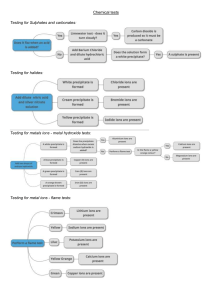
BIRLA INSTITUTE OF TECHNOLOGY MESRA RANCHI, INDIA CHOICE BASED CURRICULUM Under Graduate Programme Department of Chemistry Subject: (CH 101) Chemistry Module I: Chemical Bonding Ionic bond: Radius ratio rule, Born-Landé equation, Born-Haber cycle. Metallic Bond: valence bond and band theories, defects in solids, Werner's Theory, Bonding in Transition metal complexes, Ligands, coordination complexes, Ligand Field, Crystal Field Theory, Octahedral, Tetrahedral and square planar complexes, CFSE, Jahn Teller theorem, electronic spectra, magnetism, and isomerization in coordination compounds. [9L] TYPES OF BONDS Atoms may attain a stable electronic configuration in three different ways: by losing electrons, by gaining electrons, or by sharing electrons. Elements may be divided into: I. Electmpositive elements, whose atoms give up one or more electrons fairly readily. 2. electronegative elements. which will accept electrons. 3. Elements which have little tendency to lose or gain electrons. Three different types of bond may be formed, depending on the electropositive or electronegative character of the atoms involved. Ionic bond: An Ionic bond is formed by the transfer of one or more electron from one atom to the other. Electropositive element + electronegative element ---- Ionic bond Ionic solids: The constituent units which make up ionic solids are oppositely charged ions. The force of attraction between the ions is called an ionic bond. These crystals are formed between constituents which have very large differences in electron attracting power so as to allow complete trnsfer of electrons from one costituent to another. The force of attraction between the ions is purely electrostatic. Examples of ionic solids are: NaCl, CsCl and ZnS. Since these ions are held in fixed positions, there, ionic solids do not conduct eletricity in the solid state. They conduct eletricity in the fused state. The forces of attraction in ionic solids are very strong and therefore they exibit high melting points and cleave only if force is applied along certain directions. All ionic solids are hard and brittle. It may be observed that movement of layers of ions brings ions of the same charge near each other and this causes strong repulsions which lead to the breakdown of the crystal. The Radius Ratio Rule The ratio of radius of the cation (r+) and radius of anion (r-) is called radius ratio. Radius ratio = r+ / rThe arrangement ions in a crystal is greatly influenced by the ratio of radii of the ions. The force of electrostatic attraction between oppositely charged ions constitutes the ionic bond. Since, such bonds have Coulombic origin, each particular ion will tend to drag as many oppositely charged ions as possible around itself. The number of anions that surround a cation is called the coordination number of the cation and vice versa. Two factor appear important for an ionic structure Each ion assumes as large a coordination number as possible Attraction between opposite ions in the limiting case just leads to cation-anion contact and to anion-anion contact. Any squeezing of the ions beyond the contact positions will break down the structure. Similar AB type ionic compounds like CsCl, NaCl, ZnS do not crystallise with same structure. Stable Bonding Configurations in Ionic solids. In reality an ideal fit of a cation into the close packed anion arrangement almost never occurs. Now consider what would be the consequence of placing a cation that is (a) larger than the ideal, (b) smaller than the ideal, into the cation sites. For a stable coordination the bonded cation and anion must be in contact with each other. If the cation is larger than the ideal radius ratio value the cation and anion remain in contact, however the cation forces the anions apart. This is not a problem as there is no need for the anions to remain in contact. If the cation is too small for the site then the cation would "rattle" and would not be in contact with the surrounding anions. This is an unstable bonding configuration. Limiting radius ratio for 8-coordination (caesium chloride structure) The CsCl structure in which the Cs+ ion is surrounded by 8 Cl- ions. To get as close to the cation as possible, the anions must touch along the edge of the cube, as shown in the figure. The side of the cube has a length, a, where: a = 2rAlong the body diagonal, the Cs+ is touching the two Cl- ions on either end so its length, d, is: d = r- + 2r+ + r- = 2r+ + 2rUsing Pythagoras’ theorem, the length of the side and the body diagonal of a cube are related: d2 = a2 + a2 + a2 = 3a2 = 3 × 4r-2 = 12r-2 d = 2(3)1/2rSo, 2r- + 2r+ = 2r(3)1/2rr+ / r- = (3)1/2 – 1 = 0.732 As long as the radius of the cation is no smaller than 73% that of the anion, the CsCl structure, with its high Madelung constant, is possible. If the cation is larger than this, the structure is stable as the anions do not need to touch. If the cation is smaller than this, the cation and anion will not be in contact. A lower coordination number is then needed Limiting radius ratio for 6-coordination (sodium chloride structure) The NaCl structure in which the Na+ ion is surrounded by 6 Cl- ions. As shown in the figure, along the cube edge, the Na+ is touching two Cl- ions so its length, a, is: a = r- + 2r+ + r- = 2r+ + 2rTo get as close to the cation as possible, the anions must touch along the diagonal of a face of the cube. The diagonal has length, d, where: d = r- + 2r- + r- = 4rUsing Pythagoras’ theorem, the length of the side and face diagonal of a cube are related: d2 = a2 + a2 = 2a2 d = (2)1/2a So, 4r- = (2)1/2(2r+ + 2r-) r+ / r- = (2)1/2 – 1 = 0.414 As long as the radius of the cation is no smaller than 41% that of the anion, the NaCl structure is possible. If the cation is larger than this, the structure is stable as the anions do not need to touch but the CsCl structure is even more stable when the cation radius reaches 73% that of the anion. If the cation radius is smaller than 41% that of the anion, the cation and anion will not be in contact. An even lower coordination number is then needed. Limiting radius ratio for 4-coordination (zinc blende structure) The ZnS (zinc blende) structure in which the Zn2+ ion is surrounded by 4 S2- ions. The distance from the centre of the tetrahedron to the corner, d, is: d = r+ + rTo get as close to the cation as possible, the anions must touch along the edge of the tetrahedron. This distance, a, is a = r- + r- = 2rThe tetrahedral angle, q, is 109.5° so sin(q/2) = r- / (r+ + r-) So, sin(54.7°) = r- / (r+ + r-) r+ / r- = 0.225 As long as the radius of the cation is no smaller than 23% that of the anion, the ZnS structure is possible. If the cation is larger than this, the structure is stable as the anions do not need to touch but the NaCl structure is even more stable when the cation radius reaches 41% that of the anion. Limiting radius ratio for 3-coordination (boron oxide structure) Let say, AB is an ionic compound having coordination number 3. All the three B are in contact with A but not to each other. A limiting case arises when all the three are also come in contact with each other. Assume that the radius of cation is r + and anion is r Therefore, cos 30 ° = DC/DE or, DE = DC / cos 30 ° or, r + + r − = r − / 0.866 [ ∴ cos 30 ° = 0.866] or, r + + r − = 1.155 × r − or, r + / r − = 1.115-1.000 = 0.155 Simple geometry provides the limiting radius ration for coordination number 3 is 0.155. When the radius ratio is smaller compared to 0.155, the positive ions will not be in contact with the negative ions, and therefore, a whole will be created and the resulting structure will be unstable. If the radius ration value is more than that of 0.155, it is possible to have contact between positively charged cation and a negatively charged anion. In this case the complex will be stable. A list of limiting radius ratio is given in the following table Limiting ratio r+/r- Coordination number Structural arrangement 1 12 Close packing (ccp and hcp) 1-0.732 8 Cubic 0.732-0.414 6 Octahedral 0.732-0.414 4 Square planar 0.414-0.225 4 Tetrahedral 0.225-0.155 3 Trigonal planar Below-0.155 2 Linear By using radius ratio rule, it is possible to predict the cation/anion coordination number in any compound. So radius ratio is a useful measure in establishing the structure of ionic solids. In the following figure we have shown how the arrangement of ions in a single layer is affected by the differences in the sizes of the ions. Limitations Radius ratios provide a· useful guide to what is possible on geometric grounds, and also a first guess at the likely structure, but there are other factors involved. Radius ratios do not necessarily provide a completely reliable method for predicting which structure is actually adopted Though radius ratios indicate the correct structure in many cases, there are a significant number· of exceptions where they predict the wrong structure. It is therefore worth examining the assumptions behind the radius ratio concept, to see if they are valid. The assumptions are: 1. That accurate ionic radii are known. 2. That ions behave as hard inelastic spheres. Concept of nonpolarization of the ion. 3. That stable arrangements are only possible if the positive and negative ions touch. 4. That ions are spherical in shape. 5. That ions always adopt the highest possible coordination number. 6. That bonding is 100% ionic. 7. Overlooking the effect of crystal geometry Values for ionic radii cannot be measured absolutely, but are estimated. They are not completely accurate or reliable, Though it is possible to measure the interatomic distance between two different ions very accurately by X-ray crystallography, it is much less certain how to divide the distance between the two ions to obtain ionic radii. Furthermore the radius of an ion is not constant but changes depending on its environment. In particular the radius changes when the coordittation number changes. The radii usually quoted are for a coordination number of 6, but the radius effectively increases 3% when .the coordination number is. changed from 6 to 8. and decreases 6% when the coordination number changes from 6 to 4. Ions are not hard inelastic spheres. They are sometimes fitted into 'holes' that are slightly too small, that is the ions are compressed, and the lattice may be distorted. The assumption· that the ions touch is necessary to calculate the critical lower limit for radius ratios, In principle positive and negative ions should touch, so as to get the ions close together, and get the maximum electrostatic attraction: (Electrostatic attraction depends on the product of the charges on the ions divided by the distance between them.) A more favourable electrostatk attraction should be obtained by adopting a different geometric arrangement with a smaller coordination number' so that the ions can get closer. It has already been shown that in the alkali halides and alkaline earth oxides the NaCl structure with coordination numbers of 6:6 is sometimes adopted when other structures are predicted by radius ratios. It follows that, since the smaller ion no longer fits the site it occupies, it must either 'rattle', or be compressed. Are ions spherical? It is reasonable to consider ions with a noble gas structure as spherical. There are a small number of exceptions where the ions are not spherical. It is most unlikely that bonding is ever 100% ionic. The retention of a NaCl structure by a number of compounds which might be expected to adopt a CsCI structure is largely because there is a small covalent contribution to the bonding. The three p orbitals are at 90° to each other, and in a NaCl structure they point towards the six nearest neighbours, so covalent overlap of orbitals is possible. The geometric arrangement of the NaCl structure is ideally suited to allow some covalent contribution to bonding. This is not so for the CsCI structure. The structure adapted by the alkali metal halides may be considered to demonstrate the failure of structural prediction from the radius ratio rule. LiCl, LiBr, and LiI, the radius ratio lies around 0.3 suggesting fourfold coordination. In case of KF, KCl, RbF, RbCl, RbBr, and CsF, the radius ratio is 0.73, hence coordination number shoud be minimum 8. In fact, all the alkali metal halides adapt 6:6 (NaCl type) coordination environment except CsCl, CsBr, and CsI. It is said in the radius ratio rule that with increase of the ratio value the coordination number will increase. Let consider a AB type ionic compound, as we know that with increase of radius ration value the number of counter ions around a particular ion will increase. As the system is AB type, therefore, each cation will be surrounded by one anion. So, the gain in electrostatic attraction force will be cancelled by large repulsion force by the same charge ions as the coordination number is high. Therefore, the stability of ionic solid cannot be answered. This rule provides a rough guide to the structure of ionic solids. Ultimately, the reason why any particular crystal structure is formed is that it gives the most favorable lattice energy. Lattice Energy The stability of an ionic solid is measured in terms of its lattice energy (U0). It is defined as the energy released when on mole of an ionic crystal is formed from one mole of gaseous positive and one mole of gaseous negative ions, when these are separated from each other by infinite distance. Greater the value of lattice energy, more stable, more stable is the ionic solid. The lattice energy of sodium chloride is -185 kcal/mole. The negative sign indicates that the process is exothermic and that the energy of the system is lowered as the solid is formed. The high value suggests the stability of the solid. Qualitatively speaking, the force of attraction between oppositely charged ions is determined by their ionic radii, the charges on the ions. It is given by the relationship, where q1, q2, are the charges on ions and r is the interionic distance. It may be observed that if the radii of the ions are large, the force of attraction will be less. This is better illustrated if we compare the stability of NaCl and CsCl. The force of attraction between the Large Cs+ ions and Cl- ions will be less than that between the smaller Na+ ions and Cl- ions. As result, CsCl is less stable than NaCl. We may say that the stability of an ionic solid increases with decreasing interionic distance for crystals with similar charges. The role played by charges on the ions (on the stability of solids) is demonstrated if we compare the lattice energies of BaO and NaCl, since both have very similar internuclear distances (NaCl=2.81 Å and BaO=2.77 Å). Since BaO consists of doubly charged Ba2+ and O2- ions, the force of attraction is much larger than that in NaCl which contains singly charged ions. Hence, BaO is more stable than NaCl (melting point; NaCl=800 °C, BaO=1921 °C). The stability of an ionic solid increases with increasing nuclear charge for crystals with similar interionic distances. Determination of Lattice Energy Lattice energy can be determined by using a) Born-Lande Equation b) Born Haber Cycle a) Born-Lande Equation Theoretical values for lattice energy may be calculated. The ions are treated as point charges, and the electrostatic (coulombic) energy E between two ions of opposite charge is calculated: Where, z+ and z- are the charges on the positive and negative ions, e is the charge on an electron, r is the inter-ionic distance For more than two ions the electrostatic energy depends on the number of ions. For one mole the attractive energy is: Where N0 is Avogadro Constant, 6.023 × 1023 mol-1 The above expression express the electrostatic energy between two ions, whereas, within a crystal, the same two ions are subjected to additional coulombic forces from other ions. Because of electrostatic interactions with surrounding ions the coulomb energy of a single ion is greater than indicated in equation by a factor typical of each kind of crystal structure. This factor, known as Madelung constant and designated as A. where A is the Madelung constant and the negative sign indicates that energy is released when the two ions station themselves within the crystal. The Madelung constant (A) is a correction factor to take care of additional electrostatic forces exerted by neighbouring ions upon an ion pair and is entirely dependent upon the geometry of the crystal. When the inter-ionic distance becomes small enough for the ions, they begin to repel each other. This repulsion originates from the mutual repulsion of the electron clouds on the two atoms or ions. The repulsive forces increase rapidly as r decreases. The repulsive force is given by B/ rn, where B is a constant that depends on the structure, and n is a constant called the Born exponent. For one gram molecule the total repulsive force = (N0 B)/rn. The total energy holding the crystal together is U, the lattice energy. This is the sum of the attractive and the repulsive forces. (A is the Madelung constant and B is a repulsion coefficient, which is a constant which is approximately proportional to the number of nearest neighbours.) The equilibrium distance between ions is determined by the balance between the attractive and repulsion terms. At equilibrium. dU/dr = 0, and the equilibrium distance r = r 0 Rearranging this gives an equation for the· repulsion coefficient B. Substituting equation This equation is called the Born-Lande equation. It allows the lattice energy to be calculated from a knowledge of the geometry of the crystal, and hence the Madelung constant, the charges z+ and z-, and the interionic distance. When using. SI units, the equation takes the form: where €0 is the permittivity of free space = 8.854 x 10-12 Fm-1• Interpretation of Madelung constant The Madelung constant (A) is a correction factor to take care of additional electrostatic forces exerted by neighbouring ions upon an ion pair and is entirely dependent upon the geometry of the crystal. The force on any one ion will be determined not only by the oppositely charged ions which are directly surrounding it, but also somewhat due to other ions (both positive and negative) at greater distances. To consider the forces due to all the ions, we may take a specific example, say of sodium chloride. The potential energy of an ion will depend on the crystal structure of the solid. In a crystal of sodium chloride, each Na+ ion is surrounded by 6 nearest Cl- ions at a distance ; 12 next nearest Na+ ions at a distance ; 8 (next) nearest Cl ions at a distance ; 6 (next) nearest Na+ ions at a distance 24 (next) nearest Cl- ions at distance ; . And so on The energy expression for the energy released (attraction) in bringing a sodium ion in a crystal lattice of NaCl from infinity is given by the summation of all such terms: Or, Where A= = 1.7476 The last equation is the summation of an infinite series which is called the Madelung constant. The summation value for sodium chloride is given as 1.747558 and is the Madelung constant for sodium chloride. Since the value of this constant depends only on the geometry of the crystal, the value will be the same for all other solids which exhibit the NaCl structure. This quantity can be calculated in the same way for other crystal lattices. Values of Madelung constant for common crystal structural types which we have just discussed are given in the following table. b) Born Haber Cycle This cycle devised by Born and Haber. in 1919 relates the lattice energy of a crystal to other thermochemical data. There are several important concept to understand before the Born-Haber Cycle can be applied to determine the lattice energy of an ionic solid; ionization energy, electron affinity, dissociation energy, sublimation energy, heat of formation, and Hess's Law. Ionization Energy is the energy required to remove an electron from a neutral atom or an ion. This process always requires an input of energy, and thus will always have a positive value. Electron Affinity is the energy released when an electron is added to a neutral atom or an ion. Dissociation energy is the energy required to break apart a compound. The dissociation of a compound is always an endothermic process, meaning it will always require an input of energy. Therefore, the change in energy is always positive. Sublimation energy is the energy required to cause a change of phase from solid to gas, bypassing the liquid phase. This is an input of energy, and thus has a positive value The heat of formation is the change in energy when forming a compound from its elements. This may be positive or negative, depending on the atoms involved and how they interact. Hess's Law states that the overall change in energy of a process can be determined by breaking the process down into steps, then adding the changes in energy of each step. The Born-Haber Cycle is essentially Hess's Law applied to an ionic solid. The enthalpies of sublimation and dissociation and the ionization energy are positive since energy is supplied to the system. The electron affinity and lattice energy ate negative since energy is evolved in these processes. According to Hess’s law, the enthalpy of formation of MX should be the same whether the reaction takes place in one step or several steps. Considering the +ve sign for energy absorbed and –ve sign for energy released, we have Drawback in the concept of Born Haber Cycle The calculated values of lattice energy or electron affinity from the Born Haber Cycle are found to deviate significantly in many cases from the reported values. It is due to consideration of 100 % ionic character in constructing the cycle. Factor favouring the formation of ionic bonds i) Low sublimation energy of the metal ii) Low bond dissociation energy of the nonmetal iii) Highly electropositive character of the metal i.e. low ionization energy iv)High electron affinity of the nonmetal v) High lattice energy Application of Born Haber Cycle i) Calculation of electron affinity ii) Calculation of lattice energy iii) Stability of unknown compounds [Problem based on the above relations] Metallic Bonds Metals have several qualities that are unique, such as the ability to conduct electricity, a low ionization energy, and a low electronegativity (so they will give up electrons easily, i.e., they are cations). Their physical properties include a lustrous (shiny) appearance, and they are malleable and ductile. Metals have a crystal structure. Metals that are malleable can be beaten into thin sheets, for example: aluminum foil. Metals that are ductile can be drawn into wires, for example: copper wire. In the 1900's, Paul Drüde came up with the sea of electrons theory by modeling metals as a mixture of atomic cores (atomic cores = positive nuclei + inner shell of electrons) and valence electrons. In this model, the valence electrons are free, delocalized, mobile, and not associated with any particular atom. For example: metallic cations are surrounded by a "sea" of electrons. This model assumes that the valence electrons do not interact with each other. This model may account for: Malleability and Ductility: The sea of electrons surrounding the protons act like a cushion, and so when the metal is hammered on, for instance, the over all composition of the structure of the metal is not harmed or changed. The protons may be rearranged but the sea of electrons with adjust to the new formation of protons and keep the metal intact. Heat capacity: This is explained by the ability of free electrons to move about the solid. Luster: The free electrons can absorb photons in the "sea," so metals are opaquelooking. Electrons on the surface can bounce back light at the same frequency that the light hits the surface, therefore the metal appears to be shiny. Conductivity: Since the electrons are free, if electrons from an outside source were pushed into a metal wire at one end, the electrons would move through the wire and come out at the other end at the same rate (conductivity is the movement of charge). The electron sea model is, however, a useful qualitative model of metallic bonding even to this day. Metallic bonding in sodium Metals tend to have high melting points and boiling points suggesting strong bonds between the atoms. Even a metal like sodium (melting point 97.8°C) melts at a considerably higher temperature than the element (neon) which precedes it in the Periodic Table. Sodium has the electronic structure 1s22s22p63s1. When sodium atoms come together, the electron in the 3s atomic orbital of one sodium atom shares space with the corresponding electron on a neighboring atom to form a molecular orbital - in much the same sort of way that a covalent bond is formed. The difference, however, is that each sodium atom is being touched by eight other sodium atoms - and the sharing occurs between the central atom and the 3s orbitals on all of the eight other atoms. And each of these eight is in turn being touched by eight sodium atoms, which in turn are touched by eight atoms - and so on and so on, until you have taken in all the atoms in that lump of sodium. All of the 3s orbitals on all of the atoms overlap to give a vast number of molecular orbitals which extend over the whole piece of metal. There have to be huge numbers of molecular orbitals, of course, because any orbital can only hold two electrons. The electrons can move freely within these molecular orbitals, and so each electron becomes detached from its parent atom. The electrons are said to be delocalized. The metal is held together by the strong forces of attraction between the positive nuclei and the delocalized electrons. Figure: Delocaized electrons are free to move in the metallic lattice This is sometimes described as "an array of positive ions in a sea of electrons". Each positive center in the diagram represents all the rest of the atom apart from the outer electron, but that electron hasn't been lost - it may no longer have an attachment to a particular atom, but those electrons are still there in the structure. Sodium metal is therefore written as Na - not Na+. Metallic bonding in magnesium If you work through the same argument with magnesium, you end up with stronger bonds and so a higher melting point. Magnesium has the outer electronic structure 3s2. Both of these electrons become delocalised, so the "sea" has twice the electron density as it does in sodium. The remaining "ions" also have twice the charge (if you are going to use this particular view of the metal bond) and so there will be more attraction between "ions" and "sea". More realistically, each magnesium atom has 12 protons in the nucleus compared with sodium's 11. In both cases, the nucleus is screened from the delocalised electrons by the same number of inner electrons - the 10 electrons in the 1s2 2s2 2p6 orbitals. That means that there will be a net pull from the magnesium nucleus of 2+, but only 1+ from the sodium nucleus. So not only will there be a greater number of delocalized electrons in magnesium, but there will also be a greater attraction for them from the magnesium nuclei. Magnesium atoms also have a slightly smaller radius than sodium atoms, and so the delocalised electrons are closer to the nuclei. Each magnesium atom also has twelve near neighbors rather than sodium's eight. Both of these factors increase the strength of the bond still further. Metallic bonding in transition elements Transition metals tend to have particularly high melting points and boiling points. The reason is that they can involve the 3d electrons in the delocalization as well as the 4s. The more electrons you can involve, the stronger the attractions tend to be. The strength of a metallic bond depends on three things: 1. The number of electrons that become delocalized from the metal 2. The charge of the cation (metal). 3. The size of the cation. A strong metallic bond will be the result of more delocalized electrons, which causes the effective nuclear charge on electrons on the cation to increase, in effect making the size of the cation smaller. Metallic bonds are strong and require a great deal of energy to break, and therefore metals have high melting and boiling points. A metallic bonding theory must explain how so much bonding can occur with such few electrons (since metals are located on the left side of the periodic table and do not have many electrons in their valence shells). The theory must also account for all of a metal's unique chemical and physical properties. Valence bond model for metallic bond In the crystal lattice of lithium atom, each Li atom in this lattice is surrounded by eight nearest Li atoms and eight next to nearest Li atoms. With only one valence electron, each Li atoms is capable of forming only one normal covalent bond with one of its nearest Li atoms. This Li-Li covalent bond is assumed to resonate mainly between all the eight neighbours and to some extent between the eight next to the nearest neighbours. In this way, each Li atom would bind its neighbours. The resonance of the Li-Li bonds which is similar to the resonance of the double bonds in benzene, stabilizes the metallic bond. The resonance of Li-Li between a few Li atoms is shown below. Li can form Li+ (1s2) and Li- (1s2, 2s2). Li- can form two covalent bonds through the participation of 2p orbitals. In a three dimensional crystal, a large no of canonical forms can be considered. Greater stabilisation results from resonance of the following type: Although the valence bond model does meet some success in explaining some of the characteristics of metal, yet it is only qualitative in nature. Band Theory When solids made of an infinite number of atoms are formed, it is a common misconception to consider each atom individually. Rather, we must consider the structure of the solid as a whole. This provides the basis for the description of metals and other types of solids to account for their unique chemical and physical properties. To fully understand the properties,it is essential to start with molecular orbital theory. In the basic theory, it was assumed that if atoms were brought together, they would form bonding, non-bonding and antibonding orbitals of different energies. These molecular orbitals are described by wave functions. The most important point to come out of the theory is that for N atomic orbitals in a molecule, N molecular orbitals must be the outcome. For example, consider a molecule with two atomic orbitals. The result must be that two molecular orbitals will be formed from these atomic orbitals: one bonding and one antibonding, separated by a certain energy. I this is expanded to a molecule with three atoms, assuming 1 atomic orbital for each, then the result must be that 3 molecular orbitals will be formed: one bonding, one non-bonding and one anti-bonding. Now, let's take it to 10 atoms. This will produce 10 molecular orbitals: 5 bonding and 5 antibonding. Now lets take a close look at the separation between each set of orbitals. As the number of molecular orbitals inceases, the energy difference between the lowest bonding and the higherst antibondig increases, while the space between each individual orbital decreases. As the number of molecular orbitals increses with the number of atoms in a molecule, it will the observed that the spaciung between the lowest bonding and highest antibonding orbital will reach a maximum. Now consider a metal with an infinite number of atoms. This will form an infinite number of molecular orbitals so close together they blur into one another forming a band. This whole process is shown below. In the above image, the origin fo the band becomes quite clear because as the number of molecular orbitals incerases, they bonding and antibonding orbitals get closer together filling in the middle. This results in the band seen on the right hand side. It becomes quite clear that the molecular orbitals become blurred and hence mix with each other, which creates the delocalised cloud of electrons that metals are said to posses. By describing the molecular orbitals of certain materials as bands, it becomes much Êeasier to understand the properties of metals and semi-metals. Other materials like insulators and semiconductors will be discussed and concepts such as the valence band, conduction band and the Fermi Level will be defined using the band theory Conductors, Insulators and Semiconductors A. Conductors Metals are conductors. There is no band gap between their valence and conduction bands, since they overlap. There is a continuous availability of electrons in these closely spaced orbitals. B. Insulators In insulators, the band gap between the valence band the the conduction band is so large that electrons cannot make the energy jump from the valence band to the conduction band. C. Semiconductors Semiconductors have a small energy gap between the valence band and the conduction band. Electrons can make the jump up to the conduction band, but not with the same ease as they do in conductors. E.g. Si has a band gap 1.14 ev. Defects in solids Non-stoichiometric defects: The defects which disturb the stoichiometry of the compounds are called non-stoichiometry defects. These defects are either due to the presence of excess metal ions or deficiency of metal ions. (a) Metal excess defects due to anion vacancies: A compound may have excess metal anion if a negative ion is absent from its lattice site, leaving a ‘hole’, which is occupied by electron to maintain electrical neutrality. Anion vacancies in alkali metal halides are reduced by heating the alkali metal halides crystals in an atmosphere of alkali metal vapours. The ‘holes’ occupy by electrons are called F -centres (or colour centres). These type of defects seen because of missing of anions from regular site leaving a hole which is occupied by electron to maintain the neutrality of the comp ound. Hole occupied by electron is called F-centre and responsible for showing colour by the compound. (b) Metal excess defects due to interstitial cations: Another way in which metal excess defects may occur is, if an extra positive ion is present in an interstitial site. Electrical neutrality is maintained by the presence of an electron in the interstitial site. This type of defects are exhibit by the crystals which are likely to exhibit Frenkel defects e.g., when ZnO is heated, it loses oxygen reversibl y. The excess is accommodated in interstitial sites, with electrons trapped in the neighborhood. The yellow colour and the electrical conductivity of the non -stoichiometric ZnO is due to these trapped electrons. Zinc oxide loses oxygen on heating resulting the number of cations (zinc ion) become more than anions present in zinc oxide.Because of this zinc oxide imparts yellow colour when heated. Such defects are called metal excess defects . Consequences of Metal excess defects The crystals with metal excess defects are generally coloured due to the presence of free electrons in them. The crystals with metal excess defects conduct electricity due to the presence of free electrons and are semiconductors. As the electric transport is mainly by “excess” electrons, these are called n-type (n for negative) semiconductor. The crystals with metal excess defects are generally paramagnetic due to the presence of unpaired electrons at lattice sites. When the crystal is irradiated with white light, the trapped electron absorbs some component of white light for excitation from ground state to the excited state. This gives rise to colour. Such points are called F-centres. (German word Farbe which means colour) such excess ions are accompanied by positive ion vacancies. These vacancies serve to trap holes in the same way as the anion vacancies trapped electrons. The colour centres thus produced are called Vcentres. (c) Metal deficiency defect by cation vacancy: In this a cation is missing from its lattice site. To maintain electrical neutrality, one of the nearest metal ion acquires two positive charge. This type of defect occurs in compounds where metal can exhibit variable valency. e.g., Transition metal compounds like NiO, FeO, FeS etc. (d) By having extra anion occupying interstitial site: In this, an extra anion is present in the interstitial position. The extra negative charge is balanced by one extra positive charge on the adjacent metal ion. Since anions are usually larger it could not occupy an interstitial site. Thus, this structure has only a theoretical possibility. No example is kwown so far. Consequences of metal deficiency defects Due to the movement of electron, an ion A + changes to A +2 ions. Thus, the movement of an electron from A + ion is an apparent of positive hole and the substances are called p-type semiconductor Colour Centres Becquerel discovered that a transparent NaCl crystal was coloured yellowish when it was placed near a discharge tube. The colouration of the NaCl and other crystals was responsible for the study of colour centres. Actually, rocksalt should have an infrared absorption due to vibrations of its ions and an ultraviolet absorption due to the excitation of the electrons. A perfect NaCl crystal should not absorb visible light and so it should be perfectly transparent. This leads us to the conclusion that the colouration of crystals is due to defects in the crystals. F Centres: An F-center, Farbe center or color center (from the original German Farbzentrum; Farbe means color, and zentrum center) is a type of crystallographic defect in which an anionic vacancy in a crystal is filled by one or more unpaired electrons. Electrons in such a vacancy tend to absorb light in the visible spectrum such that a material that is usually transparent becomes colored. The greater the number of F-centers, the more intense is the color of the compound. A way of producing F-centers is to heat a crystal in the presence of an atmosphere of the metal that constitutes the material, e.g., NaCl heated in a metallic Na atmosphere. Na0 → Na+ + e− Na+ is incorporated at NaCl crystal. Cl− vacancies are generated, because of the excess of Na+. These vacancies capture available electrons, e−, to maintain charge neutrality, forming Fcenters; that is, the electrons released in this process diffuse to occupy the vacant sites. Ionizing radiation can also produce F-centers. When visible light falls over the crystal of NaCl, the unpaired electron present g ets excited because of absorption of energy and impart yellow colour. Because of similar defect if present, crystal of LiCl imparts pink colour and KCl imparts violet. V Centre: It is possible to produce halide crystals containing excess halogen ions. Thus if an alkali halide crystal is heated in a halogen vapour, a stoichiometric excess of halogen ions is introduced in it, the accompanying cation vacancies trap holes just as the anion vacancies trap electrons in F centres. Thus we should expect a whole new series of colour centres, which are produced by excess alkali metal atoms. The new centres have holes in place of electrons. The colour centres produced in this way are called V centres The formation of V centres can be explained on the same lines as for F centres. It is possible to introduce excess halogen into KBr. The excess bromine enters the normal lattice positions as negative ions. Positive holes are thus formed which are situated near a positive ion vacancy where they can be trapped. A hole trapped at a positive ion vacancy forms a V centre. Semiconductors can be divided into two categories. 1. Intrinsic semiconductors 2. Extrinsic semiconductors This classification is related to the purity of the semiconductors. Intrinsic or pure semiconductors are those that are ideal, with no defects, and no external impurities. The conductivity is temperature dependent. As opposed to intrinsic semiconductors, extrinsic semiconductors have some impurities added to modify the concentration of charge carriers and hence the conductivity. Types of Doped Semiconductors There are two different ways of adding an impurity to the semiconductor atom. The types of doped semiconductors formed after the addition of the impurity are: 1. N-type material 2. P-type material N-type Materials: If a silicon or germanium atom in its pure form is doped with an element of group five in a small amount, such as antimony, arsenic or phosphorus, these elements having 5 electrons in their outermost shell react such that they form a covalent bond with the four electrons of silicon, and one electron is left free as a mobile charge carrier, which improves the conduction ability to some extent. The resultant material is known as an n-type semiconductor. P-type Materials: If a silicon or germanium atom in its pure form is doped with an element of group three in a small amount, such as indium, gallium or boron, these elements having 3 electrons in their outermost shell react such that they form a covalent bond with the three electrons of silicon, and one hole is left free as a mobile charge carrier, which improves the conduction ability to some extent. The resultant material is known as a p-type semiconductor. WERNER’S THEORY OF COORDINATION COMPOUNDS The systematic study of coordination compounds was started by Alfred Werner whose pioneering work opened an entirely new field of investigation in inorganic chemistry. He prepared and characterized a large number of coordination compounds and studied their physical, chemical and isomeric behaviour by simple experimental techniques. On the basis of these studies, Werner, in 1898, propounded his theory of coordination compounds. Table: Series of coloured compounds obtained by the interaction of aqueous CoCl 3 and NH3 Compound Colour Name according to colour CoCl3.6NH3 Yellow Luteo Complex CoCl3.5NH3 Purple Purpureo Complex CoCl3.4NH3 Green Praseo Complex CoCl3.4NH3 Violet Violeo Complex Isomers [Co(NH3)6]Cl3 The main postulates of Werner’s theory are: metals exert two types of linkages; (i) the primary or ionizable links which are satisfied by negative ions and equal the oxidation state of the metal, and (ii) the secondary or nonionizable links which can be satisfied by neutral or negative ions/groups. The secondary linkages equal the coordination number of central metal atom/ion. This number is fixed for a metal. the ions/groups bound by the secondary linkages have characteristic spatial arrangements corresponding to different co-ordination numbers. In the modern terminology, such spatial arrangements are called coordination polyhedra. Ligands The ligands are the ions or molecules bound to the central atom/ion in the coordination entity. This is better visualized as the combination of a Lewis acid (the central atom/ion) with a number of Lewis bases (ligands). The atom in the Lewis base that forms the bond to the Lewis acid (central atom/ion) is called donor atom (because it donates the pair of electrons required for bond formation). The central atom/ ion is the acceptor atom/ion (because it receives the electron pairs from the ligands). Some of the common ligands in coordination compounds are: Br–, Cl–, CN–, OH–, O2–, CO32–, NO2–, C2O4 2– , NH3, CO, H2O, NH2CH2CH2NH2 (1,2- ethanediamine). Ligands which can ligate through two different atoms present in it are called ambidentate ligands. Examples of such ligands are the NO2– and SCN– ions. NO2– ion can coordinate through either the nitrogen or the oxygen atoms to a central metal atom/ion. Similarly, SCN– ion can coordinate through the sulphur or nitrogen atom. Such possibilities give rise to linkage isomerism in coordination compounds. Figure: Metal ion binding options for a carboxylate group, featuring various monodentate and didentate coordination modes. Types of ligands Ligands can be classified as monodentate, bidentate, polydentate etc. ligands. Monodentate ligand This will coordinate to only site of a metal ion. In other words, it can donate only one pair of electrons to the metal ion. Example: Cl-, Br-, SO42-, NH2NH3+, NH3, H2O Bidentate ligand This will occupy two sites of a metal ion. That is, it can attach itself to two positions of a metal ion. Example: NH2CH2CH2NH2 etc Polydentate ligands These ligands occupy many sites of the same metal ion. Example: EDTA etc. Isomerism in Coordination Compounds Two or more different compounds having the same formula are called isomers. Two principal types of isomerism are known among coordination compounds. Each of which can be further subdivided. 1. Stereoisomerism. a) Geometrical isomerism b) Optical isomerism 2. Structural Isomerism. a) Coordination isomerism b) Ionisation isomerism c) Hydrate isomerism d) Linkage isomerism Some terms & their definitions: Enantiomer: Stereoisomers which are not superimposable on their mirror images are called enantiomers. Diastereoisomers: Stereoisomers which do not possess mirror image relation are called diastereoisomers. Asymmetric molecule: A molecule without any symmetry (except c1) is classified as an Asymmetric molecule. Geometric Isomerism: Geometric Isomers differ in the spatial arrangement of atoms within the same structural framework. It is also called cis- trans- isomerism. Geometric Isomerism for coordination no. 4 1. Geometric Isomerism for coordination no. 6: 2. 3. Optical isomerism: A chiral complex is optically active if its structure can not be superimposed on its mirror image. The condition necessary for a molecule to exhibit Optical isomerism is the absence a rotation reflection axis(Sn). 4. Easy judgement for optical activity implies the absence of plane or centre of symmetry. 5. Q. How can you make an optically active square planer complexes? Give example. 6. Answer: Square planer complexes are generally optically inactive since the moleculer plane acts as a plane of symmetry. But optical isomerism may also appear in square planar complexes having an asymmetric ligand. Geometric Isomerism for coordination no. 6 Optical isomerism: A chiral complex is optically active if its structure cannot be superimposed on its mirror image. The condition necessary for a molecule to exhibit Optical isomerism is the absence a rotation reflection axis (Sn). Easy judgement for optical activity implies the absence of plane or centre of symmetry. Q. How can you make an optically active square planer complexes? Give example. Answer: Square planer complexes are generally optically inactive since the moleculer plane acts as a plane of symmetry. But optical isomerism may also appear in square planar complexes having an asymmetric ligand. Structural Isomerism Ionizotion Isomerism The ionization isomers [Co(NH3)5Br]SO4 and [Co(NH)5SO4]Br dissolve in water to yield different ions and thus react differently to various reagents: [Co(NH3)5Br]SO4 + Ba2+ BaSO4 (s) [Co(NH)5SO4]Br + Ba2+ No reaction [Co(NH3)5Br]SO4 + Ag+ No reaction [Co(NH)5SO4]Br + Ag+ AgBr(s) Solvate Isomerism This is a somewhat special case or the above interchange of ligands involving neutral solvate molecules. The best known example involves isomers of "chromic chloride hydrates," of which three are known: [Cr(H2O)6]Cl3, [Cr(H2O)5Cl]Cl2.H2O, and [Cr(H2O)4Cl2]Cl.2H2O, These differ in their reactions: [Cr(H2O)6]Cl3 dehydr. over H SO 2 4 [Cr(H2O)5Cl]Cl2.H2O dehydr. over H SO 2 4 [Cr(H2O)5Cl]Cl2 [Cr(H2O)4Cl2]Cl.2H2O dehydr. over H SO 2 4 [Cr(H2O)5Cl]Cl2 [Cr(H2O)6]Cl3 (no change) Coordinotion Isomerism Coordination compounds made up of cationic and anionic coordination entities show this type of isomerism due to the interchange of ligands between the cation and anion entities. Some of the examples are: (i) [Co(NH3)6][Cr(CN)6] and [Cr(NH3)6][Co(CN)6] (ii) (ii) [Cu(NH3)4][PtCl4] and [Pt(NH3)4] [CuCl4] Such isomers are expected to have significant differences in their physical and chemical properties. A special case of coordination isomerism has sometimes been given the name "polymerization isomerism" since the various isomers differ in formula weight from one another, However, the term is unfortunate since polymerization is normally used to refer to the reaction in which a monomeric unit builds a larger structure consisting of repeating units. The isomers in question are represented by compounds such as [Co(NH)4(NO2)2][Co(NH3)2(NO2)4, [Co(NH3)6][Co(NO2)6], [Co(NH3)5NO2] [Co(NH3)2(NO2)4]2, etc. These all have the empirical formula Co(NH3)3(NO2)3, but they have formula weights that are 2,2, and 3, times this, respectively. Linkage isomerism Linkage isomerism occurs with ambidentate ligands. These ligands are capable of coordinating in more than one way. The best known cases involve the monodentate ligands SCN- / NCSand NO2- / ONO-. For example: [Co(NH3)5ONO]Cl the nitrito isomer -O attached [Co(NH3)5NO2]Cl the nitro isomer - N attached. Other Types of Isomerism Ligand Isomerism Since many ligands are organic compounds which have possibilities for isomerism, the resulting complexes can show isomerism from this source. Examples of isomeric ligands are 1,2-diaminopropane ("propylenediamine," pn) and 1,3-diaminopropane ("trimethylenediamine," tn) or orlho-, meta-, and para-toluidine (CH3C6H4NH2). Bonding Theories Various theoretical approaches to the electronic structure of coordination complexes have been developed. We will discuss some of these bonding models. Valence Bond Theory From the valence bond point of view, formation of a complex involves reaction between Lewis bases (ligands) and a Lewis acid (metal or metal ion) with the formation of coordinate covalent (or dative bonds between them, The model utilizes hybridization of metal s, p. and d valence orbitals to account for the observed structures and magnetic properties of complexes, For example, complexes of Pd(II) and Pt(II) are usually four-coordinate. square planar, and diamagnetic. and this arrangement is often found for Ni(II) complexes as well. In as much as the free ion in the ground state in each case is paramagnetic (d8), the bonding picture has to include pairing of electrons as well as ligand-melal-ligand bond angles of 90°. Pauling suggested this occurs via hybridization of one (n-1)d, the ns and two np orbilals to form four equivalent dsp2 hybrids directed toward the corners of a square. These orbitals then participate in covalent σ bonds with the ligand, the bonding electron pairs being furnished by the ligands. The eight electrons that were distributed among the five d orbitals in the free ion are assigned as pairs to the four unhybridized metal d orbitals. With some ligands, such as CI-, Ni(II) forms four-coordinate complexes that are parnmagnetic and tetrahedral. For these cases, VB theory assumes the d orbital occupation of the complex to be the same as that of the free ion, which eliminates the possibility that valence-level d orbitals can accept electron pairs from the ligands. Hybrid orbitals of either the sp3 or sd3 type (the latter involving n-level d orbitals) or a combination of the two provide the proper symmetry for the σ bonds as well as allowing for the magnetic properties. The valence bond picture for six-coordinate octahedral complexes involves d2sp3 hybridization of the metal. The specific d orbitals that meet the symmetry requirements for the metal-ligand σ bonds are the dz2 and dx2-y2. As with the four-coordinate d8 complexes discussed above: the presence of unpaired electrons in some octahedral compounds renders the valence level (n-1)d orbitals unavailable for bonding. This is true, for instance, for paramagnetic [CoF6]3-. In these cases, the VB model invokes participation of n-level d orbitals in the hybridization. Examples In the diamagnetic octahedral complex, [Co(NH3)6]3+, the cobalt ion is in +3 oxidation state and has the electronic configuration 3d6. The hybridisation scheme is as shown in diagram. Six pairs of electrons, one from each NH3 molecule, occupy the six hybrid orbitals. Thus, the complex has octahedral geometry and is diamagnetic because of the absence of unpaired electron. In the formation of this complex, since the inner d orbital (3d) is used in hybridisation, the complex, [Co(NH3)6]3+ is called an inner orbital or low spin or spin paired complex. The paramagnetic octahedral complex, [CoF6]3– uses outer orbital (4d) in hybridisation (sp3d2). It is thus called outer orbital or high spin or spin free complex. Thus: In tetrahedral complexes one s and three p orbitals are hybridised to form four equivalent orbitals oriented tetrahedrally. This is illustrated below for [NiCl4]2-. Here nickel is in +2 oxidation state and the ion has the electronic configuration 3d8. The hybridisation scheme is as shown in diagram. Each Cl– ion donates a pair of electrons. The compound is paramagnetic since it contains two unpaired electrons. Similarly, [Ni(CO)4] has tetrahedral geometry but is diamagnetic since nickel is in zero oxidation state and contains no unpaired electron. In the square planar complexes, the hybridisation involved is dsp2. An example is [Ni(CN)4]2. Here nickel is in +2 oxidation state and has the electronic configuration 3d8. The hybridisation scheme is as shown in diagram: Each of the hybridised orbitals receives a pair of electrons from a cyanide ion. The compound is diamagnetic as evident from the absence of unpaired electron. It is important to note that the hybrid orbitals do not actually exist. In fact, hybridisation is a mathematical manipulation of wave equation for the atomic orbitals involved. Limitation of the VBT: 1. Fail to explain the colour & characteristics of absorption spectra of complex compounds. 2. Orbital contribution and temperature dependency on magnetic moment of coordination complex are not properly explained by VBT. 3. It is not helpful to predict the mystery of formation of outer or inner orbital coordination complex. 4. VBT fails to predict any distortion in the shapes of the coordination complexes from regular geometry. Crystal Field Theory Crystal field theory was originally developed to describe the electronic structure of metal ions in crystals, where they are surrounded by anions that create an electrostatic field with symmetry dependent on the crystal structure. The energies of the d orbitals of the metal ions are split by the electrostatic field, and approximate values for these energies can be calculated. No attempt was made to deal with covalent bonding, because covalency was assumed nonexistent in these crystals. Crystal field theory was developed in the 1930s. Shortly afterward, it was recognized that the same arrangement of electron-pair donor species around a metal ion existed in coordination complexes as well as in crystals, and a more complete molecular orbital theory was developed. However, neither was widely used until the 1950s, when interest in coordination chemistry increased. When the d orbitals of a metal ion are placed in an octahedral field of ligand electron pairs, any electrons in these orbitals are repelled by the field. As a result, the dz2 and dx2-y2 orbitals, which have eg symmetry, are directed at the surrounding ligands and are raised in energy. The dxy, dxz and dyz orbitals (t2g symmetry), directed between the ligands, are relatively unaffected by the field. The resulting energy difference is identified as Δo (o for octahedral; older references use l0 Dq instead of Δo). This approach provides an elementary means of identifying the d-orbital splitting found in coordination complexes. The average energy of the five d orbitals is above that of the free ion orbitals, because the electrostatic field of the ligands raises their energy. The t2g orbitals are 0.4 Δo below and the eg orbitals are 0.6 Δo above this average energy, as shown in Figure 2. The three t2g orbitals then have a total energy of — 0.4 Δo X 3 = — 1.2 Δo and the two eg orbitals have a total energy of +0.6 Δo X 2 = +1.2 Δo compared with the average. The energy difference between the actual distribution of electrons and that for the hypothetical configuration with all electrons in the uniform (or spherical) field level is called the crystal field stabilization energy (CFSE). The CFSE quantifies the energy difference between the electronic configurations due to (1) the d orbitals experiencing an octahedral ligand field that discriminates among the d orbitals, and (2) the d orbitals experiencing a spherical field that would increase their energies uniformly. This model does not rationalize the electronic stabilization that is the driving force for metal ligand bond formation. As we have seen in all our discussions of molecular orbitals, any interaction between orbitals leads to formation of both higher and lower energy molecular orbitals, and bonds form if the electrons are stabilized in the resulting occupied molecular orbitals relative to their original atomic orbitals. On the basis of Figure 2, the electronic energy of the free ion configuration can at best be unchanged in energy upon the free ion interacting with an octahedral ligand field; the stabilization resulting from the metal ion interacting with the ligands is absent. Because this approach does not include the lower (bonding) molecular orbitals, it fails to provide a complete picture of the electronic structure. Figure 2: Crystal Field Splitting Octahedral complexes: In an octahedral field those d orbital whose lobes are directed along the axis will experience greater interaction from the point charges placed in those directions. These are the d x2 −y2 and dz2 orbitals- the electron of these orbital will experience stronger repulsion from the point charges in the environment i.e. these orbitals will be destabilized & their energy will be raised to some extent. But the remaining three orbital dxy, dxz and dyz have their lobes between the axes (45°) and will be less effected by the point charges in the environment i.e. these orbitals will be subject less electronic repulsion and to maintain the barycentre or center of gravity, energy of these orbitals will be lowered some extent. The splitting pattern of d orbitals in octahedral field: The separation between the t2g and eg levels is conventionally represented by Δ° or 10 Dq. Tetrahedral Complexes In tetrahedral coordination entity formation, the d orbital splitting (Fig. 3) is inverted and smaller as compared to the octahedral field splitting. For the same metal, the same ligands and metal-ligand distances, it can be shown that Δt = –(4/9)Δo. Consequently, the orbital splitting energies are not sufficiently large for forcing pairing and, therefore, low spin configurations are rarely observed. Figure 3. d orbital splitting in a tetrahedral crystal field. Square Planar Complexes If two trans ligands in an octahedral ML6 complex (for example those along the z axis) are moved either towards or away from the metal ion, the resulting complex is said to be tetragonally distorted. Ordinarily such distortions are not favored since they result in a net loss of bonding energy. In certain situations, however, such a distortion is favored because of a Jahn- Teller effect (explained later). A complex of general formula trans-MA2B4 also will have tetragonal (D 4h ) symmetry. For now, we will merely consider the limiting case of tetragonal elongation, a square planar ML4 complex, for the purpose of deriving its d orbital splitting pattern. Figure 4 illustrates the effect of z-axis stretching on the eg and t2g orbitals in an octahedral complex, Orbitals having a Z component (the dxz, dyz and dz2 ) will experience a decrease in electrostatic repulsions from the ligands and will therefore be stabilized, At the same time, the "non-z" orbitals will be raised in energy, with the barycenter remaining constant. The overall result is that the eg level is split into two levels. an upper b1g (dx2–y2) and a lower a1g (dz2) and the t2g set is split into a b2g (dxy) and a doubly degenerate eg (dxz, dyz). The energy spacing between the b2g and b1g levels is defined as Δ. As in the octahedral case, this splitting is equal to 10Dq. The square planar geometry is favored by metal ions having a d8 configuration in the presence of a strong field, This combination gives low spin complexes with the eight d electrons occupying the low-energy dxz, dyz, dz2 and dxy orbitals, while the high-energy dx2–y2 orbital remains unoccupied. The stronger the surrounding field, the higher the dx2–y2 orbital will be raised. As long as this level is unoccupied, however, the overall effect on the complex will be stabilization because the lower, occupied orbitals will drop in energy by a corresponding amount. Typical low spin square planar complexes are [Ni(CN4)]2–, [PdCI 4]2–, [Pt(NH3)4]2+, [PtCl4]2–, and [AuCl4]–, all d8 species. Figure 4. An octahedral complex (a) undergoing z axis elongation such that it becomes tetragonally distorted (b) and finally reaches the square planar limit (c). Crystal field stabilization energy: The crystal field stabilization energy (CFSE) is the stability that results from placing a transition metal ion in the crystal field generated by a set of ligands. It arises due to the fact that when the d -orbitals are split in a ligand field (as described above), some of them become lower in energy than before with respect to a spherical field known as the bary-center in which all five d -orbitals are degenerate. For example, in an octahedral case, the t2g set becomes lower in energy than the orbitals in the bary-center. As a result of this, if there are any electrons occupying these orbitals, the metal ion is more stable in the ligand field relative to the barycenter by an amount known as the CFSE. Conversely, the eg orbitals (in the octahedral case) are higher in energy than in the bary-center, so putting electrons in these reduces the amount of CFSE. The only one d electron of Ti3+ will occupy the lowest available orbital & it is stabilized by the energy 4 Dq. Q. what will be the electron arrangement for d4 and more than d4 conformation? Answer: This splitting is affected by the following factors: the nature of the metal ion. the metal's oxidation state. A higher oxidation state leads to a larger splitting. the arrangement of the ligands around the metal ion. the nature of the ligands surrounding the metal ion. The stronger the effect of the ligands then the greater the difference between the high and low energy d groups. There are two possibilities for these type of cases: e.g. for d4 case Crystal field theory and molecular orbital theory were combined into ligand field theory by Griffith and Orgel. Effect of ligands on d orbital splitting (spectrochemical series): The ligands are sequencing according to their splitting ability, strong field ligand split the d orbitals more ( i . e . the separation between t 2g and e g is large) compare to weak field ligand and form low-spin complex where as weak field ligand form high spin complex. Spinels The d-block higher oxides Fe3O4, Co3O4, and Mn3O4, and many related mixed-metal compounds, such as ZnFe2O4, have very useful magnetic properties. They all adopt the structural type of the mineral spinel, MgAl2O4, and have the general formula AB2O4. Most oxide spinels are formed with a combination of A2+ and B3+ cations (that is, as A2+ B23+O4 in Mg2+[Al3+]2O4), although there are a number of spinels that can be formulated with A4+ and B2+ cations (as A4+B22+O4 as in Ge4+[Co2+]2O4. The spinel structure consists of an fcc array of O2– ions in which the A ions reside in one-eighth of the tetrahedral holes and the B ions inhabit half the octahedral holes (Fig. 5); this structure is commonly denoted A[B2]O4, where the atom type in the square bracket represents that occupying the octahedral sites. In the inverse spinel structure, the cation distribution is B[AB]O4, with the more abundant B-type cation distributed over both coordination geometries. Lattice enthalpy calculations based on a simple ionic model indicate that, for A2+ and B3+, the normal spinel structure, A[B2]O4, should be the more stable. The observation that many d-metal spinels do not conform to this expectation has been traced to the effect of crystal-field stabilization energies on the site preferences of the ions. Figure 5. A segment of the spinel (AB2O4) unit cell showing the tetrahedral environment of A ions and the octahedral environments of B ions. The occupation factor, λ, of a spinel is the fraction of B atoms in the tetrahedral sites: λ = 0 for a normal spinel and λ = ½ for an inverse spinel, B[AB]O4; intermediate λ values indicate a level of disorder in the distribution, where B-type cations occupy that portion of the tetrahedral sites. The distribution of cations in (A2+, B3+) spinels (Table 3) illustrates that for d0 A and B ions the normal structure is preferred as predicted by electrostatic considerations. Table 3 shows that, when A2+ is a d6, d7, d8, or d9 ion and B3+ is Fe3+, the inverse structure is generally favoured. This preference can be traced to the lack of crystal-field stabilization of the high-spin d5 Fe3+ ion in either the octahedral or the tetrahedral site and the ligand-field stabilization of the other dn ions in the octahedral site. For other combinations of d-metal ions on the A and B sites the relative ligand-field stabilization energies of the different arrangements of the two ions on the octahedral and tetrahedral sites need to be calculated. It is also important to note that simple ligand-field stabilization appears to work over this limited range of cations. More detailed analysis is necessary when cations of different radii are present or any ions that are present do not adopt the high-spin configuration typical of most metals in spinels (for instance, Co3+ in Co3O4, which is low-spin d6). Moreover, because λ is often found to depend on the temperature, care has to be taken in the synthesis of a spinel with a specific distribution of cations because slow cooling or quenching of a sample from a high reaction temperature can produce quite different cation distributions. Table 3. Occupation factor, λ, in some spinels. The inverse spinels of formula AFe2O4 are sometimes classified as ferrites (the same term also applies in different circumstances to other iron oxides). The compound CoAl2O4 is among the normal spinels in Table 3 with λ = 0 and thus has the Co2+ ions at the tetrahedral sites. The colour of CoAl2O4 (an intense blue) is that expected of tetrahedral Co2+. This property, coupled with the ease of synthesis and stability of the spinel structure, has led to cobalt aluminate being used as a pigment (‘cobalt blue’). Other mixed d-metal spinels that exhibit strong colours, for example CoCr2O4 (green), CuCr2O4 (black), and (Zn,Fe)Fe2O4 (orange/brown), are also used as pigments, with applications that include colouring various construction materials, such as concrete. EXAMPLE Predicting the structures of spinel compounds Q Is MnCr2O4 likely to have a normal or inverse spinel structure? Answer We need to consider whether there is a ligand-field stabilization. Because Cr3+ (d3) has a large ligand-field stabilization energy (1.2ΔO) in the octahedral site (but a much smaller one in a tetrahedral field) whereas the high spin d5 Mn2+ ion does not have any LFSE, a normal spinel structure is expected. Table 3 shows that this prediction is verified experimentally. The Jahn-Teller Effect The Jahn-Teller theorem states that degenerate orbitals (those with identical energies) cannot be unequally occupied. To avoid these unfavorable electronic configurations, molecules distort (lowering their symmetry) to render these orbitals no longer degenerate. For example, an octahedral Cu(II) complex, containing a d9 ion, would have three electrons in the two eg levels, as in the center of Figure 6, but an octahedral structure is not observed. Instead, the shape of the complex changes slightly, resulting in changes in the energies of the orbitals that would be degenerate within an octahedral ligand environment. The resulting distortion is usually elongation along one axis, but compression along one axis is also possible. In ideally octahedral complexes that experience Jahn-Teller distortion, the (formally) e* orbitals change more in energy relative to the (formally) t2g orbitals. More significant Jahn-Teller distortions occur when e* orbitals would be unequally occupied within an octahedral geometry. Much more modest distortions, sometimes difficult to observe experimentally, occur to prevent unequal occupation of t2g orbitals within an octahedral geometry. The general effects of elongation and compression on d-orbital energies are shown in Figure 7, and the expected degrees of JahnTeller distortion for different electronic configurations and spin states are summarized in the following table: Figure 7. Jahn-Teller Effect on a d9 Complex. Elongation along the z axis is coupled with a slight decrease in bond length for the other four bonding directions. Similar changes in energy result when the axial ligands have shorter bond distances. The resulting splits are larger for the eg orbitals than for the t2g orbitals.The energy differences are exaggerated in this figure. Significant Jahn-Teller effects are observed in complexes of high-spin Cr(II) (d4), high-spin Mn(III) (d4), Cu(II) (d9), Ni(III) (d7), and low-spin Co(II) (d7). Low-spin Cr(II) complexes feature tetragonal distortion. Cu(II) complexes generally exhibit significant Jahn-Teller effects; the distortion is most often elongation of two bonds. Elongation, which results in weakening of some metal- ligand bonds, also affects equilibrium constants for complex formation. For example, trans-Cu(NH3)4(H2O)2]2+ is readily formed in aqueous solution as a distorted octahedron with two water molecules at greater distances than the ammonia ligands; liquid ammonia is the required solvent for [Cu(NH3)6]2+ formation. The formation constants for these reactions show the difficulty of putting the fifth and sixth ammonias on the metal: Which factor is the cause and which the result is uncertain, but the bottom line is that octahedral Cu(II) complexes are difficult to synthesize with some ligand sets because the bonds to two trans ligands in the resulting complexes are weaker (longer) than the other bonds to the ligands. In fact, many Cu(II) complexes have square-planar or nearly square-planar geometries, with tetrahedral shapes also possible. [CuCl4]2– exhibits cation-dependent structures ranging from tetrahedral through square planar to distorted octahedral. Magnetic properties: A brief knowledge is required for understanding the electronic spectroscopy of transition metal complexes. Additional and complementary information can be provided by magnetic measurement because for the partially filled d or orbitals of transition metal, a range of magnetic properties can be expected, depending on the oxidation state, electronic configuration and coordination number of the central metal. Classification of substance according to magnetic properties: Diamagnetism: When any substance is placed in an external magnetic field, there is an induced circulation of electrons producing a net magnetic moment aligned in opposition to the applied field. This is the diamagnetic effect and it arises from paired electrons within a sample, since all compounds contain some paired electrons, diamagnetism is a universal property of matter. If a substance has only paired electrons, this effect will dominate, the material will be classified as diamagnetic, and it will be slightly repelled by a magnetic field. Paramagnetism: It is produced by unpaired electrons in a sample. The spins and orbital motions of these electrons give rise to permanent molecular magnetic moments that tend to align themselves with an applied field, Because it is much larger than the diamagnetic effect, the paramagnetic effect cancels any repulsions between an applied field and paired electrons in a sample, Thus even substances having only one unpaired electron per molecule will show a net attraction into a magnetic field. The paramagnetic effect is observed only in the presence of an external field. There are three possible modes of coupling between those components: spin-spin, orbitalorbital and spin-orbital. For some complexes, particularly those of the lanthanides, we must consider all three types of coupling. The theoretical paramagnetic moment for such a complex is given by Where J is the total angular momentum quantum number and g is the L and splitting factor for the electron, defined as The value of J depends on the total orbital angular momentum quantum number L and the total spin angular momentum quantum number S. Magnetic properties of some compound of lanthanides metals at room temperature (300 K). µ(calculated) B.M. is calculated by µ= g[J(J + 1)]½ formula For complexes in which spin-orbit coupling is non-existent or negligible but spin and orbital contributions are both significant, the predicted expression for µ is When the orbital contribution is minimal and could be ignored. Hence, L = 0 and in this condition, the previous equation reduces to µ= [4S(S + 1)]½ = 2[S(S + 1)]½ BM This is known as the spin-only formula for magnetic moment. By recognizing that S will be related to the number of unpaired electrons (II) by S = n/2, the expression may be further simplified to µ = [n(n + 2)]½ BM Special case of Copper acetate monohydrate, Cu(CH3COO)2.2H2O Copper (II) acetate monohydrate exists as a dimer and its structure is shown in fig. 6. The magnetic moment of the compound is only 1.43 BM which is less than the normal value of 1.75 – 2.2 BM for one unpaired electron. This suggests some weak coupling of spins of unpaired electrons on the two copper atoms in the dimeric structure. In its structure, the acetate groups act as bidentate bridging groups between the two copper atoms. Therefore, each copper atom is surrounded by four oxygen atoms in a square planar arrangement. The fifth position around each Cu atom is occupied by the oxygen atom from a water molecule. The other copper atom occupies the sixth of the octahedral positions. Figure 6. Structure of dimeric Cu (II) acetate monohydrate. Electronic Spectra of Transition Metal Complexes For example: If red is absorbed, the complex appears green. The variety of colors among transition metal complexes has long fascinated the observer. For example, aqueous solutions of octahedral [Co(H2O)6]2+ are pink but those of tetrahedral [CoCl4]2– are blue. The green color of aqueous [Ni(H2O)6]2+ turns blue when ammonia is added to the solution to give [Ni(NH3)6])2+. The reduction of violet [Cr(H2O)6]3+ gives bright blue [Cr(H2O)6]2+. As with all colors, these arise from electronic transitions between levels whose spacings correspond to the wavelengths available in visible light. (Of course, when a photon of visible light is absorbed. it is its complementary color that we actually see.) In complexes. these transitions are frequently referred to as d-d transitions because they involve the molecular orbitals that are mainly metal d in character (the eg and t2g or e and t2 orbitals in octahedral and tetrahedral complexes, respectively), Obviously, the colors produced are intimately related to the magnitude of the spacing between these levels, Since this spacing depends on factors such as the geometry of the complex, the nature of the ligands present, and the oxidation state of the central metal atom, electronic spectra of complexes can provide valuable information related to bonding and structure. The variation in the color of the Cr(III) complexes can be explained following similar argument. We observe the appearance of a shoulder in the case of [Ti(H2O)6]3+. Perfectly octahedral [Ti(H2O)6]3+ should give only one d-d Transition. However, distortion occurs to eliminate the degeneracy of the system. If a complex distorts from regular octahedral geometry, the t 2g and eg levels are split, the consequence of which is the appearance of a shoulder as shown in the figure right. dz 2 dx 2-y 2 dxz dyz dxy Complexes that contain metal ions of d10 electron configuration are usually colorless. Examples are [Cu(PPh3)4]+ and [Zn(H2O)6]2+. One would expect a metal complex with no deletron to be colorless as well. However, a few of such complexes are strongly colored, for example, MnO4- or [Cr2O7]2-. The origin of the color in these complexes is not the d-d transitions, rather due to ‘charge transfer’ that we will briefly discuss later. Multi-electron systems exhibit multiple transitions and the assignment of absorption bands is not straightforward. The complexity arises due to interelectron repulsions that we will not discuss in this course but will take a simpler approach to understand the color of coordination complexes. Selection rules for electronic transitions The Beer-Lambert Law A = log10(Io/I) = εcl where ε is the molar extinction coefficient ( in L cm-1 mole-1 ), c is concentration in mole L-1 and l is the path length in cm. A is known as ‘Absorbance’ and it is dimensionless. To explain the absorption spectra of coordination complexes, it is necessary to know the selection rules that govern electronic transitions. Any transition in violation of selection rule is said to be ‘forbidden’, but we will see how some rules are ‘more forbidden than others’. We shall not pursue the theoretical basis of the rules but merely outline simple tests for their application. The Laporte Rule. In a molecule or ion possessing a centre of symmetry, transitions are not allowed between orbitals of the same parity, for example d to d. In other words, there must be change in parity (∆l=±1), i.e. the orbital quantum number should differ by 1. The forbidden transitions are s → s, d → d, p → f. etc. The geometries affected by this rule include octahedral and square-planar complexes. The rule is not applicable to tetrahedral complexes as it does not contain a center of symmetry. The key element here is that there are mechanisms by which selection rules can be relaxed so that transitions can occur, even if only at low intensities. Unsymmetrical vibrations of an octahedral complex can transiently destroy its center of symmetry and allow transitions that would otherwise be Laporte forbidden. In cases where the rule applies, the colors of the complexes are usually relatively pale. As examples, consider [Cu(H2O)6] 2+ which is a rather pale blue color vs [Cu(NH3)4]2+ which is an intense dark blue. Spin Allowed - Spin Forbidden Any transition for which ∆S≠0 is strongly forbidden; that is, in order to be allowed, a transition must involve no change in spin state. Consider the case of the high spin d5 complex [Mn(H2O)6]2+. Electronic transition is not only Laporte forbidden but also spin forbidden. Absorptions that are doubly forbidden transitions are extremely weak. It is understandable, then, that dilute solutions of Mn(II) are colorless. Charge-Transfer (CT) Bands. Similar to d-d transitions, charge-transfer (CT) transitions also involve the metal d-orbitals. CT bands are observed if the energies of empty and filled ligand- and metal-centered orbitals are similar. The direction of the electron transfer is determined by the relative energy levels of these orbitals: i) Ligandto-Metal charge transfer (LMCT) like in MnO4-, CrO42- etc. For MnO4-, the d-electron count on Mn(VII) is d0. The origin of the color in this species is not due to d-d transition, rather, charge transfer from O2- to Mn(VII), described as LMCT band. ii) Metal-to Ligand charge transfer (MLCT) like in [Fe(bpy)3]2+. In this complex the charge transfer occurs from Fe(II) to the empty π* orbitals of bpy ligand.