema Herpeticum Exfoliative Dermatitis Allergic Keratoconjunctivitis Keratoconus PREVENTION Avoid triggers Chronic (>6 weeks) ▪ Not physical urticaria or recurrent urticarial with repeated exposures to a specific agent URTICARIA (Hives) And ANGIOEDEMA Hives – erythematous, pruritic raised wheal that blanches with pressure, transient and resolves without residual lesions Urticarial vasculitis – lesions that burn more than itch, last >24 hours, does not blanch, heal with scarring or associated with bleeding into the skin (purprura) PHYSICAL URTICARIA Cold Dependent Disorders ▪ Localized pruritus, urticarial/angioedema after exposure to a cold stimulus ▪ Due to the presence of cryoproteins Cholinergic Urticaria ▪ Associated with exercise, hot showers and sweating DIFFERENTIAL DIAGNOSIS ▪ Cutaneous or systemic mastocytosis ▪ Hereditary angioedema ▪ Urticaria pigmentosa ▪ Exercise induced anaphylaxis Dermatographism/Urticaria Factitia ▪ Ability to write on skin Pressure Induced Urticaria/Angioedema ▪ Occurs 4-6 hours after pressure has been applied Solar Urticaria ▪ Occurs within minutes of sun exposure ▪ Disappears within 1-3 hours after cessation of sun exposure PPS Oral Exam Reviewer 2020 Clinical diagnosis Batch Clingy Acute (<6 weeks) ▪ IgE mediated ▪ Non IgE mediated PROGNOSIS ▪ Severe and persistent in young children ▪ Spontaneous resolution after age 5 (40-60%) ▪ Adults: Hand dermatitis Poor prognostic factors: ▪ Widespread AD during childhood ▪ FLG null mutations ▪ Concomitant AR and asthma ▪ Family history of AD in parents and siblings ▪ Early age at onset of AD ▪ Only child ▪ Very high serum IgE levels ▪ Antihistamines – hydroxyzine & diphenhydramine ▪ Avoidance of triggers ▪ Epinephrine – seldom required ▪ Short course oral corticosteroids – for severe episodes ▪ Leukotriene Modifiers 156 Aquagenic Urticaria ▪ Occurs after contact with water regardless of termperature ▪ Latex Allergy - most common cause occurring in the hospital setting ▪ Food Allergy – community ▪ Mostly results from activation of mast cells and basophils via cell-bound allergen-specific IgE molecules ANAPHYLAXIS Principal pathologic features: ▪ Bronchial obstruction with pulmonary hyperinflation ▪ Pulmonary edema ▪ Intraalveolar hemorrhage ▪ Visceral congestion ▪ Laryngeal edema ▪ Urticaria ▪ Angioedema CHRONIC IDIOPATHIC URTICARIA/ANGIOEDEMA ▪ Does not appear to result from an allergic reaction ▪ Non necrotizing, perivascular, mononuclear cellular infiltration ▪ Associated with anti-thyroid antibodies ▪ 35-40% have (+) autologous serum skin test ▪ Ingested allergens have delayed onset (minutes to 2hrs) compared to injected allergens Initial symptoms ▪ Pruritus of the mouth and face ▪ Flushing, urticarial and angioedema ▪ Sensation of warmth, weakness and apprehension (sense of doom) ▪ Tightness in the throat, dry staccato cough, hoarseness ▪ Periocular pruritus ▪ Nasal congestion, sneezing ▪ Dyspnea, deep cough, wheezing ▪ Nausea, abdominal cramps, vomiting ▪ Uterine contractions (lower back pain) ▪ Faintness and loss of consciousness Labs are nonspecific ▪ Plasma histamine ▪ Plasma tryptase MEDICAL EMERGENCY! Epinephrine ▪ Most important ▪ IM on the lateral thigh ▪ 1:1000, 0.01mg/kg max 0.5mg ▪ Biphasic Anaphylaxis – occur within 4 hours of treatment Batch Clingy ▪ Cutaneous manifestations are absent in 10% of cases & the acute onset of severe bronchospasm in a previously well person with asthma should suggest the diagnosis of anaphylaxis PPS Oral Exam Reviewer 2020 157 Complex DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Chronic autoimmune disease of multisystem inflammation & (+) circulating autoAb directed against self Ag ▪ Genetic predisposition ▪ Women of reproductive age – due to hormones ● 90% are female ● Female sex – strongest risk factor (9:1) ● Estrogen → exposure promotes B-cell autoreactivity ● Estrogen containing OCPs do not appear to induce flares in quiescent SLE, risk of flares may be in in postmenopausal receiving hormone replacement therapy ▪ Viral infections, EBV may play a role ▪ UV light exposure – trigger disease activity ▪ Prevalence: 1-6/100,0000 ▪ Factors influencing risk and severity of disease: genetics, hormonal milieu, environmental exposures ▪ Genetic predisposition associated with specific genetic abnormalities: ● Congenital deficiencies of C1q, C2 and C4 ● Polymorphism (IFN regulatory factor 5, protein tyrosine phosphatase N22) ▪ Human leukocyte antigen (HLA) types occur with ↑ frequency in patients with SLE (HLA-B8, HLA-DR2, HLA-DR3) SYSTEMIC LUPUS ERYTHEMATOSUS PATHOGENESIS ▪ Hallmark: generation of autoAb against self-antigens (particulartly nucleic acid) ▪ Cell necrosis by apoptosis → antigens released ▪ ↑ Levels of apoptosis or significantly impaired ability to clear cell debris → prolonged exposure to nucleic acid antigens in the bloodstream → recognition by immune cells → B-cell stimulation and autoantibody production ▪ Circulating autoAb may form immune complexes and deposit in tissues → local complement activation, initiation of proinflammatory cascade and tissue damage ▪ Ab to double-stranded (ds) DNA form immune complexes → deposit in glomeruli → inflammation → glomerulonephritis CLINICAL MANIFESTATIONS ▪ Hallmark – multiorgan disease ▪ Most common presenting complaints in children: fever, fatigue, hematologic abnormalities, arthralgia, arthritis ▪ Renal disease – asymptomatic, monitor BP and urinalysis 4/11 ACR 1997 Criteria simultaneously or cumulative over time ▪ Malar rash ▪ Discoid rash ▪ Photosensitivity ▪ Oral or nasal ulcers ▪ Arthritis (non-erosive, > 2joints) ● Present in the 1styr of diagnosis ● Painful or painless swelling, stiffness in the morning ● Symmetric polyarthritis (large & small joints) ▪ Serositis – pleurisy, pericarditis, peritonitis ▪ Renal manifestations ● Proteinuria or casts ● Renal biopsy (dx and stage disease) ▪ Seizure or psychosis ● Neuropsychiatric complications may occur w/ or w/o apparently active SLE ▪ Hematologic manifestations ● Hemolytic anemia ● Leukopenia (<4000) ● Lymphopenia (<1500) ● Thrombocytopenia (<100,000) ▪ Immunologic abnormalities ● (+) anti ds DNA or anti-Smith AB ● False positive RPR test ● Positive lupus anticoagulant test ● Elevated anticardiolipin Ig or IgM Ab (+) ANA is not required for diagnosis, but (-) ANA in SLE is rare SLICC 2012 Criteria ▪ Inclusion of hypocomplementemia ▪ Addition of nonscarring alopecia ▪ Additional cutaneous and neurologic manifestations of lupus ▪ Positive direct Coombs test in the absence of hemolytic anemia LABORATORY FINDINGS / DIAGNOSIS ANA – 95-99% Sn; poor Sp (50%), poor screening tool TREATMENT Sunscreen and avoid direct sun exposure anti dsDNA Ab ▪ More specific (98%) ▪ May correlate with disease activity especially in nephritis, (Sn 40-64%) Hydroxychloroquine <6.5mkday (max 400mg daily) ▪ Recommended for all SLE patients if tolerated ▪ Prevents flares, improves lipid profiles, improve mortality and renal outcomes ▪ Side effects: retinal pigmentation, impaired color vision (ophtha q6-12mos) Anti-Smith ▪ Specific (98%) ▪ Does not correlated with disease activity ↓ C3 and C4 in active disease Hypergammaglobulinemia – common; nonspecific Elevated inflammatory makers ▪ ESR ▪ CRP – elevation often correlates with infection, where chronic mild elevation may indicate ↑ CV risk Antiphospholipid Ab ▪ Indicate presence of anticardiolipin Ab, prolonged phospholipid depended coagulation tests, and circulating lupus anticoagulant ▪ If it occurs, clotting + Antiphospholipid antibody → APAS (may occur ± SLE) Discoid rash biopsy: hyperkeratosis, follicular plugging, infiltration of mononuclear cells into dermal-epidermal junction (DEJ) Lupus-band test: deposition of immune complexes with the DEJ DIFFERENTIAL DIAGNOSIS ▪ Infections – sepsis, EBV, parvovirus B19, endocarditis ▪ Malignancies – leukemia, lymphoma ▪ Other rheumatic conditions – JIA, vasculitides ▪ Nephrotic Syndrome, PSGN ▪ Pleural or pericardial effusion ▪ Cardiopulmonary abnormalities ▪ New onset psychosis; movement/seizure disorder MONITORING AND PREVENTION ▪ Monitor cholesterol, BP, and BMI ▪ Smoking prevention ▪ Immunization ● Annual flu ● PCV 13 for SLE >6yo → PPSV 23 after 2 months NSAIDS – for arthralgia and arthritis Corticosteroids – mainstay ▪ Limit dose and length of exposure ▪ SE: growth disturbance, weight gain, striae, acne, hyperglycemia, HTN, cataracts, avascular necrosis, osteoporosis ▪ Severe disease: high dose IV Methylprednisone (30mkday, max1g/day) for 3d sometimes ffd by a period of weekly pulses and/or high dose oral prednisone 1-2mkday tapered overtime Steroid sparing immunosuppressive agents MTX, Leuflonamide, Azathioprine, MMF ▪ For persistent moderate disease – ↑ arthritis, significant cutaneous or hematologic involvement and pleural disease Cyclophosphamide, MMF, Azathioprine ▪ For lupus nephritis MMF and Rituximab ▪ For significant hematologic manifestations – leukopenia, hemolytic anemia, thrombocytopenia Cyclophosphamide ▪ Reserved for severe renal, neuro and cardiopulmonary disease ▪ Hydrate to prevent hemorrhagic cystitis ▪ Other SE: cytopenia, premature gonadal failure, ↑ risk for malignancy CARRA Treatment Plan ▪ 6mos induction Tx 500-1000mg/m2 IV monthly or MMF (600mg/m2 – 1500mg BID), + 1 of 3 standardized glucocorticoid regimens Lupus Nephritis maintenance ▪ Cyclophosphamide q3mos, or MMF or Azathioprine for 36mo after induction therapy Belimumab ▪ Monoclonal Ab vs Blys/BAFF FDA-approved for adult SLE → improved markers of disease severity Long-term anticoagulation ▪ SLE with APAS, with history of clot PPS Oral Exam Reviewer 2020 Batch Clingy Calcium and Vitamin D to prevent osteoporosis 158 ▪ Circulating antihistone Ab – detected in only 20% of patients with SLE ▪ Not likely to be (+) in antidsDNA, hypocomplementemia and renal or neuro disease ▪ Hepatitis more common in drug-induced lupus ▪ Annular/macular rash (face) – often appears at birth or within first 6-8 weeks of life, after exposure to UV light and lasts 3-4 months ▪ 25% may have cytopenia and hepatitis Arthritis (>6wks) present ▪ Intraarticular swelling or presence of >2: ▪ Limitation of range of motion ▪ Tenderness/pain on motion ▪ Increased heat or erythema Initial symptoms – subtle and acute ▪ Morning stiffness w/ a limp or gelling after inactivity ▪ Joints swollen, warm to touch, and painful on movement or palpation with ↓ ROM but not erythematous yet ▪ Arthritis in large joints (knees) accelerates linear growth causing limb length discrepancy ▪ Rapid and premature closure of the growth plate→ shortened bones ▪ Recovery takes several months to years COMPLICATION Congenital Heart Block ▪ Most feared complication ▪ Diagnosed in utero: fetal echo at 16 weeks, period of greatest vulnerability 18-24 weeks ▪ Fetal bradycardia from heart block → hydrops fetalis ▪ Mom with anti-RO/LA → treat with HCQ, steroids, or IVIg JIA is a clinical diagnosis without any diagnostic laboratory test Early x-ray findings ▪ Soft tissue swelling ▪ Periarticular osteoporosis ▪ Periosteal new bone apposition Late ▪ Subchondral erosions ▪ Loss of cartilage with bone destruction Changes in cervical spine ▪ C2-C3 may progress to atlantoaxial subluxation DIFFERENTIAL DIAGNOSIS ▪ SLE, JD, sarcoidosis, vasculitic syndromes ▪ Scleroderma – limited ROM because of sclerotic skin overlying a joint ▪ Acute rheumatic fever – exquisite joint pain and tenderness, remittent fever, migratory polyarthritis Goals: achieve disease remission, prevent joint damage, foster normal growth Other Management: ▪ Periodic slit lamp examinations to monitor for asymptomatic uveitis; treatment: mydriatics and corticosteroids ▪ Dietary evaluation and counseling to ensure proper calcium, vitamin D, protein, and caloric intake ▪ PT and OT PROGNOSIS Oligoarticular disease (OD) Persistent OD – majority achieve remission Extended OD – poorer prognosis ▪ OD in girls who are (+) ANA and onset of arthritis is <6yo → greatest risk for chronic uveitis ▪ Uncontrolled uveitis – posterior synechiae, cataracts, glaucoma, Batch Clingy JUVENILE IDIOPATHIC ARTHRITIS DRUG INDUCED LUPUS ▪ SLE manifestations triggered by exposure to minocycline, anticonvulsants, sulfonamides, antiarrhythmic agents ▪ Procainamide and hydralazine – promote lymphocyte hypomethylation causing lupus-like syndrome ▪ May trigger true SLE ▪ M=F NEONATAL LUPUS ▪ Not an autoimmune disease of the fetus ▪ Passively acquired autoimmunity – maternal IgG autoAb cross the placenta → fetal circulation ▪ Associated with maternal anti-RO (anti-SSA) and anti-LA (antiSSB) Ab or anti-RNP (antiribo-nucleoprotein) auto Ab ▪ 2% of offspring born to mothers (+) antiRO/LA develop neonatal lupus ▪ Most common rheumatic disease in children ▪ Peak age of onset ● Oligoarticular: 2-4yo ● Polyarthritis (bimodal): 2-4yo, 10-14yo ● sJIA: 1-5yo ▪ Unknown etiology and pathogenesis → immunogenetic susceptibility and external trigger – autoimmune, associated with alterations in both humoral and cell mediated immunity ▪ T lymphocytes release proinflammatory cytokines (TNF-α, IL6, IL1) ▪ B cell activation, immune complex formation, complement activation → inflammation ▪ sJIA – dysregulation of the innate immune system with a lack of autoreactive T cells and autoAb → inflammatory synovitis (villous hypertrophy and hyperplasia with hyperemia and edema of the synovial tissue) ▪ To prevent thrombotic events ASA 81mg/day ▪ SLE with APAS, no history of clot ▪ HCQ, NSAIDS, steroids ▪ Resolves with removal of drug PPS Oral Exam Reviewer 2020 159 ▪ Autoimmune hepatitis – may have acute arthritis ▪ Transient arthritis cause by infections – Parvovirus B19, Rubella, EBV, Hep B, HIV, Lyme disease ▪ Septic arthritis – fever with painful erythematous hot joint ▪ Isolated hip pain with limited motion: suppurative arthritis, osteomyelitis, toxic synovitis, Legg Calve Perthes, slipped capital femoral epiphysis ▪ ERA – tenderness over insertion of ligaments, tendons, and LE extremity arthritis (esp if boy) ▪ Psoriatic arthritis – limited joint involvement (small joints of hands and ankle) ▪ IBD – oligoarthritis, usually LE, with GI symptoms, elevations in ESR and microcytic anemia ▪ Growing pains – 4-12yo leg pain in the evenings with normal studies and no morning symptoms ▪ Leukemia or neuroblastoma – bone pain from infiltration of the bone, synovium or bone marrow, nocturnal pain awakens child from sleep (malignancy) blindness ORTHOPEDIC COMPLICATIONS ▪ Leg length discrepancy ▪ Flexion contractures (knees, hips, wrists) ▪ Psychosocial adaptations and problems may warrant counseling *high ESR with leukopenia and low normal platelet count may be a clue to underlying leukemia SYSTEMIC JIA ▪ Arthritis + fever + prominent visceral (hepatosplenomegaly, lymphadenopathy, and pericarditis) PPS Oral Exam Reviewer 2020 involvement serositis – ▪ Partial response to NSAIDS ▪ If no or only partial response after 4-6 weeks, or with functional limitations (joint contracture/leg-length discrepancy) → intraarticular steroids (triamcinolone) ▪ Others require DMARDS like MTX or TNF inhibitors PROGNOSIS Polyarticular JIA – prolonged course of active joint inflammation, requiring early aggressive Tx Predictors of severe disease ▪ Young age at onset ▪ RF seropositivity or Rheumatoid nodules ▪ (+) Anti-cyclic citrullinated peptide antibodies ▪ Many affected joints (hand, hip, and wrist poorer prognosis) Macrophage activation syndrome ▪ Secondary hemophagocytic syndrome or HLH ▪ Rare but fatal complication of sJIA ▪ sJIA/MAS and HLH share similar functional defects in granule- ▪ ↑ Inflammatory markers, WBC & PC ▪ ↓ Hgb (7-10), indices like anemia of chronic disease ▪ ↑ ESR (except in MAS) ▪ ANA and RF uncommon Polyarticular or sJIA ▪ DMARDS (MTX) – oldest, least toxic adjunctive treatment; 6-12 weeks to see effect ▪ Failure of MTX monotherapy, add biologic DMARD → inhibit cytokines like TNFα (Etanercept, adalimumab), IL-1, IL-6 ▪ Early aggressive Tx w/ MTX + TNFα antagonist may result in earlier achievement of clinically inactive disease ▪ Abatacept (selective inhibitor of T cell activation) & Tocilizumab (IL-6 antagonist) effective and approved for polyarticular JIA SJIA w/ dominant systemic symptoms ▪ Systemic steroids → IL-1 or IL-6 antagonist – dramatic, rapid response ▪ Anakinra may be used for severe ds activity Batch Clingy OLIGOARTHRITIS (<4 joints) ▪ Often involves single joint, affects large joints of LE (knee, ankles) ▪ Isolated involvement of the hip is almost never a presenting sign (suggests Enthesitis-Related Arthritis) ▪ (+) ANA seropositivity – ↑ risk for chronic uveitis POLYARTHRITIS (>5 joints) ▪ Both UE and LE ▪ RF (+) polyarthritis resembles the symmetric presentation of adult RA ▪ Presence of rheumatoid nodules almost exclusively occur in RF (+), associated with more severe course ▪ Micrognathia reflects chronic TMJ disease 160 ▪ Most common inflammatory myositis in children, distinguished by proximal muscle weakness and characteristic rash ▪ Genetic disposition (transfer of maternal chimeric cells) and unknown environmental trigger (hx infection, URTI or GI, 3mos before commonly reported) ▪ Peak onset 4-10yo ▪ Incidence 3/1 million/year JUVENILE DERMATOMYOSITIS PATHOGENESIS ▪ Upregulation of gene products controlled by Type I interferon (IFN) ▪ Inc susceptibility to JDM assoc with HLA alleles B8, DRB1*0301, DQA1*0501, DQA1*0301 → may have prolonged exposure to maternal chimeric cells and/or an unk environmental trigger → upregulation of MHC class I expression and maturation of dendritic cells ▪ Overexpression of MHC class I upregulates adhesion molecules → migration of lymphocytes → inflammatory infiltration of muscle dependent cytotoxic lymphocyte activity ▪ Acute onset of profound anemia, thrombocytopenia, or leukopenia with high spiking fever, lymphadenopathy, and splenomegaly ▪ Purpura and mucosal bleeding, prolonged PT and PTT, ↑ liver enzymes, LDH, ferritin, TG ▪ ESR falls because of hepatic dysfunction ▪ BM biopsy: hemophagocytosis ▪ ↓ WBC and/or platelet with active SJIA ▪ Treatment: high dose IV Methylprednisolone, cyclosporine or anakinra Criteria for sJIA-associated MAS ▪ Hyperferritinemia (>684ng/mL) + any 2 of the ffg: ▪ Thrombocytopenia (<181x109) ▪ ↑ Liver enzymes (AST >48U/L) ▪ Hypertriglyceridemia (>156mg/dL) ▪ Hypofibrinogenemia (<360mg/dL) ▪ Rash, insidious onset of weakness, or both ▪ Fever, dysphagia or dysphonia, arthritis, muscle tenderness, fatigue Rash ▪ 1st symptom in 50% cases ▪ Extreme photosensitivity to UV light exposure with generalized erythema in sun exposed areas (shawl sign → chest, neck) ▪ Erythema over knees and elbows ▪ Heliotrope rash – blue violet discoloration of eyelids associated with periorbital edema ▪ Facial erythema crossing nasolabial folds (contrast to SLE, w/o nasolabial) ▪ Gottron papules – bright pink or pale, shiny thickened atrophic plaques over PIP and DIP joints, occasionally on knees, elbows, ankle malleoli ▪ Mechanic’s hands (rare) – thickened erythematous and scaly rash over palms and soles ▪ Telangiectasia, cutaneous ulcers Weakness ▪ Insidious and difficult to differentiate from fatigue ▪ Symmetric, affecting proximal muscles (neck flexors, shoulder girdle, or hip flexors) ▪ Difficulty climbing stairs, combing hair, getting out of bed, inability to perform a sit up, head lag after infancy and Gower sign PPS Oral Exam Reviewer 2020 ▪ ↑ Ferritin (>10,000ng/mL) ▪ Canakinumab (IL-B inhibitor) & Tocilizumab – for children >2yo with sJIA PROGNOSIS sJIA’s poorer prognosis is related to: ▪ Polyarticular distribution of arthritis ▪ Fever >3mos ▪ ↑ Inflammatory markers (PC, ESR for >6mos) Standardized consensus 4 Treatment Plan for sJIA ▪ Glucocorticoids, MTX, Anakinra/tocilizumab with optional GC use in the latter 3 plans as indicated clinically COMPLICATIONS ▪ Bone mineral metabolism and skeletal maturation affected in all types JIA ▪ ↓ Bone mass (osteopenia), associated with ↑ disease activity ▪ Systemic steroids must be given only in severe systemic illness, as bridge therapy while waiting for therapeutic response to DMARDS, control uveitis. ▪ Oral Janus Kinase (JAK) inhibitors (tofacitinib, ruxolitinib) inhibit JAK signaling pathways involved in immune activation and inflammation. Diagnostic Criteria ▪ Classic rash (heliotrope rash of the eyelids, Gottron papules) ▪ Plus 3 of the ff: ● Symmetric and proximal weakness ● Muscle enzyme elevation (>1) (CK, AST, LDH, Aldolase) ● EMG changes – myopathy, denervation, fibrillations, bizarre discharges ● Muscle biopsy – necrosis and inflammation ▪ Some children present with classic rash but not apparent muscle weakness or inflammation → AMYOPATHIC JDM or dermatomyositis sine myositis – risk of progression to more severe muscle involvement with long-term sequelae if untreated (calcinosis and lipodystrophy) LABS ▪ ↑ CK, aldolase, AST, ALT, LDH – reflect muscle inflammation ▪ ALT usually ↑ on initial presentation, CK level may be normal ▪ ESR usually N, RF usually negative ▪ CBC: anemia consistent with chronic disease ▪ ANA (+) in >80% Myositis-associated antibodies (MAA) ▪ Ab to SSA, SSb, Sm, ribonucleoprotein (RNP) and dsDNA negative ▪ May ↑ likelihood of overlap disease or connective tissue myositis Esophageal and respiratory muscles also affected (aspiration and respiratory failure) ▪ Assess for dysphonia, nasal speech, palatal elevation with gag, dysphagia, GERD Myositis-specific antibodies (MSA) ▪ Positive results for anti-Jo-1, anti-Mi2, anti-p155/140, anti-NXP2 ▪ More significant, help define distinct clinical subsets, may predict development of complications Lipodystrophy and calcinosis ▪ Associated with longstanding or undertreated disease ▪ Dystrophic deposition of calcium in SC plaques or nodules MRI ▪ Identifies active sites of disease ▪ Increases sensitivity of muscle biopsy and EMG (both are Corticosteroids – mainstay ▪ Stable – oral prednisone 2mkday (max 60mg/day) ▪ Severe, with resp or oropharyngeal weakness – high dose pulse methylprednisolone (3omg/kg/day) for 3 days, max 1g/day with ongoing weekly or monthly IV dosing along w/ daily oral steroids prn ▪ Tapered over 12-24 months Methotrexate ▪ Decreases length of treatment with steroids ▪ Weekly oral, IV, or SC (the lesser of 1mg/kg or 15mg/m2, max 40mg) ▪ Given with Folic acid ▪ SE: immunosuppression, blood count dyscrasia, chemical hepatitis, pulmonary toxicity, side effects of folate inhibition (oral ulcers, nausea, anemia) Hydroxychloroquine ▪ Little toxicity, reduce rash, maintain remission ▪ 4-6mg/kg/day ▪ Monitor retinal toxicity q6mos ▪ SE – hemolysis with G6PD, GI intolerance, skin/hair discoloration IVIg – adjunct treatment for severe disease, given at 2g/kg (max 70g) every 2 weeks for 3 doses then every 4 weeks PRN For severe unresponsive disease: IVIg, MMF, cyclosporine and cyclophosphamide ▪ OGT or NGT to avoid aspiration for those with pharyngeal weakness ▪ PT and OT – integral; passive stretching → direct muscle reconditioning for strength and range of motion ▪ Avoid sun exposure and apply sunscreen ▪ Vitamin D and calcium supplements – prevent osteoporosis Batch Clingy ▪ Heat can evoke rash ▪ Koebner phenomenon – cutaneous hypersensitivity to superficial trauma ▪ Arthritis usually polyarticular and destructive and includes hip, cervical spine, and TMJ involvement 161 ▪ Lipodystrophy (face and upper body), associated with metabolic syndrome similar to PCOS with insulin resistance, hirsutism, acanthosis, hypertriglyceridemia, and abnormal glucose tolerance Vasculitis GI tract (rare) ▪ Crampy abdominal pain, pancreatitis, GI bleeding and potential for intestinal perforation or infarction nondiagnostic if not directed by MRI) Muscle biopsy ▪ Evidence of disease activity and chronicity Contrast swallow study ▪ Palatal dysfunction & risk of aspiration PFT – respiratory weakness, restrictive Radiographs – calcinosis along the fascial planes and within muscles COMPLICATIONS ▪ Due to prolonged and severe weakness ▪ Aspiration pneumonia and respiratory failure ▪ Bowel wall vasculitis ▪ Pathologic calcifications ▪ Draining lesions and nodules – cellulitis or osteomyelitis PROGNOSIS ▪ Mortality rate less than 1% DIFFERENTIAL DIAGNOSIS If weakness w/o rash ▪ Polymyositis ▪ Infection related myositis (influenza A and B, coxsackie and other viruses) ▪ Infections with prominent muscular symptoms: Trichinosis, Bartonella, toxoplasmosis, staphylococcal pyomyositis ▪ Muscular dystrophies (duchenne and becker) ▪ Myasthenia gravis, GBS ▪ Endocrinopathies (hyper/hypothy) ▪ Mitochondrial myopathies ▪ Metabolic disorders (glycogen and lipid storage diseases) ▪ TNF receptor associated periodic syndrome (TRAPS) ▪ Transient rhabdomyolysis w/ myoglobinuria in blunt trauma and crush injuries ▪ Myositis assoc w/ vaccinations, drugs, growth hormone and GHVD 2 categories: Juvenile localized scleroderma (JLS / morphea) – more common in children (>95%), largely limited to the skin Juvenile systemic sclerosis (JSSc) with multisystem organ involvement Linear scleroderma ▪ Predominantly a pediatric condition ▪ <8yo, F>M, after 8yo F=M SCLERODERMA ▪ Combination of vasculopathy, activation, and fibrosis autoimmunity, immune ▪ Triggers (trauma, infection, possibly subclinical GVH reaction from persistent maternal cells) injure vascular endothelial cells → increased expression of adhesion molecules → entrap platelets and inflammatory cells → vascular changes (Raynaud phenomenon and pulmonary hypertension) ▪ Inflammatory cells infiltrate area of vascular damage → thickened artery walls → reduction in capillary numbers → migration of macrophages and other inflammatory cells into PPS Oral Exam Reviewer 2020 LOCALIZED ▪ Insidious erythema or bluish hue around an area of waxy induration, → indurated hyper/ hypopigmented atrophic lesions ▪ Subtle erythema may be the only presenting sign ▪ Linear scleroderma – varies in size (from few cm to entire length of extremity) ▪ Sometimes with arthralgias, synovitis, or flexion contractures ▪ Limb length discrepancies as result of growth impairment of muscle and bone ▪ En coup de sabre – seizures, hemifacial atrophy, ipsilateral uveitis, learning/behavioral changes ▪ 25% have extracutaneous symptoms SYSTEMIC ▪ Insidious onset with prolonged course, periods of remission or chronic disability/death ▪ Skin – early phase of edema spreads from dorsum of hands and fingers then face ▪ ↓ Edema → induration and fibrosis of skin, loss of SC fat, sweat glands, hair follicles ▪ Atrophic skin become shiny and waxy, flexion contractures develop at elbows, knees and hips with secondary muscle weakness and atrophy ▪ Skin ulceration –with subcutaneous calcification Table 185.2 Provisional Criteria for Classification of JSSc Major (Required) ▪ Proximal skin sclerosis/induration of the skin proximal to MCP or MTP joints Minor (At least 2 Required) Cutaneous Sclerodactyly Raynaud phenomenon, nail fold capillary Peripheral abnormalities (telangiectasias), digital tip vascular ulcers GI Dysphagia, GER Cardiac Arrhythmias, heart failure renal crisis, new onset arterial Renal hypertension Pulmonary fibrosis (HRCT/ radiography), Respiratory ↓ diffusing capacity for carbon monoxide, pulmonary arterial HTN Neurologic Neuropathy, carpal tunnel syndrome MSK Serologic Tendon friction rubs, arthritis, myositis Antinuclear Ab—SSc – selective autoAb ▪ Superficial morphea – topical steroid or UV therapy ▪ Deeper structures – systemic therapy JLS ▪ Methotrexate and steroids – prevent lesion extension → skin softening, improved ROM of joints Treatment plan: ● Weekly SC MTX at 1mg/kg (max 25mg) ● Weekly SC MTX at 1mg/kg (max 25mg) + either 3mo high-dose IV CS (30mg/kg, max 1000mg) for 3 consecutive days a month or weekly CS (same dose) for 3mo OR ● High-dose daily oral CS (2mkday, max 60mg) w/ slow taper over 48 wk ▪ MMF – 2nd line agent for recalcitrant disease ▪ OT/PT JSSc – target specific disease manifestation ▪ CCBs – Nifedipine 30-60mg SR daily, amlodipine 2.5-10mg daily ▪ ACEi – HTN with renal disease ▪ MTX and MMF – for skin manifestations ▪ Cyclophosphamide and MMF for pulmonary alveolitis and prevent fibrosis ▪ Cautious use of CS in JSSc bec of association with renal crisis Batch Clingy ▪ Presence of fibrosis on skin Rash ▪ Confused with eczema, dyshidrosis, psoriaris, malar rash from SLE ▪ Biopsy helpful to confirm diagnosis ▪ New classification major PLUS 2 of 20 minor criteria 162 Autoimmunity key process in pathogenesis of both types → (+) ANA in 42% of children with localized disease, 47% of these have antihistone Ab (+) ANA in 80.7% of children with JSSc, 34% of these have anti-Scl70 Ab Prevalence: 1/100,000 LS > JSSc (10:1 ratio, linear scleradema m/c type) ▪ Sclerodactyly – severe Raynaud phenomenon ulceration of the fingertips with loss of tissue pulps and tapered fingers ▪ Acro osteolysis – resorption of the distal tufts of distal phalanges ▪ Hyperpigmented post inflammatory changes with atrophy—salt and pepper appearance ▪ Pulmonary disease – most common visceral manifestation, both arterial and interstitial involvement (alveolitis) ▪ Asymptomatic to exercise intolerance, dyspnea at rest, right sided heart failure ▪ Pulmonary HTN – poor prognostic sign ▪ Cardiac fibrosis → arrhythmia, ventricular hypertrophy & ↓ cardiac function ▪ GIT – esophageal and intestinal dysmotility – dysphagia, reflux, dyspepsia, gastroparesis, bacterial overgrowth, dilated bowel loops, malabsorption and FTT ▪ Renal – arterial disease can cause chronic or severe episodic hypertension Raynaud Phenomenon ▪ Most frequent initial symptom in pediatric systemic sclerosis, 70% of children months or years before other symptoms ▪ Classic triphasic sequence of: ● Blanching (initial arterial vasoconstriction) ● Cyanosis (venous stasis) ● Erythema of the digits (reflex vasodilation from factors released from ischemia) ▪ Caused by cold weather or emotional stress, associated with pain and paresthesia (immerse hands in cold water), may involve thumbs, toes and ears ▪ Independent of Raynaud disease, connected to SLE, Scleroderma ▪ Begins in adolescence, symmetric occurrence, no tissue necrosis and absence of periungal telangiectasia ▪ vs Acrocyanosis – vasospastic d/o resulting in cool, painless, bluish discoloration in the hands and sometimes feet despite normal tissue perfusion ▪ vs. Chilblains – episodic color changes and devt of nodules related to severe cold exposure and spasm-induced vessel and tissue damage; assoc w/ SLE ANKYLOSING SPONDYLITIS And OTHER SPONDYLOARTHRITIDES (Psoriatic Arthritis, Arthritis of IBD) ▪ May be familial, HLA-B27 strongly associated with JAS (90%) and 50% of ERA patients, 7% healthy individuals ▪ Onset in older child, boy, oligo arthritis affecting hips, knees, ankles, feet, with enthesitis, with inability to touch toes PATHOGENESIS ▪ Susceptibility is largely genetically determined ▪ 30% heritability identified ▪ HLA-B27 responsible for 2/3 of the total and >100 additional PPS Oral Exam Reviewer 2020 Raynaud disease ▪ Begins in adolescence ▪ Symmetric occurrence, absence of tissue necrosis and gangrene and lack of manifestations of an underlying rheumatic disease Distinguishing clinical manifestations in spondyloarthritis: ▪ Arthritis of the axial skeleton, sacroiliac joints, hips, enthesitis, symptomatic eye inflammation (uveitis) and GI inflammation (even in the absence of IBD) ▪ Enthesitis – inflammation at the sites of attachments of ligaments, tendons, or joint capsule to bone (anticentromere, antitopoisomerase I Scl70), antifibrillarin, anti-PM/Scl, antifibrillin, or anti-RNA plumerase I or III LABS ▪ No labs diagnostic for both types ▪ CBC, Serum Chem and UA usually normal ▪ May have ↑ ESR, Eosinophilia, hypergammaglobulinemia ▪ ↑ Muscle enzymes (aldolase) with muscle involvement ▪ JSSc – anemia, leukocytosis, eosinophilia, (+) ANA and anti-SCL 70 Ab ▪ Imaging – delineate affected area and can be used to follow disease progression MRI – en coup de sabre and Parry-Romberg syndrome (facial hemiatrophy) to determine CNS/Orbital involvement ▪ PAH – pulmo testing (↓ VC), bronchioalveolar lavage, high resolution chest CT (basilar ground glass opacities, reticular linear opacities, honeycombing and mediastinal adenopathy → for monitoring visceral involvement in JSSc Tx for Raynaud phenomenon ▪ Cold avoidance ▪ Use hand and foot warmers ▪ Avoid carrying bags by their handles ▪ Nifedipine (10-20mg TID adult dose) reduces episodes ▪ Topical nitrates for digital vasodilation PROGNOSIS ▪ LS – self-limited, ave period of stabilization 3-5yr → active disease up to 20 years ▪ Prolonged diseases – disfigurement, disability with linear and deep ds subtypes ▪ SSC – some rapidly progressive with early organ failure and death ▪ Most common cause of death – heart failure due to myocardial and pulmonary fibrosis DIFFERENTIAL DIAGNOSIS ▪ Differentiate LS from SSc ▪ Juvenile arthritis –with contractures, no skin changes ▪ Diabetic cheiroarthropathy ▪ Pseudoscleroderma – patchy or diffuse cutaneous fibrosis without the other manifestations of sclerederma ▪ PKU, syndromes of premature aging, localized idiopathic fibrosis ▪ Scleredema – transient, self-limited after febrile illness (strep) with patchy sclerodermatous lesions on neck and shoulders ▪ ↑ ESR and or CRP is variable and may or may not be present at the onset of disease ▪ Mild increase in WBC and PC ▪ RF (-) in spondyloarthropathies ▪ ANA (-) except in psoriatic arthritis (50%) ▪ HLAB27 (+) in ~90% of Px w/ JAS Conventional radiograph ▪ Detects chronic bony changes & damage but not active inflammation Goals ▪ Control inflammation, minimize pain, preserve function, prevent ankylosis ▪ Anti-inflammatory meds + PT + education SpA ▪ Monotherapy or combination with NSAIDS, DMARDS or biologic agents Batch Clingy affected tissues → secretion of cytokines that induce fibroblasts to reproduce and synthesize excessive amount of collagen = fibrosis, lipoatrophy, loss of sweat glands and hair follicles 163 ▪ Early changes in sacroiliac joint – distinct margins and erosions → joint space widening ▪ Sclerosis starts on iliac side of joint ▪ Bamboo spine – squaring of the corners of the vertebral bodies and syndesmophyte formation in advanced disease MRI ▪ Gold standard for early visualization of sacroiliitis – bone marrow edema adjacent to the joint (Short T1 inversion recovery) DIFFERENTIAL DIAGNOSIS Reactive Arthritis ▪ Onset of arthritis after recent hx of diarrhea or symptoms of urethritis or conjunctivitis See Table 181.2 Etiologic Microorganisms of Reactive Arthritis Lower back pain can be caused by any of the ff: ▪ Suppurative arthritis of the sacroiliac joint ▪ Osteomyelitis of the pelvis or spine ▪ Osteoid osteoma of the posterior elements of the spine ▪ Malignancies – osteogenic sarcoma, Ewing, or leukemia Mechanical conditions: Scheuermann disease spondylolysis, spondylo-listhesis, Fibromyalgia ▪ Pain usually affecting soft tissue of the upper back in symmetric pattern ▪ Well-localized tender points and sleep disturbance ENTHESITIS-RELATED ARTHRITIS (ERA) ▪ 60% are males > females ▪ 20% have FHx of HLA-B27 associated disease (reactive arthritis, AS, or IBD with sacroiliitis) ▪ Sacroiliac joint arthritis clinically or radiographically – in up to 40% of children ▪ 20% have evidence of sacroiliac joint arthritis at diagnosis ▪ Risk of sacroiliac joint arthritis is highest in children who are HLA-B27(+) and ↑ CRP PSORIATIC ARTHRITIS ▪ 5% of JIA ▪ Onset peaks at ● Preschool – F>M, ANA (+), at risk for asymptomatic ocular inflammation ● Early adolescent years – M=F, arthritis is asymmetric, affects <4 joints at presentation ▪ Enthesitis detectable in 20-60% of patients, more frequent in older age PPS Oral Exam Reviewer 2020 ▪ 1st 6mos of disease – arthritis is asymmetric, involves <4 joints (knees, ankles, hips) ▪ Tarsitis/inflammation of small joints of foot – highly suggestive of ERA ▪ Enthesitis usually symmetric, affecting lower limbs ▪ Sacroiliac/axial joint involvement: inflammatory back pain, hip pain, alternating buttock pain ▪ Pain with palpation of lower back or with pelvic compression ▪ Nail pitting, onycholysis, dactylitis ▪ DIP joint involvement uncommon but highly suggestive of diagnosis when present NSAIDS ▪ Naproxen 15-20mkday ▪ May slow progression of structural damage (syndesmophyte formation and growth) Intraarticular CS ▪ Triamcinolone hexacetonide ▪ Helps control peripheral joint inflammation For moderate disease and JAS ▪ Add DMARDS (Sulfasalazine up to 50mkday, max 3g/day or MTX 10mg/m2) – may be beneficial for peripheral arthritis ▪ TNF inhibitors (etanercept, infliximab, adalimumab) – ↓ symptoms, improves function ▪ PT and Low impact exercise PROGNOSIS ▪ Ongoing disease activity for >5yrs in juvenile spondyloarthritis predicts disability ▪ Disease remission in <20% of children with SpA 5 years after diagnosis Factors affecting disease progression: ▪ Tarsitis ▪ HLA-B27 positivity ▪ Hip arthritis within the 1 st 6 months ▪ Disease onset after 8yo Legg Calve Perthes disease (avascular necrosis of femoral head), slipped capital femoral epiphysis, chondrolysis ▪ Pain over inguinal ligament and loss of internal rotation of the hip joint but without SpA features (like involvement of enthuses and/joints) Children with either arthritis and enthesitis OR arthritis OR enthesitis, with at least 2 of the ff: ▪ Sacroiliac joint tenderness/inflammatory LS pain ▪ Presence of HLA-B27 ▪ Onset of arthritis in a male >6yo ▪ Acute anterior uveitis ▪ FHx of an HLA-B27-assoc disease (ERA, sacroilitis w/ IBD, reactive arthritis, or acute anterior uveitis) in a 1st deg relative *Excluded: Patient with psoriasis (or FHx of psoriasis in a 1st deg relative), (+) RF, systemic arthritis Arthritis and psoriasis OR arthritis and at least 2 of the ff: ▪ Dactylitis ▪ Nail pitting or onycholysis ▪ Psoriasis in a 1st deg relative ▪ HLA-B27 positivity – highest risk of axial arthritis Batch Clingy genetic loci accounting for only 1/3 ▪ HLA-B27 → tendency to misfold and form abnormal cell surface structures. ▪ Infection with certain GI/GU pathogen can trigger reactive arthritis ▪ Altered gut microbiota and abnormal immune response to normal microbiota may play a role ▪ Inflamed joints & entheses in spondyloarthritis contain T & B cells, macrophages, osteoclasts, proliferating fibroblasts and osteoblasts w/ activation of the IL-23/IL-17 pathway 164 REACTIVE and POSTINFECTIOUS ARTHRITIS ARTHRITIS WITH IBD ▪ Presence of erythema nodosum, pyoderma gangrenosum, oral ulcers, abdominal pain, diarrhea, fever, weight loss, anorexia in a child with chronic arthritis → suspect IBD REACTIVE ARTHRITIS ▪ Following enteropathic or urogenital infections ▪ Variable course ▪ May remit or progress to a chronic spondyloarthropathy (ankylosing spondylitis) PATHOGENESIS ▪ Follows enteric infection (Salmonella, Shigella, Yersinia, Campylobacter or GU infection with C. trachomatis or E. coli and C. difficile) ▪ 75%are HLA-B27 positive (RA represents autoimmune response involving molecular mimicry ▪ Those HLA (+) have ↑ frequency of uveitis and extraarticular features ▪ Incomplete elimination of bacteria and bacterial products such as DNA ▪ Risk factor for persistent GI inflammation → ↑ risk that individual will develop chronic spondyloarthritis POSTINFECTIOUS ARTHRITIS ▪ Occurs after illnesses not in reactive arthritis group (Group A strep, viruses), N gonorrhea, N meninigitidis, Hib type B, Mycoplasma pneumonia ▪ Pain or joint swelling transient, <6 weeks ▪ Direct infection or generation and deposition of immune complexes PATHOGENESIS ▪ Viruses (rubella, varicella, herpes simplex, CMV) have been isolated in joints of patients ▪ Antigens from Hep B and adenovirus have been identified in immune complexes from joint tissue Transient Synovitis (toxic synovitis) ▪ Affects hip, often after an URTI ▪ Boys 3-10yo ▪ Acute onset of severe pain in hip with referred pain to thigh or knee (lasts one week) ▪ ESR and WBC usually normal ▪ Radio – widening of joint space 2o to effusion ▪ Aspiration of joint fluid often necessary to r/o septic arthritis ▪ Trigger presumed to be viral PPS Oral Exam Reviewer 2020 ▪ Arthritis usually lower extremities (often hips) ▪ Axial disease and inflammatory back pain less frequent at disease onset ▪ Enthesitis and peripheral arthritis more common 2 patterns of arthritis in IBD ▪ Polyarthritis – large and small joints, reflects activity of intestine inflammation ▪ Arthritis of the axial skeleton – ↑ sacroiliac joints ▪ Presents 3 days to 6 weeks after infection ▪ Reiter syndrome – arthritis, urethritis, conjunctivitis; uncommon in children ▪ Arthritis is asymmetric, oligo, with LE predilection ▪ Dactylitis (inflammation of entire digit) may occur and enthesitis (inflammation of tendons, ligmaents and joints attached to bone) common ▪ Cutaneous manifestations – circinate balanitis, ulcerative vulvitis, oral lesions, keratoderma blenorrhagica ▪ Systemic – fever, malaise, and fatigue ▪ Less common: conjunctivitis, optic neuritis, aortic valve involvement, sterile pyuria, polyneuropathy Modified New York criteria ▪ Radiographic changes in the sacroiliac joints – takes many years to develop ▪ Clinical sequelae of axial disease – lags further behind Criteria for axial spondyloarthritis (SpA) ▪ ≥3mos back pain and sacroiliitis on imaging PLUS ▪ 1 feature of SpA: inflammatory back pain, arthritis, enthesitis (heel), uveitis, dactylitis, psoriasis, Chron disease/UC, good response to NSAIDs, FHx of SpA, HLA-B27, or ↑ CRP ▪ (+) HLA-B27 – risk factor for the development of axial disease ▪ Diagnosis: history of recent GU or GI infection ▪ No specific test ▪ Trigger organism not typically found at the time arthritis is present ▪ Imaging may be nonspecific or normal ▪ R/o other causes of arthritis ▪ ASO and anti-DNAse B (to document previous strep infection) may help in Dx of postinfectious arthritis ▪ ↑ ESR and/ CRP is variable and may or may not be present at the onset of disease Radiology: Bamboo spine ▪ ESR and CRP may be markedly elevated ▪ Rubella and Hep B – affect small joints (esp knees) ▪ Mumps and Varicella involves large joints (knees) ▪ Hepatitis B arthritis-dermatitis syndrome – urticarial rash, symmetric migratory polyarthritis, resembling serum sickness ▪ Rubella associated arthropathy – may follow natural rubella infection or immunization, common in young women, arthralgia of hands and knees within 7d of onset or rash or 10-28d after immunization ▪ Parvovirus – arthralgia, symmetric joint swelling, morning stiffness (older women) ▪ Varicella – may be complicated by suppurative arthritis, secondary to Group A strep infection ▪ Post streptococcal arthritis ● Follow infection with Group A or Group G Strep ● Oligoarticular, affecting LE joints, mild sxs can persist for months ● Differs from rheumatic fever – painful migratory arthritis of brief duration ● Sometimes with valvular lesions on 2DED so considered an incomplete form of RF ● HLADRB1*16 may predispose children to dev post-strep arthritis or ARF DIFFERENTIAL DIAGNOSIS ▪ Rheumatic fever – Group A streptococcus ▪ Non-suppurative arthritis ● Adolescent boys with severe truncal acne ● Fever, persistent infection of pustular lesions ● Pyogenic (sterile) arthritis, pyoderma gangrenosum, acne (cystic) syndrome ● Autosomal dominant, mutation in PSTPIP1 gene ● Recurrent episodes may be associated with sterile myopathy and may last for several months ▪ Infective endocarditis – associated with arthralgia, arthritis, with signs of vasculitis (Osler nodes, Janeway lesions and Roth spots) ▪ Septic arthritis – acute arthritis affecting single joint ▪ Osteomyelitis – pain and effusion in adjacent joint, focal bone pain and tenderness at the site of infection ▪ Infectious of autoimmune hepatitis – arthritis assoc with GI sx or abn liver func tests PROGNOSIS ▪ Disease progresses to involve joints of spine and sacroiliac joints and may cause fusion and disability ▪ Usually unnecessary ▪ NSAIDS for pain and functional limitation ▪ Treating offending organism not warranted unless ongoing Chlamydia infection is suspected ▪ Intraarticular steroids for refractory/severely injured joints may be given once infection ruled out ▪ Physical activity and PT – may be added ▪ If postinfectious due to Strep – Penicillin prophylaxis for at least 1 year PROGNOSIS ▪ Usually resolves w/o complication unless associated with other organ involvement such as encephalomyelitis ▪ Reactive arthritis ● Uveitis, carditis ● May progress to chronic spondyloarthritis ▪ Persistent arthritis > 6 weeks → chronic rheumatic disease – JIA or SLE Batch Clingy JUVENILE ANKYLOSING SPONDYLITIS (JAS) ▪ Onset <16yo ▪ Begins with oligoarthritis and enthesistis 165 Palpable purpura – hallmark ▪ Pink macules or wheals → petechiae, raised purpura or ecchymoses, occasionally bullae and erosions develop ▪ Occur in gravity dependent areas ▪ Last 3-10d, may recur up to 4mos after initial presentation ▪ 90% cases in children between 3-10 years old ▪ M>F (1.2-1.8:1) ▪ Follow a documented URTI PATHOGENESIS ▪ Unknown but common finding is IgA deposition, suggests IgA mediation by IgA complexes ▪ Infectious triggers: GABHS, S. aureus, Mycoplasma, adenovirus ▪ Occasionally clusters in families → genetic component (HLA-B34 and HLA-DRB1*01) ▪ Subcutaneous edema – dorsum of hands and feet, periorbital area, lips, scrotum, or scalp ▪ Musculoskeletal involvement – arthritis and arthralgias common (75%); self-limited and oligoarticular (more of LE) ▪ Gastrointestinal (80%) ● Abdominal pain, vomiting, diarrhea, paralytic ileus, melena, intussusception and mesenteric ischemia or perforation ● Purpura of intestinal tract ▪ Renal involvement (30%) ● Hematuria, proteinuria, HTN, frank nephritis, nephrotic syndrome, acute or chronic renal failure ● ESRD uncommon ● Manifestations can be delayed for several months after the initial illnsss → close ffup, serial UA ▪ Neurologic ● Due to HTN or CNS vasculitis ● Intracerebral hemorrhage, seizures, headaches, behavior changes ▪ Others – orchitis, carditis, inflammatory eye disease, testicular torsion, pulmonary hemorrhage HENOCHSCHONLEIN PURPURA TAKAYASU ARTERITIS ▪ Pulseless disease ▪ Chronic large vessel vasculitis of unknown etiology, involves aorta and major branches ▪ 10-40yo, Asians, females (2-4:1) Prepulseless phase (early) ▪ Fever, malaise, weight loss, HA, HTN, myalgia, arthralgias, dizziness, abdominal pain ▪ HTN and Headache – most common presenting symptom PATHOLOGY ▪ Inflammation of vessel wall → damaged elastic lamina and media → blood vessel dilation and aneurysm formation ▪ Progressive scarring and proliferation→ stenotic vessels ▪ Subclavian, renal, carotid arteries m/c involved ▪ Pulmonary, coronary, and vertebral arteries also affected ▪ Some report no systemic symptoms and present with vascular complications PATHOGENESIS ▪ Etiology unknown ▪ Presence of abundant T cells with a restricted repertoire of cellular immunity in TA vascular lesions → importance of cellular PPS Oral Exam Reviewer 2020 Later manifestations ▪ Diminished pulses, asymmetric BP, claudication, Raynaud phenomenon, renal failure, symptoms of pulmonary or vascular ischemia ▪ Other findings: pericardial effusion, pericarditis, pleuritis, splenomegaly, arthritis ▪ Inflammation of aortic valve – valvular insufficiency ▪ Clinical diagnosis → presence of typical rash ▪ No lab diagnostic ▪ Leukocytosis, thrombocytosis, mild anemia, elevations of CRP, ESR ▪ Platelet count normal in HSP ▪ Occult blood (+) frequently ▪ Serum albumin may be ↓ due to renal or intestinal protein loss ▪ Renal assessment: BP, UA, creatinine UTZ ▪ For GIT complaints ▪ Bowel wall edema or intussusception → barium enema (dx/tx) Skin biopsy – leukocytoclastic vasculitis of dermal capillaries and postcapillary venules Renal biopsy ▪ Endocapillary proliferative glomerulonephritis (focal segmental process to extensive crescentic involvement ▪ IgA deposition, deposition of C3, fibrin and IgM in a lesser extent DIFFERENTIAL DIAGNOSIS ▪ Other small vessel vasculitis, infection, acute PSGN, HUS, coagulopathy, other acute intraabdominal processes ▪ Papular purpuric glove and sock syndrome ▪ SLE, other vasculitides (urticarial, hypersensitivity) and thrombocytopenia ▪ Infantile acute hemorrhagic edema (AHE) ● Resembles HSP ● Cutaneous leukocytolastic vasculitis affecting infants <2yo ● Fever, tender edema of the face, scrotum, hands & feet, ecchymosis on the face & extremities ● Trunk spared ● Petechiae in mucous membranes ● PC normal or elevated ● UA abnormal ● Biopsy may help in differentiating it with HSP Table 192.5 proposed Classification Criteria for Pediatric-Onset Takayasu Arteritis Angio abnormalities (CT, MRI) of aorta and main branches and at least ONE of the ff: ▪ ↓ Peripheral artery pulse and claudication ▪ BP diff bet arms and legs >10mmHg ▪ Bruits over aorta and major branches ▪ HTN (based on childhood normative data) ▪ Elevated acute phase reactant (ESR or CRP) LABS ▪ Nonspecific ▪ ↑ ESR and CRP ▪ Other nonspecific markers of inflammation: leukocytosis, thrombocytosis, anemia of chronic inflammation, hypergammaglobulinemia ▪ Supportive ▪ Adequate hydration, nutrition, analgesia Steroids – to treat significant GIT involvement or other lifethreatening manifestations ▪ IV methylprednisolone for 1-2 weeks → taper → reduce abdominal pain and joint pain but does not alter overall prognosis ▪ Empiric use of Prednisone 1mkday for 1-2wks tapered ▪ Chronic HSP renal disease – azathioprine, cyclophosphamide, mycophenolate mofetil COMPLICATIONS ▪ GIT perforation ▪ ESRD <5% HSP nephritis ▪ Serial monitoring of BP and UA for 6mos after dx if presenting with HTN or urine abnormalities PROGNOSIS ▪ Excellent, acute self-limited course for an average of 4 weeks ▪ 15-60% with ≥1 recurrence within 4-6mos after dx ▪ Milder in each relapse ▪ Severe initial course higher risk for relapse ▪ Chronic renal disease develops in 1-2% ▪ <5% of those with HSP nephritis go on to have ESRD Glucocorticoids ▪ Mainstay of treatment ▪ High doses (1-2mkday prednisone or MP IV) → gradual dose tapering ▪ If TA progresses or recurs: MTX, Azathioprine ▪ Cyclophosphamide – for severe or refractory disease ▪ AntiHTN → control BP caused by renovascular disease COMPLICATIONS ▪ Arterial stenosis, aneurysms, occlusions → ischemic symptoms → life threatening ▪ Stroke, MI, renal impairment, limb threatening arterial disease PROGNOSIS ▪ 20% has monophasic course with sustained remission Batch Clingy ▪ Most common vasculitis in childhood ▪ Leukocytoclastic vasculitis and immune deposition in the small vessels (skin), joints, GIT, and kidney ▪ IgA vasculitis – presence of vasculitis and predominance of IgA deposits in small vessels 166 immunity → existence of specific but unknown aortic tissue antigen ▪ Expression of IL1, IL6, TNFα higher in patients with TA Subdiaphragmatic (aortic arch) disease ▪ CNS (stroke, transient ischemic attack) and cardiac (heart failure, palpitations) symptoms Infradiaphargmatic (mid-aortic syndrome) disease ▪ HTN, abdominal bruit, pain Radiology ▪ To establish large vessel arterial involvement ▪ Gold standard – Arteriography of the aorta and major branches including carotid, subclavian, pulmonary, renal, and mesenteric branches → ID luminal defects, dilatation, aneurysm, stenoses ▪ MRA and CT angio also accepted ▪ 2DED for assessing aortic valvular involvement ▪ Serial vascular imaging necessary to assess response to treatment ▪ Most suffer relapses ▪ Overall survival: 93% at 5yrs, 87% at 10yrs ▪ High risk for accelerated atherosclerosis Batch Clingy DIFFERENTIAL DIAGNOSIS ▪ Giant Cell Arteritis (temporal arteritis) – large vessel vasculitis in older adults, rare in childhood ▪ Systemic infections, autoimmune conditions, malignancies ▪ Non inflammatory conditions causing large vessel compromise – Fibromuscular dysplasia, Marfan syndrome and Ehler Danlos PPS Oral Exam Reviewer 2020 167 Acute Can be painful or painless ▪ Painful – stance phase is shortened as child decreases time on painful extremity ▪ Painless –underlying proximal muscle weakness or hip instability, stance phase is equal but the child leans or shifts the center of gravity over the involved extremity for balance Bilateral disorder produces a waddling gait Trendelenburg gait – weak abnormal hip adductors, in a single leg stance can be elicited when abductors are weak DISEASE ETIOLOGY / INCIDENCE / PATHOGENESIS CLINICAL MANIFESTATIONS Trauma – leading cause of death and disability in children >1yo → r/o fracture TRAUMA LABORATORY FINDINGS / DIAGNOSIS TREATMENT *See Fracture* CHILD ABUSE ▪ Suspected in non-ambulatory children with LE long bone fractures ▪ Transverse fractures in long bones – most prevalent ▪ Corner fractures in metaphysic – most classic S. aureus most common in all age groups Fractures that suggest non accidental injury: ▪ Femur fractures in non-ambulatory children (<18mos) ▪ Distal femoral metaphysical corner fractures ▪ Posterior rib fractures Scapular spinous process fractures proximal humeral fractures Full skeletal survey – demonstrates multiple fractures in various stages of healing ▪ Subtle, non-specific → dependent on patient age ▪ Clinical suspicion ▪ Blood CS should be performed in all suspected cases ▪ ↑ WBC and differential, ESR, CRP ▪ Leukocyte and ESR may be normal during 1st few days of infection and does not preclude diagnosis Neonates ▪ Group B Strep & gram neg (E. coli) also common), Group A Strep (<10%) Neonates ▪ Pseudoparalysis or pain with movement ▪ Half do not have fever and might not appear ill >6yo ▪ S. aureus, Strep, or Pseudomonas (puncture wounds of foot, direct inoculation from foam padding of the shoe into bone/cartilage → osteochondritis) Older infants and children ▪ Fever, pain, localizing signs (edema, erythema and warmth) ▪ Limp or refusal to walk Sickle cell patients – Salmonella and S. aureus ▪ Sickle cell <24mos - S. pneumoniae (declined incidence due to PCV vaccine) ▪ Focal tenderness over a long bone ▪ Local swelling and redness – infection has spread out of metaphysis into subperiosteal space ▪ Long bones involved – femur and tibia equally affected, UE (1/4 cases) Bartonella henselae – osteomyelitis in pelvic & vertebral bones OSTEOMYELITIS Kingella kingae – 2nd most common cause in <4yo, esp in subacute presentation, difficult to detect (PCR) Atypical mycobacteria (also S. aureus and pseudomonas) in penetrating injuries, implanted materials (orthopedic hardware) Fungal infections as part of multisystem disseminated disease (Candida osteomyelitis complicateting fungemia in neonates w/ or w/o indwelling catheters) -Blastomycosis in endemic areas (+) Confirmed microbial etiology in 60% of cases of osteomyelitis, blood culture (+) in 50% of patients ▪ Median age 6yo, boys>girls (boys’ behavior predisposing them to traumatic events) ▪ Healthy children – hematogenous ▪ Minor closed trauma (30%) – common preceding event ▪ Infection of bone following penetrating injuries or open PPS Oral Exam Reviewer 2020 Pelvic osteomyelitis – subtle findings like hip, thigh, or abdominal pain Vertebral osteomyelitis – back pain ± tenderness to palpation overlying the vertebral processes ▪ Usually single site affected except neonates (2 or more bones) Brodie abscess – children with subacute symptoms and focal finding in metaphyseal area (usually tibia) ▪ Radiographic lucency and surrounding reactive bone ▪ Contents are usually sterile ▪ Some patients with osteomyelitis (S.aureus) develop DVT adjacent to affected bone which can produce septic pulmonary emboli ▪ Mandatory reporting to social welfare agencies Radiographic findings on some systemic disease that mimic signs of child abuse: Osteogenesis imperfecta, Osteomyelitis, Caffey disease, fatigue fractures Xray ▪ Initial evaluation to exclude trauma and foreign bodies ▪ Within 72hrs, can show displacement of deep muscle planes from adjacent metaphysis caused by deep tissue edema ▪ Lytic bone changes not seen until 30-50% bony matrix destroyed ▪ Tubular long bones do not show lytic changes 7-14 days after onset of infection CT ▪ Can demonstrate osseous and soft tissue abnormalities, ideal for detecting gas in soft tissues ▪ Poor sensitivity for detecting osteomyelitis MRI ▪ Most Sn and Sp ▪ Best for identifying abscess and differentiating bone from soft tissue infection ▪ Provides precise anatomic detail of subperiosteal pus and accumulation of purulent debris (bone marrow and metaphysis) for possible surgical intervention Acute osteomyelitis – purulent debris and edema appear dark with ↓ signal intensity on T1 weighted images, fat appear bright Cellulitis and sinus tract – areas of high signal intensity on T-2 weighted images Short tau inversion recovery (STIR) MRI – rapid imaging for osteomyelitis Radionuclide Neonates ▪ MSSA, Group B strep, Gram neg bacilli ▪ Nafcillin or Oxacillin (150-200mkday q6 IV) + Cefepime (100150mkday q12) ▪ If MRSA – Vancomycin 45mkday q8 (gold standard for invasive MRSA) ▪ If preterm or with vascular catheter – cover for nosocomial (gram neg enteric, Pseudomonas or S. aureus, fungal (Candida) Older infants and children ▪ S. aureus, K. kingae, group A step ▪ MSSA – nafcillin or Cefazolin (150mkday q6) - parenteral treatment of choice, backbone of empirical treatment for acute hematogenous osteomyelitis ▪ Critically ill, high local prevalence of CA-MRSA infection, add Vancomycin to a beta-lactam ▪ Clindamycin (40mkday q6) alternative to susceptible MRSA and MSSA when beta-lactam cannot be used (allergy), anaerobic coverage ▪ S. pneumonia – Penicillin first line ▪ If resistant to penicillin, cefotaxime or ceftriaxone Sickle cell with osteomyelitis ▪ Salmonella, S. aureus ▪ Cefepime (150mkday q8) + Vancomycin/ Clindamycin Immunocompromised ▪ Vancomycin + Ceftazidime, Aminoglycoside ▪ K. kingae – Cefazolin (<4yo) Cefepime or Pip-tazo ± ▪ Duration of antibiotic individualized, based on clinical improvement, culture growth/ isolates Batch Clingy Fractures – 2nd most common manifestation of child abuse (after skin injury e.g. bruises, burns, abrasions) 168 fractures, or surgery ▪ In suspected bone infections esp in early course, alternative to MRI (esp if multiple foci suspected) ▪ Technetium 99 methylene diphosphonate accumulates in areas of ↑ bone turnover ▪ Increased uptake, 3 phase imaging ▪ Sn 84-100%, Sp 70-96% in hematogenous osteomyelitis and can detect osteomyelitis within 24-48h after onset of symptoms ▪ Sn in neonates lower bec of poor mineralization ▪ Advantage: infrequent need for sedation, can image entire skeleton for detection of multiple foci ▪ Disadvantage: radiation exposure, inability to image surrounding soft tissues, overall lack of detail PATHOGENESIS ▪ Anatomy and circulation of long bones – localization of blood borne bacteria ▪ Metaphysis has low velocity blood flow predisposing to bacterial invasion → bacterial focus, phagocyte migration → inflammatory exudate (metaphyseal abscess) → ↓ O2 tension, ↓ pH, osteolysis, tissue destruction → pressure ↑ spread via haversian system and Volkmann canals into subperiosteal space ▪ Purulence = impairment of blood supply to cortex and metaphysis NB – transphyseal blood vessels connect metaphysis and epiphyses → pus from metaphysis may enter joint space → potential for abnormal growth and bone or joint deformity Late childhood more adherent → pus decompresses through periosteum Late adolescent – hematogenous osteomyelitis begins in the diaphysis → entire intramedullary canal ▪ Septic arthritis + osteomyelitis may be seen in older children w/ S.aureus osteomyelitis → simultaneous hematogenous inoculation of bone and joint space SEPTIC ARTHRITIS ▪ Pain in groin, anterior thigh, or knee, which may be referred from the hip ▪ Able to bear weight on affected limb, usually ambulatory with antalgic gait with foot externally rotated ▪ Afebrile or with low grade fever If with venous thrombosis – anticoagulant Surgery – drainage when there is frank pus, removal of sinus tract or sequestrum in chronic osteomyelitis if present PT – preventive, start 2-3days after pain decreases, cast when there is potential for pathologic fracture PROGNOSIS ▪ CRP normalizes within 7 days after treatment starts ▪ ESR drops after 10-14 days ▪ On ff up, monitor ROM and bone length ▪ Symptomatic treatment ▪ Activity limitation and relief of weight bearing ▪ Anti-inflammatory agents and analgesics can shorten duration of pain ▪ Recovery usually within 3-6 weeks ▪ Prevalent in 3-8yo (but present in all age groups) ▪ 70% with recent URTI 7-14 days before symptoms ▪ S. aureus – most common cause in all age groups ▪ MRSA (>25%), Group A Strep, Strep pneumonia (10-20%, most likely in <2 yrs old) ▪ Kingella kingae in <4yo (culture and PCR detection) ▪ Gonococcus – sexually active adolescents, septic arthritis, and tenosynovitis usually of small joints or as monoarticular infection of a large joint (knee) ▪ N. meningitides may occur on the first few days of illness, or a reactive arthritis several days after antibiotic initiation ▪ Neonates – Group B Strep ▪ Fungal infection as part of multisystem disseminated disease Candida arthritis can complicate systemic infection in neonates with or without indwelling vascular catheters ▪ Primary viral infections of joints are rare, arthritis may accompany viral syndromes (parvovirus, mumps, rubella live PPS Oral Exam Reviewer 2020 ▪ CRP and WBC normal ▪ May have mild elevation of ESR ▪ Monoarticular ▪ Signs and symptoms depend on age of patient Neonates and infants – subtle, septic arthritis can be caused by adjacent osteomyelitis, transphyseal spread vs osteomyelitis – pseudoparalysis or pain limiting voluntary movement of affected extremity (diaper changes) Older infants and children – fever and pain ▪ Localizing signs – swelling erythema, warmth of affected joint ▪ Limp or refusal to walk ▪ Erythema and edema – seen earlier in SA than osteo because synovium more superficial, metaphyses deeper (except SA of hip, because hip joint deep) Lyme arthritis – more prominent joint swelling, disproportionate Xray (AP and Lauenstein (frog leg) lateral) of pelvis, usually normal Hip UTZ -preferred to xrays, demonstrates joint effusion ▪ ↑ WBC, differential, ESR, CRP ▪ Most children will have normal leukocyte counts and ESR at presentation → do not preclude dx of SA ▪ Blood cultures in all cases, positive in 20% ▪ Obtain cervical, anal and throat cultures when gonococcus is suspected UTZ-guided aspiration of joint fluid/pus for GS/CS – best specimen for bacteriologic culture PCR – best method to determine K. kingae in synovial fluid Synovial fluid analysis – >50k-100k cells/mm3 indicate infectious process CRP monitoring – assessment of response to treatment ▪ CRP >10-13mg/dL seen in adjacent infections complicating septic arthritis XRAY – widening of joint capsule, soft tissue edema, obliteration Neonates ▪ S. aureus, GBS, Gram neg: Nafcillin or Oxacillin (150-200mkday q6 IV) + Cefepime (100-150mkday q12) ▪ If MRSA, Vancomycin (15mkday q6) ▪ If preterm or with vascular catheter – consider Pseudomonas or fungal coverage Older children ▪ Empiric treatment (S. aureus, Strep, K. kingae): Cefazolin (100150mkday q8) or Nafcillin (150-200mday q6) ▪ Ill appearing, suspected to be bacteremic, local clindamycin resistance is 10-15% – Vancomycin ▪ CA-MRSA – Clindamycin (40mkday q6) ▪ Immunocompromised – Vancomycin + Ceftazidime, Cefepime, or Piptazo ± aminoglycoside Batch Clingy TRANSIENT MONOARTICULAR SYNOVITIS (Toxic Synovitis) ▪ Reactive arthritis that affects the hip, one of most common causes of hip pain in young children ▪ Etiology unknown ▪ Nonspecific inflammatory condition ▪ Postviral immunologic synovitis DIFFERENTIAL DIAGNOSIS ▪ Cellulitis and trauma (accidental or abuse) most common ▪ Myositis or pyomyositis –fever, warm swollen extremities and limping, tenderness more diffuse ▪ Pyomyositis – pelvis common site, distinguish from pelvic osteomyelitis, do MRI, often caused by S.aureus and group A strep ● Any child with (-) xray, (-) hip aspiration, with fever, limp and ↑ inflammatory markers should be evaluated for pyomyositis ▪ Iliopsoas abscess – thigh pain, limp, fever ▪ Pelvic osteomyelitis vs appendicitis, UTI and gynecologic disease ▪ Leukemia – bone pain and joint pain early sx ▪ Neuroblastoma – if with bone involvement can be mistaken for osteo ▪ Ewing sarcoma – no fever ▪ Diagnosis is clinical ▪ r/o Septic arthritis first! –appear more systematically ill, more severe pain, child refuses to walk or move their hip at all → high fever, ↑ CRP or ESR, WBC → UTZ-guided aspiration of the hip joint ▪ S. aureus – 21-28 days if with improvement in 5-7 days, if not total of 4-6 weeks (slower resolution of symptoms or normalization of CRP) ▪ IV to oral antibiotics if afebrile >48-72hrs, resolved bacteremia ▪ Cephalexin (100-150mkday q8) or Clindamycin (30-40mkday q8) 169 vaccines) → immune mediated pathogenesis ▪ Microbial etiology confirmed in 65% of cases ▪ Common in young children, 2-5yrs old ▪ Adolescents and neonates at risk for gonococcal septic arthritis PATHOGENESIS ▪ Most are hematogenous ▪ Hematogenous seeding of synovial space (rich vascular supply) → bacterial products within joint space stimulates cytokine production (TNF-a, IL-1) → inflammatory cascade → destruction of cartilage and synovium → ↑ pressure compromise vascular supply → necrosis ▪ Less common → follow penetrating injuries, arthroscopy, prosthetic joint surgery, intraarticular steroid injection, immunocomp pxs with rheuma joint disease to the relatively lesser degree of pain and limited range of motion when compared with suppurative arthritis ▪ Predilection for large joints (knees, hips), monoarticular or pauciarticular at presentation ▪ Joints of LE (75% of SA) ▪ Elbow wrist and shoulder (25%) of normal fat lines ▪ Hip – medial displacement of obturator into pelvis (obturator sign) UTZ – highly sensitive in detecting joint effusion and fluid in soft tissue and subperiosteal regions, help in aspiration of hip CT/MRI – useful to check joint fluid, MRI used to exclude adjacent osteomyelitis/pyomyositis Radionuclide – more sensitive than x-ray, may be positive 2d before onset of symptoms, useful for sacroiliac joint DIFFERENTIAL DIAGNOSIS Hip ▪ Toxic synovitis ▪ Legg Calve Perthes disease ▪ Slipped capital femoral epiphyses ▪ Psoas abscess ▪ Proximal femoral, pelvic osteomyelitis Knee ▪ Distal femoral or tibial osteomyelitis ▪ Pauciarticular JRA ▪ Usually 10-14 days for Streptococci, K. kingae Longer (3 weeks) for S. aureus and gram neg infections ▪ 4 weeks if with concomitant osteomyelitis, extensive disease, slow response to treatment ▪ Plain radiograph of joint before completing tx can provide evidence (periosteal new bone) of a previously unappreciated contiguous site of osteomyelitis → prolong antibiotic tx ▪ Shift to oral antibiotics for completion of tx once afebrile for 4872 hours and clearly improving Surgical ▪ Infection of hip – surgical emergency (vulnerable blood supply head of femur) ▪ Other joints – daily aspiration synovial fluid ▪ Arthrotomy or video assisted arthroscopy PROGNOSIS ▪ Recurrence and devt of chronic infection after treatment occur in <10% of patients ▪ Long term squelae (<5%): leg-length discrepancy, angular deformity from growth arrest, limitation of ROM (chrondral damage and avascular necrosis of the femoral head from septic arthritis of the hip) ▪ Long term ff up, ROM and bone length Batch Clingy ▪ r/o trauma, cellulitis, pyomyositis, Sickle cell disease, hemophilia and HSP ▪ Several joints involved – collagen vasc diseases, HSP, RF, reactive arthritis, IBD Dexamethasone for 4 days – adjunct tx w/ antibiotics to ↓ duration of fever and promote more rapid decline in inflammatory markers – not routine PPS Oral Exam Reviewer 2020 170 Complex ETIOLOGY / INCIDENCE / PATHOGENESIS CLINICAL MANIFESTATIONS Classification Acetabular dysplasia – abnormal morphology and development of acetabulum ▪ Hip sublaxation – partial contact between the femoral head & acetabulum ▪ Hip dislocation – hip w/o contact between the articulating surfaces of the hip Neonate ▪ Asymptomatic, use maneuvers ▪ Barlow – assess dislocation, adduct flexed hip push posteriorly, if positive- hip will slide out of acetabulum ▪ Ortolani – abduct flexed hip, if (+) femoral head will slip into pocket ▪ Hip click – high pitched sound at end of abduction after Barlow & Ortolani 2 groups ▪ Typical – normal pxs w/o syndromes or genetic conditions ▪ Teratologic – identifiable causes like arthrogryposis & genetic syndrome & occur before birth Infants ▪ 2nd & 3rd month—tighter tissues ▪ Limited hip abduction, shortening of thigh, proximal location of greater trochanter, asymmetry of gluteal folds, positioning of the hip ▪ Limitation of abduction – most reliable sign of dislocated hip ▪ Galeazzi sign – shortening of the thigh, both hips in 90 o of flexion vs. height of the knees, asymmetry ▪ Klisic test – place 3rd finger over the greater trochanter and index finger on the ASIS → (+) if trochanter elevated, line projects halfway between umbilicus and pubis ▪ Etiology remains unknown ▪ ↑ Laxity of joint, fails to maintain a stable femoroacetabular articulation → affected by hormonal, mechanical & genetic factors ▪ FHx of DDH in 12-33% ▪ More common in females (80%, & their greater susceptibility to maternal hormone relaxin) DEVELOPMENTAL DYSPLASIA OF THE HIP SKELETAL DYSPLASIAS RISK FACTORS ▪ Any condition that leads to a tighter intrauterine space (oligohydramnios, large BW, first pregnancy) ▪ High association w/ intrauterine molding abnormalities (torticollis, metatarsus adductus) → crowding phenomenon has a role in the pathogensis ▪ Left hip commonly affected PATHOGENESIS ▪ Fatty tissue in the depths of the socket (pulvinar) & the ligamentum teres can hypertrophy → blocked reduction of femoral head ▪ Transverse acetabular ligament thickens → narrows opening of acetabulum ▪ Shortened iliopsoas tendon becomes taut across the front of the hip → hourglass shape to the hip capsule → limited access to acetabulum ▪ Dislocated femoral head places pressure on the acetabular rim and labrum→ infolds & becomes thick ▪ Longer time hip spends dislocated → abnormal devt of acetabulum → progressively shallow, oblique acetabular roof and a thickened medial wall Genetic skeletal disorders ▪ Chondroosteodysplasias – skeletal/bone dysplasia ▪ Metabolic bone conditions – rickets, hyopophosphatasia ▪ Dysostoses – affect single bone (craniosynostosis) ▪ Other skeletal malformation Chondroosteodysplasias ▪ 1 in 4000 births ▪ Mutations of chondrodysplasia genes ▪ Types: ● Chondroosteodysplasia – genetic d/o of cartilage, deficient linear PPS Oral Exam Reviewer 2020 LABORATORY FINDINGS / DIAGNOSIS Ultrasound ▪ Diagnostic modality of choice for DDH before appearance of femoral head ossifies (4-6oms) ▪ Neonates – PE better than UTZ ( incidence of false (+) ▪ Used to monitor acetabular devt in infants in Pavlik harness ▪ Most authors recommend screening in infants at risk (breech, family hx, torticollis), regardless of clinical findings X-ray (AP) recommended at 4-6mos (after ossification) COMPLICATIONS ▪ Avascular necrosis of the femoral head – most feared ▪ Abnormal growth & devt can occur ▪ Others: redislocation, residual subluxation, acetabular dysplasia, post op complications (wound infections) TREATMENT ▪ Obtain & maintain concentric reduction of femoral head for normal devt Newborns & infants <6mos ▪ Pavlik harness as soon as dx is made ▪ Significant proportion normalize within 3-4 weeks → need to reexamine ▪ Pavlik harness manages acetabular dysplasia, subluxation, or dislocation ▪ Hip instability resolves in approx. 75% of cases if maintained on Ortolani-positive hip in a Pavlik harnesss on a full-time basis for 6 wk ▪ After 6mos Pavlik not effective anymore, >50% failure rate ▪ Set to maintain hips in flexion, if not reduced in 3-4 weeks → d/c harness Children 6mos to 2 years old ▪ Goals of treatment in late-dx dysplasia: obtain & maintain reduction of the hip w/o damaging the femoral head ▪ Closed reductions – OR under GA ▪ Maintained after in a well molded spica cast in moderate flexion and abduction ▪ Removal of cast after 12 weeks ▪ If unsuccessful → open reduction The walking child ▪ Limp or waddling gait, or leg length discrepancy ▪ Affected side shorter, child toe walks on affected side ▪ Trendelenburg sign (+) – abductor lurch observed when child walks ▪ Galeazzi sign – limited hip adduction on affected side, knees at different levels when hips are flexed ▪ Excessive lordosis – common & often presenting complaint Children > 2 years old ▪ Require open reduction ▪ Concomitant femoral shortening osteotomy performed to reduce pressure off femur and minimize risk of osteonecrosis ▪ Post op, immobilized in spica cast for 6-12 weeks Metaphyseal dysplasia – short stature, bowing of the legs and waddling gait, normal serum Ca, PO4, ALP activity and Vit D metabolites Jansen type – cupped and ragged metaphyses → mottled calcification at the distal ends of bone overtime Schmid type – less severe, radiograph resembles signs seein in familial hypophosphatemia; defects in collagen type X, alpha 1 and hip abnormal more debilitating ▪ History, PE matched with disproportionate clinical features and clinical phenotypes of well-documented d/o ▪ Radiographs → skeletal survey ▪ Molecular genetic testing – FGFR mutation in achondroplasia; used for prenatal diagnosis as well for future pregnancy planning ▪ Once correct Dx is established → prognosis and anticipation of medical and surgical problems ▪ Lifesaving measures discussion over poor prognosis cases (thanatophoric dysplasia or achondrogenesis types Ib or II) ▪ Mgt directed at preventing and correcting skeletal deformities, treating nonskeletal complications, providing genetic counseling and helping patients and families learn to cope. Batch Clingy DISEASE 171 growth → achondroplasia; generalized skeletal abnormal with nonskeletal tissue ● Osteodysplasia – abnormal bone structure → OI Table 714.2 Major Problems Associated with Skeletal Dysplasias Table 714.4 Lethal neonatal dwarfism ▪ Avoidance of contact sports or activities that cause injury or stress to joints PATHOPHYSIOLOGY Chrondrodysplasia mutations act through cartilage matrix proteins → dominant negative mechanism (protein products of mutant allele interfere w/ assembly and function of multimeric molecules containing protein products of both normal and mutant allele) ▪ Type II collagen gene COL2A1 forms the mutant & normal chains ▪ Mutations disrupt endochondral ossification affecting linear growth of skeleton Disorders involving cartilage matrix protein ▪ Spondyloepiphyseal dysplasia/type 2 collagenopathies ● Lethal spondyloepiphyseal dysplasia ● Spondyloepiphyseal dysplasia congenita – head & face normal, cleft palate common, short neck, barrel chest, club foot & hypotonia ▪ Kniest Dysplasia – flat face, prominent eyes, enlarged joints, cleft palate, club foot, hearing loss, myopia, retinal detachment common complication ▪ Late onset Spondyloepiphyseal dysplasia – mild epiphyseal & vertebral abnormality on x-ray ▪ Aggrecan-related spondyloepiphyseal dysplasias ▪ Stickler syndrome/dysplasia - associated with Pierre Robin anomaly ▪ Schmidt metaphyseal dysplasia ▪ Pseudoachondroplasia and multiple epiphyseal dysplasia ▪ Controversial on surgical limb lengthening, human growth hormone, C-natriuretic peptite to promote linear bone growth Table 714.5 Nonlethal dwarfing conditions Recognizable at Birth or within 1st few mos of life Growth ▪ Disproportionate short stature (limbs vs trunk; lt/ht below 3rd percentile) ▪ Shortening of limbs: UE do not reach mid-pelvis in infancy or upper thigh after infancy ▪ Rhizomelic shortening –greatest in proximal segments ▪ Mesomelic shortening – involves hands and feet ▪ Shortening of trunk: short neck, small chest, protruberant abdomen Non-growth-related manifestations ▪ Abn joint mobility, protruberence at and around joints and angular deformities (symmetric) ▪ Spinal cord compression in achondroplasia, shor ribs reduce thoracic volume → breathing compromise (chondrodysplasia) ▪ Cleft palate common → defective palatal growth ▪ Retinal detachment (spondyloepiphyseal dysplasia congenita), sex reversal (campomelic dysplasia), congenital heart malformations (Ellisvan Crevald Syndrome), immunodeficiency (cartilage-hair hypoplasia), renal dysfunction and asphyxiating thoracic dysplasia Family and Reproductive History ▪ Mendelian inheritance pattern ▪ Attention to mild degrees of short stature, disproportion, deformities & others (precocious osteoarthritis) that may have been overlooked ▪ Previous stillbirths, fetal losses, other abn pregnancy outcomes from skeletal dysplasia, polyhydramnios, reduced fetal movt ▪ Mostly genetic, but common to have no FH of the d/o Batch Clingy Disorders involving Transmembrane Receptors Mutations in FGFR3 & PTHR1 Achondroplasia ▪ Thanatophoric dysplasia ● Large head very short limbs, polyhydramnios, premature delivery ● Kleeblattschadel – cloverleaf skull ● Severe respiratory distress due to small thorax, poor prognosis ▪ Achondroplasia ● Short limbs, long narrow trunk, large head w/ midfacial hypoplasia, prominent forehead ● Delayed motor milestones (hypotonia, mechanical difficulty balancing → exaggerated lumbar lordosis, intelligence normal) ▪ Hypochondroplasia ● Milder than achondroplasia, not apparent until childhood, stocky build, slight frontal bossing, may have learning disability Jansen Metaphyseal dysplasia ▪ Rare, severe shortening of limbs, unusual facial appearance, sometimes with clubfoot and hypercalcemia (serum 13-15mg/dL) ▪ Normal epiphyses, enlarged joints, limited mobility → flexion contractures, bent over posture ▪ Normal intelligence, ± hearing loss ▪ Diet preventing obesity ▪ Dental care to minimize crowding and malalignment of teeth ▪ Participation in Support Groups PPS Oral Exam Reviewer 2020 172 Disorders involving transcription factors Campomelic dysplasia ▪ Bowing of long bones, short bones, respi distress, anomalies of cervical spine, Pierre-Robin sequence, CNS, heart, kidneys anomalies ▪ Karyotyping in females, SOX9 importance in gonadal differentiation Cleidocranial dysplasia – drooping shoulders, open fontanelles, prominent forehead Nail-patella syndrome ▪ Nail dysplasia, patellar hypoplasia, elbow abnormalities, spurs → osteo-onychodysostosis ▪ 30% with chronic glomerulonephritis (proteinuria ± hematuria) → ESRD (5%) Disorders involving defective bone resorption Osteopetrosis – macrocephaly, hepatosplenomegaly, deafness, blindness, severe anemia, diffuse bone sclerosis and hypocalcemia ▪ X-ray: bone within bone appearance ▪ FTT, psychomotor delay, cranial neuropathies, anemia Pyknodysostosis Osteoporosis – fragility of the skeletal system & susceptibility to fractures of the long bones / vertebral compressions from mild or inconsequential trauma Osteogenesis imperfecta – brittle bone disease ▪ Most common genetic cause of osteoporosis ▪ Structural or quantitative defects in type I collagen (1o component of extracellular matrix of bone and skin) cause full spectrum of OI OSTEOGENESIS IMPERFECTA ETIOLOGY ▪ Molecular defect in type 1 collagen ▪ Overmodified collagen due to recessive null mutations in any of the 3 components of the collagen (coded by LEPRE1 gene on chromosome 1p34.1) or its assoc protein CRTAP or cyclophilin B ▪ Defective genes w/ protein products that interact w/ type 1 collagen ▪ Rare mutations in enzyme that processes the C-propeptide of type 1 collagen causes recessive form of OI → involved in osteoblast differentiation, collagen synthesis and cross linking ▪ 1/20, 000 incidence PPS Oral Exam Reviewer 2020 Other Inherited disorders of Skeletal Development Ellis-van Creveld Syndrome ▪ Chondroectodermal dysplasia, ciliopathy ▪ Short limbs, postaxial polydactyly, knock-knee deformity, nail dysplasia, dental anomalies, ASD, CHD ▪ AR, common in Amish ▪ EVC1 or EVC2 gene mutation ▪ 30% die of cardiac or respi problems during infancy Asphyxiating thoracic dystrophy ▪ AR, Jeune syndrome, ciliopathy ▪ Long narrow thorax & respi insufficiency (pulmo hypoplasia) → death Short-rib Polydactyly Syndromes Cartilage-hair Hypoplasia-anauxetic Spectrum disorders ▪ Flaring lower rib cage, prominent sternum, bowed legs, immunodefiency, malabsorption, celiac disease, hirschprung Metatorophic dysplasia – tail like appendage base of the spine, odontoid hypoplasia, kyphoscoliosis compromising cardiopulmo function Spondylometaphyseal dysplasia, Kozlowski Type Disorders involving Filamins Juvenile Osteochondroses ▪ Localized pain and tenderness: Freiberg disease, Osgood-Schlatter disease etc) ▪ Painless limitation of joint movt (Legg-Calve-Perthes disease) Caffey Disease (infantile cortical hyperostosis) ▪ Sudden irritability, swelling of contiguous soft tissue preceding cortical thickening of bones, fever, anorexia, painful swelling w/ wood-like induration but minimal warmth & redness, no suppuration (clavicle, mandible, ulna) Fibrodysplasia ossificans progressive ▪ Progressive extraskeletal heterotopic bone formation in muscles, tendons, ligaments, fascia ▪ Normal at birth, large toe deformity w/ episodes of soft tissue swelling & inflammation Osteogenesis imperfecta Marfan Syndome Metabolic bone disease ▪ Hypophosphatasia ▪ Hyperphosphatasia ▪ Osteoporosis TRIAD: fragile bones + blue sclera + early deafness OI Type I (mild) ▪ Blue sclerae, recurrent fractures & presenile hearing loss (30-60%) ▪ Easy bruising, hyperextensible joints, thin skin, joint laxity, scoliosis, Wormian bones, henia & mild short stature OI Type II (Perinatal lethal) ▪ Stillborn or die in 1st year of life ▪ SGA, bones, and CT extremely fragile ▪ Multiple intrauterine fracture of long bones, large skull and fontanels, micromelia, & bowing of extremities, multiple rib fractures (beaded appearance), small thorax, dark blue gray sclerae ▪ Multiple neuronal defects on cerebral cortex (agyria, gliosis, PVL) Osteogenesis Imperfecta Type III (Progressive Deforming) ▪ Most severe nonlethal form → significant physical disability ▪ BW & BL low normal, fractures in utero, macrocephalic & triangular facies ▪ Disorganized bone matrix – popcorn appearance at metaphyses ▪ Pectal deformity, scoliosis & vertebral compression, extremely short DNA sequencing – test for dominant & recessive OI *mutation identification to determine type, family screening, prenatal dx -Type VI screened by determination of severe reduction in serum pigment epithelium derived factor -obtaining dermal fibroblasts can be useful for determining level of transcripts of candidate gene for collagen biochemical testing →(+) in types I-IV, IX, VII/VIII LABS -collagen biochemical studies using cultured dermal fibroblasts -must be differentiated from thanatophoric dysplasia Prenatal determination of Severe OI -detected as early as 16 wks AOG by UTZ -chorionic villus biopsy for molecular or biochemical studies Normal to elevated alkaline phosphatase in neonatal period → distinguish it OI from hypophosphatasia ▪ No cure ▪ For severe nonlethal OI – active physical rehab in early years → to attain higher functional level ▪ Type I and some type IV – spontaneous ambulators ▪ Type III, IV, V, VI and XI – benefit from gait aids and swimming and conditioning Orthopedic management – fracture management and correction of deformity to enable function Bisphosphonates (IV pamidronate or oral olpadronate or risedronate) given for several years have some benefits → bone resorption by osteoclasts → bone volume but still contain defective collagen ▪ No effect on mobility scores, muscle strength or bone pain ▪ SE: abnormal long-bone remodeling, incidence of Fx non-union, osteoperotic brittleness to bone Genetic counseling ▪ AD → 50% risk of passing to offspring ▪ Empirical recurrence risk to an unaffected couple of Batch Clingy Disorders involving ion transporters Loss of sulfate ion transporter DTDST AR Mutiple epiphyseal dysplasia ▪ Normal stature in childhood but final height slightly decreased, 50% have clubfeet & external ear abnormality ▪ Diastrophic dysplasia – very short ext, clubfoot, short hands, proximal displacement of thumb (hitchhiker), cauliflower ear deformity ▪ Achondrogenesis Type 1B and Atelosteogenesis Type 2 – rare recessive lethal dysplasias; extremely short limbs, soft head, hypoplastic pelvis, short ribs 173 PATHOGENESIS ▪ Collagen structural mutations → abnormal bone ▪ Bone matrix w/ abnormal type 1 collagen fibrils & relatively levels of types III & V collagen ▪ Abnormal osteoblast differentiation & ↑ # of active bone resorbing osteoclast ▪ Hypermineralization of the bone ▪ Classical OI (Sillence types I-IV & type V) autosomal dominant ▪ Recessive OI (7-10%) caused by null mutations in genes coding for the collagen in the ER Types of collagen structural defects ▪ Point mutation (80%) – substitution of helical glycine residues or crucial residues in the C-propeptide by other AA ▪ Single exon splicing defects (20%) – clinically mild OI type 1 – null mutations in 1 a1(I) allele → reduced collagen stature ▪ Scleral hue (white to blue) ▪ Dentinogenesis imperfecta, hearing loss & kyphoscoliosis may be present/develop OI Type IV (moderately severe) ▪ In utero fractures, bowing of bones, require ortho & rehab but can attain community ambulation skills ▪ Osteoporotic w/ metaphyseal flaring & vertebral compressions, mod short stature, sclera (blue/white) OT Type V (Hyperplastic Callus) and Type VI Hyperosteoidosis (Mineralization Defect) ▪ Similar in skeletal severity to types IV & III but have distinct bone histology ▪ Type V (<5%) – hyperplastic callus, calcification of interossesous membrane of forearm & radiodense metaphyseal band ▪ (+) Ligamentous laxity, no blue sclera/dentinogenesis imperfecta ▪ Type VI – progressive deforming that doesn’t manifest at birth. ▪ Broad osteoid seams & fish-scale lamellation under polarized light (pigment epithelium derived factor deficiency) COMPLICATIONS ▪ Cardiopulmonary M&M ▪ Recurrent pneumonias, ↓ pulmo function ▪ Cor pulmonale – adults ▪ Neuro: basilar invagination, brainstem compression, hydrocephalus & syringomyelia having a second child with OI is 5-7% (when one parent has germline mosaicism) ▪ If parent is a mosaic carrier, risk of recurrence may be 50% ▪ AR → recurrence risk is 25% per pregnancy ▪ No known patient with severe nonlethal recessive OI has had a child PROGNOSIS ▪ OI type I usually die within months to 1 year of life ▪ Occasional child w/ type II & extreme growth deficiency survives to the teen years ▪ OI type III reduced life span w/ mortality from pulmonary causes in early childhood, teen & 40s ▪ Types I, IV & V are compatible with full life span ▪ OI Type III often wheelchair dependent → aggressive rehabilitation (transfer skills & household ambulation) ▪ OI Type IV attain community ambulation skills independently or with gait aids OI Type VII, VIII and IX (Autosomal Recessive) ▪ Overlap II & III but have distinct features: white sclerae, rhizomelia, small to normal head circumference ▪ Severe osteochondrodysplasia w/ extreme short stature ▪ Type IX (very rare, only 8 cases reported) lethal to mod severe, white sclera but no rhizomelia OI Types X and XI (AR) ▪ Defects in serine-type endopeptidase inhibitor of HSP47 that helps maintain folded state of procollagen heterotrimers responsible for collagen synthesis ▪ Type XI, more prevalent, mod to severe, white sclera, normal teeth, congenital contracture of large joints ▪ Kuskokwim syndrome – deletion of single tyrosine residue w/ very mild vertebral findings and osteopenia High Bone Mass OI ▪ Mutations in cleavage of procollagen C-propeptide ▪ Normal stature, white sclera & teeth, mild to moderate OI ▪ Null mutations in BMP1 → severe skeletal phenotype w/ short stature, scoliosis & bone deformity PPS Oral Exam Reviewer 2020 → Radiographs ▪ Surgery is required only in 4-5% of pediatric fractures Indications for Operative treatment in children & adolescents ▪ Displaced physical fracture Batch Clingy FRACTURES Pediatric skeletal system vs adults predisposing them to injuries: ▪ (+) Periosseous cartilage ▪ Physes ▪ Thicker, stronger, more osteogenic periosteum that produces new bone (callus), more rapidly & in greater amounts ▪ Pediatric bone less dense, more porous OI Types XIII-XVIII ▪ Affects osteoblast differentiation & bone formation ▪ Impaired collagen posttranslational modification underhydroxylation of collagen helix Upper extremity ▪ Phalangeal fractures ▪ Forearm fractures ▪ Distal humeral fractures ▪ Proximal humerus fractures ▪ Clavicular fractures – most common site, middle and lateral third LE fractures 174 Pediatric Fracture Patterns Plastic Deformation ▪ Unique to children, commonly seen in the forearm & occasionally the fibula ▪ Fracture from force that produces microscopic failure on the tensile side of bone & does not propagate to the concave side Buckle or Torus Fracture ▪ Failure in compression of the bone at the junction of the metaphysic & diaphysis ▪ Inherently stable, heals in 3-4wks w/ simple immobilization Greenstick Fracture ▪ Occurs when the bone is bent, & failure in the tensile (convex) side of the bone ▪ Concave side shows evidence of microscopic failure with plasticdeformation Complete Fractures ▪ Propagate completely through the bone ▪ Classified as: spiral, transverse, or oblique depending on the direction & shape of fracture lines Epiphyseal Fractures ▪ Involves epiphysis, often growth plate → potential for growth disturbance leading to deformity & discrepancy ▪ Distal radial physic - m/c injured physics ▪ Salter & Harris Classification I and II – closed reduction, tend to remodel with growth III and IV – involves articular surface, require anatomic alignment (<2mm displacement) V – not diagnosed initially, manifest later with growth disturbance ▪ “Twisted neck”, not a Dx but rather a clinical manifestation TORTICOLLIS Congenital muscular torticollis (CMT) most common Dx during infancy, etiology unknown ▪ Muscle biopsies & MRI show in utero muscle compression & or stretch resulting in localized ischemia & intramuscular compartment syndrome ▪ Contracture of left SCM results in tilt to left & rotation to right ▪ Hypothesized to result from intrauterine deformation or compression PPS Oral Exam Reviewer 2020 ▪ Hip fracture ▪ Toddler fracture ▪ Tibula and fibula shaft fractures – most commonly fractured ▪ Femoral shaft fractures ▪ Triplane and tillaux fractures ▪ Metatarsal fractures ▪ Toe phalangeal fractures Hip Fracture ▪ Result from high-energy trauma, can be assoc to injury to the chest, head, or abdomen ▪ Treatment entails complication rate of up to 60%, avascular necrosis rate 50%, malunion rate 30% Femoral shaft fracture ▪ Urgent atomic reduction (open/closed), spica casting for younger AP and Lateral Xray of femur, pelvis children Femoral Shaft Fractures Tibia and Fibula Shaft Fractures ▪ Low energy twisting type injuries to high-velocity injuries in vehicular AP and lateral radiograph of the knee and ankle accidents ▪ Femur fx in <2y/o raise concern for child abuse Metatarsal fractures Proximal Tibia Fractures AP, lateral, oblique radiographs of the foot ▪ Can be physical injuries, metaphysical injuries, or avulsion injuries of the tibial spine or tubercle Toddler fracture ▪ Careful neuromuscular exam warranted (pre & post-reduction) AP and lateral views of tibia and fibula showing ▪ Proximal tibial metaphysical fracture (cozen fracture) common in 3-6 nondisplaced spiral fracture of the distal tibial y/o → can cause valgus deformity metaphysis Tibia and Fibula Shaft Fractures ▪ Tibia – m/c fractured bone of the LE in children ▪ Inflammatory markers may be requested to rule out ▪ Direct injury infectious process in dx is in doubt ▪ Most assoc with fibular fracture, mean age 8 y/o ▪ Pain, swelling, deformity of affected leg and is unable to bear weight Toddler fracture ▪ In young ambulatory children (1-4 y/o) ▪ Injury after seemingly harmless twist or fall often unwitnessed ▪ Result of torsional injury ▪ Dx by physical exam, no fracture on X-ray ▪ Refusal to bear weight (pulling up affected extremity or florid display of protest, point tenderness at the site of fracture Triplane and Tillaux Fractures ▪ Occur at the end of growth period ▪ Based on relative strength of the bone-physics junction & asymmetric closure of the tibial physis Metatarsal fractures ▪ Direct trauma to the dorsum of the foot ▪ High energy trauma or multiple fx of the metatarsal base → swelling Toe phalangeal fractures ▪ Secondary to direct blows → barefoot child ▪ Swollen, ecchymotic and tender ▪ Tilting of head & neck ipsilaterally toward the side of the contracted muscle w/ head rotation contralaterally DIFFERENTIAL DIAGNOSIS CONGENITAL Muscular torticollis, Positional deformation Hemivertebra (cervical spine), unilateral atlanto-occipital fusion Klippel-Feil syndrome Unilateral absence of sternocleidomastoid, pterygium colli TRAUMA ▪ Displaced intraarticular fracture ▪ Unstable fractures ▪ Multiple injuries ▪ Open fractures ▪ Failure to achieve adequate reduction in older children ▪ Failure to maintain an adequate reduction ▪ Certain pathologic fractures Proximal Tibia Fractures ▪ Anatomic reduction and pin fixation, preferred with unstable fractures or displaced SH III or IV fractures Tibia and Fibula Shaft Fractures ▪ Closed reduction and immobilization ▪ Open fracture → irrigation, debridement, and antibiotic treatment ▪ Any suspicion of compartment syndrome → emergent fasciotomy Toddler fracture – below knee cast for 3-4w Triplane Fracture – anatomic reduction, stabilized by internal fixation Tillaux Fractures – closed reduction; open reduction only if residual intraarticular step-off persists Metatarsal fractures ▪ Closed method (below-knee cast) ▪ Closed/open reduction e/ internal fixation for displaced fractures (using smooth Kirschner wires) Toe phalangeal fracture – buddy taping of the fractured toe *See table 703.3 common indications for External Fixation in Pediatric Fractures CMT ▪ Stretching, stimulation & positioning measures, supervised by PT ▪ Resolution in 95% cases, when tx started 1st 4mos of life Surgery ▪ If diagnosed late, surgical release of SCM Batch Clingy Unique characteristics of pediatric fractures Fracture remodeling ▪ 3rd & final phase in fracture healing, preceded by inflammatory and reparative phases ▪ 3 major factors affecting angular correction: skeletal age, distance to the physis, orientation to the joint axis ▪ Remodeling best when fx occurs close to the physics ▪ Skeletal maturity: Girls 13-15, boys 15-17 years Overgrowth – physical stimulation from the hyperemia assoc w/ fracture healing 175 ▪ Hip disorder of unknown etiology ▪ Temporary interruption of blood supply to proximal femoral epiphyses→ osteonecrosis and femoral head deformity ▪ Etiology obscure, disruption of the vascular supply to the femoral epiphysis → ischemia & osteonecrosis ▪ More common in boys, 4-8yrs old PATHOGENESIS ▪ Ischemia & necrosis of femoral head → changes during repair process ▪ Infection, trauma, & transient synovitis are unsubstantiated causative factors ▪ Thrombophilia, Factor V Leyden mutation, protein C and S deficiency, lupus anticoagulant, anticardiolipin antibodies, antitrypsin and plasminogen might play a role in the abnormal clotting mechanism LEGG-CALVE-PERTHES DISEASE 4 stages of disease Initial stage ▪ 6mos ▪ Synovitis, joint irritability, early necrosis of femoral head ▪ Revascularization → resorption of necrotic segement ▪ Necrotic bone replaced by fibrovascular tissue (not bone) which comprises structural integrity of femoral epiphysis Fragmentation stage ▪ 8mos ▪ Femoral epiphyses begin to collapse, laterally and extrudes from acetabulum Healing stage ▪ 4yrs ▪ New bone formation in subchondral region, reossification begins ▪ Deformity depends on severity of collapse and amount of remodeling that occurs Residual stage ▪ After entire head has ossified ▪ Mild amount of remodeling until child reaches skeletal maturity PPS Oral Exam Reviewer 2020 Muscular injury (cervical muscles), atlanto-occipital sublaxation Atlantoaxial sublaxation, C2-3 sublaxation Rotary sublaxation, fractures (clavicle fracture or brachial plexopathy) INFLAMMATION Cervical lymphadenitis Retropharyngeal abscess, cervical vertebral osteomyelitis Rheumatoid arthritis, Grisel syndrome (nontraumatic subluxation of the atlantoaxial joint d/t local inflammation), upper lobe pneumonia NEUROLOGIC Visual disturbances (nystagmus, superior oblique or lateral rectus paresis) Dystonic drug reaction (phenothiazines, haloperidol, metoclopramide) Cervical cord tumor, posterior fossa brain tumor, syringomyelia, Wislon disease Dystonia muscularum deformans OTHER Acute cervical disk calcification Sandifer syndrome (GERD, hiatal hernia), benign paroxysmal torticollis Bone tumors (eosinophilic granuloma), soft tissue tumor, psychogenic ▪ Most common presenting symptom is LIMPING X-ray – 1O diagnostic tool (Lauenstein/frog leg lateral) ▪ Pain if present is activity related, localized in groin or referred to thigh or knee ▪ Initial – ↓ size of ossification center, lateralization of femoral head w/ widening of medial joint space, ▪ Antalgic gait – limp characterized by shortening of gait phase on the subchondral fracture and physeal irregularity injured side to alleviate weight bearing pain, prominent after strenuous ▪ Fragmentation – fragmented epiphyses, scattered areas activity of increased radiolucency and radiodensity ▪ Reossification – bone density returns to normal by new ▪ Hip motion – limited internal rotation and abduction; initially due to bone formation synovitis but later becomes permanent ▪ Residual stage – reossification of femoral head, ▪ Mild hip flexion contracture – 10-20 degrees remodeling of acetabulum ▪ Atrophy of muscles of thigh, calf, or buttock from disuse Associated with poor prognosis when seen on radiograph: ▪ Leg length inequality – adduction contracture of true shortening on ▪ Lateral extrusion of the epiphysis, horizontal physis, involved side from femoral head collapse calcification lateral to the epiphysis, subluxation of the hip and a radiolucent horizontal V in the lateral aspect of the physics (Gage’s sign) Radionuclide bone scan – can reveal avascularity early MRI – sensitive in detecting early infarction and determine degree of impaired perfusion but cannot portray stages of healing Arthrography – assess shape of femoral head and diagnose hinge abduction Catteral Classification based on involved femoral epiphysis and a set of radiographic “head at risk” signs ▪ Group I 25% involved, no sequestrum, no metaphysial abnormality ▪ Group II 50% involved, clear demarcation b/n involved and uninvolved segments, metaphyseal cyst may be present ▪ Group III 75% involvement and a large sequestrum ▪ Group IV entire femoral head involved Herring lateral pillar classification ▪ Determine treatment and prognosis during active stage of disease ▪ Based on several radiographs during fragmentation stage ▪ Some say delay until 18mos some say 1-4yo Goal of treatment – preserve well covered femoral head and maintain hip range close to normal Non-surgical ▪ Activity limitation, protected weight bearing, NSAIDS, and PT ▪ Severe pain – bedrest ▪ Petrie casts – 2 long leg casts help hips in abduction and internal rotation (best position) Surgical ▪ Varus osteotomy of proximal femur most common procedure ▪ Pelvic osteomies (acetabular rotational osteotomyies, shelf procedures and medical displacement or Chiari osteotomyies ▪ Post op → containment to manage residual deformity PROGNOSIS ▪ If sxs develop <5yo, good prognosis, remodeling potential higher in children Batch Clingy problem & is more common in first pregnancies ▪ Fibrotic mass palpable within the muscle and disappears in first few months of life ▪ Part of molded baby syndrome – DDH, plagiocephaly, facial asymmetry & foot deformities (metatarsus adductus) ▪ Facial findings – flattening on affected side, recessed eyebrow & inferior orbital displacement Paroxysmal torticollis of infancy - uncommon & may be due to vestibule dysfunction Neurogenic torticollis – uncommon, results from tumors of the posterior fossa or brainstem, syringomyelia and Arnold-Chiari malformation 176 ▪ Occurs during period of accelerated growth, late childhood, or adolescence (10-15yrs) more common in boys, esp in athletes, due to repetitive trauma ▪ Localized soft tissue swelling, along with an eventual firm & fixed prominence at the tibial tubercle ▪ Pain aggravated by sports activities and persist with regular daily activities and even at rest ▪ Diagnosis is clinical ▪ Xray: fragmentation of tibial tubercle & soft tissue swelling ▪ Usually self-limited, resolves with rest ▪ Restriction of activities and occasionally knee immobilizer necessary with restricted weight bearing + isometric and flexibility exercise program ▪ Advise that tibial tubercle welling will not likely resolve COMPLICATIONS ▪ Rare ▪ Early closure of the tibial tubercle w/ recurvatum or hyperextension, deformity, and, rarely, patellar tendon rupture or avulsion fracture of the tibial tubercle Batch Clingy OSGOOD-SCHLATTER DISEASE PATHOGENESIS ▪ Irritation of the patellar tendon at its insertion into the tibial tubercle or a traction apophysitis of the tibial tubercle growth plate ▪ Anterior knee pain, very specifically localized point tenderness over the tibial tubercle & the distal portion of the patellar tendon PPS Oral Exam Reviewer 2020 177 Acute ETIOLOGY / INCIDENCE/ PATHOGENESIS ▪ EA: S. pneumoniae (30%), nontypeable H. influenzae (30%), M. catarrhalis (10%), S. aureus ▪ Occur at any age Sinuses Ethmoid Maxillary Sphenoid Frontal BACTERIAL SINUSITIS Present At birth At birth 5yo 7-8yo Pneumatized At birth 4yo Risk Factors ▪ Viral URTI (associated with out-of-home daycare or a school-age sibling; 0.5-2%) ▪ Allergic rhinitis, tobacco smoke exposure ▪ Immunodeficiencies, cystic fibrosis, ciliary dysfunction, abnormalities of phagocyte function ▪ GER, cleft palate, nasal polyps, cocaine abuse, nasal foreign bodies ▪ Nasotracheal intubation or NGT → sinus ostia obstruction → sinusitis with MDR organisms of the ICU ▪ Acute sinusitis – <30 days ▪ Subacute sinusitis – 1-3mos ▪ Chronic sinusitis – >3 months ▪ Viral infection → viral rhinosinusitis → inflammation and edema → blocked sinus drainage and impaired mucociliary clearance of bacteria → bacterial overgrowth CLINICAL MANIFESTATIONS ▪ Nonspecific – nasal congestion, purulent nasal discharge (unior bilateral), fever, cough ▪ Halitosis, hyposmia (↓ sense of smell), periorbital edema ▪ Headache and facial pain are rare in children ▪ Maxillary tooth discomfort, pain or pressure exacerbated by bending forward ▪ Erythema and swelling of the nasal mucosa with purulent nasal discharge ▪ Sinus tenderness in adolescents and adult ▪ Transillumination – opaque sinus that transmits light poorly LABORATORY FINDINGS / DIAGNOSIS ▪ Sinus aspirate culture – only accurate method of diagnosis but is impractical; may be necessary for the immunosuppressed ▪ Rigid nasal endoscopy – less invasive for obtaining culture material; detects a great excess of positive cultures compared with aspirates ▪ Radiographic studies (plain film, CT) – opacification, mucosal thickening, air fluid level; not diagnostic and not recommended for healthy children; can confirm the presence of inflammation but cannot differentiate among viral, bacterial, or allergic causes TREATMENT ▪ Antimicrobial treatment for those with severe onset of a worsening course to promote resolution of symptoms and prevent suppurative complications ▪ 50-60% recover without antimicrobial therapy *See Table 408.2 below ▪ If with risk factors for the presence of resistant bacterial species (antibiotic treatment in the preceding 3mos, daycare attendance, or <2yo), and with failure to respond to initial therapy with amoxicillin within 72h, or with severe sinusitis, give high-dose Co-Amox (80-90mkday) ▪ If vomiting or at risk for poor compliance, give IV Ceftriaxone followed by a course of oral antibiotics ▪ If still with treatment failure, refer to an ENT ▪ Duration of therapy: individualized; minimum of 10 days or 7 days after resolution of symptoms DIFFERENTIAL DIAGNOSIS ▪ Viral URTI – clear and usually non purulent discharge, cough, initial fever; symptoms do not usually persist beyond 10-14days ▪ Allergic Rhinitis – seasonal; nasal secretions have significant eosinophilia ▪ Persistence of nasal congestion, rhinorrhea (of any quality) and daytime cough ≥10 days, ▪ Severe symptoms of temp ≥39C with purulent nasal discharge for ≥3days, and ▪ Worsening symptoms either by recurrence of symptoms after an initial improvement or new symptoms of fever, nasal discharge, and daytime cough (double sickening) ▪ Frontal sinusitis – can rapidly progress to serious intracranial complications, hence, initiate IV Ceftriaxone until substantial clinical improvement ▪ Decongestants, antihistamines, mucolytics, intranasal corticosteroids – not enough studies hence, not recommended ▪ Saline nasal wash or nasal sprays – help liquefy secretions and act as a mild vasoconstrictor COMPLICATIONS ▪ Serious orbital/intracranial complications – due to the proximity of the paranasal sinuses to the brain and eyes ▪ Periorbital and orbital cellulitis ● Often 2o to acute bacterial ethmoiditis ● Infection spreads directly through the lamina papyracea – thin bone that forms the lateral wall of the ethmoidal sinus ● Do CT scan of the orbits and sinuses with Ophtha and ENT consults ● Treat with IV antibiotics ● Orbital cellulitis may require surgical drainage ▪ Intracranial Complications ● Epidural abscess, meningitis, cavernous sinus thrombosis, subdural empyema, brain abscess ● Altered mental status, nuchal rigidity, severe HA, focal neurologic findings, signs of ↑ ICP ● Do CT scan of the brain, orbits, and sinuses ● Treat with broad-spectrum IV antibiotics (cefotaxime or ceftriaxone combined with vancomycin) pending culture results ● In 50%, infection is polymicrobial PPS Oral Exam Reviewer 2020 Batch Clingy DISEASE 178 ● Abscesses require surgical drainage ▪ Osteomyelitis of the frontal bone (Pott puffy tumor) – edema and swelling of the forehead ▪ Mucocele – chronic inflammatory lesions in the frontal sinuses; can expand → displacement of the eye → diplopia; do surgical drainage LEPTOSPIROSIS ▪ Zoonosis caused by aerobic, motile spirochetes of the genus Leptospira ▪ Worldwide distribution, most human cases occur in tropical and subtropical countries ▪ Asymptomatic infection to severe disease (5-10%) with multiorgan dysfunction and death ▪ Abrupt; may have a mono- or biphasic course ▪ Incubation period: 2-30 days Lepstospires ▪ Survive for days to weeks in warm and damp environmental conditions – water & moist soil ▪ Infect rats, mice, moles, livestock (cattle, goats, sheep, horses, pigs), wild mammals (raccoons, opossums), domestic dogs → ANICTERIC LEPTOSPIROSIS Septicemic phase ▪ Lasts 2-7 days ▪ Leptospires can be isolated from the blood, CSF, and other tissues PPS Oral Exam Reviewer 2020 Immune phase ▪ CSF abnormalities ● Modest ↑ in pressure ● Pleocytosis ● Early PMN leukocytosis → mononuclear predominance ● N or slightly ↑ CHON levels ● N glucose values Weil Syndrome ▪ UA – hematuria, proteinuria, casts ▪ Direct and indirect hyperbilirubinemia, modestly ↑ hepatic ▪ Initiate treatment before the 7th day to shorten the clinical course and decrease the severity of the infection ▪ Parenteral penicillin G (6-8 million U/m2/day) every 4hrs for 7days ▪ If penicillin-allergic, give doxycycline (2mkday) BID (maximum of 100mg twice daily) ▪ Cefotaxime, ceftriaxone, azithromycin – alternative if doxycycline is contraindicated ▪ If mild illness, may give oral doxycycline, amoxicillin, and ampicillin Batch Clingy PREVENTION ▪ Frequent handwashing ▪ Avoid persons with colds ▪ Yearly influenza vaccine 179 excrete spirochetes in urine for prolonged periods ▪ Most human cases occur from exposure to water or soil contaminated with rat urine ▪ Enter human hosts through mucous membranes (1o eyes, nose, mouth), transdermally through abraded skin, or by ingestion of contaminated water → bloodstream → endothelial damage of small blood vessels → 2o ischemic organ damage ▪ Flulike signs – fever, shaking chills, lethargy, severe HA, malaise, n/v, severe debilitating myalgia (most prominent in the LE, LS spine, abdomen) ▪ Bradycardia and hypotension can occur ▪ Conjunctival suffusion with photophobia and orbital pain, (-) chemosis and purulent exudate ▪ Generalized lymphadenopathy ▪ Hepatosplenomegaly ▪ Transient (<24h) erythematous maculopapular, urticarial, petechial, purpuric, or desquamating rash (10%) Immune phase ▪ Recurrence of fever and aseptic meningitis ▪ 80% have abnormal CSF findings but only 5% have clinical meningeal manifestations ▪ Self-limited unilateral or bilateral uveitis – rarely results in permanent visual impairment ▪ CNS symptoms resolve spontaneously within 1wk, with almost no mortality enzymes (returns to N after recovery) ▪ Transient thrombocytopenia (50%) ▪ Diagnosis is confirmed by serologic testing ▪ Microscopic agglutination test – gold standard diagnostic method; ≥4x increase in titer in paired sera ▪ Agglutinins appear by the 12th day of illness, reach a maximum titer by the 3rd week, and low titers can persist for years ▪ Unlike other spirochetes, leptospires can be recovered from the blood or CSF during the 1st 10 days of illness and from the urine after the 2nd week by repeated culture of small inoculum (1 drop of blood or CSF in 5mL of medium) ▪ The inoculum in clinical specimens is small and growth can take up to 16 weeks ▪ If severe, give specific attention to cardiopulmonary status, renal function, coagulopathy, and fluid and electrolyte imbalance PREVENTION ▪ Rodent control measures ▪ Avoidance of contaminated water and soil ▪ Immunization of livestock and domestic dogs ▪ Use protective clothing (boots, gloves, goggles) if at risk for occupational exposure ▪ If admitted, observe contact precaution ▪ If adult and travelling to highly endemic areas for short periods, give prophylactic doxycycline 200mg PO once a week. No specific pediatric data. ICTERIC LEPTOSPIROSIS (Weil Syndrome) ▪ Severe form more commonly seen in adults (>30yo) ▪ Initial manifestations similar to anicteric leptospirosis ▪ Immune phase – jaundice, acute renal dysfunction, thrombocytopenia → pulmonary hemorrhage, CV collapse ▪ Hepatic involvement → RUQ pain, hepatomegaly ▪ Azotemia, oliguria/anuria ▪ Acute kidney failure in 16-40% ▪ Abnormal ECG (90%) ▪ Epistaxis, hemoptysis, pulmonary, GI, and adrenal hemorrhage VIRAL SYNDROMES DENGUE FEVER TYPHOID FEVER URI BRONCHIOLITIS PNEUMONIA OTITIS MEDIA UTI *see Common Viral Illnesses* *see Common Bacterial Infections* *see Cough* *see Ear Pain* *see Renal Disorders* OCCULT BACTEREMIA PFAPA SYNDROME ETIOLOGY / INCIDENCE/ PATHOGENESIS ▪ Positive blood culture for a pathogen in a well-appearing child without an obvious source of infection ▪ Prevalence: <1% in febrile, well-appearing young children ▪ At risk: Unimmunized and incompletely immunized because of pneumococcus ▪ Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Adenitis ▪ Most common recurrent fever syndrome in children ▪ Between 2-5yo PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS ▪ Recurring episodes of fever, malaise, exudative-appearing tonsillitis with negative throat cultures, cervical lymphadenopathy, oral aphthae ▪ Less often, has HA, abdominal pain, arthralgia LABORATORY FINDINGS / DIAGNOSIS ▪ CBC, blood culture TREATMENT ▪ Give IV Ceftriaxone if leukocytosis is present while awaiting blood culture results ▪ Mild leukocytosis ▪ Elevated acute-phase reactants ▪ Single oral dose of prednisone (0.6-2mg/kg) – does not prevent recurrence; shorten the interval between flares ▪ Cimetidine (20-40mkday) – effective at preventing recurrences in 1/3 of cases Batch Clingy Complex DISEASE 180 ▪ Etiology and pathogenesis are unknown ▪ Episodes last 4-6 days, regardless of antipyretic or antibiotic treatment ▪ Occur with clock-like regularity on 3-to-6-week cycles ▪ Colchicine – may extend the time between flares ▪ Complete resolution has been reported after tonsillectomy ▪ Medical management should be the first approach ▪ Mild hepatosplenomegaly ▪ Temp >38C documented by a healthcare provider and for which the cause could not be identified after at least 8days of evaluation (see Table 204.1 below) ▪ Most result from atypical presentation of common diseases *see Table 204.2 – Diagnostic considerations for FUO* ▪ JIA and SLE – CTDs most often associated with FUO ▪ IBD, Kawasaki Disease ▪ FUO >6mos – uncommon in children; suggests granulomatous, autoinflammatory, or autoimmune disease ▪ 90% have an identifiable cause – 50% infectious, 10-20% collagen vascular, 10% oncologic Fever without a source ▪ Source has not yet been identified ▪ Can progress to FUO if no cause is elicited after 7days of evaluation Drug Fever ▪ Fever is sustained with no accompanying symptoms ▪ Discontinuation of the drug = resolution of the fever, generally within 72h Factitious Fever ▪ Inoculation of pyogenic material or manipulation of the thermometer by the patient or parent Pseudo-FUO ▪ Successive episodes of benign, self-limited infection with fever that the parents perceive as 1 prolonged episode ▪ Often starts with a well-defined infection (often viral) that resolves but is followed by other viral illnesses that may be less well-defined *see Table 204.4 – examples of potential diagnostic clues to infections presenting as FUO* ▪ Do a thorough history and PE Fever History: repetitive chills and spikes – septicemia Age ▪ If >6yo, respiratory of GUT infection, localized infection (abscess/osteomyelitis), JIA, leukemia ▪ If adolescent, IBD, autoimmune processes, lymphoma, TB Animal exposure – if domestic animals, ask for vaccines Pica: ingestion of dirt – Toxocara canis (VLM) or Toxoplasma gondii (toxoplasmosis) Medication History: Including nonprescription and topical meds (eyedrops may be associated with atropine-induced fever) Family History: Familial dysautonomia (recurrent hyperthermia; common among Jews), familial Mediterranean fever ▪ Ultimate treatment is tailored to the underlying diagnosis ▪ Antimicrobial agents should only be used when there is evidence of infection, with avoidance of empirical trials of medication ▪ After a complete evaluation, may give antipyretics to control fever PROGNOSIS ▪ Better than FUO in adults ▪ Outcome depends on the primary disease process ▪ In many cases, no diagnosis can be established, and fever abates spontaenously ▪ CBC ● ANC >5000/μL – against indolent bacterial infection other than typhoid fever ● PMN >10,000/μL or a non-segmented PMN >500/μL – severe bacterial infection is highly likely ▪ Urinalysis ▪ ESR >30mm/hr – inflammation; low ESR does not eliminated the possibility of infection or JIA ▪ Blood culture (aerobic) ▪ TST – if child is >2yo, request for IGRA ▪ Imaging studies – indicated by specific history/PE findings Batch Clingy FEVER OF UNKNOWN ORIGIN ▪ The frequency and intensity of the episodes diminish with increasing age *see Table 204.3 below* PPS Oral Exam Reviewer 2020 181 *see Seizures* Batch Clingy CNS INFECTIONS PPS Oral Exam Reviewer 2020 182 Acute DISEASE ETIOLOGY / INCIDENCE/ PATHOGENESIS ▪ Acute and potentially severe illness ▪ TSS toxin – 1 – producing and some enterotoxin – producing strains of S. aureus ▪ Highest rate in menstruating women 15-25yo ▪ 1-2 cases per 100,000 menstruating women Nonmenstrual TSS ▪ Associated with S. aureus infected nasal packing and wounds, sinusitis, tracheitis, pneumonia, empyema, abscesses, burns, osteomyelitis, primary bacteremia CLINICAL MANIFESTATIONS CLASSIC TSS: ▪ Abrupt onset ▪ High fever, vomiting, diarrhea, sore throat, HA, myalgia ▪ Diffuse erythematous macular rash (sunburn-like or scarlatiniform) appears within 24hrs + hyperemia of the pharyngeal, conjunctival, and vaginal mucous membranes LABORATORY FINDINGS / DIAGNOSIS ▪ No specific laboratory tests ▪ Diagnosis is based on clinical manifestations ▪ Strawberry tongue ▪ Altered LOC, oliguria, hypotension → if severe, shock and DIC TSST-1 ▪ Act as superantigens, triggers cytokine release → massive fluid loss from the intravascular space → end-organ cellular injury ▪ Selectively produced in an environment of neutral pH, high PCO2, and an anaerobic PO2 – found in abscesses and the vagina with tampon use during menstruation TREATMENT ▪ Identify and drain/remove any focal source of infection – abscess, tampon, nasal packing ▪ β-lactamase-resistant antistaphylococcal antibiotic (nafcillin, oxacillin, cefazolin) + clindamycin ▪ If with high rates of MRSA, when hospital acquired MRSA is suspected, or when the clinical picture overlaps with staphylococcal sepsis, use vancomycin instead of a βlactam ▪ Often requires intensive supportive care ● Aggressive fluid replacement – prevent/ treat hypotension, renal failure, CV collapse ● Inotropes – treat shock ● Corticosteroids and IVIg – if severe ▪ Recovery within 7-10 days ● Associated with desquamation of the palms and soles ● Hair and nail loss after 1-2mos ▪ Recurrences can occur – inadequate antibiotic treatment and/or recurrent tampon use TOXIC SHOCK SYNDROME COMPLICATIONS ▪ ARDS, myocardial dysfunction, renal commensurate with the degree of shock failure – PROGNOSIS ▪ Overall mortality rate of treated patients – 3-5% if with early treatment KAWASAKI DISEASE (Mucocutaneous LN syndrome) ▪ Highest incidence in Asian children ▪ Leading cause of acquired heart disease in children in most developed countries ▪ Disease of early childhood (highest in those <5yo); boys > girls ▪ Cause is unknown, some say infectious in origin 3-PHASE PROCESS TO KD ARTERIOPATHY 1) Neutrophilic necrotizing arteritis – 1st 2wks of illness; begins in the PPS Oral Exam Reviewer 2020 Fever ▪ Highly spiking (≥38.3C), remitting, unresponsive to antipyretics ▪ If without treatment, lasts for 1-2wks, but may be as short as 5days or persist for 3-4wks Conjunctival Injection with limbal sparing Rash of various forms – maculopapular, erythema multiforme, scarlatiniform, psoriatic-like, urticarial, or micropustular Perineal desquamation – acute phase Periungual desquamation – begins 2-3wks after onset of the illness; fingers and toes → entire hand and foot Arthritis – small or large joints; may occur early in the illness or may develop in the 2nd or 3rd week ▪ Symptoms other than the principal criteria are common in the 10d ▪ 2DED – most useful test to monitor CAA development ● CA dimensions may be increased in the 1st 5wks after presentation ● Small Aneurysms – ≤4mm internal diameter (ID) ● Medium – >4 to ≤8mm ID PREVENTION ▪ Change tampons at least every 8 hours ▪ If (+) fever, rash, or dizziness during menstruation, remove tampon immediately and seek consult ▪ Avoidance of tampon use in subsequent menstrual cycles to reduce recurrence risk ▪ IVIG should be given within 10d of disease onset, ideally ASAP after diagnosis ▪ ASA high dose (anti-inflammatory) → low dose (antithrombotic) – continued for 6-8wks after illness onset then discontinued if normal 2DED throughout the entire course of the illness. If with CAA, continue indefinitely. ▪ Do not give other NSAIDs together with ASA Batch Clingy ▪ Systemic inflammatory disorder manifesting as vasculitis with a predilection for the coronary arteries (CA) ▪ Predominantly affects the medium-sized areteries (CA, axillary, subclavian, femoral, popliteal, brachial) DIFFERENTIAL DIAGNOSIS ▪ Streptococcal TSS – caused by GAS; severe streptococcal sepsis or a focal streptococcal infection (cellulitis, necrotizing fasciitis, pneumonia) ▪ Kawasaki Disease – resembles TSS but not as severe or rapidly progressive ▪ Scarlet fever, Rocky Mountain spotted fever, Leptospirosis, TEN, sepsis, measles ▪ No diagnostic test but have characteristic lab findings (see Table 191.1 in the next pages) ▪ Platelet is N in the 1st week → rapid ↑ by the 2nd-3rd week (sometimes >1million/mm3) ▪ Normocytic, normochromic anemia ▪ ESR may remain elevated for weeks ▪ KD is unlikely if the ESR, CRP, and platelet counts are normal after 7 days of fever 183 endothelium → coronary wall; saccular aneurysms may form 2) Subacute/chronic vasculitis – driven by lymphocytes, plasma cells, eosinophils; lasts weeks to years; results in fusiform aneurysms 3) Progressive Stenosis – the vessels affected in the 2nd phase develop smooth muscle myofibroblasts → stenosis → thrombi formation → blood flow obstruction before the diagnosis of KD ▪ GI symptoms – >60% of patients ▪ Respiratory symptoms – at least 1 in 35% Cardiac Involvement – most important manifestation of KD; myocarditis manifests as tachycardia disproportionate to fever + ↓ LV systolic function; pericarditis with a small effusion; mild MR (10-25%) Node-first KD ▪ Initially presents with only fever and lymphadenopathy ▪ Older (4yo) ▪ Have more days of fever and higher CRP levels ▪ Many also have retropharyngeal and peritonsillar inflammation on CT scans KD SHOCK SYNDROME ▪ Greatly ↓ LV function ▪ Higher risk for CA dilation ▪ Diagnosis should be made within 10 days, ideally within 7 days, of fever onset to improve coronary artery outcome ATYPICAL OR INCOMPLETE KD ▪ Persistent fever + <4 of the 5 characteristic clinical signs ▪ Labs and 2DED assist in the diagnosis ▪ Occur most frequently in infants (highest likelihood of CAA development) Any infant <6mos old with unexplained fever ≥7days should undergo 2DED to assess the coronary arteries. DIFFERENTIAL DIAGNOSIS ▪ Adenovirus – exudative pharyngitis & conjunctivitis ▪ Scarlet Fever – GAS; rapid clinical response to appropriate antibiotic therapy; rarely have ocular findings ▪ Measles, cervical lymphadenitis, rocky mountain spotted fever ▪ Leptospirosis – biphasic, few asymptomatic days between an initial period of fever and HA; late phase has renal and hepatic failure ▪ JIA – diffuse lymphadenopathy & hepatosplenomegaly ▪ Annual influenza vaccination – for patients on long-term ASA therapy to ↓ risk of Reye syndrome ▪ MMRV vaccination – defer until 11mos after IVIG administration (IVIG may interfere with the immune response to live virus vaccines d/t specific antiviral antibody) IVIG-RESISTANT KD (15%) ▪ Persistent or recrudescent fever 36h after completion of the initial IVIG infusion ▪ Increased risk for CAA Batch Clingy 3 CLINICAL PHASES 1) Acute febrile phase – fever and other acute signs of illness; lasts 1-2 weeks 2) Subacute phase – desquamation, thrombocytosis, development of CAA; highest risk of death in those who develop aneurysms; lasts 3 weeks 3) Convalescent phase – begins when all clinical signs have disappeared until the ESR returns to normal; 6-8 weeks after the onset of the illness ● Giant – >8mm ID ● Performed at diagnosis then at 1-2wks of illness - If normal, repeat 6-8wks after onset of illness - If with abnormalities or patient has recurrent fever or symptoms, more frequent 2DED - If without CAA at any time during the illness, do 2DED and lipid profile after 1year then CV follow up every 5yrs PPS Oral Exam Reviewer 2020 184 ▪ Treatment (Table 191.4): 2nd dose of IVIG (2g/kg), tapering course of corticosteroids, and/or infliximab COMPLICATIONS ▪ MI, angina, sudden death ▪ 2/3 have a history of URTI several weeks prior ▪ Almost always have serologic evidence of a recent GAS (S. pyogenes) infection ▪ Annual incidence of >50 per 100,000 children in some developing countries ▪ RHD is the most common form of acquired heart disease in all age groups worldwide ▪ 5-15yo – age of greatest risk for GAS pharyngitis ACUTE RHEUMATIC FEVER PPS Oral Exam Reviewer 2020 MIGRATORY POLYARTHRITIS (75%) ▪ Large joints – knees, ankles, wrists, elbows ▪ Hot, red, swollen, exquisitely tender joints that is dramatically responsive to even low dose salicylates ▪ Pain can precede and can appear to be disproportionate to the objective findings ▪ A severely inflamed joint can become normal within 1-3 days without treatment ▪ If suspecting RF, hold salicylates and observe for migratory progression ▪ Almost never deforming CARDITIS (50-60%) ▪ Subclinical carditis – (-) murmur of valvulitis but (+) echo evidence of valvulitis (see table 465.1) ▪ Clinical carditis – (+) murmur of valvulitis ▪ Carditis & chronic RHD – most serious manifestations of acute RF; account for all associated morbidity and mortality ▪ Pancarditis – active inflammation of the myo-, peri-, and endocardium ▪ Endocarditis (valvulitis) – universal finding in rheumatic carditis ▪ Myo- and/or pericarditis without evidence of endocarditis is almost never is rheumatic carditis ▪ Most RHD is isolated MV disease or combined AV and MV ▪ Valvular insufficiency – acute & convalescent stages ▪ MV and/or AV stenosis – years/decades after the acute illness; in *See table 210.2 below ▪ Synovial fluid: 10,000-100,000 WBC/μL with neutrophil predominance; 4g/dL CHON; N glucose; forms a good mucin clot ▪ 2DED – pericardial effusion, ↓ ventricular contractility, aortic and/or mitral regurgitation ▪ Circumstances when the diagnosis of acute RF can be made without strict adherence to the Jones Criteria: ● Chorea occurs as the only major manifestation ● Indolent carditis is the only manifestation in patients who first come to medical attention only months after the apparent onset of acute RF ● Recurrence of acute RF in particularly high-risk populations ▪ The diagnosis of acute RF should not be made in those with elevated or increasing streptococcal antibody titers who do not fulfill the Jones Criteria Recent GAS Infection ▪ Absolute requirement for the diagnosis of acute RF ▪ Acute RF develops 10-21 days after an acute GAS pharyngitis – clinical findings of pharyngitis are no longer present ▪ Evidence is based on an elevated or rising serum antistreptococcal antibody titers – ASO, anti-DNAse B, antihyaluronidase PREDICTORS OF POOR OUTCOME ▪ Young age, male gender ▪ Persistent fever, poor response to IVIG ▪ Labs – neutrophilia, thrombocytopenia, transaminitis, hyponatremia, hypoalbuminemia, ↑ CRP and N-terminalbrain-natriuretic protein ▪ Bed rest – allowed to ambulate only when the signs of acute inflammation have improved ▪ Penicillin or Amoxicillin PO for 10days OR Benzathine Penicillin G per IM for 1 dose – to ensure complete eradication of GAS from the URT ▪ Erythromycin PO for 10days OR Azithromycin for 5days OR Clindamycin for 10days if penicillin-allergic ▪ Withhold anti-inflammatory agents (salicylates, corticosteroids) if the only manifestation of presumed RF is arthralgia or atypical arthritis. May give acetaminophen to control pain and fever. ▪ Oral salicylates ● 50-70mkday QID for 3-5 days followed by ● 50mkday QID for 2-3wks → 25mkday QID for 2-4wks ● Give if (+) migratory polyarthritis and carditis, (-) cardiomegaly or congestive heart failure (CHF) ▪ Corticosteroids ● Prednisone 2mkday QID for 2-3wks → 1mkday for 23wks → taper by 5mg/day every 2-3days ● Give if (+) carditis, minimal cardiomegaly and/or CHF ● When being tapered, start ASA 50mkday QID for 6wks – to prevent rebound of inflammation ▪ Supportive therapy for mod to severe carditis: Digoxin, fluid and salt restriction, diuretics, oxygen ▪ Sedatives for Sydenham chorea – phenobarbital (16-32mg q6-8h PO) is the drug of choice; haloperidol, Batch Clingy PROGNOSIS ▪ Majority return to normal health ▪ Recurs in 1-3% of cases ▪ 20-25% of untreated children (and those treated with ASA alone) develop CA abnormalities (CAA) – aneurysms; 2nd-3rd week of illness; often asymptomatic, diagnosed by 2DED ▪ <5% treated with IVIG & ASA within the 1st 10 days of illness develop CAA ▪ 50% of CAA regress to N lumen diameter by 1-2y after the illness ▪ Almost all the morbidity and mortality occur in those with large or giant artery aneurysms (z score ≥10 or an absolute dimension of ≥8mm) – less likely to regress; greatest risk of thrombosis, stenosis, angina, MI 185 developing countries, may occur sooner ▪ Presents with tachycardia and cardiac murmurs → cardiomegaly, heart failure, hepatomegaly, peripheral & pulmonary edema ▪ Mitral regurgitation – high pitched holosystolic murmur radiating to the axilla ▪ Aortic insufficiency – high pitched decrescendo diastolic murmur at the left sternal border chlorpromazine COMPLICATIONS ▪ Arthritis and chorea resolve without sequelae ▪ Chronic, progressive valvular disease consequence of acute rheumatic carditis CHOREA (10-15%) ▪ Isolated, frequently subtle, movement disorder ▪ Emotional lability, incoordination, poor school performance, uncontrollable movement, facial grimacing, exacerbated by stress, disappears with sleep Clinical Maneuvers to elicit features of chorea ▪ Milkmaid’s grip – irregular contractions & relaxations of muscles of the fingers while squeezing the examiner’s fingers ▪ Spooning and pronation of the hands when the arms are extended ▪ Wormian darting movements of tongue on protrusion ▪ Examine the handwriting to evaluate fine motor movements ERYTHEMA MARGINATUM (1%) ▪ Characteristic rash of acute RF ▪ Erythematous, nonpruritic, serpiginous, macular lesions with pale centers ▪ Trunk and extremities; not on the face ▪ Accentuated by warming the skin major PROGNOSIS ▪ Depends on the initial manifestations, severity of the initial episode, presence of recurrences ▪ 50-70% of those with carditis recover with no residual heart disease – the more severe the involvement, the higher risk for residual ▪ Those without carditis in the initial episode are less likely to have carditis with recurrent attacks ▪ Those with acute RF are susceptible to recurrent attacks following reinfection of the URT with GAS – 50% risk with each GAS pharyngitis. Thus, requires long-term continuous chemoprophylaxis ▪ The risk of recurrence is highest in the 1st 5yrs after the initial episode and decreases with time ▪ 20% of those who present with pure chorea not given 2o prophylaxis develop RHD within 20 years PRIMARY PREVENTION ▪ Depends on identification and eradication of GAS causing acute pharyngitis ▪ Appropriate antibiotic therapy before the 9th day of symptoms of acute GAS pharyngitis SECONDARY PREVENTION ▪ Preventing acute GAS pharyngitis in patients at substantial risk of recurrent acute RF ▪ Continuous antibiotic prophylaxis beginning at the diagnosis of acute RF and immediately after a full course of antibiotic therapy has been completed Batch Clingy SUBCUTANEOUS NODULES (≤1%) ▪ Firm nodules 0.5-1cm in diameter along the extensor surfaces of tendons near bony prominences ▪ (+) Correlation with significant RHD MINOR CRITERIA ▪ CRP at least 3mg/dL in mod/high risk populations ▪ Prolonged P-R interval on ECG unless carditis is a major criterion; not an evidence of carditis nor predict long-term sequelae – PPS Oral Exam Reviewer 2020 186 *see Common Viral Illnesses* *see Common Bacterial Infections* *see Collagen and Vascular Disorders* Batch Clingy VIRAL EXANTHEMS MENINGOCOCCE MIA HENOCHSCHONLEIN PURPURA PPS Oral Exam Reviewer 2020 187 Acute ETIOLOGY/INCIDENCE/ PATHOGENESIS ▪ Humans only host ▪ Most important proteins in induction of immunity: 1) Hemagglutinin (H) Protein: neutralizing Ab are directed against 2) Fusion (F) protein: Ab limit proliferation of the virus during infection INCUBATION PERIOD ▪ 8 – 12 days ▪ Shed virus 7d after exposure to 4-6d after onset of rash ▪ Immunocompromised – shed measles during duration of disease TRANSMISSION Portal of entry ▪ Respiratory tract or conjunctivae following contact with large droplets or small droplet aerosols w/ virus ▪ Virus may remain suspended in air for an hour ▪ Infectious from 3 days before up to 4-6 days after onset of the rash MEASLES Rubeola PATHOLOGY ▪ Necrosis of resp tract epithelium and lymphocytic infiltrate ▪ Produces small vessel vasculitis on the skin and on the oral mucous membranes ▪ Rash histology: intracellular edema and dyskeratosis with formation of syncytial giant cells ▪ Fusion of infected cells → multinucleated giant cells: WARTHIN FINKELDEY GIANT CELLS (pathognomonic) 4 PHASES ▪ Incubation Phase ● Virus migrates to regional LN ● 10 viremia: spreads to RES ● 20 viremia: spreads to body surfaces ▪ Prodromal Illness ● Begins after 20 viremia ● Epithelial necrosis and giant cell formation ● Virus shedding begins ▪ Exanthematous ● With onset of rash antibody production begins, viral replication and symptoms subside ▪ Recovery Phase MUMPS ▪ Humans are the only natural host ▪ Surface glycoproteins stimulate production of protective Ab ● Hemagglutinin-neuraminidase (HN) Protein: mediate absorption of the virus to host cells ● Fusion (F) Protein: mediate penetration of virus into cells ▪ Incidence has decreased after MMR 2 dose vaccine PPS Oral Exam Reviewer 2020 CLINICAL MANIFESTATIONS / FINDINGS / DIAGNOSIS Prodrome: Mild fever, conjunctivitis w/ photophobia, coryza, cough and increasing fever Koplik Spots – enanthem ▪ Pathognomonic ▪ Appears 1-4 days prior to onset of rash ▪ Initially appears as discrete red lesions with bluish white spots in the center on the inner aspects of the cheeks at the level of premolars ▪ Symptoms ↑ in intensity for 2-4d until D1 of rash (maculopapular) ▪ Symptoms subside with onset of rash ▪ Rash appears in a cephalocaudal fashion, fades over 7 days with fine desquamation ▪ Cough lasts longest (up to 10 days) ▪ Severe: generalized LAD with prominent cervical and occipital LN MODIFIED MEASLES INFECTION ▪ Passively acquired Ab: infants & recipients of blood products → subclinical form of measles may occur ▪ Rash: indistinct, brief, or rarely, entirely absent ▪ Some who received vaccines, when exposed, may have rash but few other symptoms ▪ Not contagious DIAGNOSTICS Acute Phase: leukopenia w/ lymphocytopenia neutropenia) If without bacterial co-Infection: normal ESR & CRP (absolute Serologic Confirmation Serum IgM Ab ▪ Appears 1-2 days after onset of rash ▪ Detectable for a month ▪ If collected <72 hrs after onset of rash and (-) for measles Ab, a 2nd specimen may be obtained IgG Ab: ▪ 4-fold rise in acute and convalescent specimens collected 2-4 weeks apart ▪ Viral isolation from blood, urine, resp secretions by culture or molecular detection by PCR ▪ Prodrome lasts 1-2 days: fever, HA, vomiting, achiness → parotitis (unilateral → bilateral) ▪ Tenderness enhanced by ingestion of sour or acidic foods or liquids ▪ Ipsilateral ear pain, swelling obscuring the angle of jaw, earlobe lifted upward and outward; peaks in 3 days, subsides over 7 days MANAGEMENT ▪ Supportive VITAMIN A – QD x 2 days at ● > 12mos: 200,000 IU ● 6 – 11mos: 100,000 IU ● <6mos: 50,000 IU ▪ If with SSx of vit. A deficiency, a 3rd dose is given 2-4 weeks after 2nd dose MEASLES VACCINE (SC) Given at 9mos but may be given as early as 6mos of age in cases of outbreaks If monovalent measles vaccine is not available, then MMR/MR vaccine may be given as substitute for infants <12 months old/. In such cases, the recipient should receive 2 more MMR vaccines starting at 1 year of age, following recommended schedules. MMR (SC) Minimum age of 12mos 2 doses of MMR are recommended The 2nd dose is usually given at 4 - 6 years of age but may be given at an earlier age with minimum of 4 weeks interval between doses. MMRV (SC) Minimum age: 12 mos; Maximum age: 12 years May be given as an alternative to separately administered MMR and Varicella vaccines. The recommended minimum interval between doses is 3 months but a 2nd dose given 4 weeks from the first dose, is considered VALID. COMPLICATIONS / PROGNOSIS ▪ Morbidity & mortality highest in <5yo ▪ Serum retinol concentrations ▪ Higher rate of activation of PTB in populations infected with MTB then exposed to measles ▪ Measles during pregnancy: fetal wastage, stillbirths, congenital malformations (3%) Acute Otitis Media ▪ Most common complication Pneumonia ▪ Most common cause of death ▪ May manifest as giant cell pneumonia or as superimposed bacterial infection (S. pneumonia, S. aureus, H. influenza) ▪ Fatal outcome: bronchiolitis obliterans ▪ Infants/toddlers: croup, tracheitis, bronchiolitis Measles Encephalitis ▪ Post infectious, immune mediated ▪ Clinical onset begins during exanthem and manifests as seizures, lethargy, coma, irritability ▪ CSF: lymphocytic pleocytosis, protein ▪ Immune compromised- occurs 1-10 months after. Seen as seizures, myoclonus, stupor, and coma. PROGNOSIS Pneumonia and encephalitis were the complications in most of deaths AE (MMR): ▪ Fever (after 6-12 days), rash, transient thrombocytopenia ▪ Febrile seizure ▪ Do not give to pregnant, immunosuppressed or immunodeficient (except asymptomatic HIV-infected patients) POSTEXPOSURE PROPHYLAXIS Vaccine ▪ Effective if given within 72 hours of exposure Immunoglobulin ▪ May be given up to 6 days after exposure ▪ Indicated for susceptible household contacts of measles patients (<6mos old, pregnant and immunocompromised) ▪ Immunocompetent: 0.5ml/kg (max: 15 mL/kg) ▪ Supportive, antipyretics, isolate for 5 days after onset of parotid swelling MMR ▪ 2-dose series starting at 12-15 mos then 4-6 y/o ▪ 3rd MMR dose during outbreaks for at-risk population Most common: Meningitis + encephalitis, Gonadal involvement Meningitis + Meningoencephalitis ▪ Neurotropic, enters CNS via choroid plexus ▪ May occur before, with or after (most commonly 5 days after parotitis) Batch Clingy DISEASE 188 TRANSMISSION ▪ Via respiratory droplets ▪ Virus is present in saliva 7 days before to 7 days after parotid swelling ▪ Infectious 1-2 days before to 5 days after parotid swelling PATHOLOGY ▪ Initial viral replication: epithelium of upper resp tract, spreads to adjacent LN, viremia ensues spreading to targeted tissues (e.g. salivary glands, CNS, pancreas, testes) ▪ SALIVARY GLAND DUCTS: lined with necrotic epithelium ▪ TESTES: swelling of tissues may result in focal ischemic infarcts ▪ CSF: mononuclear pleocytosis ▪ Humans are the only known host ▪ Sensitive to heat, UV light, pH extremes ▪ Highest in pre-school & school age INCUBATION PERIOD: 14 – 21 days TRANSMISSION ▪ Period of highest communicability from 5 days before to 6 days after appearance of the rash ▪ Viral shedding from nasopharynx: 10 days after infection and up to 2 weeks of onset of rash ▪ Children with CRS excrete virus in respiratory secretions up to 1yo RUBELLA German measles 3-DAY measles PATHOLOGY Non-specific findings: ▪ Lymphoreticular inflammation, mononuclear perivascular, meningeal infiltration ▪ Virus replicates in respiratory epithelium → regional LN → viremia, most intense from 10-17 days after infection CONGENITAL RUBELLA SYNDROME (CRS) Congenital infection occurs during maternal viremia Risk for congenital defects for the ff AOG: ▪ <11 weeks: high risk ▪ >16 weeks: uncommon 85% during 1st 12 weeks of gestation 50% during 1st 13 – 16 weeks of gestation PPS Oral Exam Reviewer 2020 ▪ Opening of Stensen duct: red and edematous ▪ Submandibular salivary glands may be involved ▪ Edema over sternum as a result of lymphatic obstruction ▪ Contraindications: pregnant women, severely ID or immunosuppressed; HIV not severely immunocompromised may be given DIAGNOSTICS ▪ In highly prevalent areas: hx of exposure, appropriate incubation period, typical clinical findings (parotitis is demonstrated by serum amylase; leukopenia + relative lymphocytosis is common) PROGNOSIS Nearly always excellent even with encephalitis, although fatal cases CNS involvement or myocarditis have been reported ▪ In highly immunized populations w/ parotitis lasting longer than 2 days and of unknown cause: r/o mumps by cell culture, direct IF or rt-PCR ▪ SPECIMENS: buccal or oropharyngeal mucosa, CSF or urine ● PCR from OP mucosa should be run within 3 days (becomes negative quickly in immunized individuals) ▪ Serologic testing more convenient and available: ● serum mumps IgG b/w acute and convalescent serum specimens ● Mumps IgM (EIA): recent infection ▪ Low-grade fever, sore throat, red eyes + pain, HA, malaise, anorexia, suboccipital, postauricular, anterior CLAD ▪ 1st manifestation in children is rash: variable; beginning on the face and neck as small irregular pink macules → spreads centrifugally; fades from the face as it extends; duration: 3 days then fades without desquamation ▪ Forchheimer spots: rose colored spots or petechiae on soft palate; occurs at onset of rash DIAGNOSTICS ▪ Leukopenia, neutropenia, mild thrombocytopenia ▪ Most common test: Rubella IgM enzyme immunosorbent assay (present 4 days after appearance of rash) ▪ Confirmation: IgM capture assay, rt-PCR or viral culture For diagnosis of congenital infection: ▪ Viral isolation by culture of NP secretion, cord blood or placenta ▪ PCR of amniotic fluid during pregnancy ▪ CSF: normal or mild pleocytosis and/or protein, virus is rarely isolated ▪ Most common finding: nerve deafness ▪ Most common finding apparent at birth: IUGR ▪ Most common ocular finding: salt and pepper retinopathy ▪ Most serious ocular finding: cataracts ▪ Most freq. reported cardiac abnormality: PDA ff. by lesions of the pulmonary arteries and valvular disease ▪ 10-20% have active meningoencephalitis at birth (e.g. full anterior fontanelle, irritability, hypotonia, seizures, lethargy) ▪ Supportive ▪ Severe non-remitting thrombocytopenia: IVIg or corticosteroids ▪ Isolated for 7 days after onset of rash, standard and droplet precaution Exposed Pregnant Women Obtain blood specimen for rubella IgG-specific Ab testing, then save 1 frozen aliquot ▪ If (+), mother is immune ▪ If (-), obtain 2nd specimen after 2-3 weeks and test with saved frozen aliquot ▪ If both (-): obtain 3rd specimen 6 weeks after exposure and test with the saved frozen aliquot ▪ If 2nd and 3rd (-), infection has not occurred ▪ If (+) in either 1 – RECENT INFECTION ● Routine Ig (0.55 mL/kg IM) is considered only if termination of pregnancy is not an option VACCINE ▪ Postexposure prophylaxis within 3 days ▪ Contraindications: pregnant women, severely ID or immunosuppressed; HIV not severely immunocompromised may be given ▪ Ig may inhibit serologic response to vaccine ▪ MMR AE: fever, rash, arthralgia & arthritis (adults), peripheral neuropathies, transient thrombocytopenia ▪ Evaluate cardiac, audio, ophthalmic, and neurologic systems ▪ Contact precaution until 1yo ▪ Infants: fever, malaise, lethargy; older: headache and meningeal signs ▪ Sxs resolve 7-10 days Orchitis ▪ 2nd most common finding in adolescent and adult males ▪ Begins days ff onset of parotitis ▪ Fever, chills, exquisite pain, and swelling testes (30% bilateral) → atrophy may occur but sterility is rare Oophoritis: uncommon Pancreatitis: fever, epigastric pain, vomiting Arthritis: arthralgia, mono or migratory polyarthritis, within 3 weeks of parotid swelling Others: myocarditis, thyroiditis, conjunctivitis, optic neuritis, pneumonia, nephritis, mastitis, thrombocytopenia, transverse myelitis, ADEM, aqueductal stenosis, facial palsy, SNHL ▪ Pregnancy: fetal wastage, perinatal mumps Thrombocytopenia ▪ 2w after onset of rash – petechiae, epistaxis, GI bleeding, hematuria ▪ Self-limiting Arthritis ▪ Within 1 week of rash, self-limiting ▪ Small joints of hands Encephalitis – most serious; 2 forms Post-infectious syndrome ▪ Within 7d rash onset, HA, seizures, confusion, coma, focal neurological signs, ataxia ▪ Non-infectious pathogenesis Progressive Rubella Panencephalitis ▪ Rare, similar to SSPE, virus isolated in brain tissue ▪ Infectious pathogenesis ▪ Death after 2-5 years onset Others: GBS, peripheral neuritis, myocarditis PROGNOSIS ▪ Post-natal: excellent RE-INFECTION ▪ Significant increase in IgG and/or an IgM response in an individual with documented preexisting rubella-specific IgG above an accepted cut-off ▪ Miscarriage, fetal death, constellation of congenital anomalies ▪ CRS is one of the few known causes of autism PROGNOSIS ▪ Less favorable Batch Clingy INCUBATION PERIOD Range: 12 – 25 days Usual: 16 – 18 days 189 ▪ Tissue necrosis due to vascular insufficiency, ↓ cellular multiplication time, chromosomal breaks, production of protein inhibitor causing mitotic arrest ▪ Distinctive feature is CHRONICITY: once infected early in gestation, it persists beyond delivery ▪ dsDNA HHV-6 (6A & 6B) and HHV7 ▪ Major cause: 6B EPIDEMIOLOGY Peak of infection ▪ HHV 6B: 6-9 months old (after maternal Ab wane) ▪ HHV 7: later in childhood ROSEOLA INFANTUM Exanthem subitum/ Sixth disease TRANSMISSION HHV6 ▪ Saliva or respiratory droplets of asymptomatic adults or older children ▪ Congenital infection via vertical transmission (transplacental infection and chromosomal integration) HHV7- saliva of asymptomatic individuals Both: sexual or perinatal (as seen in cervical secretions) ▪ caused by Parvovirus B19 ▪ Rash and transient aplastic crisis: most prevalent in school – aged (5-15yo) ERYTHEMA INFECTIOSUM Fifth Disease INCUBATION PERIOD Range: 4 – 28 days Average: 16 – 17 days ▪ Transient aplastic crisis: shorter period of incubation TRANSMISSION ▪ Respiratory route – large droplet spread from NP viral shedding ▪ Blood and blood products ▪ Fomite – due to resistance to solvents but not yet established PATHOLOGY ▪ 1o target: erythroid cell line (erythroid precursors near the pronormoblast stage) → cell lysis → transient arrest in erythropoiesis PPS Oral Exam Reviewer 2020 ▪ Later neurologic manifestations: behavioral disorders, microcephaly, mental retardation ▪ Other findings: interstitial pneumonitis, dermal erythropoiesis (blueberry muffin rash), DM, thyroid dysfunction, glaucoma, hepatosplenomegaly, thrombocytopenia ▪ All infants with evidence of IUGR or stigmata suggesting CRS should undergo virologic (viral culture or PCR) and serologic study for rubella (serum IgM > 21 mg/dL during the 1 st week of life strongly indicate congenital infection) ▪ Abrupt onset of high fever (mean 39.7C) + fussiness→ resolves after 3 days (crisis) or may fade over a day (lysis) coincident with appearance of faint blanching pink or rose-colored nonpruritic morbilliform rash that lasts for 1-3 days ▪ Mild injection of pharynx, palpebral conjunctiva, or tympanic membranes (TM), enlarged suboccipital nodes, rhinorrhea, congestion, GI complaints and encephalopathy ▪ Nagayama spots: ulcers at uvulopalatoglossal junction ▪ Mean duration: 6 days ▪ Supportive ▪ Unusual/severe manifestations: ganciclovir, foscarnet, cidofovir DIAGNOSTICS ▪ Characteristic: Mean of WBC, lymphocytes, neutrophils ▪ Sporadically noted: Serum transaminase, platelet, atypical lymphocytes Encephalitis: ▪ CSF: normal or minimal pleocytosis, mild protein ▪ MRI: hyperintense signal on T2-weighted and FLAIR images of the hippocampus, uncus, and amygdala ▪ PET scan: metabolism within hippocampus ▪ 3 day hx of high fever in an otherwise nontoxic infant with a blanching maculopapular rash suggest the diagnosis ▪ Specific dx is not usually necessary except if manifestations are severe or atypical and might benefit from antiviral therapy ● Viral culture (gold standard); if not possible, detection of viral DNA by PCR or rt-PCR ▪ Prodrome: low grade fever, headache, URTI, LAD ▪ Hallmark: characteristic rash that occurs in 3 stages ● Slapped cheek: erythematous facial flushing ● Diffuse macular erythema spreading to trunk and proximal extremities ● Lacy and reticulated (central clearing) prominent on extensors, sparing palms and soles ● Resolves spontaneously without desquamation ● Waxes and wanes for 1-3 weeks ▪ Lymphadenopathy and atypical papular, purpuric, vesicular rashes are also described Arthritis/Arthralgia ▪ Older adolescents, F>M, self-limited (resolves in 2-4 weeks) ▪ Diffuse polyarthralgia with AM stiffness to frank arthritis; affects hands, wrists, knees, and ankles Transient Aplastic Crisis ▪ Most common complication: convulsions ▪ Higher frequency of partial seizures, prolonged seizures, postictal paralysis and repeated seizures ▪ Other complications: ADEM, autoimmune encephalitis, acute cerebellitis, hepatitis, myocarditis ▪ Reactivation occurred in approx. 50% of immunocompromised hosts 2-4 wks after HSCT Post-transplant acute limbic encephalitis (PALE): short-term memory dysfunction, confusion, insomnia with seizures or prolonged EEG described primarily in pts. Ff. HSCT PROGNOSIS ▪ Self-limiting PALE: ▪ Associated with increased mortality ▪ Long term neurocognitive sequelae FINDINGS / DIAGNOSIS Diagnosis of erythema infectiosum is clinical Serologic tests: ▪ B19-specific IgM: develops rapidly after infection, persists for 6-8 weeks ▪ Anti-B19 IgG: past infection or immunity ▪ Anti B19 IgM: recent/ acute infection ▪ To confirm recent infection: seroconversion of anti-B19 IgG Ab in paired sera In immunocompromised: ▪ Serologic diagnosis is unreliable ▪ Requires methods to detect viral DNA If maternal infection suspected ▪ Fetal ultrasound and peak systolic flow velocity of MCA ▪ Detection of viral DNA in fetal blood or amniotic fluid MANAGEMENT Arthropathy ▪ Arthralgias/ arthritis may persist after resolution of rash Neurologic ▪ Aseptic meningitis, encephalitis, peripheral neuropathy ▪ Stroke in children w/ SCD ff. B19V-induced transient aplastic crisis PREVENTION For (+) erythema infectiosum only, no isolation needed (+) B19V induced RBC aplasia (w/ transient aplastic crisis): ▪ Isolate for 1 week and until fever has resolved, avoid contact with pregnant women Batch Clingy 25% during the end of 2 nd trimester 190 Patients with chronic hemolytic anemia: when infected → transient arrest in RBC production → fall in Hgb (requiring transfusion) & reticulocyte count (lysis of infected erythroid precursors) → development of IgM 1-2 days after infection ff. by anti-B19 IgG → control of infection & resolution of s/sx Patients with impaired humoral immunity serious/persistent infection, chronic RBC thrombocytopenia, neutropenia, marrow failure – more aplasia, Crosses placenta → profound fetal anemia and high output cardiac failure INCUBATION PERIOD: 10 – 21 days TRANSMISSION ▪ Contagious 24-48 hrs before the rash is evident until vesicles are crusted (usually 3-7d after onset of rash) ▪ Direct contact: aerosolization in cutaneous lesions ▪ Oropharyngeal secretions Primary Infection ▪ Virus inoculates in the mucosa of URT and tonsillar lymphoid tissue → replicates in lymphoid tissue, spread to T lymphocytes → skin: innate immunity controls VZV replication → cutaneous lesions VARICELLA ZOSTER Latent Infection ▪ Develops during incubation period or the disease itself ▪ Occurs only in the ganglionic neurons ▪ Virus is transported back to sensory axons to the dorsal root ganglion throughout the spinal cord and to CN ganglia ▪ Can be established by attenuated VZV in varicella vaccine like Oka strain (similar to natural or wild type) however the risk is lower after vaccination than natural infection HERPES ZOSTER (shingles) ▪ Milder in children PPS Oral Exam Reviewer 2020 ▪ Occurs in patients with chronic hemolytic conditions (e.g. sickle cell disease, thalassemia, hereditary spherocytosis, pyruvate kinase deficiency) ▪ Fever, malaise, lethargy, ss/x of profound anemia, vasooclusive pain crisis (in px w/ SCD); rash is rarely present Immunocompromised ▪ Chronic anemia (most common) + thrombocytopenia, complete marrow suppression ▪ No specific antiviral therapy ▪ IVIg sometimes used ▪ In patients with AIDS, HAART has been reported to clear B19V infection ▪ Infected fetus: intrauterine RBC transfusions neutropenia, Fetal Infection ▪ Associated with nonimmune fetal hydrops and IUFD, 2nd trimester most sensitive period Myocarditis ▪ Fetal myocardial cells express P Ag (receptor for virus) Papular purpuric gloves-and-socks syndrome (PPGSS) ▪ Fever, pruritus, painful edema, and erythema localized to distal extremities (gloves-and-socks distribution) → acral (or generalized) petechiae and oral lesions ▪ Self-limited, resolves within a few weeks ▪ Begins 14-16 days after exposure ▪ Prodrome: fever (mean: 37.8-38.9OC; may be as high as 41OC), malaise, anorexia, headache, mild abdominal pain occurs 24-48 hrs before rash appears ▪ Systemic symptoms resolve within 2-4 days after rash onset ▪ Exanthem first at scalp, face, or trunk (CENTRAL) ▪ Simultaneous lesions in various stages of evolution: intensely pruritic erythematous macules → papules → clear fluid-filled vesicles → 24-48 hrs: clouding and umbilication of lesions → crusting FINDINGS ▪ Leukopenia (1st 72 hours) after onset of rash → Lymphocytosis ▪ Mildly LFT ▪ CSF: mild lymphocytic pleocytosis, slight to mod protein, N glucose DIAGNOSTICS ▪ Direct fluorescence assay of cells from cutaneous lesions vesicular fluid ▪ PCR (vesicular fluid and crusts): most sensitive ▪ Rapid culture with IF staining ▪ Tzanck smear: multinucleated giant cells w/ nonspecific stains, poor sensitivity ▪ Genotyping: strain identification VZV IgG Ab ▪ > 4-fold rise is confirmatory of acute infection ▪ To determine immune status if hx of varicella is unknown or equivocal ▪ Vesicular lesions clustered within 1-2 adjacent dermatomes; rash is mild, symptoms of acute neuritis are minimal, complete Acyclovir: not routinely recommended ▪ 20mkdose QID x 5 days (max 800mg) ▪ Give within 24 hours of rash onset ▪ IV form for severe or IC: 500 mg/m2 Q8 x 7-10 days until no new lesions x 48 hours If resistant (HIV-infected): IV Foscarnet or Cidofovir For older children that can tolerate tablets: Famciclovir or Valacyclovir ▪ 2 to <18yo: 20mkdose TID x 5 days (max1000 mg/dose) ▪ AE: neuro and GI illness Secondary bacterial infections ▪ S. aureus and GAS ▪ Recurrence of fever 3 days after rash Encephalitis and Cerebellar ataxia ▪ Highest: <5yo - >20yo ▪ Nuchal rigidity, altered LOC, seizures ▪ Gradual onset of gait disturbance, nystagmus, slurred speech ▪ Begins 2-6 days after onset of rash ▪ Complete clinical recovery within 24-72 hours Pneumonia: 1-6 days after onset of rash; higher risk in smokers VARICELLA VACCINE ▪ given SC for 2 doses at a minimum age of 12 mos. (12-15 mos.) and 2nd dose given at 4-6 y/o (to prevent breakthrough varicella) ▪ contraindications similar to MMR vaccine Postexposure prophylaxis ▪ vaccine given to healthy children within 3-5 days after exposure ▪ high-titer anti-VZV Ig is recommended for immunocompromised children, pregnant and newborns (dose: 1 vial per 10kg BW) given ASAP up to 10 days after exposure ▪ Anti-viral treatment is not always necessary in otherwise healthy children Progressive Varicella: severe, fatal disease ▪ Visceral organ involvement, coagulopathy, severe hemorrhage, continued vesicular lesion development after 7 days ▪ Abdominal pain: involvement of mesenteric LN or the liver ▪ Risk factors: congenital cellular ID disorders, malignancy, after organ transplant, high dose steroids, HIV PROGNOSIS ▪ Low case fatality rate in children 1-9 y/o; infants have 4x greater risk, adults have 25x greater risk ▪ Causes: pneumonia, CNS complications, 2O infections, and hemorrhagic conditions ▪ Immunocompromised children may have more severe herpes zoster similar to adults (including postherpetic neuralgia) Batch Clingy ▪ Aplastic crisis: direct result of virus ▪ Exanthem and arthritis: post-infectious phenomena (immune response) 191 ▪ Unusual in healthy children <10yo ▪ Lifetime risk of zoster for individuals w/ hx of varicella: 30%; 75% occurring after age 45 y/o ▪ Severe in immunocompromised children ▪ Reactivation of latent infection ▪ Exposure to varicella with prior infection will ↓ reactivation ▪ Suppression of cell mediated immunity to VZV will increase risk NEONATAL VARICELLA ▪ Transplacental ▪ Maternal viremia occurs up to 48hr prior to onset of maternal rash CONGENITAL VARICELLA SYNDROME ▪ High risk in moms infected <13wks AOG and 13-20wks AOG resolution within 1-2 wks ▪ Zoster sine herpete: unilateral dermatomal pain without rash ▪ Septic meningitis, enteric zoster: GI illness ▪ A large rise in VZV IgG titer in convalescent titer in atypical zoster rash is confirmatory VARICELLIFORM RASHES IN VACCINATED INDIVIDUALS ▪ Within 1-2wks after vacination: likely wild-type virus ▪ 14-42 days: either wild-type virus or vaccine strains ▪ >42 days: Breakthrough varicella caused by wild-type virus ▪ Atypical rash, <50 lesions, shorter duration, fewer complications, and little or no fever, less contagious than wild-type virus ▪ Rash appears 2 days – end of 1st wk of life ▪ Fatal if the mother has varicella 5 days prior to 2 days after delivery (no maternal antibodies formed yet) ▪ If >5 days prior to delivery and >30 weeks AOG: attenuated form of varicella ▪ Zigzag cicatricial skin scarring, dermatomal distribution, limb hypoplasia, LBW ▪ Neurologic – microcephaly, cortical atrophy, seizures, MR ▪ Eye – chorioretinitis, microphthalmia, cataracts ▪ Renal – hydroureter, hydronephrosis ▪ ANS – neurogenic bladder, swallowing dysfunction, aspiration pneumonia ▪ Oral acyclovir can shorten duration but does not prevent postherpetic neuralgia ▪ If with disseminated disease: give IV acyclovir 500mg/m2 or 10mk Q8 ▪ If immunocompromised but uncomplicated zoster (low risk for visceral dissemination): give PO acyclovir, famciclovir, or valacyclovir ▪ Steroids are not recommended PROGNOSIS ▪ Excellent ▪ Self-limited ▪ Give VZIG to NB if mother had varicella 5d prior to 2d after delivery, preterm <28wks born to mother with active varicella at delivery (even if maternal rash >1wk), hospitalized exposed preterm + mother has no evidence of immunity ▪ If no VZIG, give IVIG ▪ Symptomatic NB: Acyclovir (10 mk q8 IV) ▪ Susceptible mother exposed to varicella: give VZIG ▪ Mother with severe varicella: treat with acyclovir (class B) ▪ Although neonatal varicella may occur in about half of these infants despite administration of VZIG, it is milder than in the absence of VZIG administration DIAGNOSTICS ▪ Hx of gestational varicella combined with characteristic abnormalities in the NB ▪ Viral DNA by PCR in tissue (vesicular fluid or scabs) ▪ VZV specific IgM antibody in cord blood sample ▪ Persistently (+) VZV IgG Ab at 12-18 months old: prenatal infection ▪ Ultrasound for limb atrophy HEPATITIS TRANSMISSION ▪ Person-to-person through fecal – oral route ● Fecal excretion of virus starts late in incubation period ● Peak: just before the onset of symptoms ● Resolves by 2 weeks after onset of jaundice ● Travel to endemic areas; contact with contaminated food or water (shellfish, frozen berries, raw vegetables) ▪ Highly contagious 2wks before & 7 days after jaundice ▪ Perinatal – rare PPS Oral Exam Reviewer 2020 Serology (adapted from Nelson’s 21 st Ed., Table 385.3) Acute/Active Past/Recovered Chronic Vaccine Response (+) anti-HAV IgM, remains for 4-6 mos after infection (+) anti-HAV IgG N/A (+) Anti-HAV IgG COMMON BIOCHEMICAL PROFILES IN ACUTE INFECTION ▪ Altered synthetic function: most important marker of liver injury; prolonged PT, ↑ INR, ↓ serum albumin, hypoglycemia, lactic acidosis, ↑ NH3 ▪ Cytopathic injury: rapid ↑ AST/ALT ▪ Cholestasis: ↑ serum bilirubin, ALP, 5NT, GGT ▪ Supportive ▪ ALF (rare) ▪ Anti-pruritic agents and fat-soluble vitamins ▪ Prolonged cholestatic syndrome ▪ Handwashing ▪ Pruritus ▪ Contact and standard precautions ▪ Fat malabsorption Pre-Exposure Prophylaxis PROGNOSIS Vaccine ▪ Excellent Minimum age of 12 months Hep A Vaccine 2 doses series Inactivated (IM) Minimum interval between 1 st and 2nd dose is 6 months Minimum age of 18 Live (SC) Single dose Immunoglobulin ▪ Provides protection x 2 months ▪ Suitable for <12mo old, allergic to a vaccine component, those Batch Clingy PATHOGENESIS ▪ Direct cytopathic and/or immune mediated injury: centrilobular necrosis, bile duct proliferation, diffuse Kupffer cell hyperplasia in sinusoids ▪ Liver morphology returns to normal within 3 months after recovery from acute infection ▪ If chronic hepatitis develops, inflammatory infiltrates settle in the periportal areas → progressive scarring HEPATITIS A ▪ Causes acute hepatitis only ▪ Most prevalent ▪ Duration: 7-14 days ▪ Heat stable, limited hosts: human & other primates ▪ In young children: anicteric illness; s/sx similar to viral AGE ▪ Older adolescent or adults, underlying liver disorders, IC: acute INCUBATION PERIOD: 15- 19 days (mean 3wks) febrile illness, anorexia, nausea, malaise, vomiting, jaundice 192 who elect not to receive vaccine HAV + Ig: for older patients, immunocompromised, (+) chronic liver disease who plans to travel in endemic areas in < 2wks INCUBATION PERIOD: 45-160 days (mean: 120 days) TRANSMISSION ▪ Parenteral, sexual, perinatal ▪ Concentrations ● High: blood, serum, serous exudates ● Moderate: saliva, vaginal fluid, semen ● Inconsistent: Breast milk Risk Factors: ▪ IV drugs or blood products, contaminated needles, sex, institutional care, intimate contacts w/ carrier ▪ Most important RF: perinatal exposure to (+) HBsAg mother; high maternal HBV viral load + delivery of prior infant who developed HBV despite prophylaxis ▪ HBV is predominantly noncytopathogenic ● Causes injury mostly by immune-mediated processes ● Expression of viral antigens (i.e. HBcAg and HBeAg) + class I MHC proteins on cell surface makes it a target for cytotoxic T-cell lysis ▪ Mechanism for chronic HBV infection is less well understood ● possible non-recognition of core protein or MHC class I protein, non-activation of cytotoxic lymphocytes ACUTE HBV INFECTION ▪ Most are asymptomatic ▪ 6-8 weeks after exposure: serum ALT levels ↑ ff. by fatigue, anorexia, malaise or serum sickness like prodrome: arthralgia and skin lesions, papular acrodermatitis (Gianotti Crosti syndrome) → extrahepatic conditions may occur ▪ Jaundice – begins 8wks after exposure, lasts for 4wks ▪ Symptoms persist for 6-8 wks then resolves but some enters a chronic carrier sate CHRONIC HBV INFECTION (3 PHASES) ▪ Immune tolerant – majority; spontaneous HBeAg seroconversion (development of anti-HBe and loss of HBeAg) ▪ Immune active – active inflammation, elevated AST/ALT, progressive fibrosis ▪ Inactive HBsAg: early marker of infection, falls during recovery before s/sx wane Anti-HBc IgM: rises early after infection, remains (+) for months, replaced by Anti-HBc IgG (persists for years) Ant-HBs: marker of recovery and protection HBeAg: present in active acute or chronic infection, marker of infectivity Serology (adapted from Nelson’s 21 st Ed., Table 385.3) Acute/Active Past/Recovered Chronic Vaccine Response (+) Anti-HBc IgM (+) HBsAg (-) Anti-HBs (+) HBV DNA (PCR) (+) Anti-HBs (+) Anti-HBc IgG (+) Anti-HBc IgG (+) HbsAg (-) Anti-HBs (+) PCR (+) Anti-HBs (-) Anti-HBc Treatment ▪ Indicated for immune-active form (evidenced by ↑ AST/ALT or fibrosis on liver biopsy) ▪ Not for immune-tolerant patients (normal AST/ALT, (+) HBeAg with ↑ viral load) IFN α2b: immunomodulatory and antiviral effects; viral resistance does not develop; given SQ x 24 wks, SE: flu-like s/sx, marrow suppression, depression; C/I: decompensated cirrhosis Lamivudine: oral synthetic nucleoside analog; inhibits reverse transcriptase Adefovir: purine analog; inhibits viral replication Entecavir: nucleoside analog; inhibits replication Tenofovir: nucleotide analog; inhibits replication Peginterferon-α2: same MOA as IFN Hepatitis B Vaccine (IM) 3 doses series Administer the 1st dose of monovalent HBV to all newborns >2 kgs within 24 HOL A 2nd dose is given 1-2 months after the birth dose Final dose: administered not earlier than 24 weeks of age. Another dose is needed if the last dose was given at age <24 weeks. (see Nelson’s 21st Ed., Table 385.5 for Typical Interpretation of Test Results for HBV Infection) HEPATITIS C ▪ Most common cause of chronic liver disease in adults PPS Oral Exam Reviewer 2020 ▪ Acute: mild, insidious in onset ▪ Chronic (evidence of active viral infection with detectable HCV RNA for at least 6 months): clinically silent until with complication ▪ Acute HCV infection: due to high rate of spontaneous clearance and AE of currently available therapy, tx is not routinely given ▪ Chronic HCV infection ▪ ALF with coagulopathy, encephalopathy, cerebral edema ● Risk increased by coinfection or superinfection with HDV or in immunosuppressed host ● Liver transplant is the only effective intervention ▪ Chronic Hepatitis (HBsAG (+) for >6mos) → cirrhosis then ESLD; HCC ▪ Extrahepatic (immune mediated): PAN, GN (membranous or MPGN), aplastic anemia, polymyalgia rheumatica, leukocystoclastic vasculitis, GBS PROGNOSIS ▪ Acute: favorable ● Mortality from ALF: >30% ▪ Chronic: HCC was the most prevalent cancer-related death in young adults in Asia PREVENTION ▪ Mothers HBV DNA >200,000 IU/mL: give antiviral during 3 rd trimester ▪ Not spread by breastfeeding, kissing, hugging, or sharing water or utensils ▪ Exclude children who are prone to biting ▪ HBIG: temporary protection (3-6mo), give at separate anatomical site from vaccine Infants born to HBsAg positive mothers (preterm or term infants regardless of weight): - Administer HBV* and HBIG (0.5 mL) within 12 hours of birth. - HBIG should be administered not later than 7 days of age, if not immediately available. Infants born to mothers with unknown HBsAg status (preterm or term infants regardless of weight) - BW >2 kg, administer HBV within 12 hours of birth and determine the mother’s HBsAg as soon as possible. If HBsAg (+) administer also HBIg not later than 7 days of age. - BW <2 kgs, administer HBIG in addition to HBV* within 12 HOL *For those <2 kgs, the 1st dose received at birth is not counted as part of the vaccine series. Additional 3 HBV doses are needed. ▪ Highest risk of chronic hepatitis ▪ Cirrhosis, liver failure and primary HCC (within 20-30yo) ● Yearly liver UTZ and serum AFP Batch Clingy HEPATITIS B ▪ Risk of chronic infection ↑ in younger patients Ig Post-Exposure Prophylaxis ▪ Not effective if given > 2 weeks after exposure ▪ Preferred in <12 months old and older > 40 y/o, IC, (+) chronic liver disease or in whom vaccine is contraindicated ▪ Acute HBV infection: supportive ▪ Chronic HBV infection (goal): ↓ viral replication (undetectable HBV DNA and development of Anti-HBe) 193 Risk Factors: blood transfusion, illegal drug use, multiple sexual partners, occupational INCUBATION PERIOD: 2-24 weeks (mean: 7-9 wks) TRANSMISSION ▪ Parenteral ▪ Sexual – rare ▪ Perinatal – 5-15%, most prevalent MOT in children ● Serum aminotransferase fluctuates (sometimes normal) ● Liver fibrosis progresses slowly (see Nelson’s 21st Ed., Fig. 385.5 for Natural history of hepatitis C virus infection) Acute/Active Past/Recovered Chronic (+) Anti-HCV IgG‡* (+) HCV RNA PCR* (+) Anti-HCV (-) Blood PCR (+) Anti-HCV IgG‡ (+) Blood PCR ● Goal: achieve a sustained viral response (absence of viremia at a variable period after stopping meds; assoc. w/ improved histology & ↓ risk of morbidities) ● Tx should be considered for pts w/ evidence of advanced fibrosis or injury on liver biopsy ● Peginterferon, IFN-α2b, Ribavirin are FDA-approved for children > 3yo; (24-48 wks of peginterferon and ribavirin) ● Sofosbuvir ± Ledipasvir has FDA approval for > 12-17yo ● Side effects: influenza-like symptoms, anemia, neutropenia, lag in height ▪ Chronic HCV infection can be associated with small vessel vasculitis (Ab to smooth muscle, ANA, ↓ TH may be sometimes present) ▪ Supportive ▪ No specific HDV targeted treatments ▪ Small studies: IFN PREVENTION ▪ Immunize against HBV PROGNOSIS ▪ Children are believed to have a higher rate of spontaneous clearance (up to 45% by age 19) ● Viral titers should be checked yearly to document spontaneous remission ‡One INCUBATION PERIOD ▪ Co-infection – same as HBV ▪ Superinfection – 2 to 8 weeks TRANSMISSION ▪ Parenteral, sexual, perinatal ▪ Direct injury by cytopathic mechanisms; liver damage is usually severe HEPATITIS E INCUBATION PERIOD: 15-60 days (mean: 40 days) TRANSMISSION ▪ Fecal-oral (water-borne) ▪ Cytopathic virus; pathology similar to other hepatitis viruses ROTAVIRUS ▪ Classified by serogroup and subgroup ● Group A includes the common human pathogens; Groups B & C occassionaly reported ● Strains are species specific ▪ Disease tends to be severe in 3-24mos of age ● < 3mos old are protected by transplacental Ab and BF PPS Oral Exam Reviewer 2020 (+) Anti-HDV IgM (+) Blood PCR (+) HBsAg (-) Anti-HBs (+) Anti-HDV IgG Past/Recovered (-) Blood PCR (+) Anti-HDV IgG (-) Blood PCR Chronic (+) HBsAg (-) Anti-HBs ▪ Similar to HAV but more severe ▪ Chronic illness does not occur except in immunosuppressed patients ▪ Affects older patients (15-34yo) Acute/Active (+) Anti-HEV IgM (1 week) (+) Blood PCR (+) Anti-HEV IgG Past/Recovered (-) Blood PCR ▪ Mild to moderate fever, vomiting → frequent, loose watery stools ● Vomiting & fever abate on the 2 nd day of illness ● Diarrhea continues for 5-7 days ▪ Most severe disease: 4-36 mos. of age ▪ Major pathogen in pregnant women ● causes ALF with a high fatality incidence ▪ Decompensation of pre-existing chronic liver disease PREVENTION ▪ Recombinant vaccine is highly effective in adults Acute/Active DIAGNOSTICS ▪ Diagnosis of viral AGE is clinical ▪ Avoid & treat dehydration ▪ Maintenance of nutritional status ● Breastfeeding should be continued even during rehydration ● Hypocaloric diets (↓ CHON & fat) such as BRAT (bananas, rice, cereal, applesauce, toast) diet have not been shown to be superior to a regular diet ▪ No routine role for antivirals or antibiotics Rare: ▪ Mild encephalopathy with reversible splenium lesions → cerebellitis ▪ Necrotizing enterocolitis PROGNOSIS ▪ Subsequent reinfection decreases in severity during the 1 st 5 yrs of life Batch Clingy HEPATITIS D ▪ cannot cause infection on its own ● Co-infection: same time as initial HBV ● Superinfection: already infected with HBV disadvantage of the antibody-based tests is that they cannot distinguish acute from chronic infection; anti-HCV IgM is not useful for distinguishing b/w acute & chronic HCV infection and measuring HCV IgM is not recommended (2012 NASPGHAN Practice Guidelines) *anti-HCV IgG antibodies are usually negative during the acute phase and HCV infection during this time frame can only be determined by serum HCV RNA (2012 NASPGHAN Practice Guidelines) ▪ Symptoms are similar but more severe than those of other hepatotropic viruses ● Co-infection – acute hepatitis is more severe than HBV alone; risk of developing chronic hepatitis is low ● Superinfection – acute illness is rare, chronic hepatitis is common; risk of ALF is higher 194 INCUBATION PERIOD: <48hrs (range: 1-7 days) TRANSMISSION ▪ Fecal-oral – ↑concentration in stool before and for days after the clinical illness ▪ Isotonic dehydration with acidosis ▪ FA: no blood or WBC ▪ CBC: WBC may be moderately elevated 2O to stress (marked left shift seen with invasive bacterial enteritis is absent) PREVENTION RV (PO) Human RV1 (oral liquid formulation): 2 dose series with minimum age of 6 weeks and minimum interval of 4 weeks. The last dose should be administered not later than 24 weeks of age. ▪ Gastric mucosa is not affected ▪ Nonstructural protein 4 (NSP4) functions as an enterotoxin ▪ Infect and destroy villus tip cells in small intestine leads to ● ↓ Absorption of salt and water and an imbalance in the ratio of intestinal fluid absorption to secretion, ● Diminished disaccharidase activity and malabsorption of complex carbohydrates, particularly lactose ▪ Most common cause in older children and adults ▪ Large explosive outbreaks among older children and adults (schools, cruise ships, hospitals) ▪ Source: shellfish or water in food preparation ▪ Winter infantile AGE NORWALK (Norovirus) Human-Bovine live-attenuated reassortant (RV5) (oral liquid formulation): 3 dose series, the 1st dose is given at age 6 – 12 weeks, with minimum interval of 4 weeks. The last dose should not be administered beyond 32 weeks of age. Human-Bovine live-attenuated reassortant (RV5) (oral freezedried formulation): 3 dose series, recommended at 2, 4, and 6 months. Given at a minimum age 6 weeks and minimum interval of 4 weeks. The last dose should not be administered beyond 12 months of age. ▪ Good hygiene ▪ Vomiting and nausea tend to predominate; lasting 1-3 days ▪ Resembles food poisoning from S. aureus or B. cereus ▪ Laboratory findings similar to other viral AGE INCUBATION PERIOD: 12 hours TRANSMISSION: Fecal – oral RABIES INCUBATION PERIOD ▪ 1-3 months ▪ In severe wounds to the head, s/sx may occur w/in 5 days after exposure TRANSMISSION & PATHOGENESIS ▪ Inoculation of saliva through bite or scratch from an infected vector ● Transmission rate is higher if multiple bites and inoculation in highly innervated parts (hand or face) ▪ May enter intact mucous membranes ▪ Virus replicates slowly in muscle or skin → enters peripheral motor nerve, uses nicotinic Ach receptor ▪ Axonal transport: crosses synapses q12hrs → rapid dissemination in brain and spinal cord PPS Oral Exam Reviewer 2020 2 FORMS: Encephalitic/Furious ▪ Fever, sore throat, malaise, HA, n/v, weakness ▪ Paresthesia and pruritus at or near the site of bite → extend to the affected limb ▪ Encephalitis: agitation, sleep disturbance or depressed mentation, orofacial dyskinesias, myoclonus, seizures ▪ Periods of lucidity alternating with profound encephalopathy ▪ Cardinal sx: Hydrophobia and aerophobia (phobic spasms) ▪ Death occur in almost 100% of cases; 73% die w/in 3 days of onset; 84% die w/in 24hrs of admission (2019 DOH NRPCP) Paralytic/Dumb ▪ Uncommon ▪ Fever & ascending motor weakness involving both limbs and CN; encephalopathy (occasional) POSTEXPOSURE PROPHYLAXIS (PEP) ▪ Cell Culture & Embryonated Egg-based Vaccine (CCEEV) ● Purified Vero Cell Rabies Vaccine (PVRV) ● Purified Chick Embryo Cell Vaccine (PCECV) PREVENTION ▪ Vaccinate domestic animals ▪ Education PRE-EXPOSURE PROPHYLAXIS (PrEP) ▪ Persons traveling to a rabies-endemic region (2018 WHO Position Statement on Rabies) ▪ Regimens for individuals of all ages: ● 2-site ID vaccine (0.1 mL) on days 0 & 7 ● 1-site IM (1 vial) on days 0 & 7 PEP SCHEDULE FOR PREVIOUSLY IMMUNIZED ANIMAL BITE PATIENTS DIAGNOSTICS ▪ Often based on clinical manifestations & exposure to a rabid animal (2019 DOH National Rabies Prevention and Control Program) ▪ Fluorescent Antibody Testing (FA) – gold standard; tissue samples from brainstem, thalamus, cerebellum and the hippocampus are Batch Clingy PATHOGENESIS Similar to RV; may also cause delayed gastric emptying ▪ True animal reservoirs: terrestrial carnivores, bats ▪ Rare in small animals (mice, squirrels, rabbits) ▪ Species Affected by Rabies 2018 (2019 DOH National Rabies Prevention and Control Program) ● 96% of positives are from canines ff. by felines ● 72% of rabies cases are owned; 88% of rabies cases were either free-roaming or occasionally roaming 195 ● Histopathology: limited damage, inflammation, or apoptosis ● Pathologic hallmark: Negri bodies – cytoplasmic inclusions ▪ Virus concentrates on brainstem (autonomic dysfunction and relative sparing of cognition) ▪ Virus travels anterograde into PNS (exacerbating dysautonomia) → innervated organs → salivary glands recommended; may also be detected on skin biopsies taken from the nuchal area with hair follicles containing nerve endings ▪ rt-PCR – most sensitive; uses saliva, urine or skin ▪ Serologic (antibody) testing – only 50% will give (+) results among rabies cases; more useful to ascertain immune status of immunized animals & humans PROGNOSIS ▪ Fatal ▪ Once with symptoms, vaccine and Ig are contraindicated ▪ Presence of normal Ab response by 7d: clearance of salivary viral load and survival ● Schedule (may use either) 1-site IM, days 0-3-7 then 4th dose b/w days 14 to 28 2-site IM on day 0 and 1-site IM on days 7 and 21 TRANSMISSION ▪ Respiratory ▪ Fecal – oral: can be shed from GIT for prolonged periods ADENOVIRUS INFLUENZA ▪ Damage to epithelial mucosa → sloughing of cell debris → inflammation ▪ Host response: recruit neutrophils, macrophages, and NK cells ▪ Three genera (or types): A, B, and C ● A & B: 1o human pathogens causing seasonal epidemics ● C: sporadic ▪ Influenza A subtypes are based on 2 surface proteins: ● HA (hemagglutinin), NA (neuraminidase) PPS Oral Exam Reviewer 2020 ▪ Acute Respiratory Disease: Bronchiolitis, pneumonia (may present features more typical of bacterial disease), coryza, sore throat, fever ▪ Ocular: follicular conjunctivitis (self-limiting), epidemic keratoconjunctivitis (severe), pharyngoconjunctival fever ▪ GI: often asymptomatic but can cause acute diarrhea, mesenteric adenitis ▪ Hemorrhagic cystitis: sudden onset of hematuria, dysuria, frequency, urgency w/ (-) urine CS; resolves 1-2 weeks ▪ Myocarditis, hepatitis, meningoencephalitis ▪ Abrupt onset ▪ Predominance of systemic symptoms (i.e. fever, myalgia, chills, HA, malaise, anorexia); coryza, pharyngitis, dry cough are also usually present ● Duration of febrile illness: 2-4 days; cough may persist longer ▪ Organ failure in immunocompromised persons (esp. transplant recipients) ▪ Respiratory failure, chronic lung disease, bronchiolitis obliterans 2o to severe HAdV pneumonia ▪ Epidemic keratoconjunctivitis is vision-threatening ▪ Refractory severe anemia in hemorrhaging cystitis PROGNOSIS ▪ Risk for severe/fatal disease ▪ Reactivation in a transplant recipient MANAGEMENT ▪ Supportive care ▪ Bacterial superinfections are common; should be appropriately tx ▪ Give antivirals to those who are: ● Hospitalized PREVENTION ▪ Hygiene: handwashing and cleaning ▪ Vaccines: live attenuated HAdV-4 & HAdV-7 (for military) ▪ OM and pneumonia: most common in young children ▪ Acute myositis or rhabdomyolysis particularly calf muscles, myoglobinuria, ARF, myocarditis, sepsis ▪ CNS: encephalitis, myelitis, GBS, Reye syndrome with salicylate use, bacterial co-infection Batch Clingy ▪ Immunocompetent hosts ▪ Outbreaks: schools and military setting ▪ Rabies Ig (RIG) ● Human RIG (150 IU/mL) – Dose: 20 IU/kg ● Equine RIG (200 IU/mL) – Dose: 40 IU/kg ● Total computed dose shall be infiltrated around and into the wound as much anatomically feasible (may be diluted with NSS to increase volume) ● Given only once during the PEP course ▪ Supportive ▪ For epidemic keratoconjunctivitis: Cidofovir (highly nephrotoxic) + topical steroids or immunosuppressive agents 196 INCUBATION PERIOD: 1-4 days (average 2 days) TRANSMISSION ▪ Respiratory droplets ▪ Contact with secretions and small-particle aerosols ▪ Infectious day before until 5-7 days after onset of symptoms ▪ Children with 1o infection: higher viral loads and more prolonged viral shedding ▪ Community transmission: rapid with highest incidence within 2-3 weeks of introduction ▪ Infect respiratory epithelium → HA attaches to sialic acid → replication (10-14 days) → lytic infection of epithelium, loss of ciliary function, mucus, desquamation → permit 2O bacterial infection ▪ Enterovirus ▪ 3 serotypes (types 1, 2 and 3) ● type 1 has greatest propensity for natural polio ● type 3 has a predilection for VAPP ▪ Acute paralytic disease can be caused by: ● wild poliovirus ● vaccine-associated paralytic poliomyelitis (VAPP) – OPV viruses that cause illness in vaccine recipients (rare cause); consider if symptoms occur 7-14 days after receiving OPV ● vaccine-derived polioviruses – either OPV viruses that continuously replicate in immunodeficient persons (iVDPV) or circulating vaccine-derived polioviruses (cVDPV) that have acquired virulence properties resulting from sustained person-toperson circulation in the absence of adequate population immunity ▪ Family members/close contacts experience similar illness ▪ Abdominal pain, vomiting, diarrhea may also occur ▪ Less common: parotitis, rash ▪ Severe: underlying cardiopulmonary disease ▪ High Risk: pregnant, adolescent female, child on chemotherapy, immunodeficient DIAGNOSTICS ▪ Diagnosis is clinical in the context of an epidemic ● However, presentation is indistinguishable from other respiratory viruses ▪ Confirmation is not required for decision to prescribe antivirals (see Nelson’s 21st Ed., Table 285.2 for Influenza Virus Testing Methods) ▪ Paralysis – 3-8d after the initial symptom with extent of involvement obvious within 2-3 days possibly affecting respiration and circulation system ▪ Inapparent infection, abortive poliomyelitis and nonparalytic poliomyelitis are more commonly seen with wild type poliovirus infection POSSIBLE COURSE OF INFECTION Inapparent Infection (90-95%) – no disease/sequalae Abortive Poliomyelitis (5%) ▪ Non-specific influenza-like syndrome 1-2 wks after infection, lasting 2-3 days with complete recovery ▪ Prominent: fever, malaise, anorexia, HA, sore throat, abdominal or muscular pain, irregular vomiting POLIOVIRUS ▪ the last case of WILD poliovirus in the Philippines was in 1993; polio-free since 2000 however outbreak was declared last Sept. 2019 ▪ as of Jun 15, 2020, the Philippines is affected by both cVDPV1 and cVDPV2 (UNICEF WHO Philippines Polio Outbreak Situation Report 22) INCUBATION PERIOD: 8-12 days (range: 5-35 days) TRANSMISSION ▪ Fecal – oral ▪ GIT → CNS ▪ Infectivity is for several days at room temp. ▪ Human: only known reservoir ▪ ↑ risk of acquisition of paralysis: tonsillectomy & IM injections PPS Oral Exam Reviewer 2020 Nonparalytic Poliomyelitis (1%) ▪ Signs of abortive polio present + more intense HA, n/v; soreness and stiffness of the posterior muscles of the neck, trunk, and limbs; fleeting paralysis of bladder and constipation ● 1st phase: minor illness ● 2nd phase: CNS disease or major illness ▪ Nuchal and spinal rigidity (basis for nonparalytic polio diagnosis) ● Nuchal rigidity is present as well as head drop (normally, head follows plane of the trunk when lifting up) ▪ Changes in reflexes precede weakness by 12-24 hrs ● 1st to diminish: spinal, gluteal, abdominal, cremasteric, superficial reflexes ● Changes in DTR occur 8-24h after superficial reflex are depressed → impending paresis ● Complicated or progressive illness ● At high risk for complications (< 2yo, chronic pulmo, cardio, renal, hepa, hema, metabolic, neuro, neurodevelopmental D/O, immunosuppressed, pregnant, postpartum (2wk), < 19yo w/ longterm aspirin or salicylate medication, BMI ≥ 40, residents of long term facilities) ▪ Can also be considered for any previously healthy, symptomatic patient not at high risk if tx can be initiated within 48 hrs of onset (uncomplicated influenza) ● Oseltamivir BID x 5 days ● Clinical benefit is greatest when antiviral is given within 2 days Post exposure Prophylaxis ▪ Routine use not recommended; consider for: ● unvaccinated persons at high risk of influenza complications ● persons for whom vaccine is CI ● residents/patients in care facilities ▪ May use oseltamivir (PO) & zanamivir ▪ Can be started within 48h of exposure up to 7 days after the last known exposure ▪ CDC/IDSA: minimum 2wks – 1wk after the last known case is identified whichever is longer ▪ Chemoprophylaxis with antiviral is not a substitute for vaccination ▪ No specific antiviral ▪ Mgt is supportive and aimed at limiting progression of disease, preventing skeletal deformities and preparing the child & family for prolonged tx ▪ All IM injections & surgical procedures are contraindicated during acute phase of illness (1st wk) to prevent progression of disease ▪ Abortive Poliomyelitis – supportive, rest x 2wks, do check-up 2 months later ▪ Nonparalytic Poliomyelitis – analgesic + hot pack compress 15-30 min Q2-4hr, hot tub baths, firm bed, footboard or splint, gentle PT; check-up 2mos after recovery ▪ Paralytic Poliomyelitis ● Suitable body alignment, change position Q3-6hr, active and passive movement, opiates and sedatives, bethanecol (bladder paralysis) or bladder compression, catheter ● Head-low prone position with face to 1 side to prevent aspiration in those with bulbar polio ● Pts w/ pure bulbar polio may require tracheostomy because of vocal cord paralysis or constriction of the hypopharynx ● Impaired ventilation must be recognized early PREVENTION Polio Eradication and Endgame Strategic Plan 2013-2018 ▪ Withdrawal of trivalent OPV (tOPV) with bivalent (bOPV) in all countries by 2016 and addition of 1 dose of IPV ● risk of VDPV was highest w/ type 2 strain → switch from tOPV to bOPV ● IPV will protect against type 2 ▪ replacement of bOPV with IPV in all countries by 2019 (to prevent PROGNOSIS ▪ Excellent if uncomplicated; fatal if severe PREVENTION INFLUENZA VACCINE ▪ Trivalent (IM or SC) Quadrivalent (IM) ▪ Minimum age: 6 months For pediatric dose, follow the manufacturer’s recommendation: ▪ 6mos-8yo receiving vaccine for the 1st time should receive 2 doses separated by at least 4 weeks ▪ If only one dose was given during the previous influenza season, give 2 doses of the vaccine then 1 dose yearly thereafter ▪ 9-18yo should receive one dose of the vaccine yearly ▪ Annual vaccination should begin in February by may be given throughout the year Bulbar Poliomyelitis ▪ Hypertension and dysautonomia → associated irregular/failed respiratory effort, delirium, coma w/ Immobilized for long periods: ▪ Atrophy of the limb, failure of growth, deformity, pneumonia, renal stones (hypercalcemia/hypercalciuria sec to bone resorption) ▪ Acute gastric dilation, melena from superficial intestinal erosions (rare: perforation), HTN, cardiac irregularities, myocarditis, acute pulmonary edema, PROGNOSIS ▪ Good in inapparent, abortive and aseptic meningitis syndrome ▪ Severe bulbar poliomyelitis – 60% fatal ▪ Paralysis ● Maximum: 2-3 days after onset of paralytic phase (PP) ● Recovery: w/in 6mos (If persistent > 6mos, permanent paralysis) POSTPOLIO SYNDROME ▪ 30-40% who survived paralytic polio ▪ Occurs after 30-40yo ▪ Common in prev. wild type infection ▪ Muscle pain & exacerbation of weakness or development of new weakness or paralysis ▪ Risk: Female, presence of permanent residual impairment after recovery, length of time since acute infection POLIO OUTBREAK RESPONSE ▪ DOH initiated a Supplemental Immunization Activity to at-risk Batch Clingy Antigenic Variation ▪ Antigenic Drift – minor change within subtype → new strains of same type (influenza A & B); contributes to annual epidemics ▪ Antigenic Shift – major change in subtype → new influenza A subtype with little or no immunity (influenza A) ● Reassortment: proteins in non-human hosts could be introduced to humans → pandemic 197 PATHOGENESIS ▪ Adsorb on poliovirus receptor (CD155) mostly on the epithelial cells lining the tonsil follicle and small intestine → penetrates cell & releases viral RNA → translated to produce proteins for RNA replication & shuts off host protein synthesis ▪ Mature virus is produced in 6-8 hours ▪ Wild type virus accesses the CNS thru peripheral nerves ● primarily affects the anterior horn cells (SC) and the cranial nerve nuclei (brainstem) ▪ Perineuronal inflammation & mixed inflammatory reaction → extensive neuronal destruction ● >50% motor neurons destroyed: Limb weakness ● involvement of the reticular formation (containing vital centers) → poor outcome Paralytic Poliomyelitis (0.1%) VDPV with continued OPV use) Spinal Paralytic Poliomyelitis ▪ 2nd phase after abortive poliomyelitis ● Pt recovers for 2-5 days but followed by severe HA, fever with exacerbation of previous systematic symptoms ▪ Severe muscle pain, paresthesia, hyperesthesia, fasciculation, spasms ▪ Spotty distribution of paralysis → w/n 1-2 days asymmetric flaccid paralysis or paresis occurs (proximal > distal), nuchal stiffness, hyperactive DTRs → diminution of reflexes → paresis or flaccid paralysis ▪ Weakness of some muscle of neck, abdomen, trunk, diaphragm, thorax, or extremities ▪ Bowel & bladder dysfunction often accompany paralysis of lower limbs National Immunization Program ▪ 3 doses bOPV given starting at 6 weeks, 4 weeks apart (6-10-14) with 1 dose IPV given together with the 3 rd dose of bOPV regions using bOPV and mOPV2 ▪ No new polio cases reported after Feb 15, 2020 (UNICEF WHO Philippines Polio Outbreak Situation Report 22, June 15 2020) IPV only schedule ▪ given in combination with DPT-containing vaccines ▪ 3 doses starting at 6 weeks, 4 weeks apart ▪ 2 booster doses (either OPV or IPV): between 12-18 mos and 4-6 yo Provocation Paralysis (subset of spinal paralytic polio) ▪ occurs in 50-60% after IM injection ▪ Initially present with fever and paralysis ● Most have abrupt flaccid paralysis ● Some have spasm, ↑ muscle tone with transient ↑ DTR ▪ Once temperature is normal, progression of paralysis stops ▪ Recovery is within 6 months. ● The lack of improvement within 1st several weeks or months after → permanent paralysis ▪ CSF is often normal during minor illness; pleocytosis with PMN predominance early in the course then shift to mononuclear, CSF CHON normal to slightly elevated Bulbar Poliomyelitis ▪ Dysfunction of CN and medullary centers (CN involvement is seldom permanent) ▪ Paralysis of facial, extraocular, masticatory muscles (+) respiratory difficulty 1) nasal twang to voice/cry 2) inability to swallow smoothly 3) accumulated pharyngeal secretions 4) absence of effective coughing 5) nasal regurgitation of saliva 6) deviation of palate, uvula, tongue 7) involvement of vital centers in Medulla 8) paralysis vocal cord/s 9) rope sign: acute angulation b/w chin and larynx (weakness of hyoid muscles) ▪ HTN & other autonomic disturbances are common ▪ No apparent involvement of the SC but bulbar disease may culminate in an ascending paralysis (Landry type); uncommon Paralytic Poliomyelitis w/ Ventilatory Insufficiency ▪ Components that contribute to respiratory insufficiency: anxiety, fear → overventilation → respiratory alkalosis PPS Oral Exam Reviewer 2020 Batch Clingy Polioencephalitis ▪ Rare with higher centers of the brain severely involved ▪ Seizures, coma, spastic paralysis, increased reflexes, irritability, disorientation, drowsiness, coarse tremor, peripheral or CN paralysis ▪ Hypoxia and hypercapnia due to respiratory insufficiency 198 ▪ Pure spinal polio: tightness, weakness, or paralysis of respiratory muscles (cervical, thoracic) ▪ Pure bulbar polio: paralysis of motor CN nuclei ± vital centers (9 th, 10th, 12th → airway obstruction ▪ Virus is isolated from water sources, sewage, wet soil, contaminants from drinking water, swimming pools, ponds, hospital water reservoirs INCUBATION PERIOD: 3-6 days ▪ Acute Hemorrhagic Conjunctivitis (AHC) 1-3 days NONPOLIO ENTEROVIRUSES TRANSMISSION ▪ Fecal – oral ▪ Respiratory ▪ Airborne & eye-hand-fomite-eye transmission (AHC) ▪ Mother to neonate (vertical perinatally, peripartum, breastfeeding, intrafamilial, sporadic/epidemic transmission from nurseries) ▪ Fomites, tickborne Risk: ▪ Water contamination, diaper change ▪ Symptomatic/asymptomatic shed virus from: ● Respiratory tract <1-3wks ● Feces 7-11wks ● Mucosal sites for longer periods PATHOGENESIS ▪ Initial replication in pharynx/ intestine (w/in mucosal M cells) → multiplication in lymphoid tissue (tonsils, Peyer patches, regional LN) → minor viremia (liver, spleen, BM, distant LN) → replication is limited by host response (subclinical infection) OR proceeds → major viremia (CNS, heart, skin) ▪ Tropism to target organs is determined in part by the infecting serotype ▪ At risk for severe/chronic infection: ● Hypo/ agammaglobulinemia ● Perinatally infected neonates lacking maternal type-specific Ab to infecting virus PPS Oral Exam Reviewer 2020 Hand-Foot-and-Mouth Disease (Coxsackie A, B, Echo) ▪ Mild illness ± low-grade fever ▪ Inflamed oropharynx with scattered painful vesicles → ulcerate, 4-8 shallow lesions with surrounding erythema ▪ 3-7 mm tender maculopapular, vesicular, and/or pustular lesions on hands>feet, buttocks and groin that resolve within a week ▪ Eczema coxsackium: disseminated vesicular rashes which may complicate preexisting eczema ▪ Coxsackie A6: severe, atypical HFMD and herpangina – fever, gen rash, pain, dehydration, desquamation of palms and soles, onychomadesis (nail shedding) Herpangina (Enterovirus, Coxsackie A) ▪ Sudden fever (up to 41OC, higher in younger age, lasts 1-4 days) ▪ Sore throat, dysphagia, painful lesions in the PP, HA, backache, vomiting, abdominal pain, vesicles, and ulcers on the mouth → enlarge 2-3 days, 1-15 lesions → resolution of symptoms in 3-7d Respiratory ▪ Sore throat, coryza, wheezing, apnea, resp distress, pneumonia, OM, bronchiolitis, croup, parotitis, exudative tonsillopharyngitis ▪ Pleurodynia/Bornholm disease (coxsackie B and echovirus) – paroxysmal thoracic pain due to myositis of chest and abdominal wall muscles, malaise, myalgia, HA, sudden fever. The pain subsides w/in 3-6 days but can persist in weeks. May be assoc. with meningitis, orchitis, myocarditis or pericarditis. Ocular ▪ AHC (enterovirus D70 & coxsackie A24/A24 variant) - sudden severe eye pain, photophobia, blurred vision, lacrimation, conjunctival erythema and congestion, lid edema, preauricular LAD, ▪ Supportive ▪ Milrinone: may be used in severe enterovirus A71 cardiopulmonary disease ▪ Ig: IV and intraventricular infusion to treat hypogammaglobulinemic patients w/ chronic enterovirus meningoencephalitis ▪ IV Ig + corticosteroids: enterovirus (A71, D68) neurologic disease ▪ IF α and IF β: enterovirus myocarditis COMPLICATIONS ▪ HFMD: encephalitis, AFP, myocarditis, pericarditis, shock ▪ Herpangina: dehydration, meningitis ▪ Respiratory infection: Enterovirus A71 brainstem encephalitis → noncardiogenic pulmonary edema, hemorrhage and/or interstitial pneumonitis ▪ Ocular infection: uveitis → destruction of iris, cataracts, glaucoma ▪ Myopericarditis: sudden death (SIDS) ▪ Neurologic infection: seizures, obtundation, ICP, SIADH, ventriculitis, transient cerebral arteriopathy and coma ▪ Neonatal infections ● Endothelitis, veno-occlussive disease, placentitis ● CNS necrosis and gen or focal neurologic compromise, coagulopathy w/ intracranial or other bleeding; DIC ● Arrhythmias, CHF, MI, pericarditis, ● Hepatic necrosis, failure ● Adrenal necrosis and hemorrhage ● Rapidly progressing pneumonitis and pulmonary HTN ● Involvement of pancreas ● Rare: myositis, arthritis, NE, SIADH, HLH-like presentation, BM failure, sudden death ● Fetal demise, nonimmune hydrops PROGNOSIS ▪ Majority: excellent ▪ Morbidity and mortality assoc. w/ myocarditis, neurologic disease, severe neonatal infections, infections in IC hosts PREVENTION ▪ Good hygiene ▪ Chlorination of water ▪ Contact precaution in the hospital ▪ Droplet precaution if with respiratory syndromes ▪ Prophylactic Ig or convalescent plasma for nursery epidemic ▪ Pregnant (near term): avoid contact with ill individuals; if w/ suggestive illness, extend pregnancy to passively acquire protective Ab ▪ Maintenance of high dose IVIg hypogammaglobulinemic Batch Clingy ▪ e.g. Coxsackie virus & Echovirus/Parechovirus ▪ Humans only known natural reservoir ▪ Increased severity: young age, male, exp. to children, poor hygiene, overcrowding, low socioeconomic status DIAGNOSTICS ▪ in suspected cases of acute flaccid paralysis, 2 stool specimens should be collected 24-48 hrs apart ASAP ● Poliovirus concentrations are high in the stool in the 1 st wk after onset of paralysis ● Rectal swab may be done in children w/ constipation 2O to paralysis ▪ Once poliovirus is isolated, it is sent to CDC or WHO to distinguish b/w wild-type & vaccine-type strains Nonspecific febrile illness ▪ most common symptomatic manifestation ▪ Duration: 4-7 days (range: 1 day to >1wk) ▪ Abrupt fever (38.5-40OC) – lasts a mean of 3 days, occ. biphasic ▪ Malaise, irritability, lethargy, anorexia, diarrhea, N/V, abdominal discomfort, rash, sore throat, respiratory symptoms, mild conjunctivitis, pharyngeal injection, CLAD, meningitis ▪ Older children: HA and myalgia 199 ● Young age; immunosuppression ● Specific HLA Ag genes ● Immune response gene polymorphisms, ↓ vit A subconjunctival hemorrhages (hallmark of eD70), superficial punctate keratitis, eye discharge (serous → mucopurulent), fever, HA. Recovery complete w/n 1-2 wk. ▪ Uveitis, chorioretinitis, uveoretinitis, optic neuritis, unilateral acute idiopathic maculopathy patients Myocarditis and Pericarditis (25-35% enterovirus) ▪ CXR: cardiac enlargement ▪ 2DED: ventricular dilation, ↓ contractility, pericardial effusion ▪ ECG: ST segment, T wave & rhythm abnormality, serum myocardial enzymes GIT and GU ▪ Diarrhea, hematochezia, pneumatosis intestinalis, NEC in prematures ▪ Acute/chronic gastritis, intussusception, chronic intestinal inflammation in hypogammaglobulinemic patients, sporadic hepatitis (severe in neonates), pancreatitis (→ transient exocrine pancreatic insufficiency), nephritis, IgA nephropathy ▪ Coxsackie B is second only to mumps as a cause of orchitis Neurologic (coxsackie B, echo, entero D70 & A71) ▪ Enteroviruses are the most common cause of meningitis in mumps immunized population ● Common in < 3mos old ● Most are mild and lack meningeal signs, but nuchal rigidity is apparent in >1-2yo. Fever resolves in 3-5d and other symptoms within a week ▪ Enteroviruses are also responsible for ≥ 10-20% of encephalitis ● Nonspecific symptoms → encephalopathy ● Seizures, hemichorea, acute cerebellar ataxia, aphasia, extrapyramidal symptoms ▪ Chronic Enterovirus Meningoencephalitis: high risk in pts with Ab or combined immunodeficiencies & pts who are receiving anti-CD20 Ab therapy; presents as insidious intellectual and/or personality deterioration, altered mental status, seizures, motor weakness, ICP. ▪ Enteroviral infections may also present as acute flaccid myelitis Infections in Transplant Recipients and Patients with Malignancies ▪ Progressive pneumonia, severe diarrhea, pericarditis, heart failure, meningoencephalitis, disseminated disease PPS Oral Exam Reviewer 2020 Batch Clingy Myositis and Arthritis Neonatal Infections ▪ Majority are asymptomatic ▪ Fever, hypothermia, irritability, lethargy, anorexia, rash (MP), jaundice, respiratory symptoms, apnea, hepatomegaly, abdominal distention, emesis, diarrhea, decreased perfusion ▪ Benign course with fever resolution in 3d and other symptoms in 1 week. ▪ Some have severe disease dominated by a combination of sepsis, meningoencephalitis, mypcarditis, hepatitis, coagulopathy and/or pneumonitis 200 ▪ Bronchiolitis is a clinical diagnosis ▪ rhinorrhea: 1st sign which persists throughout the illness → 1-3 days ff by cough w/ sneezing and intermittent low-grade fever (fever is an inconsistent sign of RSV infection) ▪ PE: diffuse fine inspiratory crackles and expiratory wheezes ▪ progression of illness → air hunger, tachypnea, IC and SC retractions, hyper expansion of chest, restlessness, peripheral cyanosis ● signs of severe, life-threatening illness: central cyanosis, RR>70 bpm, listlessness, apneic spells, significantly hyper expanded chest, silent auscultation (poor air movement) ▪ Presence of ileus or other abdominal signs, high temperature and circulatory collapse → consider bacterial etiology; initiate antibiotic treatment INCUBATION PERIOD: 3-5 days DIAGNOSTICS ▪ CXR: often normal in the initial phase; 70% of hospitalized infants show hyperexpansion of the chest, peribronchial thickening, interstitial infiltrates; consolidation (unusual); pleural effusion (rare) ● Lobar consolidation without other signs or w/ PE should be considered of bacterial etiology until proved otherwise ▪ WBC normal or w/ neutrophilic or mononuclear predominance ● Neutrophilia, neutropenia in the presence of severe disease: consider bacterial etiology ▪ Hypoxemia via pulse oximeter or ABG ▪ Definitive diagnosis: viral culture, PCR or antigen testing TRANSMISSION: ▪ Droplet or through fomites inoculated in the nasopharynx PATHOGENESIS Bronchiolitis ▪ airway narrowing caused by virus-induced necrosis of bronchiolar epithelium, hypersecretion of mucus, round cell infiltration and edema of surrounding submucosa → mucus plug formation obstructing bronchioles → hyperinflation or collapse of distal lung tissue during expiration Pneumonia ▪ generalized infiltration, epithelial shedding extends to bronchi & alveoli ▪ smooth muscle hyperreactivity contributes to airway narrowing in older subjects but less typical in young infants ▪ largest of the human herpesviruses ▪ exists as genetically diverse forms w/in an individual → virus persists for a lifetime while inducing chronic immune activation INCUBATION PERIOD: highly variable CYTOMEGALO VIRUS TRANSMISSION Community ▪ horizontal (direct person to person) by intermittent shedding from mucosal surfaces (saliva, genital secretions, urine) ● CMV is considered a sexually transmitted infection; presumed cause for spike in infection for adolescents and young adults ▪ vertical (perinatal): genital secretions upon birth and breastfeeding (most common route in early childhood; 60% rate of infection) ● Infants infected through breast milk excrete virus in the saliva and urine for prolonged periods thus serve as CMV reservoir for spread to others Nosocomial PPS Oral Exam Reviewer 2020 ▪ Most common: asymptomatic Normal Host ▪ mononucleosis-like syndrome, fatigue, occ. CLAD, mild increase transaminases, thrombocytopenia Immunocompromised ▪ depends on the magnitude of the immunodeficiency ● profoundly immunocompromised hosts such as HSCT recipients can present with disseminated infection ●in less immunocompromised patients (i.e. solid organ transplant recipients), CMV infection can present with fever, hematological abnormalities and mild hepatocellular dysfunction ▪ clinical disease develops 30-60 days post-transplantation (prior to widespread antiviral prophylaxis use) Congenital Infection ▪ 90% of infected infants will have no clinical manifestations in the NB period; 10% will be symptomatic ● IUGR, hepatosplenomegaly, petechial rashes, jaundice, microcephaly, chorioretinitis ▪ Symptomatic treatment for uncomplicated bronchiolitis ▪ IV or tube feeding when suckling is difficult due to tachypnea ▪ Humidified O2 and suctioning for hypoxic infants ● high-flow nasal cannula (mostly useful for pressure support) ● nCPAP for infants w/ WOB; mechanical ventilation if with respiratory failure ▪ Heliox (helium + O2) for infants with severe respiratory distress but do not require large amount of O2 ▪ Limited use of α and β2 agonist and corticosteroids (2014 AAP Bronchiolitis Clinical Practice Guideline) ● Corticosteroid is indicated only in older children with established diagnosis of asthma (its use is assoc. with prolonged virus shedding and is of no proven clinical benefit) ▪ Ribavirin has no clear beneficial effect; no longer used for routine therapy of RSV disease ▪ Recurrent wheezing in 30-50% of children with severe RSV bronchiolitis in infancy ● Likelihood of the recurrence is increased in the presence of an allergic diathesis PROGNOSIS: ▪ Almost all deaths occur among young, premature infants or infants with underlying disease PREVENTION ▪ Contact precaution isolation to prevent nosocomial spread ▪ transplacentally acquired anti-RSV maternal IgG serum Ab appear to provide partial protection for the neonate during 1st 46 weeks of life ● Breastfeeding also provide protection on severe RSV but seen in female infants only PASSIVE IMMUNOPROPHYLAXIS ▪ Palivizumab (15 mg/kg IM monthly): neutralizing humanized murine monoclonal Ab is recommended to protect high-risk children against serious complications such as: (1) infants born < 29 wk in the 1 st year of life (2) born < 32 wk w/ hx of chronic lung disease of prematurity in the 1st yr of life (3) < 1 yo w/ significant CHD ff. cardiac transplantation (children < 2 yr old) (4) < 2 yr old w/ profound IC conditions during RSV season (5) 1 yo w/ congenital abn in airway or NM disease that compromises the handling of respiratory secretions (6) Administration in the 2nd yr of life for children who required ≥ 28 days O2 support after birth and ongoing tx for chronic pulmonary disease Immunocompromised ▪ treatment has been shown to limit both the morbidity and the mortality ▪ drugs: ganciclovir, foscarnet, cidofovir ▪ heart-lung and lung transplant recipients are at a high risk for severe manifestations ▪ most common long term sequelae in congenital infection: SNHL Congenital ▪ ganciclovir (could limit hearing loss and possibly improve developmental outcome) ▪ monitor development, NM function, audiologic, and ophthalmologic system PREVENTION Immunoprophylaxis ▪ Passive: passive transfer of anti-CMV Abs have been utilized to limit disease but not infection in allograft recipients ▪ Active: vaccine trials show that protection was very short lived, effectiveness not convincing Perinatal ▪ severe infection ff. breast milk ingestion w/ end-organ disease has been successfully treated w/ ganciclovir ▪ children who are shedding do not require exclusion or isolation; apply standard precaution and hand hygiene ▪ Prevention of transmission in blood transfusion: freezing RC in glycerol before administration, remove buffy coat or filtration to remove WBC ▪ Prevention in human milk: pasteurization will inactivate CMV, but freezing milk will only decrease viral titers Batch Clingy RESPIRATORY SYNCITIAL VIRUS ▪ Major cause of bronchiolitis and viral pneumonia in < 1 yo ▪ peak incidence of severe LRTI: 6 wks to 7 mos. ▪ infection is nearly universal by 2 yo ● reinfection occurs at least 10-20% per epidemic throughout childhood ● reinfection may occur as early as a few weeks after recovery ● severity of illness during reinfection is usually lower than in first infection (partial acquired immunity, more robust airway physiology, increased age) ▪ Medical factors associated with higher risk: ● Chronic lung disease of prematurity ● CHD ● Immunodeficiency ● Prematurity 201 ▪ blood products, allograft transplantation Congenital ▪ in-utero ▪ CMV is thought to be transferred to the developing fetus following hematogenous spread to the placenta (see Nelson’s 21st Ed., Table 282.1 for Findings in Infants with Symptomatic Congenital Cytomegalovirus Infection) ▪ Prevention in transplant recipients: prophylactic antiviral therapy ▪ Lab findings consistent with congenital infection: direct hyperbilirubinemia, hepatic transaminases, thrombocytopenia, abn. cUTZ/CT findings (periventricular calcifications) Perinatal Infection ▪ not been assoc. w/ any clinical manifestations or assoc. w/ long term sequelae ▪ in rare cases, breast milk transmission of CMV to extremely premature infants or infants born to nonimmune women can result in severe, disseminated infections assoc. w/ end-organ disease and death DIAGNOSTICS ▪ Serologic reactivity for CMV is lifelong ff. primary infection ● presence of IgG Ab to CMV does not provide evidence of acute infection ● IgM reactivity for CMV can be detected for prolonged periods after acute infection; cannot be used reliably to estimate duration of infection ▪ Recovery of virus from body fluids does not permit diagnosis of CMV infection (d/t persistent intermittent shedding) Congenital Infection ▪ Diagnosis of congenital CMV infections require recovery of replicating virus and/or viral nucleic acids w/in first 2-3 wks of life ● NBS using saliva ff. by confirmation of positive screens using urine assay INCUBATION PERIOD: 2 days – 2 weeks PPS Oral Exam Reviewer 2020 ▪ Herpes gingivostomatitis ● most often affect 6 mo-5 yr ● sudden extreme pain in the mouth, drooling, refusal to eat or drink, fever (up to 40.6OC), swollen gums, vesicles throughout the oral cavity, ulcers – may be covered w/ yellowish-gray membrane, tender submandibular, submaxillary, CLAD, foul breath, in the initial phase there may be tonsillar exudates suggestive of bacterial pharyngitis ● in older patients: tonsillitis, pharyngitis rather than herpes gingivostomatitis ● untreated, symptoms resolve in 7-14 days; LAD persists for several weeks ▪ Herpes Labialis ● fever blisters/cold sores: most common in recurrent HSV1 DIAGNOSTICS ▪ Culture: highest yield from ruptured herpetic vesicle and vigorously rubbing base of lesion to collect fluid and cells ▪ PCR: test of choice in CSF for suspected HSV encephalitis ▪ Serology: ● HSV IgM: unreliable ● HSV IgG: ≥ 4-fold rise between acute & convalescent sample is useful in retrospect ▪ Neonate: ● culture of suspicious lesions; eye and mouth swabs PCR of both CSF & blood ● elevation of liver enzymes as indirect evidence of dissemination ▪ Acute Genital Herpes: ● culture or Ag detection ● HSV-2 type specific Ab testing for sexually experienced adolescents or young adults with unexplained recurrent nonspecific urogenital s/sx ▪ Tzanck test should not be performed due to low sensitivity PROGNOSIS: ▪ most: self-limiting except in IC and newborns ▪ recurrent oral-facial herpes s/p dermobrasion or laser resurfacing can be severe and lead to scarring ▪ life-threatening: neonatal herpes, herpes encephalitis, HSV in IC and burn patients ▪ Recurrent ocular infections → blindness PREVENTION ▪ good hygienic practices, contact precaution ▪ Decreases risk: male circumcision, daily oral A, V, or F during the last 4 wk of gestation for pregnant w/ a hx of genital herpes, delivery by CS for pregnant women with active genital herpes at the time of delivery ▪ There is NO concern of transmission through milk: (+) active lesions on the breasts can feed expressed milk, but no part of pump should come in contact w/ lesions during expression Batch Clingy HERPES SIMPLEX VIRUS ▪ 2 closely related HSVs (HSV-1 & HSV-2); contains the ff. ● 12 glycoproteins: target for humoral immunity ● other nonstructural proteins: target for cellular immunity ● 2 proteins (viral DNA polymerase and thymidine kinase): target of antiviral drugs ▪ HSV 1 and 2 differ in glycoprotein G genes ● used in serologic tests to discriminate whether a patient has been infected with HSV-1, HSV-2 or both ▪ Humans are the only natural host ▪ Both viruses are equally capable of causing initial infection at any anatomic site but differ in their capacity to cause recurrent infections ● HSV-1 → recurrent oral infections ●HSV-2 → recurrent genital infections ▪ HSV is the leading cause of sporadic, fatal encephalitis in children and adults Noncongenital Infections ▪ Demonstration of CMV-specific IgG seroconversion or the presence of CMV-specific IgM Abs represent evidence of a newly acquired CMV infection ▪ Hallmarks: skin vesicles and shallow ulcers (small, 2-4 mm vesicles surrounded by erythematous base → shallow minimally erythematous ulcers) 202 Perinatal: ▪ in utero, during delivery, or neonatal period ▪ during delivery, it can enter thru conjunctiva, mucosal epithelium of nose and mouth, breaks in skin w/ scalp electrode or forceps use ▪ documented also via CS PATHOGENESIS ▪ Primary infection: HSV seronegative and have no preexisting immunity that tends to be severe ▪ Nonprimary first infection: prev. infected HSV-1 who have become infected for the 1st time with other type of HSV (less severe due to cross-protection) ▪ Recurrent infection: reactivated latent infection in regional sensory ganglion neurons (less severe and shorter duration) ▪ viral infection begins at a cutaneous portal of entry → replicates in skin and mucous membranes → spreads to neural tissue (sensory ganglia) ▪ virus replicates in some sensory neurons → progeny virions sent back to periphery → replicate further in skin or mucosal surfaces ● movement of virus through this neural arc is responsible for the development of characteristic herpetic lesions ▪some infected neurons do not support viral replication → latent infection w/ undefined changes that trigger virus to replicate ▪ Viremia/hematogenous spread: occur in neonates, individuals with eczema and severely malnourished children ● most common site: vermillion border of lip, it may also occur on nose, chin, cheek, oral mucosa ● burning, tingling, itching or pain 3-6 hr (rarely up to 2 days) before development of herpes lesion (small group of erythematous papules → vesicle → ulcers or pustules) ● lesions completely heal in 6-10 days ▪ Cutaneous Infections ● contact sports: wrestling (herpes gladiatorum), rugby (scrumpox) ● skin trauma w/ exposure to infectious secretions → herpes labialis-like lesions that completely heal 6-10 days ● regional LAD may occur; systemic symptoms are uncommon ● recurrences are assoc. w/ local edema or local neuralgia ▪ Herpes whitlow ● HSV of paronychia (fingers or toes) in infants and toddlers due to thumb sucking, adolescents due to genital secretions ● (+) LAD, lymphangitis, neuralgia ● Onset is 2-7 days after exposure (itching, pain & erythema), persists x 10 days, complete recovery x 18-20 days ● life-threatening in patients with eczema (eczema herpeticum), pemphigus, burns, Darier disease, and ff. laser skin resurfacing ▪ Genital herpes ● genital-genital (HSV-2) or oral-genital (HSV-1) ● local burning and tenderness → vesicles on genital mucosal surfaces, keratinized skin around anus, buttocks, & thighs ● urethritis, dysuria ● women may experience watery vaginal discharge; males: clear mucoid urethral discharge ● Systemic s/sx are common: fever, headache, myalgia → aseptic meningitis (15%) ● complete healing: 2-3 weeks, may recur (sometimes asymptomatic) ● For young children: consider abuse or autoinoculation ▪Ocular Infections ● kerato/conjunctivitis: usually unilateral, assoc. w/ blepharitis & tender preauricular LAD ● vesicles may be seen on the lid margins and periorbital skin ● untreated infection generally resolves in 2-3 wks ● corneal involvement is rare; retinal infections are rare, more likely in neonatal herpes & IC w/ disseminated HSV ▪ HSV Encephalitis ● Acute necrotizing infection affecting frontal/temporal cortex and limbic system. ● The cause beyond neonatal period is almost always HSV-1. ● Fever, headache, nuchal rigidity, nausea, vomiting, gen seizures, alteration of consciousness ● anosmia, memory loss, peculiar behavior, expressive aphasia, change in speech, hallucinations, focal seizure ● untreated: 75% → coma and death PPS Oral Exam Reviewer 2020 TREATMENT ▪ Acyclovir (A): poorest bioavailability, more frequent dosing ▪ Better oral bioavailability: Valaciclovir (V) prodrug of acyclovir and Famciclovir (F) prodrug of penciclovir Acute Oropharyngeal infections ▪ Gingivostomatitis: A (start within 72 hours) ▪ Herpes labialis: oral > topical; recurrence lesions in adolescents: oral A, V & F shortens duration; frequent or severe recurrences: long term use of A or V Cutaneous infections ▪ Keratitis: topical trifluridine and ganciclovir ▪ Herpes gladiatorum: oral A or V at 1st signs of outbreak can shorten the course of recurrence; if w/ hx of recurrence: chronic daily prophylaxis w/ V ▪ Herpetic whitlow: no clinical trials assessing the benefit of antiviral, high dose A started 1st signs of illness abort some recurrences and reduce duration of others in adults. ▪ Eczema herpeticum: clinical trial A (effective in adults) ▪ Oral-facial HSV: V & F in adults for prevention since HSV can reactivate after cosmetic facial laser resurfacing → extensive disease and scarring, ▪ HSV infections in burn pts: Intravenous A ▪ Daily prophylactic use of antiviral can prevent reoccurences of erythema multiforme for herpes labialis Genital Herpes ▪ A, V, or F for initial infection: reduce severity and duration but has no effect in frequency of subsequent recurrent infections ▪ Recurrences: 3 options (no treatment, episodic or long-term suppressive therapy) Ocular Infections: seek consult with an ophthalmologist CNS infections: older than neonate: IV A (10 mk Q8 over an hour for 14-21 days) Immunocompromised ▪ Intravenous A: Severe or disseminated: ▪ oral A, V or F: Less severe or suppression of recurrences during episodes of significant immunosuppression ▪ Resistance to A and Penciclovir → Foscarnet & Cidofovir Perinatal Infections ▪ proven or suspected: high-dose IV A (60mkday q8 x 14 days for SEM and 21 days for CNS or disseminated) ▪ suppressive oral A x 6 mo after completion of IV therapy improves the neurodevelopment of infants with CNS infection and prevent cutaneous recurrences, measure absolute neutrophil count at week 2, week 4 and monthly ▪ Regardless of disease classification, pxs should have an ophthalmologic exam and neuroimaging to establish baseline brain Batch Clingy TRANSMISSION: ▪ direct contact between mucocutaneous surfaces ● HSV 1: oral secretions ● HSV 2: anogenital contact ▪ all infected harbor latent infection & experience recurrent infections w/c may be symptomatic or unrecognized and thus periodically contagious 203 ▪ HIV-1 and HIV-2 are members of the Retroviridae family ▪ HIV-1 genome: ● contains 2 copies of ssRNA ● has long terminal repeats (for regulation and expression of HIV genes) ● other major regions (encodes viral part): GAG (core proteins), POL (enzymes), ENV (envelope proteins) ▪ HIV-2 genome: ● Subtypes A & B: major causes of infection in humans (rare in child) ● most prevalent in West Africa but with increasing number of cases reported in Europe and South Asia HUMAN IMMUNODEFICIENCY VIRUS ▪ Difference of HIV-2 from HIV-1: ● accessory genes: vpx gene compared to vpu gene in HIV-1 ● more difficult to dx due to major difference in genetic sequences ● longer asymptomatic stage ● slower declines in CD4+, ● less transmission perinatally ▪ HIV tropism determined by envelope glycoprotein (Env) and chemokines as co-receptors: ● gp120: contains the highly variable V3 loop which is immunodominant for neutralizing Ab; carries binding site for CD4 ● gp41: immunogenic; used to detect HIV-1 Ab in diagnostic assays ● CXCR-4: co-receptor for attachment to lymphocyte ● CCR-5: facilitates entry to the macrophages EPIDEMIOLOGY ▪ majority of HIV infections in childhood: vertical transmission ▪ 13-24 yr old constitute the growing population of newly infected (81% MSM) HIV and AIDS Estimates (UNAIDS Country Fact Sheet: Philippines, 2019) ▪ Adults and children living with HIV: 97,000 (81,000-110,000) ▪ Adults and children newly infected with HIV: 16,000 (13,00018,000) ▪ HIV incidence per 1000 population (all ages): 0.14 (0.12-0.17) PPS Oral Exam Reviewer 2020 ▪ Perinatal Infections ● INTRAUTERINE INFECTION: skin vesicles or scarring, chorioretinitis, keratoconjunctivitis, microcephaly or hydranencephaly ● POSTPARTUM INFECTION: either (1) disease localized to the skin, eyes or mouth, (2) encephalitis with or without skin, eye and mouth involvement, (3) disseminated infection ▪ At birth: PE is normal ▪ Initial s/sx: LAD, hepatosplenomegaly, FTT, chronic or recurrent diarrhea, respiratory symptoms, oral thrush ▪ S/sx commonly seen in children: recurrent bacterial infection, early onset of progressive neurologic deterioration, chronic parotid swelling & lymphocytic interstitial pneumonitis ▪ CDC Surveillance Case Definition for HIV infection is based on the age-specific CD4+ T-lymphocyte percentage of total lymphocytes ● EXCEPT when a stage 3-defining opportunistic illness supersedes the CD4 data (see Nelson’s 21st Ed., Table 302.2 for Stage 3-Defining Opportunistic Illnesses in HIV Infection) Infection ▪ 20% of AIDS-defining illnesses are recurrent bacterial infections caused by encapsulated organisms (S. pneumoniae, Salmonella) & most common serious infection: bacteremia, sepsis, pneumonia ▪ Opportunistic: usually a primary infection with a more fulminant course ● Pneumocystis jiroveci pneumonia presents as ± fever, RR, dyspnea, hypoxemia w/ CXR: int. infiltrates or diffuse alveolar disease ▪ Nontuberculous mycobacteria (NTM): most commonly caused by Mycobacterium avium-intracellulare complex (MAC) ● disseminated MAC: fever, malaise, weight loss, night sweats, diarrhea, abd. pain, rarely intestinal perforation or jaundice, focal (lymphadenitis, osteomyelitis, tenosynovitis, and pulmonary disease) ▪ Fungal: oral candidiasis as the most common that can progress to esophagus char. by anorexia, dysphagia, vomiting, & fever ▪ Parasitic: intestinal cryptosporidiosis and microsporidiosis pres. as chronic diarrhea → malnutrition anatomy (MRI: most sensitive) DIAGNOSTICS ▪ HIV Ab tests cannot be used to make a diagnosis of HIV infection in infants younger than 24 mos. ● viral diagnostic assays (i.e. HIV DNA or RNA PCR) are more useful in young infants, allowing definitive dx in most infected infants by 1-4 mos. ▪ In any child older than 24 mos., demonstration of IgG Ab to HIV by a repeatedly reactive enzyme immunoassay and confirmatory HIV PCR establishes the diagnosis ▪ Breastfed infants should have Ab testing performed 12 wk ff. cessation of BF to identify those who became infected at the end of lactation by the HIV-infected mother ▪ Testing should be done within the first 12-24 HOL for high-risk infants ● If negative at first 1-2 DOL, rpt. at 2-3 wks. of age, 4-8 wks. of age and 4-6 mos. ● for higher risk infants, additional virologic testing should be considered at 2-4 wks after cessation of ARV prophylaxis ▪ positive virologic assay should be confirmed by a rpt test on a 2nd specimen ASAP (exclude false positive) ▪ Confirmed diagnosis: 2 positive virologic test results obtained from different blood samples ▪ Presumptive exclusion (non-BF infants): 2 or more negative virologic test or 1 negative virologic test at ≥ 8 wks or 1 negative HIV Ab test at age ≥ 6 mos. ▪ Definitive exclusion (non-BF infants): 2 or more negative virologic tests with one obtained at ≥1 mo and one at age ≥ 4 mos or 2 negative HIV Ab tests from separate specimens obtained ate age ≥ 6 mos. TREATMENT ▪ to suppress the virus for extended periods of time and changes the course to a chronic process ▪ Basis of combination antiretroviral therapy (cART) ● uninterrupted HIV replication progresses to AIDS ● magnitude of viral load predicts rate of disease progression ● at least 3 drugs with at least 2 diff. MOA: restricts the selection of ART-resistant mutants ● sustainable suppression of HIV replication is best achieved by COMPLICATIONS ▪ For IRIS: continuation of ART ± NSAIDS or corticosteroids ▪ Virologic failure: repeated plasma viral load ≥ 200 copies/mL after 6 mo of therapy PROGNOSIS ▪ Best prognosis: sustained suppression of plasma viral load and restoration of normal CD4 count ▪ Worst prognosis: opportunistic infections, encephalopathy, regressing development milestones, wasting syndrome ▪ Poor outcome: Persistent fever, oral thrush, serious bacterial infection, hepatitis, persistent anemia, platelet PREVENTION ▪ All pregnant women should be tested for HIV and if (+), treat w/ cART irrespective of viral load or CD4 count ▪ All infants born to HIV-infected mother should receive zidovudine prophylaxis for 4-6 weeks ▪ HIV-infected mothers living in resource-limited settings should breastfeed their infants until at least 12 mos. of age, with exclusive BF for the first 6 mos and cART should continue ▪ All HIV-exposed and HIV-infected children should receive standard pediatric immunization ● Live OPV & BCG should NOT be given ▪ Do TST or IGRA once a year (>5 mm positive) ▪ All infants b/w 4-6 wks and 1 y/o who are proven to be HIVinfected should receive prophylaxis to prevent P. jiroveci pneumonia regardless of the CD4 count or percentage ▪ Prophylaxis: ● P.jiroveci: TMP SMX or dapsone/ atovaquone/ aerosolized pentamidine ● MAC: azithromycin, clarithromycin, rifabutin ▪ To reduce incidence of opportunistic infections: good handwashing, avoid raw or undercooked food, drinking, swimming in lake or river water or being in contact with young farm animals, risk of playing w/ pets ▪ For sexually active: condoms, male circumcision, 1% vaginal gel formulation of tenofovir, for MSM: once daily dosing of coformulated tenofovir and emtricitabine Batch Clingy ▪Immunocompromised ● The most common: mucositis & esophagitis, atypical lesions slowly enlarge, ulcerate → necrotic ● tracheobronchitis, pneumonitis, anogenital infection ● disseminated infection: sepsis-like, liver and adrenal involvement, DIC and shock 204 INCUBATION PERIOD AIDS: 8-12 yr TRANSMISSION: ▪ Sexual contact ▪ Parenteral exposure to blood: 3-6% of all pediatric AIDS cases ▪ Vertical transmission: 3 routes (1) Intrauterine: ● 20-30% detectable in culture or PCR as early as 10 wk AOG up to 1st wk of life ● most occurs in late gestation when the vascular integrity of the placenta weakens and microtransfusion across the maternalfetal circulation occurs (2) Intrapartum: ● higher percentage of HIV-infected children acquire the virus this way (70-80% of infected infants do not demonstrate detectable virus until after 1 st week of life) ● via mucosal exposure to infected blood and cervicovaginal secretions and intrauterine contractions causing late microtransfusions (3) Postpartum: ● accounts for 40% of perinatal infections in resource-limited countries ● risk of transmission through BF is increased by viremia during primary infection PATHOGENESIS ▪ Attach to dendritic cells, reach lymphatic tissue and binds to cells expressing CD4 ▪ within 3-6 weeks after infection: acute retroviral syndrome (acute HIV infection) - burst of plasma viremia that occurs when HIV replication reaches a threshold; presents like flu or mononucleosis (fever, rash, pharyngitis, LAD, malaise, arthralgia, fatigue, liver enzymes) ▪ within 2-4 months, clinical latency: ● cellular & humoral immune response established ● viral load declines, CD4 return to mod decreased levels ● asymptomatic ● very high turnover of virus and marked depletion of CD4 cells ● HIV virions and their immune complexes migrate through the LN→immune activation → promoting viral replication & depletion of CD4 cells For children, there are 3 distinct patterns: ▪ Rapid progression (15-24% of HIV-infected NB) ● onset 1st few months of life, ● median survival time 6-9 months ● most have detectable virus in the plasma in the 1st 48 HOL→ peak 2-3 mo of age→stay high at least 1st 2 years of life ● occurs if intrauterine infection coincides with the rapid expansion of CD4 cells in the fetus ▪ Slower progression (60-80%) PPS Oral Exam Reviewer 2020 ▪ Viral: HSV (recurrent gingivostomatitis), VZV, disseminated CMV, measles, molluscum contagiosum, RSV, adenovirus, HPV ▪ Immune reconstitution inflammatory syndrome (IRIS) ● increased inflammatory response from the recovered immune system to subclinical opportunistic infections ● may occur after appropriate ARV therapy; more commonly observed in pts. with progressive disease and severe CD4 + Tlymphocyte depletion ● fever + worsening CM of the opportunistic infection or new CM w/n 1st few weeks after initiation of ART CNS involvement ▪ subtle developmental delay to progressive encephalopathy, cognitive deterioration, impaired brain growth, attention problems, psychiatric disorders ▪ cerebellar atrophy, ventricular size, basal ganglia calcifications, leukomalacia ▪ focal signs and tumor may imply comorbidities: CNS tumor (lymphoma), opportunistic (toxoplasmosis, CMV, JC, HSV), stroke Respiratory ▪ recurrent OM and sinusitis → invasive sinusitis and mastoiditis ▪ Lymphocytic Interstitial Pneumonia (LIP): most common chronic process with nodular lymphoid hyperplasia leading to progressive alveolar capillary block; CXR: chronic diffuse reticulonodular pattern (lymphoproliferative response to EBV infection) ▪ Pneumonia secondary to S. pneumoniae, End-stage: P. aeruginosa & other gram negative, TB Cardiovascular - pre-cART era: LVH, LV dilation, LV fractional shortening, and/or HF; currently, cART has been shown to have a cardioprotective effect GI and Hepatobiliary ▪ Oral manifestations: candidiasis, periodontal disease, salivary gland disease, and rarely, ulceration or oral hairy leukoplakia ▪ GIT involvement is common and a variety of pathogens can cause GI disease; most severe & protracted: MAC and protozoa (Giardia, Crypto, Isospora, Microsporidia) ▪ Most common symptoms: chronic or recurrent diarrhea w/ malabsorption, abdominal pain, dysphagia, FTT (less common: wasting syndrome - loss of >10% BW w/ grave prognosis) ▪ AIDS enteropathy: syndrome of malabsorption with partial villous atrophy not assoc. w/ specific pathogen (direct HIV infection) ▪ Chronic liver inflammation ± cholestasis, cryptosporidial cholecsytitis combinations of ART that px has not been exposed prev. and not cross-resistant to drugs w/c the px. has been tx. previously ● drug interactions and toxicities should be minimal ● adherence to regimen is crucial Initiation of Therapy ▪ < 1 yo: as soon as HIV diagnosis is confirmed Age 1-6 yo > 6 yo Urgent Stage 3 opportunistic infection or Treatment CD4 <500 <200 Treatment strongly recommended Treatment recommended CD4 Moderate HIV symptoms or 500-999 200-499 Asymptomatic or w/ mild symptoms with CD4 >1000 >500 Mechanisms of Action ▪ preventing viral entrance into CD4+ T cells ● Fusion inhibitors either bind to viral gp41 causing conformational change (enfuvirtide) or blocks viral attachment to CCR5 (maraviroc) which is an essential process in the viral binding and fusion to the CD4+ cells ▪ inhibiting HIV reverse transcriptase or protease enzymes ● Nucleoside (or nucleotide) reverse transcriptase inhibitors (NRTI): act like chain terminators and block further incorporation of nucleosides preventing DNA synthesis ● Nonnucleoside reverse transcriptase inhibitors (NNRTI): attach to reverse transcriptase and cause a conformational change, reducing the activity of the enzyme ● Protease inhibitors (PI): bind to the site where the viral long polypeptides are cut into individual, mature, and functional core proteins that produce infectious virions before they leave the cell ▪ inhibiting integration of the virus into the human DNA ● Integrase inhibitors (INSTI): block the enzyme that catalyzes the incorporation of the viral genome into the host’s DNA Combination Therapy ▪ by targeting different points in the viral life cycle and stages of cell activation and by delivering drug to all tissue sites, maximal viral suppression is feasible: ● thymidine analog NRTI ● nonthymidine analog NRTI: to suppress replication in both active and resting cells ● ritonavir-boosted PI, an NNRTI, or an INSTI: to produce prolonged suppression of the virus Renal disease ▪ focal glomerulosclerosis, segmental necrotizing GN & minimal change ▪ Nephrotic syndrome: most common manifestation Skin Manifestations ▪ early nonspecific signs: severe & unresponsive seborrheic Batch Clingy ▪ Adult and child deaths due to AIDS: 1600 (1000-2400) 205 Immune changes in children: ▪ absolute CD4 cell depletion is less dramatic in infants (due to normal relative lymphocytosis) ▪ lymphopenia: rare but usually found in older children or with end-stage disease ▪ cutaneous anergy is common in HIV but also frequent in healthy children (< 1 year old) ▪ polyclonal activation occurs early (Ab response to vaccination may be abnormal) ▪ two distinct types: EBV-1 and EBV-2 ● EBV-1 more prevalent worldwide; EBV-2 more common in Africa ▪ both types lead to persistent, lifelong, latent infection INCUBATION PERIOD: 30-50 days in adolescents; shorter for children EPSTEIN-BARR VIRUS TRANSMISSION ▪ Oral secretions ● shed consistently for >6 mo after acute infection then intermittently for life ▪ Sexual contact ● penetrative sexual intercourse (esp type II), oral secretions (kissing) and sharing water bottles PATHOGENESIS ▪ after transmission, infects oral epithelial cells & tonsillar B lymphocytes → viral replication → viremia & dissemination of infected B lymphocytes into peripheral blood & lymphoreticular system (atypical CD8 T lymphocytes activation) ▪ Latent infection: ● persists in memory B lymphocytes ● EBV-determined nuclear Ag (EBNA) are produced – important in maintaining the viral episome ● reactivation is unlikely to be accompanied by distinctive clinical symptoms PPS Oral Exam Reviewer 2020 dermatitis ▪ recurrent or chronic episodes of HSV, herpes zoster, molluscum contagiosum, flat warts, anogenital warts, candidal infections, allergic drug eruptions (rare: SJS) ▪ Epidermal hyperkeratosis with dry scaling skin + sparse hair or hair loss Hematologic and Malignant Diseases ▪ Anemia (chronic infection, autoimmune factors, virus-associated: hemophagocytic syndrome, parvovirus B19 red cell aplasia or drug effect: zidovudine) ▪ Leukopenia and neutropenia (drug effect: TMP-SMX, ganciclovir, zidovudine) → treatment w/ gCSF may be necessary ▪ Thrombocytopenia (immunologic, drug toxicity or idiopathic) → cART may reverse thrombocytopenia in ART-naïve patients; IVIg in severe thrombocytopenia ▪ deficiency of F2, 7, 9 → Vitamin K ▪ most commonly reported neoplasms: NHL, primary CNS lymphoma, leiomyosarcoma (EBV associated); Kaposi sarcoma is uncommon in HIV-infected children in resource-rich countries ▪ Anterior mediastinal multilocular thymic cysts: novel disease of the thymus in a few HIV-infected children; cART may result in resolution or spon. Involution ▪ Infectious Mononucleosis (>90%) ● majority of primary EBV infection are clinically silent in infants and young children; in older patients, onset is insidious and vague ● classic triad of primary EBV infection: fatigue, pharyngitis, generalized LAD ● malaise, fatigue, acute or prolonged (> 1 wk) fever, HA, sore throat, nausea, abd. Pain, myalgia ● prodrome: 1-2 wks ● sore throat is often accompanied by moderate to severe pharyngitis with marked tonsillar enlargement, occ. with exudates (similar to Strep infection) ● classic PE findings: generalized LAD (90%; ant. & post. cervical, submandibular > axillary, inguinal & epitrochlear), splenomegaly (50%; 2-3 cm below costal margin), hepatomegaly (10%; liver enzymes, symptomatic hepatitis or jaundice is uncommon) ● rashes are maculopapular; reported in 3-15%; ampicillin rash: patients with IM who are tx. w/ ampicillin or amoxicillin, morbiliform, vasculitic rash, prob. immune-mediated, resolves w/o specific tx.) DIAGNOSTICS ▪ presumptive dx can be made by presence of classical clinical s/sx with atypical lymphocytosis ● majority have leukocytosis (10,000-20,000) w/ at least 2/3 lymphocytes (20-40% atypical) ▪ Serologic testing: for heterophile Ab or specific EBV antibodies (EBNA, EA and VCA systems) ● Heterophile Ab: cross reactive IgM Ab that agglutinate mammalian erythrocytes but are not EBV-specific (monospot test); (+) in 90% of EBV-assoc. IM in adolescent and adults; can remain positive up to 12 months ▪ No specific treatment ▪ Supportive: rest, adeq. fluid and nutrition intake, symptomatic treatment ▪ Antiviral is not recommended ● have not been shown to provide consistent clinical benefit ▪ Adoptive immunotherapy (infusion of EBV-specific cytotoxic T lymphocytes): for transplant recipients and for other patients with EBV-assoc. malignancies ▪ Splenic rupture: mild trauma or spontaneous ● avoid contact sports ▪ Airway obstruction: swelling of oropharyngeal lymphoid tissue ▪ Neurologic: ● Headache, uncommon: meningitis or encephalitis ● seizure, ataxia, Alice in Wonderland syndrome (metamorphopsia) ● some assoc. with MS ▪ Hematologic: ● mild hemolytic anemia, plt & neutrophils ● rare: aplastic anemia, severe low plt and neutrophils ▪ Other (rare): myocarditis, interstitial pneumonia, pancreatitis, parotitis & orchitis ▪ Oncogenesis ● HL, aggressive NK cell leukemia, T- & NK-cell lymphoproliferative disorder, epithelial cell malignancies (NPCA, gastric CA) ● Endemic Burkitt lymphoma: in areas endemic with P. falciparum infection and EBV ● Numerous congenital and acquired immunodeficiency syndromes are assoc. with an increased incidence of EBV-assoc. B-lymphocyte lymphoma, esp CNS lymphoma and leiomyosacroma PROGNOSIS: ▪ gradual recovery w/in 2 months of symptom onset ▪ prolonged and debilitating fatigue and malaise may wax and wane for several weeks to 6 months Batch Clingy ● median survival: 6 yrs ● (-) PCR in the 1st wk of life→viral load rapidly increases→peaks by 2-3 mos→slowly declines over a period of 24 mos. ● probably partially d/t immaturity of the immune system in NBs and infants ▪ Long-term survivors/ non-progressors: ● normal CD4 & viral loads >8 yr old ● effective humoral immunity and/or CTL responses, host genetic factors, and infection with an attenuated virus 206 ● EBV-specific Ab: if heterophile test result is (-) but EBV infection is suspected; measurement of Abs to EBV proteins (viral capsid Ag, EBV nuclear Ag, early Ag) Clinical Status VCA VCA EA EBNA IgM IgG IgG IgG Susceptible Acute Primary Infection Recent Primary Infection Past + + +/- - +/- + +/- +/- - + +/- + PREVENTION - No EBV vaccine yet VCA: Viral Capsid Ag, EBNA: EB Nuclear Ag, EA: early Ag INCUBATION PERIOD: 1-7 days (Nelsons); 3 -14 days (PIDSP) TRANSMISSION: ▪ bite of vector mosquito from viremic humans ▪ Vector: Aedes aegypti (daytime mosquito); Less commonly: Aedes albopictus DENGUE FEVER PATHOLOGY ▪ incompletely understood; DHF may be assoc. with second heterotypic infections with dengue types 1-4 or in infants born to mothers who have had two or more lifetime dengue infections ● circulation of infection-enhancing Abs at the time of infection is the strongest risk factor for development of severe disease ▪ dengue virus immune complexes attach to monocyte/macrophage Fc receptors → suppress innate immunity and enhance viral production ▪ early in the acute stage of secondary dengue infections, there is rapid activation of the complement system ● NS1: viral toxin that attaches to toll receptor 4 & activates myeloid cells, contributes to vascular permeability ▪ Mechanism of bleeding is not known but a mild degree of DIC, liver damage and thrombocytopenia may operate synergistically PHASES OF DENGUE ▪ Febrile: 2-7 days accompanied by gen. body ache, muscle, joint pains, headache, retroorbital pain, facial flushing, sore throat, hyperemic pharynx, macular or MP rash, petechiae and mild mucosal membrane bleeding ▪ Critical: During fever defervescence, D3-7 of illness, increase in capillary permeability in parallel w/ Hct levels - significant plasma leakage 24-48 hours ▪ Convalescent: Gradual improvement and stabilization of hemodynamic status PPS Oral Exam Reviewer 2020 ▪ Dengue with warning signs ● Lives in or travels to dengue-endemic area, with fever lasting for 2- 7 days PLUS ANY of the ff: ● abdominal pain or tenderness, persistent vomiting, clinical signs for fluid accumulation, mucosal bleeding, lethargy, restlessness, liver enlargement, AND laboratory test: increase in Hct and/ or decreasing platelet count. ● Confirmed diagnosis: Viral culture isolation, PCR ▪ Severe dengue ● Dengue (with/without warning signs) PLUS ANY of the following: ● severe plasma leakage, leading to: shock, fluid accumulation with respiratory distress ● severe bleeding ● severe organ impairment (liver: AST or ALT >1000, CNS: seizures, impaired consciousness, heart: myocarditis, kidneys: renal failure) ▪ Dengue Without Warning Signs: ● oral rehydration if not admitted; sports drinks should not be given ● isotonic solutions for patients who are admitted but without shock ▪ Dengue With Warning Signs/Severe Dengue: ● admit for close monitoring ● cyanotic/ labored breathing: supplemental O2 ● rapid IV replacement of fluids and electrolytes (if in compensated shock: 10-15 mL/kg/hr over 1 hr; if in hypotensive shock: 20 mL/kg in 15 mins) ● may give glucose containing IVF in the ff. conditions: HGT < 80 mg/dL; if HGT is not available and pt. is unable to eat and is weaklooking ● Transfusion to control bleeding (do not give if w/ hemoconcentration): FWB (10-20 mL/kg) or pRBC (5-10 cc/kg, if w/ fluid overload) are preferable because stored blood loses 2,3 DPG, low levels of w/c impede the O2-releasing capacity of Hgb resulting in functional tissue hypoxia ● Prophylactic platelet transfusion: do not reduce the risk of hemorrhaging or improve low platelet counts unless with significant bleeding or DIC ● Vasopressors, if necessary DISCHARGE CRITERIA ▪ No fever for at least 48-hours ▪ Improvement in clinical status ▪ Increasing trend in platelet count, stable hematocrit without IVF ▪ Hypervolemia during fluid reabsorptive phase is life threatening ( Hct & wide PP) ▪ Infants and young: ● fluid & electrolyte losses, hyper-pyrexia, febrile convulsion ● uncommon: epistaxis, petechiae, purpuric lesions ● after febrile phase: prolonged asthenia, mental depression, bradycardia, ventricular extrasystoles PROGNOSIS: ▪ Dengue Fever: Good ▪ DHF: 40-50% in shock → death but with ICU <1% PREVENTION ▪ avoid daytime mosquito using insecticides, repellents (use EPAapproved), body covering clothes, screening of house, destruction of vector breeding sites ● there is insufficient evidence to say that use of citronellabased repellents is more effective than DEET-based repellents in reducing dengue transmission (PPS PIDSP Clinical Practice Guidelines on Dengue in Children, 2017) ▪ Dengue vaccine (Dengvaxia) ● a mixture of 4 chimeras: DENV + yellow fever 17D ● 3-dose series given 6 months apart for 9-45yo in highly endemic areas ● in 2015: completed phase III w/c offered poor protection of seronegatives, good protection of seropositives w/ reduction of hospitalization and severe disease in vaccinated 9-year-old ● seronegative who were incompletely protected were sensitized to the enhanced disease of hospitalized dengue (Further reading : PPS PIDSP Clinical Practice Guidelines on Dengue in Children, 2017) DIAGNOSTICS Dengue Fever ▪ After 3-4 days → pancytopenia (neutropenia may persist), WBC <2000 µ/L, platelets rarely fall (<100 000) Batch Clingy ▪ caused by several arthropod-borne viruses ● four distinct antigenic types of dengue virus (dengue 1,2,3, and 4); members of the family Flaviviridae ● EBV DNA via PCR: Not necessary in immunocompetent patients; used for surveillance for posttransplant lymphoproliferative disease (PTLD), screen and determine prognosis for some EBV-ass malignancies (NPCA and HL) REVISED DOH/PPS CLASSIFICATION AND LEVELS OF SEVERITY 2011 ▪ Dengue without Warning Signs ● Lives in or travels to dengue-endemic area with fever PLUS ANY TWO of the ff: ● headache, body malaise, myalgia, arthralgia, retro-orbital pain, anorexia, nausea, vomiting, diarrhea, flushed skin, rash AND laboratory test, at least CBC (leucopenia with or without thrombocytopenia) and/or dengue NS1 Ag or dengue IgM Ab test (optional) ● Confirmed diagnosis: Viral culture isolation, PCR 207 ▪ (+) tourniquet test, mild acidosis, hemoconcentration, transaminase, protein ▪ ECG: sinus bradycardia, ectopic ventricular foci, flattened T waves, prolongation of P-R interval DHF/DSS ▪ 20% of Hct, plt, prolonged BT, mod. PT level (seldom <40% control), subnormal Fibrinogen, fibrin split-products, mod serum transaminase, consumption of C’, mild metabolic acidosis, Na, alb, CXR: pleural effusion (R>L) ▪ Signs of vascular permeability: thickening of GB wall, presence of perivascular fluid Batch Clingy ▪ Serology: ● NS1 (acute) requested D1-5 of illness, detectable from the beginning of febrile phase until D7-10 of illness ● IgM (recent): detectable 3-5 days after illness, disappears after 6-12 wk ● IgG (past infection), detectable 3-7 days ff infection and remain up to 6 months ▪ PCR (blood or tissue): for confirmation PPS Oral Exam Reviewer 2020 208 ETIOLOGY / INCIDENCE / PATHOGENESIS ▪ Acute toxic infection ▪ Gram (+) bacilli, aerobic, non-encapsulated, nonspore forming nonmotile, pleomorphic Corynebacterium diphtheriae ▪ Exclusive inhabitant of human mucous membranes and skin C. ulcerans ▪ Animal sources that can cause human disease, (+) Urease INCUBATION PERIOD: 1-10 days DIPHTERIA TRANSMISSION ▪ Airborne respiratory droplets ▪ Direct contact w/ respiratory secretions of symptomatic individuals or exudate from infected skin lesions ▪ For endemic areas, 3-5% of healthy individuals can carry toxigenic organisms but carriage is rare ▪ Skin infection and skin carriage are silent reservoirs of C.d. ▪ Organisms remain viable in dust or fomites up to 6 months ▪ Contaminated milk and infected food handler PATHOGENESIS ▪ Both toxigenic and non-toxigenic C. diphteriae cause skin and mucosal infection Potent polypeptide exotoxin ▪ the ability to produce diphtheritic toxin results from acquisition of a lysogenic corynebacteriophage which encodes the diphtheritic toxin gene ▪ Major virulence ▪ Inhibits protein synthesis and causes local tissue necrosis and resultant local inflammatory response ▪ Within first few days of RTI (pharynx) → WHITE dense necrotic coagulum of organisms, epithelial, fibrin, WBC, & RBC → gray-brown, leather-like adherent PSEUDOMEMBRANE (removal reveals bleeding edematous submucosa) ▪ Early effect of toxin: paralysis of palate and hypopharynx PPS Oral Exam Reviewer 2020 Acute CLINICAL MANIFESTATIONS / FINDINGS / DIAGNOSIS MANAGEMENT Respiratory tract ▪ Specific antitoxin: mainstay of therapy; should be ▪ Sites: tonsils, pharynx, nose, or larynx administered on the basis of clinical diagnosis ▪ After 2-4 days: local SSx of inflammation ▪ Antimicrobial therapy: to halt toxin production, treat localized ▪ ½ fever, fewer dysphagia, hoarseness, malaise, or infection, and prevent transmission to contacts headache ● Penicillin, erythromycin (E>P for eradication of NP ▪ Anterior nares infection: carriage) x 14 days, cutaneous x 7 days ● more common in infants ▪ Elimination should be documented by 2 successive (-) cultures ● Serosanguinous, purulent, erosive rhinitis w/ from nose and throat (or skin), 24-hour apart AFTER completion membrane formation of therapy ● Shallow ulceration of external nares and upper lip ▪ Antibiotic therapy is not a substitute for antitoxin therapy ▪ Tonsillar and pharyngeal diphtheria ▪ Supportive care ● Universal early symptom: sore throat ● droplet and contact precautions ● Mild pharyngeal injection → uni-/bilateral tonsillar ● bed rest >2wks until risk for symptomatic cardiac damage membrane formation → uvula (may cause toxin- has passed mediated paralysis), soft palate, post. oropharynx, ▪ Vaccine DTaP (IM) hypopharynx, or glottis 3 doses series with minimum age of 6 weeks and minimum interval ● Bull neck appearance: underlying soft tissue edema of 4 weeks and enlarged lymph nodes Cutaneous diphtheria ▪ Nonprogressive, superficial, ecthyma-like, nonhealing ulcer with a gray-brown membrane ▪ Pain, tenderness, erythema, or exudate ▪ Can coexist w/ strep or staph impetigo ▪ Extremities > trunk or head ▪ Assoc. with more prolonged mucosal shedding, greater contamination of environment, transmission to pharynx & skin of close contacts Other sites ▪ Ear (OE), eye (purulent and ulcerative conjunctivitis), genital (vulvovaginitis) ▪ Endocarditis (sporadic) among IV drug users, strains: nontoxigenic ▪ Pyogenic arthritis (sporadic), septicemia (rare) ▪ Diphtheroid isolated from sterile body sites should not be routinely dismissed as contaminants without careful consideration of the clinical setting DIAGNOSTICS ▪ Culture: nose, throat, mucocutaneous lesion DIFFERENTIAL DIAGNOSIS ▪ S. pyogenes, EBV 3 BOOSTER DOSE series at: 12 – 23 months, 4 – 7 years, 9 – 15 years Minimum interval between booster doses: 4 years Full dose DTP should preferably be used only until 7 years, but package inserts should be consulted for maximum age indications of specific products. Unvaccinated 5 ▪ For 1o immunization w/ 1st and 2nd doses 9-18 yrs old doses interval of 4 weeks, and the 2 nd and 3rd doses with an interval of at least 6 months ▪ 1st booster given at least 1yr after 3 rd dose, 2nd booster at least 1yr from 1st booster Rcvd 1 dose 4 0, 1, 2, 6 months With 2 doses 3 0, 1, 6 months With 3 doses 2 0, 6 months Fully 1 1 dose Td/Tdap every 10 years vaccinated Booster ▪ Adult preparation of Tdap at 11-12yo ▪ For 13-18 yo who missed Td/Tdap booster at 11-12yo OR >5y since Td booster should receive a single dose of Tdap if they completed the DTP/DTaP series. ▪ Contraindication: hx of neurologic or severe hypersensitivity reaction after a prior dose COMPLICATIONS / PROGNOSIS COMPLICATIONS ▪ Laryngeal diphtheria: suffocation – local soft tissue edema, airway obstruction ▪ Toxic cardiomyopathy ● 10-25% of patients with respiratory diphtheria ● Occurs 2nd-3rd week of illness as pharyngeal disease improves ● HR out in proportion to fever ● ECG: prolonged PR interval, changes in ST-T wave, single or progressive dysrhythmias (1st-3rd degree heart block), AV dissociation and Vtach ● 2DED: dilated/hypertrophic cardiomyopathy ● Acute or insidious HF ● AST ∝ myonecrosis ▪ Toxic Neuropathy ● Acutely or 2-3wks after onset of oropharyngeal inflammation ● Hypesthesia and local paralysis of soft palate ● Weakness of post. pharyngeal, laryngeal & facial nerves (nasal voice, difficult swallowing, risk for aspiration) ● Cranial neuropathies (5th wk) → oculomotor and ciliary paralysis (strabismus, blurred vision, difficult accommodation) ● Symmetric demyelinating polyneuropathy (10d-3mos after oropharyngeal infection) causing motor defects w/diminished DTR ● Paralysis of diaphragm ● DDX: GBS – indistinguishable nerve conduction velocity & CSF PROGNOSIS ▪ Depends on virulence of organism (gravis – highest fatality rate) ▪ Age, immunization status, site of infection or speed of administration of antitoxin ▪ Mechanical obstruction from laryngeal diphtheria or bullneck diphtheria and the complications of myocarditis account for most deaths ▪ Toxic Cardiomyopathy ● Complete recovery but if it occurs at 1st week, poor prognosis, may have permanent conduction defects ▪ Toxic Neuropathy ● Complete recovery ● Rare: vasomotor center dysfunction 2-3wks after onset of illness→ BP or HF Batch Clingy DISEASE 209 Clostridium tetani ▪ Gram (+), motile, spore-forming obligate anaerobe ▪ Natural habitat: soil, dust, alimentary tracts of various animals INCUBATION PERIOD: 2-14 days, may be months after injury TETANUS TRANSMISSION ▪ Neonatal: umbilical ▪ Maternal: postpartum, postabortal, post-surgical wound infection ▪ Non-neonatal: trauma, illicit drug injection, animal bites, abscesses (also dental), body piercing, chronic skin ulceration, burns, compound fractures, frostbite, gangrene, intestinal surgery, ritual scarification, infected insect bites, female circumcision PATHOGENESIS ▪ Toxin mediated process ▪ Forms spores resembling drumstick or tennis racket appearance ▪ Traumatic injury → spores germinate, multiply, vegetable cells produce toxin ▪ Toxin binds at NMJ and enters motor nerve by endocytosis, → retrograde axonal transport → enters spinal inhibitory neurons, prevents release of GABA and glycine → blocks normal inhibition of antagonistic muscles on which voluntary movement depends → affected muscles sustain max contraction & cannot relax (lock jaw) PPS Oral Exam Reviewer 2020 ▪ Vincent angina, Lemierre syndrome, mucositis ▪ Bacterial epiglottitis, severe viral staph/streptococcal tracheitis POSTEXPOSURE PROPHYLAXIS Asymptomatic case contacts ▪ Monitor for 7d, do culture, give antimicrobial prophylaxis regardless of immunization status ▪ Toxoid vaccine given in age-appropriate form to immunized individuals who have not received a booster dose within 5yrs LTB, Generalized ▪ Patient is conscious and in extreme pain ▪ Trismus: masseter muscle spasm (lock jaw) ▪ Headache, restless, irritability → stiffness, difficulty chewing, dysphagia, neck muscle spasm, sardonic smile (risus sardonicus) ▪ Opisthotonos: arched posture of extreme hyperextension of the body ▪ Laryngeal and respiratory muscle spasm → airway obstruction and asphyxiation ▪ Seizures: sudden, severe tonic contractions of the muscle w/ fist clenching, flexion and adduction of the arms and hyperextension of the legs ▪ Fever (as high as 400C): caused by metabolic energy consumed by spastic muscles ▪ Dysuria, urinary retention from bladder sphincter spasm, forced defecation ▪ Autonomic: HR, dysrhythmias, labile HTN, diaphoresis, cutaneous vasoconstriction ▪ The smallest disturbance by sight, sound or touch may trigger a tetanic spasm ▪ Paralysis is more severe in 1 st wk, stabilizes 2nd wk, ameliorates gradually in 1-4 wk Neonatal ▪ Manifests within 3-12 days of birth ▪ Progressive difficulty in feeding, paralysis or diminished movement, stiffness, and rigidity to touch & spasms, ± opisthotonos Localized: painful spasms of muscles adjacent to wound; may precede generalized tetanus ▪ Surgical wound excision and debridement: to remove foreign body or devitalized tissue that created the anaerobic growth ● performed promptly after administration of human tetanus Ig (HTIg) ▪ HTIg ● goal: neutralize toxin that diffuses from the wound into the circulation before the toxin can bind at distant muscle groups ● optimal dose has not been determined: 500 U/IM (range: 3,000-6,000 U) ● If unavailable: - Human IVIg (4-90 U/mL of TIG) but no optimal dose and not approved for this indication - Equine-derived tetanus antitoxin (TAT) 1,500-3,000 U, administered after appropriate testing for sensitivity and desensitization (up to 15% experience serum sickness) ▪ Antibiotic: to decrease the number of vegetative forms ● DOC: Metronidazole 30mkday q6 (max 4g/day) ● Alternative: Pen G 100,000 U/kg/day q4-6 (max 12M U/day) ▪ Muscle Relaxants: Diazepam, Midazolam, MgSO4, Chlorpromazine, Dantrolene, Baclofen (ICU setting), vecuronium, pancuronium ▪ Morphine: for autonomic instability ▪ Meticulous supportive care in a quiet, dark secluded room, sedation, ET/ tracheostomy, prophylactic SQ heparin, Enoxaparin (DVT) Asymptomatic carriers ▪ Antimicrobial prophylaxis for 10-14 days ▪ Toxoid vaccine given in age-appropriate form to immunized individuals who have not received a booster dose within 1yr ▪ Droplet or contact precautions until 2 (-) successive cultures 24hrs apart after cessation of therapy → repeat after 2 wks. If (+) → 10 days Erythromycin COMPLICATIONS ▪ Airway obstruction and asphyxiation ▪ Aspiration, pneumothorax, mediastinal emphysema ▪ Seizures: lacerations of the mouth and tongue, IM hematomas, rhabdomyolysis w/ myoglobinuria, renal failure, long bone, or spinal fractures ▪ Venous thrombosis, pulmonary embolism, gastric ulceration ± hemorrhage, paralytic ileus, decubitus ulcers ▪ Excessive use of muscle relaxants → iatrogenic apnea ▪ Autonomic NS: cardiac arrhythmias, asystole, unstable BP & labile temperature regulation ▪ Hypoxic brain injury → CP, diminished mental abilities, behavioral difficulties PROGNOSIS ▪ Recovery occurs through regeneration of synapses in spinal cord → muscle relaxation ▪ Mortality is highest: extremes of age ▪ Favorable prognosis: long incubation period, absence of fever, localized disease ▪ Unfavorable prognosis: trismus <7d after injury, gen. tetanic spasms <3 days after onset of trismus ▪ Cephalic tetanus: poor prognosis due to breathing and feeding difficulties ▪ Vaccine ● follow DTaP schedule ● For pregnant: give 1 dose of Tdap during each pregnancy Batch Clingy ▪ Toxin absorption → kidney tubule necrosis, platelet, cardiomyopathy, demyelination of nerves. Because the latter 2 occur 2-10 weeks after mucocutaneous infection, the pathophysiology is suspected to be immunologically mediated 210 ● Cephalic: rare form of localized; involves bulbar musculature, from wounds or foreign bodies in head, nostril, face, assoc. w/ chronic OM; Retracted eyelids, deviated gaze, trismus, risus sardonicus, spastic paralysis of tongue and pharyngeal musculature DIAGNOSTICS ▪ C. tetani is not always visible on GS, isolated in culture in only 30% of cases ▪ Usual setting: unimmunized patient/mother injured or born w/in the past 2 weeks presents w/ trismus, dysphagia, gen. muscle rigidity, spasm, and clear sensorium ▪ WBC: maybe d/t 2O bacterial infection or stressinduced ▪ CK and aldolase ▪ EMG: continuous discharge of motor subunits & shortening, or absence of the silent interval normally observed after an action potential ▪ Spatula test: touch the oropharynx w/ spatula or tongue blade (high Sn and Sp); if tetanus is present, (-) gag reflex (+) reflex spasm of masseter and bite the spatula PERTUSSIS ▪ usually caused by Bordetella pertussis & occasionally B. parapertussis ● Gram negative coccobacilli, small, fastidious ▪ B. holmesii: cause of bacteremia in IC w/o cough, pertussis-like cough illness in healthy persons ▪ B. bronchiseptica: common animal pathogen w/ occasional human reports in IC and young INCUBATION PERIOD: 3-12 days TRANSMISSION PPS Oral Exam Reviewer 2020 DIFFERENTIAL DIAGNOSIS ▪ Rabies – hydrophobia, marked dysphagia, chronic seizures, pleocytosis ▪ Strychnine poisoning ● Tonic muscle spasms & gen seizure, general relaxation occurs between spasms ● White, odorless, bitter crystalline powder – ingested, inhaled, or mixed in a solution → IV ● Found in Strychnos nux-vomica plant, given as “street drugs” or pesticide ▪ Hypocalcemia (no trismus) epileptic seizures, narcotic withdrawal CATARRHAL (1-2wks) ▪ Congestion, rhinorrhea, low grade fever, sneezing, lacrimation, conjunctival suffusion PAROXYSMAL (2-6wks) ▪ Dry, intermittent, irritative hack → inexorable paroxysms (hallmark) ▪ Well appearing toddler → anxious aura → machine-gun uninterrupted cough on a single exhalation – chin and chest held forward, tongue protruding maximally, eyes bulging and watering, face purple → coughing ceases with a loud whoop that follows as inspired air traverses Wound Management History of absorbed TT Uncertain or <3 doses >3 doses Clean Minor Wounds All Other Wounds DTaP, Tdap or Td TIG DTaP, Tdap or Td TIG Yes No Yes Yes No if <10 yr since last dose of tetanuscontaining vaccine Yes, if >10 yr since last dose of tetanuscontaining vaccine No No No if <5 yr since last dose of tetanuscontaining vaccine Yes, if >5 yr since last dose of tetanuscontaining vaccine No No ▪ Should always be given after a dog or other animal bite ▪ If not available → IVIg or equine-derived TAT ▪ Do not give Td more frequently if with previous Arthus reaction after a dose of TT containing vaccine ▪ Hospitalized: < 3mos old suspected case, severe paroxysms, complications ● Room: quiet, dim, undisturbed, comforting environment ● Avoid large feedings ▪ Antibiotics ● Azithromycin – drug of choice; risk for fatal heart rhythms to those at risk for CV events (esp. prolonged QT interval) ● Erythromycin – risk for infantile hypertrophic pyloric stenosis ● Alternatives: Clarithromycin & TMP-SMX ▪ Discharge Criteria: COMPLICATIONS ▪ Apnea, 2o infections (OM & pneumonia), physical sequalae of forceful coughing ▪ Acute neurologic events – 2o to hypoxemia & hemorrhage ▪ Apnea or bradycardia ▪ Seizures (hypoxemia) ▪ Hyponatremia – excessive secretion of ADH during pneumonia ▪ Intrathoracic and intraabdominal pressure: conjunctival and scleral hemorrhages, petechiae on the upper body, epistaxis, pneumothorax, SQ emphysema, umbilical or Batch Clingy Tetanospasmin ▪ 2nd most poisonous substance known (after botulinum toxin) ▪ Lethal dose 10-5mg/kg 211 PATHOGENESIS ▪ Only colonizes ciliated epithelium ▪ Only B. pertussis expresses pertussis toxin – major virulence protein → histamine sensitivity, insulin secretion, leukocyte dysfunction ▪ Aerosol acquisition → attach to resp. ciliated cells via filamentous hemagglutinin, agglutinogens, & pertactin → clearance is inhibited by tracheal cytotoxin, adenylate cyclase, PT → local epithelial damage → respiratory sx, facilitates absorption of PT the partially closed airway; post tussive emesis ▪ At the peak of the stage: 1 episode hourly CONVALESCENT (>2wks) ▪ Number, severity, duration of episode diminishes DO NOT DISPLAY THE CLASSIC STAGES: Infants < 3 months ▪ Catarrhal phase is unnoticed, cough may not be prominent, apnea and cyanosis follow coughing paroxysm or apnea as the only symptom ▪ “Exacerbations” can occur throughout the 1 styr (not a recurrence or reactivation) Adolescents & prev. immunized ▪ Foreshortening of all stages ▪ Sudden feeling of strangulation → uninterrupted coughs, feeling of suffocation, bursting headache, diminished awareness → gasping breath without a whoop ▪ 30% have nonspecific illness distinguished by duration of >21 days ● Over 48 hours, disease severity is unchanged or diminished ● Intervention is not required during paroxysms ● Nutrition is adequate ● No complication has occurred ● Parents are adequately prepared for care at home ▪ Droplet precaution & isolate immediately until 5 days after azithromycin inguinal hernia, hemorrhage in CNS & retina, laceration of lingual frenulum ▪ Severe and usual causes of death: progressive pulmonary HTN & 2O bacterial pneumonia ▪ Rare: Bronchiectasis after pertussis ▪ Children <2yo may have abnormal pulmonary function in adulthood ▪ Vaccine ● follow DTaP schedule ● Acellular pertussis vaccine: less reactogenic compared to whole-cell pertussis vaccine PROGNOSIS ▪ Severe course/Death: rapid rise and extreme leukocytosis & thrombocytosis, prematurity, pulmonary HTN & cardiogenic shock ▪ < 6mo old: excessive morbidity and mortality ● < 4mo old: 90% of cases of fatal pertussis (as well as preterm and young maternal age) ● < 2mo old: highest rates of pertussis assoc. hospitalization, pneumonia, seizures, encephalopathy, death DIFFERENTIAL DIAGNOSIS ▪ Mycoplasma – protracted episode continuous cough w/ hx of fever, systemic sx, PE: rales ▪ Adenovirus – fever, sore throat, conjunctivitis ▪ Chlamydia (staccato cough), Parainfluenza, influenza, enterovirus, RSV, FB Aspiration Typical Paroxysms that are not life threatening: ▪ duration <45 sec, red but not blue color change, HR, HR (not <60 in infants), or O2 desaturation that spontaneously resolves, whooping or strength for brisk self-rescue at the end of rescue, self-expectorated mucus plug, post tussive exhaustion but not unresponsiveness ENTERIC FEVER Typhoid Fever Salmonella enterica serovar typhi ▪ Gram negative PPS Oral Exam Reviewer 2020 DIAGNOSTICS ▪ Should be suspected in any individual who has a pure or predominant complaint of cough ▪ Clinical case definition: cough ≥ 14 days + paroxysm, whoop, or post tussive vomiting ▪ Suspect in: ● Older child whose cough is escalating at 7-10d and comes in bursts ● < 3mos old with gagging, gasping, apnea, cyanosis ▪ Leukocytosis caused by absolute lymphocytosis is characteristic in the catarrhal stage ▪ CXR: mildly abnormal ▪ PCR testing on NP wash specimens is the laboratory test of choice for B. pertussis identification ▪ Results from PCR & culture are expected (+) in unimmunized & untreated during catarrhal and early paroxysmal stage ▪ More dramatic presentation in < 5yo Primary Bacteremia: asymptomatic, blood CS (-) ▪ Adequate rest, hydration, antipyretic therapy, soft easily digestible diet POSTEXPOSURE PROPHYLAXIS Care of Household and Other Close Contacts ▪ Azithromycin should be given to ALL contacts ▪ < 7yo who had < 4 doses DTaP: give DTaP & complete recommended series ▪ < 7yo who received 3rd DTaP > 6mos before exposure or 4th dose ≥ 3yrs before exposure: give booster dose ▪ ≥ 9yo: give Tdap COMPLICATIONS ▪ Infancy – DIC Batch Clingy ▪ Extremely contagious ▪ 100% attack rate exposed to aerosol droplets at close range ▪ Rate of sub-clinical infection is high as 80% in fully immunized or previously infected ▪ No chronic carrier documented ▪ Does not survive for prolonged periods in the environment 212 INCUBATION PERIOD: 4-14 days (depending on the inoculating dose of viable bacteria) TRANSMISSION ▪ Direct or indirect contact with infected person (sick or chronic carrier) ▪ Most common: ingestion of food or water contaminated with S. typhi from human feces ▪ Water-borne outbreaks (poor sanitation or contamination): oysters, shellfish, use of night soil as fertilizer PATHOGENESIS ▪ Infective dose: 105-109 ▪ Orally ingested salmonellae survive low pH of stomach, evade SI defenses, enter epithelium (M cells) ▪ In contrast to NTS, expresses virulence factors that allows to downregulate the pathogen recognition receptor mediated host inflammatory response, colonize deeper tissues, lymphoid cells in Peyer Patches → hyperplasia, pass thru the intestinal mucosa enter the mesenteric lymphoid system then thru the blood stream causing PRIMARY BACTEREMIA → enter macrophages → disseminate in RES → necrosis and sloughing of overlying epithelium → SECONDARY BACTEREMIA → systemic symptoms is from the release of proinflammatory cytokines (IL-6, IL-1β, TNFα) from infected cells NON TYPHOIDAL SALMONELLA Typhoid toxin ▪ CdtB cytothelial distending toxin (a DNase that causes double-strand breaks in host cell DNA) ▪ PltA pertussis-like toxin ▪ Salmonellosis: a common and widely distributed food-borne disease; global major public health problem ▪ 2 most important serotypes for salmonellosis transmitted from animals to humans PPS Oral Exam Reviewer 2020 Secondary Bacteremia ▪ Onset of symptoms ▪ Marks the end of incubation period ▪ High-grade fever, coated tongue, anorexia, vomiting, hepatomegaly, diarrhea, toxicity, abdominal pain, pallor, splenomegaly, constipation, HA, jaundice, obtundation, ileus, intestinal perforation ▪ Initially, iarrhea (pea-soup) → constipation ▪ D7-10: rose spots – crops of 10-15 on the lower chest & abdomen lasting for 2-3 days ▪ No complication, resolves within 2-4 weeks DIAGNOSTICS ▪ Mainstay of diagnosis: positive culture from the blood or another anatomic site ● during the 1st week: blood and bone marrow ● after the 1st week: stool and urine ● over the 3 rd week, BM culture could remain (+) despite (-) blood CS; difficult to obtain/invasive ▪ Widal test: measures Ab O and H antigens of S typhi but lacks Sn and Sp in endemic areas ▪ Other serologic tests (i.e. Typhidot) have not been proven sufficiently robust in large-scale evaluations in community settings ▪ Leukocytosis: common in younger children ▪ Thrombocytopenia: marker of severe illness (DIC) ▪ Liver function test may be deranged, but sig. dysfunction is rare DIFFERENTIAL DIAGNOSIS ▪ Early: dengue fever or malaria ▪ Sepsis, TB, brucellosis, tularemia, leptospirosis, rickettsia, acute hepatitis Acute Enteritis ▪ most common presentation ▪ abrupt N/V, crampy abdominal pain (periumbilical area/RLQ) → watery diarrhea, sometimes w/ blood and mucus ▪ Antibiotic therapy is critical to minimize complications; influenced by the prevalence of antimicrobial resistance Uncomplicated 1st Line Sensitive MDR FLQ resistant Amox (DOH) Chloram Cipro Cefixime Azith Ceftri Alternative 14 d 14-21 d 5-7 d 7-14 d 7d 10-14 d Severe 1st Line Sensitive FLQ (Cipro) Ceftri (DOH) 10-14 d 10-14 d MDR FLQ 10-14 d FLQ Resistant Ceftri Cefotax 10-14 d 10-14 d Ampi (DOH) Cipro Azith 5-7 d 7d Cefixime 7-14 d 14 d Alternative Chloram 14-21 Amox Ceftri Cefotax Azith FLQ 10-14 d 10-14 d 7 7-14 ▪ Use of 2nd line should be reserved for suspected or proven multi-drug resistant typhoid fever: resistant to first line (Chloramphenicol, Ampicillin and TMP-SMX) ▪ Should be suspected in ANY of the following situations: ● Failure to respond after 5-7 days treatment with a first line antibiotic ● Household contact w/ documented case or during an epidemic of MDRTF and/or ● Clinical deterioration or development of complications during conventional antibiotic treatment ▪ Adults – CNS, GI bleeding ▪ GI: intestinal hemorrhage and perforation ▪ CNS: encephalopathy, cerebral edema, subdural empyema, meningitis, GBS, ventriculitis, ataxia, seizures, psychosis ▪ CV: toxic myocarditis (arrythmias, sinoatrial block, cardiogenic shock) ▪ Pulmonary: pneumonia, empyema, broncho-pleural fistula ▪ Bone and Joint: Osteomyelitis, septic arthritis ▪ Hepatobiliary system: cholecystitis, hepatitis, hepatic abscesses, splenic abscess, peritonitis ▪ GUT: UTI, renal abscess, pelvic infections, testicular abscess ▪ SSI: psoas abscess, gluteal abscess, cutaneous vasculitis ▪ Hematologic: Hemophagocytosis syndrome PREVENTION ▪ Most important risk: contamination of water supplies with sewage PROGNOSIS ▪ Depends on rapid diagnosis and treatment, age, general state of health, causative serotype, appearance of complications ▪ 2-4% can have relapse despite treatment ▪ Chronic carriers: > 3months secretion of S typhi. ● Risk increases with age ● A chronic urinary carrier state can develop in children w/ schistosomiasis ▪ Vaccine ● Oral live attenuated Ty21a strain, efficacy 5 years ● IM Vi capsular polysaccharide for ≥ 2yo, booster q2 years, efficacy 70-80% ▪ Antibiotics are not generally recommended for the treatment of isolated uncomplicated Salmonella gastroenteritis due to ● Disruption normal intestinal flora ● Prolonged excretion with ↑ risk as a chronic carrier ▪ Give antibiotics until culture is available to: COMPLICATIONS ▪ Acute dehydration can be assoc. with Salmonella gastroenteritis ▪ Osteomyelitis (in children w/ sickle cell disease) ▪ Reactive arthritis (especially in adolescents with HLA-B27 Ag) Batch Clingy ▪ other etiologies: S. enterica var paratyphi A, B, C ▪ Age-specific incidence highest in < 5yo ▪ notable for emergence of drug resistance ▪ S. typhi is highly adapted to infection of humans; lost ability to cause transmissible disease in animals ▪ Associated with malnutrition 213 INCUBATION PERIOD: 6-72 hours TRANSMISSION ▪ Food contaminated by contact w/ an infected animal product or a human carrier ▪ Contact with infected animals (e.g. domestic pets, amphibians) ▪ Animal-derived pet foods and treats PATHOGENESIS ▪ Infective dose: 106-108 ▪ Gastric acidity inhibits multiplication of salmonellae ● Achlorhydria, buffering meds, rapid gastric emptying after gastrectomy or gastroenterostomy and a large inoculum enable viable organisms to reach the SI ▪ After an infection, excreted in feces for 5 weeks ▪ During excretion, it may be transmitted by fecaloral route or indirect contamination of food ▪ Enterocolitis w/ diffuse mucosal inflammation & edema (sometimes erosions and micro abscesses) → penetrate the intestinal mucosa → lymphoid tissue and mesenteric LN enlargement & necrosis → hyperplasia of the RES → bacteremia → localized infection and suppuration to any organ ▪ Gram positive, coagulate positive ▪ Most common cause of pyogenic infection of skin and soft tissue S. AUREUS Strains that protect organism from host defenses: ▪ Produce biofilm or loose polysaccharide capsule: interfere w/ opsonophagocytosis ▪ Clumping factor interferes w/ phagocytosis ▪ Coagulase causes plasma to clot (abscess formation) ▪ Protein A: prevents antibacterial Ab from acting as opsonins → inhibit phagocytosis PPS Oral Exam Reviewer 2020 ▪ febrile ▪ symptoms last 2-7 days Bacteremia (1-5%) ▪ Transient bacteremia ▪ NB & young infants present with minimal symptoms ▪ Older infants, typically follows AGE; associated with fever, chills, septic shock ▪ Fever is present in 95% of cases but may have no apparent focus; diarrhea is NOT a prominent feature Extraintestinal Focal Infections ▪ occurs following bacteremia ▪ Most common: skeletal system, meninges (peak in infancy), intravascular sites and sites of pre-existing abnormalities Chronic Salmonella Carriage ▪ Asymptomatic, w/ colonization in the gall bladder pres. w/ intermittent (+) stool CS ● < 3 months old (risk of bacteremia) ● High risk groups w/ immune compromise ▪ Choice of antibiotics (Nelsons’ DOH NAG) ● Cefotaxime 100-200mkday q6-8 x 5-14 days or ● Ceftriaxone 75mkday QD x 7days ● Ampicillin 100mkday q6-8 x 7 days ● Cefixime 15mkday x 7-10 days ▪ AIDS: multisystem involvement, septic shock and death ▪ IBD: toxic megacolon, bacterial translocation, sepsis ▪ Schistosomiasis: may lead to chronic infection ▪ Neonates, <6mos old, children w/ 1o or 2o ID may have symptoms that lasts for weeks ▪ RES dysfunction: prolonged, intermittent bacteremia assoc. low-grade fever, anorexia, weight loss, diaphoresis, myalgia ▪ IF antimicrobial therapy in a well child with NTS gastroenteritis with no disseminated disease is initiated, give single dose of ceftriaxone → discharge with oral azithromycin pending blood CS results (alternatives: ampicillin, TMP SXT or FLQ, if susceptible) (AAP Red Book, 2018) PROGNOSIS ▪ Most recover fully ▪ Malnourished children and those with suboptimal supportive treatment: prolonged diarrhea and complications ▪ Young infants, IC: systemic involvement, prolonged course and extraintestinal foci ▪ Prolonged carrier state seen in children with biliary tract disease & cholelithiasis after chronic hemolysis PREVENTION ▪ Control transmission in the animal reservoir ▪ Judicious use of antibiotic in dairy and livestock ▪ Prevent contamination of food prepared from animals ▪ Vaccine: some are used in animals, but no human vaccine is available yet DIAGNOSTICS ▪ definitive diagnosis is based on clinical correlation of the presentation and culture from feces or other body fluids ● in children with AGE, stool CS > rectal swabs ▪ Mild leukocytosis ▪ Stools: moderate PMN and occult blood (see Nelson’s 21st Ed., Table 225.1, for Host Factors and Conditions Predisposing to Development of Systemic Disease with NTS strains) Newborn ▪ Cutaneous: omphalitis, cellulitis, mastitis, subcutaneous abscesses ▪ Staphylococcal pustulosis: large pustule filled lesions 1mm in dm and scattered around the umbilicus ▪ DDX: HSV Skin ▪ Impetigo, ecthyma, hidradenitis, folliculitis, furuncles, carbuncles, SSSS, cellulitis, necrotizing fasciitis ▪ Recurrent skin and soft tissue infections: MRSA Respiratory ▪ OM, sinusitis, suppurative parotitis DIAGNOSTICS Culture ▪ Diagnosis depends on the isolation of the organism in culture from nonpermissive sites ▪ Best: tissue samples or fluid aspirates ▪ Obtain culture of any potential focus of infection as well as blood culture PCR identify genetic patterns ass. w/ methicillin resistance Food Poisoning ▪ food may be cultured & test for exotoxin COMPLICATIONS ▪ Omphalitis → abdominal wall cellulitis or necrotizing fasciitis ▪ Osteomyelitis → DVT → septic pulmonary emboli ▪ Endocarditis → perforation of heart valves, myocardial abscesses, HF, conduction disturbances, acute hemopericardium, purulent pericarditis, sudden death PREVENTION ▪ Hand hygiene, handwash w/ chlorhexidine or alcohol ▪ Hospitals: Isolate until treated adequately ▪ Decolonization for recurrent infections ● Decontaminating baths (hypochlorite, 1 tsp common Batch Clingy ● S. enterica var enteritidis and S. typhimurium ▪ Practices that promote antibiotic-resistant bacteria ● Subtherapeutic concentration of antibiotics in animal feeds to promote growth (can also be contaminated with Salmonella) ● Previous antibiotic use among the child in the previous month 214 Strains that cause local tissue destruction: ▪ Hemolysins causes tissue necrosis ▪ Panton-Valentine leucocidin (PVL): ↑ permeability and eventual death of the cell ▪ Exfoliatin A & B: exotoxins; produce localized (bullous impetigo) or generalized (SSSS, scarlet fever) dermatologic manifestations Toxins: ▪ Enterotoxin (types A-V): ingestion of preformed enterotoxin A & B results to food poisoning ▪ Toxic shock syndrome toxin-1 (TSST-1) superantigen that induces production of IL-1 & TNF; assoc. w/ TSS, related to menstruation and focal infection (nasal packing) Expression of proteins that mediate resistance ▪ Penicillinase or ß-lactamase: produced universally by all isolates; inactivates ß-lactams ▪ Altered penicillin-binding proteins: mediates resistance to penicillinase resistant antibiotics INCUBATION PERIOD: 2 – 7 hours TRANSMISSION ▪ Person to person ▪ Autoinoculation or direct contact w/ hands of other colonized individuals ● Effective disseminators: nasal carriers ▪ Rare: Fomites (shared towels in football team) Risk factors: disruption of intact skin, corticosteroids, malnutrition, azotemia, viral infections, congenital defects in chemotaxis & phagocytosis, HIV, poor mucous clearance (cystic fibrosis) ▪ Membranous tracheitis (complication of viral croup) – high fever, leukocytosis, upper airway obstruction ▪ Pneumonia ● hematogenous pneumonia may be 2o to septic emboli from R-sided endocarditis or septic thrombophlebitis ● Inhalation Pneumonia: high fever, abdominal pain, RR, dyspnea, localized or diffuse bronchopneumonia or lobar disease ● Necrotizing pneumonitis: assoc. w/ empyema, pneumatocele, pyopneumothorax, bronchopulmo-nary fistulas Sepsis ▪ onset may be acute; nausea, vomiting, myalgia, fever, chills ▪ Organism may localize to heart valves, lungs, joints, bones muscles and deep tissue abscesses ▪ Disseminated S. aureus disease: fever, persistent bacteremia despite antibiotics, focal involvement of ≥ 2 separate tissue sites; usually have an endovascular nidus of infection Pyomyositis: localized abscess in muscle w/ out septicemia (prior trauma) Bones and joints ▪ Osteomyelitis, suppurative arthritis: pseudo paralysis (neonates) or pain w/ movement of affected extremity; older: pain, fever, localizing signs (edema, erythema, warmth, limp, or refusal to walk) CNS ▪ Meningitis (uncommon) – assoc. penetrating cranial trauma, neurosurgical procedures, endocarditis, parameningeal foci, complicated sinusitis, DM, or malignancy Heart: Acute endocarditis on native valves Kidneys: renal and perinephric abscess Intestinal Tract ▪ Enterocolitis, diarrhea + blood and mucus, peritonitis from long-term peritoneal dialysis ▪ Food poisoning: 2-7 hours after ingestion of preformed enterotoxins, sudden, severe vomiting, watery diarrhea, absent to low fever. Symptoms persist >12-24 hours. Toxic Shock Syndrome ▪ characterized by fever, hypotension, erythematous rash with subsequent desquamation on the hands and feet, multisystem involvement (see Nelson’s 21st Ed., Table 208.2 for Diagnostic Criteria of PPS Oral Exam Reviewer 2020 DIFFERENTIAL DIAGNOSIS ▪ Skin: GAS (rapid spread) ▪ Fluctuant skin & soft tissue: MTB, atypical mycobacteria, B. henselae, F. tularensis, fungi ▪ Cavitary pneumonia: K. pneumonia, MTB ▪ Bone and joint infection: GAS, Kingella kingae ▪ TSS: Kawasaki disease MANAGEMENT ▪ Incision and drainage ▪ Remove foreign bodies Initial Empiric Therapy ▪ If life threatening, Vancomycin + nafcillin or oxacillin ▪ If non-life threatening, ● No signs of severe sepsis, vancomycin or clindamycin ● Low likelihood of MRSA, cefazolin or nafcillin bleach solution per gallon of water or chlorhexidine 4% soap used weekly) ● Appropriate oral antibiotic ● Nasal mupirocin BID x 1 week ● Cleaning of household linens in hot water ▪ Food poisoning ● Exclude individuals w/ infection of the skin from preparation and handling of the food ● Prepared foods should be eaten immediately or refrigerated appropriately to prevent multiplication of S. aureus that may have contaminated the food PROGNOSIS ▪ Untreated septicemia: high fatality rate ▪ Pneumonia: fatal at any age ▪ Factors affecting prognosis: nutrition, immunologic competence, presence of other debilitating disease MSSA ▪ Nafcillin* ▪ Cefazolin ▪ Clindamycin – if with serious Pen allergy & susceptible strain ▪ Vancomycin – if Pen and Cephalosporin allergic ▪ AmpiSul – if broader gram (-) coverage is needed ▪ Less severe: Dicloxacillin, cephalexin, co-amox MRSA ▪ Vancomycin* ▪ Clindamycin (if susceptible) ▪ Daptomycin, Linezolid, TMP SXT *Add gentamycin or rifampin for endocarditis and CNS infection Batch Clingy ▪ Catalase: inactivates H2O2 for intracellular survival 215 PATHOGENESIS Staphyloccocal TSS) ▪ recovery occurs w/in 7-10 days; recurrences can occur N. GONORRHEA INCUBATION PERIOD ▪ Genital: 2-5 days in men, 5-10 days in women ▪ Peripartum: 2-5 days after birth TRANSMISSION ▪ Sexual: sustained by asymptomatic people and hyperendemic, high risk core group (CSW, MSM, adolescents w/ multiple sexual partners) ▪ Peripartum: neonates; inoculation or autoinoculation from a genital site PATHOGENESIS ▪ Gonococcal adaptations: sialylation of lipooligosaccharides (endotoxin), ↑ catalase production and high-frequency antigenic variations of surface proteins ▪ Gonococcus infects columnar epithelium → attach to host cells, adhere by pili → after 24h, invaginate epithelial cell surface → phagocytosed by neutrophils and suppress activation on CD4 + T lymphocytes → phagocytic vacuoles release gonococci → local disease or dissemination through the bloodstream and lymphatics → inguinal LAD; perineal; perianal; ischiorectal, periprostatic abscesses and disseminated gonococcal infection (DGI) PPS Oral Exam Reviewer 2020 Asymptomatic Gonorrhea ▪ Isolated in oropharynx of young children (sexually abused) ▪ Urogenital gonorrheal infection (80% female, 10% male) ▪ Rectal carriage: 26-68% females w/ urogenital infection Uncomplicated, localized gonorrhea ▪ Urethritis – males; purulent discharge & dysuria, (-) urgency/frequency, resolve spontaneously or progress ▪ Vulvovaginitis – pre-pubertal female; purulent vaginal discharge, swollen, erythematous, tender excoriated vulva, dysuria ▪ Cervicitis and urethritis – post-pubertal female; purulent discharge, suprapubic pain, dysuria, intermenstrual bleeding, dyspareunia ▪ Rectal gonorrhea – asymptomatic; may cause proctitis with anal discharge, pruritus, bleeding, pain, tenesmus, constipation ● In MSM, the rectal mucosa is painless w/ mucopurulent discharge and scant rectal bleeding to overt proctitis w/ rectal pain and tenesmus ▪ Ophthalmitis – uni/bilateral, any age group ● Ophthalmia neonatorum – 1-4 d after birth; mild inflammation and serosanguinous discharge → within 24 hours, thick purulent discharge with tense edema of eyelids and chemosis Disseminated Gonococcal Infection (DGI) ▪ 1-3% (asymptomatic>symptomatic); M>F ▪ Most common initial symptom: acute onset of polyarthralgia with fever ▪ Asymmetric arthralgia, petechial, or postural acral skin lesions, tenosynovitis, suppurative arthritis, carditis, meningitis, osteomyelitis ▪ Painful discrete 1-20mm pink or red macules → MP, vesicular, bullous, pustular, or petechial lesions and necrotic pustule on an erythematous base unevenly DIAGNOSTICS ▪ Gram stain: gram negative intracellular diplococci within leukocyte in urethral discharge ▪ NAAT ● Less stringent transport, more rapid turnaround time, flexibility in sampling; however, cannot provide antimicrobial susceptibility ● Endocervical swabs, vaginal swabs, male urethral swabs, female/male 1st catch urine ● For women, Sn is lower in urine specimen ● For MSM: annual rectal and pharyngeal sites ▪ Culture ● Swab endocervix & rectum in ALL females, regardless of history of anal intercourse; swab urethra and rectum in males ● in a suspected case of child abuse, culture remains the recommended method of detection of N. gonorrhoeae ● DGI: culture blood, pharynx, rectum, urethra, cervix, and synovial fluid ● Used to evaluate treatment failure and monitor developing resistance to current regimen MANAGEMENT ▪ Test for co-infection (syphilis, HIV, chlamydia) ● Neonates: treat for both if chlamydia results are not available or (-) in non-NAAT test ● Sexually active: abstain for 7d after treatment and until all partners are adequately treated. Sexual partners exposed in the preceding 60d should be examined, specimens collected, and presumptive treatment started ● Ophthalmia: lavage of the infected eye with saline solution once ▪ Neo: Ceftriaxone (WOF hyperbilirubinemia) or Cefotaxime ▪ Children < 45 kg: Ceftriaxone ▪ > 45 kg, adolescent and adult: Ceftriaxone + Azithromycin 1g PO (1 dose) COMPLICATIONS ▪ Ophthalmia: corneal ulceration, rupture, blindness ▪ Endometritis → salpingitis, peritonitis, tuboovarian abscess (PID) ▪ PID: vaginal discharge, suprapubic pain, cervical tenderness, fever, leukocytosis, ↑ ESR, adnexal tenderness ▪ Fitz-Hugh-Curtis syndrome/perihepatitis ± signs of salpingitis → hydrosalpinx, pyosalpinx, tuboovarian abscess, sterility ▪ Urogenital infection during the 1st trimester – high risk for septic abortion ▪ After 16wks AOG: chorioamnionitis → PROM and premature delivery ▪ Epididymitis → overt urethritis ▪ Unusual: penile edema ass. dorsal lymphangitis or thrombophlebitis, periurethral abscess or fistulas, seminal vasculitis, and balanitis (uncircumcised) PREVENTION ▪ Education ▪ Barrier protection (condoms) ▪ Screening of high-risk populations Gonococcal ophthalmia neonatorum ▪ Give erythromycin (0.5%) ophthalmic ointment ▪ If unavailable and at risk, give Ceftriaxone 25-50mg/kg IV or IM (max: 250 mg) single dose PROGNOSIS ▪ Good if w/ early appropriate therapy ▪ Treatment failure ● No resolution within 3-5d of treatment + no sex during tx ● (+) Culture > 72h or NAAT ≥ 7d after treatment + no sex during tx Batch Clingy Gonorrhea: infection of the GUT mucous membranes, mucosa of the rectum, oropharynx & conjunctiva ▪ Nonmotile anaerobic, non-spore forming, gram negative diplococcus w/ flattened adjacent surface, pili ▪ Oral sex (MSM) provided pool of asymptomatic pharyngeal infections ● account for as much as 1/3 of symptomatic gonococcal urethritis 216 Increased Risk for DGI: ▪ Asymptomatic carriers ▪ Menstruation, pregnancy & postpartum – d/t maximal endocervical shedding and ↓ peroxidase bactericidal activity ▪ Neonates due to lack of bactericidal IgM Ab ▪ Persons with terminal complement component deficiencies are at considerable risk for development of recurrent episodes N. MENINGITIDIS Meningococcus ▪ Commensal of the human nasopharynx ▪ Rarely enters the blood stream (meningitis, meningococcemia) ▪ Gram negative, fastidious, encapsulated, oxidasepositive, aerobic diplococcus ▪ Endemic disease is caused by genetically heterogenous strains ▪ Outbreaks are usually clonal, caused by a single strain ▪ Carriage peaks during adolescence and young adulthood ▪ Most cases are sporadic ▪ Highest rate: <1yo due to immunologic inexperience, immaturity of alternative and lectin complement pathway & poor response to bacterial polysaccharides INCUBATION PERIOD: 1 – 14 days TRANSMISSION ▪ Droplet: close contact thru aerosol droplets ▪ Person to person: exposure to respiratory secretions (kissing) ▪ Enhanced rates of mucosal colonization and ↑ PPS Oral Exam Reviewer 2020 distributed sparing face and scalp ▪ 2 syndromes Tenosynovitis – dermatitis syndrome ● More common ● Fever, chills, skin lesions, polyarthralgia (wrists, hands, fingers) ● (+) Blood culture 30-40%, (-) Synovial fluid cultures Suppurative arthritis syndrome ● Systemic signs less prominent ● Monoarticular arthritis common (knee); for neonates, polyarticular. ● (-) Blood culture (+) Synovial fluid cultures 45-55% consistent w/ septic arthritis Acute endocarditis: uncommon but fatal; rapid destruction of aortic valve Acute pericarditis: rare Meningitis: SSx similar to acute bacterial meningitis ▪ Most common form: asymptomatic carriage of organism in the nasopharynx Acute meningococcal septicemia ▪ Fever, irritability, lethargy, respiratory symptoms, refusal to drink, vomiting, diarrhea, sore throat, chills ▪ 7%: limb pain, myalgia, refusal to walk ▪ 10%: maculopapular rash ▪ Cold hands/feet, abnormal skin color, prolonged CRT, non-blanching, or petechial rash in > 80% ▪ Fulminant: progress rapidly over hours to septic shock – prominent petechia and purpura (purpura fulminans), poor peripheral perfusion, HR, RR, BP, confusion, coma, coagulopathy, electrolyte disturbance (hypoK), acidosis, adrenal hemorrhage, RF, myocardial failure Meningococcal meningitis ▪ Indistinguishable from other bacterial meningitis ▪ Rapidly progressive cerebral edema → death Occult meningococcal bacteremia ▪ Fever ± symptoms that suggest minor viral infection; spontaneous resolution can occur ▪ Sustained bacteremia → meningitis 60% and distant infection of other tissues ▪ If allergic to cephalosporin and uncomplicated: Gentamicin 240 mg IM + Azithromycin 2g PO ▪ If allergic to Azithromycin: Doxycycline 100mg PO BID x7d ▪ If Ceftriaxone unavailable: oral Cefixime 400mg PO, Ceftizoxime, Cefoxitin + probenecid or Cefotaxime Duration of Ceftriaxone treatment ▪ One dose – pharyngeal, anorectal, urogenital, ophthalmia neonatorum ▪ 7 days – scalp abscess, septic arthritis ▪ 10-14 days – meningitis ▪ Min. of 28 days – endocarditis ▪ If DGI, continue 24-48h after clinical improvement begins. If no organism is isolated and diagnosis is secure, continue for at least 7days ▪ Follow-up test-of-cure is not recommended for uncomplicated urogenital or rectal gonorrhea receiving recommended or alternative regimens ● Pharyngeal gonorrhea treated with alternative regimen: patient should return after 14d for CS, NAAT ▪ Once suspected, start third-generation cephalosporin until diagnosis is confirmed ▪ Once diagnosis of β lactam sensitive meningococcal disease is confirmed → switch to Penicillin Pen G 300,000 u/kg/day Q4-6 Does not clear carriage & prophylaxis is required at the end Ampicillin 200400mkday Q6 Cefotaxime 200300mkday Q6-8 Neonate Ceftriaxone 100mkday Q1224 ↓ Skin complications ▪ Supportive care: assess and correct glucose, K, Ca, Mg, PO4, clotting factors, anemia Vaccine ▪ 2 preparations ● Tetravalent (ACYW-135) conjugate vaccine: MCV4-D, MCV4-TT, MCV4-CRM; all given IM ● Tetravalent meningococcal polysaccharide vaccine: MPSV4; given IM or SQ; poorly immunogenic in infants, do not induce immunologic memory, assoc. w/ immunologic hyporesponsiveness (IH) that is why conjugated vaccines were COMPLICATIONS ▪ Acute: adrenal hemorrhage, endophthalmitis, endo/peri/myocarditis, pneumonia, lung abscess, peritonitis, renal infarcts ▪ Pre-renal failure → renal insufficiency (dialysis) ▪ Reactivation of latent herpes simplex ▪ Waterhouse-Friderichsen syndrome: adrenal insufficiency resulting from adrenal necrosis or hemorrhage 2O to severe meningococcal sepsis ▪ Immune complex vasculitis – fever, rash, iritis, carditis, pericarditis; self-limiting, occurs 1st 10 days after infection ● Arthritis: mono or oligoarticular, large joints, sterile effusions Meningococcal Meningitis ▪ Most frequent sequela: deafness ▪ Severe: cerebral arterial & venous thrombosis → infarction ▪ Subdural effusion, brain abscess, ataxia, seizures, blindness, CN palsies, hemi- or quadriparesis, obstructive hydrocephalus (3-4wks after onset) Meningococcal septicemia ▪ Most common: focal skin infarction → scarring, require skin grafting ▪ Distal tissue necrosis in purpura fulminans may require amputation Batch Clingy ▪ IgA protease inactivates IgA1 for colonization or invasion of host mucosal surfaces ▪ Estrogen-induced cornification of the vaginal epithelium of neonates and mature females resist infection 217 Chronic meningococcemia ▪ Fever, nontoxic appearance, arthralgia, headache, splenomegaly, MP or petechial rash ▪ Intermittent symptoms for 6-8 weeks ▪ May spontaneously resolve ▪ If untreated may lead to meningitis PATHOGENESIS Colonization of the nasopharynx (carriage or invasive disease) → host epithelial cells (mediated by pili) → 1st line of defense avoided by Ig A1 protease that degrades secretory IgA → close adhesion, internalization, transcytosis to the basolateral tissue → dissemination into the blood stream → resistance to complement-mediated lysis & phagocytosis is mediated by polysaccharide capsule and LPS → Multiply to high levels to cause meningococcemia → ↑ vascular permeability (hypovolemia), capillary leak syndrome (pulmonary edema & respiratory failure), pathologic vasoconstriction (pallor & cold extremities) and vasodilation (warm shock: warm extremities, bounding pulses), DIC, profound myocardial dysfunction (direct negative cytokine effect IL-6) → impairment of microvascular blood flow → multiorgan failure DIAGNOSTICS ▪ CBC: or WBC, neutrophils, bands, anemia, plt ▪ Proteinuria, hematuria, ESR & CRP ▪ If rapid onset: findings may be normal ▪ Diagnosis more likely: CRP, fever, petechiae ▪ Confirmation: isolation of organism from normal sterile body fluid (blood, CSF, synovial fluid) ● GS/CS: petechial or purpuric skin lesion or buffy coat of centrifuged blood sample. ● Culture <50% positive and isolation from the NP is not diagnostic of invasive disease ▪ PCR: High sensitivity & specificity (blood & CSF) ▪ CSF analysis: findings are the same as bacterial meningitis and showing gram negative diplococci. ▪ Meningococcal septicemia: Alb, Ca, K, Mg, PO4, glucose, met. acidosis, lactate DIFFERENTIAL DIAGNOSIS ▪ Gram (-) causing sepsis & meningitis (S. pneumonia, S. aureus, epidemic typhus) ▪ Bacterial endocarditis ▪ Petechial rash: enterovirus, influenza, measles, EBV, CMV, parvovirus ▪ Protein C or S deficiency, platelet disorders, HSP, connective tissue disorders, drug eruptions, trauma Following invasion of circulation, may enter CSF → stimulate proinflammatory cascade, upregulation of specific adhesion molecules, recruitment of leukocytes → damage directly by meningeal inflammation & indirectly by circulatory collapse → cerebral edema → herniation SHIGELLOSIS ▪ 4 species: S. dysenteriae (group A), S. flexneri (group B), S. boydii (group C), S. sonnei (group D) ▪ Gram negative bacilli, facultative, aerobic ▪ Acute invasive enteric infection ▪ Endemic to temperate and tropical climates ▪ Highest incidence: < 10yo ▪ 70% of all episodes & 60% of Shigella-related deaths occur in < 5yo ▪ Infection in the 1st 6mos of life is rare ● partially may be d/t breastmilk (contains Ab to PPS Oral Exam Reviewer 2020 ▪ Bacillary dysentery (bloody diarrhea, fever, abdominal cramps, rectal pain, mucoid stools) ● clinically similar regardless of infecting serotype ▪ Diarrhea: large volume, watery → frequent smallvolume, bloody mucoid stools (most never progress to bloody stools) ▪ High fever, severe abdominal pain, emesis, anorexia, generalized toxicity, urgency, painful defecation, dehydration ● Fever distinguishes shigellosis from EHEC invented. ▪ Indications: those at high risk for invasive disease ● persistent complement component deficiencies ● anatomic functional asplenia (including sickle cell dse) ● HIV ● Travelers to/resident of areas where it is hyper/endemic including countries in the African meningitis belt or the Hajj, ● Belonging to a defined risk group during a community or institution meningococcal outbreak ▪ Dose and Schedule CONJUGATE: ● MCV4-D - For children 9-23 months, give 2 doses 3 months apart; For children 2 yrs and above, give one dose except in cases of asplenia, HIV, persistent complement component deficiency where 2 doses given 8 weeks apart are recommended ● MCV4-TT- Infants 6-12 weeks old, give first 2 doses at least 2 months apart, the 3rd (booster) dose is at age 12 months; For children from 12 months of age to adolescence: 1 dose only ● MCV-CRM - given to children 2 years and above as single dose; revaccinate with a MCV4 Q5 years as long as the person remains at increased risk of infection POLYSACHARRIDE: ● MPSV4 - given to children 2 years and above as a single dose. if used for high risk as the 1st dose, a 2nd dose using MCV4 should be given 2 months later. No booster. (see PPS PIDSP PFV Childhood Immunization Schedule 2020 Annotations for Vaccines for High-Risk/Special Groups for further reading) ▪ Fluid and electrolyte correction and maintenance ▪ Nutrition: high protein and caloric diet enhances growth in 6mos after infection ▪ Do not use drugs that impair intestinal motility (e.g. loperamide) → can prolong the illness ▪ single dose Vitamin A 200,000 IU ▪ Zinc 20mg x 2wks ▪ Antibiotics ● 1st line: Ciprofloxacin (WHO DOC for all patients with bloody diarrhea regardless of age); Ceftriaxone (if parenteral ▪ DIC → avascular necrosis of epiphyses & epiphysealmetaphyseal defects → growth disturbance and late skeletal deformities PROGNOSIS ▪ Case fatality is 5-10% for invasive meningococcal dse ▪ Delayed initiation of supportive therapy: poor outcome ▪ Poor prognostic factors: hypothermia or extreme hyperpyrexia, hypotension/shock, purpura fulminans, seizures, WBC, plt, DIC, acidosis, endotoxin & TNF α; petechiae <12h before admission, absence of meningitis, or N ESR → rapid, fulminant progression → poor prognosis PREVENTION ▪ Droplet precaution x 24 hours after initiation of therapy POSTEXPOSURE PROPHYLAXIS ▪ Indications ● Household contact esp. < 2yo ● Childcare or preschool contact at any time during 7d before onset of illness ● Direct exposure to index patient (kissing, sharing toothbrush) during 7d before onset of illness ● Medical personnel w/ exposure to aerosols of resp. secretions (mouth to mouth resuscitation, intubation, suctioning ≤24hr after antibiotic therapy) during 7d before onset of illness ● Frequently slept in same dwelling as index px during 7d before onset of illness ● Passengers seated directly next to index case during airline flights >8hrs ▪ Most effective: Ceftriaxone 125-250mg IM x 1dose + Ciprofloxacin 20mg/kg (max 500 mg) PO x 1dose ▪ Most widely used: Rifampin 5-10mg/kg Q12 x 2days but fails to eradicate colonization in 15% of cases ▪ Alternative (not recommended routinely): Azithromycin 10mg/kg (max 500 mg) PO x 1dose COMPLICATIONS Intestinal ▪ Rectal prolapse, toxic megacolon, pseudomembranous colitis ▪ Rare: intestinal obstruction and appendicitis ± perforation Extraintestinal ▪ Most common complication: dehydration ▪ Severe hypoNa (<126), hypoglycemia, focal infections (osteomyelitis, arthritis, splenic abscess, vaginitis – typically Batch Clingy disease risk: ↑ likelihood of exposure to new strain or ↑ proximity to a carrier (kissing, bar patronage, binge drinking, night clubs, MSM, living in freshman college dorm) ▪ Risk of carriage: damage of NP mucosa – smoking, respiratory viral infection 218 INCUBATION PERIOD: 12h to several days TRANSMISSION ▪ Fecal – oral ● Vector: contaminated food (extensive handling of ingredients) and water PATHOGENESIS ▪ Shigella can survive the acid in the stomach → colon (target organ) w/ changes most intense in the distal part ▪ In the colon, it crosses the epithelium by transcytosis through M cells → encounters & kills macrophages by activating inflammasome inducing pyroptosis, apoptosis, & proinflammatory signaling → move to the basolateral side, cytoplasm and adjacent cells → activates immune response attracting NK cells and PMNs → disintegrates epithelial lining facilitating more invasion of bacteria but ultimately, phagocytosed and killed by PMNs E. COLI Shiga toxin ▪ Produced by S. dysenteriae serotype 1, Shiga toxin-producing E. coli & occ. other Shigella spp. ▪ Inhibits protein synthesis to injure vascular endothelial cells and trigger severe complications of HUS ▪ Gram negative, facultative, anaerobic, bacilli ▪ Commensal & ubiquitous among humans starting 1st mo of life, normal fecal flora ▪ Categorized by serogroup O (serotype) & H (flagellar antigen) ▪ Cause frequent infections in 1st yr of life ▪ 30-40% of all diarrheal cases worldwide ▪ Warm months in temperate climates & rainy seasons on tropical climates TRANSMISSION ▪ Fecal-oral – contaminated food or water, contact w/ infectious person, fomite, or carrier animal and its environment PPS Oral Exam Reviewer 2020 ▪ Untreated diarrhea can last 7-10 days ▪ 10% persists > 10 days (AIDS, malnourished, occ. normal children) ▪ PE: Abdominal distention and tenderness, hyperactive bowel sounds, tender rectum (DRE), rectal prolapse (esp. for malnourished) ▪ Extraintestinal findings: neurologic (convulsions, HA, lethargy, confusion, nuchal rigidity, hallucinations before or after the onset of diarrhea) therapy is warranted) DIAGNOSTICS ▪ Presumptive data supporting dx of bacillary dysentery ● Stool: leukocytes (>50-100/hpf, confirms colitis), fecal blood ● Blood: leukocytosis with shift to left, usually 500015000cells/µL, leukopenia, leukemoid reaction >40 000/ µL may occur ▪ Culture: stool & rectal swab ▪ Quantitative PCR (see Nelson’s 21st Ed., Fig. 226.1 for Management algorithm: guidelines for treatment of shigellosis) ● 2nd line: Azithromycin, Cefixime or TMP-SMX (if known to be susceptible) ● Treatment should be reserved for moderate to severe disease, IC, or to prevent or mitigate outbreaks (childcare or food handling) ● A child with typical dysentery who responds to initial treatment should be continued x 5d even if stool culture is (-) DIFFERENTIAL DIAGNOSIS ▪ Enteroinvasive E. coli (EIEC), C. jejuni, Salmonella spp., Shiga toxin-producing E. coli (O157:H7), Yersinia enterocolitica, Clostridium difficile, E. histolytica ▪ IBD PATHOGENESIS ▪ Pathogenicity: virulence characteristics (adherence factors & toxins) ▪ To produce diarrhea, adherence to glycoprotein or glycolipid receptor on intestinal cell → injure or disturb the function of intestinal cell Post-infectious ▪ HUS, reactive arthritis, IBS, protein losing enteropathy ▪ Cholestatic hepatitis, conjunctivitis, iritis, corneal ulcers, pneumonia (2-5wks after), cystitis, myocarditis ▪ Neonates: septicemia, meningitis, dehydration, colonic perforation, toxic megacolon PREVENTION ▪ Control measure: After cessation of symptoms, should avoid recreational water activities for 1 additional week ▪ Encourage breastfeeding ▪ Proper handwashing ▪ Children should be excluded in child-care facilities ▪ No vaccine, but measles immunization can reduce the incidence and severity of diarrheal disease including Shigellosis ▪ CSF: pleocytosis with minimally ↑ protein levels can be seen but meningitis caused by shigellae is rare ▪ Vertical transmission (neonatal sepsis), UTI (ascending), epididymitis (sex in men who practice insertive anal sex), pneumonia (nosocomial by aspiration or hematogenous spread) with blood-tinged discharge assoc. S. flexneri) ▪ Sepsis (malnourished or IC), leukemoid reaction ▪ Ekiri syndrome/lethal toxic encephalopathy: severe toxicity, convulsions, extreme hyperpyrexia, HA, brain edema, rapidly fatal outcome without sepsis or significant dehydration ▪ Uncommon: DIC (malnourished), bacteremia (girls or women w/ HIV, malnourished) PROGNOSIS ▪ 20% mortality: sepsis ▪ Severity of illness and risk of death greatest with S. dysenteriae type 1 and least with S. sonnei GENERAL MANAGEMENT ▪ Appropriate fluid and electrolyte therapy ▪ Early refeeding – within 6-8hours of initiating rehydration ▪ Oral zinc ▪ Other than for a child recently returning from travel in the developing world, empirical treatment of severe watery diarrhea with antibiotics is seldom appropriate DIFFERENTIAL DIAGNOSIS ▪ Presumptive diagnosis of intussusception, appendicitis, IBD, ischemic colitis PREVENTION ▪ Prolonged breastfeeding ▪ Personal hygiene ▪ Proper food and water handling ▪ Standard and contact precautions ● Outbreak in childcare center: child should not be permitted to reenter until 2 (-) stool cultures obtained at least 48h after antimicrobial therapy has been discontinued, stools contained in the diaper or if child is continent, stool frequency is ≤ 2 stools above normal Batch Clingy both virulence plasmid-coded Ag and LPS) ▪ In endemic areas, 75% of family members have asymptomatic infection ▪ In developed countries: most common cause is S. sonnei ff. by S. flexneri 219 PPS Oral Exam Reviewer 2020 ▪ Acute 3-7 days ▪ Explosive watery, nonmucoid, nonbloody diarrhea, abdominal pain, n/v, little or no fever DIAGNOSTICS ▪ Detection of enterotoxins by EIA or PCR ▪ Self-limited ▪ Resolves in 3-5 days but occ. >1 week SPECIFIC MANAGEMENT ▪ 1st line: Azithromycin 12mg/kg (day1) → 6mg/kg QD (day2-3) (AAP Red Book 2018) ▪ Acute ▪ Watery diarrhea or dysentery syndrome with blood, mucus ▪ Fever, systemic toxicity, crampy abdominal pain, tenesmus, urgency DIAGNOSTICS ▪ ↑ Peripheral blood PMN w/ left shift ▪ Fecal leukocyte examination of the stool is often positive ▪ Detection of invasion plasmid antigen of Shigella (ipaH) by PCR SPECIFIC MANAGEMENT ▪ Often initially tx as Shigellosis prior to culture ▪ If susceptible, TMP-SMX is appropriate ▪ Acute or prolonged (>7d), persistent (>14d) ▪ Profuse watery, non-bloody diarrhea with mucus, vomiting, low-grade fever DIAGNOSTICS ▪ Detection of intimin gene (eae) ± bundle-forming pili (bfpA) by PCR ▪ Absence of Shiga toxins ▪ Tissue culture assay: Hep-2 cells adherence assay (LA, LLA) ▪ Complication: Malnutrition ▪ Toxins: EspF, Map, EAST1, SPATEs SPECIFIC MANAGEMENT ▪ TMP-SMX may be effective in speeding resolution but lack of a rapid diagnostic test in resource-poor setting makes treatment decisions difficult ▪ Acute 4-7 days ▪ May be asymptomatic ▪ Mild diarrhea to severe hemorrhagic colitis ▪ Abdominal pain w/ watery diarrhea, → bloody in 1-4d ▪ Infrequent fever (unlike Shigella and EIEC) ▪ Toxins: Shiga toxins (Stx1, Stx2 & var STx2) ▪ Acute, prolonged, or persistent ▪ Watery, mucoid, secretory diarrhea ▪ Low grade fever and little or no vomiting DIAGNOSTICS ▪ ↑ Peripheral blood PMN w/ left shift ▪ Detection of Shiga toxins by EIA or PCR (Stx1, Stx2) ▪ Stool culture to detect E. coli O157 ▪ Simultaneous culture for O157 and nonculture assays to detect Shiga toxin ▪ Confirmation: latex agglutination tests SPECIFIC MANAGEMENT ▪ Do not give antibiotics – higher risk for HUS ● antibiotics induce bacterial stress → toxin production → phage-mediated bacterial lysis with toxin release DIAGNOSTICS ▪ Detection of AggR, AA (aggregative adherence) plasmid, and other virulence genes by PCR ▪ Tissue culture assay: HEp-2 cells adherence assay ▪ Most recover without complications ▪ HUS: develops in 5-10%; acute kidney failure, plt, MAHA (microangiopathic hemolytic anemia) ● at risk: young children w/ bloody diarrhea & neutrophilic leukocytosis ▪ Older: can also develop HUS or TTP ● HUS: MAHA, ARF, plt, mild CNS symptoms ● TTP: MAHA, Renal dysfunction, plt, severe CNS, fever ▪ Growth retardation, malnutrition in infants of developing world Batch Clingy ENTEROTOXIGENIC (ETEC) ▪ At risk: >1yo and travelers ▪ Most common cause of traveler’s diarrhea Watery Bloody ▪ Incubation Period: 1-5 days ▪ Secrete heat-labile enterotoxin (LT) and/or heat+++ stable enterotoxin (ST) → induces ion and water secretion into the intestinal lumen ▪ Produces type IV pilus/ longus: colonization factor ENTEROINVASIVE (EIEC) ▪ At risk: >1yo ▪ Occurs in outbreaks, endemic in developing countries ▪ Incubation: 3-4 days (range: 1-8 days) + ++ ▪ Mucosal invasion and inflammation of large bowel ▪ Shares virulence genes w/ Shigella spp (+) LPS, nonmotile (lack H or flagellar antigens), not lactose fermenting ENTEROPATHOGENIC (EPEC) ▪ At risk: <2yo ▪ Occ. cause of outbreaks in pediatric wards & daycare ▪ Small bowel adherence and effacement → blunting of villi, local inflammatory changes, sloughing of superficial mucosal cells (duodenum +++ + through the colon) ▪ Characteristic attaching and effacing histopathologic lesion: intimate attachment of bacteria to epithelial surface and effacement of host cell microvilli encoded by locus of enterocyte effacement SHIGA TOXIN-PRODUCING (STEC)/ ENTEROHEMORRHAGIC (EHEC)/ VEROTOXIN-PRODUCING (VTEC) ▪ At risk: 6mos-10yo & elderly ▪ E. coli O157:H7 – most virulent; most freq. assoc. with HUS + +++ ▪ Incubation Period: 1-9 days (often 3-4 days) ▪ Affects the colon → adhere to intestinal cells → produces Shiga toxins → systemic circulation → damage of vascular endothelial cells → activation of coagulation cascade, formation of microthrombi, intravascular hemolysis, ischemia ENTEROAGGREGATIVE (EAEC OR EGGEC) ▪ At risk: < 2yo, HIV ▪ 2nd most common cause of travelers’ diarrhea +++ + ▪ Forms characteristic biofilm on intestinal mucosa → shortening of villi, hemorrhagic necrosis, 220 inflammatory responses ▪ 3 phases of pathogenesis ● Adherence to intestinal mucosa by fimbriae ● Enhanced production of mucus ● Production of toxins that result in damage to mucosa DIFFUSELY ADHERENT (DAEC) ▪ At risk: >1 yo & travelers (adults) ▪ Express diffuse adherence pattern phenotype ▪ Loss of microvilli and decrease in the expression and enzyme activities of functional brush-borderassociated proteins ENTEROAGGREGATIVE HEMORRHAGIC E. COLI / SHIGA-TOXIN PRODUCING EAEC ▪ E. coli O104:H4 ▪ hybrid pathogen: colonization mechanisms similar to EAEC; toxin prod. typical of STEC ● stronger adherence allowing more toxin delivery to target cells ▪ Tightly spiraled motile spirochete w/ finely tapered ends ▪ Asymptomatic for years or do not recognize the early signs Pathogenic genus subspecies ▪ Venereal species – T. pallidum subsp pallidum ▪ Yaws – T. pallidum subsp pertenue ▪ Bejel or endemic syphillis – T. pallidum subsp endemicum ▪ Pinta - T. pallidum subsp carateum PRIMARY SYPHILIS ▪ Chancre: painless papule at the site of entry (usually genitals, occ. mouth or other extragenital sites) → clean, painless, highly contagious ulcer w/ raised borders, heals spontaneously w/in 4-6 weeks → thin scar ▪ Regional lymphadenitis (enlarged and nontender) ACQUIRED SYPHILIS SECONDARY SYPHILIS ▪ Untreated 1o syphilis related to spirochetemia 2-10 weeks after chancre heals ▪ Generalized non-pruritic MCP rash on the palms and soles (pustules can develop) ▪ Condyloma lata – gray white to erythematous wart-like papules in moist areas: anus, scrotum and vagina TRANSMISSION ▪ Sexual ▪ Transfusion of contaminated blood or direct contact with infected tissues PPS Oral Exam Reviewer 2020 - SPECIFIC MANAGEMENT ▪ For travelers: Ciprofloxacin or Rifaximin ▪ Acute ▪ Toxins: SPATEs (Sat)/serine-protease autotransporters of Enterobacteriaceae expresses Afa DIAGNOSTICS ▪ Detection of Dr adhesins (daaC or dad) and Dr-associated genes by PCR ▪ Tissue culture assay: Hep2-cells adherence assay ▪ Hemorrhagic colitis; some developd HUS DIAGNOSTICS ▪ Serologic testing for syphilis remains the principal means for diagnosis; involves a 2-step screening process NON-TREPONEMAL Venereal Disease Research Laboratory (VDRL) & Rapid plasma reagin (RPR) ▪ Sensitive ▪ Detects Ab against phospholipid Ag on the treponeme surface that cross react with cardiolipin-lecithin-cholesterol antigens of damaged host cells ▪ Titers increase with active disease, decline with active treatment; becomes nonreactive within 1 year of adequate therapy for 1o and within 2 years for 2o ▪ 15-25% becomes serofast (prolonged persisting low levels of titer) ▪ Mucus patches – white plaques in mucous membrane ▪ Flu-like illness w/ renal, hepatic, and ophthalmologic, neurologic symptoms ▪ Even w/o treatment, becomes latent 1-2 months after onset of rash ▪ Relapses occur during 1 st year latency (early latent period) → late syphilis follows & maybe asymptomatic (late latent) or symptomatic (tertiary) TREPONEMAL ▪ Traditionally used to confirm diagnosis ▪ Measures specific T. pallidum Abs ▪ (+) soon after initial infection; positive for life even with adequate therapy ▪ Titers do not correlate with disease activity TERTIARY SYPHILIS (see Nelson’s 21st Ed., Fig. 245.8 A, for Traditional laboratory testing MANAGEMENT ▪ Test all pregnant women, selective testing of adolescents based on lesions and risk factors ▪ Test ALL (+) SYPHILIS for HIV ▪ Pregnant women treated in the past do not require additional therapy unless w/ reinfection (4-fold ↑ in titer) ▪ Desensitize if allergic to penicillin ▪ Sexual partners should be treated if person exposed for ● ≤ 90d before the diagnosis even if seronegative ● > 90d before the diagnosis IF seropositive or if serologic tests are not available For 1O, 2O, early latent ▪ Benzathine Pen G 50,000 U/kg (pedia) up to 2.4M U (adult; max. dose) per IM x 1 dose ▪ If allergic & not pregnant: Doxycycline 100mg PO BID x14d For late latent ▪ Benzathine Pen G 50,000 U/kg (pedia) per IM x 3 doses at 1week intervals; 2.4 M U (adult) per IM each buttock x 3 doses at 1-week intervals ▪ 2nd line: Doxycycline 100mg PO BID x 4wks For tertiary: Benzathine Pen G 7.2M total administered as 3 doses of 2.4M U/IM at 1-week interval; if allergic, consult ID specialist For neurosyphilis: ▪ Pedia: Aqueous crystalline Pen G 200,000–300,000 U/kg/day Batch Clingy INCUBATION PERIOD: 2-6 weeks TREPONEMA PALLIDUM ++ ▪ Toxins: SPATEs (Pic, Pet), ShET1, EAST1 221 NEUROSYPHILIS ▪ Can occur at any stage ▪ Syphilitic meningitis, uveitis, seizures, optic atrophy, dementia ▪ CSF: pleocytosis and ↑ protein ▪ CSF VDRL test is poorly sensitive CONGENITAL SYPHILIS TRANSMISSION ▪ Transplacental or intrapartum contact with infectious lesions ▪ Can occur at any stage of pregnancy – highest during the 1st 4 years of primary infection, secondary infection and early latent disease ▪ Vasculitis → necrosis and fibrosis ▪ Untreated during pregnancy results in vertical transmission (100%) → symptoms appear w/n weeks or months Early signs (1st 2 years of life) ▪ Most are asymptomatic at birth ▪ Hepatosplenomegaly, jaundice, elevated LFT ▪ Liver involvement: bile stasis, fibrosis, extramedullary hematopoiesis ▪ Diffuse LAD resolves spontaneously (shotty nodes persist) ▪ Osteochondritis, periostitis, erythematous MP or VB → desquamation hands & feet ▪ Osteochondritis: painful → irritability and refusal to move the involved extremity (Pseudoparalysis of Parrot) ▪ Mucous patches, persistent rhinitis, condyloma ▪ FTT, nephritis, NS, HTN, hematuria, proteinuria, hypoproteinemia, hypercholesterolemia, hypocomplementemia ▪ Less common: AGE, peritonitis, pancreatitis, pneumonia, glaucoma, chorioretinitis, non-immune hydrops, testicular masses ▪ Labs: Coombs-negative hemolytic anemia, thrombocytopenia ▪ Bone: Wimberger lines – demineralization medical prox. tibial metaphysis; multiple osteochondritis (wrists, elbows, ankles, knees), periostitis of the long bones and skull (rare) Late sign (1 st 2 decades, rare) ▪ Skeletal changes: persistent or recurrent periostitis and thickening of the involved bone. ▪ Hutchinson teeth: can lead to repeated caries → tooth destruction PPS Oral Exam Reviewer 2020 algorithm for syphilis and B, for Suggested alternate testing algorithm) ▪ Darkfield or direct fluorescent Ab microscopy of scrapings from primary lesions, congenital or secondary lesions can reveal T. pallidum (confirmatory) but are usually not available in clinical practice DIFFERENTIAL DIAGNOSIS ▪ Beyond early infancy, consider ABUSE ▪ Oral lesions: aphthous ulcers, herpes ▪ Nipple: cellulitis or eczema ▪ (+) CSF VDRL warrants treatment for neurosyphilis DIAGNOSTICS ▪ For infants with proven or highly probable disease or abnormal findings ● RPR, VDRL, CBC, LFT, long-bone radiographs, ophthalmologic examination, ABR and others, as indicated ▪ Serological tests (nontreponemal and treponemal) ● In congenital infection, VDRL/RPR can become NR within a few mos. after adequate treatment ● Neonatal tests can be confounded by maternal IgG Ab transferred to fetus ● Passively acquired Ab is suggested by titer at least 4-fold less than maternal titer; can be verified by decline in Ab in the infant (undetectable 3-6mos of age) (see Nelson’s 21st Ed., Fig. 245.10 for Algorithm for evaluation and treatment of infants born to mothers with reactive serologic tests for syphilis) ▪ Examine placenta grossly & microscopically: disproportionately large placenta with focal proliferative villitis, endovascular and perivascular arteritis, focal or diffuse immaturity of the placenta villi ▪ For infant (+) VDRL or RPR, normal PE, mothers are inadequately treated: further evaluation is not necessary if 10 days of parenteral therapy is administered IV q4-6 x 10-14 days, not to exceed adult dose ▪ Adults: Aqueous crystalline Pen G 18-24M U/day administered as 3-4M U/IV q4 x 10-14 days or Pen G procaine 2.4M U/IM + Probenecid x 10-14days ▪ If allergic, desensitization most reliable strategy Jarisch Herxheimer Reaction ▪ Transient acute systemic febrile reaction caused by massive release of endotoxin like antigens in 15-20% of syphilis ▪ Do not discontinue penicillin PREVENTION ▪ Early diagnosis ▪ Timely treatment ▪ Routine prenatal screening ▪ Stillborn ≤ 20 week: test for syphilis ▪ Maternal status should be documented at least once MANAGEMENT (from DOH National Antibiotic Guidelines 2018) ▪ Aqueous Pen G 100,000-150,000 units/kg/day per IV administered as 50 000U/kg Q12 during the first 7 days of life and Q8 thereafter for a total of 10-15 days OR ▪ Procaine Pen G 50,000 units/kg/dose IM x 1 dose for 10-15 days ▪ Ff. up every 2-3 months to confirm 4-fold decrease in nontreponemal titer ▪ If (+) neurosyphilis: ff. up clinically w/ CSF evaluation every 6 months; at 2 yo: assess neurodevelopment COMPLICATIONS ▪ Obliterating endarteritis, 40% fetal or perinatal death, prematurity delivery PREVENTION ▪ Adequate maternal treatment at least 30 days prior to delivery Batch Clingy ▪ 1/3 untreated case: neurologic, cardiovascular & gummatous lesions (nonsuppurative granulomas of the skin, bone, liver; from host cytotoxic T-cell response) 222 ▪ Saddle nose: assoc. w/ perforated nasal septum ▪ Rare: soft tissue gummas & paroxysmal cold hemoglobinuria ▪ Other ocular: choroiditis, retinitis, vascular occlusion, optic atrophy (See Nelson’s 21st Ed., Table 245.1, for Late Manifestations of Congenital Syphilis) INCUBATION PERIOD Invasive: variable H. INFLUENZAE TRANSMISSION ▪ Direct contact or inhalation of respiratory droplets ▪ Neonates: intrapartum by aspiration of amniotic fluid or by contact with genital tract secretions PATHOGENESIS ▪ Hib: adherence to respiratory epithelium → colonization of the NP → once in bloodstream, Hib resists intravascular clearance mechanisms ▪ Noninvasive H. influenzae: gain access to sites by direct extension causing OM, sinusitis, and bronchitis Antibiotic Resistance ▪ 1/3 produces β-lactamase ▪ β-lactamase-negative ampicillin-resistant isolates manifest resistance by production of a β-lactaminsensitive cell wall synthesis enzyme (PBP3) Immunity ▪ Host defense: Ab against type b capsular polysaccharide polyribosylribitol phosphate (PRP) ▪ Unimmunized >6mos & young children lacked an PPS Oral Exam Reviewer 2020 MENINGITIS ▪ clinically similar with meningitis caused by other organisms CELLULITIS ▪ often have antecedent URTI – seeding to soft tissues during bacteremia ▪ Most common sites: head, neck (cheek & preseptal eye) ▪ Buccal ● Erythematous ± violaceous hue ● Dx: aspirate lead edge (seldom performed) & blood culture, consider diagnostic LP (<18mos) ▪ Preseptal ● Uncomplicated preseptal cellulitis does not imply a risk for visual impairment or direct CNS extension; However, concurrent bactermemia may be assoc. w/ meningitis ● Fever, edema, tenderness, warm lid, occ. purple discoloration ● Dx: Do blood culture & LP ● DDX: S. pneumoniae, S. aureus & GAS (latter 2: no fever & interrupted integument) ▪ Orbital ● Complications of acute ethmoid or sphenoid sinusitis ● Lid edema, proptosis, chemosis, impaired vision, limited EOM, ↓ mobility of globe or pain on movement ● Dx: CT, to delineate from preseptal cellulitis CONJUNCTIVITIS ▪ most commonly caused by nontypeable isolates Respiratory SUPRAGLOTTITIS/ACUTE EPIGLOTTITIS PNEUMONIA OTITIS MEDIA ● 29% H. influenzae SINUSITIS ▪ Initial for invasive disease: parenteral extended-spectrum cephalosporin effective in sterilizing all foci & effective against ampicillin-resistance strains ▪ DOC for susceptible isolates: Ampicillin (IV)/ Amoxicillin (oral) ● if resistant to ampicillin: ceftriaxone; if resistant to amoxicillin: amox + clavulanate or 2 nd or 3rd gen cephalosporin COMPLICATIONS ▪ Sequelae of meningitis: behavior problems, language disorders, impaired vision, mental retardation, motor abnormalities, ataxia, seizures, hydrocephalus ● 6% are left with hearing impairment (inflammation of the cochlea and labyrinth) → ▪ Meningitis ● IV antibiotics x 7-14 days ● Dexamethasone (0.6 mkday q6 x 2d) – to decrease incidence of hearing loss ▪ Buccal Cellulitis ● Parenteral antibiotic until afebrile x then switch to oral to complete 7-10 days ▪ Preseptal Cellulitis ● Parenteral antibiotics (also active against MSSA, MRSA, S. pneumoniae, GABHS) x 5d until fever & erythema have abate; complete for total of 10 days ▪ Orbital Cellulitis ● Parenteral x at least 14 days; if with sinusitis or orbital abscess, may require surgical drainage ± prolonged antibiotic POSTEXPOSURE PROPHYLAXIS ▪ Indications ● Household with at least 1 contact younger than 4 yo who is unimmunized or incompletely immunized ● Household w/ child <12mos who did not complete the primary Hib series ● Household w/ contact who is an IC child, regardless of that child’s immunization status or age ● Preschool and childcare center contact when ≥ 2 have occurred w/in 60 days ● For index patient, if < 2yo or member of a household w/ a susceptible contact and treated w/ a regimen other than cefotaxime or ceftriaxone, chemoprophylaxis at the end of therapy for invasive infection ▪ Rifampin, orally QD x 4d (CI: pregnant) ● 0 – 1 month old: 10mkdose ● >1 month old: 20mkdose (max: 600 mg/dose) ▪ Epiglottitis ● 1st line: Ceftriaxone x 7-10 days; 2nd line: Ampi-sul x 10 days (DOH National Antibiotic Guidelines 2018) ▪ Pneumonia ● < 12 mos. whom H. influenza is suspected, give parenteral antibiotics because of increased risk of bacteremia ● older children not severely ill: oral antimicrobial treat x 7-10 days ▪ Suppurative Arthritis ● Uncomplicated – parenteral 5-7d x ≥ 3wks until CRP is normal ● If GS shows G(-) organism: cefotaxime 100-200mkday or ceftriaxone 100mkday (DOH National Antibiotic Guidelines 2018) ▪ AOM ● Amoxicillin 80-90 mkday (<2yo: 10d, 2-5yo: 7d, >5yo: 5-7d) or Ceftriaxone (single dose) ● If (+) anaphylaxis: Clarithromycin 15mkday Q12 PROGNOSIS ▪ Presence of SIADH or FND during meningitis are poor prognostic features ▪ Supraglottitis is a MEDICAL EMERGENCY; potentially lethal Batch Clingy ▪ Fastidious, gram negative, pleomorphic coccobacillus ● requires Factor X (hematin) and Factor V (phosphopyridine nucleotide) for growth ▪ > 90% in children < 5yo; majority < 2yo ▪ Humans are the only natural hosts (normal flora in 60-90%) ▪ Unvaccinated infants are at increased risk of recurrence ▪ Among age-susceptible household contacts exposed to invasive Hib, increase risk for secondary cases in the 1st 30 days ▪ Most remain colonized during 1 st 24 hours of therapy 223 DIAGNOSTICS ▪ Presumptive: Gram staining (difficult to visualize) ▪ Culture: should not be exposed to drying or extreme temperatures ▪ Serotyping: slide agglutination with type specific antisera ▪ NAAT, PCR SUPPURATIVE ARTHRITIS (3%) ▪ Single large joints (knee, hip, ankle, elbow), 6% multijoint PERICARDITIS ▪ Antecedent URTI ▪ Fever, respiratory distress, tachycardia ▪ Dx: recover from blood or pericardial fluid, GS, or detection of PRP in pericardial fluid, blood or urine BACTEREMIA WITHOUT AN ASSOCIATED FOCUS ▪ Fever without any apparent focus of infection ▪ Risk factors: fever ≥ 39OC & leukocytosis ≥ 15 000 cells/µ ▪ If untreated: 25% developed meningitis ▪ Dx: reevaluate focus of infection, 2 nd blood culture, LP, CXR MISCELLANEOUS INFECTION ▪ Rare: UTI, epididymo-orchitis, cervical adenitis, acute glossitis, infected thyroglossal duct cysts, uvulitis, endocarditis, endophthalmitis, primary peritonitis, osteomyelitis & periappendiceal abscess INVASIVE DISEASE IN NEONATES ▪ Bacteremia w/ sepsis, pneumonia, RDS + shock, conjunctivitis, scalp abscess or cellulitis, meningitis, mastoiditis, septic arthritis, congenital vascular eruption GROUP B STREPTOCOCCUS Streptococcus agalactiae ▪ Major pathogen for neonates, pregnant women and nonpregnant adults ▪ Facultative anaerobic gram (+) cocci (chains or diplococci) ▪ 10 distinct GBS capsular polysaccharides identified (types Ia, Ib, II-IX) ● important virulence factors and stimulators of antibody-assoc. immunity ▪ Incidence of neonatal GBS disease is higher in premature and LBW infants although most cases occur in full-term infants ▪ Vaginal or rectal colonization in 30% of pregnant women ● maternal intrapartum chemoprophylaxis led PPS Oral Exam Reviewer 2020 ▪ Two syndromes of neonatal GBS disease Early Onset Late Onset Age at onset 0-6 days 7-90 days ↑ risk after OB Yes No complications Common Bacteremia, Sepsis, clinical meningitis, pneumonia, manifestations osteomyelitis, other meningitis focal infections Common 1a, 1b, II, III predominates serotypes III, V Case fatality 4.7% 2.8% rates ● Amoxicillin-clavulanic acid: for treatment failure or if β lactamase producing isolate (tympanocentesis or drainage fluid) ▪ Sinusitis ● Amoxicillin, Amoxicillin – Clavulanic Acid x 10d ▪ Conjunctivitis ● Tobramycin or Levofloxacin 2 drops QID x 5-7 days (DOH National Antibiotic Guidelines 2018) ● Avoid topical FQ (broad spectrum, cost, emerging resistance) Vaccine Hib Conjugate Vaccine (IM) 3 doses series with minimum age of 6 weeks and minimum interval of 4 weeks Booster dose: 12-15 mos. of age, interval of 6 mos. from the 3rd dose High Risk: - Chemotherapy recipients, anatomic/ functional asplenia including SCD, HIV, Ig or early component complement deficiency Children 12-59 months - Unimmunized * or w/ one vaccine dose received <12 months, give 2 additional doses 8 weeks apart - w/ >2 vaccine received <12 month, give 1 additional dose - <5 yrs old who received a vaccine dose during or w/in 14d of starting chemotherapy or RT, rpt the dose at least 3 months after completion - (+) Hematopoietic stem cell transplant recipients, revaccination w/ 3 doses, 4 weeks apart starting 6-12 months after transplant is recommended, REGARDLESS of vaccination history - Unimmunized* >15 months of age and undergoing elective splenectomy should be given 1 dose of Hib-containing vaccine at least 14d before the procedure - Unimmunized* 5-18 yr + anatomic or functional asplenia (SCD) or HIV, should be given 1 dose * Unimmunized: without primary series and booster those or those w/o at least one dose of vaccine after 14 months of age ▪ Drug of choice: Penicillin G Neonatal Sepsis ▪ Empiric therapy: Ampicillin + Aminoglycoside (Nelson’s) / Cefotaxime or Ceftriaxone (avoid ceftri in jaundiced NB) + Genta or Amik ± Oxacillin or Vanco (if w/ SSTI) (DOH National Antibiotic Guidelines 2018) ● once identified as GBS & with good clinical response, therapy may be completed with penicillin alone ▪ Treat for 10 days Uncomplicated meningitis ▪ High dose of Penicillin or Ampicillin (Nelson’s) / Ampicillin or Cefotaxime (may use Ceftriaxone if cefotaxime is not available and NB is not jaundiced) + Amik or Genta (DOH National Antibiotic COMPLICATIONS ▪ Bacteremia, SSI, bone and joint infections, endocarditis, pneumonia, meningitis ▪ Meningitis: developmental delay, spastic quadriplegia, microcephaly, seizure disorder, cortical blindness, or deafness, PVL ▪ Shock in premature infants PREVENTION ▪ Chemoprophylaxis – effective only during labor ▪ Vaginorectal GBS screening of all pregnant women at 35-37 wks AOG, except for those w/ GBS bacteriuria during current or a previous infant w/ invasive GBS disease ▪ Intrapartum prophylaxis: (+) prenatal screening culture, GBS bacteriuria during pregnancy or previous infant w/ invasive Batch Clingy anti-PRP Ab conc. reflecting maturational delay in the immunologic response to thymus-independent type 2 Ag such as unconjugated PRP → increased susceptibility to disease 224 INCUBATION PERIOD ▪ Early onset <7 days, Late-onset & late-late onset: unknown TRANSMISSION ▪ Early onset: ascending infection or during passage through the birth canal, fetal aspiration of infected amniotic fluid ▪ Late onset: vertical transmission, horizontal acquisition from nursery or other community sources PATHOGENESIS ▪ Type specific capsular polysaccharide (CP): protects from opsonophagocytosis in the nonimmune host and downregulates leukocyte activation ● also contains sialic acid w/c prevents activation of the alternative complement pathway in the absence of type-specific Ab; also dampen inflammatory gene activation ▪ Type specific virulence attributes: ● Type III strains are implicated in most cases of late-onset neonatal GBS disease and meningitis (taken up by brain endothelial cells more efficiently vs. other serotypes) ● also produces hypervirulent GBS adhesin (HvgA): adherence to intestinal and endothelial cells and mediates invasion to CNS ▪ GBS surface proteins: adhesion to host cells ▪ C5a peptidase: inhibit recruitment of PMN ▪ β hemolysin: cell injury in vitro ▪ Hyaluronidase: spreading factor ENTEROCOCCUS ▪ Invasive disease: GBS gain access into the bloodstream by invasion from alveolar space ff. aspiration of infected fluid ● Except Type III: induce TNF α release in vitro ▪ formerly classified as Lancefield group D streptococci PPS Oral Exam Reviewer 2020 Early Onset: ▪ In utero → septic abortion or immediate distress after birth ▪ Non-specific signs, prominent respiratory signs, persistent fetal circulation may develop Late Onset: ▪ Focal: bone and joints, SSI, UTI, lungs, cellulitis & adenitis (submandibular or parotid regions) ▪ Invasive GBS disease beyond early infancy is uncommon ● Bacteremia without a focus is the most common syndrome DIAGNOSTICS ▪ Isolation from blood, urine, or CSF ● GS (CSF, pleural or joint fluid): (+) in cocci in pairs or short chains ● Culture (CSF, blood, suppurative focus) ● Latex agglutination: Lancefield group B CHO Ag ● PCR: CSF, DNA probe assays, NAAT ▪ CBC: ↑ or ↓ neutrophil, ↑ band count, ↑ ratio of bands to total neutrophils or leukopenia ▪ CRP: potential early marker for sepsis (unreliable) ▪ CXR: indistinguishable from RDS Guidelines 2018) ▪ Start immediately after LP or if delayed, after blood culture ▪ Suggest doing additional CSF sample at 24-48h to determine when sterility is achieved ▪ Persist GBS: unsuspected intracranial focus or an insufficient antibiotic dose ▪ Treat for 14 days ● Ventriculitis: at least 4 weeks Recurrent neonatal GBS disease ▪ standard IV antibiotics ff. eradiation of mucosal colonization ● Rifampin is freq. used for eradication Septic arthritis or osteomyelitis: ▪ Vancomycin + Cefotaxime or Ceftriaxone (DOH National Antibiotic Guidelines 2018) ▪ Treat for 3-4 weeks Pneumonia ▪ Ampicillin or Pen G; add Aminoglycosides (for severe infections) (DOH National Antibiotic Guidelines 2018) UTI ▪ Cefotaxime + Amikacin (DOH National Antibiotic Guidelines 2018/PNSP Consensus on UTI in Children 2018) ▪ Treat for 10-14 days GBS disease ● give prophylaxis as well to those with unknown culture and who deliver prematurely, PROM, intrapartum fever ≥ 38OC, (+) NAAT ▪ Preferred agent: Penicillin (alternative: Ampicillin) ● if w/ amnionitis: broad spectrum with activity against GBS ▪ Cefazolin for PCN-intolerant but if at high risk for anaphylaxis, may give Clindamycin ▪ Vancomycin for resistant isolates Maternal immunization ▪ Transplacental transfer of naturally acquired maternal Ab to the GBS capsular polysaccharide protects NB from invasive GBS and that efficient transplacental passage of vaccine induced GBS Ab occurs ▪ Conjugated vaccines (type III polysaccharide coupled to tetanus toxoid): protective in >90% of recipients ▪ GBS vaccine: to induce specific Ab both in the serum and at mucosal surfaces ▪ 2 levels of protection: 1) passive immunity 2) prevent colonization in the reproductive tract that would protect the neonate in utero or childbirth ▪ Due to antigenic variation – trivalent (1a, 1b, III) polysaccharide- protein conjugate vaccine has been developed & under human trials in early phase PROGNOSIS ▪ Mortality is higher in premature infants ▪ Meningitis: 19% neurologic impairment, 25% mild to moderate impairment at long term follow-up ▪ Favorable prognosis: bone or soft tissue infections Neonatal Infections ▪ Early onset: <7d, mimicking early-onset GBS septicemia ANTIMICROBIAL RESISTANCE ▪ Beta-lacmtams PREVENTION ▪ Removal of urinary and IV catheters, debridement of Batch Clingy to a decrease in incidence of early-onset neonatal GBS ▪ Risk factors for early onset sepsis: prematurity, LBW, PROM, intrapartum fever, maternal bacteriuria during pregnancy or previous delivery with GBS 225 ▪ Members ● Enterococcus faecalis: predominant organism; accounts for 80% of infections; colonization in 1st week of life ● Enterococcus faecium: colonization less consistent (25% in adults); accounts for the rest of infections ● E. gallinarum & casseliflavus: rare causes however notable for their low-level intrinsic vancomycin resistance ▪ Disruption of normal intestinal microbiota by antibiotic exposure or HSCT greatly enriches for fecal enterococcal abundance and dramatically increases risk for subsequent blood stream infection TRANSMISSION ▪ Person to person or from contaminated medical devise (e.g. nurseries, NICU) ▪ Contiguous spread (NTD, or in assoc. intraventricular shunt) PATHOGENESIS ▪ Cause disease in children w/ damaged mucosal surfaces or impaired immune response ▪ Hospital-associated enterococci: lack CRISPR (clustered regularly interspaced short palindromic repeat) elements → diverse antimicrobial resistance ▪ Adhesion promoting factors likely account for the propensity of these organisms to cause endocarditis & UTI ▪ Can also form biofilms, facilitating colonization of urinary and vascular catheters) ▪ Other virulence factors: cytolysin, aggregation substance, gelatinase, & extracellular superoxide PPS Oral Exam Reviewer 2020 but milder, often occurring in healthy full-terms ▪ Late onset: ≥ 7d; Risk factors: prematurity, (+) intravascular catheter, NEC, or s/p intraabdominal surgery ▪ More severe: apnea, bradycardia, deteriorating respiratory function, ass. focal infection (scalp abscess & catheter infection) Infections in older children ▪ nosocomial infections (i.e. UTI, bacteremia) ● Risk factors: (+) indwelling catheter, intestinal perforation or s/p surgery, immunodeficiency, cardiovascular abnormalities, residence on Hematology/Oncology unit, prolonged MV, immunosuppression, recent broad-spectrum antibiotic exposure ▪ CLABSI: catheter-associated bloodstream infections DIAGNOSTICS ▪ CSF: minimal abnormality ▪ Culture: sterile body sites or abscess w/ appropriate biochemical and serologic analysis ▪ Rapid automated molecular assay: for direct detection of vanA ● Highly resistant to cephalosporins and semisynthetic penicillins (e.g. nafcillin, oxacillin, methicillin) ● Moderately resistant to extended-spectrum penicillins (e..g ticarcillin, carbenicillin) ● Ampicillin, imipenem, penicillin: most active against these organisms ● Some strains have decreased resistance due to mutations in PBP5 ● Some strains produce a plasmid-encoded B-lactamase → completely resistant to penicillins (thus, use penicillin + Blactamase inhibitor or imipenem or vancomycin) ▪ Aminoglycosides ● all have intrinsic low-level resistance (d/t poor transport across cell wall) → combination of cell wall active agent plus aminoglycoside results in synergestic killing ▪ Vancomycin ● resistance to this and ampicillin prevalent among E. faecium ▪ Resistance to almost all other antibiotic classes warrant individual susceptibility testing necrotic tissue ▪ Decolonization are generally ineffective in eradicating skin or GI carriage of VRE ▪ Standard & Contact precautions: discontinue if with 3 consecutive negative cultures from multiple sites (stool or rectal swab, perineal area, axilla or umbilicus, wound, indwelling urinary catheter or colostomy sites) ▪ Culture obtained after cessation of antimicrobial therapy w/ each culture 1 week apart ▪ Proper diet, oral health, (dental sealants, adequate fluoride), prevention or cessation of smoking) PROGNOSIS Mortality: 6% early-onset, 15% late onset + NEC (see Nelson’s 21st Ed., Table 213.1 for Intrinsic Resistance Mechanisms Among Enterococci and Table 213.2 for Acquired Resistance Mechanisms Among Enteroccoci) MANAGEMENT ▪ Ampicillin – minor localized infection ▪ Nitrofurantoin – uncomplicated UTI ▪ PCN or Ampicillin – invasive (sepsis, meningitis, endocarditis), if susceptible ● Vancomycin + aminoglycoside – PCN substitutes ▪ Endocarditis – may relapse after prolonged therapy due to high level of aminoglycoside resistance ▪ Catheter-associated enterococcal bacteremia – remove catheter, Ampicillin or Vancomycin + Aminoglycoside VANCOMYCIN- RESISTANT ENTEROCOCCI: Oxazolidinone (e.g. linezolid, tedizolid) ▪ Inhibits protein synthesis, bacteriostatic E. faecium & E. faecalis; Linezolid preferred agent for VRE ▪ Causes reversible BM suppression after prolonged use & assoc. rare lactic acidosis & irreversible peripheral neuropathy ▪ Tedizolid – better in vitro activity, favorable pharmacokinetic & toxicity profiles compared to linezolid Daptomycin ▪ cyclic lipopeptide; rapidly bactericidal VS gram positives Quinupristin-Dalfopristin ▪ Streptogramin; inhibits bacterial protein synthesis at 2 Batch Clingy ▪ Gram (+), catalase (-), facultative anaerobes that grow in pairs & short chains ▪ Non-hemolytic (γ-hemolytic), some have α & β hemolysis ▪ Normal inhabitants of GIT in humans & animal kingdom ● also colonize: oral secretions, dental plaque, URT, skin, vagina 226 INCUBATION PERIOD: 3 days (1-7 days) CAMPYLOBACTER JEJUNI TRANSMISSION ▪ Food borne: consumption of raw or undercooked meat, cross contamination w/ food (chicken – classic source); ingestion of raw (unpasteurized) milk ▪ Water borne: shedding of animals contaminating water & food ▪ Airborne droplets: poultry workers (antimicrobials in animal foods, ↑ the prevalence of antibioticresistance) ▪ C.jejuni: person to person (fecal-oral), perinatally, childcare centers (diapered toddlers), shed for weeks – months ▪ Fecal shedding in untreated lasts for 2-3wks (daysmonths) ▪ Nosocomial infection in nurseries PATHOGENESIS ▪ Transit stomach → adhere to intestinal mucosal cells → initiate intestinal lumen fluid accumulation (direct damage to mucosal cells from invasion, enterotoxin and other cytotoxins) ▪ Differ from other enteric pathogens: presence of N- (surface) & O- (flagellae) linked glycosylation ▪ Slipped-strand mispairing in glycosylation loci: antigenic variation PPS Oral Exam Reviewer 2020 Acute Gastroenteritis ▪ Fever, HA, dizziness, myalgias → 1-3 days later, cramping periumbilical pain, loose watery stools, or mucus + bloody stools, n/v ▪ Severe: bloody stools 2-4 days after onset of the symptoms ▪ >1yo: fever may be the only manifestation ▪ Diarrhea lasts 7d & resolves spontaneously ▪ DDx: appendicitis, colitis, or intussusception, IBD (persistent C. jejuni infection) ▪ (+) C. jejuni s/p appendectomy: acute appendicitis, mesenteric lymphadenitis & ileocolitis Bacteremia ▪ Transient asymptomatic to rapidly fatal, prolonged bacteremia (lasting 8-13wk) ▪ Fever, HA, malaise, abdominal pain ▪ Relapsing or intermittent fever, night sweats, chills, weight loss ▪ Lethargy, confusion, but focal neurologic signs are unusual w/o CVD or meningitis Focal Extraintestinal ▪ mostly caused by C. jejuni; rare, mainly neonates and IC ▪ Meningitis, pneumonia, thrombophlebitis, pancreatitis, cholecystitis, UTI, arthritis, peritonitis, pericarditis, endocarditis Perinatal ▪ Maternal C. fetus & C. jejuni: asymptomatic → abortion, stillbirth, premature delivery or neonatal infection with sepsis and meningitis. ▪ C. jejuni rare: severe type ▪ C. jejuni associated with diarrhea that may be bloody COMPLICATIONS ▪ Most common: GBS, reactive arthritis ▪ Severe prolonged infection in patients with ID (i.e. hypogammaglobulinemia, malnutrition, AIDS) DIFFERENTIAL DIAGNOSIS ▪ Shigella, Salmonella, E. coli, Y. enterocolitica, Aeromonas, V. parahaemolyticus, amebiasis GBS ▪ Acute demyelinating disease of PNS – acute flaccid paralysis ▪ Miller-Fischer variant: affects CN; ataxia, areflexia, ophthalmoplegia, cross-reacting Ab to the GQ1b ganglioside found in CN myelin MANAGEMENT Acute gastroenteritis ▪ Fluid replacement, correction of electrolyte imbalance, supportive care ▪ Antibiotics ● may result in shortened duration of symptoms and shedding ● Recommended: bloody stools, high fever, severe course, immunosuppressed, or have underlying disease, high risk for developing severe (pregnant), extra intestinal infections ● DOC: Azithromycin 10mkday PO x 3days, Erythromycin 40mkday q6 x 5days Sepsis ▪ parenteral meropenem or imipenem ± aminoglycoside GBS ▪ supportive care, IVIg, and plasma exchange Reactive Arthritis (3%) ▪ Adolescents & adults esp. (+) HLA B27 ▪ Appears 1-2wks (5-40 days) after the onset of diarrhea ▪ Migratory, large joints, afebrile, resolves without sequelae ▪ Responds to NSAIDS, resolved after 1wk – months ▪ Less common: conjunctivitis, urethritis, & rash (erythema nodosum) Other ▪ IgA nephropathy, immune complex GN ▪ Hemolytic anemia, HUS PREVENTION ▪ Cook meat thoroughly, prevent recontamination ▪ Avoid unpasteurized dairy products ▪ Prevent contamination of water (chlorination) ▪ Avoid infected animals ▪ Breastfeeding – ↓ symptoms but does not ↓ colonization ▪ Standard precaution, in hospital: contact precaution PROGNOSIS ▪ Self limited ▪ Protracted or severe: immunosuppressed (AIDS) ▪ Poor: septicemia in NB, IC (mortality 30-40%) ▪ GBS: more in axonal form; worse prognosis with slow recovery and more neurologic disability Mortality rate: 2%, Batch Clingy ▪ Gram negative, non-spore-forming rods, microaerophilic, anaerobic, oxidase positive ▪ One of the most common causes of human intestinal infections; a leading cause of acute diarrhea ▪ Peak in early childhood and young adulthood (1544 yr) ▪ Repeated infection → increased immunity & rare in adulthood different stages Tigecycline ▪ Inhibits protein synthesis by binding to the 30S ribosome, bacteriostatic ▪ Broad: gram (+/-), anerobic, MRSA, VRE ▪ Avoid <8 yr old Ceftarolline ▪ 5th generation cephalosporin VS MRSA ▪ Highly synergistic w/ daptomycin w/ poor activity vs E. facium DIAGNOSTICS ▪ CBC: moderate leukocytosis with left shift ▪ Stool: leukocytes 75%, blood 50% ▪ Gram stain: less sensitive ▪ Stool culture >90% sensitive, apparent 1-2 days ▪ Blood culture: not apparent until 5-14 days – can grow more slowly which can result to failure due to overgrowth of other enteric bacteria → selective culture media for C. jejuni ▪ To confirm campylobacter enteritis: rectal swabs or stool culture ▪ Rapid diagnosis: direct carbolfuschin test, indirect fluorescence Ab, dark field microscopy, latex agglutination, PCR ▪ Do drug sensitivities: unresponsive, invasive, extraintestinal 227 YERSINIA ENTEROCOLITICA INCUBATION PERIOD : 4-6 days (range 1-14d) TRANSMISSION ▪ Food, water, animal contact, contaminated blood products ▪ Mother to newborn ▪ Excreted in stool for 1-4 weeks PATHOGENESIS ▪ Adherence, invasion, and toxin production ▪ Enter alimentary tract → mucosal ulcerations in the ileum → necrotic lesions of Peyer patches and mesenteric lymphadenitis → septicemia ▪ Suppurative lesions can trigger reactive arthritis and erythema nodosum ▪ Gram negative envelope, obligate intracellular ▪ Most significant human pathogens encode proteins forming a pathway for synthesis of peptidoglycan, including PBP ▪ Military recruits & nursing homes INCUBATION PERIOD: 21 days CHLAMYDIA PNEUMONIAE TRANSMISSION ▪ Person to person through respiratory droplets ▪ NP shedding occurs for months even after treatment PATHOGENESIS ▪ All has major outer membrane protein, but it is the major determinant for C. trachomatis & C. psittaci isolates ▪ Elemental body and reticulate body PPS Oral Exam Reviewer 2020 20% has neurologic sequelae Enterocolitis ▪ Diarrhea, fever, abdominal pain, pharyngitis (20%) ▪ Younger children: Acute enteritis ▪ Older: mesenteric lymphadenitis ▪ Watery stools ± WBC, frank blood, mucus ▪ Duration: 12-22 days Septicemia ▪ Less common ▪ <3mos and immune compromised ▪ Splenic & hepatic abscesses, osteomyelitis, septic arthritis, meningitis, endocarditis & mycotic aneurysms ▪ Infrequent: exudative pharyngitis, pneumonia, empyema, lung abscess, ARDS ▪ Antibiotic for systemic infection and very young children: TMP-SMX, aminoglycosides, 3rd generation cephalosporin, quinolones ▪ Enterocolitis: TMP SXT x 5 days ▪ Severe infections: 3rd generation cephalosporin ± aminoglycosides x 3wks ▪ If on deferoxamine, discontinue iron chelation therapy esp. complicated GI and extraintestinal infection COMPLICATIONS ▪ Erythema nodosum, reactive arthritis, erythema multiforme, hemolytic anemia, thrombocytopenia, systemic dissemination of bacteria, rarely uveitis ▪ Younger: septicemia ▪ Older: reactive arthritis, PREVENTION ▪ Reduce contact with environmental sources ▪ Break or sterilize chain from animal reservoirs to humans PROGNOSIS ▪ Self-limiting disease DIAGNOSTICS ▪ Stool: leukocytes, mucus, blood ▪ Culture: normal sterile sites but if stool (CIN, cefsulodinirgasan-novobiocin) ▪ PCR DIFFERENTIAL DIAGNOSIS ▪ Salmonella, Shigella, Campylobacter, C. difficile, EIEC, Y. pseudotuberculosis, Vibrio-related diarrheal disease ▪ Amebiasis, appendicitis, Crohn disease, ulcerative colitis, diverticulitis, pseudomembranous colitis CLINICAL MANIFESTATIONS ▪ Cannot be differentiated w/ M. pneumoniae ▪ Atypical pneumonia: ● Mild: pertussis-like ● Mild to Moderate: fever, malaise, HA, cough, and pharyngitis ● Severe pneumonia + pleural effusions and empyema ● PE: rales, wheezing, nonexudative pharyngitis ● CXR: worse than the clinical status – mild diffuse or lobar infiltrates with small pleural effusions ▪ Trigger asthma exacerbations ▪ Acute chest syndrome + SCD ● CXR: New consolidation + ≤1 of the following: fever, hypoxia, tachypnea, ↑ work of breathing, chest pain, wheezing, new cough ▪ showed In vitro activity: Tetracycline (not <8yo), macrolides, and quinolones; resistant to sulfonamides ▪ Optimum dose and duration: uncertain because of recrudescent symptoms; usually 2 wks or longer d/t persistent positive cultures ff. 2wks erythromycin & 30d of tetracycline & doxycycline ▪ Studies for eradiation of nasopharynx in 80%: ● Erythromycin 40mkday PO BID x10d, ● Clarithromycin 15mkday PO BID x10d, ● Azithromycin 10mg/kg D1, 5mkday D2-5 ● Duration 10-14d (except Azithromycin x5d) PREVENTION ▪ Standard & Droplet Precaution PROGNOSIS ▪ Clinical response varies ▪ Coughing persists even after therapy Batch Clingy ▪ Complications such as GBS, reactive arthritis, erythema nodosum are due to molecular mimicry ▪ Most common Yersinia sp. causing human disease ▪ Large, gram negative coccobacillus, ferments glucose & sucrose but not lactose, oxidase negative, reduces nitrate to nitrite, motile ▪ Relies on other bacteria for iron uptake and conditions associated with iron overload ▪ Cold months, M>F ▪ Natural reservoirs: pigs, rodents, rabbits, sheep, cattle, horses, dogs, cats (undercooked pork, chitterlings/pig intestines/ “chitlins”), an occupational threat to butchers 228 ▪ AOM (co-infection w/ other bacteria) ▪ Asymptomatic respiratory infection 2-5% and persists for a year TRACHOMA ▪ Most important preventable cause of blindness ▪ Primarily caused by serotypes A, B, Ba, C; in areas endemic for trachoma, it can also be caused by serotypes for genital infections (D – K) ▪ Poverty and lack of sanitation INCUBATION PERIOD ▪ Variable depending on the type of infection, usually 1 week ▪ Azithromycin COMPLICATIONS ▪ Blindness ▪ Bacterial superinfection ▪ 1st line for uncomplicated infections in men & nonpregnant women: Azithromycin or Doxycycline COMPLICATIONS ▪ Inclusion conjunctivitis ▪ Proctocolitis (rectal infection, LGV strain) ▪ Perihepatitis, salpingitis ▪ PID: chronic pelvic pain, 17% infertility, 9% ectopic pregnancy ▪ 50% neonates born to mother with untreated infection will DIAGNOSTICS ▪ Culture & staining during active stage: Confirmation ▪ Serologic: not helpful due to long duration & high seroprevalence TRANSMISSION ▪ Eye to eye (common vector: flies) PATHOGENESIS ▪ Follicular conjunctivitis → heal → scar → entropion (eyelid turning inward w/ lashes abrading cornea) → corneal ulceration → scarring & blindness GENITAL TRACT INFECTIONS ▪ Major cause of epididymitis ▪ Co-infection: N. gonorrhea TRANSMISSION ▪ Perinatally acquired rectal and vaginal ▪ Autoinoculation from the genital tract to the eyes PPS Oral Exa
 0
0
advertisement
Related documents







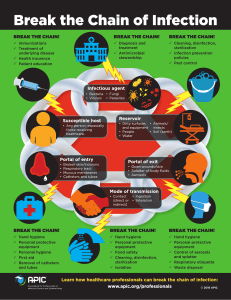
Download
advertisement
Add this document to collection(s)
You can add this document to your study collection(s)
Sign in Available only to authorized usersAdd this document to saved
You can add this document to your saved list
Sign in Available only to authorized users