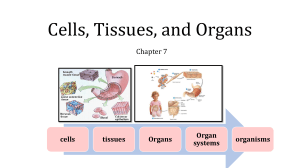
Cells, Tissues, and Organs Chapter 7 cells tissues Organs Organ systems organisms GENERAL CYTOTOXICITY AND HISTOPATHOLOGY The study of effects at the cell, tissue, and organ level of organization is invaluable for several reasons. In the context of the hierarchical "relay race" just described, these effects are consequences of biochemical mechanisms that provide interpretative insight about potential harm to individuals. They integrate damage done at the molecular level (Hinton and Lauren, 1990a), perhaps modulated by higher level processes such as those of the endocrine or immune systems. These biomarkers can be used as an early warning system for potential effects at the level of individual and, sometimes, population. Histopathology, the study of change in cells and tissues associated with communicable or noncommunicable disease, provides a cost-effective way to verify toxicant effect as well as exposure. A. Necrosis and Apoptosis A wide range of lesions (pathological alterations of cells, tissues, or organs) can indicate exposure to toxicants and suggest mechanisms of action. Often, they are associated with a specific target organ as a result of preferential toxicant transport to, accumulation in, or activation within that organ. As an important example of xenobiotic activation within a specific organ, the high biotransformation activity and consequent activation of carbon tetrachloride in the liver is responsible for carbon tetrachloride's hepatoxic nature. Cytotoxicity (toxicity causing cell death) may be detected in a tissue or target organ as localized or diffuse necrosis, cell death from disease, or injury. Pyknosis, one of the most obvious evidence of necrosis, involves the cell nucleus. With pyknosis (pycnosis), the distribution of chromatin in the nucleus changes with this material condensing into a strongly staining, basophilic mass. The nucleus becomes irregular in shape. Often pyknosis is followed by karyolysis, the disintegration of the nucleus. The cytoplasm of necrotic cells tends to become more acidophilic (eosinophilic) than that of viable cells. Mitochondria swell and more granules can appear in the cytoplasm. Necrosis might also be indicated by the displacement or separation of the cell from its normal location in a tissue, for example, cell sloughing from the gill epithelium or arterial wall. Necrosis is often associated with inflammation, which will be described shortly. Different types of necrosis manifest after various insults. Stop here Coagulation necrosis is characterized by extensive coagulation of cytoplasmic protein, making the cell opaque. The cell's outline and position within its tissue are retained for some time after cell death. As instances of its appearance after poisoning, coagulation necrosis occurs in the mammalian alimentary tract after ingestion of phenol or after acute inorganic mercury exposure because both poisons denature and precipitate proteins. Renal failure may result from accumulation and eventual spillover of metallothionein-associated metals in kidneys (the target organ) and subsequent coagulation necrosis of kidney cells. Liquefactive (cytolytic) necrosis occurs with rapid breakdown of the cell as a consequence of the release of cellular enzymes. Fluid-filled necrotic spaces in tissues might appear if many cells undergo liquefactive necrosis, especially if the involved tissue has considerable enzymatic activity. With caseous necrosis, cells disintegrate to form a mass of fat and protein. Gangrenous necrosis is a combination of coagulation and liquefactive necrosis often resulting from puncture (with an associated lack of blood supply to the damaged tissue') and subsequent infection. Meyers and Hendricks (1985) describe fat necrosis, which involves deposits of saponified fats in dead fat cells. Zenker's necrosis occurs only in skeletal muscle and is similar to coagulation necrosis. All reflect cell death, but coagulation necrosis seems to best reflect toxicant effect. Apoptosis, often called programmed cell death (PCD), is another useful biomarker of toxicant effect. The appearance of cells that have undergone apoptosis can be distinct from necrotic cells. With apoptosis, the chromatin condenses and the cytoplasm shrinks. The shrinking cell detaches from adjacent cells and its shape becomes irregular. The apoptotic cell can break into membrane-bound fragments called apoptotic bodies. In contrast to necrosis, apoptosis is not normally accompanied by inflammation. Apoptosis occurs for single cells, but as already detailed, necrosis can involve clusters or foci of many dead cells in affected tissue. Apoptosis occurs if a damaged or otherwise compromised cell begins a programmed sequence of biochemical steps intended to result in its death and removal from the tissue. Central to this process is activation of a particular class of proteases, caspase enzymes. Apoptosis begins by activation of caspases and the disruption of the mitochondrial membrane potential. According to Sweet et al. (1999), the cell then undergoes changes in calcium, potassium and water fluxes, and the loss of intercellular junctions and surface extensions. Finally, the cell's chromatin condenses, the DNA fragments, and apoptotic bodies form. Apoptosis can appear at lower toxicant concentrations than necrosis and, as a consequence, might be a very good biomarker for detecting low-level toxicant effects. B. Inflammation Inflammation can be useful as a biomarker of toxicant effect It is a response to cell injury or necrosis that isolates and destroys the offending agent or damaged cells (Sparks 1972). As such, inflammation often is associated with toxicant-induced necrosis. For example, hepatic necrosis attributable to toxicant action can be accompanied by inflammation. The process of inflammation continues through to lesion healing. The net result of inflammation is healed tissue with damaged cells being replaced by functioning cells. The four cardinal signs of inflammation are heat, redness, swelling, and pain, although heat is irrelevant for poikilotherms and redness is relevant to redblooded animals. Regardless, the underlying processes remain universal in all animals. C. Other General Effects Several other general effects of toxicants are observed in cells and tissues. Swelling of mitochondria has been correlated with metal contamination or poisoning. A few of the more prominent effects will be highlighted in this section. III. GENE AND CHROMOSOME DAMAGE Contaminants can produce changes in DNA and, in so doing, affect cells and tissues. The changes can increase both somatic and genetic risk. Somatic risk is risk of an adverse effect to the exposed individual resulting from genetic damage to somatic cells (e.g., damage leading to cancer), and genetic risk is the risk to progeny of an exposed individual as a consequence of heritable genetic damage (e.g., damage to germ cells or gametes leading to a nonviable fetus or an offspring with a birth defect). These effects are manifested in a variety of cellular structures or processes. Several effects are routinely used as biomarkers and provide a mechanism for consequences at the individual level, such as mutation or cancer. Structural chromosomal aberrations (CAs) include breakage and loss of segments of DNA, or chromosomal rearrangements. Fragments of chromosomes might also be apparent. Also visible may be "gaps" in chromosomes, small discontinuities in the chromosome. A chromatid aberration occurs if only one strand (chromatid) is broken. https://www.youtube.com/watc h?v=LEpTTolebqo 2 min https://www.youtube.com/watc h?v=46Xh7OFkkCE 12 min IV. CANCER Normal cells have the capacity to multiply and increase in tissues. This increase in the numbers of cells in a tissue or organ via mitosis that results in an increase in tissue or organ size is called hyperplasia. Hyperplasia in response to a variety of normal stimuli such as that involved in the tissue repair process is one type of physiologic hyperplasia. This form of physiologic hyperplasia is called compensatory hyperplasia and occurs to compensate for lost or damaged tissue, for example, the enlargement of a remaining kidney after removal of a diseased second kidney. A second form of pathologic hyperplasia is neoplastic hyperplasia, which is distinct from other forms because it involves a hereditary change to cells such that they no longer respond properly to chemical signals that would normally control their growth. Neoplastic hyperplasia provides a mechanism for cancerous growth. Neoplasia that grow slowly and tend to remain differentiated in their morphology are not as invasive of neighboring tissues as other neoplasia and are classified as benign neoplasia. Those that take on undifferentiated forms, tend to grow rapidly, and invade other tissues are classified as malignant neoplasia. Malignant neoplasia or cancers are more life-threatening than benign cancers. Pieces of the original malignant cancerous growth can dislodge and move to another tissue via the circulatory or lymphatic system to establish other foci of cancerous growth. This process of metastasis spreads a malignant cancer from the site of origin throughout the body. Cancer is a result of heritable change in cells. A chemical (e.g., numerous carcinogens), physical (e.g., ionizing radiation), or biological (e.g., a retrovirus) agent modifies a gene or its normal relation to other genes, which results in neoplasia. This may occur through point mutations, deletions, additions, or rearrangements as described above, or by gene insertion by a virus. A retrovirus might insert DNA into a cell's chromosome and transform that cell so it responds improperly to growth-regulating signals. A chemical or physical agent may interact with the cell's genome such that a gene involved with cell signaling or the regulation of normal growth and differentiation of cells (proto-oncogene) is changed to an oncogene, an altered gene causing cancer. Change to an oncogene results in loss of normal growth dynamics and/or differentiation of the cell. Cancer results from the inappropriate activation or activity of a gene that would normally be involved in cellular growth and/or differentiation. Other genes called suppressor genes function normally to suppress cell growth and might inhibit abnormal growth. Like proto-oncogenes, suppressor genes are thought to function in the process of growth and differentiation in normal cells but are most often explored in the context of suppressing growth, and even stimulating apoptosis, of cells with a mutated gene. A cancerous tumor can also develop if some agent or event affects the functioning of the suppressor gene. Modifications to proto-oncogenes and/or suppressor genes facilitate carcinogenesis. It follows from the above discussion that some agents initiate the neoplastic process and others promote the development of the tumor. This fact has lead to the classification of two groups of agents, initiators that convert normal cells to latent tumor cells and promoters that enhance the growth and continued expansion of latent tumor cells. Cancers are thought to pass through an initiation stage in which the DNA is permanently changed and then a promotional stage in which the modified cell then undergoes clonal increase. Cancer can be promoted by cell proliferation consequent to lesion formation (e.g., those associated with asbestos fiber in the lung), high hormone levels associated with hyperplasia (hormonal oncogenesis), and chemical agents. Often, inappropriate inactivation of suppressor genes is associated with the promotion process, resulting in unregulated growth. After initiation and promotion, the final step of carcinogenesis occurs, that is, cancer progression. Cancer progression is the change in the biological attributes of neoplastic cells over time that leads to a malignancy: it involves an additional genetic mutation to produce cancer cells able to sustain themselves in the body. There can be a long latent (or latency) period between exposure to a carcinogen and the appearance of an observable cancer because carcinogenesis involves several stages, each having an associated probability of occurring. This makes assignment of cause and effect difficult because the exposure might have occurred years before effect manifestation. The latent period and multistage process leading to cancer also make prediction of the exact shape of the dose-cancer response curve challenging. Two theories exist and are supported by different data. The threshold theory holds that there is no effect below a certain low dose. Above this threshold, the slope of the response versus dose curve increases rapidly. The dose-response curve takes on the appearance of a hockey stick. The linear-no threshold theory is based on several radiation-induced cancer studies where no apparent threshold exists for effect. Cancer initiation being the mechanism for radiation-induced carcinogenesis lends support to this theory because even a single mutation has a chance of producing a cancer. This theory carries the assumption that any lack of detected cancer below a certain dose reflects our inability to measure low incidences of cancers at these exposure levels. This dose-response model describes a line with no threshold. Chemical and physical agents can act by initiation or promotion of the process leading to cancer Relevant mutagenic mechanisms are those described previously for alterations of DNA and chromosomes. Furthermore, agents that inhibit DNA repair may also increase the probably of cancer Differences in DNA repair fidelity (accuracy in repairing and returning the DNA to its original state after damage) associated with various agents can also result in differences in carcinogentcity For example, Robison et al. (1984) suggested that, although chromate, nickel, and mercury damage DNA, there is a difference in the nature of the damage and its repair. Generally, mercury produces more singlestrand breaks than nickel and chromate, which cause more protein-DNA crosslinking.