Chimica degli Alimenti: Reattività dei Carboidrati
advertisement

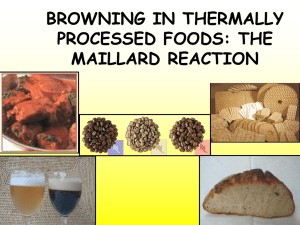
3° LEZIONE DI CHIMICA DEGLI ALIMENTI (20/10/2020) REATTIVITA’ DEI CARBOIDRATI La nucleofilicità delle funzioni OH a livello della catena saccaridica non è la stessa, sebbene si tratti di funzioni tra di loro similari. Ad essere maggiormente nucleofilo è l’OH a livello del C anomerico (OH emiacetalico) il quale si troverà ad essere più nucleofilo rispetto all’OH legato al C2 e lo è particolarmente se occupa posizione equatoriale rispetto all’ assiale. Quindi l’OH emiacetalico del beta-D-glucosio è più nucleofilo di OH emiacetalico dell’alfa di glucosio. Si è osservato che la POSIZIONE VICINALE va ad influenzare la reattività e quindi la nucleofilia, infatti se OH equatoriale si trova prossimo in posizione vicinale ad un OH assiale, è più nucleofilo di un OH equatoriale in assenza di una posizione assiale. L’ordine stimato di nucleofilia va a verificare che -6OH>20H>3OH=4OH QUANDO LA POSIZIONE 1 è ALFA -6OH>3OH>2OH>4OH QUANDO LA POSIZIONE è BETA Nel caso in cui ipotizziamo la reattività dei carboidrati, bisogna ricordare che non tutti gli OH reagiscono allo stesso modo e dove c’è possibilità di utilizzare un OH come nucleofilo, la sua posizione a livello di una struttura a sedia va ad influenzare anche in considerazioni delle posizioni vicinali, la sua reattività. Una delle forme dominanti di presenza degli zuccheri a livello di matrici alimentari, in particolare dei vegatali, è dei GLICOSIDI, che sono derivati di zuccheri in cui lo zucchero rappresenta una parte chiamata RESIDUO GLICONICO, la parte non zuccherina può essere rappresentata da composti diversi, quindi i glicosidi sono variabili e molti dei glicosidi presenti in natura agiscono come sostanza bioattiva, ovvero nutraceutica. La sintesi dei nucleotidi tiene conto della reattività, della nucleofilia degli OH a livello della porzione saccaridica. E’ coinvolto un Beta-glucopiranosio il quale deve essere attivato a livello della funzione emiacetalica grazie ad un catalisi acida, che fa sì che quell’OH emiceatalico venga converito in H2O che è una base stabile e quindi un ottimo gruppo uscente, tale che la delocalizzazione di una coppia di elettroni da parte dell'O eterociclico ne promuove la fuoriuscita. Quando H2O esce si forma C elettrofilo, ciò si osserva dalla struttura di risonanza dove O richiama elettroni pgreco lasciando sul C vicinale una deficienza elettronica netta con la forma di carica positiva. Questo C con carica positiva netta è elettrofilo forte e può essere attaccato anche da nucleofilo debole come un alcol semplice come il metanolo. Il metanolo attacca l'elettrofilo formando intermedio O-protonato che subisce astrazione del H acido a livello di O con carica positiva ad opera di una base come H20. Siamo in presenza di una spezie con ibridazione sp2, quindi la planarità dell'intermedio fa sì che l'attacco del nucleofilo possa avvenire da entrambi i lati, con formazione di due derivati glicosidici (il metilbeta-glucopiranoside e il metil-alfa-glucopiranoside). Il gruppo metossi nei prodotti costituisce la parte agliconica della molecola (priva di zucchero) che è deputata alla bioattività delle sostanze glicosidiche presenti in natura; l'attività antipiretica della salicina è dovuta al fenolo che in orto ha funzione idrossimetilica. La formazione di glicosidi, quando si parte dall' anomero beta porta ad una miscela di prodotti alfa, beta-glicosidici ma l'alfa si forma in quantità superiore a causa dell' EFFETTO ANOMERICO, che si basa sul fenomeno dell'iperconiugazione tale che l'ingresso del nucleofilo tenda ad essere accolto in maniera più favorevole in virtù di quella che è l' interazione orbitalica perchè l'orbitale che contiene la coppia di elettroni non condivisa tende a sovrapporsi all'orbitale sigma star del legame C-Z(Z è l'eteroatomo formante il legame con il C anomerico) se Z si trova in assiale. La GLICOSIDAZIONE è sfruttata a livello sintetico, lo zucchero è forma di protezione rispetto alla tossicità intriseca della porzione agliconica. Questa reazione avviene anche in laboratorio e si basa su una SN a carico del C anomerico dove ci deve essere specie che deve aggire da gruppo uscente tale che l'attacco del nucleofilo possa favorire la fuoriuscita del gruppo uscente. -l' agente glicosilante è il glicosil donatore -il nucleofilo è il glicosil accettore In laboratorio si può sentitezzare residuo acetalico al C1, prima cosa: proteggere fx OH che non devono reagire seconda cosa: se vogliamo che reagisca solo C anomerico, questo C deve legare una specie che funge da miglior gruppo uscente come gli alogenuri(grazie alla base stabile che ne deriva, grazie all'ottetto raggiunto) terza cosa: l'addizione di una qualsiasi specie che funge da nucleofilo come alcol ci porta alla dintesi di un O-glicoside, ciò che favorisce in modo peculiare la reazione di Knoegs-Knorr è la sintesi dei beta-glicosidi, che in natura non si formano perchè prevale la formazione dell' anomero alfa. La reazione procede in più step nei quali la formazione di un eterociclo intermedio favorisce l'attacco preferenziale della specie che deve fungere da nucleofilo in posizione beta. Si parte da nucleoside protetto con gruppi acetilici, (l'acetilazione avviene tra OH a carico del saccaride che fungono da nucleofili e attaccano l'anidride acetica o un alogenuro acilico come cloruro di acetile, in entrambi i casi si ha sostituzione nucleofila acilica e si hanno ottimi gruppi uscenti ovvero ione acetato stabilizzato per risonanza e uno ione alogenuro che è base stabile). La molecola intermedia protetta a livello delle fx OH già presentante un atomo di Br in posizione alfa a livello del C anomerico, può in virtù della delocalizzazione elettronica ad opera di una coppia solitaria presente sull'O eterociclico causare la fuoriuscita del gruppo uscente, condizione che fa sì che il C anomerico diventi positivo e subisca attacco nucleofilo interno da una delle coppie solitaria della fx acetilica presente a livello del C2, cosa possibile perchè nell'attacco nucleofilo ad opera di questo O, ciò che si chiude è un ciclo a 5 termini con minimizzazione della tensione angolare. Con questa chiusura la posizione alfa si trova stericamente impedita all' attacco di qualsiasi specie nucleofila che possa attaccare il C anomerico quindi questa specie può essere attaccata solo in beta dal nucleofilo. A partire da intermedio con ciclo a 5 termini si possono scrivere strutture di risonanza che evidenziano perchè si ha la reazione al Canomerico. Il nucleofilo scelto è all'O, come qualsiasi alcol, l'attacco al C anomerico favorisce il distacco C-O, l'O ripristina quella che è la sua configurazione elettronica ottimale, ottenendo la molecola di interesse, in realtà per ottenerla è necessaria la rimozione dei gruppi acetilici, che sono rimossi con idrolisi basica. Alla stregua degli O-GLICOSIDI O-> perchè l'O fa da ponte, di estrema importanza negli alimenti la hanno gli N-glicosidi in cui il legame glicosidico è mediato da N, a fungere da nucleofilo sarà un gruppo amminico che agisce sul C aldeidico del glucosio stipulando nella forma aperta, la sintesi di un immina che si può riarrangiare formando struttura ciclica che ricorda quella degli emiacetali nella quale a livello del C1 c'è residuo azotato. Si possono formare anche gli S-glicosidi in cui il nucleofilo è un tiolo. Ed esistono in natura anche C-glicosidi dove il legame glicosidico è legato dal C, sebbene la promozione sia mediata dalla presenza in posizione vicinale di una funzione fenolica I GLICOSIDI POSSONO ESSERE IDROLIZZATI Un glicoside è dal punto di vista organico un acetale e può essere idrolizzato solo in ambiente acido, in ambiente basico gli acetali sono stabili. In ambiente acido la reazione procede in modo inverso alla sintesi del glicoside, sarà l'O a livello del C anomerico, quello della funzione acetalica che si protonerà in modo da stipulare un buon gruppo uscente che grazie alla delocalizzazione della coppia solitaria a livello dell'O eterociclico, si ha fuoriuscita del gruppo uscente con formazione di C anomerico elettrofilo forte che può essere attaccato da nucleofilo debole come H2O, si ha linea ondulata, si ha attacco di H20 da entrambi i lati sintetizzando i due anomeri. IMBRUNIMENTO NON ENZIMATICO: SINTESI DI MAILLARD Avviene senza l'ausilio di enzimi, nel momento in cui si ha cottura, esposizione, conservazione prolungata degli alimenti perchè lo zucchero può interagire con un'ammina come gruppo ammidico degli aa presenti in forma libera o nella forma di peptidi e proteine negli alimenti formando polimeri condensati a base eterociclica. AD ES: MELA, che viene tagliata bella bianca grazie alla caratterizzazione della sua polpa, ne è lasciata una parte e in pochi minuti la polpa bianca diventa scura, avviene ox ed è reazione di imbrinimento ENZIMATICA, ci sono nella mela degli enzimi, le fenol ossidasi che interagendo con l'area e la luce sono atte alla degradazione del polifenoli della mela con imbrunimento della specie. Maillard si rese conto che durante la cottura possono avvenire reazioni di imbrunimento che non coinvolgevano enzimi, con interazione tra zuccheri riducenti (piccoli monosaccaridi come glucosio) e gruppi amminici primari. Maillard era un medico, che si interessò di ciò che accadeva nella cottura casalinga soprattutto durante l'arrostimento della carne perchè queste reazioni di imbrunimento non enzimatico avvengono anche a livello dell' organismo portando ai prodotti di glicazione avanzata correlati all'instaurarsi di alcune patologie che di loro sono correllate a condizioni di stress ossidatico. La reazione di Maillard è stat ripresa più volte, ancora oggi studiata, il primo ad occuparsene dopo Maillard fu OGE, dividendo la reazione in 3 stadi -uno iniziale in cui non c'era imbrunimento -uno intermedio in cui si hanno saturazioni con formazione del colore bruno -uno finale dove non si ha piena caratterizzazione delle molecole che si formano, sono MELANOIDINE, distinte sulla composizione in termini di derivati amminoacidici e saccaridici e sono sfruttati nell'industria alimenare e in farmaceutica per dare il colore bruno. Siccome questa reazione sfrutta residui amminoacidici di proteine non è assimilabile alla reazione di caramellizzazione. La reazione di M è favorita dal calore ma può avvenire anche a T ambiente a 25° durante la conservazione degli alimenti. Queste T sono incontrate durante la frittura o l'arrostimento oppure quando inforniamo, come la crosta durata del pane o la preparazione delle patatine fritte (accompagnata alla formazione di molecole piccine ma tossiche, la cui introduzione nell'organismo causa mutagenità e tossicità). OGE: con Maillard aldosi e chetosi reagiscono con le ammine in condizioni di calore si formano composti che aumentano l'appetibilità di un composto, forniscono aroma, colore e definiscono la caratterizzazione di un dato alimento. Nella suddivisione di Oge in 3 stadi 1) condensazione tra zucchero riducente ed ammina per formare a seconda della natura dello zucchero, aldoso o chetoso, dal nome dello scopritore...il prodotto di Amadori e il prodotto di Heyns 2) Questi prodotti subiscono riarrangiamenti, perdite di H20, con sintesi di molecole con numero inferiore di atomi di C, da molecole con nuclei aromatici come furfurale, o a piccole aldeie aromatiche come quelle che derivano dalla reazione di Streicker 3)Questi piccoli composti possono anche essere caratterizzate da 2C possono complessare tra di loro formando molecole che danno realmente colore bruno agli alimenti. MECCANISMO CHE PORTA ALLA SINTESI DI AMADORI E' definita condensazione, tra zucchero riducente ed ammina, è il risultato di reazione di addizione nucleofilla in cui il nucleofilo è l'amminogruppo e il C aldeidico dello zucchero che è il glucosio è l'elettofilo. La reazione di addizione nucleofila che porta alle immine o basi di Schiff, prevede che il gruppo azotato "N" attacchi C carbonilico promuovendo la delocalizzazione degli elettroni sull'O aldeidico con formazione di intermedio in cui C che ha subito l'attacco varia la sua ibridazione da sp2 a sp3 e la protonazione dell'O che ha ricevuto elettroni pgreco (l'O che è diventato alcossidico) porta alla formazione di un residuo di natura carbinolamminica, cioè la carbinolammina, che è instabile perchè a livello del C sono geminali un OH e un gruppo azotato, entrambi capaci di operare da gruppo uscente ma la basicità intrinseca di questi due gruppi fa si che se la reazione che è reversibile, sia proiettata verso l'immina, sarà l'OH a fungere da gruppi uscente e per farlo deve diventare una base stabile, a questo punto si modula PH tra 4-5/ 5.5 la protonazione converte OH in H2O(si ha catalisi acida) e la fuoriuscita è promossa dalla coppia solitaria presente sull'N. Quando l'N delocalizza la sua coppia solitaria, si forma specie N positiva che poi subirà deprotonazione del H presente sull'N da parte dell'H2O appena fuoriuscita in modo che si abbia il ripristino del catalizzatore acido e formazione del doppio legame tipico dell'immina che può avere duplice destino: -OH legato al C5 funge da nucleofilo data l'elettrofilia del C imminico rispetto al C carbonilico con ciclizzazione e formazione di ciclo a 5 termine , si forma GLICOSILAMMINA cioè un N-glicoside -Se è sfruttata l' acidità del H in alfa al C immidico, ciò che si ha sono reazioni di tautomeria che si spingono verso la sintesi dei prodotti di Amadori. L'atrazione di questo protone può favorire la delocalizzazione di elettroni pgreco all'N, formulando tautomeria di tipo IMMINA/ENAMMINA, l'N può acquisire H, si forma enammina particolare con fx enolica al C2 che può essere tautomerizzata nella forma più stabile del tautomero chetonico formando chetoso che è il composto di Amadori che lega al C1 il residuo azotato. La tautomeria I/E come quella cheto/enolica può realizzarsi se c'è la promozione ad opera di catalisi acida e basica (nell' es scegliamo una delle due) SVILUPPARE QUESTO MECCANISMO Il prodotto di Amadori rispetto al glucosio da cui sono partita ha numero di elettroni pgreco che è lo stesso, nel glucosio c'è fx carbonilica di natura aldeidica con una insaturazione, nel P di Amodori si ha fx chetonica con una insaturazione. Con una insaturazione quando arriva la luce UV, gli elettroni pgreco possono essere solleticati ma non c'è acquisizione del colore (per la quale sono necessarie più insaturazioni che tra di loro devono essere coniugate perchè quando si riceve fotone e si ha l'eccitazione di elettroni pgreco, se c'è coniugazione l'eccitazione favorisce anche delocalizzazione. Il prodotto di Amadori avendo un solo doppio legame è incolore e inodore. ALLO STESSO MODO, SE SI PARTE DA UN CHETOSO, come fruttosio e lo si fa reagire con ammina, si forma prodotto incolore e indore detto prodotto di Heyns, si ha sempre dopo addizione nucleofila prima formazione di intermedio carbinolamminico al C2 che per fuoriuscita di una molecola di H2O mi da un gruppo ammidico al C2. Anche qui ci sono posizioni alfa al C imminico, ne sono 2: -l'astrazione di un H al C in alfa della posizione idrossimetilica, favorisce delocalizzazione elettronica, formazione di intermedio enamminico enolico che tautomerizza nel corrispondente composto aldeidico. Con Amadori si partiva da aldoso e si arrivava al chetoso, qui si parte da chetoso e si arriva ad aldoso ovvero prodotto di Heyns sostituito al C2 con gruppo azotato, ma la reazione è la stessa. PUO' SUCCEDERE CHE UN PRODOTTO DI HEYNS PUO'ESSERE CONVERTITO IN PRODOTTO DI AMADORI O VICEVERSA. Se consideriamo ad es il prodotto di Amadori(che deriva da zucchero esoso e reagisce con ammmina, l'ammina attaca C carbonilico con i meccanismi dell'addizione nucleofila formando solito intermendio carbinolamminico, si perderà H2O, con fomazione di residuo imminico), nella seconda struttura c'è immina al C2 che però deriva da un prodotto di Amadori e quindi a livello del C in alfa, in particolare al C1 della catena, presenta un residuo di derivazione ammidica, questo intermedio può tautomerizzare(T I/E). L'enammina che si forma può di suo ancora tautomerizzare coinvolgendo nella formazione di un'immina il C1 che se diventa C imminico diventa elettrofilo, cioè attaccabile da H20 nella reazione di idrolisi che porta alla sintesi dell' aldeide corrispondente(siccome derivava da reazione di addizione nucleofila al C2 è il prodotto di Heyns). La reazione è reversibile e cioè si puo arrivare da H. ad A. 2 STEP Nel secondo step, il prodotto di Amadori e di Heyns subiscono disidratazioni che portano a riarrangiamneti che definiscono la peculiarità della reazione di Maillard. La perdita di molecole di H2O porta a formare intermedi reattivi che sono detti OSONI, a seconda del C coinvolto nel meccanismo di disidratazione si può avere il 3-deossiosone, 1-deossiosone, 4-deossiosone Ciò che è certo è che il ph del mezzo incide molto sul sito in cui avremo perdita di H2O, in particolare altro fattore che distingue reazione di Maillar dalla caramellizzazione è che il ph giace in range limitato tra 4 e 7 e ci sono reazioni di perdite di H2O che vengono favorite a ph acido laddove altre sono favorite a condizioni di ph della neutralità. SI PARTE DAL PRODOTTO DI AMADORI, che deriva dalla reazione tra zucchero che è glucosio e ammina secondaria(dimetilammina che reagisce con C aldeidico del glucosio)-> sviluppare meccanismo...Ottenuto imminio è convertito in composto enamminico. Partendo da prodotto di Amadori, con tautomeria da chetone ad enolo corrispondente la presenza della coppia solitaria sull'N in posizione vicinale agli elettroni pgreco della fx enolica fa sì che si possa avere un riarrangiamento elettronico tale da far fuoriuscire a livello del C3 una fx OH con meccanismo dell'eliminazione base/coniugata E1CB secondo il quale si ha l'eliminazione di una fx OH(grazie a delocalizzazione elettronica su C1)che di suo è base forte e quindi pessimo gruppo uscente laddove c'è passaggio di elettroni pgreco con fuoriuscuta di questo gruppo che può protonarsi nel mezzo di reazione. A seguito della delocalizzazione elettronica si formano due gruppi fx -gruppo imminico in cui N ha carica positiva che può essere rimossa solo con attacco nucleofilo perchè l'N manca di elettroni, sul C2 invece c'è fx enolica. Grazie all N positivo a livello imminico si può scrivere struttura di risonanza che videnza che è il C ad essere positivo, elettrofilo forte che può essere attaccato da nucleofilo debole. L'attacco forma intermedio nel quale per traferimento inetrno di H, è generato gruppo aldeidico al C1 con fuoriuscita della dimetilammina ma la formazione di questo intermedio deve ancora subire il processo di tautomeria cheto-enolica in cui gruppo enolico al C2 che si instaura tra C2 e C3 viene converito in gruppo chetonico. Con questo processo di disidratazione siamo passati dal prodotto di Amadori ad un composto di natura aldeideica che è un alfa-ossoaldeide xk in posizione alfa al gruppo aldeidico c'è fx cheonica che si trova in posizione vicinale C3 di natura metilenica che è il C al quale era stato rimosso in virtù della prima reazione di delocalizzazione elettronica la fx OH. LA FORMAZIONE DEI DEOSSIESOSONI può coivolgere diverseposizioni, prima abbiamo visto 3-deossiesosone da intermedio che ha natura 1,2-eneaminolica, si può avere sintesi di 1- deossiesosone e di 4-deossiesosone a partire dall'intermedio 2,3eneaminolica, cioè la prima reazione di astrazione del H se nella sintesi del 3deossiesosone avveniva a livello del C1 che di suo è legato anche all'N, qui per avere 2,3-eneaminolo deve avvenire astrazione del H al C3. Sia C1 che C3 presentano H in alfa alla fx chetonica in posizione 2 del prodotti di Amadori e quindi sono H inusualmente acidi. La protonazione si spinge alla formazione di una fx enolica la quale in posizione 1 ha fx amminometilica la cui protonazione a livello dell'N porta in modo esclusivo alla sintesi dell'1deossiesosone, infatti se quando arriva H, è l'N a protonarsi, l'N configura meglio il residuo come un buon gruppo uscente e determina delocalizzazione elettronica da OH in 3 ottenendo eliminazione del residuo amminico con formazione di una nuova fx enolica tra C1 e C2 che tautomerizza nel corrispondente chetone, anche in questo caso come per la sintesi del 3deossiesosone, avremo prodotto in cui due fx carboniliche sono tra di loro vicinali ma ad essere privo di fx con eteroatomi è il C1, quinsi si avrà 1deossiesoesone. SE a partire dal 2,3-eneaminolo, la protonazione avviene a livello del C4, quando parte la delocalizzazione da OH su C2 della fx di-enolica la delocalizzazione promuove fuoriuscita di H20 che si è generata al C4 con formazione di una fx enolica tra C3 e C4 che tautomerizza nella corrispondente fx chetonica con formazione di fx metilenica CH2 a livello del C4.