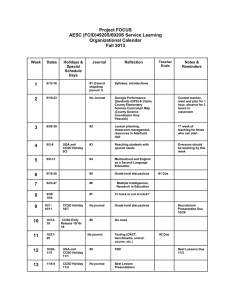
Chemical Physics Letters 585 (2013) 37–41 Contents lists available at ScienceDirect Chemical Physics Letters journal homepage: www.elsevier.com/locate/cplett Which isomeric form of formaldehyde dimer is the most stable – a high-level coupled-cluster study Grygoriy A. Dolgonos Computational Centre of Chizevsky’s Regional Scientific Library, 24 V. Perspektyvna str., Kirovograd 25006, Ukraine a r t i c l e i n f o Article history: Received 24 July 2013 In final form 17 August 2013 Available online 23 August 2013 a b s t r a c t Frozen-core and all-electron CCSD(T) calculations have been performed in order to derive accurate geometrical characteristics as well as dimerization energies of the two lowest-lying isomers of formaldehyde dimer (H2CO)2: dimer I (Cs) and dimer II (C2h). Contrary to early MP2 calculations, it has been unambiguously determined on the basis of CCSD(T) complete-basis-set extrapolations that dimer I is the true global-minimum structure, which lies 0.3–0.4 kcal/mol lower in energy than the dimer II structure at 0 K. The obtained equilibrium geometries and dimerization energies can serve as a benchmark for testing the performance of other, less computationally demanding methods. Ó 2013 Elsevier B.V. All rights reserved. 1. Introduction Weak interactions in molecular systems are known to play a significant role in stabilizing of many chemically and biologically relevant systems [1–3]. In particular, C–H O interactions involving formyl hydrogen atoms have recently attracted much attention due to their role in crystal packing [4], in stabilizing and designing supramolecular systems [5,6], in molecular recognition [7] and even in selectivity control of organic reactions [8]. The strength of C–H O interactions typically lies midways between that of systems with strong hydrogen bonds and of systems stabilized by nonbonding van der Waals interactions and is generally in the range of 2.4–2.8 kcal/mol per one C–H O interaction involving formyl hydrogen donor [9]. This strength is however sufficient to perturb the equilibrium geometries of interacting monomers in such a way that C–H bond lengths become shorter whereas carbonyl C@O bonds lengthen, which can easily be detected spectroscopically. This shortening of C–H bond lengths and, consequently, a blue shift of corresponding C–H stretching vibrational frequency makes C–H O interactions completely different from their conventional hydrogen-bonded (e.g., O–H O) counterparts [10,11] (although examples of blue-shifting bifurcated O– H O hydrogen bonds are also known [12]). Nevertheless, these interactions belong formally to the framework of hydrogen bonds due to similar driving forces involved in their formation [10,13]. Formaldehyde dimers represent one of the simplest examples which involve C–H O interactions. Theoretical investigations predict an existence of two lowest-energy isomers of (CH2O)2 [14–18]. The first isomer of Cs symmetry (hereafter referred to as: dimer I) has a perpendicular arrangement of oppositely oriented CH2O monomers leading to only one C–H O contact whereas the secE-mail address: dolgonos@gmail.com 0009-2614/$ - see front matter Ó 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.cplett.2013.08.073 ond isomer (hereafter called dimer II) is characterized by a completely planar arrangement of all atoms leading to C2h symmetry and, consequently, to the appearance of two C–H O contacts (Figure 1). Only the first isomer has been detected experimentally by means of microwave spectroscopy [19]. However, on the theoretical side it has not been unambiguously determined up to now which one of these two isomers is energetically the most stable. Leaving aside historically first but the least accurate Hartree– Fock and semiempirical calculations, early MP2 results of Ford and Glasser [14] indicated that dimer I should be the lowest-energy structure according to the uncorrected MP2/6-31++G⁄⁄ energies, but the stability order has reversed after including corrections for the basis-set incompleteness (basis set superposition error or BSSE) and for the zero-point energy (ZPE). As pointed out by Hermida–Ramón and Ríos [15], although the reported by Ford and Glasser [14] dimerization energies were inconsistent with their total MP2 energies, the stability trend remained the same if the correct values of dimerization energies of both dimers were obtained – the dimer II was by approximately 0.1 kcal/mol more stable than the dimer I at 0 K. According to MP2/6-31++G (2d,2p) results of Hermida–Ramón and Ríos [15], the BSSE-only corrected value of dimerization energy of dimer I lies by 0.62 kcal/mol lower in energy than that of dimer II. A similar dimerization energy difference of 0.66 kcal/mol in favor of dimer I has been recently reported by Holt et al. [16] on the basis of MP2/ANO-L calculations (where ANO-L stands for the large (-L) version of generally contracted Atomic Natural Orbital (ANO) basis sets [20]). On the other hand, single-point calculations at the coupled-cluster level of theory including single, double and quasiperturbative triple excitations (CCSD(T)) of Smith et al. [17] combined with the ZPE corrections calculated at the MP2 level resulted in numerically equal total energies of both dimers. In addition, ZPE-only corrected dimerization energies obtained with MP2/6-311++G⁄⁄ led to only a 38 G.A. Dolgonos / Chemical Physics Letters 585 (2013) 37–41 H3 H 4 C2 O2 H1 H1 O1 C1 C1 H2 O1 H2 Dimer I (Cs) limit using a scheme of Peterson et al. [33] that relies on the usage of a mixed exponential/Gaussian formula: O2 H4 C2 σ ðX1Þ EX ¼ ECBS þ be H3 σ Dimer II (C2h) Figure 1. Schematic view of the two most stable structures of (CH2O)2. Atom numbering follows that of Ford and Glasser [14]. slight stabilization of dimer I compared to dimer II (by 0.16 kcal/ mol) [21]. These findings clearly indicate an importance of ZPE correction on the resulting stability order of formaldehyde dimers since most of electron-correlation methods typically yield dimer I as the most stable isomer on the basis of respective uncorrected total energies [14–18]. Since these two dimers can serve as benchmark structures for testing other, less computationally demanding approaches (i.e., for testing the performance of different DFT functionals) it is essential that the geometric and energetic properties of these systems are very accurately determined. Recent attempts in this direction have been made by Remya et al. [22] who assessed the accuracy of many DFT functionals against reference CCSD/aug-cc-pVTZ geometry and dimerization energy of the dimer II reported earlier by Mackie and DiLabio [23]. The latter authors also investigated the effects of basis set size and electron correlation at MP2, CCSD and CCSD(T) levels on the dimerization energies of ten weakly bound systems including the formaldehyde dimer II and proposed a composite scheme to accurately calculate these energetic properties. Only very recently, Řezáč et al. [24] performed composite CCSD(T) complete basis set (CBS) extrapolations to yield accurate dimerization energy of dimer I (as a part of their set of 24 model complexes) and estimated the effects of core-valence, relativistic and higher-order excitations on its value. However, the very important ZPE correction was not considered in their study [24]. Hence, one can conclude that the accurate data for both dimers evaluated using the same high-level model chemistry are still missing in the literature. Therefore, the main aim of this Letter is not only to report accurate geometries and CBS-extrapolated CCSD(T) values of dimerization energies for these two dimers but also to consider, at the highest possible level of theory, the effects of the most significant ZPE and anharmonicity as well as core-valence corrections on the resulting values. 2. Methodology and computational details Geometries of formaldehyde dimers (dimer I and dimer II) and formaldehyde molecule itself have been fully optimized at the frozen-core CCSD(T) level of theory [25–28] employing Dunning’s augmented correlation-consistent polarized valence basis sets, aug-cc-pVXZ (X = T, Q, 5). [29,30] To estimate the influence of core-valence correlation effects, analogous geometry optimizations have been also performed at the CCSD(T) level with all electrons correlated (denoted hereafter as ae-CCSD(T)) but with nonaugmented, weighted core-valence basis sets, cc-pwCVXZ (X = T, Q, 5) for carbon and oxygen atoms [31]. The choice of this family of basis sets is based on the fact that the ae-CCSD(T)/CBS extrapolated equilibrium geometries of H2CO molecule are more accurate and close to experimental values than those obtained after CCSD(T)/ CBS extrapolations using aug-cc-pVXZ basis sets (cf. Ref. [32]). On the other hand, analogous ae-CCSD(T) calculations with the use of aug-cc-pwCVXZ (X > 4) basis sets are still beyond the reach of today’s computational resources. In all cases, CCSD(T) total energies for the optimized structures have been further extrapolated to the complete-basis-set (CBS) 2 þ ceðX1Þ ; where EX and ECBS are the total energies for a given basis set (with its cardinal number X defined as previously) and for the CBS, respectively; b and c are fitting parameters. This formula has been shown by Feller et al. [34] to yield very accurate CBS atomization energies of small molecules, especially, in the case of a small range of considered cardinal numbers X (i.e., up to 5). For a more detailed discussion on the performance of different high-quality ab initio approaches to obtain accurate thermochemical and spectroscopic properties of molecules the interested reader is referred to recent reviews [35–37]. Dimerization energy DE of a given dimer at the CBS limit has been defined in a standard way as ECBS ðdimerÞ 2 ECBS ðH2 COÞ. Harmonic vibrational frequencies have been calculated with CCSD(T)/aug-cc-pVQZ and with ae-CCSD(T)/cc-pwCVQZ model chemistries for the respectively optimized geometries, from which the corresponding zero-point energies (ZPEH) were obtained. The ZPE correction to dimerization energy, DZPEH, has been calculated then as ZPEH ðdimerÞ 2 ZPEH ðH2 COÞ. Since obtained this way harmonic ZPEs are generally larger than the real ones (and traditionally require a scaling factor for a given model chemistry to be employed), an explicit calculation of anharmonicity corrections to ZPEH has been performed instead based on the formula: ZPE ¼ 0:5 ðZPEH þ ZPEF Þ þ v0 0:25 X vii ; where ZPEH and ZPEF represent harmonic and fundamental zeropoint energies, respectively, and v0 and vii are anharmonic constants [38,39]. Anharmonic corrections to ZPEH, Danharm have been obtained as a difference ZPE-ZPEH at the frozen-core second-order Møller–Plesset (MP2) [40] level of theory utilizing the aug-cc-pVTZ basis set. Consequently, anharmonicity correction to dimerization energy, DDanharm is defined as a difference Danharm (dimer) 2Danharm (H2CO). As demonstrated by Feller et al. [32] for the case of formaldehyde molecule, anharmonicity corrections evaluated with MP2 are quite insensitive to the basis set size and agree within 0.01– 0.05 kcal/mol with those obtained using either CCSD(T) quartic force fields [41] or with those based on variational vibrational computations [42]. Therefore, the usage of MP2/aug-cc-pVTZ model chemistry for anharmonicity correction determination in this work is well justified. Finally, the corrected dimerization energies DEcorr of both dimers have been obtained as a sum: DE + DZPEH + DDanharm + BSSE, where the last (BSSE) term is applied only in the case of calculations with finite basis sets and vanishes upon estimating the CBS extrapolated values. Calculations of MP2/aug-cc-pVTZ anharmonic corrections have been performed using the GAUSSIAN 09 suite of programs [43] whereas all other calculations have been performed with the MOLPRO [44] software package. 3. Results and discussion 3.1. Equilibrium geometries Tables 1 and 2 contain the equilibrium geometric parameters of dimer I and dimer II, respectively, obtained with different MP2 and coupled-cluster approaches. According to the present frozen-core and all-electron CCSD(T) results with quintuple-zeta basis sets, the C@O bond lengths in both dimers become longer than that in the isolated monomer – by 0.003 and 0.004 Å for the dimer I and dimer II, respectively. These findings are in agreement with 39 G.A. Dolgonos / Chemical Physics Letters 585 (2013) 37–41 Table 1 Geometric characteristics of optimized dimer I structure of (CH2O)2 obtained with electron-correlation methods. a b c Model chemistry/geometric parameter ae-CCSD(T)/ccpwCV5Za CCSD(T)/aug-ccpV5Zb Composite CCSD(T)/CBS [24] Earlier MP2 data 6-31++G⁄⁄ [14] 6-31++G (2d,2p) [15] 6-311++G⁄⁄ [21] r(C1O1) r(C1H1) r(C1H2) a(H1C1O1) a(H2C1O1) a(H1C1H2) r(C2O2) r(C2H3)=r(C2H4) a(H3C2O2)=a(H4C2O2) a(H3C2H4) r(H1 O2) r(C2 O1) 1.207 1.099 1.099 121.3 121.1 117.6 1.207 1.098 121.8 116.4 2.391 2.718 1.210 1.101 1.100 121.3 121.1 117.6 1.210 1.100 121.8 116.4 2.380 2.708 1.211 1.102 1.101 121.3 121.1 117.6 1.211 1.101 121.8 116.4 2.383 2.709 1.226 1.096 1.096 121.3 121.1 117.5 1.226 1.096 121.8 116.4 2.434c 2.741 – – – – – – – – – – – – – – – 1.215 1.103 – 2.406 2.702 2.480 – ae-CCSD(T)/cc-pwCV5Z equilibrium geometry of CH2O is characterized by r(CO) = 1.204 Å, r(CH) = 1.100 Å and by angle(HCO) = 121.7°. CCSD(T)/aug-cc-pV5Z equilibrium geometry of CH2O is characterized by r(CO) = 1.207 Å, r(CH) = 1.102 Å and by angle(HCO) = 121.7°. The original value of 2.831 Å reported by Ford and Glasser [14] is incorrect. Table 2 Geometric characteristics of optimized dimer II structure of (CH2O)2 obtained with electron-correlation methods. a b Model chemistry/geometric parameter ae-CCSD(T)/ccpwCV5Za CCSD(T)/aug-ccpV5Zb CCSD/aug-ccpVTZ [23] r(C1O1) = r(C2O2) r(C1H1) = r(C2H3) r(C1H2) = r(C2H4) a(H1C1H2) = a(H3C2H4) a(H1C1O1) = a(H3C2O2) a(H2C1O1) = a(H4C2O2) r(O1 H4) = r(O2 H2) 1.208 1.100 1.098 117.5 121.0 121.4 2.465 1.211 1.102 1.099 117.6 121.0 121.4 2.461 1.208 1.101 1.098 117.4 121.1 121.4 2.512 Earlier MP2 data 6-31++G⁄⁄ [14] 6-31++G (2d,2p) [15] 6-311++G⁄⁄/aug-cc-pVTZ /aug-ccpVQZ [21] 1.226 1.098 1.095 117.6 121.1 121.3 2.535 – – – – – – 2.530 1.216/–/– 1.101/–/– – – – – 2.579/2.491/2.483 See also the footnote a to Table 1. See also the footnote b to Table 1. previously reported MP2 data [14,21]. One can also see in Tables 1 and 2 that the lowest value of the bond length is given normally by the ae-CCSD(T) approach and the largest one – by MP2 with a double-zeta 6-31++G⁄⁄ basis set. In the case of dimer II, the ae-CCSD(T)/cc-pwCV5Z C@O bond length value fortuitously coincide with the CCSD/aug-cc-pVTZ one reported by Mackie and DiLabio [23]. Interestingly, the C–H bond lengths which involve H atoms not participating in the formation of a C–H O contact in dimer I demonstrate a shortening of about 0.002 Å, which is slightly larger (though by not more than 0.001 Å) than the bond length shortening observed for the C–H bond forming the C–H O contact. On the contrary, the respective C–H bond lengths containing noninteracting with the other monomer H atoms in dimer II remain rather unchanged whereas the C–H bonds involved in the formation of C– H O contacts become by 0.002–0.003 Å shorter than those of a free CH2O molecule. Almost identical MP2 values corresponding to the bond length shrinking in the latter case have been also reported earlier in the literature [14,21]. Typically, the smallest values of C–H bond lengths are obtained with the MP2/6-31++G⁄⁄ model chemistry and the largest ones – with either MP2/6311++G⁄⁄ (for dimer I) or with CCSD(T)/aug-cc-pV5Z (for dimer II). One notices again in Table 2 that the corresponding bond length values obtained with the ae-CCSD(T)/cc-pwCV5Z model chemistry and with the CCSD/aug-cc-pVTZ one are in excellent agreement with each other. On the other hand, the C@O and C–H bond distances for the dimer I (Table 1) obtained with the composite CCSD(T)/CBS approach by Řezáč et al. [24] are typically by 0.001 Å larger than the CCSD(T)/aug-cc-pV5Z ones obtained in this work and correspond roughly to the current CCSD(T)/aug-cc-pVQZ values (not shown in Table 1) . The most sensitive parameters to the model chemistry employed are the intermolecular O H and C O distances in both dimers. As can be inferred from Tables 1 and 2, the MP2 usually overestimates the values of O H distances – unless the basis set of (at least) a quadruple-zeta quality has been utilized (as has been investigated in detail for the dimer II by Kovács et al. [21]). All CCSD(T) approaches generally agree within 0.011 Å with each other – leading to ae-CCSD(T)/cc-pwCV5Z values which are slightly larger than those obtained with the frozen-core CCSD(T). However, the CCSD/aug-cc-pVTZ value [23] of the O H distances in dimer II is by 0.05 Å larger than those predicted by CCSD(T) approaches employed in this work. Interestingly, the C O nonbonded distance in dimer I calculated by Hermida-Ramón and Ríos using MP2/631++G (2d,2p) model chemistry [15] lies very close to the CCSD(T) ones although the MP2 values with smaller, double-zeta quality basis set of Ford and Glasser [14] are much larger. As in the case of O H distances, an agreement within 0.01 Å for the C O distance type has been found for all considered CCSD(T) approaches. HCO bond angles in dimer I for the perpendicularly oriented monomer whose hydrogen atoms do not form the C–H O contacts have been found to be larger by only 0.1° compared to the same angle in CH2O but these angles in the other monomer exhibit a decrease by 0.6° and 0.4° for the angles containing C–H O noninteracting and interacting H atoms, respectively. In dimer II all HCO bond angles lie in the same plane leading to only two distinct HCO angle types and, according to both CCSD(T) approaches, the respective decrease in their values equals to 0.7° and 0.3° for the 40 G.A. Dolgonos / Chemical Physics Letters 585 (2013) 37–41 Table 3 Dimerization energies (in kcal/mol) of the two lowest-lying formaldehyde dimers calculated with electron-correlation methods. Model chemistry/energetic characteristic ae-CCSD(T)/ CBSa CCSD(T)/ CBS Composite CCSD(T)/ CBS [24] Dimer I DE BSSE DZPEH DDanharm DEcorr 4.43 0.00 1.51b 0.21c 3.13 4.48 0.00 1.52d 4.55, 4.47e 0.00 – – – Dimer II DE BSSE DZPEH DDanharm DEcorr 3.58 0.00 1.05b 0.16c 2.69 3.68 0.00 0.96d 3.17 2.88 – – – – – CCSD/aug-ccpVTZ [23] – – – – 3.66f 0.36f – – – Earlier MP2 data 6-31++G⁄⁄ [14] 6-31++G (2d,2p) [15] 6-311++G⁄⁄ / aug-cc-pVTZ / augcc-pVQZ [21] 4.40g 1.07 1.37 – 1.96 – – – – 3.68h,i 2.53j/–/– – – – – 3.66g 0.78 0.85 – 2.03 – – – – 3.06h,i 2.37j/2.92j/2.82j 0.75/0.41/0.19 Taken from MP2/6-31++G⁄⁄ – 1.62/2.51/2.63 a Using core-valence cc-pwCVXZ (X = T, Q, 5) basis sets. Calculated with ae-CCSD(T)/cc-pwCVQZ. c Calculated with MP2/aug-cc-pVTZ. d Calculated with CCSD(T)/aug-cc-pVQZ. e Including core-valence, relativistic and quadruple-excitation corrections. f For a detailed analysis of the influence of single-point MP2, CCSD and CCSD(T) calculations as well as aug-cc-pV(D,T,Q)Z basis sets on resulting dimerization energies see Ref. [23]. g Since the reported by Ford and Glasser [14] dimerization energies were in error, the recalculated values based on total MP2 energies from Table 2 of their paper [14] are shown here. h BSSE-corrected value only (no DZPE reported). i Slightly smaller absolute BSSE-corrected values (3.35 and 2.69 kcal/mol for dimer I and dimer II, respectively) have been calculated with MP2/ANO-L by Holt et al. [16]. j Corrected for ZPE (obtained with MP2/6-31++G⁄⁄) but not corrected for BSSE value. b angles not involving and involving hydrogen atoms of C–H O contacts, respectively. The bond angle values obtained with the considered electron-correlated methods generally agree with each other within 0.2°. 3.2. Dimerization energies Table 3 summarizes the uncorrected and corrected values of dimerization energies obtained with different electron-correlation methods. Uncorrected DE values extrapolated using ae-CCSD(T) and CCSD(T) methods agree within 0.05 and 0.1 kcal/mol for dimer I and dimer II, respectively. This energy difference cannot be attributed to the core-valence contribution alone since the basis sets used for respective geometry optimizations are different. The core-valence contribution to DE is supposed to be much smaller (i.e., 0.004 kcal/mol for dimer I [24]). Indeed, if one performs geometry optimizations using ae-CCSD(T) and CCSD(T) using the same cc-pwCV5Z basis set in both cases, the core-valence contribution becomes equal only to +0.02 kcal/mol (dimer I) and +0.01 kcal/ mol (dimer II). It should be mentioned that the composite CCSD(T)/CBS DE value of 4.55 kcal/mol obtained by Řezáč et al. [24] for the dimer I lies slightly lower in energy than the corresponding current CBS value, being midway between our CCSD(T)/aug-cc-pVQZ and CCSD(T)/aug-cc-pV5Z results, on account of different optimized geometrical parameters obtained for the dimer I with the composite and conventional CCSD(T) methodologies (see also Section 3.1) Nevertheless, an inclusion of core-valence, relativistic and quadruple-excitation corrections to the composite CCSD(T)/CBS DE value brings the dimerization energy by 0.08 kcal/mol up [24] leading to a better agreement with the current results. Somehow surprisingly, the (recalculated) MP2 values of uncorrected DE of Ford and Glasser [14] for both dimer I and dimer II agree very well with the current CCSD(T)/CBS results due to a fortuitous cancellation of errors. However, MP2 values of dimerization energies corrected for the BSSE and for the DZPE become, in general, much smaller in absolute values than those obtained with high-level CCSD(T)/CBS approaches. In a similar manner the uncorrected DE value of 3.66 kcal/mol obtained with CCSD/aug-cc-pVTZ model chemistry for dimer II [23] agrees perfectly (within 0.1 kcal/mol) with the current (ae-)CCSD(T)/CBS uncorrected values since the effects associated with BSSE and with an inclusion of perturbative triple excitations on the resulting CCSD/aug-cc-pVTZ dimerization energy almost completely cancel out. After inclusion of all significant corrections to DE, the CCSD(T)/ CBS corrected values (DEcorr) for dimer I and dimer II at 0 K were found to be 3.17 and 2.88 kcal/mol, respectively, i.e. the dimer I is now by 0.29 kcal/mol more stable than the dimer II. Corresponding core-valence ae-CCSD(T)/CBS results yield even better stabilization of the dimer I versus dimer II – by 0.44 kcal/mol. Therefore, it is unlikely that the usage of other extrapolation schemes or the inclusion of other correction types (i.e., higher order correlation, relativistic or diagonal Born–Oppenheimer corrections) could invert the stability order in favor of dimer II. It is also desirable to include the present high-level energetic and geometric characteristics of both dimer I and dimer II into the benchmark sets (like the A24 set [24]) to test the performance of different density-functional theory (DFT) functionals (and other computational approaches). This will definitely help to elucidate the predicted differences in the location of both stationary points on the formaldehyde dimer potential energy surface. For instance, according to the recent M062X/6-311++G(3df,3pd) results [4], dimerization energies (corrected for BSSE only) resulted in a more pronounced stabilization of the dimer I (5.09 kcal/mol) over the dimer II (3.73 kcal/mol) indicating that the former dimer is probably less accurately described with this model chemistry than the latter. Therefore, a more extended DFT benchmark study than already available [22] is still needed before recommending a particular group of DFT functionals to investigate the properties of structurally similar (but larger) structures. 4. Conclusions Accurate geometric and energetic characteristics of the two lowest-lying isomers of formaldehyde dimer, dimer I of Cs G.A. Dolgonos / Chemical Physics Letters 585 (2013) 37–41 symmetry and dimer II of C2h symmetry, have been calculated employing high-level CCSD(T) computations with CBS extrapolations. In agreement with earlier MP2 calculations, both dimers have been found to exhibit C@O bond length elongation (by 0.003–0.004 Å) and shrinking of most C–H bonds (up to 0.003 Å) compared to a free formaldehyde molecule. Nonbonded C O and O H distances in both dimers agree within 0.011 Å depending on whether core correlation is, or is not, taken into account in respective (ae-)CCSD(T) geometry optimizations. After including the very important ZPE and anharmonicity corrections, the calculated ae-CCSD(T)/CBS (CCSD(T)/CBS) dimerization energies are equal, correspondingly, to 3.13 (3.17) kcal/mol for dimer I and 2.69 (2.88) kcal/mol for dimer II, indicating that dimer I is the true global-minimum structure on the potential-energy surface. Much less significant corrections to dimerization energies (including higher order correlation, relativistic or diagonal Born–Oppenheimer corrections) seem unlikely to invert the obtained stability order. It is recommended to include both formaldehyde dimeric structures in reference datasets and in benchmark studies of other methods to be able to choose the most appropriate (but less expensive) model chemistry suitable for an accurate description of this type of weakly bound complexes. Acknowledgments The author gratefully acknowledges the hospitality of the Bremen Center for Computational Materials Science (Bremen, Germany) and the use of its computational facilities at the early stage of this work. Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cplett.2013. 08.073. References [1] P. Hobza, R. Zahradnik, K. Müller-Dethlefs, Collect. Czech. Chem. Commun. C 71 (2006) 443. [2] H.-J. Schneider, Angew. Chem. Int. Ed. 48 (2009) 3924. 41 [3] G.R. Desiraju, T. Steiner, The Weak Hydrogen Bond, IUCr Monographs on Crystallography 9, Oxford University Press, Oxford, 1999. [4] T.S. Thakur, M.T. Kirchner, D. Bläser, R. Boese, G.R. Desiraju, Phys. Chem. Chem. Phys. 13 (2011) 14076. [5] L. Brammer, in: G.R. Desiraju (Ed.), Crystal Design: Structure and Function, Perspectives in Supramolecular Chemistry, vol. 7, John Wiley & Sons, Ltd., Chichester, West Sussex, England, 2003, p. 1. [6] R.K. Castellano, Curr. Org. Chem. 8 (2004) 845. [7] T. Neuheuser, B.A. Hess, C. Reutel, E. Weber, J. Phys. Chem. 98 (1994) 6459. [8] R.C. Johnston, P.H. Cheong, Org. Biomol. Chem. 11 (2013) 5057. [9] R. Vargas, J. Garza, D.A. Dixon, B.P. Hay, J. Am. Chem. Soc. 122 (2000) 4750. [10] Y.L. Gu, T. Kar, S. Scheiner, J. Am. Chem. Soc. 121 (1999) 9411. [11] S. Scheiner, T. Kar, J. Phys. Chem. A 106 (2002) 1784. [12] G. Litwinienko, G.A. DiLabio, P. Mulder, H.G. Korth, K.U. Ingold, J. Phys. Chem. A 113 (2009) 6275. [13] K. Hermansson, J. Phys. Chem. A 106 (2002) 4695. [14] T.A. Ford, L. Glasser, J. Mol. Struct. (Theochem) 398 (1997) 381. [15] J.M. Hermida-Ramón, M.A. Ríos, J. Phys. Chem. A 102 (1998) 10818. [16] A. Holt, J. Bostrom, G. Karlstrom, R. Lindh, J. Comput. Chem. 31 (2010) 1583. [17] D.M.A. Smith, J. Smets, Y. Elkadi, L. Adamowicz, Chem. Phys. Lett. 305 (1999) 169. [18] A. Vila, A.M. Graña, R.A. Mosquera, Chem. Phys. 281 (2002) 11. [19] F.J. Lovas, R.D. Suenram, L.H. Coudert, T.A. Blake, K.J. Grant, S.E. Novick, J. Chem. Phys. 92 (1990) 891. [20] P.-O. Widmark, P.-Å. Malmqvist, B.O. Roos, Theor. Chim. Acta 77 (1990) 291. [21] A. Kovács, A. Szabó, D. Nemcsok, I. Hargittai, J. Phys. Chem. A 106 (2002) 5671. [22] K. Remya, C.H. Suresh, J. Comput. Chem. 34 (2013) 1341. [23] I.D. Mackie, G.A. DiLabio, J. Chem. Phys. 135 (2011) 134318. [24] J. Řezáč, L. Šimová, P. Hobza, J. Chem. Theory Comput. 9 (2013) 364. [25] C. Hampel, K.A. Peterson, H.-J. Werner, Chem. Phys. Lett. 190 (1992) 1. [26] M.J.O. Deegan, P.J. Knowles, Chem. Phys. Lett. 227 (1994) 321. [27] P.J. Knowles, C. Hampel, H.-J. Werner, J. Chem. Phys. 99 (1993) 5219. [28] P.J. Knowles, C. Hampel, H.-J. Werner, J. Chem. Phys. 112 (2000) 3106. [29] T.H. Dunning Jr., J. Chem. Phys. 90 (1989) 1007. [30] D.E. Woon, T.H. Dunning Jr., J. Chem. Phys. 98 (1993) 1358. [31] K.A. Peterson, T.H. Dunning Jr., J. Chem. Phys. 117 (2002) 10548. [32] D. Feller, K.A. Peterson, D.A. Dixon, J. Chem. Phys. 129 (2008) 204105. [33] K.A. Peterson, D.E. Woon, T.H. Dunning Jr., J. Chem. Phys. 100 (1994) 7410. [34] D. Feller, K.A. Peterson, J.G. Hill, J. Chem. Phys. 135 (2011) 044102. [35] D.A. Dixon, D. Feller, K.A. Peterson, in: R.A. Wheeler (Ed.), Annual Reports in Computational Chemistry, vol. 8, Elsevier, Amsterdam, 2012, p. 1. [36] D. Feller, K.A. Peterson, D.A. Dixon, Mol. Phys. 110 (2012) 2381. [37] K.A. Peterson, D. Feller, D.A. Dixon, Theor. Chem. Acc. 131 (2012) 1079. [38] V. Barone, J. Chem. Phys. 120 (2004) 3059. [39] V. Barone, J. Chem. Phys. 122 (2005) 014108. [40] C. Møller, M.S. Plesset, Phys. Rev. 46 (1934) 618. [41] J.M.L. Martin, T.J. Lee, P.R. Taylor, J. Mol. Spectrosc. 160 (1993) 105. [42] G. Czakó et al., Int. J. Quant. Chem. 109 (2009) 2393. [43] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, et al., GAUSSIAN 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010. [44] H.-J. Werner, P. J. Knowles, R. Lindh, F. R. Manby, M. Schütz, et al., MOLPRO, version 2010.1, a package of ab initio programs. See http://www.molpro.net.