Journal of Electroanalytical Chemistry 655 (2011) 23–31
Contents lists available at ScienceDirect
Journal of Electroanalytical Chemistry
journal homepage: www.elsevier.com/locate/jelechem
How well does simple RC circuit analysis describe diffuse double layer
capacitance at smooth micro- and nanoelectrodes?
Edmund J.F. Dickinson, Richard G. Compton ⇑
Department of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QZ, United Kingdom
a r t i c l e
i n f o
Article history:
Received 9 November 2010
Received in revised form 7 February 2011
Accepted 16 February 2011
Available online 20 February 2011
Keywords:
Diffuse double layer
Nernst–Planck–Poisson equations
Capacitive charging
RC circuit
Electrochemical impedance spectroscopy
Nanoelectrode
a b s t r a c t
The capacitive charging of a diffuse double layer is discussed. Results from simple RC circuit analysis (an
ideal resistor and capacitor in series) are compared with results from a more complete model in which
the Nernst–Planck–Poisson equations are solved in a hemispherical space, both analytically and by simulation. This complementary approach allows an assessment of certain conditions which are required in
order for RC circuit analysis to be suitable to describe the diffuse double layer. In particular, deviations are
noted for nanoscale electrodes. Additionally, RC circuit behaviour breaks down for applied overpotentials
greater than 25 mV (RT/F), such that values for solution resistance and double layer capacitance inferred
from impedance spectroscopy may not apply to other experimental techniques. These conclusions apply
to a smooth electrode and so are not associated with ‘‘constant phase angle’’ effects.
Ó 2011 Elsevier B.V. All rights reserved.
1. Introduction
circuit is at all physically meaningful for describing the diffuse
double layer and its interface with a charged electrode.
1.1. Preamble
It has been well understood since the early days of solutionphase electrochemistry that the excess charge on the surface of
an electrode, an unmeasurable property, induces a ‘double layer’
with opposite charge. This exists in the form of a charge separation
due to unequal concentrations of cations and anions in the electrolyte solution close to the electrode interface. The model due to
Stern [1] and Grahame [2] considered this double layer to consist
of a compact layer containing adsorbed solvent, and possibly adsorbed ionic species, and a diffuse layer in which charge separation
decays smoothly into bulk solution.
Charging processes, particularly when occurring in tandem with
electrolysis, are commonly analysed using the RC equivalent circuit
(Fig. 1), in which an ideal resistor and capacitor are considered in
series with a voltage source [3]. The solution is therefore considered to have an ideal, constant and homogeneous resistance, Rs,
and the double layer is considered to have an ideal, constant and
homogeneous capacitance, Cd.
In the following discussion we shall use the Nernst–Planck–
Poisson equations (Eqs. (1.1) and (1.2)) in conjunction with the
law of conservation of mass (Eq. (1.3)) to describe both equilibrium
and charging problems, to determine under what conditions the RC
⇑ Corresponding author. Tel.: +44 (0) 1865 275413; fax: +44 (0) 1865 275410.
E-mail address: richard.compton@chem.ox.ac.uk (R.G. Compton).
1572-6657/$ - see front matter Ó 2011 Elsevier B.V. All rights reserved.
doi:10.1016/j.jelechem.2011.02.016
zi F
C i r/
Ji ¼ Di rC i þ
RT
X
F
r2 / þ
zi C i ¼ 0
s 0
@C i
¼ r Ji
@t
ð1:1Þ
ð1:2Þ
i
ð1:3Þ
For species i, Ji is the flux vector, Ci is concentration, Di is diffusion
coefficient, and zi is charge number in unit electron charges. / is potential, F is the Faraday constant, R is the gas constant, T is temperature and s0 is the permittivity of the solvent medium. t is time.
The compact layer will not be discussed, but its incorporation into
the arguments ought not be problematic.
Here our arguments will concern ideally smooth electrodes, and
so we are not concerned with deviations from ideal RC behaviour
associated with ‘‘constant phase angles’’ due to electrode surface
roughness [4–6]. Rather, we are concerned with defining regimes
in which the charging behaviour of an ionic solution differs from
the ideal circuit sometimes used to describe it. In particular we
shall discuss the effects of size, such that double layers at nanoelectrodes behave differently from those at microelectrodes, due
in part to the pronounced curvature of the double layer [7,8]. Additionally the dependence of the meaning of certain parameters
upon the experiment used to determine them will be underlined.
24
E.J.F. Dickinson, R.G. Compton / Journal of Electroanalytical Chemistry 655 (2011) 23–31
Ecap(t)
V
V¼
I¼
C
Z¼
i (t )
ð1:11Þ
E 1 þ jRs C d x
1 1
1
¼ Rs þ
¼ Rs j
jx
C d jx
Cd x
EC d
ð1:12Þ
0
E (t )
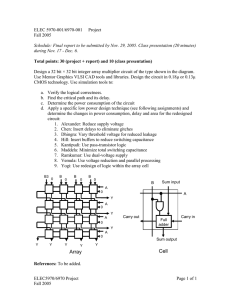
Fig. 1. Schematic of the RC equivalent circuit.
1.2. Results from the RC equivalent circuit
In the RC circuit (Fig. 1), we may use Kirchhoff’s voltage law to
take the sum of voltages as zero. If the applied voltage is E, the
charge on the capacitor is q, and the current is i:
q
¼0
Cd
EC d
1 þ jRs C d x
so
A
E iRs ð1:10Þ
and
R
V
E
jx
ð1:4Þ
00
Therefore Z = Rs and Z = 1/Cdx, such that the resistive and capacitive components of charging are separated into the real and imaginary components of the impedance, respectively.
The problems above, and others, have been reviewed in the
textbook of Bard and Faulkner [11]. In all of the above q = 0 has
been taken to be zero when t = 0, i.e. the step or sweep is beginning
at the potential of zero charge (PZC) of the system. This can rarely
be guaranteed in practice.
In this paper we shall consider the merits of this idealised RC
circuit description as compared to a more complete analysis on
the basis of transport equations, still assuming a smooth electrode.
In particular the physical meaningfulness of the parameters Rs and
Cd will be assessed.
Noting that i = dq/dt, and rearranging:
dq
q
E
þ
¼
dt Rs C d Rs
ð1:5Þ
such that if a potential step is applied from initial conditions of
q = 0:
t
q ¼ EC d 1 exp
Rs C d
ð1:6Þ
and
i¼
dq
E
t
¼
exp
dt Rs
Rs C d
ð1:7Þ
It is evident from the above results that q = ECd at t ? 1, thus confirming that the capacitor is ideal, and that the charging current decays according to a characteristic time constant RsCd, such that more
resistive or more capacitive interfaces will charge more slowly.
The RC circuit falls under a class of problems in circuit analysis
which are described as linear time-invariant [9]. For such problems,
the output (current) is a linear function of the input (voltage) and
therefore the current for any voltage waveform can be determined
from the current for a single example. Therefore the solution for
the potential step experiment can be converted into a solution
for the problem with swept potential, E = vt, by the mathematical
process of convolution. The result is that:
t
i ¼ EC d v 1 exp
Rs C d
ð1:8Þ
Therefore a constant charging current is attained which is only a
function of the capacitance; after a certain delay, the system attains
the condition of ideal capacitance.
In impedance spectroscopy [10], the impedance spectrum
0
00
Z(jx) = Z (x)jZ (x) may be inferred from Ohm’s law for the linear
system by Laplace transformation under a Laplace frequency jx
[9]:
ZðjxÞ ¼
VðjxÞ
IðjxÞ
ð1:9Þ
where V and I are the Laplace (frequency) space transforms of the
potential step transient for charging, j is the imaginary unit and x
is the spectroscopic frequency. It follows that:
2. Theory
2.1. Development of a self-consistent model based on transport
equations
The major alternative interpretation of the double layer is in
terms of the Nernst–Planck (electrodiffusion) equation for the
mass transport of a species, together with the Poisson equation
relating the charge density and the electric field. At steady state,
the Nernst–Planck equation reduces to the thermodynamic Boltzmann equation, and solution of the combined Poisson–Boltzmann
equation was first achieved for a planar geometry by Gouy [12,13]
and Chapman [14] independently, yielding the Gouy–Chapman
equation for the capacitance of the double layer. A great deal of
work has since been done in terms of considering diffuse double
layers, both at steady state and dynamically, and how their formation varies with applied voltage and solution composition. This
work has been thoughtfully reviewed by Bazant et al. [15] and further study by Beunis et al. [16] introduced a variety of perceived
limiting cases. Bazant et al. [15] additionally extended the range
of analytical solution, providing an exact Laplace transform for
the transient capacitive current for low applied voltages where
the Nernst–Planck equation can be linearised, as well as applying
asymptotic analysis to non-linear cases.
Common to almost all of these studies, however, is the adoption
of a linear geometry for the study. The argument is that since the
size of the electrode greatly exceeds the typical extent of the double layer, the effect of double layer curvature only arises for very
small electrodes, as reported by Dickinson et al. [7], and so the
double layer is indeed physically planar, to all intents and purposes
for electrodes larger than nanoscale. This argument applies exclusively at equilibrium, however. Immediately following a potential
step, the electric field will extend in all directions – and so has
spherical symmetry, assuming that the cell is large with respect
to the electrode. Unless the solution is confined to a capillary leading directly to the reference electrode, there is very little linearity
in the system, and an attempt to consider a simple model problem
underlines this.
Let us suppose that the cell is being potentiostatically held at its
potential of zero charge Epzc, such that solution is everywhere electroneutral. Then suppose the cell potential is rapidly stepped to a
E.J.F. Dickinson, R.G. Compton / Journal of Electroanalytical Chemistry 655 (2011) 23–31
new potential E. If we assume that reorientation of the polar solvent is much faster than the motion of solvated ions, an electric
field is established across solution instantaneously, with the solution remaining roughly electroneutral. If the cell is very large compared to the electrode, a typical situation, we must solve:
r2 / ¼ 0
ð2:1Þ
such that / = E Epzc at the electrode, and / = 0 at an infinite distance from the electrode. If the geometry is linear (with coordinate
x), we immediately encounter a problem since there exists no solution consistent with these boundary conditions. If we introduce an
arbitrary double layer length x = L where / = 0, we may write
/ ¼ ðE Epzc Þ 1 xL , but L must then be a strong function of the displacement of the reference and working electrodes. We then expect
the initial capacitive current to depend strongly on this displacement, even when it is very large, which is a rather nonsensical situation and entirely ignores bulk solution which is equally distant
from both electrodes, in which range electroneutrality is also
retained.
In a hemispherical space (with coordinate r), however, the Cou lombic solution / ¼ ðE Epzc Þ rre arises straightforwardly, giving a
simple dependence on electrode radius, re, and no dependence at
all on the exact position of the reference electrode, provided it is
sufficiently distant and bulk solution is plentifully available. This
conforms to expected behaviour and has the added advantage that
all length scales are quantified easily and in a manner that is not
arbitrary. We shall therefore consider all transport in a hemispherical space. In the limit re rD, where rD is the Debye length, the linear result should be recovered at steady state.
2.2. Choice of transformed coordinates for analysis
To generalise the process of solution as much as possible,
dimensionless variables and coordinates are employed. We note
that the work of Bazant et al. [15] clarified the presence of mixed
diffusional and Debye timescales. In a spherical space, these scales
are:
Dt
sdiff ¼ 2
re
Dt
sD ¼ 2
rD
ð2:2Þ
ð2:3Þ
The former is widely applied in voltammetric problems. In the
latter, the Debye length rD is a representation of the screening
length of electric fields, which may be determined by dimensional
analysis of the Poisson equation (Eq. (1.2)). Potential is normalised
to the thermal volt RT/F and represented h, and concentration is
normalised to bulk concentration C⁄ and represented ci:
r2 h þ
1 X
zi c i ¼ 0
2r 2D i
ð2:4Þ
where
rD ¼
sffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
RT s 0
2F 2 C ð2:5Þ
A factor of two is incorporated at this stage to simplify future
equations. It is therefore clear that a space normalisation by rD
will render the Poisson equation ideally dimensionless, i.e. without extraneous coefficients. However, a space normalisation by
re and time normalisation by r 2e is standard for diffusional
processes and renders a Coulombic field ideally dimensionless.
Following Bazant et al. [15] we shall compromise by normalising
the coordinates according to a geometric mean of the two
scales:
r
R ¼ pffiffiffiffiffiffiffiffiffi
re rD
D t
s¼ A
re rD
25
ð2:6Þ
ð2:7Þ
where DA is a chosen normal species. With the incorporation of conservation of mass (Eq. (1.3)) we achieve a dimensionless equation
set:
@ci Di
¼
r ðrci þ zi ci rhÞ
@ s DA
1 X
r2 h þ R2e
zi c i ¼ 0
2
i
ð2:8Þ
ð2:9Þ
where Re is the value of R where r = re, and hence R2e ¼ re =r D , the ratio of the electrode size to the Debye length. For most systems,
Re 1 will therefore hold.
3. Differential capacitance
The capacitance of an interface is a measure of how much
charge can be separated across it for a given potential difference.
Differential capacitance considers this property for different potentials under an equilibrium condition. Therefore i = 0, corresponding
to the t ? 1 condition of the RC circuit. Hence steady-state problems are not strictly RC circuit problems, but physical theories at
steady state can provide a value for Cd and therefore it is entirely
relevant to discuss steady-state problems in order to find the physical meaning of this value in the RC circuit problem, if any.
In the absence of electrolysis, the fluxes of all species are zero
and hence the equilibrium Boltzmann equation, ci = exp(zih), will
apply, as may confirmed by integration of Eq. (1.1) when Ji = 0. This
may be substituted into the Poisson equation to yield the Poisson–
Boltzmann equation, which can be solved exactly in linear space
(Gouy–Chapman theory) and has been considered using simulation
in cylindrical and spherical spaces [7,8]. The Gouy–Chapman equation in the limit of low potential is:
C 0d;GC ¼
s 0
rD
ð3:1Þ
where C 0d is a capacitance per unit electrode area, in units F m2, or
more typically lF cm2.
Here we shall find a spherical solution in the low potential (Debye–Hückel) limit. For an inert monovalent binary electrolyte AX,
our problem is:
1
2
r2 h þ R2e ðexpðhÞ expðhÞÞ ¼ r2 h R2e sinh h ¼ 0
ð3:2Þ
subject to h = h0 at R = Re and h ? 0 as R ? 1. The Debye–Hückel
approximation takes h 1 (i.e. / RT/F) which is a common situation for impedance problems but much less so for traditional voltammetric study. In this limit, sinh(h) h, thus linearising the
equation. Linear problems in spherical symmetry of this type are often solved by a substitution of the form v = R h. By this method we
can solve the problem (see Supporting information) and we find, in
real variables:
C 0d ¼
s 0
rD
rD
rD
¼ C 0d;GC 1 þ
1þ
re
re
ð3:3Þ
where C 0d indicates a capacitance per unit electrode area. Therefore
there is an ‘excess’ double layer capacitance due to the hemispherical nature of the diffuse double layer. At low applied potentials, the
deviation from classical Gouy–Chapman theory is given by the
equation above. It is evident that the Gouy–Chapman theory is
obeyed very closely where re rD, i.e. it is only in error for nanoscale electrodes. These results show very close agreement with
those predicted by simulation in past work for h0 1. [7]
26
E.J.F. Dickinson, R.G. Compton / Journal of Electroanalytical Chemistry 655 (2011) 23–31
4. Construction of a spherical Debye–Falkenhagen equation
that the Laplace coordinate s corresponds roughly to
write:
We may consider dynamic processes under the same condition
of low applied potential. The largest deviation of any concentration
from bulk will be the surface excess, at equilibrium, of the ion
which has the opposite charge as the electrode. This ion has a normalised surface concentration of exp(h0) according to the Boltzmann equation. So, if h0 1, we can write ci 1 + dci where
dci 1 everywhere. Hence:
1
R2
pffiffiffi < e
s an
zi ci rh zi rh
ð4:1Þ
thus linearising the Nernst–Planck equation.
For the case of inert binary monovalent electrolyte, if we initially assume DA = DX (but see below, Section 6) then the Nernst–
Planck equation for X may be subtracted from that for A, such that:
@q
¼ r ðrq þ 2rhÞ ¼ r2 q þ 2r2 h
@s
ð4:2Þ
where q = cA cX and is a measure of charge separation. The Poisson equation, similarly, gives:
1
2
r2 h þ R2e q ¼ 0
ð4:3Þ
Therefore, combining these, we achieve the Debye–Falkenhagen
equation [17]:
@q
¼ r2 q R2e q
@s
ð4:4Þ
which is a linear equation describing dynamic mass transport of
charged species at low potential and hence low deviation from
electroneutrality.
The properties of the Debye–Falkenhagen equation following a
potential step were discussed in detail for linear space by Bazant
et al. [15]. In the spherical case, the complexity of r2 makes direct
solution more challenging. The spherical equation, however, has
the estimable advantage of being entirely independent of the reference electrode position and involving no arbitrary length scales.
The technique for solution is the removal of radial dependence
by introducing a variable u = R q, such that the resulting equation
is amenable to Laplace transformation by analogy to the linear
case. The Poisson equation may be transformed similarly, using
v = R h as before.
5.1. The charging transient following a potential step
It can be demonstrated that the theory of linear time-invariant
systems is applicable to charging of a solution with equal diffusion
coefficients (see Supporting information). Defining capacitive current [18] such that:
jcap ¼
icap
2 @ 2 h ¼
Re @ s@R
2pFC Dre
ð5:1Þ
ð5:3Þ
and hence
a2n
s>
!
ð5:4Þ
R4e
Substituting back into dimensional coordinates, this gives a range of
accuracy of
rD
t > a2n tD re
1
1
qffiffiffiffiffiffiffiffiffiffiffiffiffi s
1
þ
s
s þ 1 þ R2
jcap ðsÞ ¼ 2h0 L1
s!s
1
s þ Rke
!
0
and
L1
s!s
1
¼ expðsÞ
1þs
B
¼ 2h0 L1
s!s @
sþ
1
C
qffiffiffiffiffiffiffiffiffiffiffiffiffiA
1 þ Rs2
ð5:2Þ
e
qffiffiffiffiffiffiffiffiffiffiffiffiffi
where s is the Laplace transform coordinate, k ¼ s þ R2e , and L1
represents an inverse Laplace transform. This inversion has no exact
solution, but it may be usefully approached under certain limits.
qffiffiffiffiffiffiffiffiffiffiffiffiffi
Let us approximate 1 þ Rs2 by its Taylor series. To order n, this
e
is accurate to 1% when
s
R2e
< a1n , where a0 50 and a1 3. Noting
ð5:7Þ
i.e. at long time and low voltage, and with equal diffusion coefficients,
an exponential decay analogous to the RC circuit, with a decay constant exactly equal to 1 in our choice of a mixed time coordinate.
This approximation is accurate to 1% for t > (2500 tD (rD/re)).
This inequality is satisfied for any experimentally measurable timescale, except when the electrode is nanoscale and so re ? rD.
Less stringently, the second term of the Taylor series may be
included:
1
1
qffiffiffiffiffiffiffiffiffiffiffiffiffi ¼
s
1 þ Rs2 s þ 1 þ 2R2e þ . . .
ð5:8Þ
e
This is 1% accurate for s < ð3 R2e Þ which is equivalent to
t > (9 tD (rD/re)). Further terms of this Taylor series may reasonably be neglected: we are excluding the contribution of large values of s as corresponding to (vanishingly) small values of s. So:
1
1
qffiffiffiffiffiffiffiffiffiffiffiffiffi s
1
s þ 1 þ R2
1 þ 2R2 s þ 1
e
ð5:9Þ
e
and so rearranging, we find that to this next order of approximation
jcap ðsÞ 2h0
1
ð5:6Þ
e
R¼Re
we find that, for the potential step problem:
ð5:5Þ
where tD is the characteristic Debye time and is defined r2D =D. For
typical electrolyte concentrations and diffusion coefficients, the Debye time is at most 1 ls and more typically is tens of nanoseconds.
It is clear, then, that the requirement of ‘long time’ is in fact not too
stringent; since Re is of the order 1 – 103 for most real systems, the
available t range is large even for a low order of approximation.
qffiffiffiffiffiffiffiffiffiffiffiffiffi
Considering the zeroth order Taylor series, we take 1 þ Rs2 ! 1,
e
so:
sþ
5. Low potential charging for equal diffusion coefficients
s12 , we can
2R2e
2R2e þ 1
!
exp 2R2e
2R2e þ 1
! !
s
ð5:10Þ
Again we recover an exponential decay at long time, with a
slightly altered decay constant; for large Re (large electrodes) this
further term rapidly tends to unity and so the first order correction
is unnecessary. It should be noted here that since the additional
term in the decay constant also appears as a multiplying factor
to the current as a whole, it can be identified with a change in
the resistance, Rs, and not of the capacitance.
Converting back into real units, we note that for the charging
problem at low potential, to the first approximation:
E.J.F. Dickinson, R.G. Compton / Journal of Electroanalytical Chemistry 655 (2011) 23–31
Rs;0 ¼
C 0d ¼
RT
1
1
r 2D
¼
F 4pFDC r e 2ps 0 r e D
s 0
ð5:11Þ
ð5:12Þ
rD
Therefore, the capacitance associated with the charging process
is the same as that given for differential capacitance by the Gouy–
Chapman theory. This strongly suggests that this capacitance is
physically meaningful, and in the limit of a large electrode and
low applied potential, charging is well described by an RC circuit.
To the second approximation, the capacitance is unchanged, but:
rD
Rs;0
Rs;1 ¼ 1 þ
2r e
ð5:13Þ
i.e. a slightly elevated resistance over that predicted by Gouy–Chapman theory is seen as re ? rD.
At first sight our determination here is entirely contradictory
with the steady state. The inclusion of a spherical term in our analysis has introduced a correction to the capacitance at steady state,
but an unaltered capacitance and a correction to the resistance for
the dynamic charging process. In both cases the correction is only
relevant when re is a few Debye lengths or less. The resolution of
this problem lies in appreciating that although the initial electroneutral condition for the dynamic solution requires that the solution charge [19], qsoln, is zero at s = 0, it does not constrain that
the electrode charge, qe = 0. Indeed, since the applied potential is
well defined, the enclosed charge on the electrode immediately following the potential step, before any ionic migration, is easily calculated, since this follows simply from Coulomb’s law:
qe ¼ 2ps 0 re /0
ð5:14Þ
and therefore
q0e ¼
s 0
re
/0
Since we have developed an exact result (under the low potential limit) for the Laplace space solution for the spherical system,
the impedance spectrum follows directly [20] by substituting jx
for s and taking ZðjxÞ ¼ h0 ðjxÞ=iðjxÞ (see Supporting information).
If re rD, i.e. the electrode is not nanoscale, and x R2e :
1
1
j
2
2x
ZðjxÞ ZðjxÞGC ð5:16Þ
R2e
!
R2e þ 1 þ jx
ð5:17Þ
which is similar to the first order correction noted for the transient
resistance. It must be noted, however, that in this case the resistance is slightly reduced. Therefore the nanoscale correction to
the complex impedance is opposite to the correction to the transient resistance, underlining that properties due to an ‘‘electrical
circuit analogy’’ cease to be meaningful at the nanoscale.
6. The case of unequal diffusion coefficients
When DA – DX, the above analysis may not apply. If one ion is
more mobile than the other, it will react more quickly to a step
in potential and so the capacitive properties of the double layer
are altered. We approach the problem analytically by choosing to
normalise time more generally by the arithmetic mean of the diffusion coefficients:
s¼
5.2. Impedance for low applied potential
ZðjxÞ ¼
which corresponds to the ideal (zeroth order) resistance as real
impedance and the Gouy–Chapman capacitance as imaginary
impedance respectively, in complete agreement with the predictions of RC circuit analysis.
The condition x R2e corresponds to frequencies less than the
Debye frequency D=r2D , typically 1 MHz–1 GHz, which is not an
experimentally achievable range with typical electrochemical
impedance spectroscopy equipment, and is not a function of electrode size. Hence even when Re is small we can take x R2e ;
applying this but including the nanoscale terms achieves a more
complicated result which we will not report here (but see Supporting information). The result is a correction to the impedance of the
form:
ð5:15Þ
which is the correction term noted at steady state. Therefore the
correction to the steady state capacitance is associated with the
‘‘Coulomb charge’’ on the electrode. This charge density becomes
increasingly significant for nanoelectrodes, as compared to the double layer charge density which is only a function of Debye length.
Since no ionic motion is required to achieve this charge, it occurs
effectively immediately (at s = 0 in tandem with the potential step).
It is not measured by the charging transient since this measures the
change of electrode charge after s = 0 and so does not alter the
apparent capacitance associated with the decay constant for this
process. However, this charge must still be compensated by the
development of the double layer.
Therefore, for a nanoelectrode, the change of charge in solution
over the charging transient is not exactly opposite to the change of
charge on the electrode. This marginally lengthens the time required to form the double layer and arises in the RC expression
as an excess resistance. The Cd predicted at steady state does not,
however, dictate the charging capacitance for a nanoelectrode
and so there is no single ‘system capacitance’ as is normally understood. The ‘solution’ resistance, Rs, is a strong function of re and so
is as much a property of the electrode as it is of the solution.
27
ðDA þ DX Þt Dmean t
¼
2r e rD
re rD
ð6:1Þ
The solution of the NPP equations for unequal diffusion coefficients is feasible in the Laplace space under the Debye–Falkenhagen approximation (see Supporting information). It arises that
the effect of unequal diffusion coefficients is parameterised by a
constant d = (DA DX)/(DA + DX) which for less than infinite disparity in rates of diffusion takes values jdj < 1. For the equal diffusion
coefficients case above, d = 0.
Unfortunately, both the analysis and the result are extremely
cumbersome for unequal diffusion coefficients. Under the approximation that s2 R4e ð1 d2 Þ2 =4d2 , long time values for Rs and Cd
can be derived, with the charging transient again taking an ideal
exponential form. The calculation is detailed in the Supporting
information, and has the result that, for a large electrode at low
overpotential:
Rs ¼
C 0d
¼
1
re
ps 0 ðDA þ DX Þ r2D
C 0d;GC
¼
2DA
Rs;0
DA þ DX
ð6:2Þ
ð6:3Þ
i.e. the elevated diffusion coefficients increase the conductance of
the solution without affecting the capacitance, which remains
equivalent to its value as inferred from the system equilibrium.
Therefore RC circuit analysis is still relevant at low potential even
in the case of unequal diffusion coefficients; the resistance is altered
such that the rate of charging is proportional to the arithmetic mean
of the diffusion coefficients of the ions involved. In effect, the component behaves as two resistances in series, one associated with
each ion.
A ‘‘very long time’’ solution exists (see Supporting information)
which predicts that the resistance will increase so as to behave as
though the component resistances from each ion are in parallel
28
E.J.F. Dickinson, R.G. Compton / Journal of Electroanalytical Chemistry 655 (2011) 23–31
rather than in series. This regime is not significant until long after
the charging current has decayed to zero, however, except in the
case of very small electrodes, when more terms are required for
an accurate expression in any case. This appears to be a mathematical curiosity of the system without any real physical relevance
since it only concerns behaviour once the double layer is indistinguishable from its equilibrium state.
7. Simulation results
7.1. The effect of size
The analysis above was confirmed and extended using numerical simulation techniques. The Nernst–Planck–Poisson equations
were solved dynamically in a transformed space from R = Re to
R ? 1, using a fully implicit finite difference method. A normalised space is used and the simulations are formulated to solve
the Nernst–Planck–Poisson equations with ln ci as the variables in
place of ci, as this was found to provide better convergence for dynamic simulation, by taking advantage of natural exponential relations within the system and reducing the dominance of non-linear
terms.
Two representative capacitive transients are shown at Figs. 2
and 3: one for re/ rD = 10 and one for re/rD = 103, which are typical
nano- and microelectrode situations respectively. A very small
applied potential of h0 = 0.01 is assumed, from an initially uncharged electrode. According to the first-order theory developed
above, the correction from the classical resistance is 5% in the first
case and 0.05% in the second.
In the simulations presented we note the strong agreement of
only the first-order corrected case for the nanoelectrode transient,
whereas the zeroth order theory is entirely adequate for the microelectrode where the Debye length is vanishing on the electrode
scale. Fig. 2 also contains a comparison of the nanoelectrode and
microelectrode cases, since the latter is given accurately by the zeroth order treatment; a distinction in the decay constant of charging
is evident between the two electrode sizes. Simulation confirms
the prediction that for microelectrodes an RC description using
Gouy–Chapman theory to describe capacitance and a classical
resistance as shown above is an excellent description of the diffuse
Fig. 3. Simulated capacitance transient for a step from the PZC to h0 = 0.01, with re/
rD = 1000. The zeroth order analytical treatment is shown for comparison.
double layer, noting, however, that this resistance is a property of
the electrode as well as of the solution. For the nanoelectrode, the
curve can still be described in terms of Rs and Cd, but as demonstrated above these parameters are dependent on the experiment
in question and cannot be directly associated with those inferred
from impedance spectroscopy or differential capacitance, thus
undermining the physical validity of the model.
7.2. The effect of large overpotential
The analysis above does not apply to overpotentials similar to or
greater than RT/F. The low potential range is typical for impedance
studies where small overpotentials are preferred, but is not typical
in chronoamperometry or cyclic voltammetry procedures where
ranges of many units RT/F are explored in order to access different
thermodynamic regimes of an electrolysis reaction. The linear
range of the Nernst–Planck–Poisson equation set has recently been
discussed for the related problem of an ion exchange membrane by
Moya [21].
The Gouy–Chapman theory, which is applicable to the limit of
large electrodes, gives the form of the double layer surface charge
density as:
Q 0DL ¼
Fig. 2. Simulated capacitance transient for a step from the PZC to h0 = 0.01, with re/
rD = 10. Zeroth and first-order analytical treatments are shown for comparison. The
zeroth order transient is an accurate approximation of the case re/ rD = 1000,
normalised appropriately, so a comparison of micro- and nanoelectrode transients
may also be seen.
q0DL
h0
¼ 4 sinh
FC r D
2
ð7:1Þ
which clearly is approximately linear only in the limit of small h0.
For higher overpotentials the double layer charge is predicted to
vary exponentially with applied potential. Since proportionally
more charge has to be assembled on the double layer, more migration of material is required, and so capacitive charging takes longer.
In particular, the significance of non-linear terms in the high
overpotential limit implies deviation from an ideal exponential
behaviour. This is indeed observed; a comparison between charging
transients for h = 0.05 and h = 5 is shown for jcap vs. s and ln jcap vs. s
at Figs. 4 and 5 respectively, at a microelectrode where re/rD = 1000.
The significant feature is the much longer non-exponential ‘tail’ of
the high potential transient, corresponding to the assembly of excess charge at the double layer. More specifically, in this case a
greater accumulation of anion is required than the depletion of cation, and so the formation of the double layer requires the establishment of a significant ionic strength gradient in addition to the
separation of charge. This process clearly requires more time than
the ideal ‘linear’ charging of a low potential double layer in
E.J.F. Dickinson, R.G. Compton / Journal of Electroanalytical Chemistry 655 (2011) 23–31
Fig. 4. Simulated capacitance transient for a step from the PZC to h0 = 0.05 and to
h0 = 5, with re/rD = 1000, comparing the low and high potential limits.
Fig. 5. Simulated capacitance transient for a step from the PZC to h0 = 0.05 and to
h0 = 5, with re/rD = 1000, comparing the low and high potential limits. In this scale,
an exponential decay is a straight line.
which the variation in surface excess is roughly equivalent for both
ions.
The extent of deviation from exponential decay (RC behaviour)
may be explored by considering the ‘‘apparent’’ exponential decay
of a charging transient. Since an ideal exponential has @ ln jcap/
@ s = k where k is the decay constant, we can plot the apparent
k, kapp @ ln jcap/@ s as a s-dependent function. In regions where
it is constant, the RC circuit is a good description, but not where it
is variable. The results are shown for a series of applied h0 values, at
different times s, in Figs. 6 and 7. The predicted constant k from the
spherical Debye–Falkenhagen equation is also plotted for clarity. It
is clear that while the RC description is excellent at all times for
h0 1, it becomes slightly inaccurate at longer times for h0 1
and is wholly inaccurate for h0 > 1.
We may otherwise remark that at very short times, all transients deviate from exponential behaviour for a nanoelectrode,
since the asymptotic requirement that s R2
e is no longer so well
obeyed. What is more, all transients show similar behaviour, exponential or otherwise, at short times – only once the surface excesses of the two ions are no longer symmetric does the rate of
29
Fig. 6. kapp @ ln jcap/@ s, for various values s in a capacitance transient and at
various applied h0 from h0 = 0.01 to h0 4, with re/rD = 10.
Fig. 7. kapp @ ln jcap/@ s, for various values s in a capacitance transient and at
various applied h0 from h0 = 0.01 to h0 4, with re/rD = 1000.
the charging process become potential-dependent. Of course, a
higher applied potential induces this situation more rapidly.
In the high potential range, capacitive charging is a non-linear
process. Consequently low potential analysis does not apply and
since the form of the transient is not exponential, RC circuit analysis is also inappropriate as the charging ‘constant’ RsCd is a strong
function of time. Therefore RC circuit analysis is not a good
description of the diffuse double layer for techniques in which applied potential varies by much more than RT/F, such as, for instance, conventional chronoamperometry or cyclic voltammetry.
7.3. The effect of unequal diffusion coefficients
A range of numerical simulations were conducted to consider
the effect of unequal diffusion coefficients on capacitive charging
transients. It was suggested by the analysis above that the characteristic decay time would be reduced according to the arithmetic
mean of the diffusion coefficients. Additionally, some deviation
from pure exponential behaviour may be expected for unequal diffusion coefficients, particularly the observation of a reduced decay
constant at very long time.
30
E.J.F. Dickinson, R.G. Compton / Journal of Electroanalytical Chemistry 655 (2011) 23–31
Three normalised transients are shown at Fig. 8 on a constant
time scale (s = DAt/rerD), for modest differences in diffusion coefficients; DX and DA are in the ratios 2:3, 1:1 and 3:2, respectively.
From this it is clear that while the decay constant is inversely proportional to the mean diffusion coefficient, the deviations from
exponential behaviour are in practice negligible.
A more complete study at Fig. 9 shows the variation of kapp, as
defined above, in time and with varying diffusion coefficient ratio.
It is clear that for more unequal diffusion coefficients, a greater
acceleration of charging at short time is balanced by a greater
deceleration at long time; this is associated with the formation of
the symmetric double layer (at low potential) requiring the system
to ‘‘wait’’ for the slower ion to migrate. Since the diffusion coefficients do not affect the charge on an equilibrated double layer,
the integral of the charging transient must be constant and hence
faster charging at some time will correspond to slower charging at
some other time.
In general, however, the observation of kapp very close to unity
in a timescale normalised to the mean of the diffusion coefficients
Fig. 8. Simulated normalised capacitance transients for a step from the PZC to
h0 = 0.05, with re/rD = 104 and DX/DA = 0.666. . ., 1, and 1.5. In this scale, an
exponential decay is a straight line.
confirms the dominance of the long (but not ‘‘very long’’) time regime cited above, in which the two ions behave as two component
resistances in series. This indicates that the effect of the charge
interaction between the ions on their migration is to rapidly balance the rates of migration to a mean transport rate. The rate of
double layer formation is hence the average of the contributions
of each individual ion.
8. Conclusions
In the arguments and results presented above, we have developed a self-consistent approach for the analysis of diffuse double
layer charging that avoids the traditional use of arbitrary length
scales. Simulations and mathematical analysis were used in a complementary manner and with strong mutual agreement. The exponential charging transient and impedance spectrum predicted by
RC circuit analysis and those based on a complete treatment using
the Nernst–Planck–Poisson equations were compared. RC circuit
analysis was found to agree with the more complete model only
subject to certain conditions. These conditions are: an electrode
larger than nanoscale; an applied potential not larger than RT/F
(25 mV); and moderately similar diffusion coefficients. Where
these conditions are violated, either ideal RC behaviour breaks
down altogether, as is the case for the non-exponential transients
observed at high applied potential, or the meanings of the ‘constants’ Rs and Cd change depending on the experiment under
consideration.
In particular we highlight that the condition of low applied potential, common in electrochemical impedance spectroscopy, is
strictly necessary for equivalent circuit analysis to be meaningful
to describe a diffuse double layer. Common experimental techniques in Faradaic analysis, such as chronoamperometry or cyclic
voltammetry, regularly probe much larger potential ranges, and
so the applicability of simple models will break down for the double layer in these situations. Consequently, values of Rs and Cd inferred from impedance spectroscopy must be treated with
caution, since they are not representative universal properties of
the system. They are not applicable to descriptions of the double
layer under potential conditions where a Faradaic reaction is
strongly driven by high overpotential. Equally, they are uncertain
for nanoelectrodes due to their variation depending on the experiment in question.
Acknowledgement
E.J.F.D. thanks St. John’s College, Oxford, for support via a graduate scholarship.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in
the online version, at doi:10.1016/j.jelechem.2011.02.016.
References
Fig. 9. The effect of the diffusion coefficient ratio DX/ DA on kapp @ ln jcap/@ s, at
different times during a capacitance transient with h0 = 0.05 and re/rD = 104.
[1] O. Stern, Z. Elektrochem. 30 (1924) 508–516.
[2] D.C. Grahame, Chem. Rev. 41 (1947) 441–501.
[3] Note that while the capacitance C describes the ratio of charge on the capacitor
to voltage across it (Ecap(t)), the applied potential E(t) is the potential difference
associated with the voltage source. These are equivalent only when the current
is zero. In the discussion hereafter, capacitance will be understood to refer to
the circuit (and the physical system it represents) as a whole, and so E refers to
the applied voltage. As shall be demonstrated, this circuit capacitance equals C
at zero current.
[4] W. Scheider, J. Phys. Chem. 79 (1975) 127–136.
[5] R.D. Armstrong, R.A. Burnham, J. Electroanal. Chem. 72 (1976) 257–266.
[6] T.C. Halsey, M. Leibig, Ann. Phys. (San Diego) 219 (1992) 109–147.
[7] E.J.F. Dickinson, R.G. Compton, J. Phys. Chem. C 113 (2009) 17585–17589.
E.J.F. Dickinson, R.G. Compton / Journal of Electroanalytical Chemistry 655 (2011) 23–31
[8] M.C. Henstridge, E.J.F. Dickinson, R.G. Compton, Chem. Phys. Lett. 485 (2010)
167–170.
[9] R.J. Smith, R.C. Dorf, Circuits, Devices and Systems, fifth ed., John Wiley & Sons,
New York, 1992.
[10] E. Barsoukov, J.R. Macdonald (Eds.), Impedance Spectroscopy: Theory,
Experiment, and Applications, John Wiley & Sons, New York, 2005.
[11] A.J. Bard, L.R. Faulkner, Electrochemical Methods: Fundamentals and
Applications, 2nd ed., John Wiley & Sons, New York, 2001.
[12] L.G. Gouy, C.R. Hebd, Séances Acad. Sci. 149 (1909) 654–657.
[13] L.G. Gouy, J. Phys. Theor. Appl. 9 (1910) 457–467.
[14] D.L. Chapman, Philos. Mag. 25 (1913) 475–481.
[15] M.Z. Bazant, K. Thornton, A. Ajdari, Phys. Rev. E 70 (2004) 021506.
[16] F. Beunis, F. Strubbe, M. Marescaux, J. Beeckman, K. Neyts, A.R.M. Verschueren,
Phys. Rev. E 78 (2008) 011502.
31
[17] P. Debye, H. Falkenhagen, Phys. Z. 29 (1928) 121–132.
[18] The use of j for dimensionless current is standard in the voltammetric
literature and is used here to emphasise the correlation between Faradaic and
capacitive currents, and their relative magnitudes. We are conscious of the
possible confusion with the imaginary unit j in other parts of the manuscript
and its Supporting Information and therefore consistently ‘‘cap’’ as a subscript
when current is being discussed.
[19] We use the notation q with appropriate subscripts here for the charges on the
electrode and in the solution in the double layer system, in order to emphasise
the analogy with the charge q on the capacitor in the discussion of the RC
circuit in Section 1.2.
[20] A. Bezegh, J. Janata, J. Electroanal. Chem. 215 (1986) 139–149.
[21] A.A. Moya, Electrochim. Acta 55 (2010) 2087–2092.