' !
$ % #
&
' !
* + ,
- . - (
)
".$
* + ,
" . " $
* + ,
1 + $
/
0
".$
".$
* + ,
* + ,
3 2
, $
, $
* + ,
* + ,
!
.
#
- &
4 3 " 6
$ $
%& &'&()
$
! ! *
! " # (
% (27 /27
227 5+(276
4 $
%& &'&( )
5 - 8 8$
9 &7 9 27 8 8
8 8 3 " : +
;
3 # 1 # <
3 $ 5 6 : 8= 8 3 > ? >$
@ 8 >
A 5 >6
> 8 8 >
+
/
: + - ! B C
1 <
! + : + 56 +
1 <
* + $
; D .. - * + " : + : " D > + $
> A 5>(2E6
+ A 5/2E6
!$
F ! 5+ +! 6$
+ " + F #
&
!$
! 3 5 6
" (
)
- % ; D$
/
0
: % $ 3 - % $
D ; $
# !% 5+ +! 6$
. +
+
%FG+. 2
: % $ 3 : % $ 3 5
6 5 6
- *. $
#
: % $ 3 : % $ 3 H 5 6
$
. ! $
#
: $ A &'%(%
#
&
; $= " 56
: % $ 3 #
! 5#6 $
(
" I+ 8*:8$ J = 8+*+:8 . B K+
3 B + "
2
/
L #2
#
* L $FF . F . F* M #
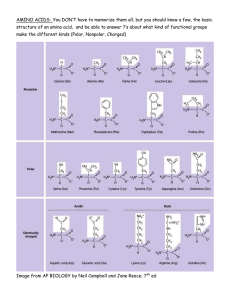
Amino acid nomenclature
• What do we call the pieces?
?
R
?CD
Covalent Structure of Proteins
R-group
R
-gr
group side chain,
specific for each
amino acid type
CH
?
Amino group
N
C
H
O
Carbonyl
FDUERQ &·
?
Carbonyl
oxygen
?
3URWHLQV+HWHURSRO\PHUVRI$PLQRDFLGV DD
A simple mnemonic for correct L-form is
&251ZKHQWKH&ĮDWRPLVYLHZHGZLWKWKH+
LQIURQWWKHUHVLGXHVUHDG&2-R-1LQD
FORFNZLVHGLUHFWLRQ
Primary Structure of Protein
• 20 natural amino acids
• They are D- amino acids. They are derivatives of straight
chain acids
• C-C-C-C-COOH
• į-Ȗ-ȕ-Į-COOH
• All are chiral, except Glycine
• Generated billions of years before (!!!)
NH2
:LUHIUDPHVWLFNEDOODQGVWLFNVSDFHILOOPRGHOV
of L- DODQLQH
Amino acid
Amino acid
R-
Representation
COOH
Residues - 1
$OLSKDWLF H[FHSW*O\ – Non-=ZLWWHULRQLFVWDWH
ESS: Essential
$ODQLQH $OD $
9DOLQH 9DO 9
0 VD 5HV9RO &U\'HQ 0 VD 5HVYRO &U\'HQ ESS
Amino acid
Residues - 2
1RQSRODU– Non-=ZLWWHULRQLFVWDWH
/HXFLQH /HX /
,VROHXFLQH ,OH ,
0 VD 5HVYRO &U\GHQ ESS
0 VD 5HVYRO ESS
*O\FLQH *O\ *
&\VWHLQH &\V &
0 VD 0 VD 5HVYRO G
"
5HVYRO G Amino acid
Residues –3
$URPDWLF– Non-=ZLWWHULRQLFVWDWH
0HWKLRQLQH 0HW 0
3UROLQH 3UR 3
0 VD 5HVYRO G ESS
0 VD 5HVYRO
G "
+LVWLGLQH +LV +
0 VD 5HV9RO G "
3KHQ\ODODQLQH 3KH )
0 VD 5HVYRO G " ESS
Amino acid
Residues - 4
3RODU– Non-=ZLWWHULRQLFVWDWH
7\URVLQH 7\U <
7U\SWRSKDQ 7US :
0 VD 5HVYRO G 0 VD 5HVYRO G "ESS
$VSDUDJLQH $VQ 1
*OXWDPLQH *OQ 4
0 VD 0 VD 5HVYRO G "
5HV9RO G Amino acid
Residues – 5
&KDUJHG– Non-=ZLWWHULRQLFVWDWH
6HULQH 6HU 6
7KUHRQLQH 7KU 7
/\VLQH /\V .
$UJLQLQH $UJ 5
0 VD 5HVYRO G 0 VD 5HVYRO G "ESS
0 VD 0 VD 5HVYRO G 5HV9RO G "ESS
Hydrophobicity Scales
$VSDUWLFDFLG $VS '
*OXWDPDWH *OX (
0 VD 5HVYRO G
0 VD 5HVYRO G Information about amino acids
-DQLQ :ROIHQGHQHWDO2 .\WHDQG
'RROLWWOHDQG 5RVHHWDO
+\GURSKRELF
KWWSSURZOURFNHIHOOHUHGXDDLQIRFRQWHQWVKWP
KWWSZZZUHDOWLPHQHWDQUDPLQRDFGKWPO
3RODU
+\GURSKRELFLW\RIDPLQRDFLGV
•
--DQLQ1DWXUH -
•
5:ROIHQGHQ/$QGHUVVRQ3&XOOLVDQG&6RXWKJDWH%LRFKHPLVWU\
-
•
-.\WHDQG5'RROLWH-0RO%LRO -
•
*5RVH$*HVHORZLW]*/HVVHU5/HHDQG0=HKIXV6FLHQFH
-
•
-&RUQHWWH.%&HDVH+0DUJDOLW-/6SRXJH-$%HU]RIVN\DQG&
'H/LVL-0RO%LRO -
•
0&KDUWRQDQG%,&KDUWRQ-WKHRU%LRO -
$FLGLF
%DVLF
Amino Acids – Ionization properties
• $PLQRDFLGUHPDLQVLQ]ZLWWHULRQLF IRUPDWS+ ,QDONDOLQHPHGLXP
DQLRQGRPLQDWHV
NH2-CHR-COO+1+–
,Q$FLGLFPHGLXP
FDWLRQGRPLQDWHV
• )RXUDPLQRDFLGVKDYHDGGLWLRQDOFKDUJHDWQHXWUDOS+
• Asp, *OX1HJDWLYH/\V$UJ3RVLWLYH
CHR-COO-
+1+-CHR-COOH
II
I
III
,QDQHOHFWULFILHOG HOHFWURSKRUHVLV WKHQHW
PLJUDWLRQIRUDPLQRDFLGVLQ,,VWDWHWRZDUGVWKH
DQRGHDQGZKHQLQVWDWH,,,WKH\ZLOOPLJUDWH
WRZDUGVWKHFDWKRGH
/HW$+EHDQDWRPJURXSLQDPROHFXOH $+FRXOGEHQHXWUDO
RUFKDUJHG $IWHU$+ORVHVDSURWRQLWLVGHQRWHGE\$- The
SURWRQDWLRQGHSURWRQDWLRQUHDFWLRQPD\EHZULWWHQDV
[A-@>$+@ S+-S.D
•7KHKLJKHUWKHS+YDOXHWKHPRUHOLNHO\DPROHFXOHZLOO
ORVHDSURWRQ
Q. :KDWLVWKHS+ZKHQRIWKH+LVWLGLQHLVSURWRQDWHG"
7KHVLGHFKDLQRIWKHDPLQRDFLG+LVWLGLQHKDVDS.D RI
Amino Acids – Ionization properties
Acid-Base Titration curve of Alanine
+NHCHRCOOH
NH2CHRCOO-
+1+&+5&22-
pH
S.2 +1+&+5&22NH2CHRCOO-
pH=6.1
+NH3CHRCOO-
S. +NH3CHRCOOH
7RWDOSURWRQVGLVVRFLDWHG
Titration Curve of D and K
Amino Acids – Ionization properties
• +NH3 – CHR-COO:KHQWKHFDWLRQDQGDQLRQVDUHH[DFWO\EDODQFHGWKHUHLV
QRQHWPLJUDWLRQDQGWKDWS+LVFDOOHGisoelectric pH of
DPLQRDFLG
&DWLRQÅÆ Zwitterion + H+
.
. >=ZLWWHULRQ@>+@>&DWLRQ@
Zwitterion Å Æ Anion + H+
.2
.2 >$QLRQ@>++]/[Zwitterion]
Ionic equilibrium constants are :
K1 = [Zwitterion][H+]/[Cation]
K2 = [Anion][H+]/[Zwitterion]
K1K2 = [H+]^2 / [Anion]/[Cation]
At isoelectric point, [Anion] { [Cation]; by definition
Isoelectric [H+] = K1K2
pH(isoelectric) = (-logK1 – logK2)/2
= (pK1 + pK2)/2
Example: pH (alanine) = (2.3 + 9.9)/2 = 6.1
It is easy to measure the pK1, pK2 by titration and one can know
the pH when one can have zwitterion state of that amino acid.
Primary Structure-Amino Acids
• So far we are discussing electrical
properties of amino acids
• How simple experiments can tell us their
ionic state
• pH and ionization measurements can tell
about the nature of their ionic state
• We can apply this knowledge into
predicting the ionic interaction in higher
level (secondary or tertiary structure)
• We need to know ionic interaction because
that is by far the strongest interaction
which determines the structure
Coulomb Force
+
-
,RQLFLQWHUDFWLRQVEHWZHHQIXOO\RUSDUWLDOO\FKDUJHGJURXSVFDQEH
DSSUR[LPDWHGE\&RXORPE¶VODZIRUHDFKDWRPSDLU^LM`
)FRXO Uij ןqiqj /rij2
) )RUFH
qi (IIHFWLYHFKDUJHRQDWRPL
PHGLXP
rij 'LVWDQFHEHWZHHQDWRPLDQGM
Coulomb Interaction
-
+
Ei , j
qi q j
SHH rij
HR 3HUPLWWLYLW\RIIUHHVSDFH × - &2 /Jm
H 5HODWLYHSHUPLWWLYLW\RUGLHOHFWULFFRQVWDQWRIWKH
PHGLXP
T HOHPHQWDU\FKDUJH × -F
Coulomb Interaction
Ei , j
Kcoul qi q j
H rij
.FRXO LVWKHFRQYHUVLRQIDFWRUQHHGHGWRREWDLQ
HQHUJLHVLQXQLWVRINFDOPROZLWKWKHFKDUJHXQLWVXVHG
H GLHOHFWULFFRQVWDQWRIWKHPHGLXP
Coulomb interaction- How strong it is?
Consider, two monovalent charges separated by 0.3 nm,
Ei ,i
u 2
u u 8 u u u
u J YDFXXP
(per ion pair in vacuum at 300 K )
Why the amino acid are Zwitterionic ?
• Due to the presence of more than one ionic group
in the same molecule, one influence the ionization
behavior of other.
• By Coulomb interaction of two opposite charges:
Q1.Q2/4SHH r
• So we qualitatively understand that there would
be a free energy of stabilization due to
Coulombic interaction
Why the amino acid are Zwitterionic ?
Suppose, this simple ionization is coupled with some other
related process
Related free energy is 'GC (coupling)
'GTot = 'Gioniz + 'GC
= 'Go+ RT ln {[H+][A-]/[H-A]} + 'GC
At equilibrium 'GTot = 0 and the [H+] concentration at
which the acid is half- ionized.
Now consider
0 = 'Go + RT ln [H+]1/2 + 'GC
[H+](1/2) = exp{-('Go + 'GC )}/RT
This gives the pH at which the molecule has coupling free
energy (have zwitterionic state) as well as half ionized.
Consider first the simple ionization reaction
l H+ + AH-Al
Ka = [H+][A-]/[H-A]
(Ka is acid dissociation constant)
pKa = -log Ka
o
'G = - RT ln Ka = 2.303 RT pKa
o
'G is the free energy difference between products
(H+, A- ) and reactant (H-A) when both are in their
standard states (say, 1M in aq. Solution)
At some other condition for ionization:
'G ionz
= 'Go + RT ln {[H+][A-]/[H-A]}
Whenever (H+), (A-) and (H-A) satisfy the condition of
the equilibrium constant Ka , the 'G ionz = 0
[H+](1/2) = exp{-('
'Go + 'GC )}/RT
pK’a = -log [H+](1/2)
= ('Go + 'GC )/2.303RT
When there is no coupling
pKa = 'Go /2.303RT
'GC = 2.303 RT (pK’a – pKa)
The above expression gives an estimate of Coulombic
interaction due to presence of opposite charges present in same
molecule / Coupling.
How to calculate?
Needs pK’a and pKa
The pK’a is just the pKa value of an amino acid in Zwitterionic
state.
The pKa is the pKa value of same amino acid in same solution
condition but without Coulombic interaction/no coupling
How much is the stability of Zwitterion?
'GC= 2.303 RT (pK’a – pKa)
Use to analyze the electrostatic interaction between
COO- and NH3+ in zwitterion. Consider alanine and
its oligomers:
Peptide bond formation – condensation
reaction
-$-1++
FRPELQHVZLWK+A2-COOWRJLYH
- $1+-COA2+
2QO\WHUPLQDOFKDUJHVDW$DQG$UHPDLQ
'GC= 2.303 RT (pK’a – pKa)
S. S.2
$OD- ÅÆ $OD2.34 9.69
+$OD-$ODÅÆ +$OD-$OD- ÅÆ $OD-$OD3.12 8.30
+$OD-$OD-$ODÅÆ +$OD-$OD-$OD- ÅÆ $OD-$OD-$OD3.39 8.03
+$OD-$OD-$OD-$ODÅÆ +$OD-$OD-$OD-$OD- ÅÆ $OD-$OD-$OD-$OD3.42 7.94
+$ODÅÆ
In (Ala)4, the ionized groups are far apart and (one
can approximate that) no interaction is present
between them. Thus, unperturbed pKa (from (Ala)4
data) is 3.42 (the ionizable groups are far apart).
Perturbed pKa (arising from electrostatic interaction
between COO- and NH3+, due to coupling) is 2.34
How much is the stability of Zwitterion?
'Gc= 2.303 RT (pK’a – pKa)
+Ala ÅÆ +Ala- ÅÆ Ala- ; pk1 = 2.34
+AlaAlaAlaAla ÅÆ +Ala-Ala-AlaAla- ÅÆ AlaAla-AlaAla- ;
pk 3.42
Using RT = 0.6 kCal/mole at room temp.
'G (C) = 2.303 (0.6) ( 2.34-3.42) kcal /mole
= - 1.49 kcal /mole
This is the stabilization energy or coupling energy for
zwitterionic state and electrostatic in nature.
,WZRXOGYDU\IRUDPLQRDFLGWRDPLQRDFLG
Uncertainty in understanding ionization
Uncertainty in understanding side
chain ionization
A potentiometric WLWUDWLRQGRHVQRWGLUHFWO\UHYHDOZKLFK
SRVLWLRQLVWLWUDWLQJ
7KLVFRXOGEHXQGHUVWDQGE\VSHFWURVFRSLFVWXGLHVRU
WKURXJKWKHXVHRIFKHPLFDODQDORJ/LNH
&DUER[\OGLVVRFLDWLRQE\
CH-CO-NH-&+ &+ -COOH
CH-CO-NH-&+ &+ -COOS. $PLQRGLVVRFLDWLRQE\
NH+-&+ &+ -CO-NH2
NH2-&+ &+ -CO-NH2
S.2 Possibility of ionization pathways
Uncertainty in side chains
DDPLQR
+
NH3 – CH –COOH
|
(CH2 4
|
NH3+
HDPLQR
Uncertainty in side chains
• 3ULPDU\DON\ODPLQHKDYHKLJKHUS.D WKDQD-FDUERQ\O
VXEVWLWXWHGPHWK\ODPLQH(JS.D of n-EXW\ODPLQHLV
ZKHUHDVDPLQHRID-DPLQRDFLGWLWUDWHVQHDUS+
,IWKHDERYHVFKHPHLVFRUUHFWWKHQ
• S.D of H-DPLQRJURXSRQO\VLQHVKRXOGEHKLJKHUWKDQ
WKDWRIQ-EXW\ODPLQHEHFDXVHRIFRXSOLQJHIIHFWWRWKH
COO• The D-1++ WLWUDWHVLQSUHVHQFHRIH-1++VRLWVS.D
ORZHUWKDQWKHD-DPLQRLQ$OD-$OD • ,ISDWK E LVFRUUHFWWKHQRQHKDYHWRH[SODLQZK\WKHDDPLQRLQ$OD-$ODWLWUDWHVZLWKDS.D RIZKHUHDV$OD/\VZLWKQRFKDUJHLQWKHVLGHFKDLQWLWUDWHVZLWKDS.D
RI
How the concept of polarity of amino acid is useful?
• Where a particular amino acid could be located?
• Is it placed in the interior or exterior of that protein?
• It is useful to classify or categorize the amino acids.
• One method of classifying is as charged or uncharged amino
acids at any pH. However, this classification is rather broad
and not useful for detailed analysis of location of residues.
Environmental Preference of Amino Acids
• ,QWKHIROORZLQJZHVKDOOGHVFULEHVRPHDSSURDFKHV
XVHGWRTXDQWLI\WKHSRODULW\RIDPLQRDFLGV8VHRI
VXFKFODVVLILFDWLRQLVWKDWRQHFDQ KRSHIXOO\ SUHGLFW
WKHORFDWLRQRIDUHVLGXHLQDSURWHLQ-GLPHQVLRQDO
VWUXFWXUH RUDWOHDVWWKHSURSHQVLW\RILWWRJRHLWKHU
WRLQWHULRURUH[WHULRURISURWHLQVWUXFWXUH RUWKH
ORFDWLRQRIDSURWHLQ NQRZLQJWKHSRODULW\RIDOO
FRQVWLWXHQWDPLQRDFLGV LQDPHPEUDQH
How the concept of polarity of amino acid is
useful?
• Second method is to define them as polar (higher solubility
in water and strongly interacting) and nonpolar (lower
solubility in water)
• Polar residues are: Glu, Asp, Arg, Lys, Gln, Ser, Thr.
• Nonpolar residues are Ala, Val, Ile.
• The residues Cys and His can not be unambiguously
classified.
• However, it is important to note that the concept of
polarity is also relative because the polarity is dependent
on the solvent used to estimate it.
How the concept of polarity of amino acid is
useful?
• One can use to predict the location of a residue
inside the protein 3- dimensional structure or at
least its tendency to go either inside or outside the
protein core.
Scales of hydrophobicity
•Measurement of solubility of different amino acids in
two different solvents (Ethanol and water or Octanol
and water). One solvent has almost no hydrophobic
effect)
•Difference in solubility can be used to calculate free
energies of solvation in two solvents (ȝ = ȝ 0 + RT ln a).
• Tanford used this idea to locate the side chain of
amino acids in either interior or exterior of protein
core.
•Partition coefficient (K) is a parameter measuring it.
In dilute solutions K=a1/a2= C1/C2 (ratio of
concentrations).
COONH3+
H
How the concept of polarity of amino acid is useful?
• Solubility of amino acid is considered in water
(polar) and ethanol (nonpolar) solvent.
• Solubility data give an estimate of transfer free
energy for ethanol
water or polar to
nonpolar environment and vice versa
Side chain
Ethanol (non-polar)
Water (Polar)
Ethanol (nonpolar)
Water (Polar)
ǻ*
–1.98
Kcal/mole
Ethanol (nonpolar)
Water (Polar)
ǻ*
-4.63 Kcal/mole
Ethanol (nonpolar)
Water (Polar)
Subtract !
Side chain’s transfer free energy from nonpolar to polar
medium
Side chain transfer energy for ethanol Æ
water; (nonpolar Æ ZDWHU
If this energy turns out to be positive then the side
chain should prefer to go reverse: water Æ Nonpolar
Trp + 3.00 kCal/mole ; Prefer nonpolar
Ile + 2.95 kCal/mole ; Prefer nonpolar
Tyr + 2.85 kCal/mole ; Prefer nonpolar
Phe + 2.65 kCal/mole ; Prefer nonpolar
These side chains prefer to go to interior region of protein.
How much is the tendency?
Depends on magnitude of free energy.
Trp has most tendency among the set; Phe is least among the
set
How the concept of polarity of amino acid is
useful?
• Solubility data give an estimate of transfer free
energy for ethanol
water or polar to
nonpolar environment and vice versa
• Such difference for Phenyl alanine and Glycine is
–1.98 – (-4.63) = + 2.65 kCal/mole
• Two structures differ by phenyl alanine side chain.
• The side chain prefer to go to nonpolar solvent
(ethanol) – like to stay interior of protein
Classification of PROTEINS based on polarity of
primary structure
• Protein can be located inside or outside the
membrane
3RODUDTXHRXV
1RQSRODU
• Knowledge of polarity of residues of a protein can
be used to location of protein inside or outside the
membrane.
Classification of PROTEINS based on polarity of primary structure
• Scale of average hydrophobicity
+ij Ȉǻ*t L ;Ȥ L
Different Nonpolar amino acids
Different polar
amino acids
More nonpolar; Intrinsic membrane protein
More polar; External membrane protein
How good be the prediction? Depends on how good
one can identify an amino acid as non-polar (shades of
red) and polar (shades of blue). This will be used as
Scale. Success of prediction depends on how good a
scale is
?
Classification of PROTEINS based on polarity of primary structure
• Ratio of frequency of occurrence
5 ȈȤ N ȈȤ M
ǻ*t (i) is transfer free energy of i-th residue.
:KHUHȤ L LVWKHPROHIUDFWLRQRIL-th residue.
Ȥ N DQGȤ M FRXOGEHK\GURSKLOLF
and hydrophobic residues
+ijFOXVWHUDURXQG,WLVQRWVXFFHVVIXOLQ
classifying the proteins into polar preferring or
nonpolar preferring.
• R3 scale selects k as Arg, Lys,His,Gly,
Glu,Asp,Asn,His and j as Ile,Tyr,Phe,Leu,Val,Met
• R3 is turned out 0.6 for internal membrane
proteins and 1.4 for external membrane proteins
Classification of PROTEINS based on polarity of primary structure
Classification of PROTEINS based on polarity of primary structure
• Discriminant function
Z= -5+ij
$FRPSDULVRQRI+ijDQG=
• Internal membrane proteins : 0.52±0.11
• External membrane proteins 0.12±0.16
• Nonmembrane membrane proteins: 0.16±0.17
• Chance of misclassification is only 8%
Protein
+ij
Acetyl choline
Receptor
(Subunit 1-4)
1.12 - 1.18
0.29 - 0.38
Bovine
Rhodopsin
(Subunit 1-4)
1.21
0.51
Inside
0.56
Inside
Purple
membrane
Z
1.25
Location
Outside
Polypeptides
O
H2N
PROTEIN SECONDARY
STRUCTURE
O
R
NH
CH
OH
CH
CH
N
C
CH
R
H
O
R
?
R
NH
O
Peptide bond
N-terminus
1
i-1
Numbering
2
i
3
i+1
C-terminus
4
i+2
Geometry - bond angles
Atom
Valence
Hybridization
3HSWLGHERQGJHRPHWU\
Coodinat- Bond
ion
angle
Nitrogen
3?
sp
?2
Trigonal
?
planer
Carbon - CD
4?
sp?3
?
tetrahedral
?
109°
?
Trigonalplanar
?
120°
Carbon
carbonyl
–
?
4
?2
sp
120°
?
The distances and the angle
determine the structure
Trigonometric
Representation
GLKHGUDODQJOH
D
E
c
2
2
N
c
D
ERQGDQJOH
c
D E DE FRV DA E
2
C
E
CĮ
N
Covalent structure of p
peptide
p
unit
7RUVLRQ$QJOHV 'LKHGUDO
• ~ "Free" rotation about single covalent bonds
• Demonstrate with model.
• ~ Only conformational variables
– Bond angles, lengths are ~ constant.
– Torsion angles are the primary determinant of
protein & nucleic acid structure.
Backbone Torsion Angles - Importance
• Describe overall fold
– Almost completely
– Remember…
• Bond lengths ~fixed
• Bond angles ~fixed
• Only torsion angles
variable
• Only 2 variables /
amino acid
– IRUWKHEDFNERQH
Torsion Angles: Protein Nomenclature
non-standard rule for proteins:
• polypeptide backbone (N, CD, C') always
heaviest!
– &·!!5- even if R = CH3OH
Values of Torsion Angles: Z
• Consider a peptide bond...
– Where are any lone pair electrons?
– How might this change the covalent bonding?
Loss of lone
SDLUPDNHV1
WULJRQDOSODQDU
N
N
C
C
O
O
G-
3DUWLDO
GRXEOHERQGLQJ
restricts
URWDWLRQWR
r ° DERXW
Z, i.e.
SODQDU
SHSWLGH
ERQG
Planar Peptide Bond
5HVLGXHL
5HVLGXHL
H
CH
N
C
CH
O
7KHVHDWRPVEHWZHHQ
WKHUHVLGXHVDUHQHDUO\LQ
RQHSODQH
Dihedral angle
% Trans and cis peptide bonds
7KHWUDQVFRQILJXUDWLRQLVDGRSWHGIRUDOPRVWDOO
SHSWLGHERQGV
Values of torsion angles: Z
• Usually trans
– with Z | 180q r 6q rmsq.
• Occasionally cis
– with Z | 0q:
– ~ 1/4 of prolines
– very infrequently glycines
– almost never other amino
acids
– Z is not very important to
protein conformation.
H
N
CH
Fisher Projection
CH
C
trans
O
CH
N
CH
C
O
cis
H
7KHSHSWLGHEDFNERQHFRQIRUPDWLRQFDQEHGHVFULEHGLQ
WHUPVRIWZRGLKHGUDODQJOHV3KL ) DQG3VL < Anatomy of a I\ plot
• Where are the axes?
– X vs. Y plot:
• Which is plotted horizontally?
• Which comes 1st in the alphabet?
– Now I vs \ plot:
• Which comes 1st in the DOSKDEHW?
• So which is plotted horizontally?
ĭ PhL LVWKHGLKHGUDODQJOHIRUWKH1-CD ERQG hHWHUR
Ȍ PsL LVWKHGLKHGUDODQJOHIRUWKH&Į-&ERQG sDPH)
Ramachandran Plot
3URI*15DPDFKDQGUDQ
• What are the intercept values?
– Usually -180°, -180°
– With center point at 0°, 0°
The Ramachandran Plot
*15DPDFKDQGUDQDQGKLVFROOHDJXHVXVHGIRUFHILHOG
FDOFXODWLRQVRIVPDOOSRO\SHSWLGHVWRV\VWHPDWLFDOO\YDU\
SKLDQGSVLZLWKWKHREMHFWLYHRIILQGLQJVWDEOH
FRQIRUPDWLRQV
$WRPVZHUHWUHDWHGDVKDUGVSKHUHVZLWKGLPHQVLRQV
FRUUHVSRQGLQJWRWKHLUKDUGVSKHUHUDGLL LQSUDFWLFHYDQ
GHU:DDOVUDGLL– ZHOOGRFXPHQWHG
9
RÆ
SKLDQGSVLDQJOHVZKLFKFDXVHVSKHUHVWRFROOLGH
FRUUHVSRQGWRVWHULFDOO\GLVDOORZHGFRQIRUPDWLRQVRIWKH
SRO\SHSWLGHEDFNERQH
I, \ Ramachandran plots
• Ramachandran calculated the potential
energy of peptides according to I, \:
– $OD n *O\ n
– Dominated by van der Waals
interactions between atom n & n + 3
6WHULFHQFRXQWHUV UHSXOVLYHSDUWRILQWHUDFWLRQ EHWZHHQQRQERQGHGDWRPVDQGJURXSVKDVPRUH
LPSRUWDQW
Shown here,
) < FRPELQDWLRQLV
IRUELGGHQ
• Ramachandran in fact approximated
that all interactions except vDW were
zero
• Plotted so that contours surround an
area where E{I\} < Econtour.
6WHULFDOLQKLELWLRQRISHSWLGHEDFNERQHPRWLRQ
6DPSOH5RWDWLRQ:
+RZWRJHQHUDWHWKH5DPDFKDQGUDQSORW"
5RWDWLRQWRGHJUHH
5RWDWLRQWRGHJUHH
) < Z Ramachandran Details
\
• If same values repeated:
– 5HJXODU VHFRQGDU\ structure
I
,VRHQHUJ\VXUIDFHV
3KL3VL(QHUJ\
most favored region
allowed region
generously allowed region
disallowed region
Peptide Conformation
A Ramachandran Plot for Polyglycine
Glycine is highly flexible
Fully allowed
• Observed I\ values for each amino acid of a protein
always fall near the calculated energy minima
– Well, nearly always
– Why?
Non-glycine
Glycine
At limits of
allowability
Branden & Tooze
© 1999 Garland
Basics of Protein Structure
• Primary
• Secondary
• Tertiary
primary structure
ACDEFGHIKLMNPQRSTVWY
Protein Secondary Structure
• The secondary structure is the periodic
structure formed from primary structure.
• The major types are alpha helix and beta
sheet and turns.
• Pauling and Corey first proposed these
two structures by using experimental
bond angles and bond distances for amino
acids and peptides and building periodic
model structures.
Primary structure
Protein Secondary Structure
3URWHLQ6WUXFWXUHKLHUDUFK\
$OSKDKHOL[
%HWDVKHHW
6HFRQGDU\VWUXFWXUDOHOHPHQWV
• &ODVVLFDOD- KHOL[
• DQGS-KHOL[
•/HIWKDQGHGD-KHOL[
• E-VKHHWV SDUDOOHODQGDQWLSDUDOOHO
•7XUQV
%HWDWXUQ
E-sheets
$OSKD-+HOL[
D-+HOL[
3DXOLQJ VGLVFRYHU\ 3DXOLQJ V
FODVVLFSDSHU 'LPHQVLRQV
JHRPHWU\ +-ERQGV
UHVLGXHVWXUQ
SLWFK cWXUQ
ULVHUHVLGXH c
)\ angle
value determine
secondary
structure
Main chain CD
Ribbon
Amide plane
)=-57Û
\=-47Û
D-carbon i+4
LWRLK\GURKHQERQG
Hydrogen bond
i,i+4
I DQG\ is –º, -º
D-carbon i
Secondary structure
involves hydrogen
bonding between
atoms of the backbone
Side chains R outside the Helix
Secondary Structure – Alpha helix
•The alpha helix is rod like periodic unit.
•The tightly coiled polypeptide main chain forms the inner
part of the rod.
•Side chains are protruded outside
•In the helix the residues are held by hydrogen bond
between NH and CO units (all the main chain NH and CO
are h-bonded) and van der Waals interactions.
Handedness of helix
5LJKWKDQGHGKHOL[)URPDSDUWLFXODUUHVLGXHYHFWRUVDUH
GUDZQWRVKRZWKHFHQWHUVRIRWKHUUHVLGXHFHQWHUV
3URMHFWHGKHOLFDO
ZKHHOGLDJUDP
FRQQHFWLQJWKH
UHVLGXHV
Secondary Structure – Alpha helix
•Coulombic interactions
interactions playing roles
and
other
Development of helix
polar
•Each residue is related to the next one by a rise of
1.5 Å along the helix axis and a rotation of 100°
which gives 3.6 residues per turn of helix.
•Amino acids which are three-four residues away
in linear sequence are spatially close in helical
structure.
ij
Pitch
Secondary Structure – Alpha helix
•The distance a point moves in the direction of its
axis per revolution is called pitch (P).
•The gradient angle, ij is given by the relation,
P/2ʌU = WDQij where, r is the radius of helix.
•Pitch is proportional to r. As 3.6 residues per turn
exist,Translation along the helix axis is 1.5 Å per
residue. Pitch = (3.6 x 1.5 = ) 5.4 Å.
•Typical radius of alpha helix is ~1Å to 1.4Å
+HOLFDO:KHHOV
- DWRROWRYLVXDOL]HWKHSRVLWLRQRI
DPLQRDFLGVDURXQGDQDOSKD-KHOL[
- DOORZVIRUTXLFNYLVXDOL]DWLRQRI
ZKHWKHUDVLGHRIDKHOL[SRVVHV
VSHFLILFFKHPLFDOSURSHUWLHV
- H[DPSOHVKRZQLVDKHOL[WKDW
IRUPVDLeucine-Zipper
Hydrophobic residues
on one side interact with helix
displaying same pattern
Conformational features
Helix Dipole
•
•
•
•
6WDELOLW\IDFWRUVRI$OSKDKHOL[
(OHFWURVWDWLFLQWHUDFWLRQEHWZHHQFKDUJHG5JURXSV
%XONLQHVVRI5
,QWHUDFWLRQEHWZHHQUHVLGXHVUHVLGXHDSDUW LRQSDLU
• 2FFXUUHQFHRI3UR*O\ PRUHIOH[LEOH– FDQFRLOLQ
DGLIIHUHQWZD\WKDQDOSKDKHOL[
• 'LSRODULQWHUDFWLRQ
D-Helix Breakers
0RVWDPLQRDFLGVOLNHWREHLQDQD-KHOL[
1RWDEOHH[FHSWLRQV
*/<&,1(
352/,1( ,PLQR$FLG
1R+\GURJHQ
On this N to
H-%RQG
O
C-O
N
H
Proteins with D-helices
0DMRUVWUXFWXUDOFRPSRQHQWLQPDQ\SURWHLQVVRPHJOREXODU
SURWHLQVFRQWDLQPRVWO\D-KHOLFHVFRQQHFWHGE\WXUQV
i.e.KHPRJORELQD-KHOLFHV
Some Interesting D-Helices
- VPDOO'1$ELQGLQJKHOLFHV
- PHPEUDQH– VSDQQLQJKHOLFHV
- SUROLQHUHVLGXHVRIWHQVHUYHDV‘D-+HOL[%UHDNHUV¶
- RIWHQIRXQGDWWKHERXQGDULHVRID-+HOLFHVDQGLQWXUQV
- DPSKLSDWKLFKHOLFHV
- FRLOHG&RLOV
Amphipathic a-helix
$PSKLSDWKLF+HOLFHV
Amphipathic: hydrophilic & hydrophobic
- these helices posses
K\GURSKLOLFDPLQRDFLGV
RQRQHVLGHDQGK\GURSKRELF
UHVLGXHVRQWKHRWKHU
+\GURSKRELF
- these D-KHOLFHVLQVRPHFDVHVFDQ
EHXVHGWRDVVRFLDWHDSURWHLQWR
DPHPEUDQH
hydrophilic head group
aliphatic carbon chain
+\GURSKLOLF
lipid
bilayer
Protein secondary structure:: helices
alpha
310
pi
- µURG¶OLNHULJKW-KDQGHG
INTRA-FKDLQ+-ERQGV
EHWZHHQ!& 2JURXSRI
HDFKSHSWLGHUHVLGXHDQG
WKH!1-+JURXSRIWKH 4th
DPLQRDFLGDZD\
- DOSKDKHOLFHVDUHDERXW
UHVLGXHVRQDYHUDJH
H-ERQGLQJLQ
D-KHOL[
amino acids
per turn:
3.6
frequency
~97% ~3%
3.0
4.4
rare
- VLGHFKDLQVRIDOSKDKHOLFHVDUHZHOO
VWDJJHUHGSUHYHQWLQJ
VWHULFKLQGUDQFH
- KHOLFHVFDQIRUP
EXQGOHVFRLOHGFRLOVHWF
Conformational features
• $OSKDKHOL[KDYH
SVL -WR–DQGSKL -WR-
• 1RWDOOSRO\SHSWLGHVIRUPDOSKDKHOL[
• /RQJEORFNRI*OX QHJDWLYHFKDUJHUHSHO ZLOOQRW
form
• /RQJEORFNRI/\V$UJ FKDUJH ZLOOQRW
• S+GHSHQGHQW
• $VQ6HU7KU&\VFDQGHVWDELOL]H
• 3UROLQHLVUDUHO\IRXQGLQDOSKDKHOL[ 1LVDSDUWRI
ULJLGULQJDQG1-CD URWDWLRQLVQRWSRVVLEOH1R
VXEVWLWXHQWK\GURJHQWRIRUP+-ERQG
Nter
)=-57Û
\=-47Û
)=-139Û
\=+135Û
DCarbon
$OSKD-+HOL[
E-Pleated Sheets
• Polypeptide chain is almost fully extended i.e.
not tightly coiled like a-helix.
• 3.5 Å between residues.
• H-bonds occur between directly opposed
strands.
• Antiparallel or parallel: parallel sheets are
less stable since H-bonds are distorted.
• R-groups alternate above and below the plane
of the sheet.
Cter
Cter Nter
Carbon
(C=0)
DCarbon
%HWD-6KHHW DQWLSDUDOOHO
H-bonding scheme of parallel and antiparallel
E-sheets
ȕ-pleated sheet
Protein structure:: beta
ta-sheets
N
O
C
parallel
- the basic unit of a
beta-sheet is called a
beta-strand
- repeating unit like the
alpha helix
- beta-sheets can form
various higher-level
structures, supersecondary
structure such as a betabarrel
The Beta-Sheets
- strands of amino acids held together in sheets by INTER-STRAND
H-Bonding
- bonding between backbone >C=O and >N-H on different strands
-strands of the E-sheets tends to be twisted and inclinated in a E-barrel
- the R-groups lie perpendicular to the sheets; stick out on either face
of the sheet
‘twisted’
anti-parallel
Green
Fluorescent
Protein
(GFP)
R
R
R
R
R
R
R
R R
R
R
In a E-barrel the amino
acids side chains inside the
barrel are very often Ebranched or hydrophobics
An example of
complex beta-sheets:
Silk Fibroin
- multiple pleated
sheets provide
toughness & rigidity
to many structural
proteins.
Structure
I
\
n
p (Å)
Atom
H-bond
3.613helix
-57º
-47 º
3.6
5.4
13
i to i+4
310helix
-50º
-25 º
3
6
10
i to i+3
S-helix
-60º
-70 º
4.4
5.2
16
i to i+5
Parallel bstrand
-120
115
2.0
6.4
Inter
strand
Antiparallel
-140
135
2.0
6.8
Inter
strand
Beta Turns/loops (Common structural motif
of proteins
B
A
C
D
Nomenclature for
residues in hairpin
beta-turns
(a) type I, (b) type II, (c) type I', and (d) type II' turns.
Protein structure:: turns/loops
Beta - Turns
alpha-helix
There are two classes of beta-turns:
- type I
- type II
Note: the position of R2 and R3 in both
cases
Type I turns have the amino acids side
chains on the same side.
Type II turns have the amino acids side
chains on the opposite sides.
- there are various types of
turns, differing in the
number of residues and
H-bonding pattern
- loops are typically longer;
they are often called coils
and do not have a
‘regular’,
or repeating, structure
Note: H-bonding between backbones
of residue 1 & 4
Gamma-Turns
Proline
A 3 amino acid turn utilizing
proline at the turn.
H-bonding with C=O of
residue 1 and N-H of residue 2
beta-sheet
ribonuclease A
loop
(usually exposed on
the surface of proteins)
Super secondary and tertiary
structure
• D-proteins
• E-proteins
• D/E proteins
• D+ E proteins
Secondary structure
•A fold is defined by the arrangement of
major elements of secondary structure
and the connection between them.
Secondary structure
All alpha class
•In most proteins the alpha helices and
beta sheets pack together in small
number of different ways – studied by
SCOP
•Due to the underlying interactions:
hydrophobic, H-bonding, electrostatic,
vdW, entropic factors
Secondary structure
All alpha class
Secondary structure
Multihelical assemblies
Secondary structure
Beta Sandwich
Secondary structure
Beta Propeller
Secondary structure
Beta Barrel
Secondary structure
Beta Prism
Secondary structure
Beta helix
38221 PDB Entries (23 Feb 2009).
110880 Domains
Class
Number of folds
Number of families
All alpha proteins
284
507
871
All beta proteins
174
354
742
147
244
803
376
552
1055
66
66
89
58
110
123
90
1195
129
1962
219
3902
Alpha and beta
proteins (D/E)
Alpha and beta
proteins (D+E)
Multi-domain
proteins
Membrane and
cell surface
proteins
Small proteins
Total
Tertiary Structure
Number of
superfamilies
Myoglobin Tertiary Structure
•More than one units of secondary
structure give rise to form the tertiary
structure.
•It is the structure formed by amino acid
sequence those are far in the linear
sequence
•Pattern of disulfide bonds plays a role.
This is the first protein to be seen in
atomistic detail. This is the oxygen
carrier in muscle and have single
polypeptide chain with 153 amino acids
and a mass of 18 kilo Dalton (1 Dalton is
equal to 1.0000 on atomic mass scale).
Myoglobin is built of eight helices.
•core is exclusively nonpolar
•polypeptides chains spontaneously fold to have the
nonpolar, hydrophobic side chains inside and polar or
charged side chains on the surface.
•unpaired NH and CO main chain accompanying the
hydrophobic side chain could prefer water rather
than nonpolar core.
•They are buried in core by forming H-ERQGVLQĮ
KHOL[RUȕVKHHW
Tertiary Structure
•Proteins like Ribonuclease A, a pancreatic enzyme
contains largely beta strands.
•Most ambitious goal of sequence information is to
predict the tertiary structure and perhaps
function.
•small variations in the size and shape of amino acids
gives perfect space filling
•van der Waals interaction plays a crucial role.
Tertiary Structure
• Simulations using energy functions also aimed at the
same goal.
• X-ray crystallographic studies are also aimed at that.
• It is necessary to identify the interactions to find the
stable tertiary structure
Interactions in tertiary structure
The major interactions involved in tertiary structure
formation are:
1. Hydrophobic effect
2. Hydrogen bonding
3. Electrostatic interactions involving ionizable
groups
4. Close packing due to van der Waals interactions
5. Entropic contribution due to structural ordering
1XFOHLFDFLG
1XFOHLF$FLGV
+LVWRU\\
±'1$LVWKHPDLQFRQVWLWXHQWRIJHQHV
$YHU\
±)LUVW;UD\SLFWXUHVRI'1$ )UDQNOLQ
±'1$VWUXFWXUHUHYHDOHG :DWVRQDQG
&ULFN
RQZDUGV0XOWLSOHFRQIRUPDWLRQVDQG
VWUXFWXUHV
;UD\VWUXFWXUHFRQILUPVGRXEOHKHOL[ 5LFK
W51$VWUXFWXUH .LP
±6WUXFWXUHRIILUVWFRPSOHWHWXUQRI%'1$
'LFNHUVRQ
'1$ 'HR[\ULER1XFOHLF$FLG
a6WRUHJHQHWLFLQIRUPDWLRQ&DOOHGWKHEOXH
SULQWRIOLIH
51$ 5LER1XFOHLF$FLG
a+HOSVLQSURWHLQV\QWKHVLVVRPHWLPHDOVR
DFWDVHQ]\PH
6WUXFWXUH
7KH'1$0ROHFXOHFRQVLVW
RIWZRXQEUDQFKHG
SRO\QXFOHRWLGHVFKDLQV
VWUDQGV KHOGWRJHWKHULQ
DQDQWLSDUDOOHO KHOL[
PDQQHUE\K\GURJHQ
ERQGVIRUPHGEHWZHHQ
VSHFLILFSDLUVRIEDVHV
>$GHQLQH7K\PLQH@
>*XDQRVLQH&\WRVLQH@
'1$
3ODQRIOHFWXUHV
1RWDWLRQ
1XFOHLFDFLGFRQIRUPDWLRQDOIOH[LELOLW\
1XFOHLFDFLGV
&RPSOH[SRO\PHULFPROHFXOHVZKLFKVWRUH
DQGWUDQVIHULQIRUPDWLRQZLWKLQDFHOO
&RQVWUXFWHGIURPEDVLFPRQRPHULFXQLWV
NQRZQDV1XFOHRWLGHV
1XFOHRWLGH VXJDUSKRVSKDWH
RUJDQLFQLWURJHQRXVEDVHV
6XJDUV5LERVH'HR[\ULERVH
6WUXFWXUHRI'1$51$
%DVHVDQG6XJDUVDQG3KRVSKDWHV
S\ULPLGLQHV
&
7
8
SXULQHV
*
$
5LERVH
VXJDUV
6WDQGDUG :DWVRQ&ULFN W\SHEDVH
SDLULQJ
.HWRHQROWDXWRPHULVP
$PLQRLPLQRWDXWRPHULVP
&RQVHTXHQFHRIWDXWRPHULVP
.HWR (QRORU$PLQR ,PLQRWDXWRPHULVP
FKDQJHV$FFHSWRUDQGGRQRUSURSHUWLHV
,QHQROIRUPV* $8 &
,QLPLQRIRUPV
1²+ $ 8*
& 8
$GLYHUVLW\FRXOGDULVHLILPLQRDQGHQRO
JURXSURWDWHV
2QO\.HWRDQGDPLQRIRUPVRFFXU!
XQGHUSK\VLRORJLFDOFRQGLWLRQV
S.DYDOXHVIRUEDVHVLQQXFOHRVLGHV
DQGLQQXFOHRWLGHV
&RPSRXQG 1XVLGH
6LWH
$1
¶SKRV
¶SKRV
&1
*1
81
6WDQGDUG :DWVRQ&ULFN W\SHEDVH
SDLULQJ
&KDUJDII¶VUXOHV
Ȥ$ Ȥ7DQGȤ* ȤF
)RUIXOO\GRXEOHVWUDQGHG'1$RQO\VLQJOH
YDULDEOHLVQHHGHGWRGHVFULEHFRPSRVLWLRQ
6RPHEDFWHULD*&H[LVWV
,QPRVWPDPPDOV*&LVDURXQG
*HRPHWU\RI
:DWVRQ&ULFN
%DVH3DLUV
$7DQG*&SDLUVDUH
VSDWLDOO\VLPLODU
+ERQGVYV
6XJDUJURXSVDUHDWWDFKHG
DV\PPHWULFDOO\RQWKHVDPH
VLGHRIWKHSDLU
/HDGVWRDPDMRUDQGPLQRU
JURYH
%DVHVDUHIODWEXWWKH
K\GURJHQERQGLQJOHDGVWR
FRQVLGHUDEOHIOH[LELOLW\
%DVHVWDFNLQJLVIOH[LEOH
'HILQLWLRQRI0DMRUDQG0LQRU
*URRYH
+\GURJHQ
ERQGLQJRI
:&EDVH
SDLU
V
0HFKDQLVPV
RI
UHFRJQLWLRQ
%DFNERQH
&RQIRUPDWLRQ
7KH5LERVH
5LQJLV
1HYHU)ODW
&KDQJHLQ
VXJDU
FRQIRUPDWLRQ
DIIHFWVWKH
EDFNERQH
&¶(QGR
&¶(QGR
'1$GRXEOHKHOL[
:K\KHOL["
:K\ULJKWKDQGHGKHOL["
:K\DQWLSDUDOOHOKHOL["
:K\:DWVRQ&ULFN*&DQG$7
SDLULQJ"
:K\KHOL["
+\SRWKHWLFDO/DGGHU
6NHZHG/DGGHUYV+HOLFDO7ZLVW
6NHZHG/DGGHU
Molecular Mechanics and Molecular
Interaction
•Biophysics looks for principles that describe
patterns. If the principles are powerful, they make
detailed predictions that can be tested.
ș DUFVLQ
Basic Principles
•Physics looks for mathematical laws of nature and
makes detailed predictions about the forces that drive
idealized systems
Basics of quantum mechanics
y Wave-functions and operators
y The best prediction of structure and physical
properties for a molecule come from exact
quantum mechanical treatment of every atom of
molecules.
y However analytically it is possible only for
Hydrogen atoms
The wave-function is a single-valued squareintegrable function of the system parameters and
time which provides a complete description of the
system. Linear Hermitian operators act on the wavefunction and correspond to the physical observables,
those dynamical variables which can be measured,
e.g. position, momentum and energy.
Schrödinger equation
Quantum Vs Classical Mechanics
y Describes the motion of electrons and nuclei
y H<n=E<n
y H(P,X)=Ek(P)+Ep(X)
y P and X is momentum and position of all the
electrons and nuclei
Classical Mechanics
y The behavior of object describe by two
equation
1. Total energy is conserved in absence
of external force
2. The other express the response of
particles to the forces acting on them
y The Schrodinger equation plays the role
of Newton's laws and conservation of
energy in classical mechanics - i.e., it
predicts the future behavior of a
dynamic system.
Force Field
yThe forcefield is a collection of
equations and associated constants
designed to reproduce molecular
geometry and selected properties of
tested structures.
The Anatomy of a Force-Field
Energy = Stretching Energy + Bending Energy + Torsion
Energy + Non-Bonded Interaction Energy
These equations
together with the data
(parameters) required to
describe the behavior of
different kinds of atoms
and bonds, is called a
force-field
FORCE FIELD
Vn is barrier height, n=
multiplicity (number of
minimum points in the
function as the bond is rotated
through 360), J = phase factor
(torsion angle passes its
minimum value)
Stretching Energy
Harmonicc Oscillator
y The harmonic oscillator consists of a
particle that experience a restoring
force proportional to its displacement
from its equilibrium position
A deviation of 0.2 Å from the
reference value can lead to
12kcal/mol change of energy of
a system with a 300 kcal/mol-1Å2 force constant
v(l)= De{1-exp[-a(l-l0)]}2
Bending Energy
n" reflects the type symmetry in the dihedral angle, The
parameter phi can be used to synchronize the torsional
potential to the initial rotameric state of the molecule whose
energy is being computed
Torsion Energy
Non-Bonded Energy
The "A" and "B" parameters control the depth and position (interatomic
distance) of the potential energy well for a given pair of non-bonded
interacting atoms (e.g. C:C, O:C, O:H, etc.). In effect, "A" determines the
degree of "stickiness" of the van der waals attraction and "B" determines
the degree of "hardness" of the atoms
Lennard
d Jones Interaction
V (r )
ª§ V ·12 § V · 6 º
4H «¨ ¸ ¨ ¸ »
© r ¹ »¼
«¬© r ¹
repulsive
r
V is collision diameter and
H is the depth of potential
attractive
Lennard
d Jones Interaction
Minimum has the well depth as -İ which can be
shown easily as IROORZV
dV (r )
dr min
ª 12ı 12 6ı 6 º
4İ «
»
13
r
r7 ¼
¬
Solving this for r we will JHW
r
21/ 6 V
0
Interaction Energy
gy
Lennard
d Jones Interaction
y For two different types of atoms or molecules 1 and 2:
q1q2
4SHH 0 r
1
U i ,i
y Berthelot rule: İ = İ1 İ2)1/2
y Reliable estimates of the LJ diameter and potential
parameter are available for several compounds.
Uind
Coulomb Interaction
-
+
U d ,d
QP cos T
4SH 0Hr 2
P1P 2 (3 cos 2 T 1) 1
4SH 0H
r3
U d ,d
y ı = ı1 + ı2)/2
U i ,d
P1P 2
^2 cos T1 cos T 2 sin T1 sin T 2 cos I `
4SH 0Hr 3
>
1 P 2 4 cos 2 T sin 2 T
D
2
2
4SH 0H r 6
@
>
@
1 DP 2 3 cos 2 T 1
2 4SH 0H 2 r 6
Coulomb interaction- How strong it is?
&RQVLGHU two monovalent charges separated by 0.3
QP
U i ,i
q1q2
4SHH 0 r
1
Ho = Permittivity of free space (8.854 × 10-12 ) C2 /Jm
H = Relative permittivity or dielectric constant of the
medium
q = elementary charge (1.602 × 10-19c)
U i ,i
(1.602 u10 19 ) 2
4 u 3.14 u 8.854 u10 12 u 0.3 u10 9
7.69 u10 19 J (vacuum)
200 k BT
(per ion pair in vacuum at 300 K )
Hydrogen
y
g
Bonding
g
“Hydrogen bonds are weak DWWUDFWLRQV with binding
strength less than one-tenth that of a normal covalent
HYDROGEN BONDING
bond.
hydrogen
bonds
are
of
extraordinary
importance: without them
• all wooden structures would collapse !
• cement would crumble !
• oceans would vaporize!
• living things would disintegrate into random dispersions of
inert matter!
Hydrogen
y
g
Bonding
g
Hydrogen
y
g
Bonding
g
y Bond between the donor covalent pair X-H in which
a
hydrogen
atom
H
is
bound
to
a
more
electronegative atom ; 1 2 ) Cl) and other
noncovalently
bound
nearest
neighbor
electronegative acceptor atoms.
į-
į
į-
į
X
H---------------A
Y
y The interaction of the dipole with the excess electron
density at the acceptor atom(s) is responsible.
y Hydrogen bonds are rarely linear in biomolecules.
y They are weak in biomolecules.
Hydrogen
y
g
Bonding
g
Hydrogen
y
g
and covalent bond
y Hydrogen bonds are about 15 times weaker than
covalent bonds.
y The bond geometry can be compressed or expanded
by up to 20% relative to the equilibrium values.
y The H–bond length and angles are dependent on
structure as well as environment in which they occur.
y “Most probable” hydrogen bond length can be think
by statistical survey over a large number of structures
• H–bonds are not atom-pair properties like covalent
ERQGV but are group pair properties.
• Covalent bond (or ionic bond) can be decomposed
into atomic properties like covalent (or ionic) radii
and are additive. They are unaffected ( or affected by
less than 2% in general) by the structure or
environment of the molecules.
in which they occur.
Hydrogen
y
g
and covalent bond
Hydrogen
y
g
and covalent bond
• The properties of hydrogen bonds are dependent not
• Covalent bonds and energies differ little from one
only on the first neighbor atoms but also on the
sequential nature of the total pattern of bonding.
• One can not break up hydrogen bond length to the
‘hydrogen bond atom radii’.
• It is best to model the hydrogen bond interaction by
donor and acceptor group potential rather than
atom-pair function.
type of molecule to the other.
• This is not the case of H-bonds. H-bond lengths are
statistical properties and the values observed for a
particular donor-acceptor combination varies by
±10% from the mean value.
Hydrogen
y
g
bonding
g
Hydrogen
y
g
bonding
g
Substance
Tm , K
Tb , K
Heat of
vaporization
Molar
volume
on the physical and physio-chemical properties of
HF
181
292
30.2
20.2
substances.
H2O
273
373
40.8
18.0
NH3
195
240
23.4
20.8
CH4
89
112
9.3
34.0
C2H5OH
(ethanol)
161
351
42.8
(CH3 )2O
(dimethyl ehter)
135
249
18.7
y The presence of hydrogen bonds exerts a strong effect
y The intermolecular hydrogen bonds determine the
association of molecules.
y Let us compare six substances and their properties.
Hydrogen
y
g
bonding
g
Hydrogen
y
g
bonding
g
y Due to absence of hydrogen bond in PHWKDQH its
y The structural studies of crystals containing hydrogen
melting as well as boiling point is low.
bonds show that when two electronegative atoms A &
y Even in two isomeric VXEVWDQFHV ethanol has higher
B are bound by a hydrogen DWRP the equilibrium
melting and boiling point because of presence of OH
separation rAB between them is smaller than the sum
which forms a hydrogen bond.
of their van der Waal’s radii.
y The dielectric constant of the substances having
hydrogen bonding is high.
y For eg: this is 80 for H2O while 15.5 for NH3
y For eg: for two oxygen atoms this is 0.28 QP in
presence of hydrogen atoms this reduces to 0.255 QP
for an oxygen and nitrogen atoms this is 0.30 nm
while it reduces to 0.288 nm.
Hydrogen
y
g
bonding
g
y The free energy of formation of hydrogen bond is
y Normal or weak
y Bond energy is less than
given E\
'G = 'H - T 'S = -RT ln K
where, 'H is the enthalpy 'S is the entropy of bond
formation. K represents the equilibrium constant.
y The values of
Types
yp of Hydrogen
y
g
Bonding
g
'H are of the order of 12-30 N-PROH
for water this is 11.8 for ice 25.6 for ammonia 15.518.5 kJ/mole.
20
kJ/mole
(~5
kcal/mole).
y Bond angle is weakly
directional.
y H---A ranges 1.5 A to
3.0
Hydrophobic
y
p
Effect
y Water is quite choosy in its affinities with some
other molecules.
HYDROPHOBIC EFFECT
y While H2O2 can mix IUHHO\ sugar can GLVVROYH yet
others hardly dissolve at all (oils).
y This is due to difference in free energy of formations.
y Hydrophobicity
Hydrophobic
y
p
Effect
y When a hydrocarbon is added to ZDWHU it develops a
structure “clathrate cage” in which the hydrocarbon
is surrounded by water molecules.
y Actually water has to choose between free energy
cost for breaking a hydrogen bond or to lose entropic
energy.
Clathrate
e cage
More strained water
molecules – less entropy
In Liquid certain
disorder is there –
entropy!
Interatomic
c Potentials
Less strained
water
molecules –
Randomness is
more – more
entropy
Energetically
more favorable
Potential Energy Description of Structure
A molecule changes from higher potential energy form to lower potential
energy form.
• Potential energy is determined by inter-molecular, intra-molecular, and
environmental forces
• Molecular structural “evolution” can be performed by systematic
variation of the atom positions towards the lower energy directions.
This procedure is called “structure optimization” or “energy
minimization”
Energy Minimization for Structural Optimization
Structure “evolution” can be performed by systematical variation of the atom
positions towards the lower energy directions. This procedure is called
“structure optimization” or “energy minimization”
Energy minimization
Population of Minima
y The problem of minimizing the energy of a
model macromolecular system fall into
general area of non-linear optimization
problem
y Variable X=(x1, x2, x3,….,xn)
y Objective function V=f(x)
• Gradient Methods (First derivative
method):These are currently the most popular
methods in molecular mechanics e.g conjugated
gradient, Steepest descent
• Utilizes values of a function and its gradients
• They offer a much better convergence rate than
search methods and do not require a lot of
computer memory
• First the decent direction is chosen (3N
dimensional vector of length sk)
• Descent step size is chosen (OK)
• Descent step is taken according to
Xk=xk-1 + OKsk
Active Structure
Most populated minimum
Global minimum
Most minimization method can only go downhill and so locate
the closest (downhill sense) minimum.
No minimization method can guarantee the location of the
global energy minimum.
No method has proven the best for all problems.
Overview
Molecular Minimization
• Process Overview
• Conjugate Gradient (Powell)
modify steepest descent to increase efficiency
¾ Initial steps are steepest descent
current step vector is not similar to previous step vectors
accumulates information about the energy function from one iteration to the next
One of two factors determines when a
minimization calculations is completed:
a) Number of defined steps (Gn) have
been calculated.
b) a predefined value of the gradient (g)
has been reached. (gradient very
rarely actually reaches exactly zero)
Molecular dynamics (MD)
y The evolution of the molecular system is studied as a series of
snapshots taken at close time intervals (in the range of 10-15
Sec)
y The time dependent behaviour of atomic and molecular
system.
y Solve newtonian equation of motion
How do you run a MD simulation?
For each time step:
Compute the force on each atom:
wE
X: cartesian vector
F ( X ) E ( X ) of the system
wX
Solve Newton’s 2nd law of motion for each atom, to
get new coordinates and velocities
xx
MX
F(X )
Store coordinates
Stop
Newton’s equation cannot be solved analytically:
Use stepwise numerical integration
Hydrophobic
Polar
Acidic
Basic